
Reatividade do grupo carbonila:Construção do esqueleto carbônico
por condensacondensacondensaçççõesõesõesArmin Isenmann
Parte I: os princípios da homocondensação e retro-síntese

�Reatividade do grupo carbonila (incl. equilíbrio ceto-enol).
�O alvo 1,3-difuncional, a partir dos componentes
pseudo-ácido e aceitador (incl. descarboxilações).
�Manipulação das reatividades em condensações mistas:
1. Utilização de um pseudo-ácido com baixa tendência de condensação
consigo mesmo e/ou um aceitador sem α-H. (incl. Aminometilação de Mannich)
2. Desativação do pseudo-ácido por formação de uma imina.
3. Ativação do pseudo-ácido pela estabilização da forma enólica.
4. Emprego de um pseudo-ácido duplamente ativado (condensações de
Knoevenagel).
5. Com bases muito fortes se consegue desprotonar o pseudo-acido
quantitativamente em uma etapa preliminar.

Reatividade do grupo carbonilaDefinição:
Condensação é a unificação de dois compostos orgânicos sob formação de uma nova ligação carbono-carbono.
Ampliação do esqueleto carbônico a partir
de um substrato A com carbono eletrofílico
e um substrato B com carbono nucleofílico.
Caso A = B : “Homocondensação” ou “autocondensação”
A ≠ B : “Condensação mista” ou “condensação cruzada”

Um carbono positivado (= eletrófilo) reage com um carbono carbanóide (= nucleófilo).
O grupo carbonila pode providenciar os dois componentes:
CH
O
C
OH
Tautomeria
Base, - H+
C
O-
MesomeriaC
O
Enolato
Enol
Carbono nucleofílicoem posição αao grupo carbonila
Carbono positivado:Efeito do oxigênio –I >> +M
C
O
δ+

TautomeriaDeslocamento reversível de um próton e uma ligação π ao mesmo tempo.Não é uma mesomeria, mas sim, um verdadeiro equilíbrio entre dois compostos isoméricos.
Tautomerias são bastante comuns na química orgânica•ceto - enol •azometina (= imina = base de Schiff) – enamina•amida (ou lactama) – ácido imido carboxílico•endiol - α-hidroxicetona (na vitamina C)•nitro - aci•nitroso – oxima (aldoxima ou cetoxima) •ácido hidroxâmico – carbiminodiol

Formas da catálise da tautomeria ceto-enólica
R CH C
H O
R CH C
OR CH C
O
R CH2 C
OHR CH2 C
OH
R CH C
OH
E
δ+
δ−
+ H+
- H+
- H+
- H+
- H+
+ H+
+ H+
+ H+
forma ceto forma enol
Cátion carbênio (oxônio)
Ânion enolato
BaseNu-
H+(ou E+)
Base
E+

O enolato e também o enol são carbanóides em posição α ao grupo C=O. São sujeitos de ataque por eletrófilos.
O equilíbrio ceto-enol é acelerado por ácidos e por bases. Em qualquer caso a tautomeria passa por uma forma intermediária carregada. Portanto, a polaridade do ambiente (solvente, teor de água, presença de sais) acelera esta reação.
Em compostos carbonilados simples a forma ceto édominante. Acetona: apenas 0,00025% em propenol.
Comprovantes para a forma enólica:�A espectroscopia r.m.n. revela uma posição especial dos sinais 1H (δ de até17ppm)�Deslocamento das bandas de absorção no espectro infra vermelho�O composto carbonilado troca H por D em posição α, ao colocá-lo em D2O ou C2H5OD
�Pode-se adicionar bromo na dupla ligação da forma enólica

A velocidade da troca de hidrogênio, entre oxigênio e carbono α é bastante baixa. Titulação com Br2: A reprodução do enol pelo equilíbrio dinâmico dentro do tempo experimental édesprezível, desde que se trabalha abaixo da temperatura ambiente.
CH C
O H
Br Br
- Br-- H+CH
Br
C
O H
CH
Br
C
O
CH2 C
O+ Br2
forma enólica:
forma ceto:
Titulação

Observação: no ácido carboxílico também pode-se discutir uma tautomeria. Porém, a adição de bromo não funciona por que a densidade eletrônica deste “enol” é muito baixa. Portanto, a α-bromação precisa da presença de um catalisador.
(Reação de Hell-Volhard-Zelinsky)

O que estes três compostos têm em
comum?
OHOH OAc
NH2
O
OOHOH OAc
NH2
O
O
11
12
2 233 3
Grupos funcionais nos carbonos 1 e 3.

O alvo 1,3-difuncionalRetro-síntese (símbolo ⇒):
1. Analisar o esqueleto carbônico da molécula alvo (TM = Target Molecule)
2. Transformar todos grupos funcionais em oxigênios de NOX certo usando transformações entre grupos funcionais (FGI = Functional GroupInterconversion) ou desconexões C-X.
3. Desconectar dois carbonos do esqueleto, geralmente os mais ramificados e mais centralizados, usando uma reação de condensação.

•1,3-bifuncionais e compostos carbonilados α,β-insaturados (= sistema de Michael)•1,5-bifuncionais•1,2-bifuncionais•1,4-bifuncionais•1,6-bifuncionais
1,3 e 1,5 podem ser feitos a partir de sintons com polaridade natural.
R1
O
R2
OH
R1 CH2
O
R2
OH
+
R1
O
CH2
Alvo 1,3-bifuncionalAldol Aldeído (protonado) =
sinton eletrófilo
Enolato = sinton nucleófilo

Sintons naturais e reagentes para condensações
Nucleófilo: enolato ou enol
R
O
X
X = H, OEt, Alq, Ar
Sinton Reagente
R
O
X

Eletrófilo:
X = Cl, OEt
Sinton Reagente
RCH
OH
RCH
O
R1
R2
OH
R1
R2
O
RC
O
RC
X
O

Sinton Reagente
O
R
O
R
Sistema conjugado(sistema Michael)
Sintons não-naturais que levam aos alvos 1,2 e 1,4-difuncionais:
R
O
R
O
R
O
⇐ Alvo 1,5-difuncional

Segue: Tabela de ácidos C-H e as bases necessárias para sua desprotonação
Fonte: S. Warren, Organic Synthesis - The Disconnection Approach, Wiley & Sons, Chinchester 1982, p. 143
Nucleófilo: Enolato, ou mais em geral, um carbono desprotonado.


Compostos 1,3-difuncionais: �1,3-dicarbonila�β-hidroxicarbonila (= aldol) �Carbonila α,β-insaturada
Nucleófilo em todos: enolato
vêm da acilação do enolato
→ Condensação de Claisen
Catalisador = base (muitas vezes EtO-); Reagente acilante (= eletrófilo): éster ou cloreto do ácido carboxílico
1,3-Dicarbonilas

Exemplos
• Como fazer
OEt
O O
?

C
O O
OEt
Alvo
a
ba
b
O
C
O
OEtEtO+
OEt
O
C
O
OEt
Base
CC
OO
OEtEtO
Éster adípico
ruim: separou dois fragmentos desiguais
bom: só uma molécula de partida, simétrica = limpociclização, intramolecular = fácil, rápido

Exemplo 2: inclui decomposição do composto 1,3-dicarbonila
R C
O
OEt EtO-
R
CO OEt
O
R2 x
hidróliseH2O / OH-
descarboxilação H+,
R
O
R
∆
Rota padrão para cetonas simétricas.
Excurso:Como funciona a descarboxilação térmica?1) Liberar o ácido carboxílico livre (= hidrólise do éster)2) Aquecer e desprender o gás carbônico.

O
CC
C
OH
O
- CO2
C
OH
C C
O
C H
Enol
∆
Regra geral:Um ciclo de 5 ou 6 membros é uma conformação favorável. Caso o estado de transição pode ser formulado assim,A energia de ativação é baixa, então a reação é fácil e rápida!

β-hidroxicarbonila
eletrófilo = aldeído ou cetona
Mais reativo é o aldeído (Porquê? Duas respostas certas.)
Exemplo: • Como fazer
OHO
?

O HO
Alvo
O HO
Alvo
12
3O HO
Alvo
12
3
O O
+
Síntese: O
Alvo2Base

Base: Ba(OH)2 = insolúvel; retirada imediata do alvo da zona reativa
A condensação é realizada favoravelmente no extrator Soxhlet:

Remoção de um dos produtos = Técnica mais importante
de levar reações equilibradas até altos rendimentos!
Controle termodinâmico
Em condensações:
1. Tirar H2O (subproduto de enonas, feitas por condensação aldólica, cat. ácida ou por condensação de Knoevenagel)
2. Desprender CO2 , a partir de intermediários β-cetoácidos e 1,3-diácidos
3. Depositar o produto 1,3-dicarbonila em forma de complexo num cátion duro:
O OBase
O O
Sal, insolúvel

Composto carbonilado α,β-insaturado
É muito fácil a desidratação da β-hidroxicarbonila (= aldol):
1) o próton a ser removido em posição α é enólico
2) o produto é estabilizado por conjugação (∆H (conjugação) ≈ 15 - 17 kJ⋅mol-1)
OH
CH
O
R
H
H+OH2
CH
O
R
H- H2O
- H+
CH
O
R

Reação anterior, esta vez sob catálise ácida:
não pára na β-hidroxicarbonila
H3C
C
O
CH C
CH3
CH3
"Óxido de mesitila"
2 H3C C
O
CH3
[ H+ ]
- H2O
Análise: O
O
O
+

Outro exemplo: O O
O
Onde quebrar?
O O
O
OO
O
O
+

Condensações mistas
Nucleófilo (= componente metilênico = pseudo-ácido) é diferente do eletrófilo (= carbonílico = aceitador).
Objetivo principal é direcionar as reatividades, isto é, marcar o papel do eletrófilo em um composto carbonilado e do nucleófilo, seletivamente, em outro composto carbonilado.
Assim se forma somente um produto.
Problema: Os dois compostos carbonilados têm reatividades muito semelhantes!

PhO
Alvo problema
PhO
PhO
+CHO
CH3
Alvo problemaSíntese não funciona bem!
1. Quem enoliza?
2. No caso da cetona, em que lado enoliza?
3. Quem é eletrófilo?
(Esses problemas se anulam ao usar dois componentes simétricos e/ou idênticos.)

As reatividades como nucleófilo e eletrófilonão se opõem, mas geralmente aumentam juntos:
Amida Éster
Cetona Aldeído
Anidrido Cloreto de acila
Eletrofilia
Enolizabilidade

Régio-seletividade em cetonas assimétricas
Controle cinético vs. Controle termodinâmico
Exemplo: autocondensação da etilmetilcetona
O Base
ÁcidoOH
O O OH
O
OH
O
O
Ambiente básico Ambiente ácidoVia única Sistema equilibradoTemperatura baixa Temperatura altaProduto mais rápido Produto mais estável
Porém:A escolha das condições certas é delicada! Melhor usar uma das...

Reatividade do grupo carbonila:Construção do esqueleto carbônico
por condensacondensacondensaçççõesõesõesParte II
Técnicas de manipulação das reatividades em
CondensaCondensaçções Mistasões Mistas

Técnicas de manipulação das reatividades em condensações mistas
1. Utilização de um pseudo-ácido com baixa tendência de condensação consigo mesmo e/ou um aceitador sem α-H.
2. Desativação do pseudo-ácido por formação de uma imina.
3. Ativação do pseudo-ácido pela estabilização da forma enólica (enoléter / enamina).
4. Uso de um pseudo-ácido duplamente ativado (= condensações de Knoevenagel).
5. Com bases muito fortes se consegue desprotonar o pseudo-acido
quantitativamente em uma etapa preliminar.
Nitroalcanos são bastante usados em condensações mistas: só podem ter o papel do pseudo-ácido.

Técnica 1: usar compostos carbonilados não-enolizáveis
R1
O
R2R1,2 = H, OEt, Cl, Ar, Bu-t, COOEt
Exemplos:
O
H EtO
O
Cl
O
EtO
O
OEt

Condensação de Claisen-Schmidt
Cetona + aldeído aromático
H3C C CH2
O
+
O
C
H
ArH+
- H2O
H3C C CH2
O
CH
O-
Ar H3C C CH2
O
CH
OH
Ar
H3C C CH
O
CH Ar
Cetona α,β− insaturada
Estratégia principal para os derivados do ácido cinâmico.

Síntese do ácido cinâmico segundo Perkin
Anidrido + benzaldeído
Base: AcetatoSem solvente adicional o acetato age como base forte!(em soluções aquosas, por outro lado, os sais de ácidos carboxílicos são bases fracas, devido a camada de solvente em volta dos ânions.)
H3C C O C
O O
CH3
+ AcO-
- AcOH
O
C
H
C6H5
H3C C O C
O O
CH2C6H5 CH
CH2
CO
OC
OH3C
O-
- AcOHC6H5 CH CH COO-
Sal do ácido cinamômico-Eliminaçãoβ
C6H5 CH
OC
CH
H
O
H3C
C
O-
O
∆

Condensação com o éster do ácido fórmico
Ph C
O
CH3 + H C
O
OC2H5
Aceitador Acetofenona= pseudo-ácido
[ C2H5O- Na+ ]
Ph C
O
CH2 CH O + HOC2H5
Ph C
O
CH2 CH
O-
OC2H5

Condensação com o éster do ácido oxálico
O éster do ácido oxálico, além de não ter um grupo metileno em posição α, é um aceitador mais forte do que o éster simples. Isto se deve à presença do segundo grupo acila em posição vizinha = retirador de elétrons. Assim o carbono do grupo carboxílico fica mais positivado (= mais eletrofílico) ainda. Portanto, ele pode ser usado em condensações mistas com cetonas ou outros ésteres – o produto sempre fica definido.
H3C CH2 C
O
OC2H5
+ C2H5O C
O
C
O
OC2H5
AceitadorPseudo-ácido
+ C2H5O- Na+
- C2H5OH CH C
O
C
O
OC2H5H3C
COOC2H5

Ao aquecer o produto primário desta síntese acima de 100 °C acontece uma descarbonilação(essa degradação é típica somente para os compostos 1,2-dicarbonilados).
Produto: Derivado do ácido malônico 2-substituído
Esta estratégia serve para produzir β-oxoéstere ésteres malônicos mono-substituídos - que não podem ser obtidos de maneira satisfatória via alquilação do próprio éster malônico. (Porquê?)
CH C
O
C
O
OC2H5H3C
COOC2H5
120 °C - CO
Éster 2-metilmalóico
H5C2O C
O
CH
CH3
C
O
OC2H5

Condensação com o éster do ácido carbônico
Com cetonas para β-oxoésteres, com nitrilas para ésteres do ácido cianoacético.
Atenção: os ésteres do ácido carbônico são cancerígenos!
+ C2H5O- Na+
- C2H5OH
O
α + C2H5OC
O
OC2H5
δ+
OH
COC2H5
ODietilcarbonato

Outras condensações mistas com ésteres
Mais sensível às condições reacionais (solvente, temperatura, base), mas também viável é a maioria das condensações mistas entre ésteres e cetonas e entre ésteres e nitrilas. Nestes casos, o éster tem predileção de ser o componente carbonílico:
R1 C
O
OR2
R3 CH2C
R4
O
R1
C
O
CH
R3
C
O
R4+ R2OH
-dicetona
R1 C
O
OR2
R3 CH2 C NR1
C
O
CH
R3
CN
+ R2OH
-oxocarbonitril
β
β

Restrições:Rendimentos típicos em todas essas condensações: cerca de 50%. Porquê?
O produto da condensação é quase sempre um composto duplamente ativado (= 1,3-dicarbonilado) ⇒ reagem com muita facilidade como pseudo-ácidos.
Consequência: desprotonação, condensação com mais um componente carbonílico, “supercarbonilação”.
Para reprimir esta reação paralela deve-se usar:1) pequena quantidade de base (no exemplo acima: etóxido)2) evitar um excesso do componente carbonílico na mistura

Além disso:Todos os compostos 1,3-dicarbonilas podem sofrer “clivagem de ácido”(= reversa da condensação; ocorre sob catálise básica)
e
“clivagem de cetona”(= descarboxilação de ácidos carboxílicos β-substituidos),
especialmente durante um tratamento prolongado a temperaturas altas e sob influência de uma base (que é essencial na condensação de Claisen).

Caso especial: Formaldeído
• F. é reativo demais• podem ligar-se mais de um F. (= aceitador) ao componente metilênico• F. polimeriza• F. disproporciona aos poucos (Cannizzaro)
Muitas desvantagens, mas existe uma condensação mistaque aproveita da reatividade do formaldeído:
Síntese do pentaeritritol
OH
OH
HO
HO

Síntese de pentaeritritol
Acetaldeído + formaldeído + base
H3C C
O
H
+ H2C O
Aceitador(excesso)
[ Base ] + B-
- HB
H2C O
- HCOOH
CH2
CH2
C
O
H
HOCH
CH2
C
O
H
HO
H2C O(HO CH2)3C C
O
H RedoxPentaeritrita
Pseudo-ácido
Base
C
CH2OH
CH2OHHOCH2
CH2OH

Existe um N-derivado do F. ainda mais reativo!
Aminometilação de Mannich
R C
O
CH3H+
+ H+
- H2O
+ Base
R C
OH
CH2
H3C
NH + H2C
H3C
O
H3C
N
H3C
CH2
δ−
δ+
R C
O
CH
H
CH2 N
CH3
CH3
-EliminaçãoβCiclo catalítico - (CH3)2NH
R C
O
CH CH2
Base de Mannich
∆"Imônio"

Além dos pseudo-ácidos mencionados podem também ser usados compostos aromáticos que são ativados no sentido substituição eletrofílica. Isto podem ser fenóis ou hompostos heterocíclicos. Um exemplo seja a síntese da gramina, por aminoalquilação de indol:
N
HIndol
+ HCHO + HN(CH3)2- H2O N
H
CH2 N(CH3)2
Gramina
Todas as etapas reacionais ocorrem sob condições extremamente brandas. Algumas condensações de Mannich funcionam sob condições “in vivo”!

• A base de Mannich pode ser isolada e armazenada, especialmente em forma do seu hidrocloreto.
• Quando for preciso, a base de Mannich pode ser degradada formandocetona α,β-insaturada com grupo metileno final que é
• muito reativa e instável; difícil preparar por outro método (Wittig) (= eletrófilo em uma adição de Michael ou dienófilo em ciclizaçõesde Diels-Alder,...)
• A degradação é bastante facilitada pela quaternização do N da basede Mannich. Assim sai uma amina terciária.
(= variação da eliminação de Cope).
O
CH2
H
NR2
MeI
Quaternização
O
CH2
H
MeNR2
- MeNR2
O
CH2
Grupo metilenoexocíclico

Técnica 2: Desativação do componente metilênico por aminas primárias
(Wittig 1964)Aldeído + amina primária + base forte + cetona
A transformação do aldeído em uma base de Schiff reduz drasticamente a sua capacidade como componente carbonílico. Esta função agora pode ser preenchida por uma cetona que em si não é tão eletrofílica.
Etapa 1: desprotonar a base de Schiff = ativar o componente metilênico.
Etapa 2: adição do eletrófilo (que pode ser fraco).
H3C CHO + H2N H3C CH N+ LDA
- LDAH+
1) (C6H5)2CO
2) H2O
- LiOH
+ H+ / H2O
- C6H11 NH2
H2C CH N
C6H5
C
OH
C6H5 CH2 CH N
- H2O
(C6H5)2C CH CHO

Técnica 3: Estabilização da forma enólica / enolatoPrincípio: Estabilizar o enol = fortelecer o papel metilênico do composto
Enolálcool = instável ⇒ Transformação do componente nucleofílicoem enamina ou (silil)-enoléter
Papel da amina secundária / do cloreto de silila: Ativadores!O
Me3SiClO
SiMe3
Amina sec.
NR2
Nucleófilo ruim!
Enamina Silil-enoléter

Exemplo 1:Componente metilênico: cetona; componente carbonílico: aldeído.
O
NH
O
H+
N
O
CHO
N
O
Hidrólise
O
Morfolina= amina sec.

Exemplo 2:Silil-enoléteres reagem com alta regiosseletividade e sob condições brandas, na presença de ácidos de Lewis (neste exemplo: TiCl4).
C6H5 CH2 C
O
H
+
O Si(CH3)3
δ−δ+
+ TiCl4
- (CH3)3SiCl
H2OC6H5 CH2 CH
OOTiCl3
C6H5 CH2 CHOOH
68%
(Segundo efeito do grupo trimetilsilil: hidrofobização.)

Técnica 4: Pseudo-ácidos com grupo metileno duplamente ativado (Condensação de Knoevenagel)
H2C
retirador 1
retirador 2
Dois vizinhos retiradores de elétrons ⇒ elevada acidez C-H, pois a carga negativa da base conjugada pode ser distribuída, através de mesomeria, em cima dos dois vizinhos.
Três efeitos contribuem à reatividade elevada deste grupo metileno:1) O grupo –CH2 – torna-se muito mais ácido:
pKa(carbonila simples) ≈ pKa(1,3-dicarbonila) - 10.
2) A troca entre a forma ceto e enólica é acelerada.
3) Mais estabilidade termodinâmica do enol, então mais enol no equilíbrio.

Ponto 1 e 2: Acidez C-H e rapidez da enolização
R3C Hk1
k -1R3C + H+ Ka =
k1
k -1
Composto duplamente ativado pKa k1 (s-1)
H2C(NO2)2 Dinitrometano 4 8,3 ⋅ 10-1
H2C(COCH3)2 Acetilacetona 8,8 1,7 ⋅ 10-2
H3C-NO2 Nitrometano 10,2 4,3 ⋅ 10-8
H3CCO-CH2-COOC2H5 Acetoacetato de etila 10,7 1,2 ⋅ 10-3
H2C(CN)2 Dinitrila do ácido malóico 12 1,5 ⋅ 10-2
H2C(COOC2H5)2 Dietilmaloato 13,3 2,5 ⋅ 10-5
Em comparação:H3C-CO-CH3 Acetona 20 4,7 ⋅ 10-10
Todos esses compostos (menos a acetona) podem ser desprotonados, quase quantitativamente, por NaOH. (pKa da água (ácido do OH-) ≅ 15).

Possíveis retiradores ao lado do grupo –CH2 – :
� carbonila
� carboxila (ácido, éster, amida)
� ciano
� nitro
� também apóiam o carbânion: sistemas aromáticos

3a Explicação da alta reatividade como pseudo-ácido: A forma enólica é estabilizada, podendo até prevalecer no equilíbrio com a forma ceto.
Isso se deve à ponte de hidrogênio intramolecular:O
CCH
C
OH
Forma enólica do composto dicarbonila
O
CCH2
C
O
Composto % enol (na fase líquida, ausência de solventes)
H3C-CO-CH3 Acetona 1,5 ⋅ 10-4
H2C(COOC2H5)2 Dietilmalonato 7,7 ⋅ 10-3
NC-CH2-COOC2H5 Cianoacetato de etila 2,5 ⋅ 10-1
C6H10O Ciclohexanona 1,2
H3CCO-CH2-COOC2H5 Acetoacetato de etila 8,0
H3CCO-CH(C6H5)-COOC2H5 30,0
H2C(COCH3)2 Acetilacetona 76,4
C6H5CO-CH2-COCH3 Fenilacetilacetona 89,2

O teor da forma enólica depende da temperatura e sensivelmente do solvente. Observa-se a estabilização da forma enólica por solventes apolares. (O efeito de desaceleração por estes solventes é desprezível)
Acetilacetona:
Solvente % enol
H2O 15
CH3CN 58
sem solvente, em fase líquida 80
C6H14 92
sem solvente, em fase gasosa 92

Os seguintes compostos têm relevância na síntese:
N C CH2 C N
C2H5 O C
O
CH2 C N
C2H5 O C
O
CH2 C
O
OC2H5
C6H5 CH2 C N
H3C C CH2
O
C
O
CH3
H3C C CH2
O
C
O
OC2H5
(RO)2P
O
CH2 C
O
OR
Dinitrila do ácido malônico
Nitriloacetato de etila
Dietilmaloato
Cianeto de benzila
Acetilacetona
Acetoacetato de etila
Éster do ácido fosfônico
O último composto tem importância na síntese de olefinos a partir de compostos carbonila, segundo Horner e Emmons, variação da reação de Wittig.
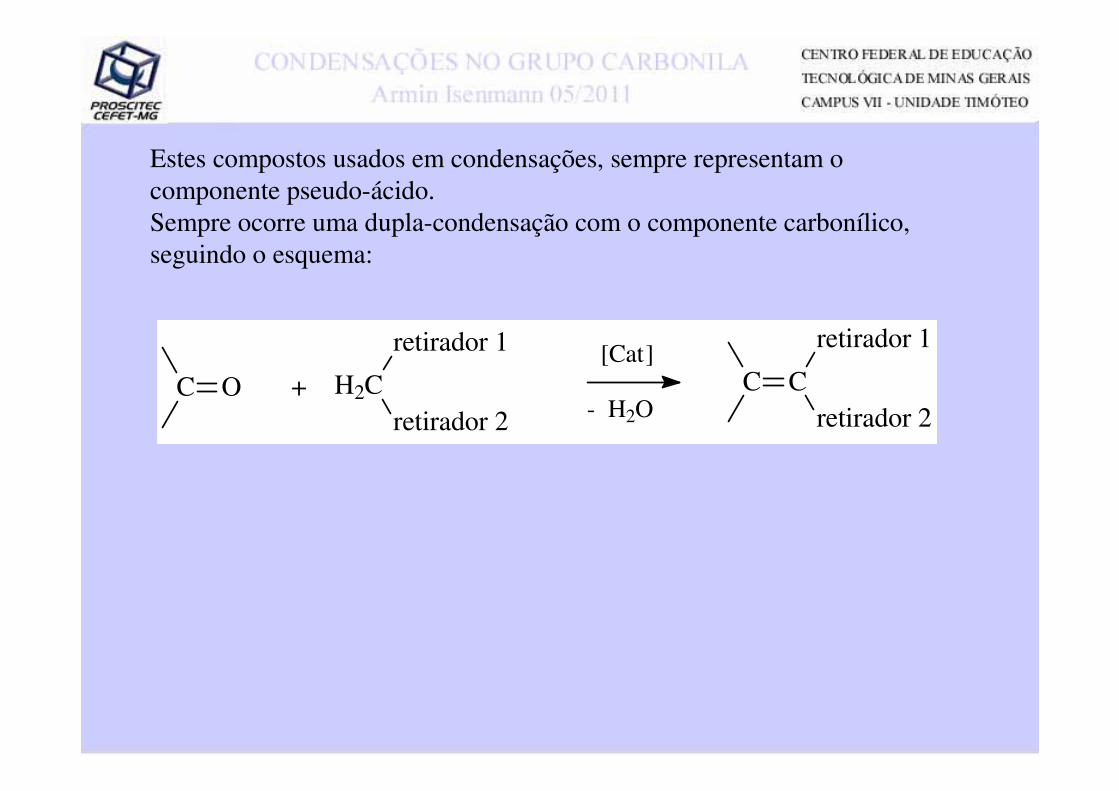
Estes compostos usados em condensações, sempre representam o componente pseudo-ácido. Sempre ocorre uma dupla-condensação com o componente carbonílico, seguindo o esquema:
C O + H2C
retirador 1
retirador 2 - H2OC
retirador 1
retirador 2C
[Cat]

Em comparação:
Condensação aldólica verso condensação de Knoevenagel
1) A condensação de Knoevenagel é uma condensação mista, portanto o espectro de produtos acessíveis é muito mais amplo.
2) A escolha do catalisador é mais sútil: enquanto na condensação aldólica qualquer ácido ou base de Brønsted serve, a reação de Knoevenagel usa catalisadores brandas e específicas, tais como acetato de amônio, β-alanina, piridina ou piperidina *.
* G. Jones, Org.Reactions 15 (1967) 204

Exemplo 1: Condensação com éster malônico
X = Y = C
O
OC2H5
H5C2 CH C
COOC2H5
COOC2H5
[ H+ ]
- CO2H5C2 CH C
COOH
COOH
H5C2 CH CH COOH∆
Comp. metilênico: Comp. carbonílico: H5C2 CHO
Shortcut!Usar diretamente o ácido malônico, em vez do seu éster. A descarboxilação ocorre automaticamente.Catalisador: HOAc / NaOAc = 1 : 1; pouco DMSO ajuda solubilizar o sal
Não pode ser ácido forte nem base forte, mas o mais suave possível, para promover a reação mais rápida (controle cinético)
Solvente: benzeno ⇒ remoção azeotrópica da água liberada. Dean Stark

Estratégia de síntese:Um grupo éster (ácido carboxílico) somente tem o papel de ativador (= oposto de
protetor), para reforçar o papel do componente metilênico.
Exemplo 2:Condensação com acetoacetato de etila
X = C
O
OC2H5
Y = C
O
CH3
H3C CH C
COOC2H5
C
O
CH3
[ H+ ]
- CO2H3C CH C
COOH
C
O
CH3
H3C CH CH C CH3
O
Cetonas α,β− insaturadas
∆

Exemplo 3:Alvos 1,3-dicarbonilas, via acilação do éster malônico ou derivados
Base: Mg(OH)2 : Mg2+ complexa e bloquéia os centros duros (= oxigênios)!
O- O
OEt
Nu- macio
Nu- duro
R COCl
E+ duro
O- O
OEt
Na+
R COCl
R COClO O
OEt
Mg
OH
O O
OEt
O
R
O O
OEt
OR
Hidrólise
DescarboxilaçãoR
O
O

Técnica 5: Desprotonação quantitativa do pseudo-ácido
A desprotonação completa e rápida de um composto carbonilado impede sua atividade como eletrófilo (= componente carbonílico)!
� Na maioria dos casos: nucleófilo = composto com grupo metileno duplo-ativado (discutidos no último item) a ser desprotonado com bases comuns (alcóxidos).
� Em outros casos: base bastante forte (NaH ou Ph3C- Na+)
� Formação quantitativa do enolato numa etapa prévia (controle mediante indicador!)
� Depois acrescentar o componente carbonílico, aos poucos.
� Aplicar os participantes, pseudo-ácido : base : eletrófilo = 1 : 1 : 1.
Rendimentos mais altos do que as condensações mistas “comuns”.

Atenção!
Falta de base ⇒ condensação do composto 1 entre si mesmo. Isto é, uma parte funciona como componente metilênico, outra parte como componente carbonílico.
Excesso de base ⇒ condensação do composto 2 entre si mesmo, pela mesma razão.

Condensação de Stobbe
Éster succínico + aldeído / cetona + base forte
CH2
CH2
COOEt
COOEt
(CH3)3CO- K+R2C O
H+ / H2O
- CO2
CH
CH2
COOEt
COOEt
CH
H2C
C
CR2
O-
CO
EtO
O OEt
CR2
O
COOEt
H
O
COOH
C
CH2
COOH
CR2
Lactona
R2C CH CH2 COOH
Ácidos β,γ− insaturados1)
2)
COOEt
C
CH2
COO-
CR2 H+ / H2O
1)
2)R2C C
COOH
CH3
Base
∆
Enolatoquantitativo

Síntese do éster glicídico, segundo Darzens
Cloracetato + aldeído + alcóxido (base)
Cl CH2 COOCH3+ CH3O-
- CH3OH
R C
O
H
- Cl-H2O / OH- H+
- CO2
Cl CH COOCH3
R
CH
O-CH COOCH3
Cl
R
CH
O
CH COOCH3
Éster glicídico
R
CH
O
CH COO-
R
CH
O
CH
C O
OH
R
C
H
C
H
OH
Enol
R CH2 C
O
H
∆
Resultado: prolongação da cadeia carbônica do aldeído por um grupo metileno.

Síntese de Reformatzky
Bromoacetato de alquila + composto carbonila + zinco
Br CH2 COOCH3 + Zn [ Br Zn CH2 COOCH3 ] CH2 COOCH3ZnBr +
quantitativo!
R C
O
H
CH2 COOCH3+ R C
H
O-
CH2 COOCH3
[ H3O+ ]
- H2O
R C
H
OH
CH2 COOH
R CH CH COOH
Outras sínteses que aproveitam deste efeito: quase todas as reações organo-metálicas – especialmente aquelas com metais muito eletropositivas (preparo do reagente de Grignard: R-X + Mg → R-MgX).
Inversão da polarização no carbono α

FIM !!!!
Leia mais, no capítulo 6: Condensações, do livro "Princípios da Síntese Orgânica"
Recommended














![Adição Nucleofílica na Carbonila (impressão) [Modo de Compatibilidade]](https://img.pdfslide.net/doc/110x75/54f7d2724a7959303c8b490c/adicao-nucleofilica-na-carbonila-impressao-modo-de-compatibilidade.jpg)

