
Pneumologia Paulista | Setembro 2018 1
ISSN (on-line): 2448-0533

2 Pneumologia Paulista | Setembro 2018
SOCIEDADE PAULISTA DE PNEUMOLOGIA E TISIOLOGIABIÊNIO 2018/2019
Diretoria
Presidente: Roberto Rodrigues JúniorVice-Presidente: Frederico Leon Arrabal FernandesSecretário Geral: Willian Salibe Filho1ª Secretária: Roberta Pulcheri RamosDiretor de Finanças: José Gustavo Barian RomaldiniDiretor de Assuntos Científicos: Rodrigo Abensur AthanazioDiretora de Divulgação: Eloara V. M. Ferreira Alvares da Silva CamposDiretora de Assuntos do Interior: Suzana Erico Tanni MinamotoDiretor de Informática: Marcos Naoyuki Samano
COMISSÕESDefesa Profissional: Lilian Serrasqueiro Ballini Caetano
Ensino: Gustavo Faibichew Prado
Promoções: Evelise Lima
Assuntos da Grande São Paulo: Adriano Cesar Guazzelli
Publicações: Regina Célia Carlos Tibana
DEPARTAMENTOS
Cirurgia Torácica:Luis Carlos LossoAlessandro MarianiAndré Miotto
Endoscopia Respiratória:Diego Henrique RamosFelipe Nominando Diniz OliveiraViviane Rossi Figueiredo
Pediatria:Karina Pierantozzi VerganiAdyleia Aparecida Dalbo Contrera ToroClaudine Sarmento da Veiga
Fisioterapia Respiratória:Luciana Dias ChiavegatoAdriana Claudia LunardiLara Maris Nápolis Goulart Rodrigues
CONSELHO FISCAL:Efetivos:Ricardo Mingarini TerraMaria Raquel SoaresMaria Vera Cruz de Oliveira Castellano
Suplentes:Silvia Carla S RodriguesLiana Pinheiro SantosCiro Botto
CONSELHO DELIBERATIVO
Regina Maria de Carvalho PintoOliver Augusto NascimentoMônica Corso PereiraJaquelina Sonoe Ota ArakakiJosé Eduardo Delfini CançadoRafael StelmachRoberto StirbulovAna Luisa Godoy FernandesMário Terra FilhoEliana Sheila Pereira da Silva MendesJorge NakataniAlberto CukierCarlos Alberto de Castro PereiraMiguel BogossianFrancisco Vargas SusoManuel Lopes dos SantosNelson Morrone

Pneumologia Paulista | Setembro 2018 3
REGIONAIS
Regional do ABCPresidente: Mônica Silveira LapaSecretária: Franco Chies Martins
Regional de Araraquara / Bauru / BotucatuPresidente: Marcos Abdo ArbexSecretário: José Eduardo Bergami Antunes
Regional de CampinasPresidente: Paulo Roberto TonidandelSecretário: Mauricio Sousa de Toledo Leme
Regional de MaríliaPresidente: Gisele César de Rossi AgostinhoSecretária: Maria de Lourdes Marmorato Botta Hafner
Regional de Ribeirão PretoPresidente: Luis Renato AlvesSecretária: Andrea de Cassia Vernier Antunes Cetlin
Regional de SantosPresidente: Alex Gonçalves MacedoSecretário: Thiago Fernandes Leomil
Regional de São José dos CamposPresidente: José Eduardo de OliveiraSecretária: Márcio Adriano Leite Bastos
Regional de São José do Rio PretoPresidente: Leandro Cesar SalvianoSecretário: Clélia Margarete Trindade Borralho
SUB-COMISSÕES
Câncer - Ilka Lopes Santoro
Circulação - Jaquelina Ota
Doenças Intersticiais - Regina Célia Carlos Tibana
D.P.O.C. - Flavio Arbex
Epidemiologia - André Nathan
Infecções Respiratórias e Micoses - Mauro Gomes
Pleura - Roberta Sales
Doenças Ambientais e Ocupacionais - Ubiratan de PaulaSantos
Tabagismo - Aldo Agra de Albuquerque Neto
Terapia Intensiva - Ricardo Goulart
Tuberculose - Marcia Telma Guimarães Savioli
Função Pulmonar - Marcelo Macchione
Imagem - Gustavo de Souza Portes Meirelles
Doença Pulmonar Avançada - Ricardo Henrique deOliveira Braga Teixeira
Exercício e Atividade Física - André Luis Pereira deAlbuquerque

4 Pneumologia Paulista | Setembro 2018
Apresentação
Prezados (as) leitores (as),
As doenças pulmonares órfãs caracterizam-se por serem raras, em muitos casos de causadesconhecida e com poucas opções terapêuticas. Neste grupo estão inseridas as doençascísticas pulmonares, cujo diagnóstico diferencial é desafiador, devido ao grande número deetiologias, e de extrema importância, devido à sua implicação terapêutica.
No PP deste mês, temos a apresentação de um caso menos comum de histiocitose pulmonar decélulas de Langerhans.
Aproveitem a leitura.
Dra. Regina Célia Carlos TibanaEditora-chefe do Pneumologia Paulista
Pneumologia Paulista | Setembro 2018 5
José Roberto Megda [email protected]
Histiocitose pulmonar de células deLangerhans em tabagista passivoJosé Roberto Megda Filho1; Amanda Maria Reis dos Santos1; Laura Fadel Monteiro dos Santos1;Vera Luiza Capelozzi2
1Hospital Universitário de Taubaté - SP2Hospital das Clínicas - USP
Caso clínico
Paciente do sexo feminino, 61 anos, branca, do lar,previamente hígida, nunca fumou, casada há 40 anos comesposo tabagista de três maços/dia. Relatava que o mesmomantém o hábito de fumar mesmo em ambientes fechados,como o quarto e a sala de estar. Negava uso de medicações,exposições ambientais ou domiciliares (além dotabagismo passivo) e comorbidades familiares relevantes.Referiu dispneia progressiva desde agosto de 2016,atualmente aos mínimos esforços, como tomar banho. Emjulho de 2017 foi diagnosticada com “bronquite” e iniciadotratamento com Budesonida 400mcg + Formoterol 12mcgduas vezes ao dia, porém sem melhora do quadro. Nessaépoca, iniciou também quadro de tosse seca com chiadono peito e predomínio noturno. Como não houve melhoracom o tratamento, foi orientada a procurar umpneumologista para condução do caso. Em consultainicial, o que chamava atenção era ausculta pulmonarcom quadro de sibilos difusos e bilaterais além de SpO2
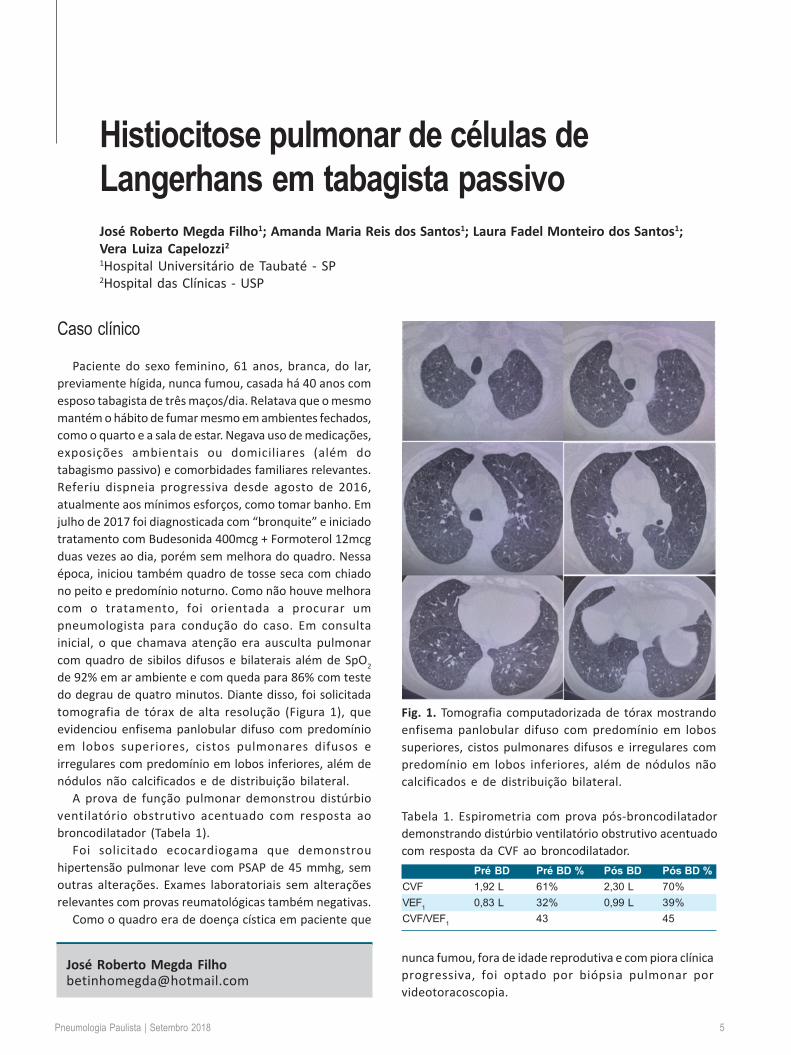
de 92% em ar ambiente e com queda para 86% com testedo degrau de quatro minutos. Diante disso, foi solicitadatomografia de tórax de alta resolução (Figura 1), queevidenciou enfisema panlobular difuso com predomínioem lobos superiores, cistos pulmonares difusos eirregulares com predomínio em lobos inferiores, além denódulos não calcificados e de distribuição bilateral.
A prova de função pulmonar demonstrou distúrbioventilatório obstrutivo acentuado com resposta aobroncodilatador (Tabela 1).
Foi solicitado ecocardiogama que demonstrouhipertensão pulmonar leve com PSAP de 45 mmhg, semoutras alterações. Exames laboratoriais sem alteraçõesrelevantes com provas reumatológicas também negativas.
Como o quadro era de doença cística em paciente que
Pré BD Pré BD % Pós BD Pós BD %CVF 1,92 L 61% 2,30 L 70%VEF1 0,83 L 32% 0,99 L 39%CVF/VEF1 43 45
nunca fumou, fora de idade reprodutiva e com piora clínicaprogressiva, foi optado por biópsia pulmonar porvideotoracoscopia.
Tabela 1. Espirometria com prova pós-broncodilatadordemonstrando distúrbio ventilatório obstrutivo acentuadocom resposta da CVF ao broncodilatador.
Fig. 1. Tomografia computadorizada de tórax mostrandoenfisema panlobular difuso com predomínio em lobossuperiores, cistos pulmonares difusos e irregulares compredomínio em lobos inferiores, além de nódulos nãocalcificados e de distribuição bilateral.

6 Pneumologia Paulista | Setembro 2018
Na avaliação histológica, os cortes evidenciaramnódulos inflamatórios, de forma irregular e limites poucoprecisos, com extensão secundária aos septos alveolares.O infiltrado celular era composto por linfócitos,histiócitos, plasmócitos e raros eosinófilos. As célulashistiocitárias eram escassas e exibiam núcleosvesiculosos com dobras ou endentações e citoplasmaabundante eosinofílico. Fibrose estava presente sobretudono interstício peribronquiolar ou formando “bridgings”com o interstício subpleural, e obliterando ouaprisionando alvéolos revestidos por pneumócitoshiperplásicos. Os bronquíolos mostravam-secomprometidos por proeminente hipertrofia muscular(“cirrose muscular ”), metaplasia respiratória ebronquioloectasias. A presença de tais parâmetroshistológicos sugeriu correlação clínico-tomográfica parahistiocitose de células de Langerhans (HCL) em fasecicatricial, corroborada pela imuno-histoquímica positivapara marcadores específicos (Figura 2).
Por tratar-se de doença de possível acometimentosistêmico, após o diagnóstico foram solicitadosradiografia de crânio e ossos longos, além de tomografiacomputadorizada de abdome – todos sem alterações.
Discussão
A HCL é uma doença rara e de etiologia desconhecida,caracterizada pela proliferação clonal de células de
Fig. 2. Histiocitose de células de Langerhans. (A) Nódulo inflamatório alveolar e (B) bronquiolar contendo linfócitos,histiócitos, plasmócitos (seta dupla) e Células de Langerhans (seta única). (C) Cicatriz peribronquiolar com fibrose ehipertrofia muscular. (D) Imunoexpressão de células de Langerhans S100 positivas. (E) Imunoexpressão de células deLangerhans CD1a positivas. (E) Imunoexpressão de actina de músculo liso (AML) (*) nas áreas de cicatriz peribronquiolar.(A,B,C- hematoxilina&eosina; D,E,F- imuno-histoquímica).
Langerhans com genótipo e fenótipo alterados, queocasiona o acúmulo e formação de lesões semelhantesaos granulomas1,2. É mais comum em crianças e pode levarao comprometimento de diversos órgãos e sistemas3.Quando um único órgão é acometido, a doença édenominada HCL de único sistema. Nos casos em queocorre o acometimento de dois ou mais órgãos esta éconhecida com HCL multissistêmica, sendo que osprincipais sítios envolvidos são os ossos, a hipófise e, emmenor frequência, a pele. Além disso, algumas localizaçõescomo baço, fígado e sistema hematopoiético sãoconhecidos como “órgãos de risco” por apresentarem piorprognóstico2,4.
Em pacientes adultos, a HCL pode acometer os pulmõesde forma isolada ou como característica predominanteda doença, sendo chamada de histiocitose pulmonar dascélulas de Langerhans (HPCL)3. Esta está intimamenterelacionada ao tabagismo (90-100% dos casos), sendomais comum entre os 20 e 40 anos de idade3.
Clinicamente a doença se caracteriza pela presença desintomas respiratórios (tosse e dispneia), associados ounão a sintomas constitucionais, como perda ponderal efebre. Pneumotórax espontâneo pode ocorrer em 15-20%dos casos e inclusive ser a manifestação inicial da doença.
A hipertensão pulmonar pode estar presente, comoconseqüência da destruição progressiva do parênquimapulmonar e/ou da hipoxemia, sendo classificada no grupocinco (hipertensão pulmonar com mecanismos

Pneumologia Paulista | Setembro 2018 7
multifatoriais ou desconhecidos), e pacientes com tal achadoapresentam uma pior morbimortalidade. Até o momento,nenhum tratamento específico para hipertensão pulmonarnestes casos (inibidores do receptor de endotelina, inibidoresda fosfodiesterase 5 e análogos da prostaciclina) se mostrouefetivo2,3.
A radiografia de tórax evidencia infiltrado reticulo-nodularbilateral, sendo raros os casos em que se apresenta normal. Atomografia computadorizada de tórax demonstra nóduloscentrolobulares, com aspecto de “árvore em brotamento” ecistos de paredes grossas, isolados ou confluentes. Com aevolução da doença os cistos se tornam maiores e com paredesfinas. Parênquima pulmonar aparentemente normal separaas lesões, sendo o acometimento radiológico mais evidenteem ápices e terço médio2,3.
Quanto à função pulmonar, o padrão obstrutivo é observadoem uma parcela considerável de pacientes. Cerca de metadedas pessoas diagnosticadas com HPCL vão apresentardeterioração de função pulmonar em cinco anos3,5. Aobstrução ao fluxo aéreo associada à idade avançada estárelacionada a piores desfechos da doença5. A pletismografiapulmonar mostra um volume residual aumentado e a difusãode CO está baixa nos casos mais avançados e naqueles comhipertensão pulmonar.
O diagnóstico definitivo de HPCL é mais comumente obtidopor toracoscopia com a biópsia de áreas nodulares, sendo amarca histológica a presença dos antígenos CD1a+ ou CD207na amostra examinada2,3.
A interrupção do tabagismo é o principal pilar notratamento da HPCL, resultando em melhora clínica, funcionale radiológica na grande maioria dos casos3,6. Drogas comoVinblastina e Cladribina, usadas no tratamento da HCL, vemganhando espaço também no tratamento da HPCL3.
A Vinblastina associada a esteroides é hoje o principalmedicamento usado para tratamento da HCL em crianças, mascom eficácia e tolerância ainda limitadas na HPCL, alta taxade eventos adversos e necessidade de interrupção detratamento2,4. Tazi et al. compararam o uso de Vinblastina empacientes sem acometimento de órgãos de risco durante umtempo médio de 7,6 meses, com resposta em 70% dos casosao final do tratamento. Recorrência da doença em cinco anosocorreu em 40%, sendo que apenas 20% destas ocorreram noprimeiro ano após o uso da Vinblastina4.
A Cladribina trata-se de análogo nucleosídeo da purina etem forte efeito imunossupressor2,7. Consiste na segunda linhano tratamento da HCL e tem sido usada para promover aremissão ou melhorar sintomas da HPCL. Em um estudo,Lorillon et al.7 testaram a eficácia da Cladribina em trêspacientes fumantes, que já tinham feito uso de Vinblastina e/ou corticosteroides. Notou-se melhora clínica e radiológica,com diminuição do número de cistos e nódulos em tomografiade alta resolução. Em estudo mais recente, Nasser et al.6
documentaram o uso de Cladribina como primeira linha detratamento em paciente jovem do sexo feminino, havendodramática melhora dos testes de função pulmonar, diminuição
do número de cistos, com redução na espessura daparede dos remanescentes – alguns, paradoxalmente,aumentaram em tamanho. A Figura 3 mostra um casode um paciente que usou Cladribina e apresentou umamelhora clínica e radiológica importantes6.
Fig. 3. Tomografia de tórax de paciente com HPCL etratado com Cladribina. a,b-alterações pré tratamento;c,d - seis meses depois do início do tratamento; e,f -quatro anos depois do tratamento com cladribina6.
Estes achados demonstram haver opções emquadros avançados de HPCL, todavia, necessita-se demaiores estudos para avaliar risco-benefício do usode drogas com alto potencial de toxicidade.
Em pacientes com doença avançada o transplantepulmonar é indicado3.
HCL é uma neoplasia?
A resposta traz implicações importantes. Há vinteanos, Arceci e colegas especularam que a incertezasobre se a HCL era uma neoplasia ou um distúrbioimunológico levou à abordagem “ambivalente”imunoquimioterápica da HCL. O tratamento deprimeira linha com vinblastina e prednisona continuasendo o padrão-ouro, proporcionando melhorsobrevida global, mas ainda com altas taxas de falhado tratamento. Acredita-se que a presença de mutaçõessomáticas de MAPK em células precursoras mieloidesresilientes forneça a base para a definição de HCLcomo um distúrbio neoplásico mieloide. Essareinterpretação da HCL abre oportunidades paraestudos clínicos adicionais conduzidos pororganizações de pesquisa cooperativa em oncologiapediátrica8.

8 Pneumologia Paulista | Setembro 2018
A paciente aqui descrita apresenta exposição passivaimportante ao tabagismo de seu cônjuge. Na investigaçãoa mesma não apresentou quadro tomográfico típico deHPCL, sendo o diagnóstico confirmado por biópsia. Apósexcluídos acometimentos de outros sítios, o cônjuge foiorientado sobre cessação do tabagismo. A paciente inicioutratamento inalatório com Indacaterol 110 mcg +Glicopirrônio 50 mcg uma vez ao dia, devido ao distúrbioventilatório obstrutivo na função pulmonar. Seguirá emavaliação periódica e se a doença se mostrar progressivafuncionalmente e clinicamente, deverá ser considerado ouso de Cladribina.
Referências Bibliográficas
1. Magaton IM, Tzankov A, Krasniqi F, Rottenburger C, Zanetti-Daellenbach R, Grendelmeier P, et al. Spontaneous remission ofsevere systemic langerhans cell histiocytosis with bladder involvement:A case study. Case Rep Oncol. 2017;10(3):876–84.
2. Feuillet S, Giroux-Leprieur B, Tazi A. [Pulmonary Langerhans cellhistiocytosis in adults]. Presse Med. 2010;39(1):107–15.
3. Lorillon G, Tazi A. How i manage pulmonary langerhans cellhistiocytosis. Eur Respir Rev [Internet]. 2017;26(145):1–14. Availablefrom: http://dx.doi.org/10.1183/16000617.0070-2017
4. Tazi A, Lorillon G, Haroche J, Neel A, Dominique S, Aouba A, et al.Vinblastine chemotherapy in adult patients with langerhans cellhistiocytosis: a multicenter retrospective study. Orphanet J Rare Dis.Orphanet Journal of Rare Diseases; 2017;12(1):1–10.
5. Tazi A, De Margerie C, Naccache JM, Fry S, Dominique S, JouneauS, et al. The natural history of adult pulmonary Langerhans cellhistiocytosis: A prospective multicentre study. Orphanet J Rare Dis.2015;10(1):1–10.
6. Nasser M, Traclet J, Cottin V. Effect of cladribine therapy on lungcysts in pulmonary Langerhans cell histiocytosis. ERJ Open Res[Internet]. 2018;4(1):00089–2017. Available from: http://openres.ersjournals.com/lookup/doi/10.1183/23120541.00089-2017
7. Lorillon G, Bergeron A, Detourmignies L, Jouneau S, Wallaert B,Frija J, et al. Cladribine is effective against cystic pulmonary langerhanscell histiocytosis. Am J Respir Crit Care Med. 2012;186(9):930–2.
8. Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. NEngl J Med. 2018 Aug 30;379(9):856-868.
Recommended

















