
Stabilization of poly-L-lysine/DNA polyplexes for in vivo gene delivery tothe liver
Deborah Y. Kwoh 1;*, Christopher C. Co¤n, Charles P. Lollo, Jocelyn Jovenal,Mariusz G. Banaszczyk, Patricia Mullen, Alison Phillips, Arjang Amini, Joanne Fabrycki,
Richard M. Bartholomew, Steven W. Brosto¡, Dennis J. CarloGene Therapy Department, The Immune Response Corporation, 5935 Darwin Court, Carlsbad, CA 92008, USA
Received 26 August 1998; received in revised form 7 December 1998; accepted 8 December 1998
Abstract
We are developing a self-assembling non-viral in vivo gene delivery vehicle based on poly-L-lysine and plasmid DNA. Wehave characterized poly-L-lysines of different chain lengths for DNA condensation and strength of DNA binding. Poly-L-lysine chains s 20 residues bound DNA efficiently in physiological saline, while shorter chains did not. Attachment ofasialoorosomucoid to PLL increased the PLL chain length required for efficient DNA binding in saline and for efficientDNA condensation. By electron microscopy, poly-L-lysine/DNA polyplexes appeared as toroids 25^50 nm in diameter orrods 40^80 nm long; conjugation of asialoorosomucoid to the polylysine component increased the size of resulting polyplexesto 50^90 nm. In water, poly-L-lysine and asialoorosomucoid^PLL polyplexes have effective diameters of 46 and 87.6 nm,respectively. Polyplexes containing only poly-L-lysine and DNA aggregated in physiological saline at all charge ratios andaggregated at neutral charge ratios in water. Attachment of asialoorosomucoid lessened, but did not eliminate, theaggregation of PLL polyplexes, and did not result in efficient delivery of polyplexes to hepatocytes. Conjugation ofpolyethylene glycol to poly-L-lysine sterically stabilized resulting polyplexes at neutral charge ratios by shielding the surfaces.For efficient in vivo gene delivery, polyplexes will need to be sterically stabilized to prevent aggregation and interaction withserum components. ß 1999 Published by Elsevier Science B.V. All rights reserved.
Keywords: Polyplex; Poly-L-lysine; Asialoorosomucoid; Polyethylene glycol ; DNA condensation; Steric stabilization
1. Introduction
In vivo gene therapy has been achieved with viralvectors, albeit with limited success. Even thoughthese vectors were designed speci¢cally for gene ther-apy, they produce unacceptable immune responses tothe viral structural proteins when delivered at doses
required for successful gene therapy [1]. Viral systemsalso have the potential for generating replication-competent virus by recombination of vector se-quences with either endogenous viral sequences inthe host or sequences in viral packaging systems.These systems also pose large-scale controlled man-ufacturing challenges [2,3]. For these reasons, non-viral vectors composed of non-immunogenic, self-assembling components are attractive alternatives toviral systems for gene therapy. Several such systemsare currently under development including anionicor cationic liposome/DNA complexes (lipoplexes),
0167-4781 / 99 / $ ^ see front matter ß 1999 Published by Elsevier Science B.V. All rights reserved.PII: S 0 1 6 7 - 4 7 8 1 ( 9 8 ) 0 0 2 7 4 - 7
* Corresponding author. Fax: +1 (760) 431-8636;E-mail : [email protected]
1 Present address: 2404 Jacaranda Ave. Carlsbad, CA 92009,USA.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
Biochimica et Biophysica Acta 1444 (1999) 171^190

cationic polymers or peptides/DNA complexes (poly-plexes) and naked DNA [4^15].
We have been developing a stable parenteral poly-plex gene delivery vehicle based on the self-assemblyof poly-L-lysine (PLL) and plasmid DNA that is tar-geted to the liver [16]. Attachment of asialoorosomu-coid (AsOR) to PLL has been used to direct genedelivery to hepatocytes via asialoglycoprotein recep-tors (ASGPr) [17,18]. The in vivo e¤ciency of thepolyplex delivery system is dependent upon its abilityto overcome several extracellular and intracellularbarriers that hinder transport to hepatocyte nuclei.Extracellular barriers include the innate immune sys-tem of the host (opsonization and complement acti-vation), Kup¡er cells (resident macrophage) in liversinusoids, the limited size of fenestrations betweenendothelial cells within sinusoids, and ¢nally, the ex-tracellular matrix in the space of Disse [3,19,20]. Fol-lowing binding to hepatocyte receptors, polyplex up-take may be restricted by the size of endosomes(V100 nm) [21]. Once internalized, polyplexes mustescape the endosome prior to endosome/lysosomefusion and traverse the cytoskeletal matrix to reachthe nuclear membrane where polyplexes must then beactively transported through the outer and innermembrane. Finally, the transgene must be tran-scribed and translated to produce signi¢cant levelsof the therapeutic protein [3,22^24].
We have made improvements in the formulation ofPLL and AsOR^PLL polyplexes over those reportedby Wu and coworkers which led to improved report-er gene expression in vivo after intravenous injectionof polyplexes into mice (2000 ng luciferase/g liver)[16]. The improved formulation resulted in less ag-gregation of the starting components and therebyproduced polyplexes which were more soluble. Bio-distribution studies with labeled AsOR^PLL poly-plexes prepared using the enhanced formulationdemonstrated that 67% of total label (90% of therecovered label) was delivered to the liver after intra-venous injection in a 0.2-ml volume. With larger in-jection volumes, greater than 99% of the reportergene expression occurred in the liver. However, ex-pression diminished with decreasing injection vol-umes and was undetectable when polyplexes wereinjected in 0.2 ml. In vivo gene expression also de-creased when polyplexes were mixed with mouse se-rum prior to injection, even when large injection vol-
umes were used. These results indicated that thestability of polyplexes was adversely a¡ected by in-teractions with blood components and possibly re-sulted in uptake by the reticuloendothelial systemssuch as the non-parenchymal cells within the liversinusoids.
Polyplex interactions with serum and cells aremost likely determined by the polyplex size, shapeand surface characteristics. Plank and coworkershave shown, using in vitro assays, that PLL poly-plexes can activate serum complement [25]. The de-gree of complement activation was dependent onPLL chain length and on the (+/3) charge ratiosof the PLL polyplexes. Longer chained PLL andpolyplexes with large (+/3) charge ratios producedmore activation. Longer-chain PLLs also show great-er cytotoxicity both in vitro [26] and in vivo (unpub-lished results). Although many successful in vitrogene expression studies use polyplexes with high (+/3) charge ratios, most studies reporting successfulgene expression in vivo utilized polyplex composi-tions that had a calculated negative net charge[16,17,27^29].
Particle shape, deformability and surface charac-teristics may help de¢ne the polyplex size limitationsfor e¡ective delivery to hepatocytes. Accordingly,smaller polyplexes are expected to pass more easilythrough hepatic fenestrations (100^200 nm) and beinternalized via receptors into clathrin-coated pits(100 nm) [3]. The small size of viruses is, in part,due to the compaction of their genomes. Virusescan achieve unimolecular DNA condensation withinthe con¢nes of a small de¢ned shell (e.g. SV40 andpolyoma with 45-nm virions and 5243-base pair,double-stranded DNA genome) [30]. In vitro, how-ever, DNA is condensed in a multimolecular fashionby polycations, such as polyamines. DNA of s 400base pairs is condensed into 35^100 nm coacervatesirrespective of condensing agent [31^34]. Bloom¢eldand coworkers suggest that DNA molecules of ap-proximately 40 kilobases (kb) in length are needed toobtain unimolecular polyplexes, whereas multiplemolecules of DNA shorter than 40 kb are requiredto achieve condensation [33,35]. Most plasmids de-veloped for gene therapy are currently in a rangefrom 5 to 10 kb, and thus multiple DNA moleculesare expected to condense into one PLL or AsOR^PLL polyplex. Perales and coworkers, however, have
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190172

reported unimolecular condensation of short plasmidDNA using galactosylated-PLL (V50 kDa poly-L-lysine, 236-mer) and high concentrations of NaCl[36]. Also, 30 nm PLL/DNA polyplexes have beendescribed recently for plasmid DNAs using smallerPLL 4K (4 kDa poly-L-lysine) while longer PLLchains produced larger polyplexes [26]. Conjugationof a ligand to the PLL moiety may also increase thesize of the resulting polyplexes. Wagner and co-workers have reported that transferrin^PLL (200-mer) polyplexes form 80^100 nm toroid (donut-shaped) particles when prepared by direct mixing ofconjugate and DNA at low concentrations (3 nMDNA) [37].
Using a systematic approach to the developmentof a stable in vivo gene delivery system, we haveexamined whether the size of the PLL used inAsOR^PLL or PLL polyplexes alters DNA bindingand condensation. The size and stability of poly-plexes made with the di¡erent sizes of PLL havebeen characterized both in water and at physiologicalionic strength. We have also examined how the poly-plex biophysical characteristics are altered after con-jugation of PLLs to an AsOR ligand or with poly-ethylene glycol. The relationship between in vivodistribution of the polyplexes and the biophysicalcharacteristics is examined.
2. Materials and methods
2.1. Reagents
Protamine, poly-L-lysine (4, 10, and 26 kDa; meanMW) and ethidium bromide (EtBr) were purchasedfrom Sigma, St. Louis, MO. 1-[3-(Dimethylamino)-propyl]-3-ethylcarbodiimide (EDC) was purchasedfrom Aldrich, Milwaukee, WI. Synthetic poly-L-ly-sines were purchased from Research Genetics(Huntsville, AL) or Dr. Schwabe (Protein ChemistryFacility at the Medical University of South Caroli-na). Polyethylene glycol (PEG) derivatives were pur-chased from Shear-Water Polymers (Huntsville, AL).Orosomucoid (OR) was purchased from Alpha Ther-apeutics, Los Angeles, CA. AsOR was prepared fromorosomucoid (15 mg/ml) by hydrolysis with 0.1 Nsulfuric acid at 76³C for 1 h. AsOR was puri¢edfrom the reaction mixture by neutralization with
1.0 N NaOH to pH 5.5 and exhaustive dialysisagainst water at room temperature. AsOR concen-tration was determined using an extinction coe¤cientof 0.931 ml mg31 cm31 at 280 nm. The thiobarbituricacid assay of Warren [38] or of Uchida was used toverify desialylation of the OR [39]. AsOR preparedby the above method was determined to be 98% de-sialylated. Labeled nucleotides [K-33P]dCTP and [K-33P]dATP were obtained from New England Nuclear(Boston, MA). NTB-3 autoradiography emulsion,developer and ¢xer were purchased through EastmanKodak (Rochester, NY). Isopentane, 10% formalinand reagent grade sucrose were from Sigma (St.Louis, MO).
2.2. Poly-L-lysine conjugate preparation
AsOR^poly-L-lysine conjugate (AP26K) was pre-pared by carbodiimide coupling similar to that re-ported by McKee et al. [40]. AsOR, 26 kDa poly-L-lysine and EDC in a 1:1:0.5 mass ratio were reactedas follows. EDC was added directly to a stirringaqueous AsOR solution. Poly-L-lysine (26 kDa) wasthen added and the reaction mixture was adjusted topH 5.5^6.0 and stirred for 2 h at ambient temper-ature. AsOR concentration was 5 mg/ml in the ¢nalreaction conditions. The reaction was quenched byaddition of Na3PO4 (200 mM, pH 11) to a ¢nalconcentration of 10 mM. The AP26K conjugatewas ¢rst puri¢ed on a Fast Flow Q Sepharose anionexchange chromatography column (Pharmacia)eluted with 50 mM Tris, pH 7.5; and then dialyzedagainst water.
PLL4K^PEG5K (1:2) was prepared by reactingPLL4K (100.0 mg, 0.032 mmol) dissolved in 0.05M sodium borate pH 9.0 (25 ml) with PEG5K^Tre-sylate (320 mg, 0.064 mmol) at room temperature.After stirring for 24 h, the reaction mixture was dia-lyzed to water (3500 MWCO; 3U20 l) and thenlyophilized. PLL10K^PEG5K (1:2) was prepared inthe same manner as PLL4K^PEG5K using PLL10K(100.0 mg, 0.01 mmol) in 50 ml 0.05 M sodiumborate pH 9.0 and PEG5K^Tresylate (100 mg,0.02 mmol).
2.3. Plasmid DNA
Plasmid DNA (pCMVL, Clontech, Palo Alto, CA
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 173

and pCMV^luciferase [41]) was prepared by BIO 101(San Diego, CA). Plasmid DNA used for experi-ments contained at least 90% covalently closed circu-lar DNA by agarose gel electrophoresis and EtBrstaining. DNA used for in vivo experiments con-tained 6 10 U/mg endotoxin.
2.4. Polyplex charge ratio calculations (+/3)
Puri¢ed protein^polylysine conjugates (AsOR^PLL or OR^PLL) were exhaustively dialyzed againstultra-pure water. An aliquot of the dialyzed conju-gate solution was lyophilized, weighed and dissolvedin ultra-pure water at a speci¢c concentration (w/v).Since poly-L-lysine has minimal absorbance at 280nm, the AsOR component of AsOR^polylysine(w/v) was quanti¢ed using the extinction coe¤cientat 280 nm. The composition of the conjugate wasestimated by comparison of the concentration ofthe conjugate (w/v) with the concentration ofAsOR (w/v) as determined by UV absorbance. Thedi¡erence between the two determinations was attrib-uted to the polylysine component of the conjugate.The composition of OR^polylysine was calculated inthe same manner. The ratio of conjugate to DNA(w/w) necessary for speci¢c charge ratios was thencalculated using the determined conjugate composi-tion. Charge ratios for polyplexes made with poly-lysine or protamine were calculated from the aminoacid composition. Charge ratios for PLL^PEG con-jugates were calculated using a PEG to PLL ratiothat was derived from 1H-NMR spectra [42].
2.5. Polyplex formation
Polyplexes were prepared by rapidly adding anequal volume of plasmid DNA to a volume of poly-cationic species unless noted otherwise. DNA wasprepared in water and conjugates were dissolved inthe 2U diluent (salt, glycine or mannitol) prior tomixing. Polyplex concentrations are reported byDNA content and were 10 Wg/ml unless stated other-wise. All biophysical characterizations were donewith polyplexes prepared with pCMV^Luc DNA;polyplexes containing pCMVL DNA were used forcryoautoradiography. All biophysical measurementsand EM grids were made 15^30 min after polyplexformation unless stated otherwise. Polyplexes pre-
pared for cryoautoradiography were used within 1 hof preparation.
2.6. Gel electrophoresis of PLLs
Tris^borate urea gels were obtained through No-vex (San Diego, CA). The gels were run in 1UTBEbu¡er [43]. The samples were mixed with an equalvolume of sample bu¡er containing 40% sucrose,0.1% methyl green dye (Sigma, St. Louis, MO) and7.2 M urea in 1UTBE. The gels were run at 180 Vwith polarity reversed for V2 h.
2.7. Fluorescence quenching assay
The relative binding e¤ciencies of polycationicpolymers were examined using an ethidium bro-mide-based quenching assay. Solutions were pre-pared containing 2.5 Wg/ml EtBr and 10 Wg/mlDNA (1:5 EtBr:DNA phosphates, molar ratio) ina total volume of 1 ml. The polycation was addedincrementally with £uorescence readings taken ateach point using a Sequoia-Turner 450 £uorometerwith excitation and emission wavelengths at 540 and585 nm, respectively. Fluorescence readings were ad-justed to compensate for the change in volume dueto the addition of polycation which never exceeded3% of the original volume. Results are reported asthe percentage of £uorescence relative to that of un-complexed plasmid DNA (no polycation).
2.8. Particle size analysis by laser light scattering
Light scattering measurements were determined ona Brookhaven Instruments Corporation 90 Plus par-ticle size analyzer equipped with a 50-mW laserwhich emits light at a wavelength of 532 nm. Re-agents were passed through a Nalgene 200 nm sur-factant-free cellulose acetate ¢lter prior to polyplexformation. Results are reported as e¡ective diam-eter de¢ned as the average diameter which isweighted by the intensity of light scattered by eachparticle.
It should be noted that the equations used to de-termine the e¡ective diameter assume the particlesbeing measured are spherically shaped. Since DNAcondensed with PLL forms toroidal or rod-shapedparticles, the measured e¡ective diameters should
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190174

be considered an approximation of the actual size ofthe polyplexes [44].
2.9. Electron microscopy
Carbon-coated copper grids with formvar support¢lm (Ted Pella, Redding, CA) were glow-dischargedfor 30 s just prior to sample preparation. Sampleswere negatively stained with uranyl acetate by oneof the following methods. (1) The grid was £oatedon a 15-Wl droplet containing polyplexes (at 10 Wg/mlunless otherwise stated) for 3 min, then wicked to¢lter paper. The grid is then placed on a droplet of1% uranyl acetate (w/v in water) for 1 min andwicked to ¢lter paper. The grid was washed 2U byplacing on distilled water droplet for 15 s followedby thoroughly removing liquid by wicking to ¢lter.(2) The grid was £oated on a droplet containingequal volumes of sample and 1.5% uranyl acid stain(1 min) followed by two washes in water. The gridswere inspected under a Zeiss EM 10b microscope at10 000U and 40 000U magni¢cation.
2.10. Serum protection assay
Free plasmid DNA or DNA (1 Wg) complexedwith AsOR^PLL conjugates at di¡erent charge ratioswere incubated at 37³C for 30 min in fresh humanserum. The plasmid DNA was dissociated from se-rum and AsOR^PLL by digestion with an equal vol-ume of 10U trypsin (Life Technologies, Gaithers-burg, MD) at 37³C for 1 h. The plasmid DNA wasthen extracted with phenol/chloroform and ethanolprecipitated. The DNA was resuspended in 10 mMTris, pH 7.4, 1 mM EDTA and run on 0.8% agarosegels in Tris^borate bu¡er. The gels were stained withethidium bromide and photographed under UV light[43].
2.11. In vivo cell disposition
pCMVL plasmid DNA was labeled with 33P usinga modi¢ed replacement synthesis procedure [43]. Thetemperature for both T4 DNA polymerase exonu-clease digestion and replacement synthesis was 14³Cfor 60 min. The ¢ll-in reaction contained K-33P-la-beled dCTP and dATP and cold dGTP and dTTP.Labeled polyplex was administered to 10-week-old
female Balb/c mice (Charles River, Wilmington,MA) via tail vein injection. The dose per mousewas 13.3 Wg of 33P-labeled AP26K polyplex (0.9 +/3) containing 5U106 cpm in 0.8 ml of 220 mMglycine or 10 Wg 33P-labeled PLL10K polyplex (1.35+/3) containing 6U105 cpm in 0.8 ml of 287 mMmannitol. Isotonic solutions of mannitol and glycinewere used in place of water in the formulation ofpolyplexes for in vivo studies to prevent shock inanimals. Polyplexes were as stable in both glycineand mannitol as in water. Polyplexes prepared inglycine and mannitol were also similar in size topolyplexes prepared in water (see Section 3). Animalswere sacri¢ced 5 min after injection by cervical dis-location. The livers were excised, washed with PBS(phosphate-bu¡ered saline, Life Technologies, Gai-thersburg, MD) and ¢xed for 4 h in 10% formalin.Fixed liver pieces were washed in PBS, infused with20% sucrose/PBS solution for 2 h, and then frozen incryomolds using liquid nitrogen chilled isopentane.Liver pieces were sectioned (5 Wm) on a Leica Cryo-cut 1800 at 318³C and mounted onto Vectabond-coated slides. The slides were dipped in NTB-3 emul-sion diluted 1:1 with water and air dried for 4 h. Theslides were exposed for 10 days at 4³C before devel-opment and subsequent counterstaining with hema-toxylin and eosin. Slides were viewed using an Olym-pus BH-2 microscope under oil immersion at 600Umagni¢cation.
3. Results
3.1. Commercial poly-L-lysines
The sizes of de¢ned length PLL peptides and poly-disperse PLLs of varying degrees of polymerizationwere analyzed by reversed polarity electrophoresis ondenaturing tris^borate^urea polyacrylamide gels(Fig. 1). The peptides of de¢ned length were usedas size markers for the polydisperse PLLs. SyntheticCK48 peptide containing an N-terminal cysteine (lane6) shows two bands in the stained gel. The lowerband represents the monomer peptide, while theupper band represents a dimer formed upon oxida-tion of cysteine to cystine, i.e. (CK48)2. The peptideK48, without a cysteine residue, (lane 5) shows onlyone band that correlates with the monomer band
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 175

seen with CK48 (lane 6). Similarly the peptideY(K)35YGYSC (lane 4) gave two bands in the gelindicating dimerization; the dimer form was the ma-jor species in this particular sample. A dimer band ofCK24 was observed on some gels which migrated tothe same position as K48, (data not shown).
The polydisperse PLLs from Sigma had a muchsmaller average size than reported by Sigma whencompared to the de¢ned length peptides. SigmaPLL10K (lane 2) migrated slightly farther than thesynthetic CK24 (lane 3), while Sigma PLL4K (lane 1)migrated much farther than CK24 after electrophore-sis. The average degree of polymerization forPLL10K and PLL4K should be 48 and 19, respec-tively. Sigma PLL26K (lane 8) should have an aver-age degree of polymerization of 123 but moved as a96-mer ((CK48)2, lane 6). PLL20.7K (lane 7) shouldhave a degree of polymerization of 99, but appears tohave two populations; one with an average degree ofpolymerization of approximately 90 and a secondwith an average degree of polymerization of 45.
3.2. DNA binding characteristics of PLL
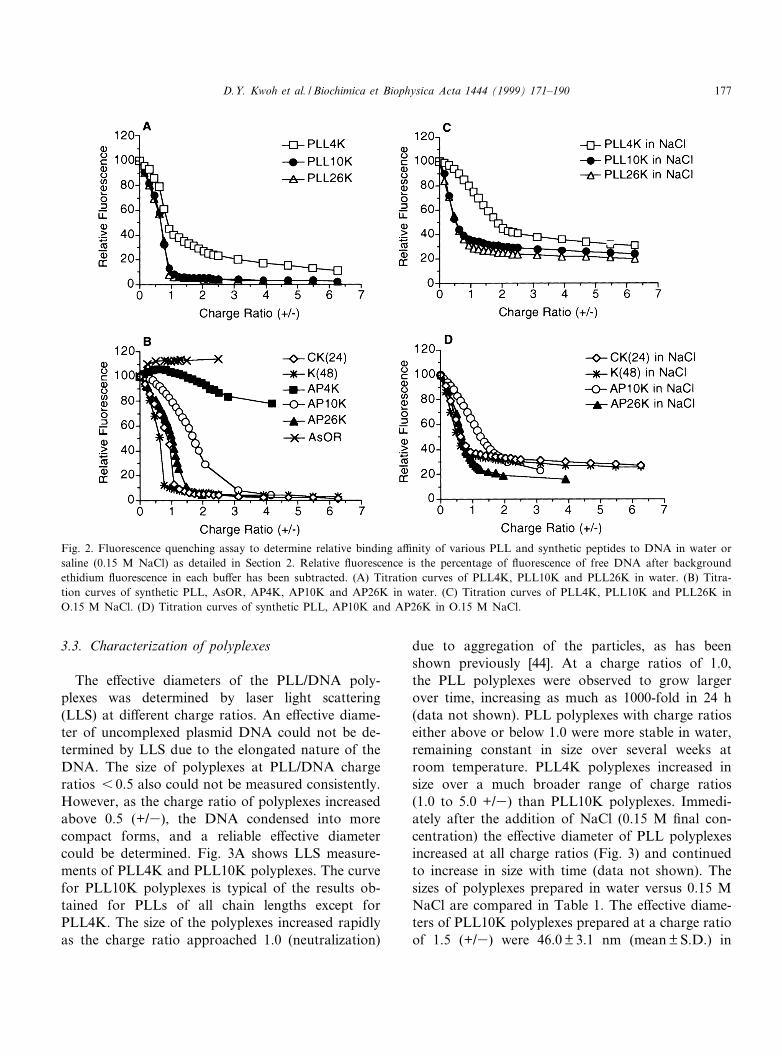
Binding of the various PLLs to plasmid DNA wasstudied using a £uorescence quenching assay. Brie£y,addition of increasing amounts of PLL to a solutionof ethidium bromide/DNA results in a rapid decreasein £uorescence. A relative binding a¤nity of the
PLLs can be inferred from the residual £uorescenceof ethidium bromide/DNA in excess PLL [45]. Fig.2A shows the £uorescence quenching obtained inwater using PLL 4K, 10K and 26K compared tothe quenching obtained with peptides K48 andCK24 (Fig. 2B). Maximum quenching is ¢rstachieved near the neutralization point where the pos-itive lysine charges of the PLL balance the negativephosphate charges on the DNA, i.e. the charge ratio(+/3) equals 1.0. Very little di¡erence in quenchingwas observed among the di¡erent polydisperse andde¢ned length PLL at high charge ratios except forPLL4K. PLL4K required a much higher charge ratioto achieve a level of quenching approaching the oth-er PLLs tested. The PLL4K quenching curve had notreached a plateau at the highest charge ratios tested.Longer PLL chains are favored for DNA binding,and a signi¢cant number of chains in the PLL4Kpreparation are likely too short to bind DNA anddisplace ethidium bromide. At high charge ratios, theconcentration of longer PLL chains in the PLL4Kpreparation was high enough to quench £uorescence.PLL binding to DNA was also studied in isotonicsolutions (287 mM mannitol or 220 mM glycine)which gave comparable results to those observed inwater (data not shown).
Fig. 2C shows the £uorescence quenching obtainedfor the various PLLs dissolved in 0.15 M NaCl. Thetotal £uorescence observed in saline was lower thanin water, in agreement with a previous report [46].Residual £uorescence values of 30, 25 and 20% max-imal £uorescence were observed for PLL4K,PLL10K and PLL26K, respectively, showing thatPLL chains bind less tightly to DNA in saline thanin water. The residual £uorescence values for pepti-des CK24 and CK48 are 25% of maximal £uorescencein saline (Fig. 2D).
Covalent attachment of a bulky ligand, such asAsOR, to the PLL chain shifted the maximalquenching to higher charge ratios in both waterand saline (Fig. 2B,D) compared to PLLs. However,the levels of residual £uorescence for AP10K(AsOR^10 kDa poly-L-lysine) and AP26K poly-plexes were the same as those observed forPLL10K and PLL26K polyplexes. AP4K showedvery little £uorescence quenching when assayed inwater, suggesting very weak binding of this conjugateto DNA.
Fig. 1. Sizing of PLL and synthetic peptides by gel electropho-resis on Tris^borate EDTA/urea gels using reverse polarity asdescribed in Section 2. Lanes: 1, PLL4K; 2, PLL10K; 3,CK24; 4,Y(K)35YGYSC; 5, K48; 6, CK48; 7, PLL20.7K; 8PLL26K; 9, PLL38K.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190176
3.3. Characterization of polyplexes
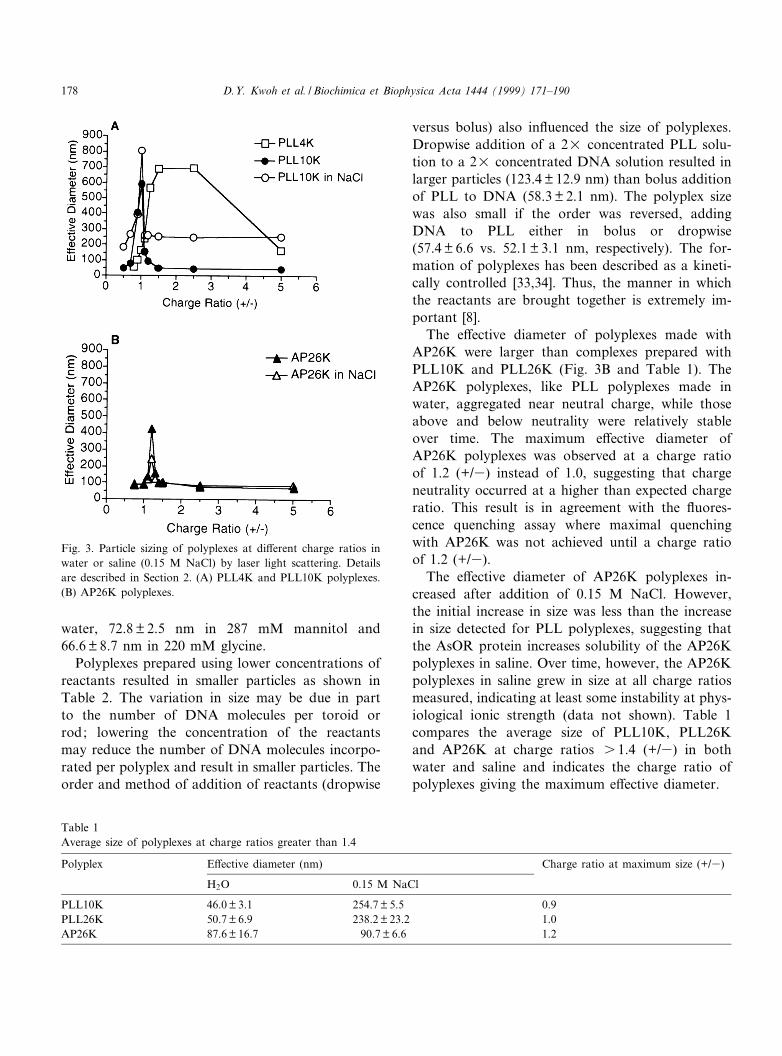
The e¡ective diameters of the PLL/DNA poly-plexes was determined by laser light scattering(LLS) at di¡erent charge ratios. An e¡ective diame-ter of uncomplexed plasmid DNA could not be de-termined by LLS due to the elongated nature of theDNA. The size of polyplexes at PLL/DNA chargeratios 6 0.5 also could not be measured consistently.However, as the charge ratio of polyplexes increasedabove 0.5 (+/3), the DNA condensed into morecompact forms, and a reliable e¡ective diametercould be determined. Fig. 3A shows LLS measure-ments of PLL4K and PLL10K polyplexes. The curvefor PLL10K polyplexes is typical of the results ob-tained for PLLs of all chain lengths except forPLL4K. The size of the polyplexes increased rapidlyas the charge ratio approached 1.0 (neutralization)
due to aggregation of the particles, as has beenshown previously [44]. At a charge ratios of 1.0,the PLL polyplexes were observed to grow largerover time, increasing as much as 1000-fold in 24 h(data not shown). PLL polyplexes with charge ratioseither above or below 1.0 were more stable in water,remaining constant in size over several weeks atroom temperature. PLL4K polyplexes increased insize over a much broader range of charge ratios(1.0 to 5.0 +/3) than PLL10K polyplexes. Immedi-ately after the addition of NaCl (0.15 M ¢nal con-centration) the e¡ective diameter of PLL polyplexesincreased at all charge ratios (Fig. 3) and continuedto increase in size with time (data not shown). Thesizes of polyplexes prepared in water versus 0.15 MNaCl are compared in Table 1. The e¡ective diame-ters of PLL10K polyplexes prepared at a charge ratioof 1.5 (+/3) were 46.0 þ 3.1 nm (mean þ S.D.) in
Fig. 2. Fluorescence quenching assay to determine relative binding a¤nity of various PLL and synthetic peptides to DNA in water orsaline (0.15 M NaCl) as detailed in Section 2. Relative £uorescence is the percentage of £uorescence of free DNA after backgroundethidium £uorescence in each bu¡er has been subtracted. (A) Titration curves of PLL4K, PLL10K and PLL26K in water. (B) Titra-tion curves of synthetic PLL, AsOR, AP4K, AP10K and AP26K in water. (C) Titration curves of PLL4K, PLL10K and PLL26K inO.15 M NaCl. (D) Titration curves of synthetic PLL, AP10K and AP26K in O.15 M NaCl.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 177
water, 72.8 þ 2.5 nm in 287 mM mannitol and66.6 þ 8.7 nm in 220 mM glycine.
Polyplexes prepared using lower concentrations ofreactants resulted in smaller particles as shown inTable 2. The variation in size may be due in partto the number of DNA molecules per toroid orrod; lowering the concentration of the reactantsmay reduce the number of DNA molecules incorpo-rated per polyplex and result in smaller particles. Theorder and method of addition of reactants (dropwise
versus bolus) also in£uenced the size of polyplexes.Dropwise addition of a 2U concentrated PLL solu-tion to a 2U concentrated DNA solution resulted inlarger particles (123.4 þ 12.9 nm) than bolus additionof PLL to DNA (58.3 þ 2.1 nm). The polyplex sizewas also small if the order was reversed, addingDNA to PLL either in bolus or dropwise(57.4 þ 6.6 vs. 52.1 þ 3.1 nm, respectively). The for-mation of polyplexes has been described as a kineti-cally controlled [33,34]. Thus, the manner in whichthe reactants are brought together is extremely im-portant [8].
The e¡ective diameter of polyplexes made withAP26K were larger than complexes prepared withPLL10K and PLL26K (Fig. 3B and Table 1). TheAP26K polyplexes, like PLL polyplexes made inwater, aggregated near neutral charge, while thoseabove and below neutrality were relatively stableover time. The maximum e¡ective diameter ofAP26K polyplexes was observed at a charge ratioof 1.2 (+/3) instead of 1.0, suggesting that chargeneutrality occurred at a higher than expected chargeratio. This result is in agreement with the £uores-cence quenching assay where maximal quenchingwith AP26K was not achieved until a charge ratioof 1.2 (+/3).
The e¡ective diameter of AP26K polyplexes in-creased after addition of 0.15 M NaCl. However,the initial increase in size was less than the increasein size detected for PLL polyplexes, suggesting thatthe AsOR protein increases solubility of the AP26Kpolyplexes in saline. Over time, however, the AP26Kpolyplexes in saline grew in size at all charge ratiosmeasured, indicating at least some instability at phys-iological ionic strength (data not shown). Table 1compares the average size of PLL10K, PLL26Kand AP26K at charge ratios s 1.4 (+/3) in bothwater and saline and indicates the charge ratio ofpolyplexes giving the maximum e¡ective diameter.
Fig. 3. Particle sizing of polyplexes at di¡erent charge ratios inwater or saline (0.15 M NaCl) by laser light scattering. Detailsare described in Section 2. (A) PLL4K and PLL10K polyplexes.(B) AP26K polyplexes.
Table 1Average size of polyplexes at charge ratios greater than 1.4
Polyplex E¡ective diameter (nm) Charge ratio at maximum size (+/3)
H2O 0.15 M NaCl
PLL10K 46.0 þ 3.1 254.7 þ 5.5 0.9PLL26K 50.7 þ 6.9 238.2 þ 23.2 1.0AP26K 87.6 þ 16.7 90.7 þ 6.6 1.2
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190178

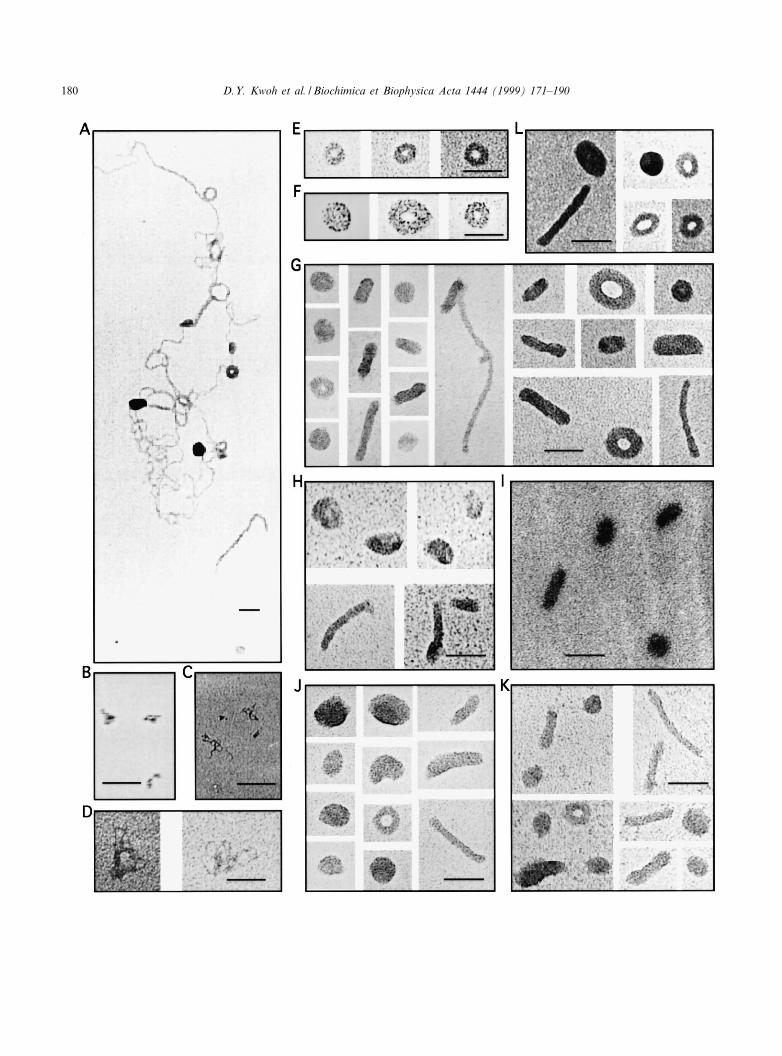
3.4. Size and shape of polyplexes
PLLs of all sizes condensed plasmid DNA intotoroidal and rod-shaped structures at charge ratiosabove 0.5 (+/3) as shown by electron microscopy(EM) (Fig. 4). Many large structures, containingmultiple molecules of plasmid DNA and showingmultiple points of condensation along the DNAstrands, were observed at charge ratios of 6 1 (+/3) (Fig. 4A). At higher PLL/DNA charge ratios,individual polyplexes were observed ranging in sizefrom 25 to 50 nm for toroids and from 40 to 80 nmfor rods (Fig. 4E^K). PLL10K/DNA toroids had amean ( þ S.D.) outer diameter of 30.1 þ 2.9 nm(n = 24) and rods had a mean length of 61.3 þ 20.3nm (n = 28) when prepared in water at a charge ratioof 1.2 (+/3) and a DNA concentration of 10 Wg/ml.Approximately 60^65% of the observed particleswere toroids (66 toroids of 108 particles and 133toroids of 203 particles observed on two grids pre-pared independently); the remaining particles wererods. Generally, the polyplexes made with PLL4K(Fig. 4E,F) gave toroids more uniform in size withvisible holes in the center compared with toroidsformed by either PLL10K (Fig. 4G,H) or PLL26K(Fig. 4J,K). Center holes were observed less fre-quently at higher PLL/DNA charge ratios withPLL4K. Polyplexes prepared at higher charge ratios(s 1.0 +/3) appear to be positively stained with ur-anyl acetate which resulted in less discernible struc-ture and may cause the particles to appear smaller[47]. DNA condensed with synthetic peptides (CK48,Y(K)35YGYSC and CK24) showed similar toroidand rod structures (data not shown). Toroids ob-tained with PLLs are similar to the structures de-tected when DNA is condensed with protamine(Fig. 4L) [32].
Grids of uncondensed DNA that were prepared by
the same method as those of polyplexes are shown inFig. 4B^D. No toroids or rods were observed inelectron micrographs of PLL alone, although therewas an increase in background staining. Polyplexesprepared in mannitol or glycine (Fig. 4I) had similarstructures to those prepared in water, although theyappeared positively stained with uranyl acetate. PLLpolyplexes prepared in NaCl at concentrations rang-ing from 10 to 150 mM did not adhere well to gridsand gave unreliable EM results.
The shapes observed by EM of the AP4K, AP10Kor AP26K polyplexes demonstrated that attachmentof bulky groups along the backbone of the PLLchain interferes with condensation of the DNA. Asshown in Fig. 5A, AP10K conjugates did not fullycondense DNA into small toroids or rods even atcharge ratios as high as 5.0 (+/3). The thicknessand total length of the DNA strands observed withAP10K polyplexes con¢rms that AP10K is bound toDNA and binding results in partial DNA condensa-tion. Orosomucoid^PLL26K (OR26K, Fig. 5B,C)and AP26K (Fig. 5D) condensed DNA into asym-metrical toroids, rods and other irregular shapes atcharge ratios v 2.0 (+/3). Toroids were observed atlower charge ratios, but appeared more compact atratios s 1.25 (+/3). The toroids observed withAP26K polyplexes were larger and had a greater in-ner diameter than toroids observed with PLL poly-plexes. Many of the AP26K polyplexes appeared tohave one thinner section along the toroid (indicatedwith arrows in Fig. 5D^F). Unlike PLL polyplexes,AP26K polyplexes prepared in 0.1 M NaCl adheredto EM grids (Fig. 5E and F). Small spherical par-ticles, 20^25 nm in diameter (indicated with closedarrowheads) and ring structures (open arrowheads)were observed in electron micrographs of AP26Kpolyplexes prepared in saline, but were not observedwhen AP26K polyplexes were prepared in water. Thering structures appeared to be intermediate structuresbetween the small spheres and the toroids.
3.5. DNA condensation and serum protection
To determine whether DNA was protected fromnucleases when complexed with AP4K, AP10K orAP26K, polyplexes were prepared at di¡erent chargeratios and exposed to human serum. The integrity ofplasmid DNA was determined by electrophoresis
Table 2E¡ective diameter of PLL10K polyplex (+/3= 1.35) made atdi¡erent concentrations and sized at 10 Wg/ml
Starting concentration (Wg/ml) E¡ective diameter (nm)
10 58.050 76.8
100 111.3250 144.2825 498.8
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 179
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190180

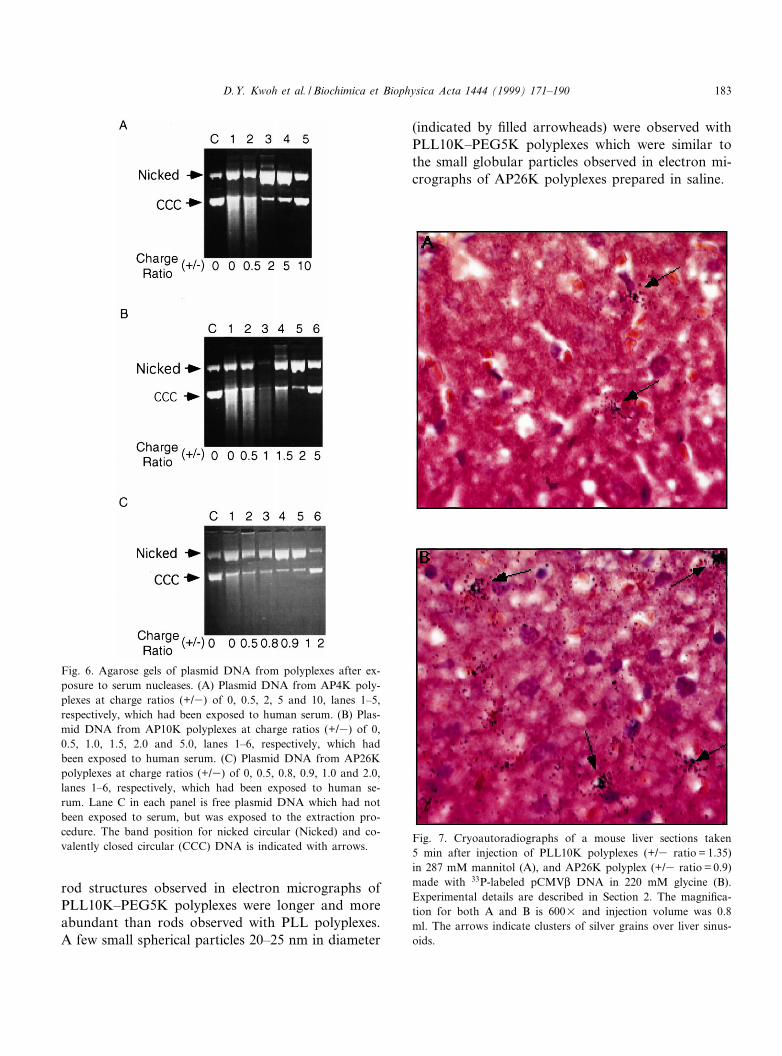
through agarose gels after the DNA was dissociatedfrom polyplexes and serum proteins by digestionwith trypsin and phenol extraction (Fig. 6). Uncom-plexed plasmid DNA, which had not been exposed toserum, but was taken through the extraction proce-dure, served as control (lane C). DNA bands corre-sponding to nicked circular and covalently closedcircular forms are labeled for each gel. AP4K andAP10K did not protect the plasmid DNA from nick-ing even at charge ratios of 10 and 5 (+/3), respec-tively. DNA in AP26K polyplexes at a charge ratioof 2 (+/3) was completely protected from nucleasedigestion, while DNA in AP26K polyplexes at acharge ratio of 1.0 (+/3) was still susceptible to nick-ing in this assay. Occasionally, the extraction proce-dure failed to remove all protein from DNA, andbands running above the nicked DNA were detectedin gels (Fig. 6A, lane 3). DNA from AP10K poly-plexes (1.0 +/3) was lost during the extraction pro-cedure resulting in reduced £uorescence (Fig. 6B,lane 3). These results, combined with the resultsfrom EM, suggest that DNA is protected from serumnucleases when fully condensed by PLL conjugates(AP26K polyplexes at charge ratios v 2 (+/3), whileDNA is not protected in polyplexes where DNA isonly partially condensed, as in AP4K and AP10Kpolyplexes.
3.6. In vivo destination of AP26K polyplexes
The intrahepatic disposition of AsOR^PLL andPLL polyplexes containing 33P-labeled DNA was ex-amined after intravenous injection into the tail veinof Balb/c mice to determine whether AsOR alteredthe destination of PLL polyplexes. Cryoautoradiog-raphy of liver sections taken 5 min after injectionshow that much of the labeled polyplex remainedin liver sinusoids for both PLL10K and AP26K poly-plexes (Fig. 7A and B, respectively). Since both PLL
and AP26K polyplexes show sinusoidal clumping ofsilver grains, the partial stabilization of PLL poly-plexes achieved with the attachment of AsOR isnot su¤cient to prevent aggregation or opsonizationof the polyplexes during delivery. The distribution oflabel was similar with both small (0.2 ml) or large(0.8^1 ml) injection volumes (data not shown) sug-gesting that the increased expression of AP26K poly-plexes observed after injection in a large volume re-sults from a minor portion of polyplexes escapingentrapment in the liver sinusoids [16].
3.7. Steric stabilization of polyplexes by addition ofpolyethylene glycol
Polyethylene glycol has been used to coat surfacesof various types of particles to reduce colloidal ag-gregation and to minimize interaction with serumproteins [3,48]. Therefore, we covalently attachedpolyethylene glycol MW 5000 (PEG5K) to bothPLL4K and PLL10K using tresylate chemistry toexamine the e¡ects of PEG on polyplexes. The de-gree of derivatization of the PLL^PEG5K conjugateswas determined by 1H-NMR and purity by electro-phoresis through denaturing Tris^borate^urea poly-acrylamide gels.
PLL^PEG polyplexes were signi¢cantly di¡erentfrom PLL polyplexes in size and shape as determinedby EM. Electron micrographs of the PLL^PEG poly-plexes showed toroids of variable sizes with increasedinner diameters compared to toroids observed withPLL polyplexes (Fig. 8). The size variation likelyresulted from the distribution of PEG on polydis-perse PLL, as well as from di¡erent ratios of PLL^PEG and PLL bound to DNA in these polyplexes.The PLL4K^PEG5K conjugate had detectable levelsof underivatized PLL on gels; the underivatized PLLis likely responsible for the smaller polyplexes (tor-oids) detected at high charge ratios (Fig. 8E). The
6
Fig. 4. Electron micrographs of free DNA and polyplexes made with di¡erent length PLL polymers or protamine sulfate. (A) PLL4Kpolyplexes (+/3= 0.62) in water, scale bar: 50 nm. (B) Free pCMV-Luc DNA in water, scale bar: 200 nm. (C) Free pCMV-LucDNA in 0.15 M NaCl, scale bar: 200 nm. (D) Free pCMV-Luc DNA in 0.15 M NaCl, scale bar: 50 nm. (E) PLL4K polyplexes (+/3= 0.77) in water, scale bar: 50 nm. (F) PLL4K polyplexes (+/3= 2.5) in water, scale bar: 50 nm. (G) PLL10K polyplexes (+/3= 1.2) in water, scale bar: 50 nm. (H) PLL10K polyplexes (+/3= 1.55) in water, scale bar: 50 nm. (I) PLL10K polyplexes (+/3= 2.5) in 227 mM glycine, scale bar: 50 nm. (J) PLL26K polyplexes (+/3= 0.9) in water, scale bar: 50 nm. (K) PLL26K polyplexes(+/3= 1.5) in water, scale bar: 50 nm. (L) Protamine polyplexes (+/3= 1.25) in water, scale bar: 50 nm. Grids were prepared bystaining method 1 for A^F, K and L and staining method 2 for G^J as described in Section 2.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 181
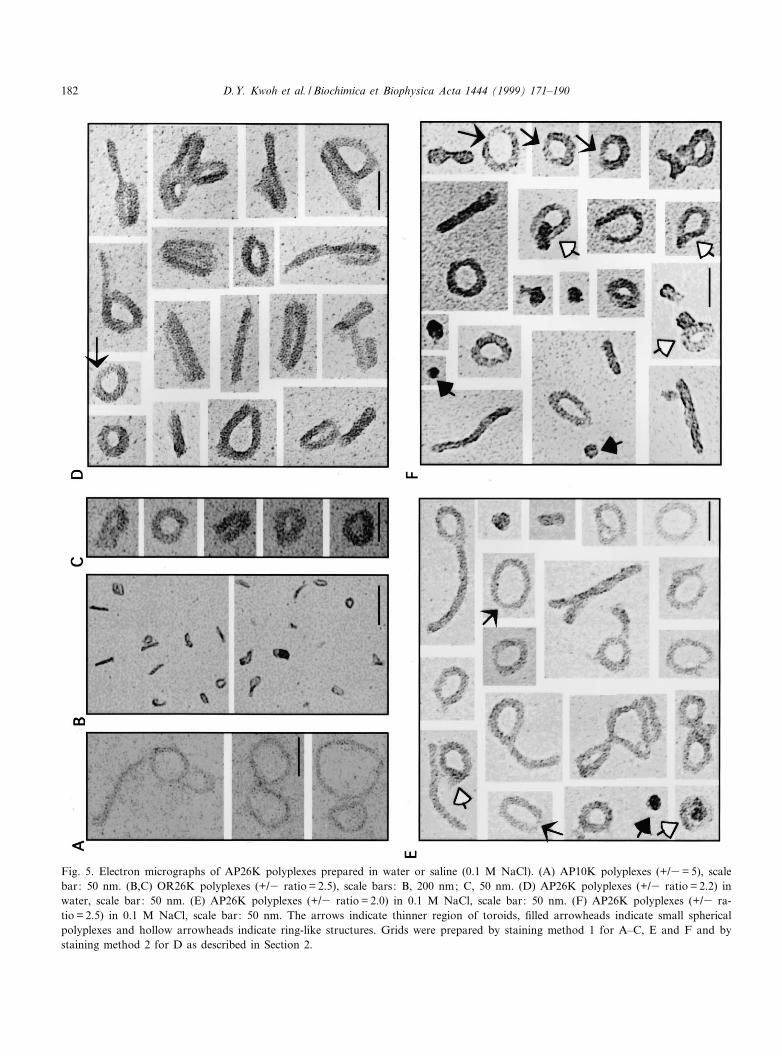
Fig. 5. Electron micrographs of AP26K polyplexes prepared in water or saline (0.1 M NaCl). (A) AP10K polyplexes (+/3= 5), scalebar: 50 nm. (B,C) OR26K polyplexes (+/3 ratio = 2.5), scale bars: B, 200 nm; C, 50 nm. (D) AP26K polyplexes (+/3 ratio = 2.2) inwater, scale bar: 50 nm. (E) AP26K polyplexes (+/3 ratio = 2.0) in 0.1 M NaCl, scale bar: 50 nm. (F) AP26K polyplexes (+/3 ra-tio = 2.5) in 0.1 M NaCl, scale bar: 50 nm. The arrows indicate thinner region of toroids, ¢lled arrowheads indicate small sphericalpolyplexes and hollow arrowheads indicate ring-like structures. Grids were prepared by staining method 1 for A^C, E and F and bystaining method 2 for D as described in Section 2.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190182
rod structures observed in electron micrographs ofPLL10K^PEG5K polyplexes were longer and moreabundant than rods observed with PLL polyplexes.A few small spherical particles 20^25 nm in diameter
(indicated by ¢lled arrowheads) were observed withPLL10K^PEG5K polyplexes which were similar tothe small globular particles observed in electron mi-crographs of AP26K polyplexes prepared in saline.
Fig. 6. Agarose gels of plasmid DNA from polyplexes after ex-posure to serum nucleases. (A) Plasmid DNA from AP4K poly-plexes at charge ratios (+/3) of 0, 0.5, 2, 5 and 10, lanes 1^5,respectively, which had been exposed to human serum. (B) Plas-mid DNA from AP10K polyplexes at charge ratios (+/3) of 0,0.5, 1.0, 1.5, 2.0 and 5.0, lanes 1^6, respectively, which hadbeen exposed to human serum. (C) Plasmid DNA from AP26Kpolyplexes at charge ratios (+/3) of 0, 0.5, 0.8, 0.9, 1.0 and 2.0,lanes 1^6, respectively, which had been exposed to human se-rum. Lane C in each panel is free plasmid DNA which had notbeen exposed to serum, but was exposed to the extraction pro-cedure. The band position for nicked circular (Nicked) and co-valently closed circular (CCC) DNA is indicated with arrows.
Fig. 7. Cryoautoradiographs of a mouse liver sections taken5 min after injection of PLL10K polyplexes (+/3 ratio = 1.35)in 287 mM mannitol (A), and AP26K polyplex (+/3 ratio = 0.9)made with 33P-labeled pCMVL DNA in 220 mM glycine (B).Experimental details are described in Section 2. The magni¢ca-tion for both A and B is 600U and injection volume was 0.8ml. The arrows indicate clusters of silver grains over liver sinus-oids.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 183
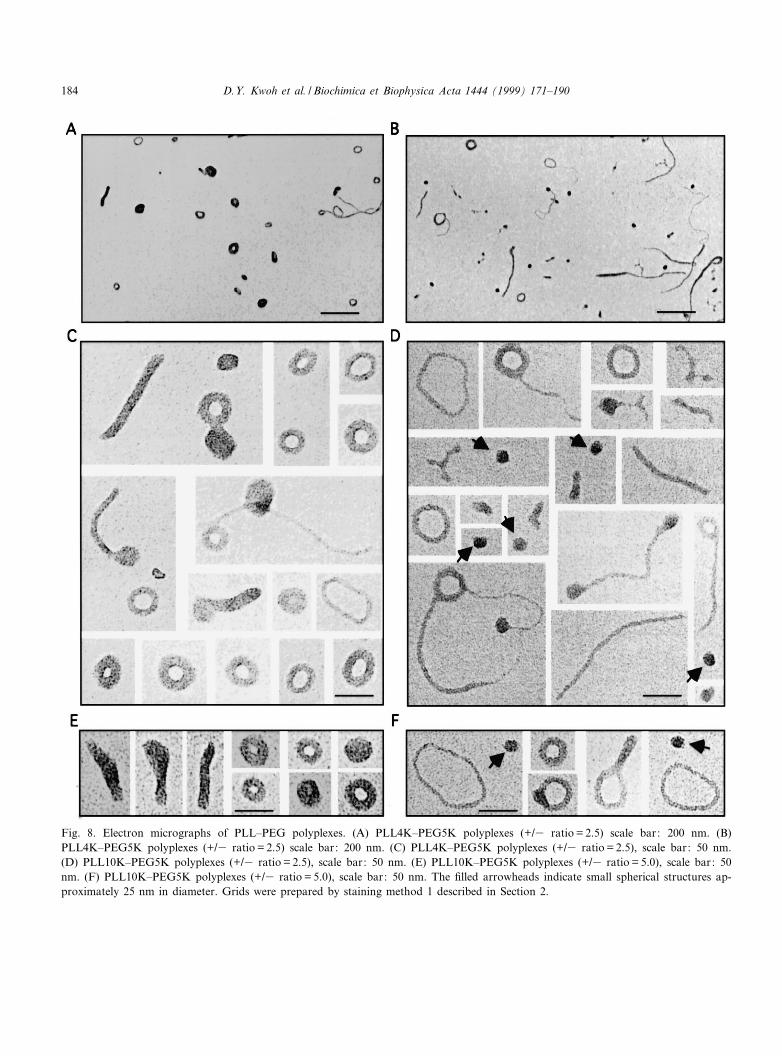
Fig. 8. Electron micrographs of PLL^PEG polyplexes. (A) PLL4K^PEG5K polyplexes (+/3 ratio = 2.5) scale bar: 200 nm. (B)PLL4K^PEG5K polyplexes (+/3 ratio = 2.5) scale bar: 200 nm. (C) PLL4K^PEG5K polyplexes (+/3 ratio = 2.5), scale bar: 50 nm.(D) PLL10K^PEG5K polyplexes (+/3 ratio = 2.5), scale bar: 50 nm. (E) PLL10K^PEG5K polyplexes (+/3 ratio = 5.0), scale bar: 50nm. (F) PLL10K^PEG5K polyplexes (+/3 ratio = 5.0), scale bar: 50 nm. The ¢lled arrowheads indicate small spherical structures ap-proximately 25 nm in diameter. Grids were prepared by staining method 1 described in Section 2.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190184
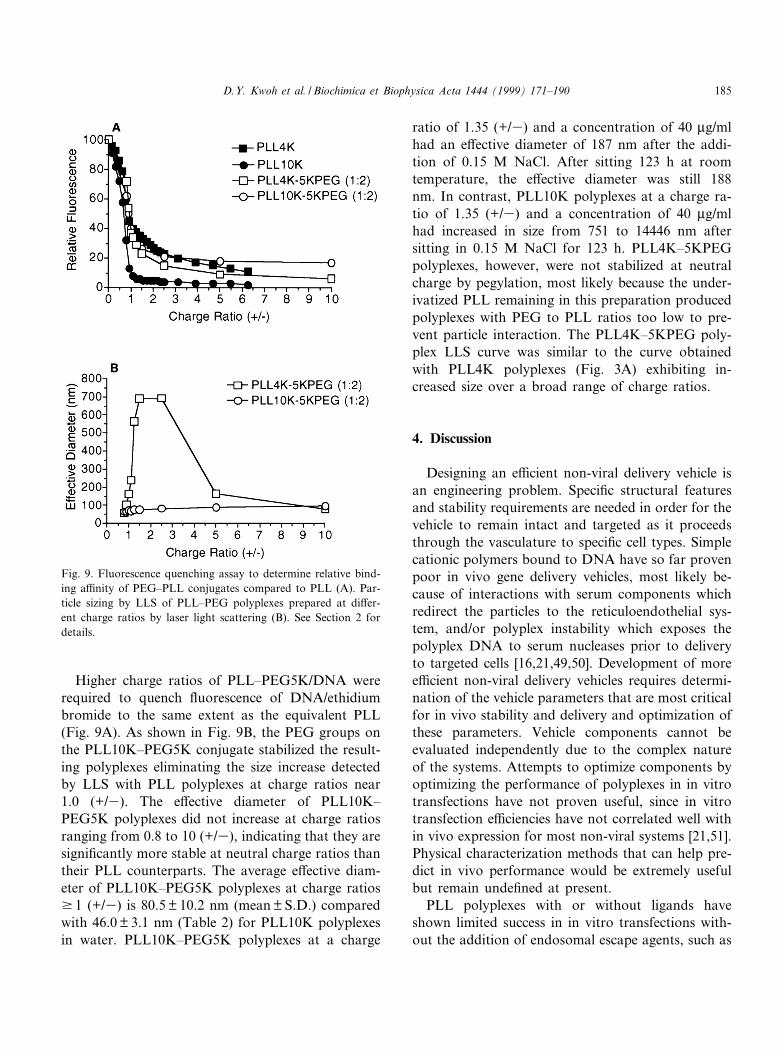
Higher charge ratios of PLL^PEG5K/DNA wererequired to quench £uorescence of DNA/ethidiumbromide to the same extent as the equivalent PLL(Fig. 9A). As shown in Fig. 9B, the PEG groups onthe PLL10K^PEG5K conjugate stabilized the result-ing polyplexes eliminating the size increase detectedby LLS with PLL polyplexes at charge ratios near1.0 (+/3). The e¡ective diameter of PLL10K^PEG5K polyplexes did not increase at charge ratiosranging from 0.8 to 10 (+/3), indicating that they aresigni¢cantly more stable at neutral charge ratios thantheir PLL counterparts. The average e¡ective diam-eter of PLL10K^PEG5K polyplexes at charge ratiosv 1 (+/3) is 80.5 þ 10.2 nm (mean þ S.D.) comparedwith 46.0 þ 3.1 nm (Table 2) for PLL10K polyplexesin water. PLL10K^PEG5K polyplexes at a charge
ratio of 1.35 (+/3) and a concentration of 40 Wg/mlhad an e¡ective diameter of 187 nm after the addi-tion of 0.15 M NaCl. After sitting 123 h at roomtemperature, the e¡ective diameter was still 188nm. In contrast, PLL10K polyplexes at a charge ra-tio of 1.35 (+/3) and a concentration of 40 Wg/mlhad increased in size from 751 to 14446 nm aftersitting in 0.15 M NaCl for 123 h. PLL4K^5KPEGpolyplexes, however, were not stabilized at neutralcharge by pegylation, most likely because the under-ivatized PLL remaining in this preparation producedpolyplexes with PEG to PLL ratios too low to pre-vent particle interaction. The PLL4K^5KPEG poly-plex LLS curve was similar to the curve obtainedwith PLL4K polyplexes (Fig. 3A) exhibiting in-creased size over a broad range of charge ratios.
4. Discussion
Designing an e¤cient non-viral delivery vehicle isan engineering problem. Speci¢c structural featuresand stability requirements are needed in order for thevehicle to remain intact and targeted as it proceedsthrough the vasculature to speci¢c cell types. Simplecationic polymers bound to DNA have so far provenpoor in vivo gene delivery vehicles, most likely be-cause of interactions with serum components whichredirect the particles to the reticuloendothelial sys-tem, and/or polyplex instability which exposes thepolyplex DNA to serum nucleases prior to deliveryto targeted cells [16,21,49,50]. Development of moree¤cient non-viral delivery vehicles requires determi-nation of the vehicle parameters that are most criticalfor in vivo stability and delivery and optimization ofthese parameters. Vehicle components cannot beevaluated independently due to the complex natureof the systems. Attempts to optimize components byoptimizing the performance of polyplexes in in vitrotransfections have not proven useful, since in vitrotransfection e¤ciencies have not correlated well within vivo expression for most non-viral systems [21,51].Physical characterization methods that can help pre-dict in vivo performance would be extremely usefulbut remain unde¢ned at present.
PLL polyplexes with or without ligands haveshown limited success in in vitro transfections with-out the addition of endosomal escape agents, such as
Fig. 9. Fluorescence quenching assay to determine relative bind-ing a¤nity of PEG^PLL conjugates compared to PLL (A). Par-ticle sizing by LLS of PLL^PEG polyplexes prepared at di¡er-ent charge ratios by laser light scattering (B). See Section 2 fordetails.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 185

chloroquine, amphipathic peptides (such as in£uenzavirus hemagglutinin) or conjugated adenovirus[13,15,52^54]. There are several reports of successfulin vivo expression using PLL based non-viral vec-tors; however, the e¤ciency is well below the e¤-ciency of viral based vectors [17,27^29]. The use ofchloroquine or partial hepatectomy to increase endo-somal escape has improved the levels of in vivo ex-pression with PLL based vectors, but the levels arestill well below those achieved with some viral vec-tors [27,55^57].
We have improved reporter gene expression inmice after intravenous injection of AsOR^PLL poly-plexes by improving the solubility of the polyplexes[16]. Increasing the pH during preparation of AsOR^PLL polyplexes decreased aggregation of the conju-gate and increased solubility of the resulting poly-plexes. After intravenous injection of the AsOR^PLL polyplexes into mice, 99% of reporter gene ex-pression occurred in the liver. When L-galactosidasegene expression was examined by X-gal (5-bromo-4-chloro-3-indolyl-L-D-galactoside) staining of sec-tioned tissue, hepatocytes were the predominantcell-type expressing in liver tissue; however, someL-galactosidase expression was also detected in non-parenchymal cells such as Kup¡er and endothelialcells (unpublished results). Although this enhancedformulation protocol resulted in high levels of re-porter gene expression after injection into mice(V2000 ng luciferase protein/g liver), the level ofexpression decreased over 200-fold if the sameamount of polyplex (10 Wg DNA) was injected inhalf the volume. The in vivo expression also de-creased over 100-fold when the polyplexes weremixed with mouse serum (25% ¢nal) prior to injec-tion. These results suggested that interaction withblood components adversely a¡ected the polyplexes.It also indicated that signi¢cant levels of in vivo ex-pression are achievable with PLL-based vectors.However, to achieve high levels of expression usinginjection volumes that are clinically relevant will re-quire formulation improvements which stabilizepolyplexes in the presence of blood components.
In this study, we have systematically examinedmethods of formulation, polylysine chain length,and the charge ratio of PLL and AsOR^PLL poly-plexes using in vitro assays to characterize the size,shape and stability of polyplexes in an e¡ort to ¢nd
components and formulations which will lead to im-proved stability within the vasculature. The size ofPLL polyplexes did not increase signi¢cantly withincreasing PLL chain length shown by EM andLLS. Wolfert and Seymour have reported a largerdi¡erence in the average size of polyplexes with in-creasing PLL chain length [26]. This discrepancy mayre£ect di¡erences in the concentration of reactantsduring polyplex formulation, or in the method usedfor size determination (EM and LLS versus atomicforce microscopy). We found that shorter chain PLL(PLL4K) did not bind DNA e¤ciently and the poly-plex aggregation observed at charge ratios near 1 (+/3) with longer chain PLL polyplexes was extendedto a broader range of charge ratios for PLL4K poly-plexes.
Polyplex size was a¡ected by the attachment ofasialoorosomucoid, but AP26K polyplexes remainedsmaller than 100 nm. AP4K and AP10K polyplexes,however, were not compact, most likely because thebulky AsOR protein interferes with DNA bindingwhen attached to short PLL chains. The number ofAsOR groups per polyplex increases with decreasingPLL chain length which likely increases constraintson DNA condensation. AP26K polyplexes wouldhave an average of 16 AsOR molecules per 1000base pairs of DNA at a charge ratio of 1 comparedto 42 molecules per 1000 base pairs for AP10K poly-plexes. This is assuming one AsOR molecule perPLL chain, and an average of 123 and 48 lysineresidues per chain for PLL26K or PLL10K, respec-tively.
DNA in the more compacted AP26K polyplexes(AP26K/DNA ratios v 2 +/3) was completely pro-tected from serum nucleases, while DNA in AP4Kand AP10K polyplexes was susceptible to nickingeven at high AsOR^PLL/ DNA ratios. Althoughthe AP10K conjugate quenched DNA/ethidium bro-mide £uorescence to the same extent as PLL10Ksuggesting e¤cient DNA binding, AP10K did notcondense DNA to the same extent as PLL10K orAP26K. These results suggest that DNA condensa-tion is the important factor in conferring nucleaseresistance to DNA in polyplexes.
Attachment of AsOR to PLL increased theamount of conjugate required to obtain maximalbinding to DNA (determined by £uorescencequenching). Charges on the PLL may be masked
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190186

by the bulky nature of the AsOR ligand (32.5 kDa).In addition, the net negative charge of AsOR maycounterbalance some of the positive charges on thePLL. These e¡ects may account for the shift to high-er charge ratios in the £uorescence quenching ofAP10K and AP26K polyplexes. The residual £uores-cence both in water and saline was not signi¢cantlyincreased for AP10K or AP26K suggesting that theseconjugates bind to DNA as e¤ciently as PLL10Kand PLL26K.
The cellular distribution of AP26K polyplexes ap-peared no di¡erent from PLL polyplexes, althoughthe solubility in physiological saline was slightlygreater for AP26K polyplexes than for PLL poly-plexes if examined at times 9 30 min. DNA inAP26K polyplexes remained protected from nucle-ases suggesting that polyplexes remained intact inthe presence of serum. The cell distribution patternof AP26K polyplexes was not detectably di¡erent forAP26K polyplexes at di¡erent charge ratios or afterdelivery with di¡erent injection volumes. We hadpreviously shown that the expression of AP26Kpolyplexes deliver in larger volumes was increasedby up to a factor of 200 [16]. These results suggestthat factors other than serum nucleases and the sizeof AsOR^PLL polyplexes are interfering with deliv-ery to hepatocytes. We suspect that polyplexes areinteracting with other blood components, such asopsonins, which are responsible for the entrapmentin liver sinusoids. Although the cell disposition is thesame for AsOR^PLL polyplexes and PLL poly-plexes, they may interact with di¡erent blood com-ponents.
Increasing the charge ratio of polyplexes, whichincreases the surface charge of polyplexes [58], didnot alter the cell distribution pattern. More positivelycharged liposomes, however, have been shown to in-teract with blood components more than neutral orslightly negatively charged liposomes [59,60]. The na-ture of PLL or AsOR^PLL binding to the polyplexesat charge ratios greater than 1 (+/3) must changefrom just electrostatic binding between lysine resi-dues and the phosphate groups on the DNA. Ifthis binding is weaker than the PLL or AsOR^PLLbinding to DNA, then interaction with polyanions inthe blood could rapidly remove any excess PLL orAsOR^PLL leaving only that bound to the DNA.This could explain why polyplexes with charge ratios
greater than 1 (+/3) do not have altered cell dispo-sition from that of polyplexes with neutral chargeratios. Polyanions may also in£uence the binding ofPLL or PLL-conjugates to DNA, especially thoseshowing weaker DNA binding, such as PLL4K andAP10K.
PLL and AsOR^PLL polyplexes exhibited colloi-dal instability which accounts for the drastic increasein size at neutral charge ratios of PLL polyplexes.This instability was thought to play a signi¢cantrole in non-speci¢c uptake of polyplexes in vivo.Electrostatically neutral polyplexes may aggregatedue to van der Waals attractive forces which becomemore signi¢cant as the ionic forces are neutralized[61]. Polyplexes with minimum surface charge aredesirable to avoid complement activation or opsoni-zation in vivo [25]. Colloidal aggregation has beenreduced for liposomes and other drug delivery ve-hicles by introduction of an inert polymer (poly-ethylene glycol) to the surface of the particles. PEGsterically stabilizes particles by preventing close con-tact between individual particles and by increasingthe overall hydrophilicity [3,48]. Wolfert and co-workers have shown that the addition of poly-ethylene glycol to PLL changed the zeta potentialof the polyplexes made with PEG^PLL comparedwith PLL [58,62]. Furthermore, the addition ofPEG to liposomes has increased their circulationtime in vivo presumably by shielding the liposomesfrom blood components [63].
Our initial attempts to make the surface of thepolyplexes more innocuous using PEG show thatpolyplex aggregation can be eliminated by incorpo-ration of minimal amounts of PEG. The PLL10K^PEG5K (1:2) polyplexes were more stable over timein physiological saline than PLL10K or AP26K poly-plexes; however, this did not result in signi¢cantchanges in in vivo circulation times or in vivo ex-pression (data not shown). We have observed thatchanging the chemistry of attachment of PEG toPLL and increasing the ratio of PEG to PLL inpolyplexes improves the circulation times and in-creases in vivo gene expression (D.Y. Kwoh andM.G. Banaszczyk unpublished observations).PLL10K^PEG5K (1:14.3) polyplexes show a 54-fold and 4.3-fold improvement in expression of luci-ferase in vivo compared with PLL10K and AP26Kpolyplexes, respectively (80.5 ng luciferase/g liver for
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 187

PLL10K^PEG5K (1:14.3) polyplexes versus 1.5 ngluciferase/g liver for PLL10K and 18.7 ng lucifer-ase/g liver for AP26K polyplexes; polyplexes weremade at a charge ratio of 1.35 (+/3) in 0.15 MNaCl at 10 Wg/ml and injected slowly, in a 1 mlvolume, into the tail vein of Balb/c mice with fourmice per group). The in vitro stability of the poly-plexes is important not only for improvement in theshelf-life of the polyplexes, but also necessary to en-sure that polyplexes can overcome the in vivo sizebarriers. However, it is not only the size of the poly-plexes that is changed by the addition of PEG, butalso the surface characteristics. We are continuing tooptimize the amount, as well as the length of thePEG attached to the polyplexes using in vivo clear-ance and biodistribution studies to determine theminimal amount of PEG required to increase circu-lation times and prevent uptake by the reticuloendo-thelial systems.
PEG addition to the surface of polyplexes ad-dresses the problem of non-speci¢c uptake of thepolyplexes; however, it may also minimize interac-tions between polyplexes and cell surfaces decreasingheptocyte-speci¢c uptake. We are using a systematicapproach to examine di¡erent length PEG chainsand di¡erent ratios of PEG on the surface of theligand^PLL polyplexes in order to maximize ligand-speci¢c receptor binding of the polyplex while main-taining steric stabilization through the PEG moieties.Although our goal is to target polyplexes to the liver,once polyplex components are optimized for stabili-zation in the vasculature, they could be directed toother tissues and cell types using ligands other thanAsOR.
Acknowledgements
We are grateful for the technical help of AbbyAnnSisk, Patricia Reed and Kelly Soules in the Depart-ment of Research Electron Microscopy, VeteransAdministration Hospital, La Jolla, CA. We alsowould like to thank C.M. Venne for technical assist-ance with the serum protection assay and R. Alemanand M. Guido for help with the animal studies. Allanimal studies were done in accordance with USDAguidelines outlined in the Animal Welfare Act underregistration number 93-R-278.
References
[1] J.P. Yang, L. Huang, Direct gene transfer to mouse mela-noma by intratumor injection of free DNA, Gene Ther. 3(1996) 542^548.
[2] A.E. Smith, Viral vectors in gene therapy, Annu. Rev. Mi-crobiol. 49 (1995) 807^838.
[3] S.S. Davis, Biomedical applications of nanotechnology ^ im-plications for drug targeting and gene therapy, Trends Bio-technol. 15 (1997) 217^224.
[4] J.A. Wol¡, R.W. Malone, P. Williams, W. Chong, G. Acsa-di, A. Jani, P.L. Felgner, Direct gene transfer into mousemuscle in vivo, Science 247 (1990) 1465^1468.
[5] C. Nicolau, A. Cudd, Liposomes as carriers of DNA, Crit.Rev. Ther. Drug Carrier Syst. 6 (1989) 239^271.
[6] P.L. Felgner, Nonviral strategies for gene therapy, Sci. Am.276 (1997) 102^106.
[7] P.L. Felgner, Y. Barenholz, J.P. Behr, S.H. Cheng, P. Cullis,L. Huang, J.A. Jessee, L. Seymour, F. Szoka, A.R. Thierry,E. Wagner, G. Wu, Nomenclature for synthetic gene deliverysystems, Hum. Gene Ther. 8 (1997) 511^512.
[8] E. Wagner, M. Zenke, M. Cotten, H. Beug, M.L. Birnstiel,Transferrin^polycation conjugates as carriers for DNA up-take into cells, Proc. Natl. Acad. Sci. USA 87 (1990) 3410^3414.
[9] E. Wagner, M. Cotten, K. Mechtler, H. Kirlappos, M.L.Birnstiel, DNA-binding transferrin conjugates as functionalgene-delivery agents: synthesis by linkage of polylysine orethidium homodimer to the transferrin carbohydrate moiety,Bioconjug. Chem. 2 (1991) 226^231.
[10] J. Haensler, F.C.J. Szoka, Polyamidoamine cascade poly-mers mediate e¤cient transfection of cells in culture, Bio-conjug. Chem. 4 (1993) 372^379.
[11] J. Fominaya, W. Wels, Target cell-speci¢c DNA transfermediated by a chimeric multidomain protein ^ novel non-viral gene delivery system, J. Biol. Chem. 271 (1996) 10560^10568.
[12] J.D. Fritz, H. Herweijer, G.F. Zhang, J.A. Wol¡, Genetransfer into mammalian cells using histone-condensed plas-mid DNA, Hum. Gene Ther. 7 (1996) 1395^1404.
[13] S. Gottschalk, J.T. Sparrow, J. Hauer, M.P. Mims, F.E.Leland, S.L.C. Woo, L.C. Smith, A novel DNA^peptidecomplex for e¤cient gene transfer and expression in mam-malian cells, Gene Ther. 3 (1996) 448^457.
[14] B.A. Sosnowski, A.M. Gonzalez, L.A. Chandler, Y.J.Buechler, G.F. Pierce, A. Baird, Targeting DNA to cellswith basic ¢broblast growth factor (FGF2), J. Biol. Chem.271 (1996) 33647^33653.
[15] D.T. Curiel, S. Agarwal, E. Wagner, M. Cotten, Adeno-virus enhancement of transferrin-polylysine-mediated genedelivery, Proc. Natl. Acad. Sci. USA 88 (1991) 8850^8854.
[16] C.P. Lollo, D.Y. Kwoh, T.C. Mockler, P.M. Ley, M.S. Gui-do, C.C. Co¤n, R. Aleman, R.M. Bartholomew, D.J. Carlo,Nonviral gene delivery: vehicle and delivery characteriza-tion, Blood Coagul. Fibrinol. 8 (1997) S31^38.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190188

[17] G.Y. Wu, C.H. Wu, Receptor-mediated gene delivery andexpression in vivo, J. Biol. Chem. 263 (1988) 14621^14624.
[18] C. Plank, K. Zatloukal, M. Cotten, K. Mechtler, E. Wagner,Gene transfer into hepatocytes using asialoglycoprotein re-ceptor mediated endocytosis of DNA complexed with anarti¢cial tetra-antennary galactose ligand, Bioconjug.Chem. 3 (1992) 533^539.
[19] G. Scherphof, R.F.J. Dijkstra, H. Ellens, R. de Zanger, E.Wisse, Uptake of liposomes by rat and mouse heptocytesand Kup¡er cells, Biol. Cell 47 (1983) 47^58.
[20] E. Wisse, R.B. De Zanger, K. Charels, P. Van Der Smissen,R.S. McCuskey, The liver sieve: considerations concerningthe structure and function of endothelial fenestrae, the sinus-oidal wall and the space of Disse, Hepatology 5 (1985) 683^692.
[21] R.I. Mahato, Y. Takakura, M. Hashida, Nonviral vectorsfor in vivo gene delivery: physicochemical and pharmacoki-netic considerations, Crit. Rev. Ther. Drug Carrier Syst. 14(1997) 133^172.
[22] F.D. Ledley, Nonviral gene therapy: the promise of genes aspharmaceutical products, Hum. Gene Ther. 6 (1995) 1129^1144.
[23] F.D. Ledley, Pharmaceutical approach to somatic gene ther-apy, Pharm. Res. 13 (1996) 1595^1614.
[24] J. Smith, Y.L. Zhang, R. Niven, Toward development of anon-viral gene therapeutic, Adv. Drug Deliv. Rev. 26 (1997)135^150.
[25] C. Plank, K. Mechtler, F.C. Szoka Jr., E. Wagner, Activa-tion of the complement system by synthetic DNA com-plexes: a potential barrier for intravenous gene delivery,Hum. Gene Ther. 7 (1996) 1437^1446.
[26] M.A. Wolfert, L.W. Seymour, Atomic force microscopicanalysis of the in£uence of the molecular weight of poly-(L)lysine on the size of polyelectrolyte complexes formedwith DNA, Gene Ther. 3 (1996) 269^273.
[27] C.H. Wu, J.M. Wilson, G.Y. Wu, Targeting genes: deliveryand persistent expression of a foreign gene driven by mam-malian regulatory elements in vivo, J. Biol. Chem. 264 (1989)16985^16987.
[28] J.C. Perales, T. Ferkol, M. Molas, R.W. Hanson, An eval-uation of receptor-mediated gene transfer using syntheticDNA^ligand complexes, Eur. J. Biochem. 226 (1994) 255^266.
[29] J. Stankovics, A.M. Crane, E. Andrews, C.H. Wu, G.Y. Wu,F.D. Ledley, Overexpression of human methylmalonyl CoAmutase in mice after in vivo gene transfer with asialoglyco-protein/polylysine/DNA complexes, Hum. Gene Ther. 5(1994) 1095^1104.
[30] K.V. Shah, Polyomaviruses, in: B.N. Fields (Ed.), Virology,Raven Press, New York, 1990, pp. 1609^1623.
[31] P.G. Arscott, A.-Z. Li, V.A. Bloom¢eld, Condensation ofDNA by trivalent cations. 1. E¡ects of DNA length andtopology on the size and shape of condensed particles, Bio-polymers 30 (1990) 619^630.
[32] M.J. Allen, E.M. Bradbury, R. Balhorn, AFM analysis of
DNA^protamine complexes bound to mica, Nucleic AcidsRes. 25 (1997) 2221^2226.
[33] V.A. Bloom¢eld, Condensation of DNA by multivalent cat-ions: considerations on mechanism, Biopolymers 31 (1991)1471^1481.
[34] V.A. Bloom¢eld, DNA condensation, Curr. Opin. Struct.Biol. 6 (1996) 334^341.
[35] V.A. Bloom¢eld, S. He, A.-Z. Li, P.B. Arscott, Light scat-tering studies on DNA condensation, Biochem. Soc. Trans.19 (1990) 496.
[36] J.C. Perales, G.A. Grossmann, M. Molas, G. Liu, T. Ferkol,J. Harpst, H. Oda, R.W. Hanson, Biochemical and function-al characterization of DNA complexes capable of targetinggenes to hepatocytes via the asialoglycoprotein receptor,J. Biol. Chem. 272 (1997) 7398^7407.
[37] E. Wagner, M. Cotten, R. Foisner, M.L. Birnstiel, Trans-ferrin^polycation^DNA complexes: the e¡ect of polycationson the structure of the complex and DNA delivery to cells,Proc. Natl. Acad. Sci. USA 88 (1991) 4255^4259.
[38] L. Warren, The thiobarbituric acid assay of sialic acids,J. Biol. Chem. 234 (1959) 1971^1975.
[39] Y. Uchida, Y. Tsukada, T. Sugimori, Distribution of neu-raminidase in Arthobacter and its puri¢cation by a¤nitychromatography, J. Biochem. 82 (1977) 1425^1433.
[40] T.D. McKee, M.E. DeRome, G.Y. Wu, M.A. Findeis, Prep-aration of asialoorosomucoid-polylysine conjugates, Biocon-jug. Chem. 5 (1994) 306^311.
[41] J.R. Merwin, G.S. Noell, W.L. Thomas, H.C. Chiou, M.E.DeRome, T.D. McKee, G.L. Spitalny, M.A. Findeis, Tar-geted delivery of DNA using YEE(GalNAcAH)3, a syntheticglycopeptide ligand for the asialoglycoprotein receptor, Bio-conjug. Chem. 5 (1994) 612^620.
[42] J.M. Dust, Z. Fang, J.M. Harris, Proton NMR character-ization of poly(ethylene glycols) and derivatives, Macromo-lecules 23 (1990) 3742^3746.
[43] J. Sambrook, E.F. Fritsch, T. Maniatis, Molecular Cloning:A Laboratory Manual, 2nd edn., Cold Spring Harbor Lab-oratory Press, Cold Spring Harbor, NY, 1989.
[44] R.J. Lee, L. Huang, Folate-targeted, anionic liposome-en-trapped polylysine-condensed DNA for tumor cell-speci¢cgene transfer, J. Biol. Chem. 271 (1996) 8481^8487.
[45] M.X. Tang, F.C. Szoka, The in£uence of polymer structureon the interactions of cationic polymers with DNA and mor-phology of the resulting complexes, Gene Ther. 4 (1997)823^832.
[46] S. Bhattacharya, S.S. Mandal, Interaction of surfactantswith DNA. Role of hydrophobicity and surface charge onintercalation and DNA melting, Biochim. Biophys. Acta1323 (1997) 29^44.
[47] M.A. Hayat and S.E. MIller, Negative Staining, McGraw-Hill, New York, 1990.
[48] B. Ceh, M. Winterhalter, P.M. Frederik, J.J. Vallner, D.D.Lasic, Stealth0 liposomes: from theory to product, Adv.Drug Deliv. Rev. 24 (1997) 165^177.
[49] D. Lew, S.E. Parker, T. Latimer, A.M. Abai, A. Kuwahara-Rundell, S.G. Doh, Z.Y. Yang, D. Laface, S.H. Gromkow-
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190 189

ski, G.J. Nabel et al., Cancer gene therapy using plasmidDNA: pharmacokinetic study of DNA following injectionin mice, Hum. Gene Ther. 6 (1995) 553^564.
[50] A.V. Kabanov, I.V. Astafyeva, M.L. Chikindas, G.F.Rosenblat, V.I. Kiselev, E.S. Severin, V.A. Kabanov, DNAinterpolyelectrolyte complexes as a tool for e¤cient celltransformation, Biopolymers 31 (1991) 1437^1443.
[51] E.R. Lee, J. Marshall, C.S. Siegel, C. Jiang, N.S. Yew, M.R.Nichols, J.B. Nietupski, R.J. Ziegler, M.B. Lane, K.X.Wang, N.C. Wan, R.K. Scheule, D.J. Harris, A.E. Smith,S.H. Cheng, Detailed analysis of structures and formulationsof cationic lipids for e¤cient gene transfer to the lung, Hum.Gene Ther. 7 (1996) 1701^1717.
[52] E. Wagner, C. Plank, K. Zatloukal, M. Cotten, M.L. Birn-stiel, In£uenza virus hemagglutinin HA-2 N-terminal fuso-genic peptides augment gene transfer by transferrin^polyly-sine^DNA complexes: toward a synthetic virus-like gene-transfer vehicle, Proc. Natl. Acad. Sci. USA 89 (1992)7934^7938.
[53] E. Wagner, K. Zatloukal, M. Cotten, H. Kirlappos, K.Mechtler, D.T. Curiel, M.L. Birnstiel, Coupling of adenovi-rus to transferrin^polylysine/DNA complexes greatly enhan-ces receptor-mediated gene delivery and expression of trans-fected genes, Proc. Natl. Acad. Sci. USA 89 (1992) 6099^6103.
[54] C. Plank, B. Oberhauser, K. Mechtler, C. Koch, E. Wagner,The in£uence of endosome-disruptive peptides on gene trans-fer using synthetic virus-like gene transfer systems, J. Biol.Chem. 269 (1994) 12918^12924.
[55] N.R. Chowdhury, C.H. Wu, G.Y. Wu, P.C. Yerneni, V.R.Bommineni, J.R. Chowdhury, Fate of DNA targeted to theliver by asialoglycoprotein receptor-mediated endocytosis invivo. Prolonged persistence in cytoplasmic vesicles after par-tial hepatectomy, J. Biol. Chem. 268 (1993) 11265^11271.
[56] G.Y. Wu, J.M. Wilson, F. Shalaby, M. Grossman, D.A.Shafritz, C.H. Wu, Receptor-mediated gene delivery invivo. Partial correction of genetic analbuminemia in Nagaserats, J. Biol. Chem. 266 (1991) 14338^14342.
[57] M. Yamamoto, N. Hayashi, Y. Miyamoto, T. Takehara, E.Mita, M. Seki, H. Fusamoto, T. Kamada, In vivo transfec-tion of hepatitis C virus complementary DNA into rodentliver by asialoglycoprotein receptor mediated gene delivery,Hepatology 22 (1995) 847^855.
[58] M.A. Wolfert, E.H. Schacht, V. Toncheva, K. Ulbrich, O.Nazarova, L.W. Seymour, Characterization of vectors forgene therapy formed by self-assembly of DNA with syntheticblock co-polymers, Hum. Gene Ther. 7 (1996) 2123^2133.
[59] D.C. Litzinger, A.M. Buiting, N. van Rooijen, L. Huang,E¡ect of liposome size on the circulation time and intraor-gan distribution of amphipathic poly(ethylene glycol)-con-taining liposomes, Biochim. Biophys. Acta 1190 (1994) 99^107.
[60] M. Foradada, A. Manzano, T. Roig, J. Estelrich, J. Bermu-dez, Serum^liposome interaction is an oxygen-dependentprocess, Biochim. Biophys. Acta 1345 (1997) 43^55.
[61] P.C. Hiemenz, Principles of Colloid and Surface Chemistry,2nd edn., Marcel Dekker, New York, NY, 1986.
[62] V. Toncheva, M.A. Wolfert, P.R. Dash, D. Oupicky, K.Ulbrich, L.W. Seymour, E.H. Schacht, Novel vectors forgene delivery formed by self-assembly of DNA with poly-(L-lysine) grafted with hydrophilic polymers, Biochim. Bio-phys. Acta 1380 (1998) 354^368.
[63] T.M. Allen, C. Hansen, Pharmacokinetics of stealth versusconventional liposomes: e¡ect of dose, Biochim. Biophys.Acta 1068 (1991) 133^141.
BBAEXP 93238 8-2-99 Cyaan Magenta Geel Zwart
D.Y. Kwoh et al. / Biochimica et Biophysica Acta 1444 (1999) 171^190190
Recommended

















