Embed Size (px)
Citation preview

PATOLOGIA OSTEOARTICULAR
JUAN ALBERTO PEREZ CARDONA

El aparato locomotor transmite forma y movimiento al
cuerpo humano. Además de proporcionar puntos de
apoyo y palancas contra los que se contraen los
músculos para permitir que se produzca el movimiento
El esqueleto desempeña una función critica para la
homeostasia mineral (en especial del calcio) y protege
las vísceras proporcionando un ambiente para el
desarrollo
Las patologías osteoarticulares abarcan un gran numero de
afecciones, que van desde lesiones benignas y localizadas del
hueso como el osteocondroma, hasta trastornos generalizados
como la osteoporosis , la ontogénesis
Imperfecta , etc.
A continuación hablaremos al respecto

EMBRIOGENESIS
Los huesos del cuerpo humano se desarrollan a partir de las placas paraxial y lateral (división parietal) del mesodermo y a partir de la cresta neural
El mesodermo paraxial forma unas series segmentadas de bloques tisulares en cada lado del tubo neural que en la región cefálica se conoce como Somitomeros y de la región cefálico caudal como Somitas
Los Somitas se diferencian en una parte ventromedial (Esclerotoma) y una Dorsolateral (Dermomiotoma)
Al final de la 4 semana las células del esclerotoma se vuelven polimorfas y forman el mesénquima embrionario
Las células mesenquimatosas de diferencian en fibroblastos, condroblastos u osteoblastos
Existen dos maneras de formación osea: OSIFICACION INTRAMEMBRANOSA Y OSIFICACION ENDOCONDRAL
Osificacion intramembranosa: Mesenquima se diferencia directamente en hueso
Osificacion Endocondral: Celulas mesenquimatosas primero originan moldes de cartílago hialino, que en su momento se osifican
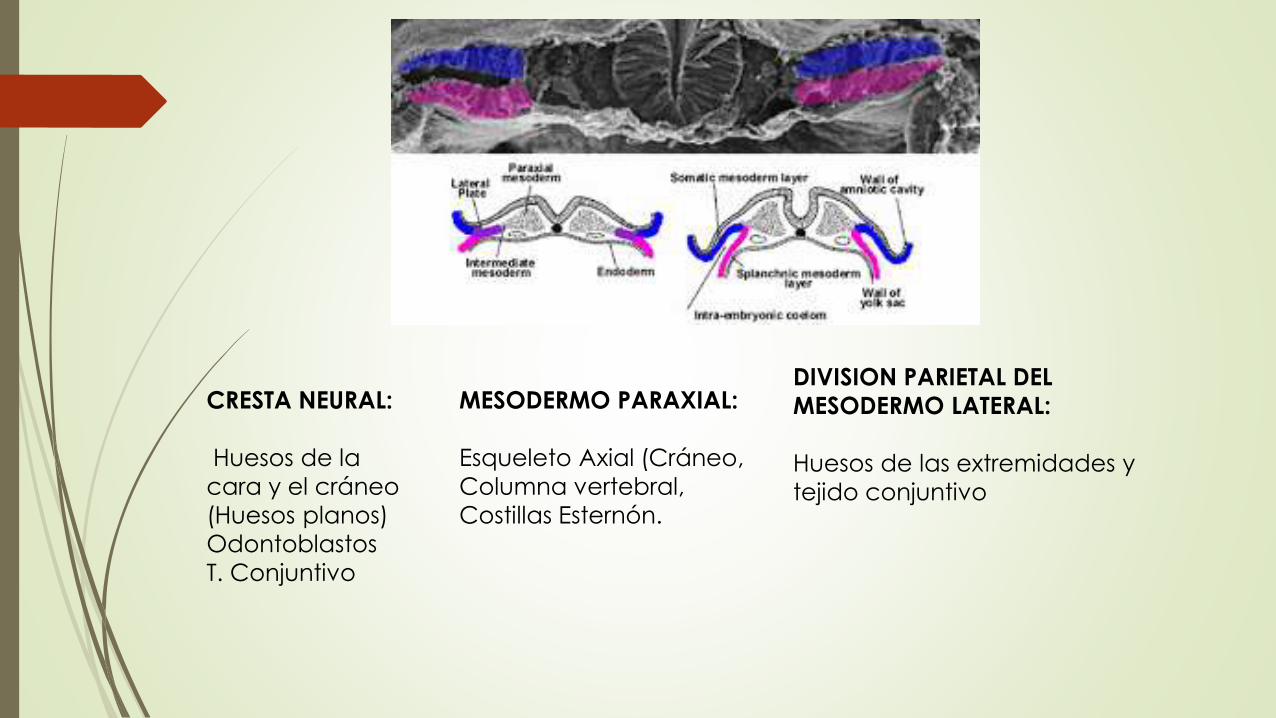
CRESTA NEURAL:
Huesos de la
cara y el cráneo
(Huesos planos)
Odontoblastos
T. Conjuntivo
MESODERMO PARAXIAL:
Esqueleto Axial (Cráneo,
Columna vertebral,
Costillas Esternón.
DIVISION PARIETAL DEL
MESODERMO LATERAL:
Huesos de las extremidades y
tejido conjuntivo
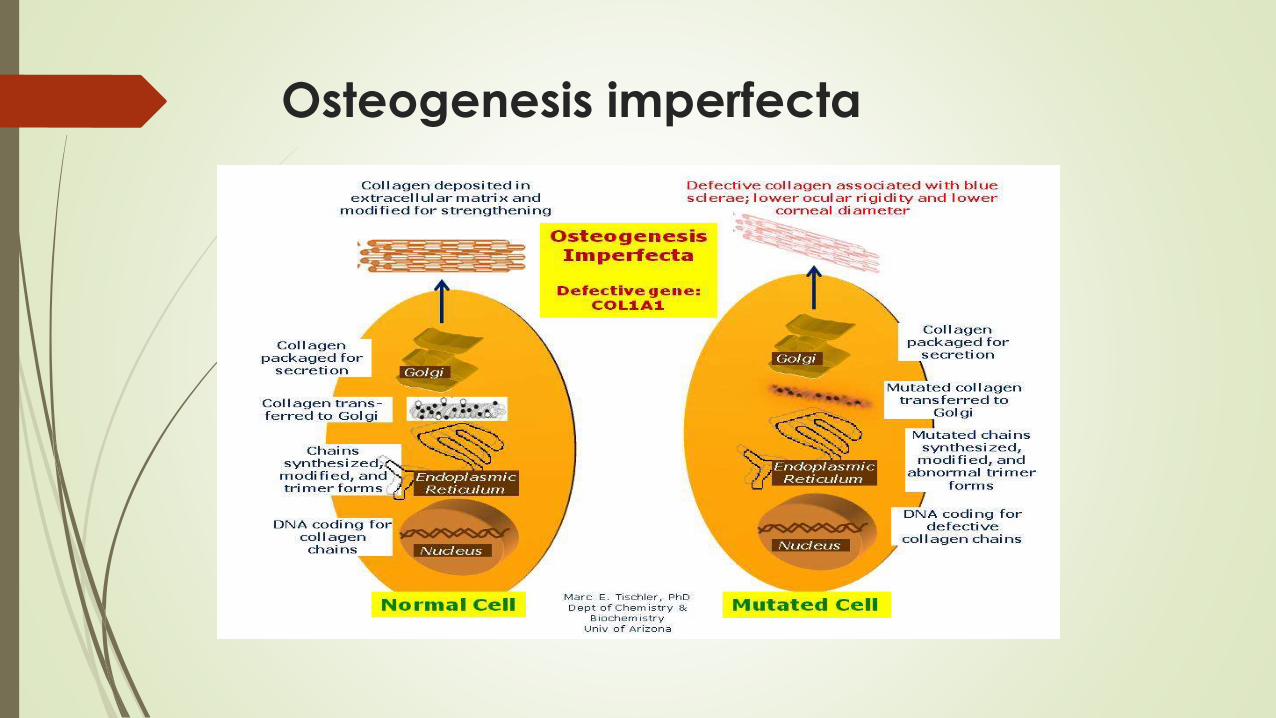
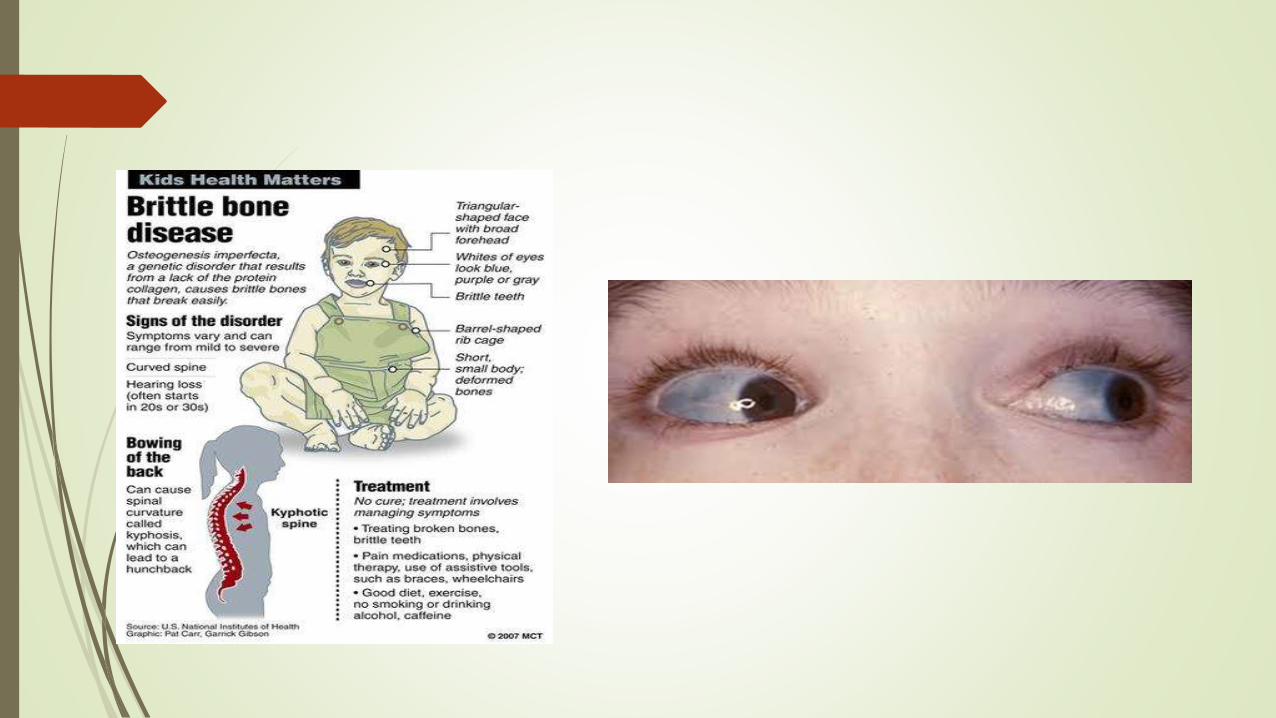
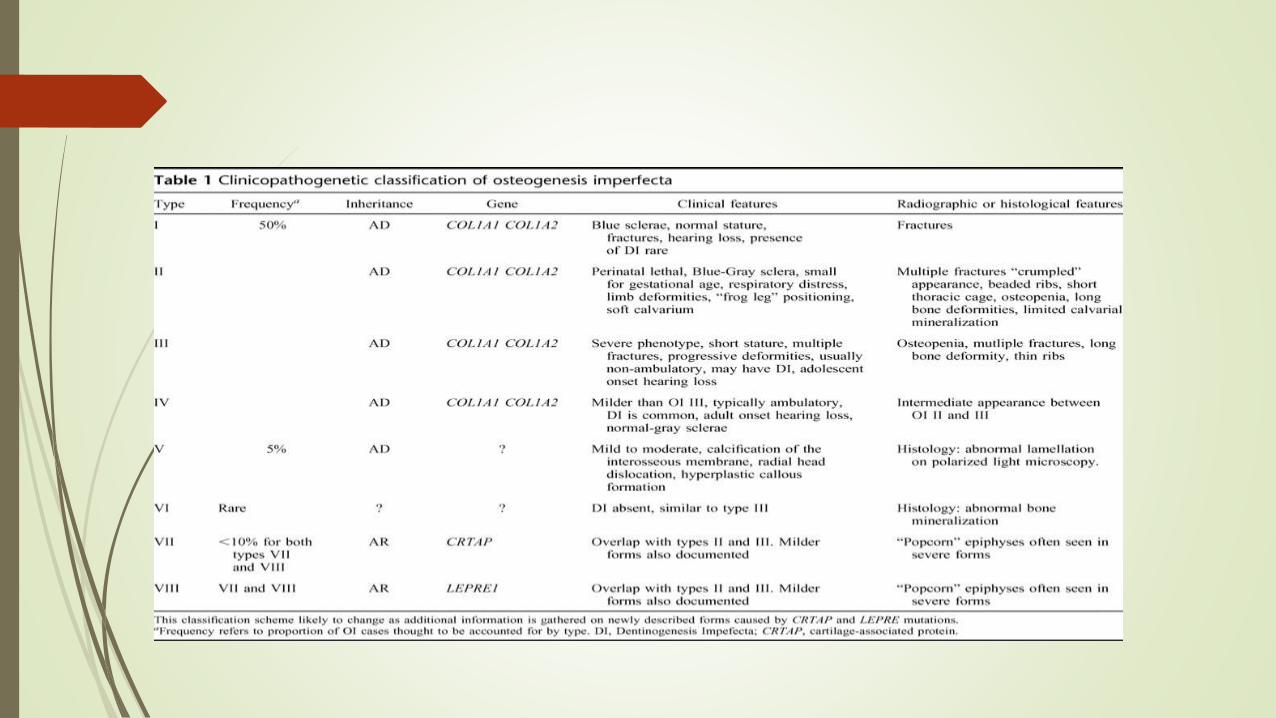
Osteogenesis imperfecta
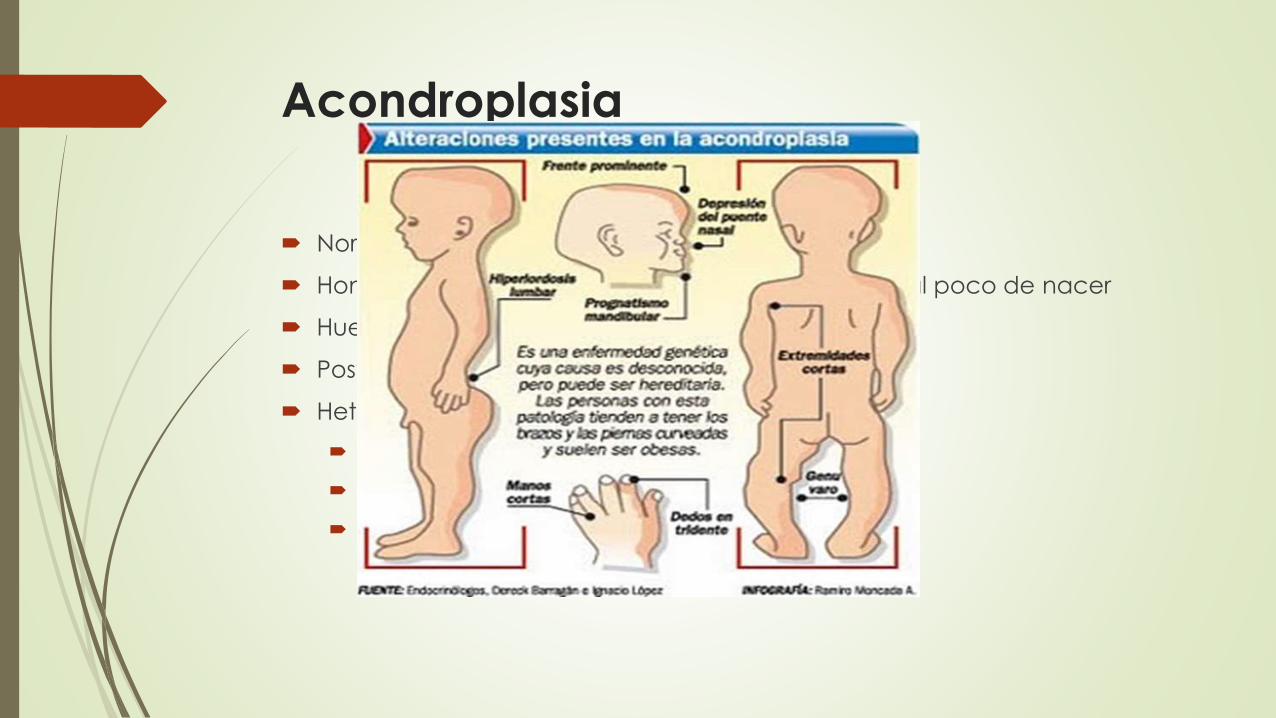
Acondroplasia
Normalmente autosómico dominante
Homocigotos: muerte por insuficiencia respiratoria al poco de nacer
Huesos de origen cartilaginosa
Postura lordótica (lomo hundido)
Heterocigotos afectados
Acortamiento extremo de las extremidades
Protrusión frontal del cráneo
Insuficiencia respiratoria mortal (período perinatal)

Placas de crecimiento del cartílago están desorganizadas e hipoplásicas
Enanismo tanatofórico : 1 de cada 20.000 nacidos vivos
Mutaciones del FGFR3 (receptor 3 del factor de crecimiento de fibroblastos
G->A Glicina-> Agrinina
Osteopetrosis
disminución de la resorción ósea mediada por los osteoclastos y, por lo
tanto, por una remodelación ósea defectuosa
hueso afectado de aspecto pétreo
La resorción ósea
Matriz se descalcifica
El espacio extracelular se acidifica mediante una bomba de protones
La matriz de hidroxiapatita inorgánica se disuelve.
anhidrasa carbónica II
excreción de iones hidrógeno
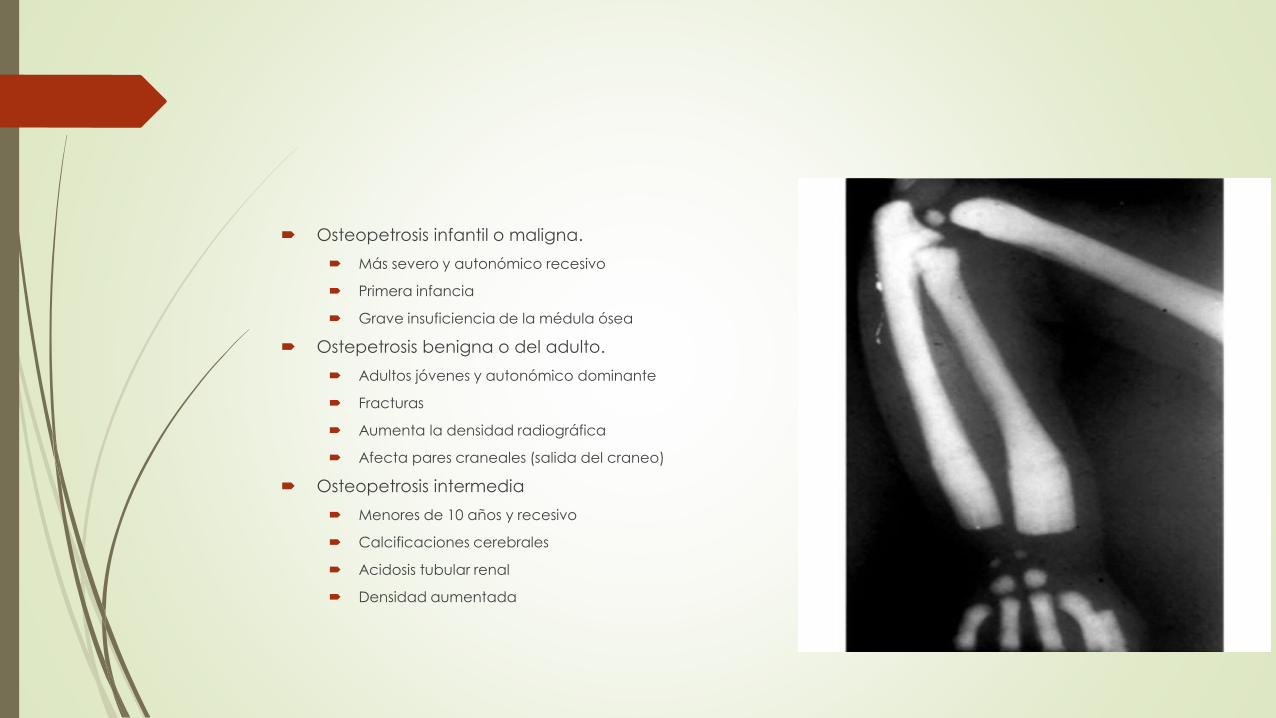
Osteopetrosis infantil o maligna.
Más severo y autonómico recesivo
Primera infancia
Grave insuficiencia de la médula ósea
Ostepetrosis benigna o del adulto.
Adultos jóvenes y autonómico dominante
Fracturas
Aumenta la densidad radiográfica
Afecta pares craneales (salida del craneo)
Osteopetrosis intermedia
Menores de 10 años y recesivo
Calcificaciones cerebrales
Acidosis tubular renal
Densidad aumentada

Evolucion clinica
Aumento difuso de la densidad
Huesos del cráneo, pelvis ósea, manos y pies
Fracturas múltiples espontáneas
Anemia mieloptísica (anemia, leucopenia y trombocitopenia)
Infecciones recurrentes y hemorrágicas
Esplenomegalia por metaplasia mieloide
Ausencia o dentición anormal
Presión intracraneal incrementada
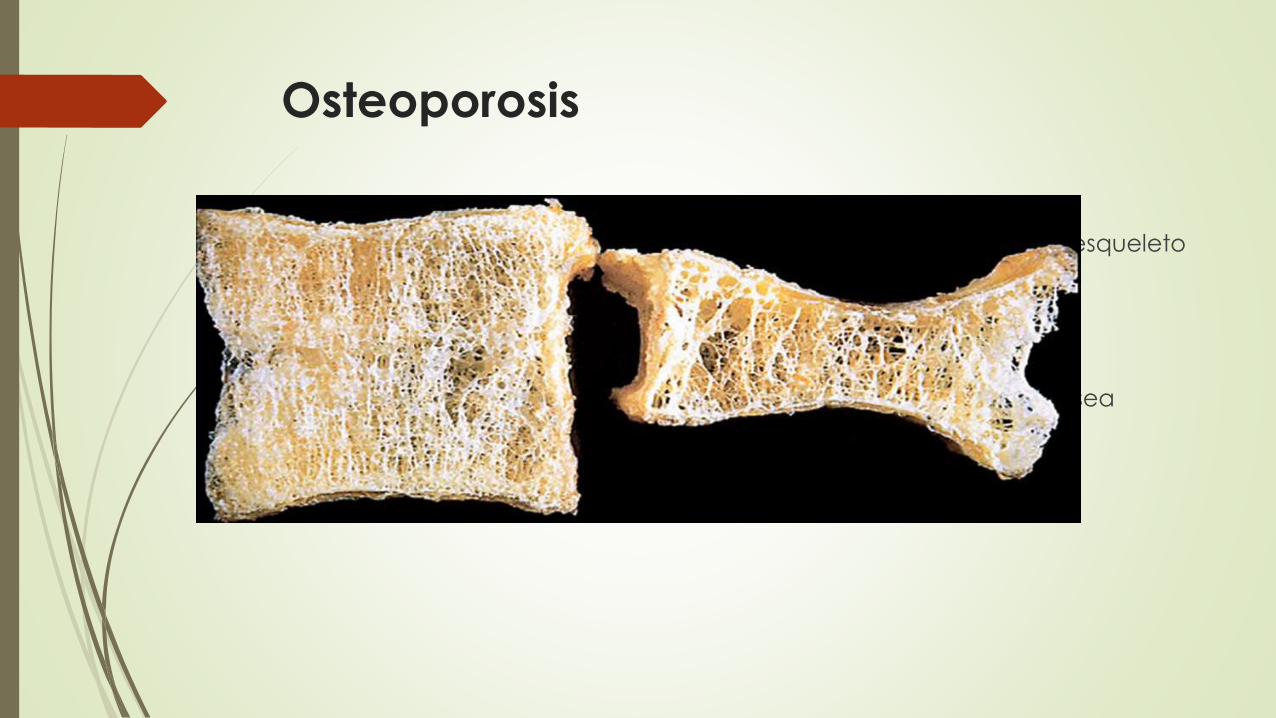
Osteoporosis
Enfermedad caracterizada por un aumento de la porosidad del esqueleto
secundaria a una disminución de la masa ósea.
Región o a un hueso
Senil y la posmenopáusica
Tercera o cuarta décadas de vida en ambos sexos la resorción ósea
empieza a superar al depósito de hueso
Promedio del 0,7%
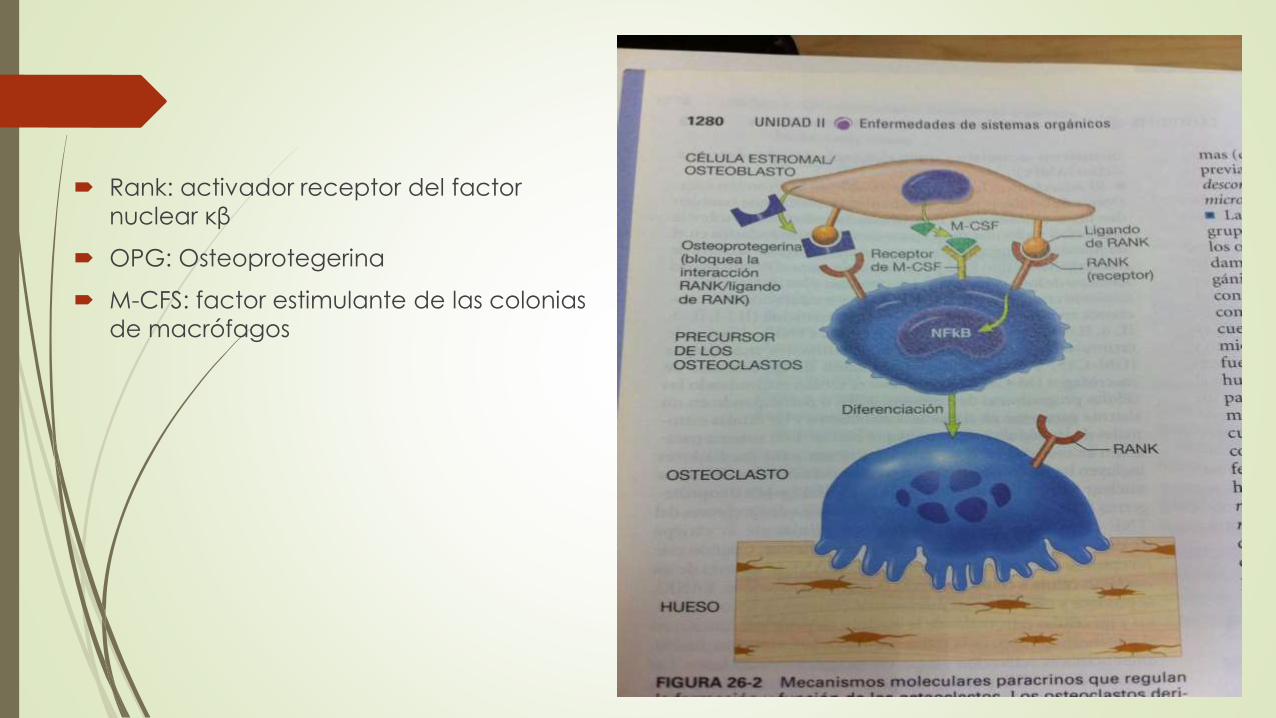
Rank: activador receptor del factor
nuclear κβ
OPG: Osteoprotegerina
M-CFS: factor estimulante de las colonias
de macrófagos


Cambios relacionados con edad
Los osteoclastos siguen conservando su vigor juvenil mientras decrece la síntesis
de hueso nuevo al envejecer
Factores genéticos
Los polimorfismos del receptor de la vitamina D son los responsables de,
aproximadamente, el 75% del valor máximo de masa ósea que se alcanza en
un individuo.

Estado nutricional del calcio
En la mayoría de las adolescentes la ingesta dietética es insuficiente.
Desafortunadamente, este déficit de calcio se produce durante un período de
crecimiento óseo rápido. Como consecuencia, no alcanzan el valor óseo
máximo que deberían lograr y, por lo tanto, muestran propensión a desarrollar
una osteoporosis clínicamente significativa a una edad más temprana.

Influencias hormonales
Los efectos hipoestrogénicos son atribuibles, en parte, a una hiperproducción de citocinas
(especialmente de interleucina-1 y TNF).
Aumento de la actividad del conjunto del receptor RANK con su ligando y una disminución de la
OPG
Actividad física
Las fuerzas mecánicas estimulan la remodelación del hueso, por lo que una disminución de la
actividad física incrementa la pérdida ósea.

Evolución clínica
Fracturas vertebrales torácicas y lumbares
Pérdida de altura y diferentes deformidades como cifoscoliosis
Las embolias pulmonares y las neumonías son complicaciones frecuentes de las
fracturas del cuello del fémur, la pelvis o la columna

• La prevención y el tratamiento:
Ingesta dietética adecuada de calcio, complemetos de vitamina D y un régimen de ejercicio regular instaurado antes de los 30 años
• Bifosfonatos: disminuir la resorción ósea.
• Los agonistas selectivos de los receptores de estrógenos
Sin los efectos adversos asociados a tratamiento con estrógenos.
• Hormona paratiroidea
No pueden tolerar un tratamiento con estrógenos.
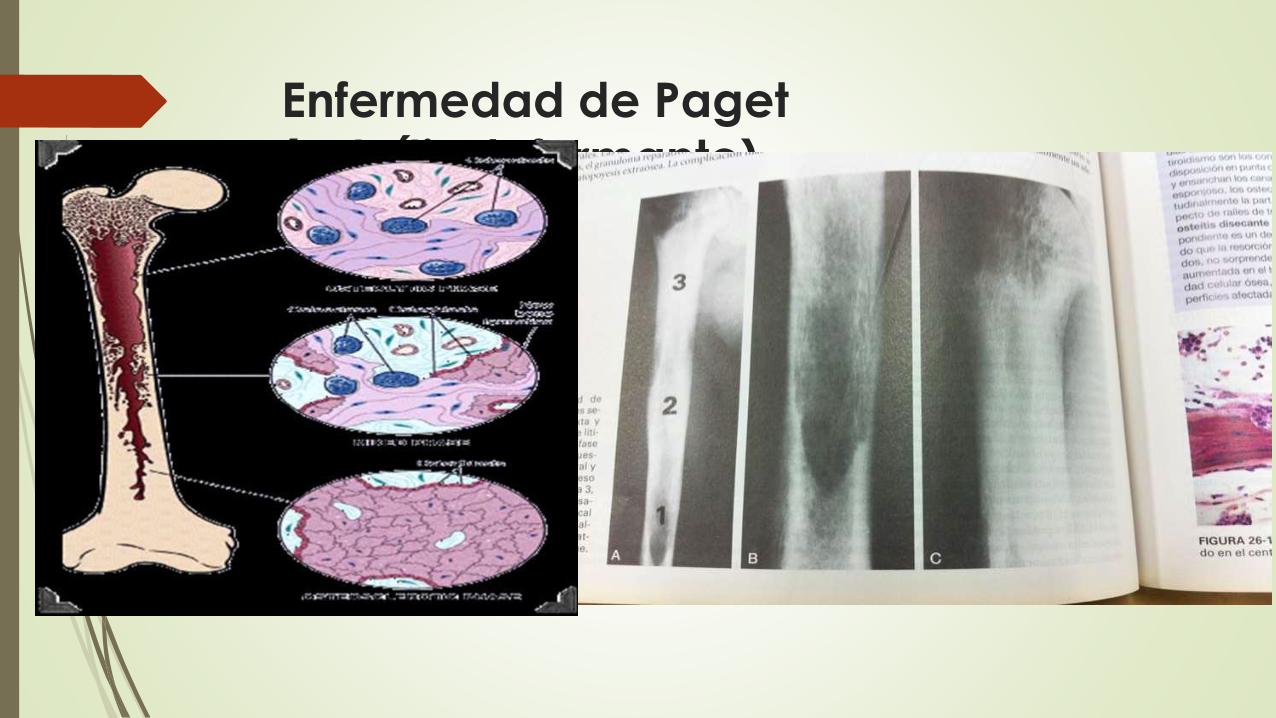
Enfermedad de Paget(osteítis deformante)
Es un tipo de osteopatía en la que se produce una renovación acelerada,
aunque defectuosa, del tejido óseo unida a hipervascularización ósea. Es
también llamada osteítis deformante. Esta singular enfermedad se
caracteriza por regiones de resorción ósea osteoclástica frenética,
seguidas de un período de formación ósea intensiva y, por último, una
disminución notable de la actividad de las células óseas. Esta secuencia
repetitiva y superpuesta es la base para dividir la enfermedad de Paget en
una fase osteolítica inicial, seguida de una fase mixta osteoclástica-
osteoblástica, que finaliza con un predominio de actividad osteoblástica y
finalmente evoluciona a una fase osteoesclerótica quiescente. El efecto
neto de este proceso es una ganancia de masa ósea, sin embargo, el
hueso recién formado está alterado y es de arquitectura poco sólida.
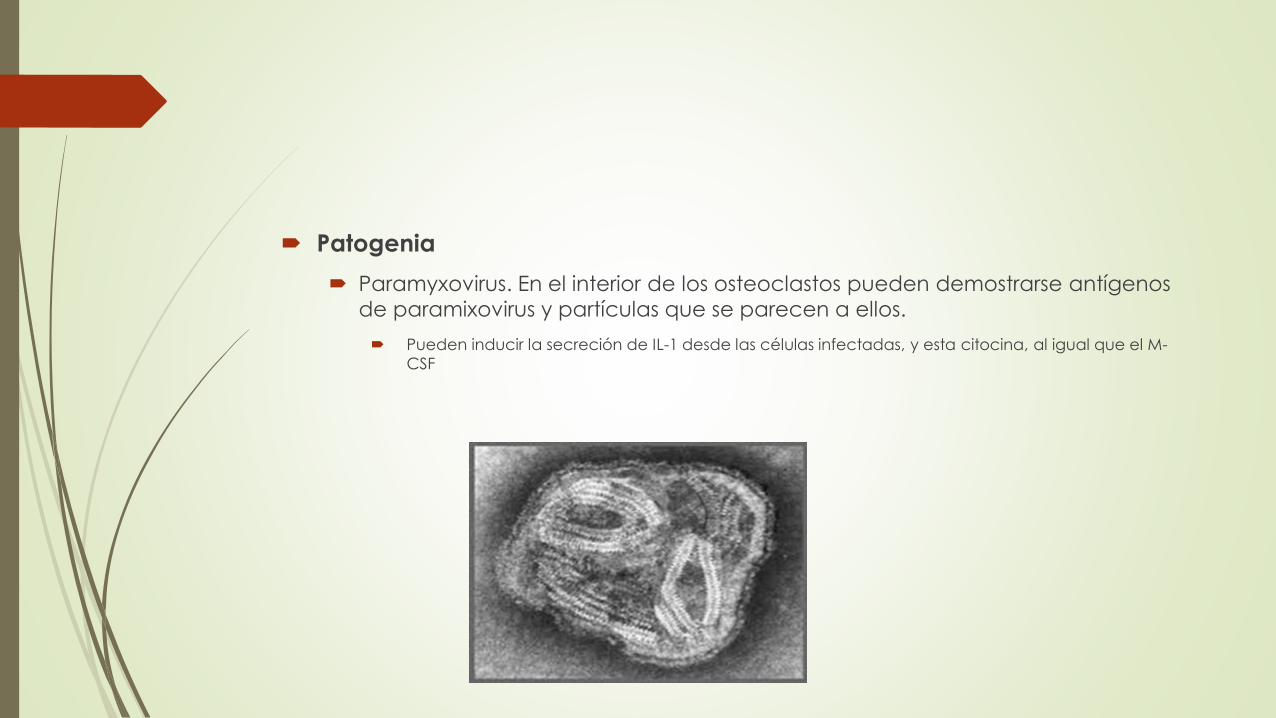
Patogenia
Paramyxovirus. En el interior de los osteoclastos pueden demostrarse antígenos
de paramixovirus y partículas que se parecen a ellos.
Pueden inducir la secreción de IL-1 desde las células infectadas, y esta citocina, al igual que el M-
CSF
Herencia autosómica recesiva
Gen SQSTM1: las mutaciones de este gen aumentan la actividad de NFκβ
mediante señalización RANK, con aumento de la actividad del osteoclasto y
aumento de la propensión a la enfermedad.

La elevación en las concentraciones de fosfatasa alcalina y el aumento de la
excreción urinaria de hidroxiprolina reflejan un recambio óseo exuberante.
Las lesiones óseas hipervasculares precoces provocan calor de la piel y el tejido
subcutáneo que las recubre.
Hipervascularidad puede dar lugar a una insuficiencia cardíaca de gasto alto.
En la fase proliferativa de la enfermedad con afectación del cráneo, los
síntomas más frecuentes y que pueden atribuirse a la compresión nerviosa
consisten en cefalea y trastornos visuales y auditivos.
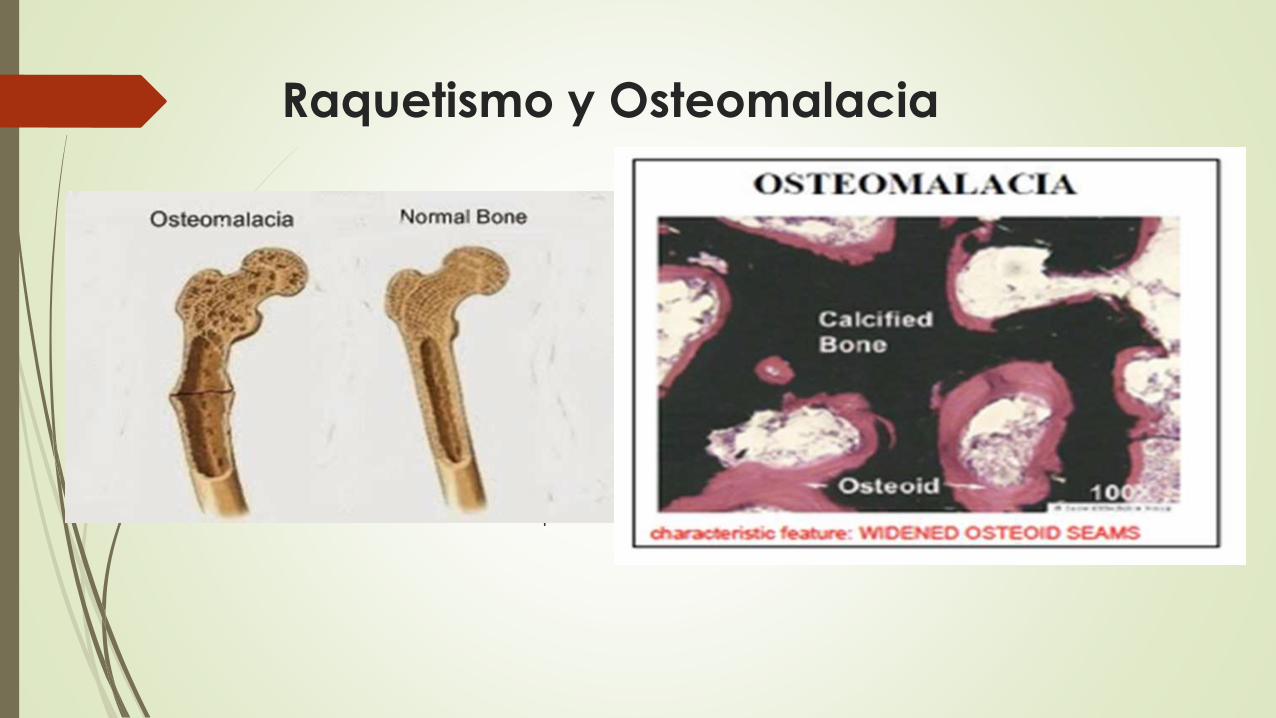
Raquetismo y Osteomalacia
Son manifestaciones del déficit o de un metabolismo anormal de la
vitamina D
Es una mineralización ósea defectuosa que da lugar a una
sobreabundancia de osteoide no mineralizado.
El raquitismo hace referencia a un trastorno infantil en el que un
crecimiento óseo desordenado y da origen a deformidades esqueléticas.
La osteomalacia es el equivalente en el adulto; la mineralización del hueso
que se forma durante el proceso de remodelación es inferior a la normal,
ocasionando osteopenia y predisposición a las fracturas.
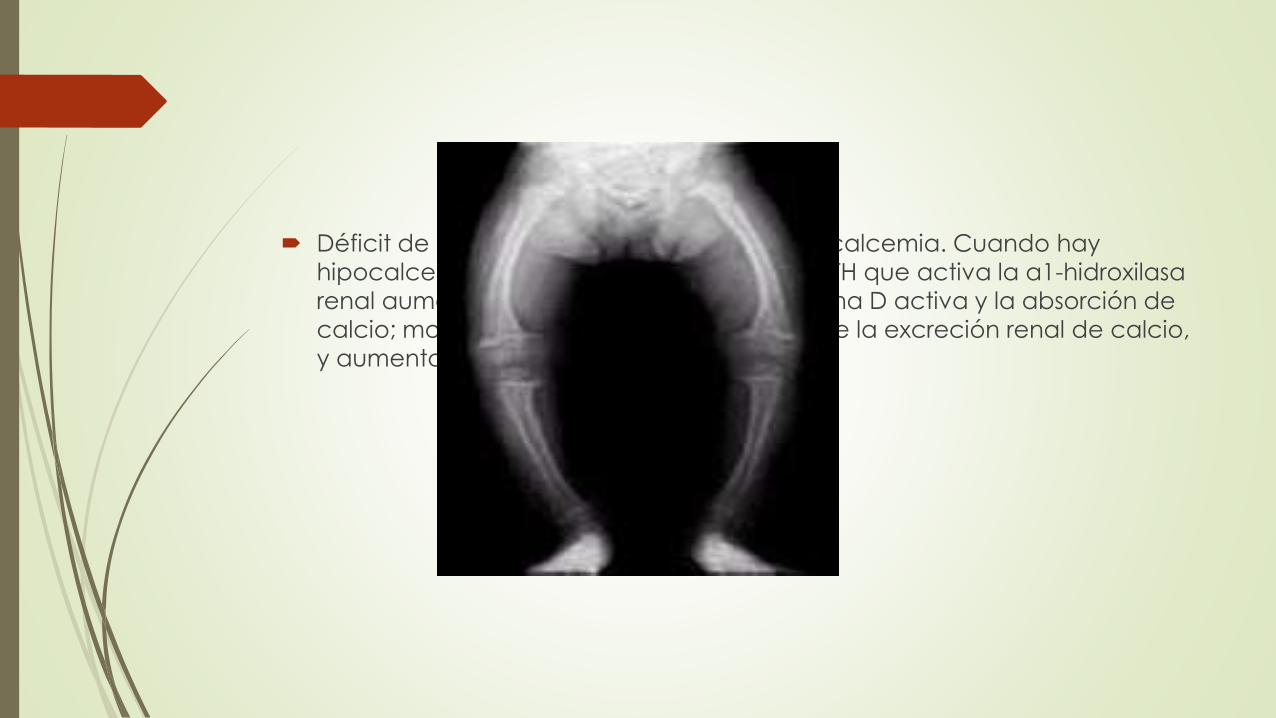
Déficit de vitamina D tiende a producir hipocalcemia. Cuando hay
hipocalcemia, aumenta la producción de PTH que activa la α1-hidroxilasa
renal aumentando así la cantidad de vitamina D activa y la absorción de
calcio; moviliza el calcio del hueso; disminuye la excreción renal de calcio,
y aumenta la excreción renal de fosfato.
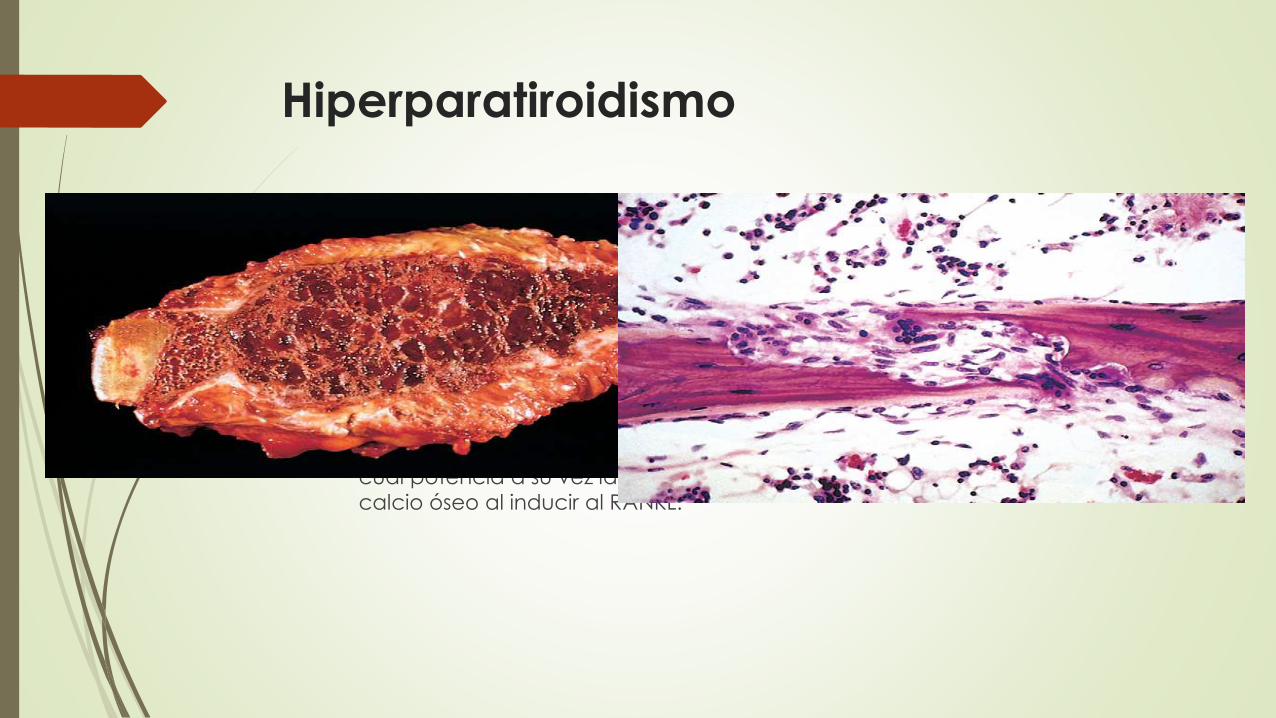
Hiperparatiroidismo
Efectos sobre el sistema oseo
La activación de los osteoclastos, aumentando la resorción ósea y la
movilización del calcio. Este efecto está mediado indirectamente por un
aumento en la producción de RANKL por parte de los osteoblastos.
Aumento de la resorción del calcio en los túbulos renales.
Aumento de la excreción urinaria de fosfatos.
Aumento de la síntesis de vitamina D activa, calcitriol, por parte de los riñones, lo
cual potencia a su vez la absorción de calcio desde el intestino y moviliza el
calcio óseo al inducir al RANKL.

Insuficiencia renal crónica, hay una síntesis inadecuada de calcitriol que
afecta, en último término, a la absorción gastrointestinal de calcio
La hiperfosfatemia de la insuficiencia renal suprime la α1-hidroxilasa renal,
deteriorando aún más la síntesis de vitamina D; otras influencias son la
acidosis metabólica y el depósito de aluminio en el hueso
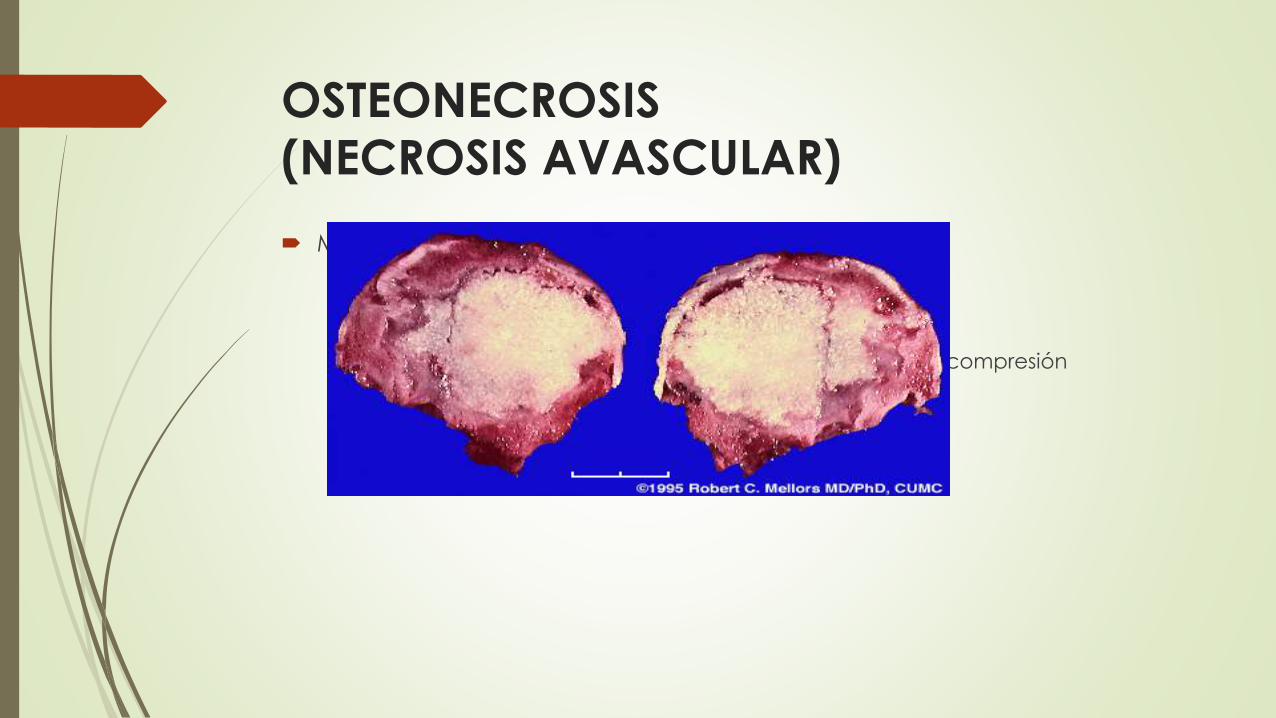
OSTEONECROSIS (NECROSIS AVASCULAR)
Mecanismos que contribuyen a la isquemia del hueso
Compresión o desorganización vascular
Administración de corticoides.
Enfermedad tromboembólica en la enfermedad por descompresión
Vasculopatía primaria

El hueso muerto con lagunas vacías se entremezcla con áreas de necrosis
grasa y jabones de calcio insolubles.
Los osteoclastos pueden reabsorber muchas de las trabéculas óseas
necróticas; cualquier fragmento de hueso muerto que permanezca puede
actuar a modo de andamiaje para la formación de hueso nuevo, un
proceso que recibe el nombre de sustitución progresiva.

Evolución clínica
Los infartos subcondrales debutan inicialmente con dolor durante la actividad
física, adquiriendo un carácter más persistente con el paso del tiempo.
Los infartos medulares suelen ser clínicamente silentes, salvo los de gran tamaño
(p. ej., enfermedad de Gaucher, enfermedad por descompresión o
drepanocitosis).
Los infartos medulares suelen ser estables; sin embargo, los subcondrales a
menudo se hunden y pueden dar lugar a una artrosis intensa.

Osteomielitis purulenta
Vias de llegada de microorganismo
1) Diseminación hematógena
más frecuente
2) Extensión desde una infección en una articulación o partes blandas
adyacentes
3) Implantación traumática después de fracturas abiertas o procedimientos
ortopédicos.

Las bacterias se proliferan, inducen una reacción inflamatoria aguda y provocan muerte celular.
El hueso sufre una necrosis precoz
El hueso muerto en los focos infectados se denomina secuestro.
Las bacterias y la inflamación pueden filtrarse a través de los sistemas de Havers para alcanzar el periostio.
La unión del periostio a la cortical es laxa y pueden formarse abscesos subperiósticos grandes y extenderse a distancia a lo largo de la superficie del hueso
En niños
La elevación adicional del periostio deteriora aún más la irrigación de la región afectada
Lesiones purulentas como las isquémicas pueden ocasionar una necrosis ósea segmentaria.
La rotura del periostio puede dar lugar a la aparición de abscesos en los tejidos blandos circundantes y a la formación de fístulas.
En ocasiones, el secuestro disminuye y da lugar a la formación de cuerpos extraños libres que viajan a través del trayecto del seno.

Staphylococcus aureus
Microorganismo mas frecuente
La expresión de proteínas de superficie que permiten su adhesión a la
matriz ósea propensa a infectar el hueso
Escherichia coli y los estreptococos del grupoB
Msteomielitis aguda en recién nacidos
Salmonella
Individuos con drepanocitosis

En lactantes, la infección epifisaria puede diseminarse hacia la articulación
adyacente para dar lugar a una artritis purulenta
La citocina leucocitaria liberada estimula la resorción ósea osteoclástica,
el crecimiento de tejido fibroso hacia el interior y la formación de hueso en
la periferia.

Características clínicas
Manifiesta clásicamente como una enfermedad generalizada aguda
Malestar, fiebre, leucocitosis y dolor pulsátil sobre la región afectada.
Características radiológicas
Foco lítico destructivo rodeado de un reborde esclerótico
Normalmente se necesita una biopsia y cultivos de hueso para identificar al
microorganismo causante.

Tratamiento
Combinación de antibióticos y drenaje quirúrgico
25% de los casos permanece en forma de infección crónica
Cronicidad
Demora el diagnóstico
Si la necrosis ósea es extensa
Si se acorta el tratamiento antibiótico
Si el desbridamiento quirúrgico es inadecuado
Si las defensas del huésped están debilitadas.
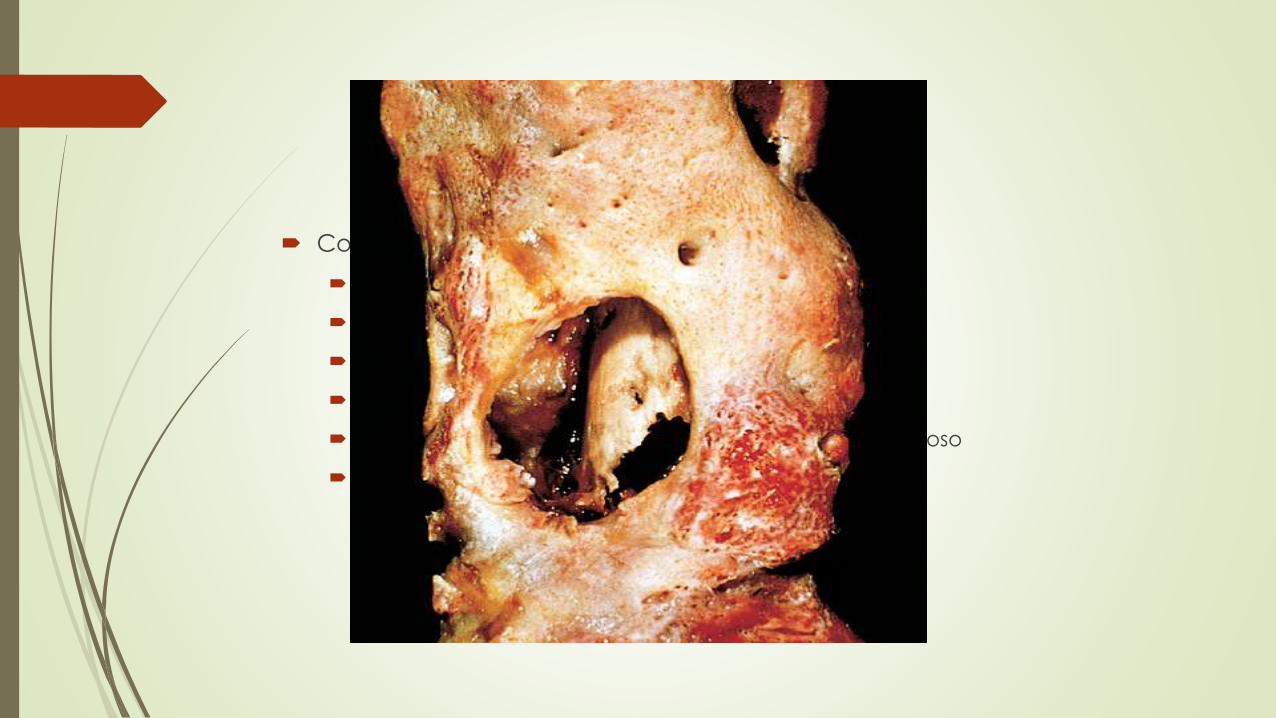
Complicaciones Osteomielitis cronica
Fracturas patológicas
Amiloidosis secundaria
Endocarditis
Sepsis
Aparición de carcinoma escamoso en el trayecto fistuloso
Osteosarcoma
Rara vez

Osteomielitis tuberculosa
La infección ósea complica entre un 1 y un 3% de los casos de tuberculosis
pulmonar.
Los microorganismos alcanzan el hueso por vía hematógena o por
diseminación directa desde un foco infeccioso contiguo (p. ej., desde
ganglios mediastínicos hasta las vértebras).
Via Hematogena
Localizaciones más frecuentes son los huesos largos y las vértebras.

Primoinfección es en la sinovial, debido a que sus presiones de oxígeno son
más altas.
Infección se disemina hacia las epífisis adyacentes donde da lugar a una
inflamación granulomatosa típica, con necrosis caseosa y destrucción
ósea extensa.
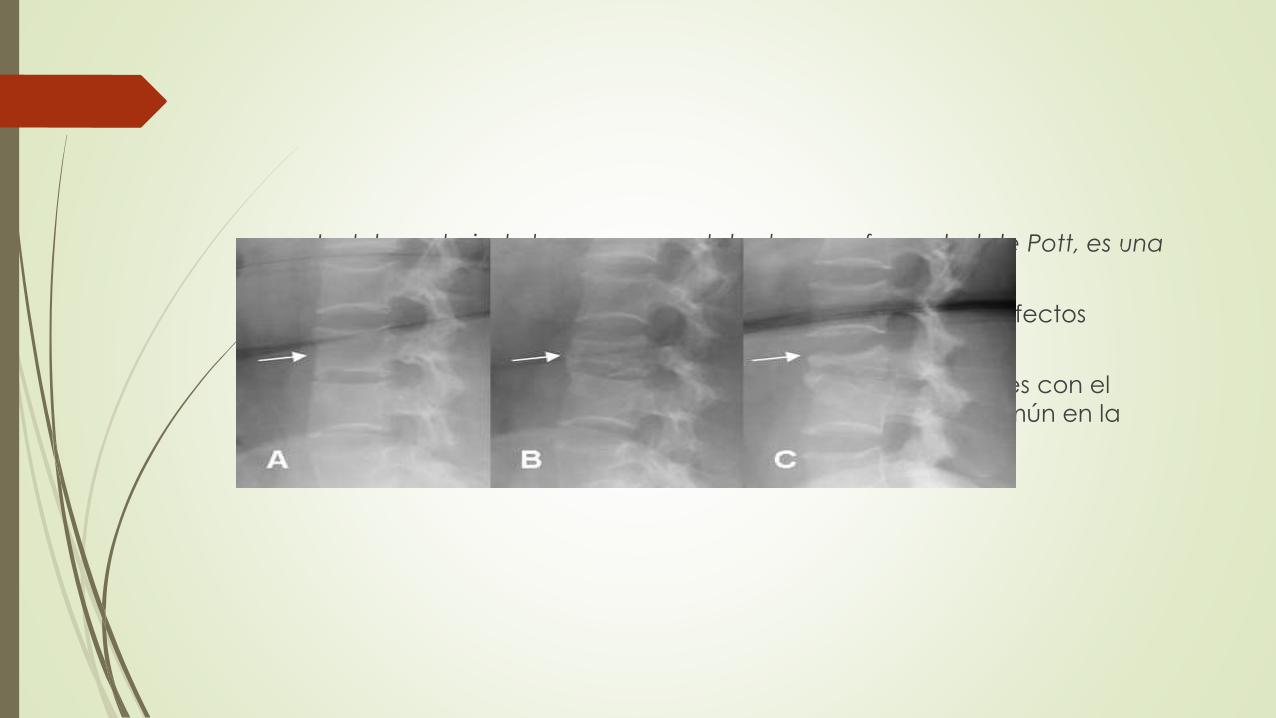
La tuberculosis de los cuerpos vertebrales, o enfermedad de Pott, es una
variante importante de osteomielitis.
Provoca deformidad y hundimiento de las vértebras con defectos
neurológicos secundarios.
La extensión de la infección a los tejidos blandos adyacentes con el
desarrollo de abscesos en el músculo psoas es bastante común en la
enfermedad de Pott.

TUMORES OSEOS
CLASIFICACIÓN DE LOS TUMORES BENIGNOS DEL HUESO
I. Tumores formadores de hueso1. Osteoma
2. Osteoma osteoide
3. Osteoblastoma
II. Tumores formadores de cartílago1. Condroma
2. Osteocondroma
3. Condroblastoma
4. Fibroma Condromixoide
III. Tumores de células gigantes
IV. Tumores de la médula (ninguno)
V. Tumores vasculares1. Hemangioma
2. Linfangioma
3. Tumor glómico
VI. Otros tumores del tejido conjuntivo1. Fibroma de Desmoplástico
2. Lipoma
3. Histiocitoma fibroso

VII. Otros tumores
1. Neurilemoma
2. Neurofibroma
VIII. Tumores Inclasificados (ninguno)
IX. Lesiones Tumor-Like
1. Quiste solitario del hueso
2. Quiste óseo aneurismático
3. Defecto fibroso metafisario
4. Granuloma de eosinófilo
5. Displasia fibrosa
6. Displasia osteofibrosa
7. Miositis osificante
8. Tumor pardo de hiperparatiroidismo
9. Quiste epidermoide intra óseo
10. Granuloma (reparador) de células gigantes

ESTADIOS DE LOS TUMORES BENIGNOS DEL HUESO
Estadio I (latente)Fibroma no osificante
Encondroma
Quiste óseo unicameral
Osteocondroma
Osteoma Osteoide
Displasia fibrosa
Granuloma Eosinófilo
Estadio 2 (activo)Encondroma
Osteocondroma
Osteoma Osteoide
Osteoblastoma
Tumor de células gigantes
Fibroma Condromixoide
Displasia fibrosa
Granuloma de Eosinófilo
Quiste óseo aneurismático
Quiste óseo unicameral
Displasia osteofibrosa
Estadio 3 (agresivo)Tumor de células gigantes
Osteoblastoma
Condroblastoma
Quiste óseo aneurismático


Osteoma
Lesiones benignas del hueso
Representan aberraciones del desarrollo o excrecencias reactivas más
que neoplasias verdaderas
Normalmente aparecen en la cabeza y el cuello, incluyendo los senos
paranasales
Osteomas suelen ser solitarios y se presentan como masas exofíticas, duras,
localizadas y de crecimiento lento sobre la superficie ósea.
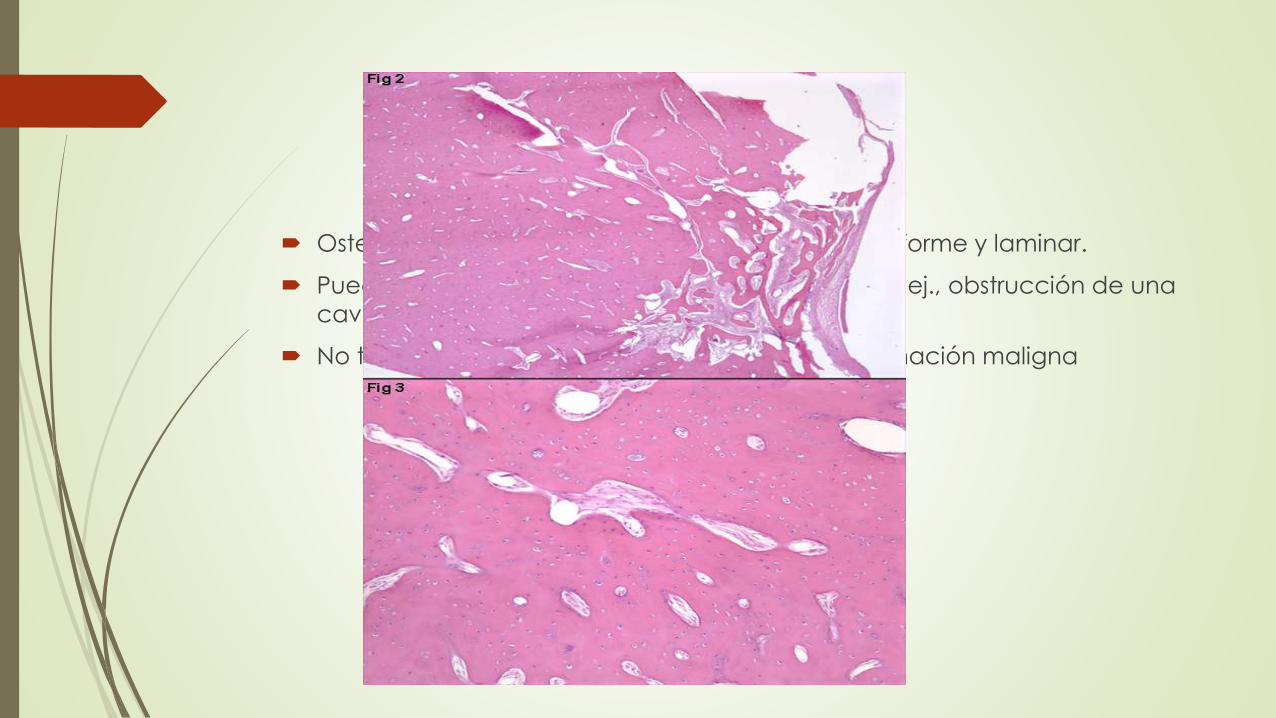
Osteomas son una mezcla irregular de hueso plexiforme y laminar.
Pueden generar problemas mecánicos locales (p. ej., obstrucción de una
cavidad sinusal) y deformidades estéticas
No tienen un carácter invasor y no sufren transformación maligna
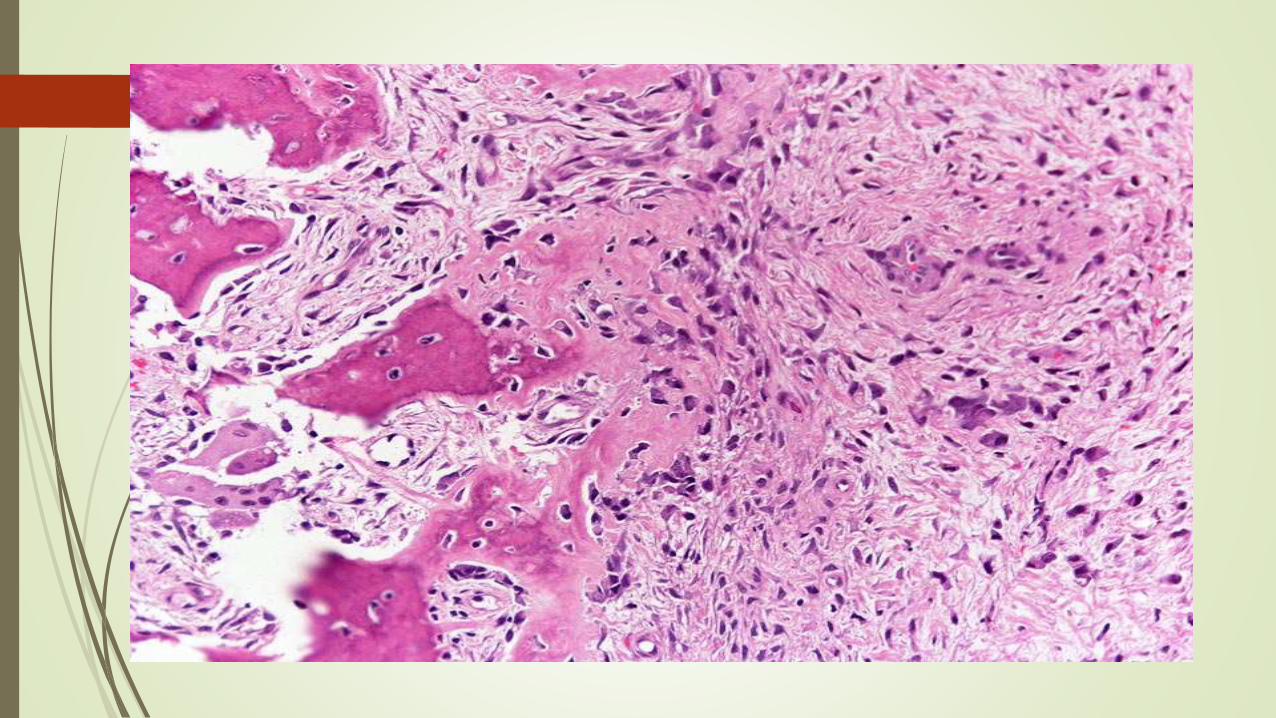
Osteoma osteoide y Osteoblastoma
Son neoplasias benignas con características histológicas muy similares.
Adolecencia y segunda decada
Los osteomas osteoides surgen con mayor frecuencia en las porciones
proximales del fémur y la tibia, y por definición su tamaño es menor de 2
cm
Los osteoblastomas se originan sobre todo en la columna vertebral;
también generan dolor pero difícil de localizar
La resección local es el tratamiento de elección; las lesiones resecadas
incompletamente pueden recidivar. La transformación maligna es rara a
menos que la lesión se trate con radiación.
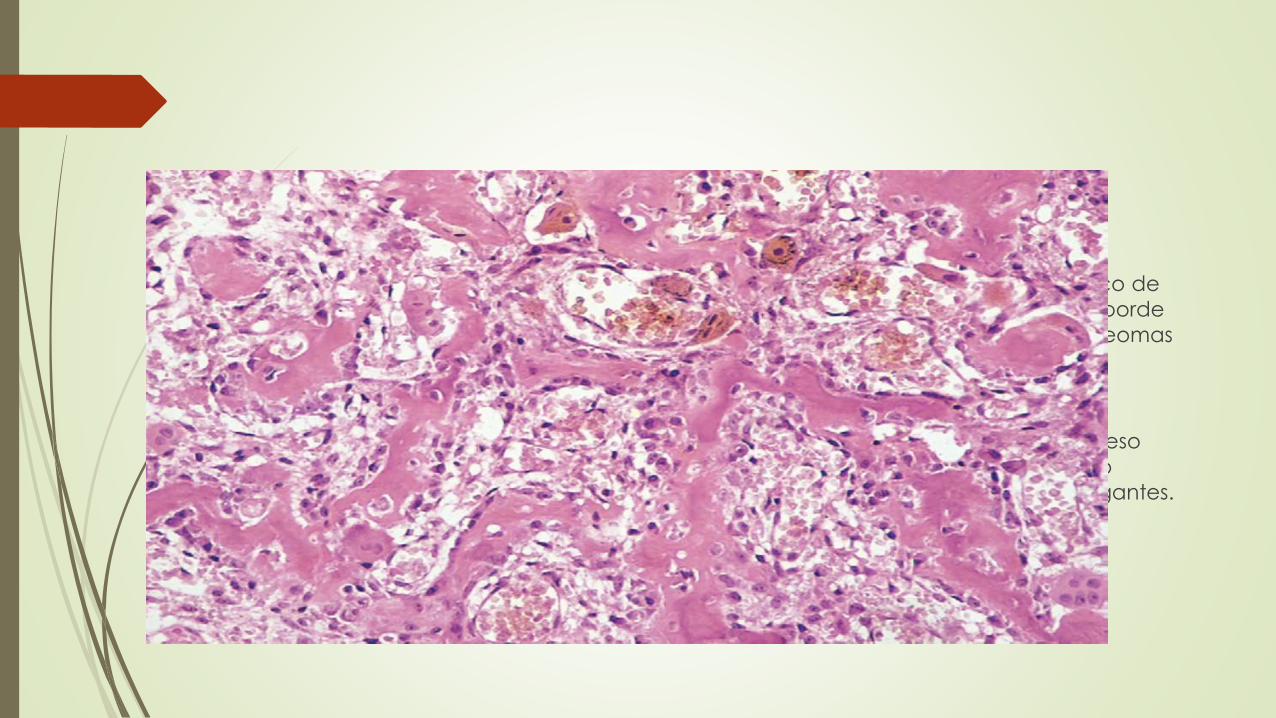
Macroscópicamente
Las lesiones son masas entre redondeadas y ovales de tejido hemorrágico de
color marrón arenoso. En el borde de ambos tipos de tumores hay un reborde
de hueso esclerótico; sin embargo, es mucho más prominente en los osteomas
osteoides.
Microscópicamente
Ambas neoplasias están compuestas por trabéculas entrelazadas de hueso
plexiforme rodeadas de osteoblastos. La estroma interpuesta es un tejido
conjuntivo laxo vascular que contiene un número variable de células gigantes.

Osteosarcoma
Tumor mesenquimal maligno productor de hueso
Neoplasia ósea maligna primaria más frecuente, siendo el responsable de,
aproximadamente, el 20% de los cánceres óseos primarios
Cualquier grupo de edad
75% en menores de 20 años

La mayoría de las afecciones surgen en la región metafisaria de los huesos largos de las
extremidades
60% en la zona de la rodilla
15% alrededor de la cadera
10% en el hombro
8% en la mandíbula.

Osteosarcoma primario
Es solitario y escasamente diferenciado que produce predominantemente
matriz ósea.
Osteosarcoma secundario
Aparece apartir de complicaciones de la enfermedad de Paget poliostótica;
histológicamente similar al osteosarcoma primario
Edad mas avanzada
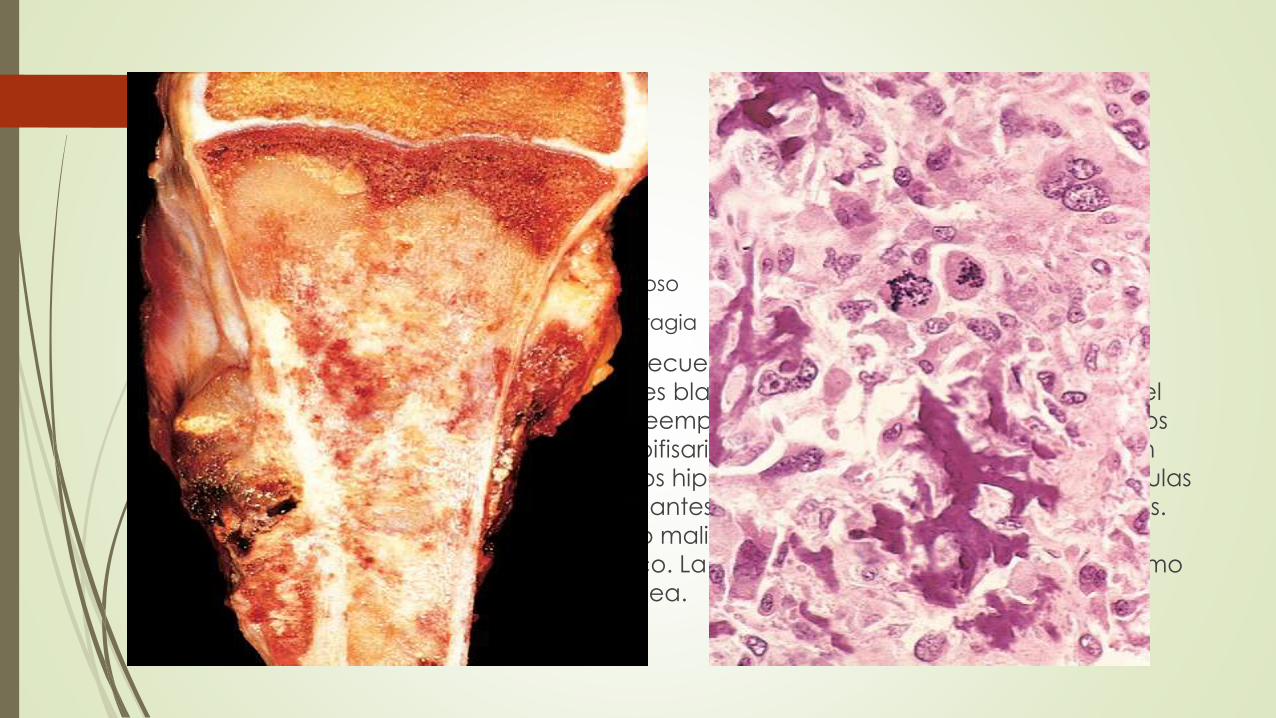
Macroscópicamente
Color blanco grisáceo arenoso
A menudo muestran hemorragia y degeneraciones quísticas.
Los tumores destruyen con frecuencia las corticales circundantes y
producen masas en las partes blandas. Se diseminan ampliamente en el
canal medular infiltrando y reemplazando la médula, pero sólo en pocos
casos atraviesan la placa epifisaria o acceden al espacio articular. Con
frecuencia presentan núcleos hipercromáticos de gran tamaño; las células
gigantes tumorales extravagantes son frecuentes, al igual que las mitosis.
Cuando abunda el cartílago maligno el tumor recibe el nombre de
osteosarcoma condroblástico. La invasión vascular es frecuente, así como
la necrosis tumoral espontánea.

Patogenia
Las mutaciones del gen RB aparecen en el 60 al 70% de los tumores esporádicos
y los individuos con retinoblastomas hereditarios (secundarios a mutaciones de
la línea germinal en el gen RB) tienen un riesgo mil veces mayor de desarrollar
osteosarcoma.
Los osteosarcomas espontáneos también muestran con frecuencia mutaciones
en los genes que regulan el ciclo celular
p53, las ciclinas, las cinasas dependientes de las ciclinas y los inhibidores de la cinasa

Características clínicas
Empiezan como masas dolorosas que aumentan de tamaño
Primer síntoma: fractura patológica
Radiografías muestran una masa mixta lítica y blástica, destructiva, de gran
tamaño, con márgenes infiltrantes poco definidos.
El tumor se rompe a través de la cortical y eleva el periostio, dando lugar a la
formación de hueso perióstico reactivo.
Triángulo de Codman es característica de los osteosarcomas
La diseminación típica es por vía hematógena
10 y un 20% de los pacientes tiene metástasis pulmonares

Tratamiento
Quimioterapia y cirugía con preservación de la extremidad
Supervivencias entre el 60 y el 70%
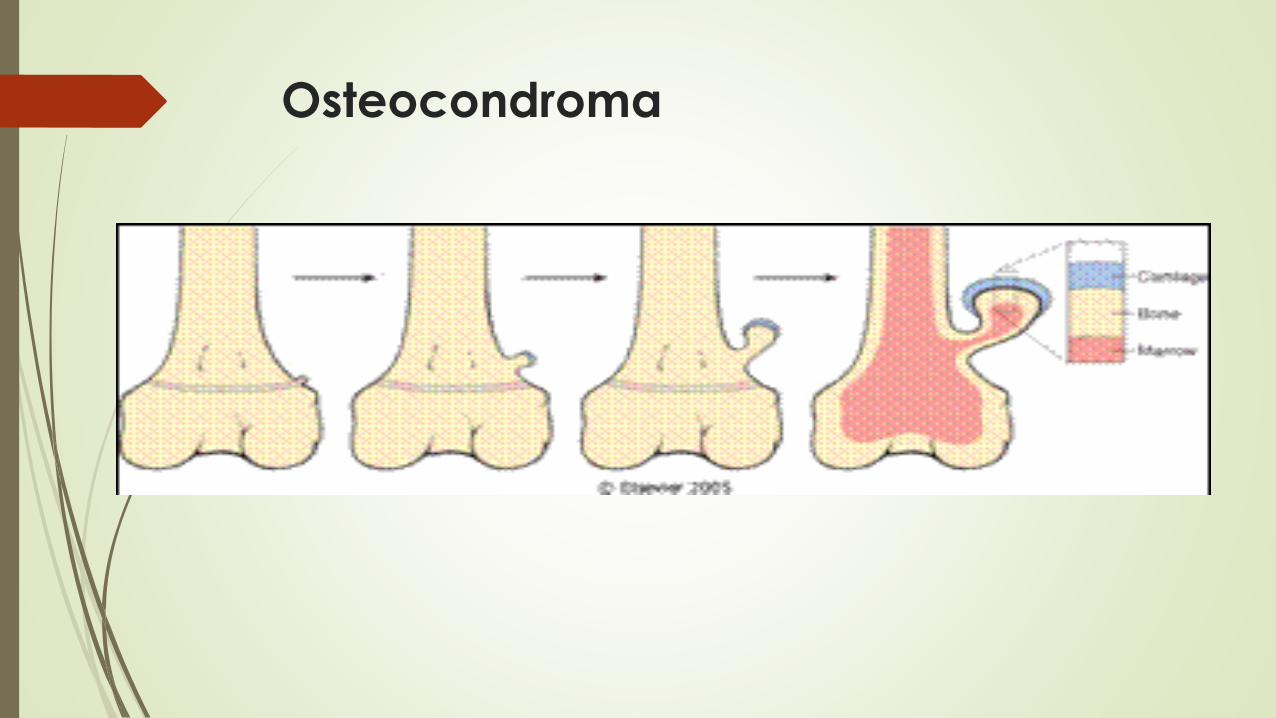
Osteocondroma
Exostosis
Son excrecencias benignas recubiertas de cartílago que están unidas por
un tallo óseo al esqueleto subyacente
Osteocondromas solitarios
Diagnostican: Final de la adolecencia
Osteocondromas múltiples
Se manifiestan durante la niñez (exostosis hereditarias múltiples)
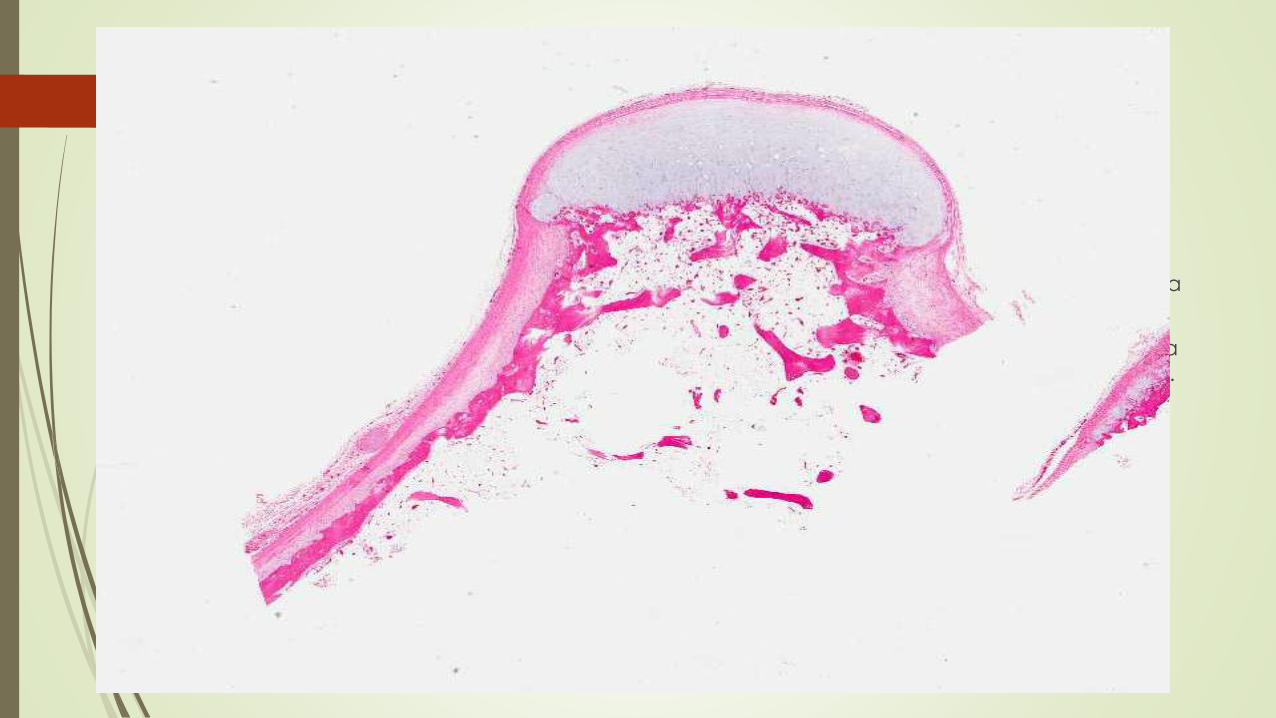
Patogenia
La inactivación de las dos copias del gen EXT en los condrocitos está implicada
tanto en los osteocondromas esporádicos como en los hereditarios.
Gen supresor tumoral codifica una serie de glucosiltransferasas esenciales para
la polimerización del heparín sulfato, un componente importante del cartílago.

Afecta huesos de origen endocondral y surgen en las metáfisis, cerca de la placa de crecimiento de los huesos cilíndricos largos, en especial alrededor de la rodilla.
Morfologia
Entre 1 y 20 cm
La cubierta
Es cartílago hialino benigno que imita una placa de crecimiento desorganizado que sufre una osificación endocondral.
La porción interna de la cabeza y el tallo
Es hueso neoformado,
La cortical del tallo se mezcla con la cortical del hueso donde se aloja.

Características clínicas
Masas de crecimiento lento
Generan dolor
Si comprimen un nervio o si el tallo se fractura
Exostosis hereditaria múltiple
Deformidad del hueso subyacente sugiere un trastorno asociadoen el crecimiento epifisario.
Rara vez progresan a condrosarcomas u otros sarcomas

Condroma
Tumores benignos de cartílago hialino
Endocondromas: medula
20 a 50 años
Lesiones solitarias localizadas en la región metafisaria de los huesos cilíndricos
Manos y pies
Condromas yuxtacorticales: superficie osea

Enfermedad de Ollier
Condromas múltiples que afectan preferentemente a un lado del cuerpo
Síndrome de Maffucci
Condromas múltiples asociados a angiomas benignos de partes blandas.
Se asocia a mayor riesgo de desarollar otros tipos de neoplasias maginas
Carcinomas de ovario y gliomas cerebrales.
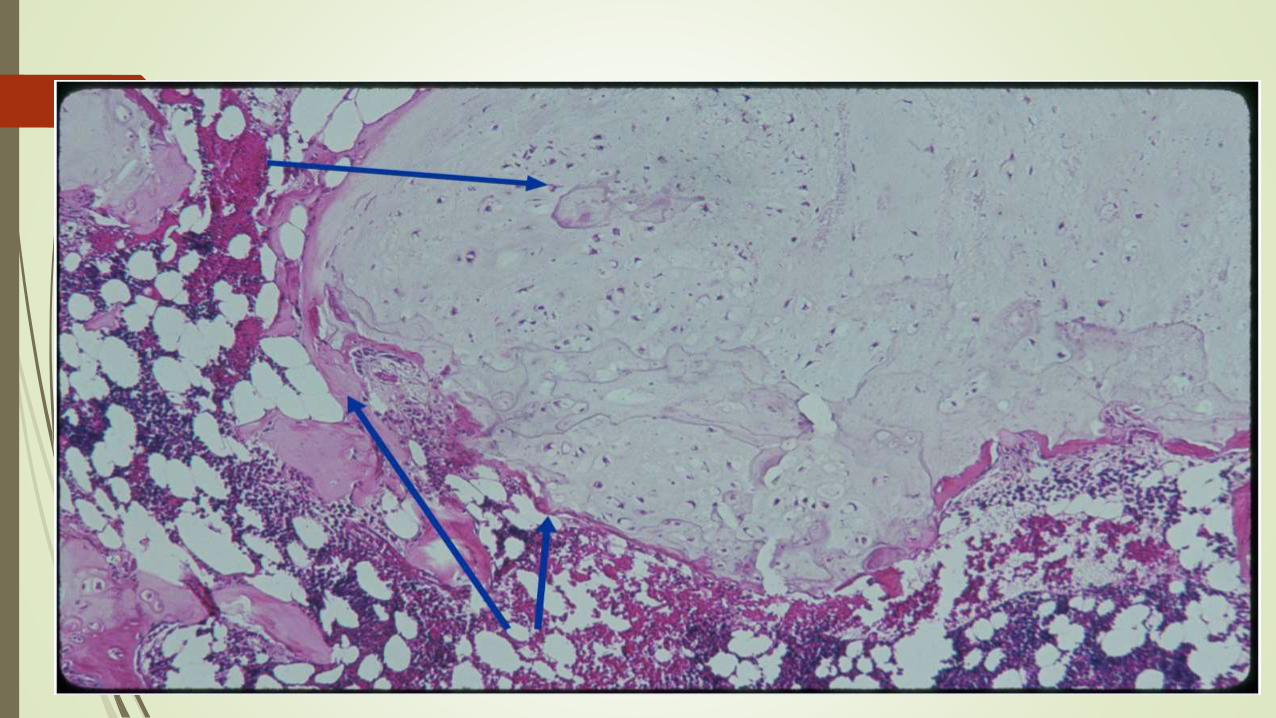
Patogenia
Se desarrollan a partir de restos lentamente proliferativos de cartílago de la
placa de crecimiento
Morfología
Nódulos traslúcidos de color azul grisáceo
Inferior a 3 cm
Matriz hialina bien delimitada y condrocitos citológicamente benignos.
En la periferia hay osificación endocondral, mientras que el centro suele
calcificarse y morir.
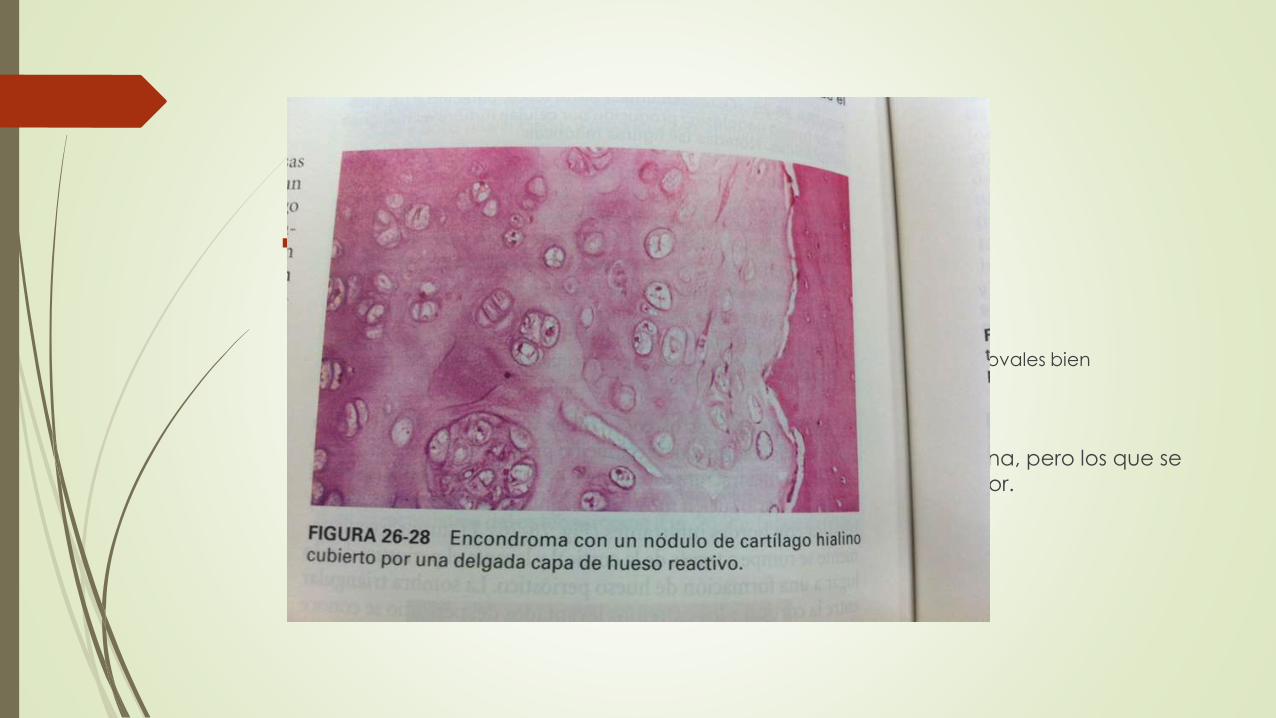
Características clínicas
dolorosos o provocan fracturas patológicas.
Radiografia: signo del anillo en O
nódulos desmineralizados de cartílago producen transparencias ovales bien delimitadas rodeadas de rebordes finos de hueso radiopaco
Crecimiento de condroma es limitado
Los condromas solitarios rara vez sufren transformación maligna, pero los que se
asocian a encondromatosis están expuestos a un riesgo mayor.

CONDROSARCOMA
Abarcan una amplia gama de tumores que comparten la capacidad de
producir cartílago neoplásico
Se clasifican según su localización (intramedular o yuxtacortical)y las
variantes histológicas
Mayor parte de los pacientes tiene 40 años o mas y afecta el doble a
varones respecto de mujeres
Condrosarcoma convencionales surgen en el interior de la cavidad
medular del hueso para formar una masa expansiva que frecuentemente
erosiona la cortical. Muestran cartílago hialino y mixoide maligno
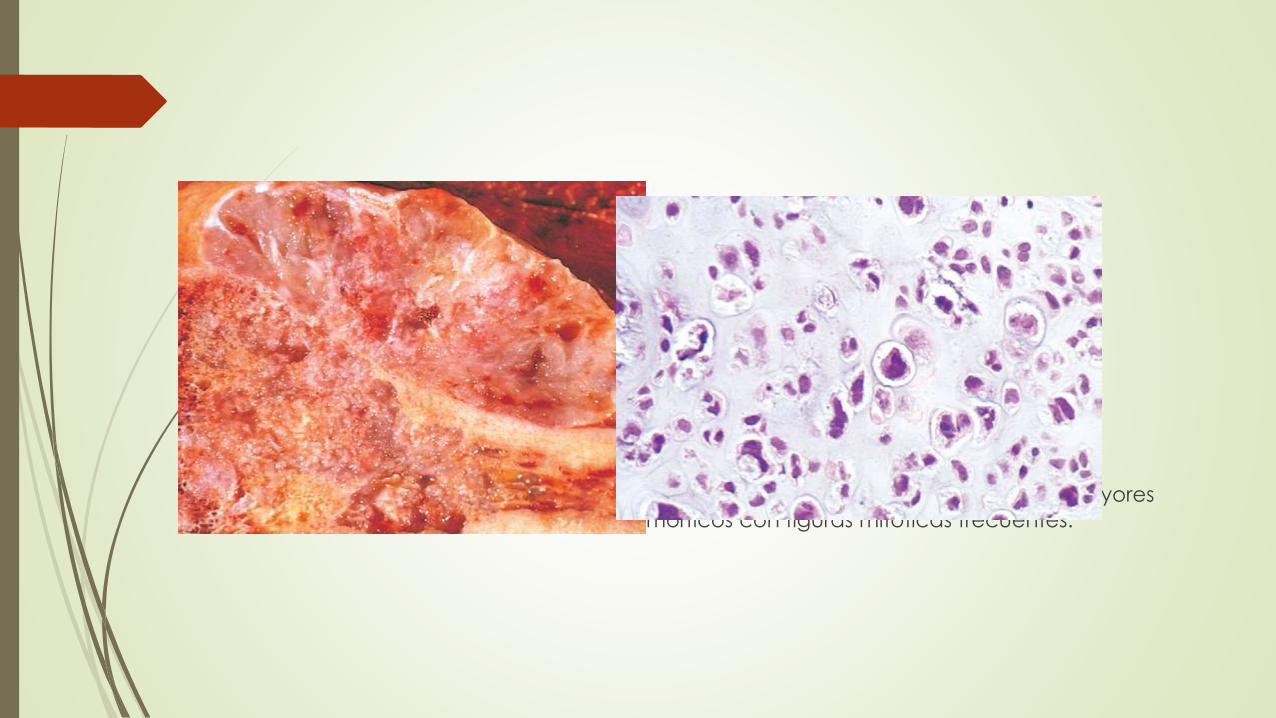
Condrosarcomas mixoides
viscosos y gelatinosos
la matriz protruye sobre la superficie de corte
típica la presencia de calcificaciones moteadas,
necrosis central puede dar lugar a espacios quísticos.
cortical adyacente está engrosada y erosionada
Tumor crece hacia los espacios medulares y las partes blandas circundantes
Los de bajo grado imitan al cartílago normal. Las lesiones de grados mayores
contienen condrocitos pleomórficos con figuras mitóticas frecuentes.

Características clínicas.
Pelvis, los hombros y las costillas
Rara vez afectan a las extremidades distales
Empiezan como masas dolorosas de crecimiento progresivo
Bajo grado de crecimiento lento
Da un engrosamiento reactivo de la corteza
Alto grado más agresiva
destruye la corteza y da lugar a una masa en el tejido blando
cuanto más radiotransparente sea el tumor, mayor será la probabilidad de que sea de alto
grado

Condrosarcomas convencionales son
Mayoria son indolentes y de bajo grado,
Índice de supervivencia a los 5 años entre el 80 y el 90%
43% para los tumores de grado 3
los tumores de grado 1 rara vez metastatizan
70% de los tumores de grado 3 diseminan.
Tumores mayores de 10 cm muestran una agresividad notablemente mayor
Metastatizan por vía hematógena,
hacia los pulmones y el esqueleto.
Tratamiento
Los condrosarcomas convencionales se tratan con una resección quirúrgica amplia
Ademas quimioterapia para las variantes mesenquimales y desdiferenciadas
debido a la agresividad de su evolución clínica.

Sarcoma de Ewing y tumor
neuroectodermico primitivo TNEP
Tumores malignos del hueso y de partes blandas de células redondas pequeñas.
Comparten una translocación cromosómica idéntica y por ende se considera como un mismo tumor, distinguiéndose únicamente por el grado de diferenciación.
TNEP
Diferenciación nerviosa
Sarcom de Ewing
Indiferenciado
Responsables del 6 al 10% de los tumores óseos malignos primarios.
Segundos sarcomas óseos pediátricos más comunes
Entre 10 y 20 años
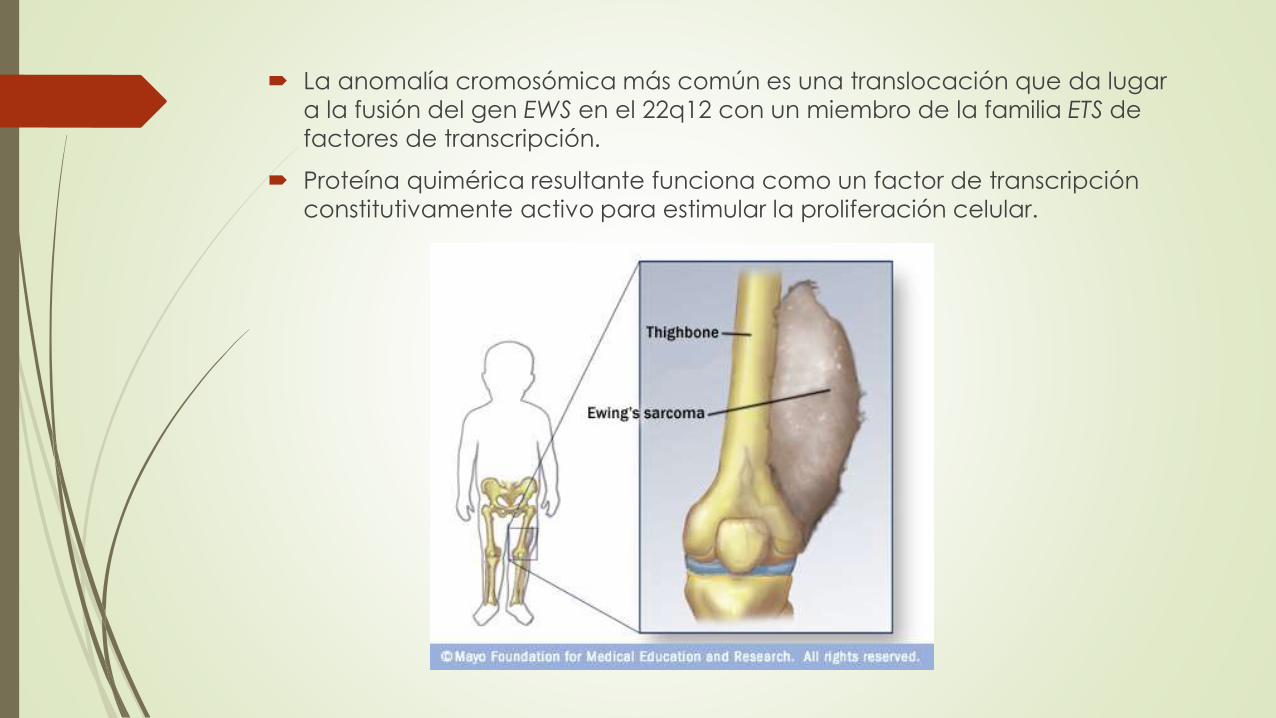
La anomalía cromosómica más común es una translocación que da lugar
a la fusión del gen EWS en el 22q12 con un miembro de la familia ETS de
factores de transcripción.
Proteína quimérica resultante funciona como un factor de transcripción
constitutivamente activo para estimular la proliferación celular.

Morfologia
surgen en la cavidad medular e invaden la cortical y el periostio para producir
una masa en el tejido blando.
Color marfil
Se asocia a hemorragias y necrosis
sábanas de células redondas, pequeñas y uniformes
Citoplasma escaso rico en glucógeno
rosetas de Homer-Wright: células tumorales alrededor de un espacio fibrilar
central
indica la presencia de diferenciación nerviosa.

Características clínicas:
masas dolorosas en las diáfisis de los huesos cilíndricos largos
fémur y los huesos planos de la pelvis.
fiebre, velocidad de sedimentación alta, anemia y leucocitosis parecidos a los
de las infecciones.
Tratamiento
Quirúrgica con o sin radiación.
La supervivencia a los 5 años se sitúa actualmente en el 75%, y el 50% logra un
índice de remisiones duradero a largo plazo.
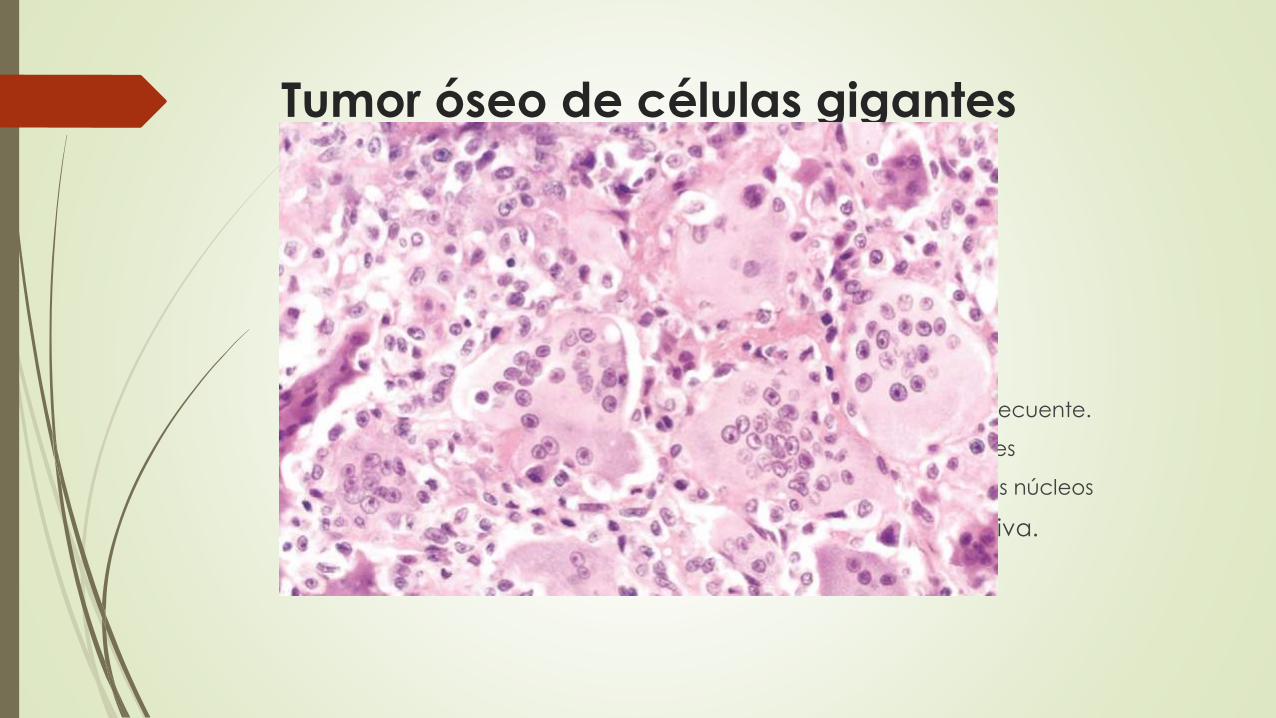
Tumor óseo de células gigantes
Osteoclastoma
benigno pero localmente agresivo
20 a 40 años
Morfologia
grandes y de color marrón rojizo con degeneración quística frecuente.
células mononucleares ovales uniformes con mitosis frecuentes
Células gigantes de tipo osteoclástico dispersas con 100 o más núcleos
presencia de necrosis, hemorragia y formación ósea reactiva.

Evolución clínica
surge en la epífisis de los huesos largos alrededor de la rodilla
extremo distal del fémur y proximal de la tibia
Sintomas seudoartríticos, fracturas patológicas.
cortical que los recubre está destruida produciendo una masa en el tejido
blando que sobresale con una cubierta delgada de hueso reactivo.

ARTICULACIONES
Las articulaciones están sometidas a una amplia variedad de trastornos
como degeneración, infecciones, lesiones de causa inmunitaria,
desordenes metabólicos y neoplasias.
A continuación describiremos algunas de las variedades de artritis mas
frecuentes, como la artropatía degenerativa (artrosis), la gota t la artritis
infecciosa. Así como los tumores articulares benignos mas comunes.
También haremos noción en la artritis reumatoide otra de las causas
importantes y devastadoras de las artropatías

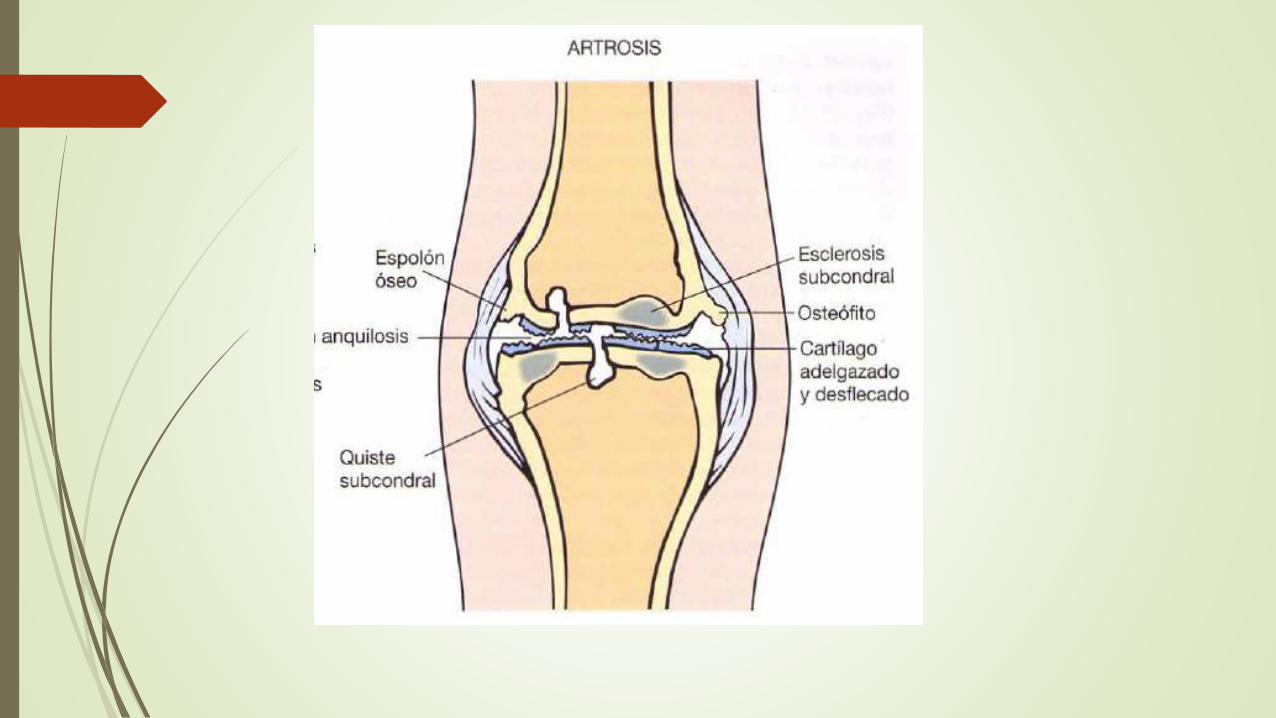
ARTROSIS
Trastorno articular mas común
Componente frecuente del envejecimiento y cauda importante de discapacidad física en individuos mayores de 65 años
La artrosis es la degeneración del cartílago articular , cualquiera de los cambios estructurales del hueso subyacente es secundario
En la mayoría de casos la artrosis comienza insidiosamente con la edad y sin una causa precipitante aparente = Artrosis Primaria
En la minoría de casos en que la artrosis incide en la juventud suele haber una afección predisponente como una lesión traumática, una deformidad del desarrollo o una enf generalizada subyacente como la diabetes, obesidad = Artrosis Secundaria
Rodillas y manos mas afectadas en mujeres y las caderas en los hombres
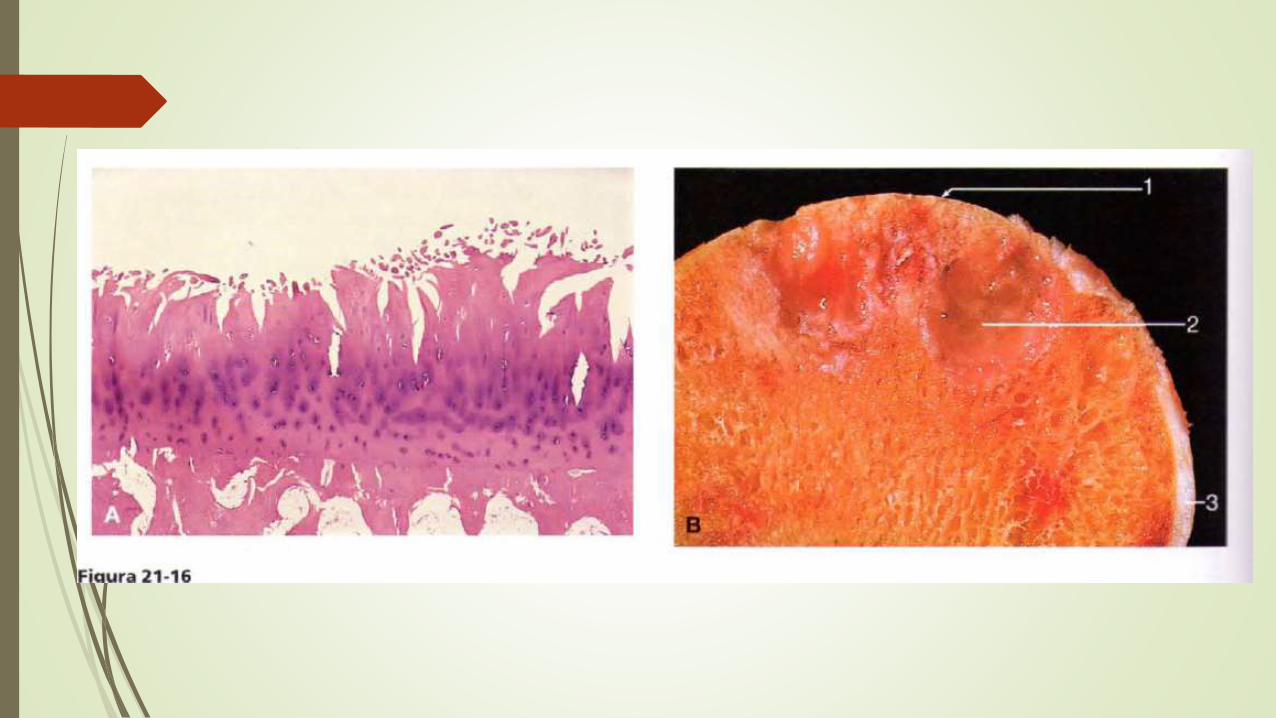

MORFOLOGIA
Los cambios estructurales mas precoces en la artrosis consisten en un
agrandamiento, proliferación y desorganización de los condrocitos en la parte
superficial del cartílago articular
Se produce una fibrilación y agrietamiento de la matriz en vertical y horizontal a
medida que las capas del cartílago van degradándose
La fricción alisa y pule el hueso expuesto dándole un aspecto de marfil pulido lo cual
se denomina condensación ósea.
EVOLUCION CLINICA
La artrosis afecta a pacientes de 50 años en adelante, sus síntomas son dolor
profundo que se exacerba con el uso, rigidez matutina, crepitación (sensación de
rechinamiento) y limitación del arco del movimiento
Aparte de la inactividad completa, no hay ninguna forma de prevenir o frenar la
progresión de la artrosis primaria, puede estabilizarse durante años pero por lo
general progresa lentamente
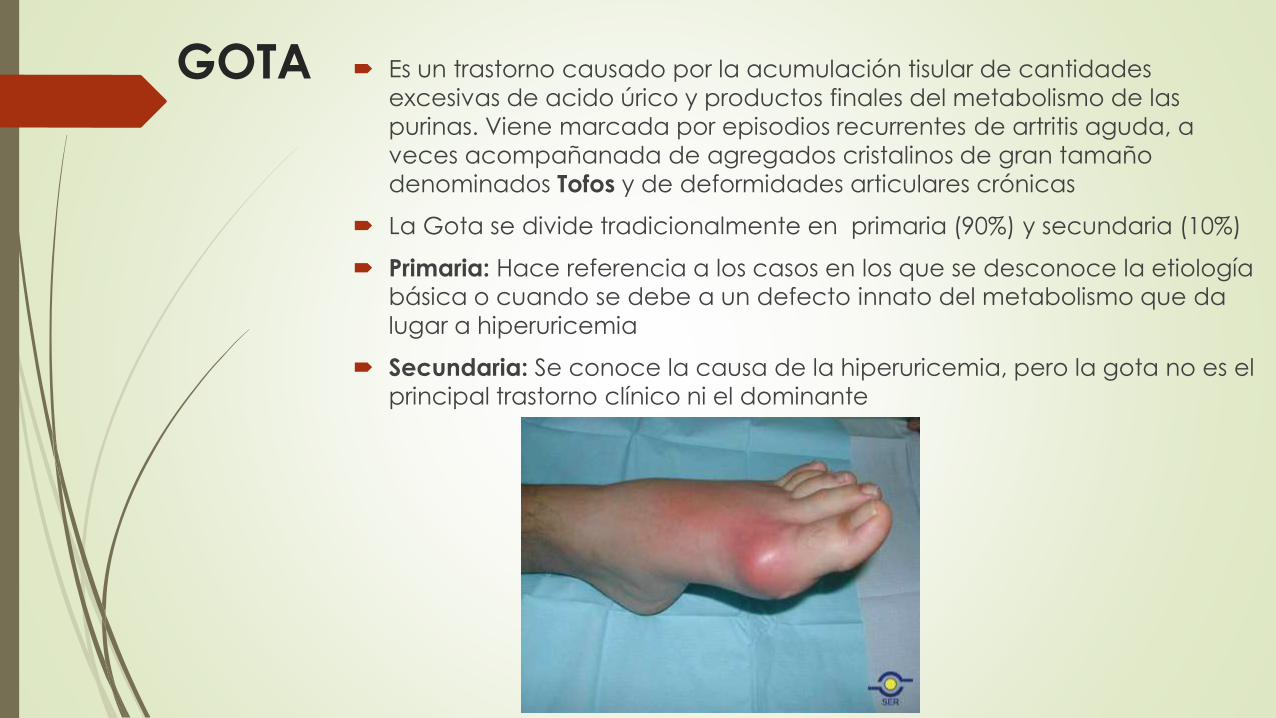
GOTA Es un trastorno causado por la acumulación tisular de cantidades
excesivas de acido úrico y productos finales del metabolismo de las
purinas. Viene marcada por episodios recurrentes de artritis aguda, a
veces acompañanada de agregados cristalinos de gran tamaño
denominados Tofos y de deformidades articulares crónicas
La Gota se divide tradicionalmente en primaria (90%) y secundaria (10%)
Primaria: Hace referencia a los casos en los que se desconoce la etiología básica o cuando se debe a un defecto innato del metabolismo que da
lugar a hiperuricemia
Secundaria: Se conoce la causa de la hiperuricemia, pero la gota no es el
principal trastorno clínico ni el dominante


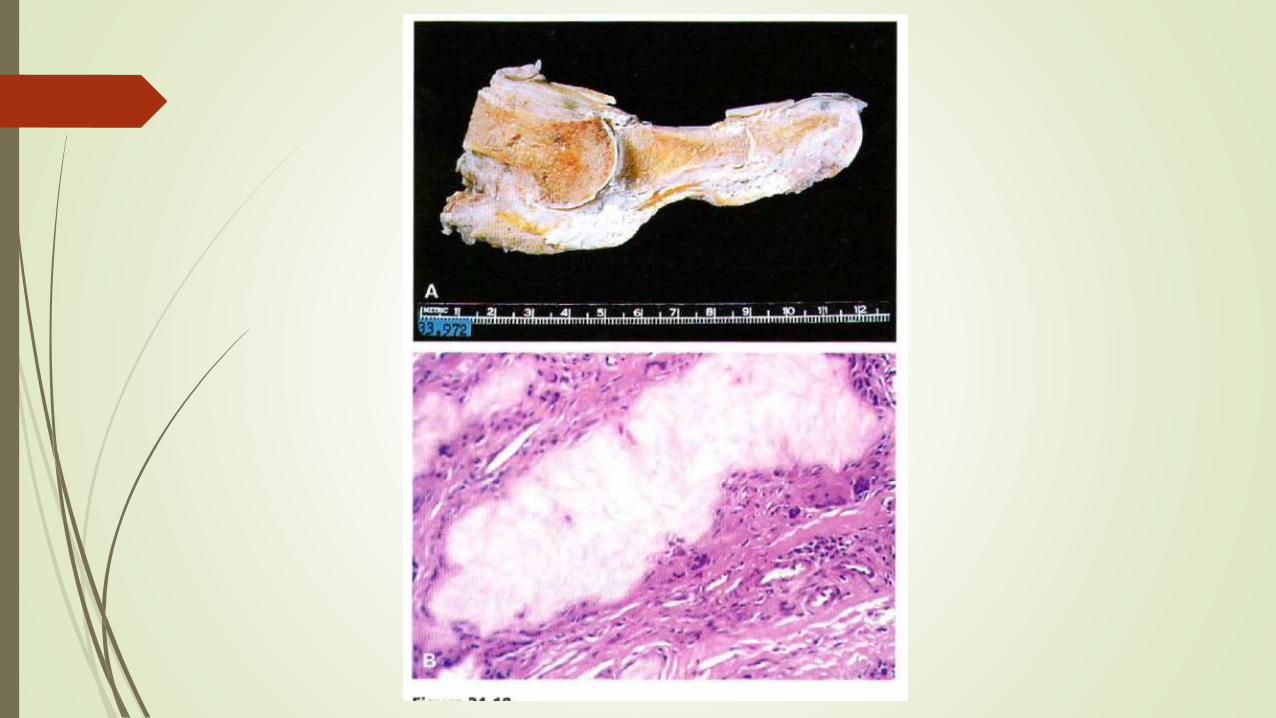
MORFOLOGIA
Sus manifestaciones morfológicas principales son artritis aguda, artritis tofacea crónica,
tofos en diferentes localizaciones y nefropatía gotosa
La Artritis Aguda se caracteriza por un infiltrado neutrófilo denso que invade la sinovial
y el liquido sinovial. Con frecuencia se aprecian cristales de urato monosodico largos y
delgados en forma de aguja en racimos pequeños. La sinovial esta edematosa y
congestionada
La artritis tofacea crónica evoluciona a partir de la precipitación de cristales de urato
durante las crisis agudas , los uratos se incrustan en las superficies articulares y forman
depósitos visibles en la sinovial , volviéndola hiperplasica, fibrotica y engrosada
Los Tofos son la característica patognomónica que define la gota , formados por agregados de cristales de urato de gran tamaño rodeados de una reacción
inflamarotia intensa de linfocitos macrófagos y células gigantes
La nefropatía gotosa hace referencia a diversas complicaciones renales diferentes
que se asocian al deposito de uratos que forman tofos medulares, precipitaciones
intrtubulares y cálculos renales.
Características Clínicas:
La gota es mas frecuente en hombres, no suele ocasionar síntomas antes de los 30 años
Se han descrito clásicamente 4 etapas
1) Hiperuricemia asintomática (aparece alrededor de la pubertad en varones,
menopausia en mujeres)
2) Artritis gotosa aguda (tras años aparece con dolor articular asociado a eritema y
calor)
3) Gota intercritica (Periodo asintomático )
4) Gota tofacea crónica (lesión incapacitante crónica )

SEUDOGOTA
Conocida también como condrocalcinosis o mas formalmente como enfermedad
por deposito de cristales de pirofosfato cálcico.
Debuta típicamente el personas de 50 años o mas, aumentando su frecuencia con
la edad . No existe una predilección por sexos o razas
No es muy conocida las vías de formación de cristales, pero lo mas probable es que
se deba en enzimas defectuosas que producen o degradan pirofosfatos lo que
produce acumulación y finalmente cristalización en calcio
La afectación articular puede durar varios días o semanas y puede ser tanto
monoarticular o poliarticular. Las zonas mas afectadas son Rodillas, muñecas ,
codos, hombros , tobillos
El tratamiento es sintomático, no se conoce ningún fármaco que retrace impidan
la formación de cristales

ARTRITIS INFECCIOSA ARTRITIS PURULENTA
Las bacterias pueden sembrar las articulaciones durante los episodios de bacteremia; la infección articular con dichos microorganismos da lugar uniformemente con una artritis purulenta. Cualquier bacteria puede convertirse en agente etiológico.
Haemophilus influenzae: Predomina en niños menores de 2 años
S. Aureus: Predomina en niños mayores y adultos
Gonococos: Prevalece en los últimos años de adolescencia y primeros de la madurez
Existe un dolor de comienzo súbito con rubor, tumefacción de la articulación con limitación del movimiento. La fiebre, la leucocitosis y la elevación de la velocidad de sedimentación son frecuentes . La aspiración articular es purulenta y permite identificar el microorganismo causante
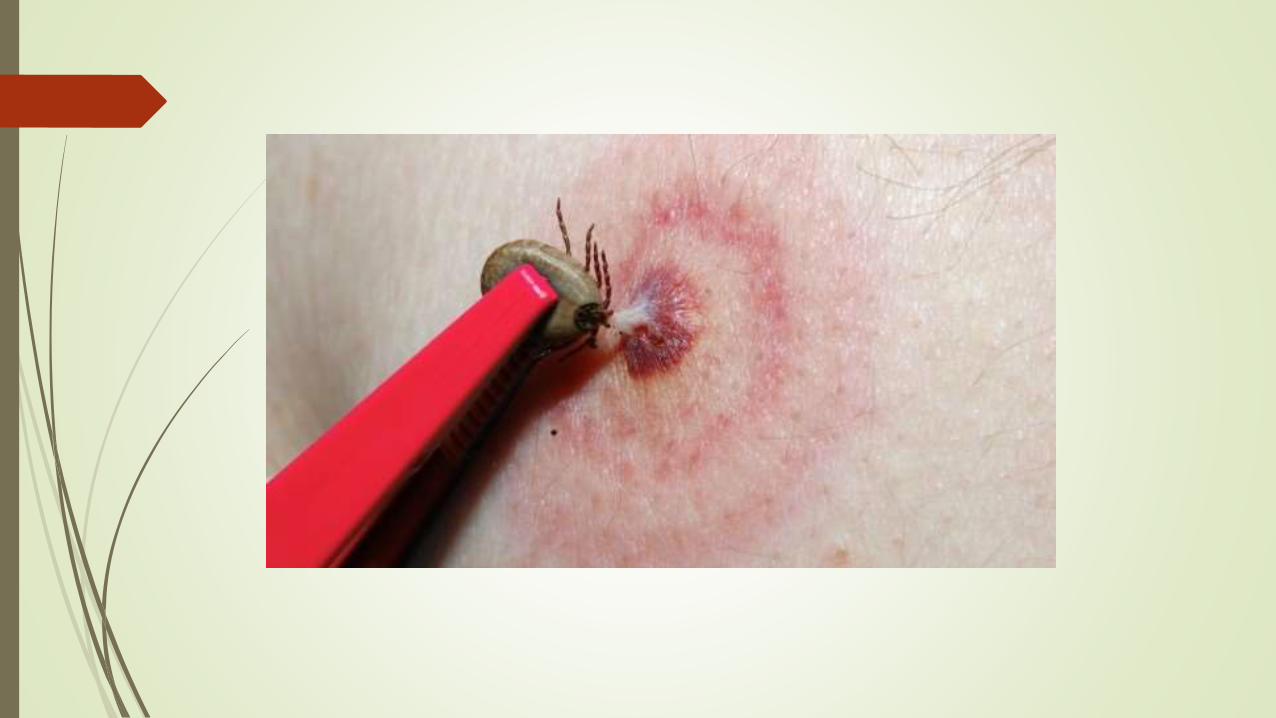
ARTRITIS DE LYME
Se debe a una infección por la espiroqueta BORRELIA BURGDORFERI, que se transmite a través de las garrapatas de los ciervos. En estados unidos es la enfermedad trasmitida por artrópodos mas frecuente. Existen tres etapas de la enfermedad de Lyme:
Etapa 1, llamada enfermedad de Lyme temprana y localizada. La infección aún no se ha propagado por todo el cuerpo.
Etapa 2, llamada enfermedad de Lyme de diseminación temprana. La bacteria ha comenzado a propagarse por todo el cuerpo.
Etapa 3, llamada enfermedad de Lyme de diseminación tardía. La bacteria se ha diseminado por todo el cuerpo.
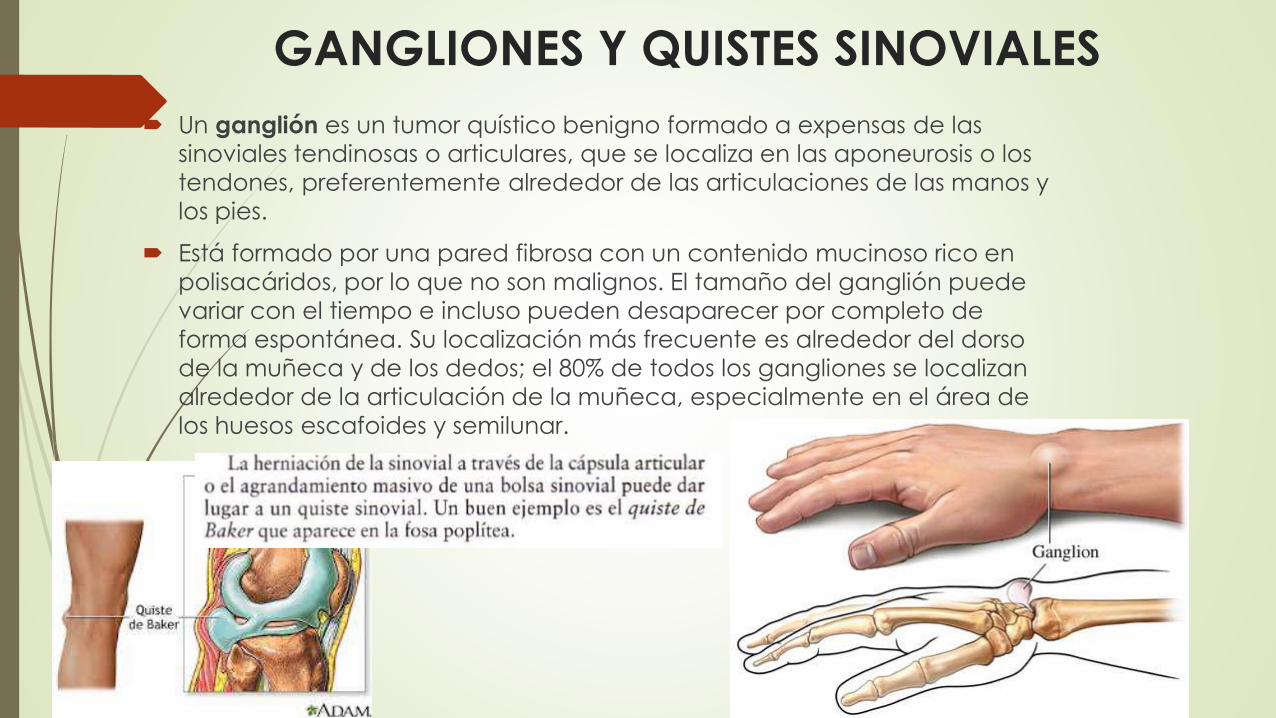
GANGLIONES Y QUISTES SINOVIALES
Un ganglión es un tumor quístico benigno formado a expensas de las
sinoviales tendinosas o articulares, que se localiza en las aponeurosis o los
tendones, preferentemente alrededor de las articulaciones de las manos y
los pies.
Está formado por una pared fibrosa con un contenido mucinoso rico en
polisacáridos, por lo que no son malignos. El tamaño del ganglión puede
variar con el tiempo e incluso pueden desaparecer por completo de
forma espontánea. Su localización más frecuente es alrededor del dorso
de la muñeca y de los dedos; el 80% de todos los gangliones se localizan
alrededor de la articulación de la muñeca, especialmente en el área de
los huesos escafoides y semilunar.

BIBLIOGRAFIA
KUMAR, V., A. K. ABBAS, N. FAUSTO y J. C. ASTER Robbins y Cotran -
Patología estructural y funcional Ed. Elsevier, 8ª ed., 1464 págs., 2010
RUBIN, E., F. GORSTEIN, R. RUBIN, R. SCHWARTING y D. STRAYER Rubin -
Patología estructural. Fundamentos clinicopatológicos en Medicina Ed.
McGraw-Hill, 4ª ed., 1440 págs., 2006
Rafael Andrade P; Juan Manuel González C; Rodrigo Restrepo M;
Alejandro Vélez H-Patología. Ed Corporación para Investigaciones
Biológicas, 2 ed., 598pag., 2006
IVAN DAMJANOV , PATOLOGIA (3ª ED.) (SERIE SECRETOS): PREGUNTAS
ESENCIALES S.A. ELSEVIER ESPAÑA, 2010























![SEMIOLOGIA OSTEOARTICULAR [Autoguardado]](https://img.pdfslide.net/doc/110x75/55cf859d550346484b8ffe4c/semiologia-osteoarticular-autoguardado.jpg)





