Embed Size (px)
Citation preview

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2015
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1109
Validation of antibodies for tissuebased immunoassays
SANDRA ANDERSSON
ISSN 1651-6206ISBN 978-91-554-9258-8urn:nbn:se:uu:diva-251344

Dissertation presented at Uppsala University to be publicly examined in Fåhreussalen,Rudbecklaboratoriet, hus C5, Dag Hammarskjölds väg 20, Uppsala, Saturday, 13 June 2015at 12:30 for the degree of Doctor of Philosophy (Faculty of Medicine). The examinationwill be conducted in English. Faculty examiner: Dr Mike Taussig (Babraham BioscienceTechnologies, Cambridge).
AbstractAndersson, S. 2015. Validation of antibodies for tissue based immunoassays. DigitalComprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1109.45 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-9258-8.
In situ protein detection in human tissues using antibodies reveals the cellular proteinlocalization, and affinity-based proteomic studies can help to discover proteins involved in thedevelopment of diseases. However, antibodies often suffer from cross-reactivity, and the lack ofpositive and negative tissue controls for uncharacterized proteins complicates the mapping ofthe proteome. The aim of this thesis is thus to improve the methodology for validating antibodiesused for immunostaining on formalin-fixed paraffin-embedded tissues.
Two of the papers include comparisons between mRNA-expression and immunostainingof corresponding protein. In paper I, ISH and IHC staining patterns were compared onconsecutive TMA-slides. The study of well-characterized genes showed that ISH could beused for validation of antibodies. ISH was further used for antibody evaluation, and couldvalidate four out of nine antibodies showing potentially interesting staining patterns. In paperIII, transcriptomic data generated by RNA-sequencing were used to identify tissue specificexpression in lymphohematopoietic tissues. An increased expression in one or more of thesetissues compared to other tissue types was seen for 693 genes, and these were further comparedto the staining patterns of corresponding proteins in tissues.
Antibody labeling is necessary for many immunoassays. In paper II, two techniques forantibody-biotinylation were compared, aiming to find a stringent labeling method for antibodiesused for immunostaining on TMAs. The ZBPA-method, binding specifically to Fc-part ofantibodies, was found to be superior to the Lightning Link-biotinylation kit targeting aminegroups, since labeling of amine groups on stabilizing proteins in the antibody buffer causesunspecific staining.
The localization of the estrogen receptor beta (ERβ) in human normal and cancer tissues wasstudied in paper IV. Thorough evaluation of 13 antibodies using positive and negative controlcell lines showed that only one antibody, PPZ0506, is specific for ERβ in all three immunoassaysused. Contradictory to previously published data, tissue profiling using PPZ0506 showed thatERβ is expressed in a limited number of normal and cancer tissues.
In conclusion, the present investigations present tools for validation of antibodies used forlarge-scale studies of protein expression in tissues.
Keywords: Antibody validation, Conjugation, Estrogen receptor-beta, IHC, ISH,Lymphohematopoietic tissues, Proteomic, RNAseq, TMA, Transcriptomic
Sandra Andersson, Department of Immunology, Genetics and Pathology,Rudbecklaboratoriet, Uppsala University, SE-751 85 Uppsala, Sweden.
© Sandra Andersson 2015
ISSN 1651-6206ISBN 978-91-554-9258-8urn:nbn:se:uu:diva-251344 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-251344)

Det är vad vi tror att vi redan vet somhindrar oss från att lära oss nytt
Claude Bernard

Main supervisor: Anna Asplund, Associate Professor Department of Immunology, Genetics and Pathology Uppsala University Uppsala, Sweden
Assistant supervisor: Fredrik Pontén, MD, Professor
Department of Immunology, Genetics and Pathology Uppsala University Uppsala, Sweden
Assistant supervisor: Kenneth Wester, Associate Professor
Department of Immunology, Genetics and Pathology Uppsala University Uppsala, Sweden
Faculty opponent: Michael Taussig, Dr
Protein Technology Group Babraham Institute Cambridge, United Kingdom
Examining committee: Amelie Eriksson Karlström, Professor
Division of Protein Technology, School of biotechnology Royal Institute of Technology Stockholm, Sweden
Per-Arne Haldosén, MD, Associate Professor Department of Biosciences and Nutrition Karolinska Institutet Stockholm, Sweden
Per Westermark, MD, Professor
Department of Immunology, Genetics and Pathology Uppsala University Uppsala, Sweden
Chairperson: Erik Larsson, MD, Professor
Department of Immunology, Genetics and Pathology Uppsala University Uppsala, Sweden

List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Kiflemariam S, Andersson S, Asplund A, Pontén F, Sjöblom T. (2012) Scalable in situ hybridization on tissue arrays for validation of novel cancer and tissue-specific biomarkers. PLoS One, 7(3):e32927.
II Andersson S, Konrad A, Ashok N, Pontén F, Hober S, Asplund A. (2013) Antibodies biotinylated using a synthetic Z-domain from pro-tein A provide stringent in situ protein detection. J Histochem Cyto-chem, 61(11):773-84.
III Andersson S, Nilsson K, Fagerberg L, Hallström BM, Sundström C, Danielsson A, Edlund K, Uhlén M, Asplund A. (2014) The tran-scriptomic and proteimic lanscapes of bone marrow and secondary lymphoid tissues. PLoS One, 9(12):e115911.
IV Andersson S, Sundberg M, Pristovsek N, Clausson C-M, Zieba A, Ramström M, Söderberg O, Pontén F, Williams C, Asplund A. Es-trogen receptor beta profiling in human tissues following extensive antibody validation. Manuscript.
Reprints were made with permission from the respective publishers.

Front-page image: IHC staining of estrogen receptor alpha in breast cancer, using antibody CAB000037 in the Human Protein Atlas. Back page drawing: “Cell and antibodies” by Dameon Andersson, 4 years old.

Contents
Populärvetenskaplig sammanfattning ........................................................... 11
Introduction ................................................................................................... 14Proteins ..................................................................................................... 14
The central dogma ................................................................................ 14Proteomics ............................................................................................ 16Affinity-based proteomics ................................................................... 16The Human Protein Atlas ..................................................................... 17
Antibodies ................................................................................................. 18Utilization of antibodies in immunoassays .......................................... 19Immunohistochemistry ........................................................................ 20Antibody validation ............................................................................. 22Antibody labeling ................................................................................. 24
Tissue microarrays .................................................................................... 25Transcriptomic studies of lymphohematopoietic tissues .......................... 26Estrogen receptors .................................................................................... 27
Present investigation ..................................................................................... 29Aim ........................................................................................................... 29Methodological considerations ................................................................. 29Results and discussion .............................................................................. 30
Paper I: ................................................................................................. 30Paper II: ................................................................................................ 31Paper III: .............................................................................................. 32Paper IV: .............................................................................................. 33
Concluding remarks and future perspectives ................................................ 34
Acknowledgements ....................................................................................... 37
References ..................................................................................................... 39


Abbreviations
AP alkaline phosphatase DAB diaminobenzidine DNA deoxyribonucleic acid ELISA enzyme-linked immunosorbent assay ER estrogen receptor ESI electrospray ionization FFPE formalin-fixed paraffin-embedded FPKM fragments per kilobase of exon model per million
mapped reads GCT granulosa cell tumor HIER heat-induced epitope retrieval HRP horseradish peroxidase HSA human serum albumin Ig immunoglobulin IHC immunohistochemistry IP immunoprecipitation ISH in situ hybridization mAb monoclonal antibody MALDI matrix-assisted laser desorption-ionization mRNA messenger ribonucleic acid MS mass spectrometry pAb polyclonal antibody PLA proximity ligation assay PrEST protein epitope signature tag recAb recombinant antibody RNAseq RNA sequencing siRNA small interfering RNA TMA tissue microarray TOF time of flight WB Western blot


11
Populärvetenskaplig sammanfattning
Många relaterar ordet protein till näringsinnehållet i livsmedel och något man måste äta för att bygga muskler. Men proteiner är inte bara ett ämne med en och samma funktion. Alla levande organismer, till och med virus, har många olika proteiner. Vi människor har runt 20 000 olika typer av pro-teiner som skiljer sig åt både i utseende och funktion. Proteiner är upp-byggda som en kedja av aminosyror, av vilka det finns ca 20 varianter som kan kombineras på olika sätt för att bygga upp de typer av proteiner som behövs i en cell. Några av aminosyrorna, de s.k. essentiella aminosyrorna, kan kroppen inte själv bilda och måste fås från kosten.
DNA består bl.a. av byggstenar som kallas baser. Det finns fyra olika typer av baser, A, T, C och G, som kan kombineras på många sätt. Vissa sekvenser i DNA utgör gener, och det är ordningen av baser i en gen som bestämmer aminosyraordningen i ett protein. Man kan säga att varje gen är mallen till ett protein, och denna mall kan användas många gånger om för att producera mycket av samma protein. På detta sätt produceras många olika proteiner i varje enskild cell i kroppen. DNAt är exakt lika i alla celler i en individ, men vilka gener som genererar proteiner och hur mycket av varje protein som tillverkas skiljer sig åt mellan olika celltyper. Det är därför en nervcell ser annorlunda ut än en levercell eller en blodcell. Det krävs dock ett mellansteg från DNA till protein. Det är en molekyl som liknar DNA och som heter mRNA. mRNA innehåller också fyra baser, A, U, C och G, och man kan säga att en gen i DNAt kopieras till ett mRNA. Det är sedan mRNA-molekylens bassekvens som översätts till aminosyrasekvensen som bildar proteinet, se figur 1.
Proteinerna utför många viktiga funktioner i en cell, men ibland kan de or-saka uppkomsten av sjukdomar också. Det händer t.ex. när det blir fel i re-gleringen av en gen så att det bildas för mycket eller för lite av ett protein, eller att aminosyrasekvensen blir fel p.g.a. mutationer i genen. Ibland kan sådana fel leda till att cellen börjar dela sig ohämmat och en tumör bildas. Därför är det av intresse att kunna ta reda på vilka proteiner som finns i olika celltyper både i normal vävnad och i cancer. Om man vet vilka proteiner som är inblandade i uppkomsten av en viss cancerform, kan läkemedel så småningom utvecklas som är riktade mot dessa proteiner. För att kunna de-tektera ett protein i många vävnader samtidigt kan en ”tissue microarray”

12
(TMA) användas. En TMA består av en paraffinklots med vävnadsstansar som är 1 mm i diameter. Dessa stansar kommer från olika organ och tumörer som erhålls från patologiska arkiv. Sedan kan 4 m tunna snitt skäras från en TMA och läggas på objektglas, se figur 5.
Ett sätt att hitta proteinerna i TMA-snitt är att använda sig av antikroppar, se figur 3. Antikroppar är också ett protein, och de bildas och utsöndras av en typ av celler i immunförsvaret som kallas B-lymfocyter. Antikropparnas funktion är att binda till proteiner på ytan av invaderande mikroorganismer som bakterier och virus, så att de kan ätas upp av andra immunceller. Att immunförsvaret kan känna igen så många olika mikroorganismer beror på att det kan bildas nästan oändligt många typer av antikroppar som känner igen olika proteiner. Denna förmåga hos antikroppar kan användas även labora-tivt. Genom att tillverka antikroppar som kan binda till ett visst protein, och sedan koppla antikroppen till något som kan ge en färg, kan man detektera proteinet i ett TMA-snitt. En vanlig metod för att göra detta heter immunhis-tokemi (IHC). Det innebär att ett objektglas med vävnadssnitt ”färgas” med en antikropp. Sedan kan man med hjälp av mikroskop se en infärgning i de celler i vävnaden där antikropparna har bundit till sitt protein, se figur 4.
Det är tyvärr långt ifrån alla antikroppar som bara binder till ett slags pro-tein. Många binder även till en del andra proteiner som liknar målproteinet. Den här avhandlingen handlar därför om att utvärdera hur bra antikropparna fungerar för att hitta rätt protein. Det är viktigt att veta att antikropparna 1) binder till det proteinet man vill hitta och 2) inte binder till något annat pro-tein. Först när man har utvärderat antikroppen kan man använda den till att undersöka proteinets lokalisation och funktion. De fyra arbeten som avhand-lingen bygger på handlar om att utveckla bättre valideringsmetoder av anti-kroppar som ska användas i storskaliga studier av människans alla proteiner.
I det första arbetet har antikroppsfärgningar jämförts med var mRNA för respektive protein finns. mRNA-molekyler kan färgas in med en metod som heter in situ hybridisering, och på så vis studeras i TMA-snitten med mikro-skop, precis som antikroppar. Eftersom mRNA är mallen för ett protein, borde både mRNA och protein från samma gen kunna detekteras. Om man jämför färgning av mRNA och protein på två parallella snitt från samma vävnad kan man se om de finns i samma celltyper. Om mRNA för en viss gen hittas i samma celltyper som antikroppen är det troligt att antikroppen har bundit rätt protein.
En annan metod för att utvärdering av antikroppar är att jämföra färgnings-mönstren från två eller flera antikroppar. Man kan t.ex. färga två antikroppar mot samma protein på samma TMA-snitt, men för att man ska kunna skilja på vilken antikropp som binder var, måste man koppla dem med olika mole-

13
kyler som ger olika färg. Det finns många sätt att binda molekyler till anti-kroppar, men alla fungerar inte lika bra. Det andra arbetet i avhandlingen syftar därför till att hitta en passande metod för att koppla en molekyl, biotin, till antikroppar som ska användas för IHC på TMA-snitt. Två metoder jäm-fördes; en snabb och enkel metod, och en som tar längre tid, men som är mer specifik. Slutsatsen blev att den mer specifika metoden fungerar bättre för de flesta antikroppar då inget reningssteg behövs för att ta bort andra proteiner.
I det tredje arbetet användes en storskalig metod, RNA-sekvensering (RNA-seq), i kombination med IHC för att identifiera vävnadsspecifika uttryck av gener i fyra vävnader (benmärg, lymfkörtel, mjälte och blindtarm) som alla innehåller celler som är involverade i immunförsvaret. RNAseq utfördes på 27 vävnadstyper inklusive de fyra immuncellsrika vävnaderna. RNAseq-analysen resulterar i ett värde för hur mycket mRNA som uttrycks av varje gen i en viss vävnad, och man kan på så vis jämföra nivåerna av genuttryck mellan vävnader. I detta arbete identifierades gener som hade ett högre ut-tryck i en eller flera av de fyra immuncellsrika vävnaderna än i övriga väv-nader. Flera av dessa gener var inte tidigare beskrivna och kan därför vara intressanta att forska vidare på. För de gener som visade sig ha ett vävnads-specifikt uttryck användes också antikroppsfärgning på TMA för att kunna se i mikroskop vilka celler i vävnaden som uttrycker motsvarande protein.
Det fjärde arbetet syftade till att validera antikroppar mot östrogenreceptor beta (ERβ), ett protein som är potentiellt intressant för bröstcancerforskning, för att kunna ta reda på i vilka vävnader och celltyper detta protein finns. Det finns sedan tidigare många vetenskapliga publikationer om ERβs lokalisat-ion och funktion, men i dessa studier har antikroppar som inte är tillräckligt utvärderade använts, och därför visar studierna olika resultat. I den här stu-dien utvärderas därför antikropparna på positiva och negativa kontrollceller, d.v.s. celler som uttrycker mycket eller inget ERβ, med flera olika metoder. Endast 1 av de 13 utvärderade antikropparna visade sig vara specifik för ERβ. När den specifika antikroppen användes med IHC på TMAer innehål-lande många vävnadstyper visade det sig att ERβ bara finns i ett fåtal väv-nadstyper.

14
Introduction
Proteins The human genome harbors 20,300 protein-coding genes (Ensembl, release 79) (1), which would give rise to the same number of protein types if one gene was translated into only one isoform of the protein. However, the hu-man body is complex and can generate subtypes of proteins from the same gene. Different isoforms of a protein can be formed due to mRNA splicing and post-translational modifications (2) such as glycosylation and phosphor-ylation, which greatly increases the diversity of the proteome. However, it has been shown that for most protein coding genes, one transcript is the dominant form (3). Each protein performs special functions in the cell, for example many proteins serve as enzymes that catalyze reactions. Others have a structural function, such as tubulin that makes the cell’s microtubules, while some proteins are involved in movement, e.g. myosin in muscle cells. Yet another type of proteins, the antibodies, plays a role in the immune sys-tem as a defense against invading microorganisms.
The DNA sequence is identical for all cells in the body, so what actually distinguishes one cell type from another is due to which genes that are ex-pressed and translated into proteins, and the quantity they are expressed in. Many proteins perform housekeeping functions required for the maintenance of basic cellular functions, and are therefore common for numerous cell types, whereas some proteins are cell specific, such as insulin, expressed exclusively in beta cells of the pancreas, or antibodies that are only produced by B-lymphocytes. Tissue specific gene expression in lymphohematopoietic tissues is discussed in Paper III.
The central dogma Proteins constitute most of a cell’s dry weight (4). It is the composition of different proteins that determines the cell’s shape and function. Proteins are polymers of amino acids, of which the order is determined by the nucleotide gene sequence. The amino acid order varies between different proteins and decides the shape of the protein. Amino acids have side chains with different properties. They can e.g. be polar, nonpolar, acidic or basic, and the poly-peptide folds into the conformation of lowest energy depending on the prop-
15
erties of the side chains. Specialized proteins, molecular chaperones, often assist in the protein folding. The full three-dimensional shape of a protein is called tertiary structure, and when several proteins form a complex, the complete structure is denoted quaternary structure. Furthermore, some pro-teins have substructures called protein domains that are often associated with certain functions (4).
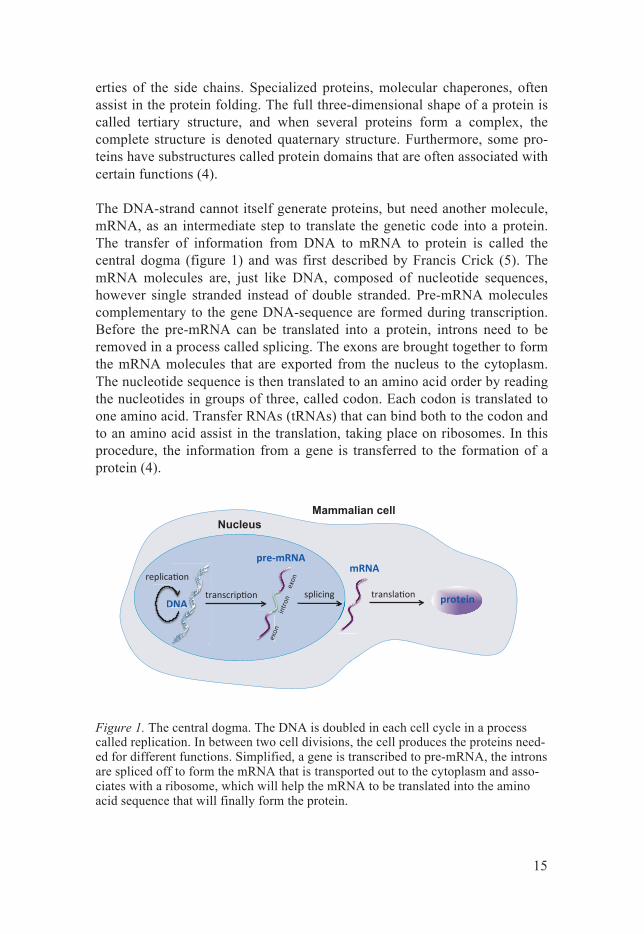
The DNA-strand cannot itself generate proteins, but need another molecule, mRNA, as an intermediate step to translate the genetic code into a protein. The transfer of information from DNA to mRNA to protein is called the central dogma (figure 1) and was first described by Francis Crick (5). The mRNA molecules are, just like DNA, composed of nucleotide sequences, however single stranded instead of double stranded. Pre-mRNA molecules complementary to the gene DNA-sequence are formed during transcription. Before the pre-mRNA can be translated into a protein, introns need to be removed in a process called splicing. The exons are brought together to form the mRNA molecules that are exported from the nucleus to the cytoplasm. The nucleotide sequence is then translated to an amino acid order by reading the nucleotides in groups of three, called codon. Each codon is translated to one amino acid. Transfer RNAs (tRNAs) that can bind both to the codon and to an amino acid assist in the translation, taking place on ribosomes. In this procedure, the information from a gene is transferred to the formation of a protein (4).
Figure 1. The central dogma. The DNA is doubled in each cell cycle in a process called replication. In between two cell divisions, the cell produces the proteins need-ed for different functions. Simplified, a gene is transcribed to pre-mRNA, the introns are spliced off to form the mRNA that is transported out to the cytoplasm and asso-ciates with a ribosome, which will help the mRNA to be translated into the amino acid sequence that will finally form the protein.
Nucleus Mammalian cell

16
Proteomics Publishing of the first draft of the human gene sequence in 2001 (6, 7) paved the way for studying the proteins expressed from our genome. The term pro-teome refers to all proteins expressed by the genome in an organism. The study of the proteome is consequently called proteomics, referring to the large-scale studies of proteins (8, 9). The DNA sequence and transcripts from a gene act as templates of protein molecules, however perform no func-tions them selves. Instead, the proteins are the functional units that perform processes in a cell. For example, it is not the insulin gene or the mRNA ex-pressed from this gene, but the protein insulin that regulates the blood sugar level. It is consequently of great interest to study the localization and func-tion of proteins.
Proteomics can roughly be divided into mass spectrometry (MS)-based (9-11), and affinity-based proteomics (12-15). For analyzing complex samples with MS, the proteins are often first separated using gel-electrophoresis, and the sample of interest is extracted from the gel and enzymatically digested into peptides. There are several different types of MS-methods, but the basic principle for all is to generate a mass spectrum of peptides, for identification of the proteins in a sample. First, an ionization system, e.g. electrospray ion-ization (ESI) (16) or matrix-assisted laser desorption-ionization (MALDI) (17), is used to convert the peptides into ions. Then, the ions are separated by mass-to-charge ratio with e.g. and ion trap or time-of flight (TOF) analyzer to produce a mass spectrum (18). The data set can then be matched against databases to identify which proteins the sample contains (10, 19). MS-based and affinity-based proteomics can further be combined. For example, in pa-per IV, MS was used to analyze the immunoprecipitation (IP)-products of control cell lysates incubated with ERβ antibodies.
Affinity-based proteomics For affinity-based proteomics, affinity-reagents, most often antibodies, in-stead detect the proteins (reviewed in (20)). There are several widely used antibody based protein detection methods, such as ELISA (21, 22), flow cytometry (23), western blot (WB) (24) and immunohistochemistry (IHC) (25). The advantage with IHC over the other methods is that the proteins can be visualized in a tissue context (26). The IHC method is described below, but in short, an enzymatic reaction generates a color indicating where the antibody has bound its antigen. This makes it possible to determine in what cell types the protein is located, and even in what subcellular localization it is sited. Neither of the other antibody-based, nor the MS-based methods can offer this type of spatial information. Pathologists use sections of tissue samples collected from surgical specimen for diagnostics. Before the IHC

17
technique was discovered, histological dyes were the only method to prac-tice. Nowadays, antibodies are used as protein biomarker tracers for diseas-es, and panels of antibodies are employed for IHC staining to help pathologists to set a diagnosis, prognosis or to decide about treatment (27, 28). The method of using IHC on tissues can also be applied in a large-scale approach for affinity-based proteomics. This is currently being done by the Human Protein Atlas project, described below.
The Human Protein Atlas The Human Protein Atlas (www.proteinatlas.org) project is a large-scale proteomic effort aiming to map the human proteome using antibodies for IHC on tissue micro arrays (TMAs) (14, 29-31). The project generates affini-ty-purified polyclonal antibodies towards all non-redundant human proteins (20,300; Ensembl, release 79) (1). The antibodies are used for IHC staining of 44 different normal human tissues, 20 different cancer types, and 46 dif-ferent human cell lines to explore the human proteome. The workflow for generation of the antibodies, as well as the applications for which they are used, is shown in figure 2. Briefly, a gene segment, so called protein epitope signature tag (PrEST), with low homology to other genes is selected and cloned into bacteria that will express the peptide. The peptide is used for immunization to produce a polyclonal antibody serum that is purified to-wards its own antigen, the PrEST, for generation of affinity-purified antibod-ies. The antibodies are thoroughly validated using protein array, WB, IHC, immunofluorescence and siRNA knock down. Furthermore, the next genera-tion RNA sequencing (RNAseq) is performed on both tissues and cell lines, which allows for comparison between relative RNA-levels and the antibody staining pattern. The results are published on the Human Protein Atlas web-site (www.proteinatlas.org). This proteomic effort can greatly increase our understanding of where proteins are located in normal and diseased tissues. Furthermore, by using the antibodies in other set ups, such as in-situ proxim-ity ligation assay (PLA) (32), protein-protein interactions, phosphorylations etc. can also be studied on TMAs.

18
Figure 2. Work flow for generation of affinity-purified antibodies from Protein Epitope Signature Tags (PrEST). A PrEST is selected using computer software. The sequence, including a his-tag, is synthesized and cloned into bacteria for expression of the peptide. The amplified peptide is purified using the his-tag, and used for im-munization to produce antibodies. The antibodies are purified against their own antigen, the PrEST, to generate affinity-purified antibodies. These are tested in dif-ferent applications; protein array, western blot, immunohistochemistry and immuno-fluorescence. Approved antibodies are published on the Human Protein Atlas (www.proteinatlas.org). Figure from reference (33), used with permission.
Antibodies Antibodies, also called immunoglobulins (Ig), are proteins produced by B-lymphocytes as part of the adaptive immune system. They are the secreted form of the B-cell antigen receptor, and have two functions: to bind mole-cules of invading pathogens, and to recruit other cells and molecules to elim-inate pathogens (34). Five isoforms of antibodies exist, IgM, IgD, IgG, IgA and IgE (35). IgG is by far the most abundant antibody isotype in plasma. IgG antibodies are large proteins with a molecular weight of around 150 kDa. The structure of an antibody contains two heavy chains and two light chains. The heavy chains are linked to each other, and to one light chain each, with disulfide bonds (figure 3). Furthermore, it is divided into variable regions for binding of a specific antigen, and a constant region (figure 3). The variable regions differ between antibodies targeting different antigens.

19
However, the amino acid variability is not evenly distributed over the whole region, but focused into three hypervariable regions denoted HV1, HV2 and HV3 in both the heavy and the light chains. Those are the regions that form a hypervariable binding site (figure 3) that determine the antigen specificity (34). The enormous variety of antibodies targeting different antigens is due to special antibody gene segments that can be combined in numerous ways, and somatic hypermutations, mainly in the hypervariable regions, that takes place during the maturation of B-lymphocytes (36).
Figure 3. The antibody consists of two heavy (H) and two light (L) chains. It is further divided into a constant region, similar for all antibodies, and a variable re-gion with three hypervariable regions (HV1, HV2 and HV3) for antigen binding. The heavy chains are linked to each other, and to one light chain each, with disulfide bonds.
Utilization of antibodies in immunoassays Antibodies as an affinity-reagent for protein detection can be of different types: polyclonal antibodies (pAbs; including affinity-purified pAbs), mono-clonal antibodies (mAbs) and recombinant antibodies (recAbs). Which type to use depends on the application the antibody is intended for. The produc-tion of pAbs starts by injecting a purified protein/peptide into an animal, which starts an immune response against the foreign antigen. Each antigen mostly contains many epitopes that can be recognized by numerous lympho-cytes. The total production of antibodies from these lymphocytes results in a serum with pAbs (37). The advantage of pAbs is thus that it comprises anti-bodies towards several different epitopes. If a few epitopes are not accessible

20
due to e.g. modification or folding, there are still antibodies that can target other epitopes. Moreover, pAbs are not very sensitive for variation in pH and salt concentration (37). They can therefore be used in many different appli-cations, independently of the antigens conformation (13). However, pAbs lack the ability to be reproduced. A second immunization with the same antigen in the same species will not result in the exact same composition of pAbs.
Köhler and Milstein developed a method for generating mAbs, and for this they were awarded the Nobel prize (38). B-cells and myeloma cells were fused to generate hybridomas producing unique mAbs. These antibodies have the advantages of being more pure, they can be produced with much higher concentration than pAbs, and above all, they can be generated as a constant and renewable resource. However, producing mAbs is much more time consuming and expensive than production of pAbs. Also, whereas pAbs are quite stable, mAbs are sensitive to changes in the structure of an epitope. Consequently, for generation of mAbs, the state of the antigen that the anti-body should bind need to be taken into consideration (37).
In polyclonal sera, 99 % of all antibodies target other, unknown antigens, which can cause unspecific staining (13). The pAbs can therefore be purified against its own antigen to generate affinity-purified antibodies (39). With this approach, the antibodies will exhibit advantages from both pAbs and mAbs. They are more pure than regular pAbs, but still have the ability to recognize several epitopes on the antigen.
Antibodies can also be produced in vitro e.g. by using the phage display technology, first introduced by G. Smith in 1985 to generate recAbs (40, 41). The gene of interest is inserted into a bacteriophage that is used to infect E. coli bacteria. The antibody will be expressed as a fusion protein together with a phage coat protein on the phage surface. By expressing many variants of fusion proteins, a phage antibody library can be produced (42, 43). The antibodies can then be selected and purified.
Immunohistochemistry IHC is a method that visualizes protein localization in situ in tissue sections using antibodies (26, 44). The method was first described by Albert H Coons in 1941, who used fluorescently labeled antibodies for immunostaining (25). Since then, the technique has been refined in several ways, e.g. by develop-ing better methods for production and purification of antibodies, antibody labeling and detection, as well as for tissue fixation and retrieval (45).
21
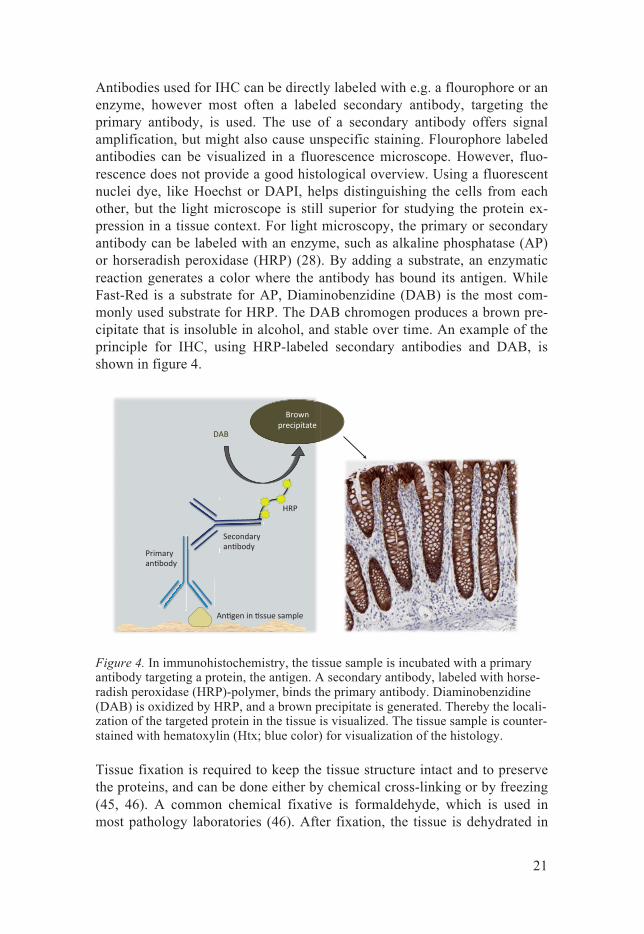
Antibodies used for IHC can be directly labeled with e.g. a flourophore or an enzyme, however most often a labeled secondary antibody, targeting the primary antibody, is used. The use of a secondary antibody offers signal amplification, but might also cause unspecific staining. Flourophore labeled antibodies can be visualized in a fluorescence microscope. However, fluo-rescence does not provide a good histological overview. Using a fluorescent nuclei dye, like Hoechst or DAPI, helps distinguishing the cells from each other, but the light microscope is still superior for studying the protein ex-pression in a tissue context. For light microscopy, the primary or secondary antibody can be labeled with an enzyme, such as alkaline phosphatase (AP) or horseradish peroxidase (HRP) (28). By adding a substrate, an enzymatic reaction generates a color where the antibody has bound its antigen. While Fast-Red is a substrate for AP, Diaminobenzidine (DAB) is the most com-monly used substrate for HRP. The DAB chromogen produces a brown pre-cipitate that is insoluble in alcohol, and stable over time. An example of the principle for IHC, using HRP-labeled secondary antibodies and DAB, is shown in figure 4.
Figure 4. In immunohistochemistry, the tissue sample is incubated with a primary antibody targeting a protein, the antigen. A secondary antibody, labeled with horse-radish peroxidase (HRP)-polymer, binds the primary antibody. Diaminobenzidine (DAB) is oxidized by HRP, and a brown precipitate is generated. Thereby the locali-zation of the targeted protein in the tissue is visualized. The tissue sample is counter-stained with hematoxylin (Htx; blue color) for visualization of the histology.
Tissue fixation is required to keep the tissue structure intact and to preserve the proteins, and can be done either by chemical cross-linking or by freezing (45, 46). A common chemical fixative is formaldehyde, which is used in most pathology laboratories (46). After fixation, the tissue is dehydrated in

22
graded alcohols and xylene, and finally embedded in paraffin blocks. This procedure results is formalin-fixed paraffin-embedded (FFPE) tissue sam-ples, which can be sectioned and placed on glass slides to be used for IHC. Formaldehyde introduces cross-links between proteins, which can mask epitopes and thereby prevent the antibody from binding its target (46). Therefore, antigen retrieval is necessary to break the cross-links and unmask hidden epitopes. Proteolytic treatment and heat-induced epitope retrieval (HIER) are frequently used antigen retrieval methods (45, 47).
Antibody validation An antibody produced for immunoassays holds a certain affinity to its in-tended antigen, but usually displays varying affinity to other proteins as well, a phenomenon referred to as cross reactivity. The bigger the difference is between antigen affinity and affinity towards other proteins, the higher the antibody specificity and signal-to-noise ratio is in an immunoassay. To find out the specificity of an antibody, stringent antibody validation is of great importance (48-50). For commercially available antibodies, far from all ven-dors provide sufficient information about validation (49). Consequently, the customer has to validate the antibody before using it. Since 2008 there is a web-based portal, www.antibodypedia.com, for commercially available an-tibodies, aiming to present information about how an antibody performs in different applications (51, 52). This can greatly help users to find suitable antibodies for the application of interest.
Most importantly when validating an antibody is that the antibody should work for the application it is intended for. WB data is often required for pub-lishing antibody results in scientific journals. This technique can confirm that the antibody is capable of binding its antigen in a linear conformation (24, 53), but cannot assure that the antibody will detect the folded protein in a tissue section for IHC. In the Human Protein Atlas project, protein arrays are used as a first screening step to decide if an antibody is specific enough to go through to the next validation step (30). The antigen, along with 384 other peptides, is spotted onto a slide and incubated with the antibody. A fluorescently labeled secondary antibody is used for detection. Another fluo-rescent dye verifies the presence of the spotted proteins. This assay shows whether the antibody binds its antigen and if there is any cross reactivity to other peptides on the array.
Another method sometimes used for antibody validation is peptide blocking with the peptide used for immunization when generating the antibody (54). The antibody is pre-incubated with the peptide in excess before staining. Blocking the antibody with this peptide should optimally result in loss of staining on the tissue if the antibody is specific. While this strategy shows if

23
the antibody has an affinity for its antigen, it does not, however, prove that the antibody is selective for one protein since pre-adsorption with the peptide will prevent off-target binding as well (55).
Optimally, there should be positive and negative controls when validating an antibody. To exclude the primary antibody in an immunoassay is a technical negative control that shows whether the other reagents give rise to unspecific staining. However, this control does not give any information about the pri-mary antibody’s specificity. Instead, for antibody validation, biological posi-tive and negative controls are more suitable, e.g. tissues known to express or to lack expression of a certain protein. Alternatively, the protein expression in cell lines can be knocked down by using small interfering RNA (siRNA) (56, 57). A drastic decrease of the signal in gene specific siRNA-treated cells compared to cells treated with scrambled siRNA indicates that the antibody targets the intended protein. This can be studied with e.g. WB of the cell lysates and immunostaining of the cells. As a positive control, a cell line overexpressing the protein may be used. However, even this positive control is suboptimal since the antibody might show specific staining only when the protein of interest is highly expressed compared to other proteins. In a tissue sample, the endogenous expression of the target protein might be low com-pared to the expression of off-target proteins, resulting in a low signal-to-noise ratio.
For well-characterized proteins, tissues known to express or not express the protein can be used as biological controls. Furthermore, the staining pattern of the antibody can be compared to information from gene (www.ensembl.org) or protein (www.uniprot.org) databases, as well as other published data. However, for uncharacterized proteins, there is no literature to rely on. Therefore, in the Human Protein Atlas, the staining patterns of two or more antibodies targeting non-overlapping epitopes, so called paired antibodies, for each protein are compared on parallel slides (30, 31). Anti-bodies showing the same staining pattern validate each other. Co-localization can also be studied using two antibodies targeting the same pro-tein simultaneously on one TMA-slide. This can be done with double IHC (58) or in situ-PLA (59). Co-localization of paired antibodies implies identi-fication of the true protein expression pattern with a high probability.
Another antibody validation strategy is comparing the staining patterns from IHC with RNA-data, based on the fact that mRNA-transcripts is a prerequi-site for making the protein. The RNA can either be detected in situ on a TMA-slide using in situ hybridization (ISH), or by extracting mRNA from tissue samples followed by RNAseq. Using the first method, IHC and ISH are stained on consecutive TMA-sections and the staining patterns compared on a cellular level (Paper I). Protein- and mRNA-levels do not always corre-

24
late due to different turnover and post-translational modification. Therefore, lack of correlation between IHC and ISH results cannot disapprove the stain-ing of an antibody. Conversely, when similar staining patterns are obtained, ISH can validate the antibody (60). This method has the advantage of com-paring antibody staining and RNA-expression in cells of the same tissue samples on two consecutive TMA-sections. RNAseq of complex tissue ho-mogenates cannot offer information about what cell type, or the distribution of cells in the tissue that are accountable for the gene expression. This is instead a high throughput method that has the capacity to analyze the whole transcriptome of tissues, and can therefore be useful for transcriptomic stud-ies. RNAseq data can also validate an antibody when the staining pattern follows the mRNA expression in tissues. Version 13 of the Human Protein Atlas, released in November 2014, includes transcriptomic data from RNAseq analyzes on 32 tissues and 44 cell lines, as a complement to the antibody based proteomic results (14, 61).
Antibody labeling Most immunological methods, e.g. ELISA, IHC, IF and WB, require labeled antibodies for detection of the antigen. The primary antibody can be directly labeled, or a labeled secondary antibody can be used to target the primary antibody. The advantage with secondary antibodies is that they can be used for many different primary antibodies as long as the primary antibodies are generated in the same species, however they cannot be used simultaneously. Moreover, the use of a secondary antibody can greatly amplify the signal from the primary antibody binding to its target. On the other hand, secondary antibodies prolong the staining protocol and might also cause cross reactivi-ty. Instead, using a directly labeled primary antibody removes the need for a secondary antibody, and therefore shortens the staining procedure. Most importantly, directly labeled primary antibodies enable simultaneous detec-tion of two (or more) proteins regardless of antibody species, which is cru-cial in many studies of proteins. Moreover, labeled paired antibodies can be used for double IHC to validate each other and to show the true protein lo-calization with a higher credibility if overlapping staining is obtained.
Flourophores, enzymes and biotin are commonly used conjugates used for antibodies. Flourophore-labeled antibodies can be used for e.g. ELISA or for studying IF-stained cells in high resolution with confocal microscopy (62), whereas enzyme and biotin-labeled antibodies are often used for IHC and WB. The binding between biotin and avidin, or its analogue streptavidin, is among the strongest known naturally occurring interactions (63, 64). This enables biotinylated antibodies to be detected with HRP- or fluorescently labeled streptavidin, which specifically binds biotin.

25
There are several different ways of labeling antibodies, as well as other pro-teins. Reactive groups on the antibody that can be targeted for labeling in-cludes primary amines, sulfhydryls, carbonyls, carbohydrates and carboxylic acids (65). Conjugation at amine sites of antibodies in solution is the most common commercial labeling method (66). It is a relatively easy procedure but may result in labeling near the antigen-binding site, which could alter the antibodies binding properties. There are many commercial labeling kits on the market, although they often require a high amount of purified antibody, free from albumin or gelatin. Some efforts have been done to develop label-ing techniques, such as a solid-phase labeling (67, 68), to avoid these prob-lems. Another newly developed conjugation method uses the immunoglobu-lin binding Z-domain from staphylococcal protein A, specifically labeling the Fc-part of antibodies, and thereby also avoid labeling near the antigen-binding site (69-71) (Paper II). Regardless of conjugate and labeling tech-nique used, it is important to validate the labeled antibody, to make sure its antigen binding properties is maintained.
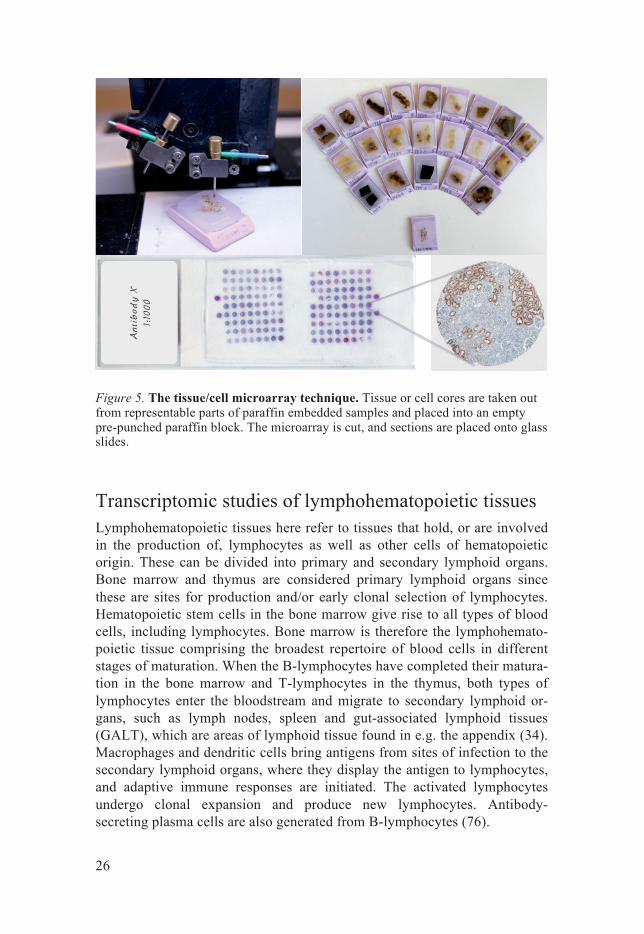
Tissue microarrays To localize the expression of all human proteins in a large set of tissues us-ing antibodies requires a time and reagent efficient system. Immunostaining of one tissue section per glass slide is both time consuming and involves a great amount of reagents. If instead numerous tissue samples could be put together and stained on one glass slide, a great deal of time and reagents would be saved. This groundbreaking technique is called tissue microarray (TMA; figure 5), and was first described by Kononen et.al (72, 73). In brief, tissues cores are taken from each sample of FFPE tissue donor blocks, ex-tracted from surgical specimen, and put in an empty paraffin block (recipient block). It is of great importance that the cells of interest are represented in the extracted tissue core. Therefore, the first step is to cut a section of the donor block, put it on a glass slide and stain with histological dyes, hema-toxylin and eosin. An area containing a representative region is marked out on the slide, and a punch of e.g. 1 mm diameter is used to extract a tissue core within the corresponding area of the donor block and placed in pre-punched holes in a recipient block. In this way, hundreds of tissue samples can be analyzed on one TMA glass slide (74). Duplicate cores from each patient assure a good representation of all tissue samples (75).
26
Figure 5. The tissue/cell microarray technique. Tissue or cell cores are taken out from representable parts of paraffin embedded samples and placed into an empty pre-punched paraffin block. The microarray is cut, and sections are placed onto glass slides.
Transcriptomic studies of lymphohematopoietic tissues Lymphohematopoietic tissues here refer to tissues that hold, or are involved in the production of, lymphocytes as well as other cells of hematopoietic origin. These can be divided into primary and secondary lymphoid organs. Bone marrow and thymus are considered primary lymphoid organs since these are sites for production and/or early clonal selection of lymphocytes. Hematopoietic stem cells in the bone marrow give rise to all types of blood cells, including lymphocytes. Bone marrow is therefore the lymphohemato-poietic tissue comprising the broadest repertoire of blood cells in different stages of maturation. When the B-lymphocytes have completed their matura-tion in the bone marrow and T-lymphocytes in the thymus, both types of lymphocytes enter the bloodstream and migrate to secondary lymphoid or-gans, such as lymph nodes, spleen and gut-associated lymphoid tissues (GALT), which are areas of lymphoid tissue found in e.g. the appendix (34). Macrophages and dendritic cells bring antigens from sites of infection to the secondary lymphoid organs, where they display the antigen to lymphocytes, and adaptive immune responses are initiated. The activated lymphocytes undergo clonal expansion and produce new lymphocytes. Antibody-secreting plasma cells are also generated from B-lymphocytes (76).

27
The lymph node can be divided into cortex, paracortex and medulla. The B-lymphocytes, along with follicular dendritic cells (FDCs) are mainly local-ized in follicles in the cortex, while the T-lymphocytes are located in the paracortex. Some of the follicles contain germinal centers where B-cell pro-liferation takes place. The medulla mainly consists of macrophages and plasma cells (34, 76). While the antigens are transported to lymph nodes through the lymph, the antigens enter the spleen from the blood. The spleen is divided into red pulp, were erythrocyte destruction takes place, and white pulp containing the lymphocytes. Similarly to lymph node, B-cells are found in follicles, and T-cells are located in the periarteriolar lymphoid sheath (PALS). The distribution of lymphocytes in GALTs is comparable to lymph node and spleen, with T-cell areas and B-cell follicles containing germinal centers. However, in GALTs, the antigens are instead retrieved from the intestinal epithelium (34).
Besides cells of hematopoietic origin, the lymphohematopoietic tissues also comprise other cell types, such as adipocytes in bone marrow, cells of blood vessels in the highly vascularized spleen, and glandular epithelium in appen-dix. Therefore, when performing high throughput transcriptomic studies using RNAseq on lymphohematopoietic tissue lysates, the gene expression is not solely representing transcription from blood cells, but from all cells with-in the tissue. Furthermore, cells of hematopoietic origin, especially lympho-cytes, are additionally found in peripheral tissues, i.e. outside the primary and secondary lymphoid organs. Genes expressed by these cells are conse-quently detected in many tissue types. Interesting findings from RNAseq analyses can be further analyzed with IHC on tissues to provide information about what cell types that express the protein (61, 77-80). The strategy of combining transcriptomic and proteomic profiling of lymphohematopoietic tissues is discussed in paper III.
Estrogen receptors Two types of estrogen receptors (ERs) exist, ERα (595 amino acid) and ERβ (530 amino acids), of which genes are located on chromosomes 6 and 14 respectively (81, 82). ERα was identified in 1986 (81, 83), and ERβ in 1996 (82, 84). Like all steroid hormone receptors, ERs are composed of six do-mains, denoted A-F. These include the N-terminal A/B-region, the DNA-binding C-domain, the hinge D-domain, the ligand-binding E-domain and the C-terminal F-domain. The DNA-binding domain is a highly conserved region with an amino acid homology of approximately 97 % between ERα and ERβ, while the ligand-binding domain shows 56 % similarity, and the N-terminus only share 24 % amino acid identity (85). The A/B region con-tains the transcription activation function 1 (AF1), which is a ligand-

28
independent transcription activator, responsible for recruitment of co-regulatory proteins. The ligand-dependent AF2 is located in the N-terminal part of the receptor (86).
Estrogens are steroid hormones and ligands for ERs. They are mainly pro-duced in ovaries, and contribute to giving female characteristics. When a ligand has bound to the ligand-binding domain of the estrogen receptor, lig-and-specific conformational changes take place, and two ligand-bound re-ceptors dimerize as homo- or heterodimers, and bind to estrogen response elements (EREs) on DNA (87). Co-regulatory protein complexes (co-activator or co-repressor multiprotein complexes) are recruited to the AF1 domain, and these influence the activity of the receptors to either activate or repress gene transcription (88).
ERα is known to be involved in the tumorigenesis of e.g. breast cancer, and approximately 70 % of all breast cancers overexpress the receptor. ERα is therefore a therapeutic target in the treatment of the disease using the anti-estrogen drug, tamoxifen. However, all ERα positive breast cancers do not respond to tamoxifen therapy and some develop resistance to tamoxifen (89). Much effort has therefore been put into studying ERβ to elucidate whether this second ER can be used as a complementary treatment-predictive marker in breast cancer. Studies of breast cancer cell lines indicate that ERβ has an anti-proliferative function (90-92). Furthermore, the expression of ERβ de-creases with increasing tumorigenesis (93). However, contradictory results about the proteins function and localization are being reported, which partly depends on the choice of antibody, the two most frequently used being the mAbs 14C8 that recognizes all ERβ isoforms (94-96), and PPG5/10, directed towards the C-terminal region of only ERβ1 (wild type) (97-100). Therefore, it is of great interest to develop a standardized assay with well-validated antibodies (101). This topic is discussed further in paper IV.

29
Present investigation
Aim The overall aim was to identify suitable strategies for validation of antibod-ies used for large-scale studies of protein expression in situ. The particular aims for each paper were:
Paper I: To investigate the specificity of ISH on FFPE tissues, and to evaluate if ISH could be used for validating novel antibodies. Paper II: To compare two antibody-labeling assays for protein detec-tion in situ. Paper III: To identify genes with elevated expression in four lym-phohematopoietic tissues, using a combined strategy of RNAseq and antibody-based proteomics. Paper IV: To systematically validate 13 ERβ antibodies on control cells and tissues, and to characterize the expression profile of ERβ in human tissues.
Methodological considerations In Paper I, the staining patterns of ISH (RNA expression) and IHC (protein expression) for 17 genes were compared on consecutive sections of a TMA. The correlation between RNA and protein expression for each gene was evaluated by manual annotation of the digitalized images from the stained slides.
In Paper II, two antibody-labeling methods, the ZBPA-technique and the commercially available Lightning-Link kit, were examined and compared on 14 different antibodies. Each antibody was biotinylated using the two meth-ods, and then stained on TMA sections using IHC with streptavidin-HRP detection. IHC of unconjugated antibodies, stained using a secondary anti-body coupled to HRP, served as references to which the biotinylated anti-bodies were compered.

30
In paper III, next generation mRNA sequencing was performed on 27 tissue types, including bone marrow, spleen, lymph node and appendix. The gene expression, in terms of FPKM (fragments per kilobase of exon model per million mapped reads) values, in the four lymphohematopoietic tissues was compared to all analyzed tissues for identification of genes with elevated expression in any or several of these tissues. For tissue enriched genes, the RNA-data was complemented with protein expression, using IHC on tissues, which provides information about the expression pattern on a cellular level.
In paper IV, 13 antibodies directed towards varying sequences of the ERβ protein were evaluated using IHC on a specially designed TMA containing well-validated FFPE-treated control cells and eight different tissue types. Three of the antibodies were further validated, using Western blot, and IP followed by MS to examine whether they bind ERβ. The expression profile of ERβ was then assessed using IHC, with the only specific antibody, on extended TMAs.
Results and discussion Paper I: Scalable in situ hybridization on tissue arrays for validation of novel cancer and tissue-specific biomarkers.
In paper I, mRNA and protein expressions were analyzed and compared on TMAs consisting of FFPE normal and malignant tissues. First, validated antibodies towards well-characterized proteins CHGA, KRT17, LYN, MKI67, PDE6A, PECAM1, PTPRC and VIL1 were stained with IHC and compared to antisense probe ISH on consecutive sections for analysis of corresponding transcripts. Sense probe ISH was used as a negative control. Concordance between IHC and ISH was found for CHGA, KRT17, MK167, PECAM1 and VIL1, and semi-correlation for PDE6A, suggesting that ISH is specific at the cell type level, and could therefore be used for validation of novel antibod-ies. The antibody staining for LYN and PTPRC respectively was supported by literature. Hence, the lack of ISH staining for LYN and PTPRC can be due to technical matter, or explained by biological reasons, such as mRNA de-cay.
Next, novel antibodies towards the proteins BRD1, EZH2, FAM174B, GAD1, JAK3, JUP, MIXL1, ZNF473 and the potential colorectal cancer bi-omarker SATB2 were analyzed. Here, ISH could validate antibodies for BRD1, EZH2, JUP and SATB2. GAD1 showed semi-correlation and FAM174B, JAK3, MIXL1 and ZNF473 no correlation. The staining patterns

31
of these antibodies thus need to be further evaluated with other methods. SATB2 was moreover analyzed on a colorectal cancer array containing tissue from 59 primary tumors in duplicate. Protein and mRNA expression were found to correlate in 56 out of 59 samples, which further support the speci-ficity of the SATB2 antibody.
These results demonstrate that ISH can be used for in situ transcriptomic analyses and antibody validation on FFPE tissues.
Paper II: Antibodies biotinylated using a synthetic Z-domain from protein A provide stringent in situ protein detection.
In paper II, two antibody biotinylation methods were evaluated and com-pared, ZBPA and the commercially available kit, Lightning Link, for IHC with Streptavidin-HRP detection on TMAs. ZBPA consists of the Z-domain of protein A which has been synthesized with biotin and BPA, and binds covalently and specifically to the Fc-part of antibodies. The Lightning Link-kit biotinylates all amine groups of antibodies and other proteins. IHC stain-ing patterns for unconjugated antibodies were used as references when eval-uating the two alternative biotin conjugation methods. All Z-biotinylated antibodies (n=14) showed a staining pattern concordant to that of the uncon-jugated antibodies. Only four out of the 14 Lightning Link-conjugated anti-bodies showed a staining pattern similar to the corresponding unconjugated antibody, and five antibodies showed expected staining but with additional unwanted staining, which was often extensive. The immunostaining of the remaining five Lightning Link-conjugated antibodies was conflicting as compared to the unconjugated antibodies.
Twelve of the antibodies examined contained albumin of different concen-trations, and one contained gelatin as antibody stabilizer. Albumin is known to be sticky and can therefore bind to tissues. Furthermore, all proteins con-tain amine groups that can be biotinylated by the Lightning Link-kit and cause unspecific staining. This was shown when labeling human serum al-bumin (HSA) and gelatin using the Lightning Link-kit. The biotinylated proteins gave rise to a staining pattern similar to the unwanted staining seen for many Lightning Link-biotinylated antibodies. No off-target staining was seen for Z-biotinylated antibodies, however the staining intensity for one Z-conjugated antibody was lower compared to the unconjugated antibody. ZBPA-biotinylation resulted in a specific staining pattern similar to uncon-jugated antibody for all tested antibodies, and is therefore the method to prefer for in situ protein detection.

32
Paper III: The transcriptomic and proteomic landscapes of bone marrow and second-ary lymphoid tissues.
Paper III describes the transcriptomic profile in four lymphohematopoietic tissues (bone marrow, lymph node, spleen and appendix) and identifies genes that show an increased expression in these four tissues compared to 23 other tissue types. Furthermore, the RNAseq results are validated with IHC on tissues from the Human Protein Atlas, which also provide information about the protein localization on a cellular level.
Out of the four lymphohematopoietic tissues, bone marrow shows the lowest fraction of expressed genes (57 %), and appendix the highest (69 %), which reflects that the ficoll separated bone marrow samples represent a relatively pure population of hematopoietic cells, while appendix samples harbor many cell types including glandular epithelium, smooth muscle and lymphoid cells. Bone marrow has the highest number of tissue enriched genes (80), while only 6 genes are tissue enriched in spleen, 5 in lymph node and 2 in appendix. Bone marrow, which is a primary lymphoid organ, furthermore shows the highest gene expression levels, with FPKM values up to 25,947, almost five times higher than for any of the three secondary lymphoid tis-sues. Genes expressed at high levels in bone marrow include e.g. defensins expressed by neutrophils, and hemoglobins expressed by erythrocytes. These cells reach maturity in the bone marrow, and express high levels of proteins needed for the function they will perform when released into the blood stream as effector cells.
An increased expression in one or more lymphohematopoietic tissue was found for 693 out of 20,050 genes. Most of these were well-characterized genes, known to have a function in lymphohematopoietic cells. Uncharacter-ized genes with an increased expression were also found, e.g. C19ORF59 that was found to be group enriched in bone marrow, appendix and lung, and RP11-685N3-1 that showed an enhanced expression in e.g. lymph node and appendix. The combination of high throughput transcriptomics and prote-omics thus enables identification of tissue specific gene expression and visu-alization of the protein with cellular resolution.

33
Paper IV: Estrogen receptor beta profiling in human tissues following extensive anti-body validation.
Paper IV aims to characterize the expression pattern of the steroid hormone receptor, ERβ, in human normal and malignant tissues. First, 13 ERβ anti-bodies were evaluated using a screening TMA with positive and negative control cell lines and selected tissues. Based on the antibody staining pat-terns, three of the antibodies were further analyzed using WB and IP/MS. Only one antibody, the mouse mAb clone PPZ0506, was proven to be spe-cific for ERβ in all assays. The two other antibodies that were examined in this procedure included the two most commonly used ERβ antibodies, clone PPG5/10 and 14C8. PPG5/10 showed a distinct nuclear staining in tissues and were therefore selected for further evaluation, however did not show specificity for ERβ in the control cells with either of the methods IHC, WB or IP/MS. 14C8 showed specific staining of control cells, but not in applica-tions IP/MS or WB. In this thorough antibody evaluation procedure, PPZ0506 showed nuclear IHC staining in positive control cells, but not in negative controls, and gave a band of expected molecular weight in the ly-sate from positive control cells only. Furthermore, the IP/MS analysis result-ed in a hit for ERβ in the positive, but not negative control.
The well-validated antibody, PPZ0506, was further used to characterize the localization of ERβ with IHC on TMAs including a wide range of normal tissues and multiple cases of 20 different cancer types. Nuclear staining was found in selected cells of secondary lymphoid organs, as well as peripheral lymphocytes in other tissues. Weaker nuclear staining was also seen in testis and in a few cells in breast. Low levels of ERβ were further detected in 2/12 cases of melanoma. However, the most prominent staining was seen in gran-ulosa cell tumors (GCTs), which showed moderate to strong staining in a high fraction of the cells.
In conclusion, comprehensive evaluation of 13 ERβ antibodies resulted in the finding of one highly specific ERβ antibody. IHC staining of normal and cancer tissues using this antibody revealed ERβ expression in a limited set of human tissues.

34
Concluding remarks and future perspectives
This thesis presents tools to validate antibodies for in situ protein detection on TMAs. Correlating the IHC staining pattern to mRNA-expression is a strategy that can be applied for antibody validation. The mRNA expression can either be examined by ISH on consecutive slides that are consecutive to slides stained with IHC, or by RNAseq on tissue lysates to generate an FPKM-value for each gene, which can be related to the IHC staining. Two antibodies towards different epitopes on the same protein can also validate each other when showing congruent staining patterns. The ZBPA-labeling technique was found to be suitable for antibodies used for IHC on TMAs, which enables simultaneous dual staining of antibodies from the same spe-cies. Furthermore, cell lines overexpressing or lacking expression of a pro-tein is a valuable tool when validating antibodies and can be used in several assays. One out of 13 antibodies towards the nuclear receptor ERβ was prov-en to be specific in three different assays using positive and negative control cell lines.
The high degree of concordance between IHC and ISH for well-characterized proteins suggests that ISH could be used to validate antibodies when correlation is seen between the two methods. Therefore, ISH was used to test the specificity of novel antibodies that are interesting due to a tissue selective staining pattern or as potential biomarkers. ISH was able to confirm the staining pattern for four out of nine antibodies, and thereby validating these novel antibodies. The potential colorectal cancer biomarker antibody for SATB2 was further compared to ISH on a colorectal cancer array and correlated in 56 out of 59 cases, which strengthens the use of this antibody as a biomarker. The remaining antibodies have to be further evaluated, e.g. by using dual in situ protein detection, or by comparing the antibody staining pattern to large-scale transcriptomics data. The Human Protein Atlas in-cludes RNAseq results for all genes in a wide range of tissues and cell lines, and has become a great resource for validating the antibodies used in this large proteomic project. The transcriptomic data has also been further exam-ined to identify tissue specific gene expression, including four lymphohema-topoietic tissues. For this purpose, the digitalized images of immunostained tissues and cell lines from the Human Protein Atlas offered spatial infor-mation about the protein expression. The potentially interesting tissue specif-ic proteins discovered using this strategy might be further studied in bigger

35
cohorts of patient samples, and their function examined in functional studies in cell lines.
Dual detection with paired antibodies from the same species requires anti-body labeling. Many labeling-kits offer a fast and convenient laboratory procedure. Furthermore, as for the Lightning Link-kit, it also eliminates the need for purification steps, and no antibody will be wasted. Although, for antibodies used for IHC on TMAs, it requires a protein free antibody buffer to avoid unspecific staining as biotinylation of amine groups causes unspe-cific staining in tissues sections. The Z-biotinylation technique is a specific conjugation method, only targeting the Fc-part of antibodies. Therefore, independently of stabilizing proteins in the buffer, Z-biotinylation does not cause any unspecific staining for IHC. The Z-biotinylation technique is thus to be preferred for in situ protein detection. However, only one biotin is in-corporated in the Z-molecule, and maximum two Z-molecules can bind to each antibody, which offers no signal amplification and thus requires quite a high antibody concentration for immunostaining. For future studies, the Z-biotinylation method needs to be optimized to increase the staining intensity. A study on Z-molecules with two incorporated biotins has already been initi-ated. Preliminary IHC results show that double biotin Z is superior to single biotin Z. Furthermore, simultaneous protein detection using two antibodies requires labeling with dissimilar conjugates for each antibody. Consequent-ly, other conjugates than biotin need to be tested for incorporation in the Z-molecule.
Not only can dual detection of a protein be used for validation of antibodies, but also for detecting a protein with a higher probability. This is especially important for proteins with unknown localization. However, dual detection requires that two specific antibodies exist for the protein of interest. The expression of the nuclear receptor ERβ in tissues is debated, which is partly due to the use of unspecific antibodies and lack of well-validated antibodies. To elucidate the true localization of ERβ in human normal and cancer tis-sues, 13 antibodies were thoroughly evaluated in positive and negative con-trol cells in different assays. Only one antibody, PPZ0506, was proven to be highly specific in all assays, and was further used for tissue profiling. ERβ was shown to be expressed in a limited set of tissues and cell types, with the highest expression in GCTs, while no expression was seen in normal breast or the 12 cases of breast cancer. However, to fully assess whether ERβ ex-pression may be used as a complement to ERα as a treatment predictive bi-omarker in breast cancer, larger cohorts of breast cancer cases need to be immunostained using the specific ERβ-antibody PPZ0506.
Systematic validation of antibodies is crucial for accurate protein detection, and this thesis presents a selection of validation strategies. A combination of

36
two or more validation methods is often desirable, and the protein’s confor-mational status should be taken into consideration.

37
Acknowledgements
This work was performed at the Department of Immunology, Genetics and Pathology, Rudbeck Laboratory, Uppsala University, and was financially supported mainly by grants from Knut and Alice Wallenberg Foundation.
I would like to thank all patients who donated tissue samples for medical research, and all people who contributed to sample collection, making this work possible!
I would like to express my sincere gratitude to all of you who have support-ed and encouraged me during the work with this thesis. Special THANKS to,
My supervisor, Anna Asplund, for all your support, help and inspiration. You are an excellent scientist and supervisor, and a wonderful person and friend!! I really appreciate our scientific discussions, the small talk and our long walks to get “lyx-kaffe” ;-)
My supervisor, Fredrik Pontén, for sharing your great knowledge in every-thing from pathology to scientific writing, and also for giving me the oppor-tunity to do my PhD in HPA! Keep up your enthusiasm and positivity.
My co-supervisor, Kenneth Wester, for teaching me IHC, antibody-validation, histology and much more! It is partly thanks to you that I started my PhD studies in the HPA group.
Tobias Sjöblom and Sara Kiflemariam for a nice collaboration, and for sharing your knowledge about the automated ISH technique. Sophia Hober and Anna Konrad for lovely discussions and fruitful meetings, and for in-troducing me to the ZBPA-labeling technique. Kenneth Nilsson and Chris-ter Sundström for your great knowledge in the hematology field. Linn Fagerberg and Björn Hallström for patiently answering all my questions and helping out with the figures. Cecilia Williams for your ENORMOUS knowledge about estrogen receptors, your significant contribution to the ER-beta paper, and for our nice Skype-meetings! Margareta Ramström and Mårten Sundberg for your mass spec-expertise. All other co-authors and collaborators for your contribution. I have learned a lot from all of you!!

38
All present and former colleagues in the HPA-group, for help and support, interesting discussions about really everything, nice “fika-fikas” and for creating a good atmosphere at work!
Present and former room-mates, for all the fun we’ve had, and for being wonderful friends both at Rudbeck and outside work. You are the best room-mates ever! ☺
New, old, close and far away Friends, for making me smile, laugh, relax and have fun! Whether we see each other every day or just once in a while, you are all important to me. Now I will hopefully have more time to do things ☺
Och sist men inte minst, min familj…
Mamma, pappa och Mickan, för att ni alltid finns där som ett tryggt stöd! Det är alltid lika roligt att komma “hem” och hänga i trädgården eller hitta på något kul tillsammans. Älskar er! Och ni vet ju att ni även är Dameons och Leonels stora favoriter ☺
Kazeem, for encouraging me to do doctoral studies, and for supporting me during the writing of this thesis. Mo ni ife re! Our lovely children, Dameon and Leonel, for all the joy you bring to my life. Words cannot describe how much I love you. You are the best!

39
References
1. Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2015. Nucleic acids research. 2015;43(Database issue):D662-9.
2. Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nature biotechnology. 2003;21(3):255-61.
3. Gonzalez-Porta M, Frankish A, Rung J, Harrow J, Brazma A. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome biology. 2013;14(7):R70.
4. Alberts B. Molecular biology of the cell. Sixth edition. ed. New York, NY: Garland Science, Taylor and Francis Group; 2015. p. p.
5. Crick F. Central dogma of molecular biology. Nature. 1970;227(5258):561-3. 6. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al.
Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921.
7. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291(5507):1304-51.
8. Wilkins MR, Pasquali C, Appel RD, Ou K, Golaz O, Sanchez JC, et al. From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology (N Y). 1996;14(1):61-5.
9. Naaby-Hansen S, Waterfield MD, Cramer R. Proteomics--post-genomic cartography to understand gene function. Trends in pharmacological sciences. 2001;22(7):376-84.
10. Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312(5771):212-7.
11. Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annual review of biomedical engineering. 2009;11:49-79.
12. Agaton C, Galli J, Hoiden Guthenberg I, Janzon L, Hansson M, Asplund A, et al. Affinity proteomics for systematic protein profiling of chromosome 21 gene products in human tissues. Molecular & cellular proteomics : MCP. 2003;2(6):405-14.
13. Uhlen M, Ponten F. Antibody-based proteomics for human tissue profiling. Molecular & cellular proteomics : MCP. 2005;4(4):384-93.
14. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419.
15. Solier C, Langen H. Antibody-based proteomics and biomarker research - current status and limitations. Proteomics. 2014;14(6):774-83.

40
16. Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246(4926):64-71.
17. Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Analytical chemistry. 1988;60(20):2299-301.
18. Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198-207.
19. Baldwin MA. Protein identification by mass spectrometry: issues to be considered. Molecular & cellular proteomics : MCP. 2004;3(1):1-9.
20. Stoevesandt O, Taussig MJ. Affinity reagent resources for human proteome detection: initiatives and perspectives. Proteomics. 2007;7(16):2738-50.
21. Engvall E, Perlmann P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8(9):871-4.
22. Van Weemen BK, Schuurs AH. Immunoassay using antigen-enzyme conjugates. FEBS letters. 1971;15(3):232-6.
23. Fulwyler MJ. Electronic separation of biological cells by volume. Science. 1965;150(3698):910-1.
24. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(9):4350-4.
25. Coons AH, Creech HJ, Jones RN. Immunological Properties of an Antibody Containing a Fluorescent Group. Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine (New York, NY). 1941;47(2):200-2.
26. Warford A, Howat W, McCafferty J. Expression profiling by high-throughput immunohistochemistry. Journal of immunological methods. 2004;290(1-2):81-92.
27. Robinson G, Dawson I. Immunochemical studies of the endocrine cells of the gastrointestinal tract. II An immunoperoxide technique for the localization of secretin containing cells in human duodenum. Journal of clinical pathology. 1975;28(8):631-5.
28. Nakane PK, Pierce GB, Jr. Enzyme-labeled antibodies: preparation and application for the localization of antigens. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1966;14(12):929-31.
29. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. Towards a knowledge-based Human Protein Atlas. Nature biotechnology. 2010;28(12):1248-50.
30. Berglund L, Bjorling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Molecular & cellular proteomics : MCP. 2008;7(10):2019-27.
31. Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Molecular & cellular proteomics : MCP. 2005;4(12):1920-32.
32. Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, et al. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45(3):227-32.

41
33. Pontén F, Schwenk JM, Asplund A, Edqvist PHD. The Human Protein Atlas as a proteomic resource for biomarker discovery. Journal of internal medicine. 2011;270(5):428-46.
34. Murphy K, Travers P, Walport M, Janeway C. Janeway's immunobiology. 8th ed. New York: Garland Science; 2012. xix, 868 p. p.
35. Kracker S, Durandy A. Insights into the B cell specific process of immunoglobulin class switch recombination. Immunology letters. 2011;138(2):97-103.
36. Diaz M, Casali P. Somatic immunoglobulin hypermutation. Current opinion in immunology. 2002;14(2):235-40.
37. Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR journal / National Research Council, Institute of Laboratory Animal Resources. 2005;46(3):258-68.
38. Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. 1975. J Immunol. 2005;174(5):2453-5.
39. Agaton C, Falk R, Hoiden Guthenberg I, Gostring L, Uhlen M, Hober S. Selective enrichment of monospecific polyclonal antibodies for antibody-based proteomics efforts. Journal of chromatography A. 2004;1043(1):33-40.
40. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315-7.
41. Azzazy HM, Highsmith WE, Jr. Phage display technology: clinical applications and recent innovations. Clinical biochemistry. 2002;35(6):425-45.
42. Bradbury AR, Marks JD. Antibodies from phage antibody libraries. Journal of immunological methods. 2004;290(1-2):29-49.
43. Marks JD, Bradbury A. Selection of human antibodies from phage display libraries. Methods Mol Biol. 2004;248:161-76.
44. Ramos-Vara JA. Technical aspects of immunohistochemistry. Veterinary pathology. 2005;42(4):405-26.
45. Yamashita S. Heat-induced antigen retrieval: mechanisms and application to histochemistry. Progress in histochemistry and cytochemistry. 2007;41(3):141-200.
46. Werner M, Chott A, Fabiano A, Battifora H. Effect of formalin tissue fixation and processing on immunohistochemistry. The American journal of surgical pathology. 2000;24(7):1016-9.
47. Shi SR, Cote RJ, Taylor CR. Antigen retrieval techniques: current perspectives. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2001;49(8):931-7.
48. Howat WJ, Lewis A, Jones P, Kampf C, Ponten F, van der Loos CM, et al. Antibody validation of immunohistochemistry for biomarker discovery: recommendations of a consortium of academic and pharmaceutical based histopathology researchers. Methods. 2014;70(1):34-8.
49. Bordeaux J, Welsh A, Agarwal S, Killiam E, Baquero M, Hanna J, et al. Antibody validation. BioTechniques. 2010;48(3):197-209.
50. Ivell R, Teerds K, Hoffman GE. Proper application of antibodies for immunohistochemical detection: antibody crimes and how to prevent them. Endocrinology. 2014;155(3):676-87.
51. Bjorling E, Uhlen M. Antibodypedia, a portal for sharing antibody and antigen validation data. Molecular & cellular proteomics : MCP. 2008;7(10):2028-37.

42
52. Alm T, von Feilitzen K, Lundberg E, Sivertsson A, Uhlen M. A chromosome-centric analysis of antibodies directed toward the human proteome using Antibodypedia. Journal of proteome research. 2014;13(3):1669-76.
53. Renart J, Reiser J, Stark GR. Transfer of proteins from gels to diazobenzyloxymethyl-paper and detection with antisera: a method for studying antibody specificity and antigen structure. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(7):3116-20.
54. Skliris GP, Rowan BG, Al-Dhaheri M, Williams C, Troup S, Begic S, et al. Immunohistochemical validation of multiple phospho-specific epitopes for estrogen receptor alpha (ERalpha) in tissue microarrays of ERalpha positive human breast carcinomas. Breast cancer research and treatment. 2009;118(3):443-53.
55. Holmseth S, Zhou Y, Follin-Arbelet VV, Lehre KP, Bergles DE, Danbolt NC. Specificity controls for immunocytochemistry: the antigen preadsorption test can lead to inaccurate assessment of antibody specificity. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2012;60(3):174-87.
56. Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286(5441):950-2.
57. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494-8.
58. van der Loos CM. Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2008;56(4):313-28.
59. Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nature methods. 2006;3(12):995-1000.
60. Kiflemariam S, Andersson S, Asplund A, Ponten F, Sjöblom T. Scalable in situ hybridization on tissue arrays for validation 2 of novel cancer and tissue-specific biomarkers. In press.
61. Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & cellular proteomics : MCP. 2014;13(2):397-406.
62. Fagerberg L, Stadler C, Skogs M, Hjelmare M, Jonasson K, Wiking M, et al. Mapping the subcellular protein distribution in three human cell lines. Journal of proteome research. 2011;10(8):3766-77.
63. Heitzmann H, Richards FM. Use of the avidin-biotin complex for specific staining of biological membranes in electron microscopy. Proceedings of the National Academy of Sciences of the United States of America. 1974;71(9):3537-41.
64. Green NM. Avidin. Advances in protein chemistry. 1975;29:85-133. 65. Mattson G, Conklin E, Desai S, Nielander G, Savage MD, Morgensen S. A
practical approach to crosslinking. Molecular biology reports. 1993;17(3):167-83.
66. Haugland RP. Antibody conjugates for cell biology. Current protocols in cell biology / editorial board, Juan S Bonifacino [et al]. 2001;Chapter 16:Unit 16 5.

43
67. Lundberg E, Sundberg M, Graslund T, Uhlen M, Svahn HA. A novel method for reproducible fluorescent labeling of small amounts of antibodies on solid phase. Journal of immunological methods. 2007;322(1-2):40-9.
68. Strachan E, Mallia AK, Cox JM, Antharavally B, Desai S, Sykaluk L, et al. Solid-phase biotinylation of antibodies. Journal of molecular recognition : JMR. 2004;17(3):268-76.
69. Konrad A, Karlstrom AE, Hober S. Covalent immunoglobulin labeling through a photoactivable synthetic Z domain. Bioconjugate chemistry. 2011;22(12):2395-403.
70. Andersson S, Konrad A, Ashok N, Ponten F, Hober S, Asplund A. Antibodies biotinylated using a synthetic Z-domain from protein A provide stringent in situ protein detection. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2013;61(11):773-84.
71. Perols A, Karlstrom AE. Site-specific photoconjugation of antibodies using chemically synthesized IgG-binding domains. Bioconjugate chemistry. 2014;25(3):481-8.
72. Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nature medicine. 1998;4(7):844-7.
73. Kallioniemi OP, Wagner U, Kononen J, Sauter G. Tissue microarray technology for high-throughput molecular profiling of cancer. Human molecular genetics. 2001;10(7):657-62.
74. Kampf C, Olsson I, Ryberg U, Sjostedt E, Ponten F. Production of tissue microarrays, immunohistochemistry staining and digitalization within the human protein atlas. Journal of visualized experiments : JoVE. 2012(63).
75. Kampf C, Andersson A-C, Wester K, Björling E, Uhlen M, Ponten F. Antibody-based tissue profiling as a tool for clinical proteomics. Clinical Proteomics. 2004;1(3):285-99.
76. Willard-Mack CL. Normal structure, function, and histology of lymph nodes. Toxicologic pathology. 2006;34(5):409-24.
77. Andersson S, Nilsson K, Fagerberg L, Hallstrom BM, Sundstrom C, Danielsson A, et al. The transcriptomic and proteomic landscapes of bone marrow and secondary lymphoid tissues. PloS one. 2014;9(12):e115911.
78. Kampf C, Mardinoglu A, Fagerberg L, Hallstrom BM, Edlund K, Lundberg E, et al. The human liver-specific proteome defined by transcriptomics and antibody-based profiling. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2014;28(7):2901-14.
79. Lindskog C, Fagerberg L, Hallstrom B, Edlund K, Hellwig B, Rahnenfuhrer J, et al. The lung-specific proteome defined by integration of transcriptomics and antibody-based profiling. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2014;28(12):5184-96.
80. Gremel G, Wanders A, Cedernaes J, Fagerberg L, Hallstrom B, Edlund K, et al. The human gastrointestinal tract-specific transcriptome and proteome as defined by RNA sequencing and antibody-based profiling. J Gastroenterol. 2015;50(1):46-57.

44
81. Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. 1986;231(4742):1150-4.
82. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(12):5925-30.
83. Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, et al. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. 1986;320(6058):134-9.
84. Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS letters. 1996;392(1):49-53.
85. Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, et al. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacological reviews. 2006;58(4):773-81.
86. Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, et al. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. 1989;59(3):477-87.
87. Cowley SM, Hoare S, Mosselman S, Parker MG. Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem. 1997;272(32):19858-62.
88. Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11(8):597-608.
89. Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22(47):7316-39.
90. Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(6):1566-71.
91. Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer research. 2004;64(1):423-8.
92. Sotoca AM, van den Berg H, Vervoort J, van der Saag P, Strom A, Gustafsson JA, et al. Influence of cellular ERalpha/ERbeta ratio on the ERalpha-agonist induced proliferation of human T47D breast cancer cells. Toxicological sciences : an official journal of the Society of Toxicology. 2008;105(2):303-11.
93. Roger P, Sahla ME, Makela S, Gustafsson JA, Baldet P, Rochefort H. Decreased expression of estrogen receptor beta protein in proliferative preinvasive mammary tumors. Cancer research. 2001;61(6):2537-41.
94. Skliris GP, Leygue E, Curtis-Snell L, Watson PH, Murphy LC. Expression of oestrogen receptor-beta in oestrogen receptor-alpha negative human breast tumours. British journal of cancer. 2006;95(5):616-26.
95. Fuqua SA, Schiff R, Parra I, Moore JT, Mohsin SK, Osborne CK, et al. Estrogen receptor beta protein in human breast cancer: correlation with clinical tumor parameters. Cancer research. 2003;63(10):2434-9.

45
96. Skliris GP, Carder PJ, Lansdown MR, Speirs V. Immunohistochemical detection of ERbeta in breast cancer: towards more detailed receptor profiling? British journal of cancer. 2001;84(8):1095-8.
97. Honma N, Horii R, Iwase T, Saji S, Younes M, Takubo K, et al. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(22):3727-34.
98. Novelli F, Milella M, Melucci E, Di Benedetto A, Sperduti I, Perrone-Donnorso R, et al. A divergent role for estrogen receptor-beta in node-positive and node-negative breast cancer classified according to molecular subtypes: an observational prospective study. Breast cancer research : BCR. 2008;10(5):R74.
99. Shaaban AM, Green AR, Karthik S, Alizadeh Y, Hughes TA, Harkins L, et al. Nuclear and cytoplasmic expression of ERbeta1, ERbeta2, and ERbeta5 identifies distinct prognostic outcome for breast cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(16):5228-35.
100. Reese JM, Suman VJ, Subramaniam M, Wu X, Negron V, Gingery A, et al. ERbeta1: characterization, prognosis, and evaluation of treatment strategies in ERalpha-positive and -negative breast cancer. BMC cancer. 2014;14:749.
101. Haldosen LA, Zhao C, Dahlman-Wright K. Estrogen receptor beta in breast cancer. Mol Cell Endocrinol. 2014;382(1):665-72.

Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1109
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-251344
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2015





























