Embed Size (px)
Citation preview

Variabilità genetica

Genoma• L'intera sequenza del
genoma umano, scritta in Times New Roman, dimensione 12, avrebbe una lunghezza di 5000 km
• Consiste di una sequenza di tre miliardi di questi nucleotidi

Fenotipo normale WT
• Wild type ceppo selvatico versione più comune di un tratto genetico nella popolazione
• Non sempre si può individuare WTFatta eccezione fenotipo patologico
– Accoppiamento selettivo nelle piante e negli animali per aumentare o diminuire la frequenza di un carattere

Eugenetica
• Descrivere gli studi delle caratteristiche umane e applicazioni dell’ accoppiamento selettivo per aumentare la frequenza di alcune caratteristiche.
• Eugenetica: strumento politica sociale razzista• CREARE UNA RAZZA PERFETTA (campagna NAZISTA
ANNI 30-40)attraverso sterminio di massa degli individui inferiori
• Attualmente lo scopo degli studi è di lenire le sofferenze dei soggetti malati e non di migliorare la razza

VARIAZIONE DEI
CARATTERI
•
…nel bene…nel male

• Il genoma umano contiene variazioni nella sequenza di basi tra un individuo e l’altro.
– Sequenze codificanti
– Sequenze non codificanti
– Effetto sul fenotipo
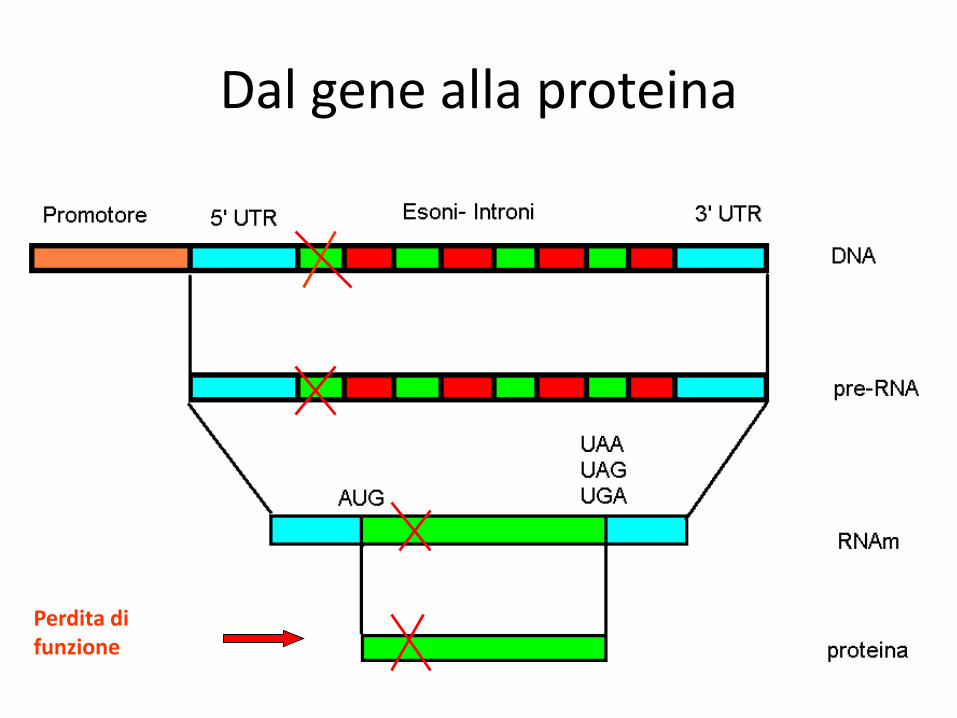
Dal gene alla proteina
Perdita di funzione





Un esempio di mutazione non patologica…

Mutazioni
• Cambi nella normale sequenza del DNA
• Origine:– Spontanea (errori di replicazione o riparazione del
DNA)
– Indotta (da agenti fisici o chimici)
• Sede:– Germinali (ereditabili)
– Somatiche (non ereditabili)

Germinali
Figura 2.11 Differenze
nella maturazione degli
spermatozoi (sinistra) e
delle cellule uovo (destra).
Gli spermatozoi vanno
incontro a molteplici cicli di
mitosi, che iniziano alla
pubertà e continuano per
tutta la vita adulta. Le
cellule uovo, invece,
completano le divisioni
mitotiche durante la vita
fetale.

Agenti mutageni

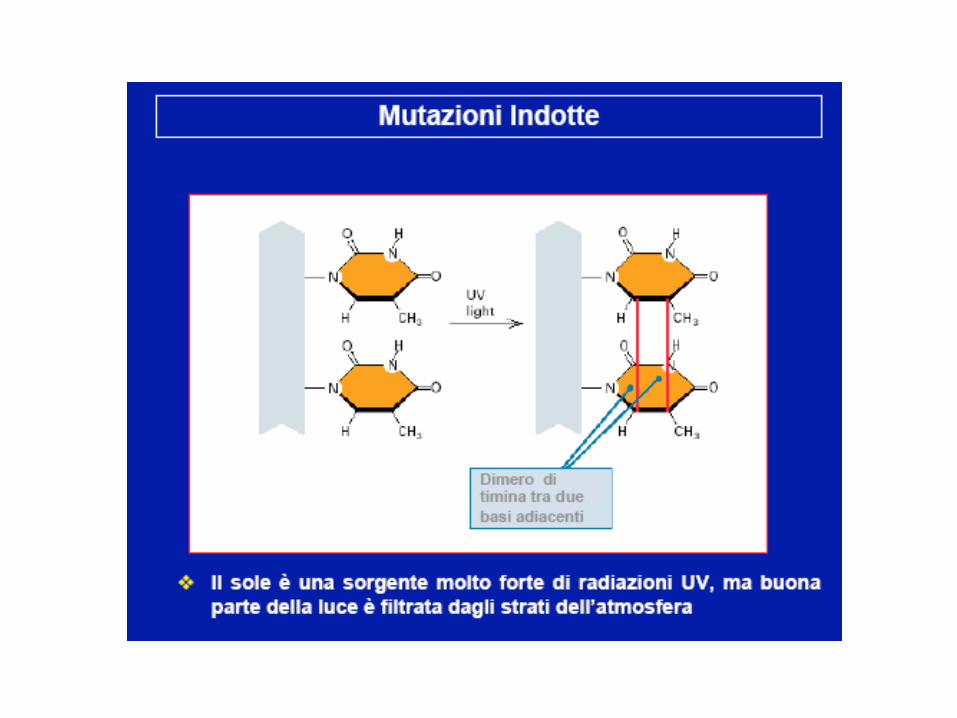



Effetti delle mutazioni
• Mutazioni letali– Provocano la morte del 100% dei portatori prima della pubertà
• Mutazioni subletali– Portano a morte più del 50% (ma non il 100%) dei portatori prima
della pubertà
• Mutazioni neutrali– Non modificano la salute dell’individuo in senso negativo o positivo
• Mutazioni vantaggiose– Sono favorevoli per la specie, aumentando la fitness riproduttiva dei
portatori
Una stessa mutazione può avere effetto diverso in base all’ambiente in cui il portatore vive

Cosa può essere colpito da una mutazione?
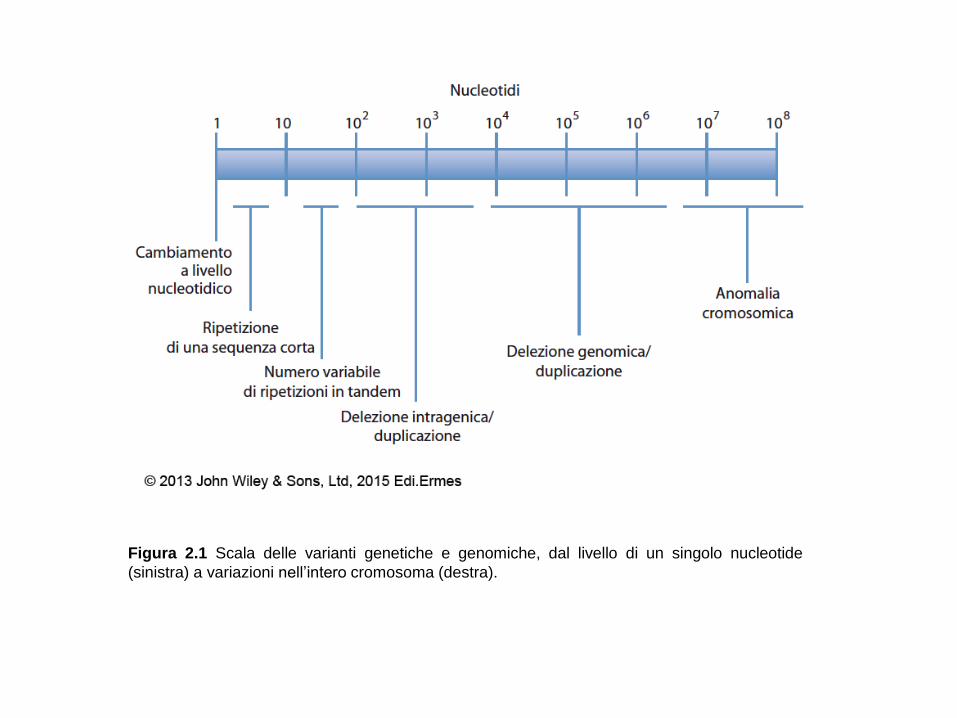
Figura 2.1 Scala delle varianti genetiche e genomiche, dal livello di un singolo nucleotide
(sinistra) a variazioni nell’intero cromosoma (destra).

Cosa può essere colpito da una mutazione?
• Una singola base del DNA (mutazioni puntiformi)

Tipi di mutazioni puntiformi
• Mutazioni “missenso”: produzione di un diverso aminoacido
– GAC->GAA = Asp->Glu
• Mutazioni “sinonime”: produzione di uno stesso aminoacido
– GGT->GGC = Ser->Ser
• Mutazioni “non senso”: formazione di un codone di stop
– TAT->TAA = Tyr->STOP

Mutazioni frameshift
GAT CCG TAT CTG GTC GTA TCT…
Asp Arg Tyr Leu Val Val Ser
G/AT CCG TAT CTG GTC GTA TCT…
ATC CGT ATC TGG TCG TAT CT…
Ile Ala Ile Trp Ser Tyr …

Come agisce una mutazione puntiforme?
• Produzione di una proteina tronca (mutazione non-senso, formazione di un codone di stop)
• Produzione di una proteina di sequenza diversa da quella attesa (mutazioni frameshift, da scivolamento della cornice di lettura)
• Produzione di una proteina di struttura diversa da quella attesa (mutazione missenso, da cambio di un aminoacido importante per la struttura tridimensionale della proteina).

Figura 2.4 Conseguenze di mutazioni di una singola base all’interno di un esone. Se l’aminoacido non è
cambiato, la mutazione è silente. Le mutazioni missenso provocano la sostituzione di aminoacidi che potrà avere
conseguenze sulla funzione della proteina.
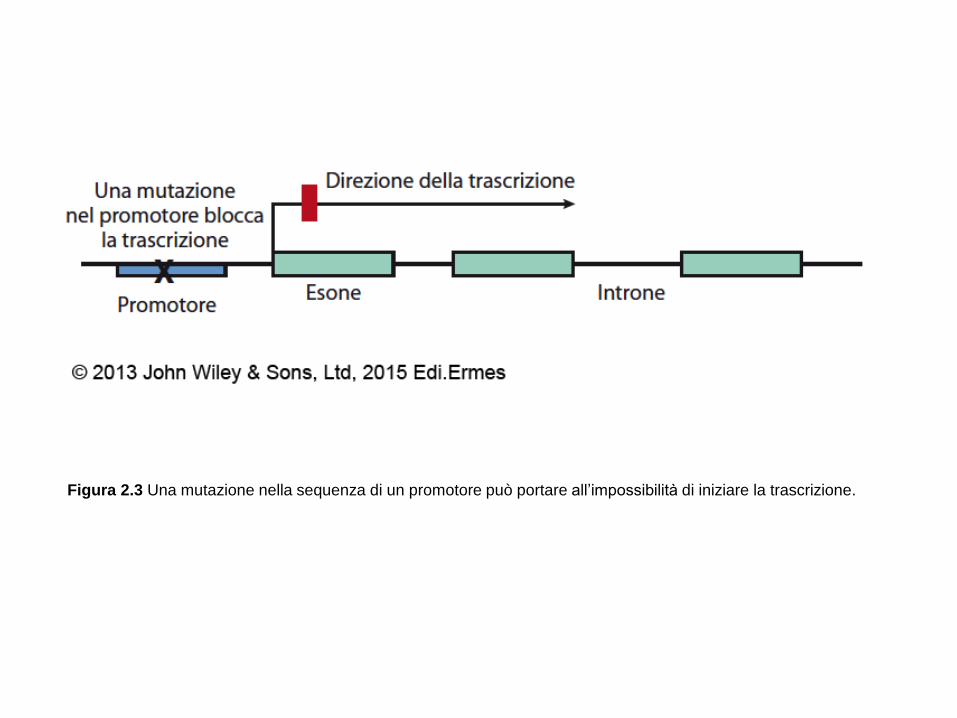
Figura 2.3 Una mutazione nella sequenza di un promotore può portare all’impossibilità di iniziare la trascrizione.
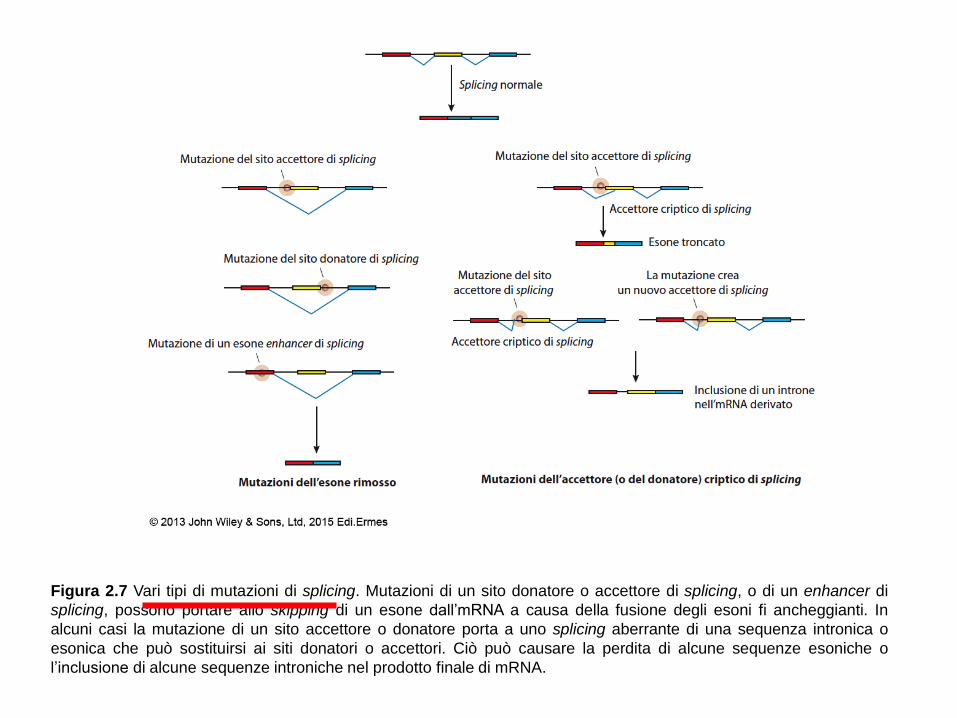
Figura 2.7 Vari tipi di mutazioni di splicing. Mutazioni di un sito donatore o accettore di splicing, o di un enhancer di
splicing, possono portare allo skipping di un esone dall’mRNA a causa della fusione degli esoni fi ancheggianti. In
alcuni casi la mutazione di un sito accettore o donatore porta a uno splicing aberrante di una sequenza intronica o
esonica che può sostituirsi ai siti donatori o accettori. Ciò può causare la perdita di alcune sequenze esoniche o
l’inclusione di alcune sequenze introniche nel prodotto finale di mRNA.

Nomenclatura

Cosa sappiamo oggi?
Progetto genoma




Tratti complessi
• Nonostante le alte capacità di questa nuova tecnologia vanno presi in considerazione numerosi fattori che rendono ancora complessa la interpretazione delle informazioni ottenute.
• La complessità del nostro genoma….

La complessità del nostro genoma
• Il genoma umano contiente4 milioni sequenze variabilidel DNA(DSVs)– 3.5 milioni sono singoli
nucleotidi (SNVs)
• Migliaia di varianti strutturali inserzioni delezioni duplicazioni
(SVs)(Kidd et al., 2008; Korbel et al., 2007).
• In ogni individuo la metà dei geni è polimorfica ( le due copie del gene non sono identiche Levy et al., 2007).
• genoma/exoma contienecirca 10,000 varianti non sinonime (nsSNVs) che per definizione alterano la sequenza amminoacidicanella sequenzacodificante e potrebbero avere un effetto biologico (Levy et al., 2007; Ng et al., 2008; Wang et al., 2008; Wheeler et al.,

Progetto 1000 genomi
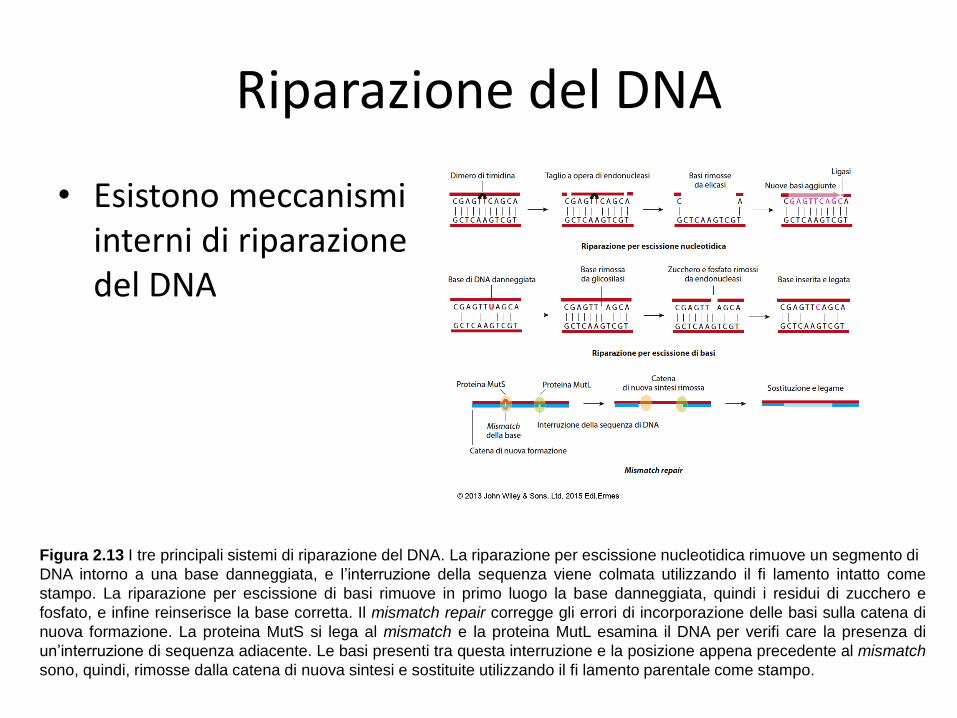
Riparazione del DNA
• Esistono meccanismi interni di riparazione del DNA
Figura 2.13 I tre principali sistemi di riparazione del DNA. La riparazione per escissione nucleotidica rimuove un segmento di
DNA intorno a una base danneggiata, e l’interruzione della sequenza viene colmata utilizzando il fi lamento intatto come
stampo. La riparazione per escissione di basi rimuove in primo luogo la base danneggiata, quindi i residui di zucchero e
fosfato, e infine reinserisce la base corretta. Il mismatch repair corregge gli errori di incorporazione delle basi sulla catena di
nuova formazione. La proteina MutS si lega al mismatch e la proteina MutL esamina il DNA per verifi care la presenza di
un’interruzione di sequenza adiacente. Le basi presenti tra questa interruzione e la posizione appena precedente al mismatch
sono, quindi, rimosse dalla catena di nuova sintesi e sostituite utilizzando il fi lamento parentale come stampo.

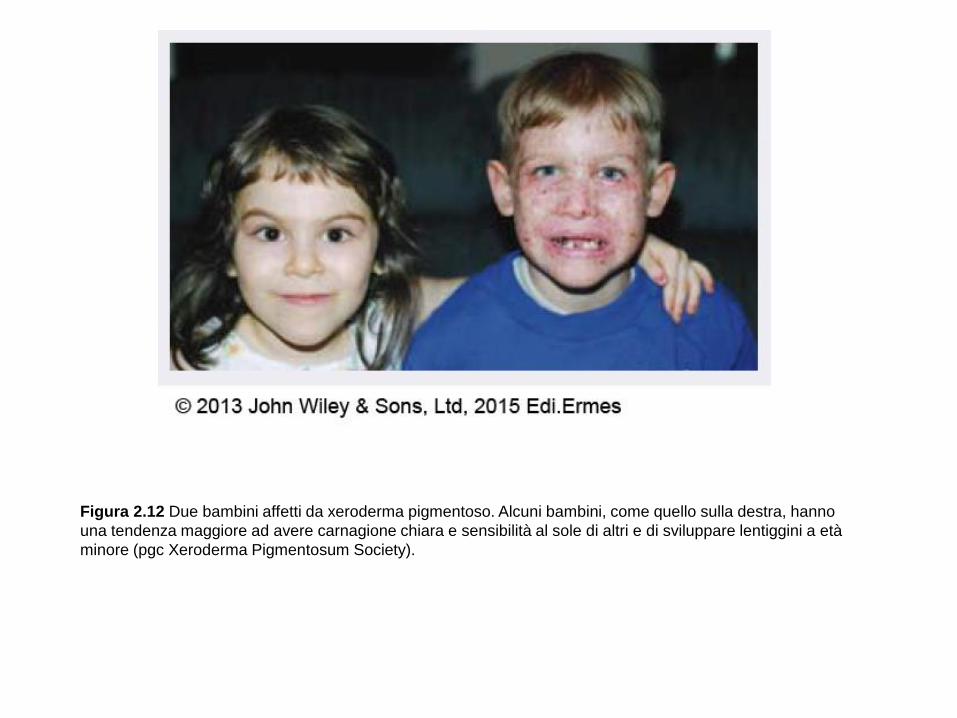
Figura 2.12 Due bambini affetti da xeroderma pigmentoso. Alcuni bambini, come quello sulla destra, hanno
una tendenza maggiore ad avere carnagione chiara e sensibilità al sole di altri e di sviluppare lentiggini a età
minore (pgc Xeroderma Pigmentosum Society).



Polimorfismo genico
Presenza in una popolazionedi due o più alleli di un locuscon una frequenza uguale osuperiore a 0,01

Polimorfismi genici e polimorfismi del DNA: distinzione
• Polimorfismi genici:
– varianti all’interno di un gene che possono o meno modificare la funzionalità dello stesso
• Polimorfismi del DNA
– Varianti in regioni esterne ai geni, che non hanno mai un ruolo funzionale

Polimorfismi genici
Presenti in più dell’1% degli alleli di una popolazione
Creano dei cosiddetti loci di suscettibilità (es. Alzheimer, Diabete non-insulino dipendente)
Si trasmettono con eredità complessa, multifattoriale (intervento dell’ambiente)

S.N.P. (Single Nucleotide polymorphisms)
Presenza di un cambio diuna singola base in puntispecifici del genoma
Possono essere presentiin regioni non codificantima anche all’interno deigeni

Mutazioni
Cambi nella sequenza delDNA presenti in menodell’1% degli alleli di unapopolazione
Legate con rapportocausale a malattieereditarie ad altapenetranza (Es. FibrosiCistica, Distrofia diDuchenne, Corea diHuntington, ecc)
Trasmesse
con eredità mendeliana

Polimorfismi Sequenza
– Mutazione
– Delezione
– Inserzione
• ATTTCGCCTAA
• ATTTCTCCTAA
• ATTTCGCCTAA
• ATTTCCCTAA
• ATTTCGCCTAA
• ATTTCGACCTAA
Polimorfismi a singolo nucleotide (SNP)
APOE
Presenta 3 alleliAPOEe2, APOEe3,APOEe4
L’allele APOEe4 èassociato a unaumento di rischio disviluppare il morbo diAlzheimer
Questo rischio èrelativo in particolareall’età di insorgenza

Analisi di specifiche regioni
• Tutte le applicazioni della biologia molecolare utilizzate nella genetica forense sfruttano la possibilità di identificare con precisione le forme alternative di queste zone polimorfiche.
• Per identificare una traccia lasciata da un individuo non è necessario analizzare tutto il DNA, ma studiare solo alcune zone particolarmente variabili nella popolazione.

Polimorfismi di lunghezza
• In queste zone del DNA, particolari sequenze delle basi ACGT vengono ripetute più volte determinando nelle varie forme alleliche, lunghezze variabili del tratto medesimo.
• A seconda della lunghezza della sequenza ripetuta sono ulteriormente suddivisi in:

Polimorfismi del DNA
Variazioni interindividuali dellacomposizione o della lunghezza dispecifici tratti di DNA che permettono,tramite analisi molecolare, didistinguere ciascun singolo individuo(tranne i gemelli monovulari)

• Satelliti da 1 a diverse migliaia di basi
• VNTR da 9 a 100 basi
• Microsatelliti o STR da 1 a sei basi
• La classificazione attuale, utilizza dei numeri che indicano il numero delle ripetizioni presenti in ogni forma allelica

VNTR
Ripetizioni in tandem di 15-30basi di DNA all’interno dispecifici loci del genoma
Un individuo sarà omozigote peruno specifico locus se i due allelicontengono lo stesso numero diripetizioni, sarà eterozigote se,al contrario, il numero diripetizioni nei due alleli èdiverso
Il numero di queste ripetizionipuò variare da individuo aindividuo, producendo deiframmenti di DNA di dimensionidiverse
(Variable Number of Tandem Repeats)

Short Tandem Repeats (STRs)
the repeat region is variable between samples while the flanking regions where PCR primers bind are constant
7 repeats
8 repeats
AATG
Homozygote = both alleles are the same length
Heterozygote = alleles differ and can be resolved from one another

Short Tandem Repeats (STRs)
Sono come delle impronte digitali
Alto grado di polimorfismo ed alta capacità informativa
Utilizzati nei paternity testing e nelle analisi forensi

Figura 2.21 Ripetizione di sequenza semplice. In questo caso il dinucleotide CA è ripetuto diverse
volte. L’allele mostrato ha sette ripetizioni diverse, ma altri alleli possono avere cinque, sei, sette,
otto, nove o anche più copie della sequenza CA. Il numero di copie può essere identifi cato da
un’amplificazione in PCR della regione (le frecce indicano i primer della PCR fiancheggianti la
sequenza di DNA) e le dimensioni dei prodotti di reazione possono essere rilevate mediante
elettroforesi su gel di poliacrilamide.

Figura 2.22 Un polimorfismo di numero variabile di ripetizioni in tandem consiste di ripetizioni multiple di un
blocco di DNA. Gli alleli possono essere identificati per mezzo di una PCR che interessa l’intera regione (le
frecce indicano i primer di PCR).
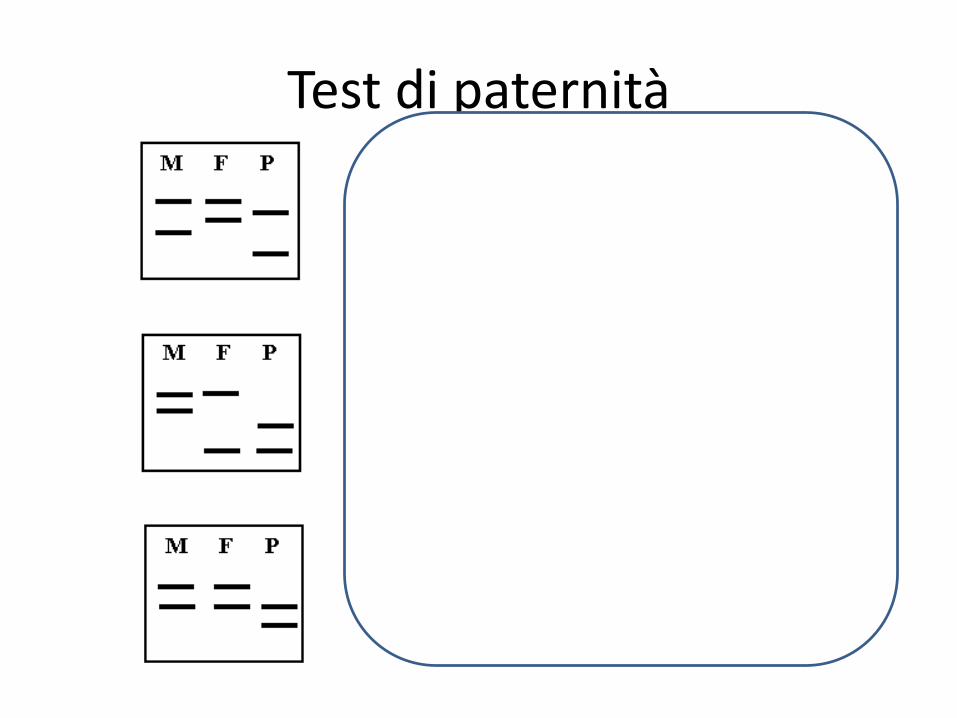
Test di paternità
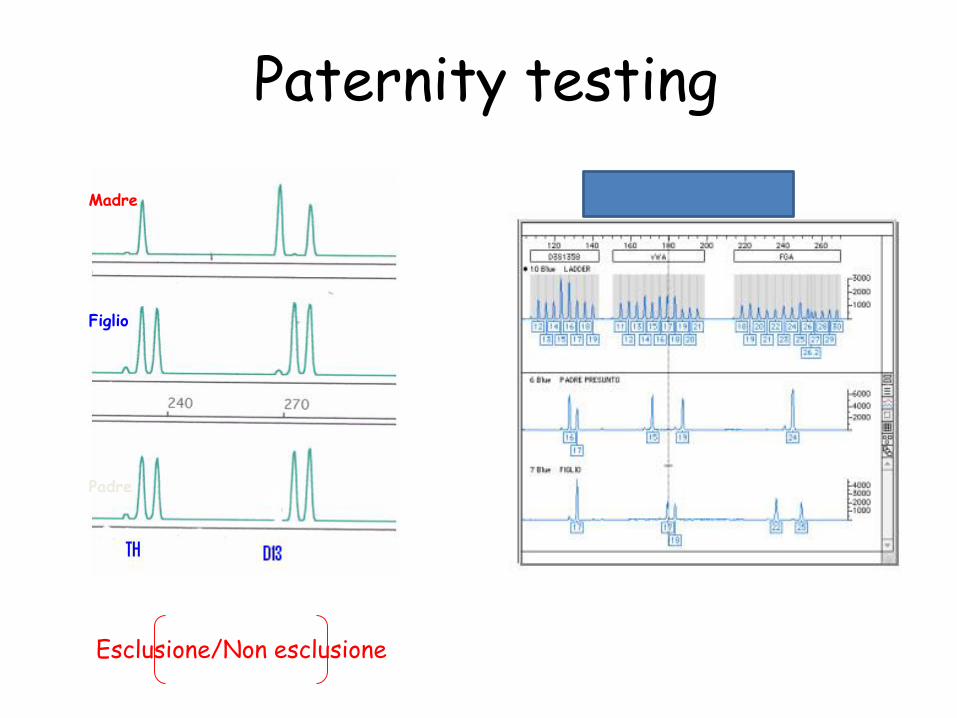
Paternity testing
Madre
Figlio
Padre
Esclusione/Non esclusione

tecniche

Figura 2.20 Diagramma della reazione di polimerizzazione acatena (PCR). I fi lamenti di DNA target sono separati (1) e
i primer si legano sui due fi lamenti opposti (2). Parte quindi una reazione di sintesi del DNA che copia la sequenza
target di DNA (3). Questo processo è ripetuto ciclicamente (4) e porta aun’amplificazione esponenziale della sequenza
target.
























![[Co(NH3 6 - units.it · LGO di [Co(NH 3) 6]3+ 3 MO solo interazioni s . 4 p-donatore p-accettore . 5 LGO p in un piano di un ottaedro p z p x combinazione legante combinazione antilegante](https://img.pdfslide.net/doc/110x75/5f7f912729e2262737266599/conh3-6-unitsit-lgo-di-conh-3-63-3-mo-solo-interazioni-s-4-p-donatore.jpg)


