Embed Size (px)
Citation preview

Complex metagenome
assembly, + bonus career thoughts.
C. Titus BrownUC Davis


Hello!Research background:
Computing, modeling, and data analysis: 1989-2000
(high school & undergrad+)
Molecular biology, genomics, and data analysis: 2000-2007
(grad school + postdoc)
Bioinformatics, data analysis, and Comp Sci: 2007-present
(assistant professor)
Genomics and Veterinary Medicine (?)

Two topics for this talk:
1. Metagenome assembly.
2. Careers & a “middle class” of bioinformaticians.

Shotgun metagenomics
Collect samples;
Extract DNA;
Feed into sequencer;
Computationally analyze.
Wikipedia: Environmental shotgun sequencing.png

To assemble, or not to assemble?
Goals: reconstruct phylogenetic content and predict functional potential of ensemble.
Should we analyze short reads directly?
OR
Do we assemble short reads into longer contigs first, and then analyze the contigs?
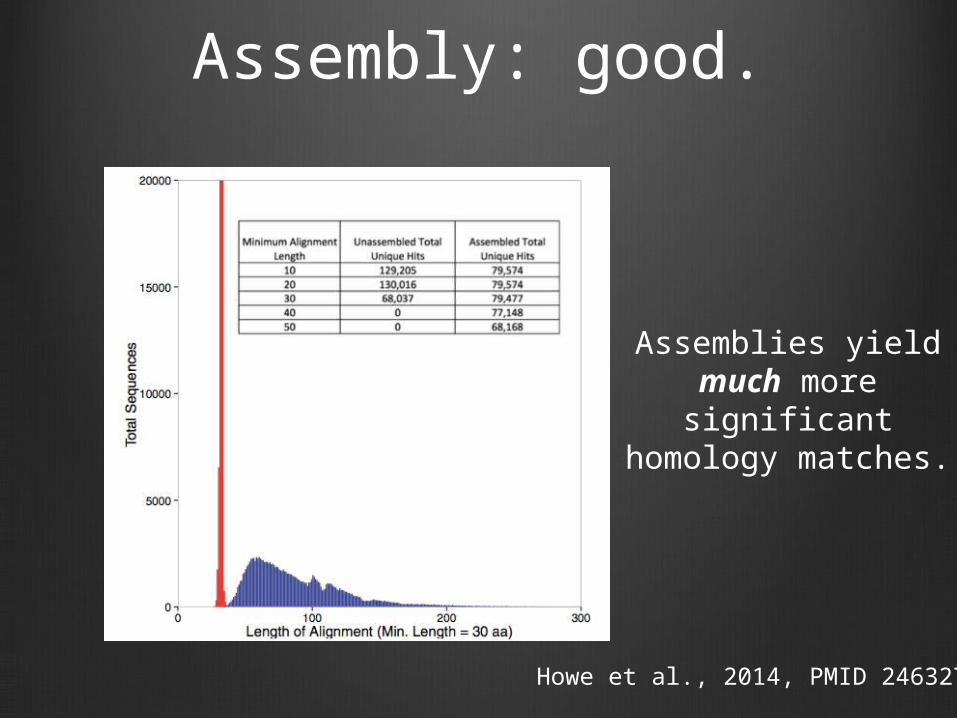
Assembly: good.
Howe et al., 2014, PMID 24632729
Assemblies yield much more
significant homology matches.

But!
Does assembly work well!?
(Short reads, chimerism, strain variation, coverage, compute
resources, etc. etc.)

Yes: metagenome assemblers recover the majority of known
content from a mock community.
Velvet IDBA Spades
Total length (>= 0 bp) 1.6E+08 2.0E+08 2.0E+08
Total length (>= 1000 bp) 1.6E+08 1.9E+08 1.9E+08
Largest contig 561,449 979,948 1,387,918
# misassembled contigs 631 1032 752
Genome fraction (%) 72.949 90.969 90.424
Duplication ratio 1.004 1.007 1.004
Results: Dr. Sherine AwadReads from Shakya et al., 2013; pmid 23387867

But!
A study of the Rifle site comparing long read (Moleculo/TruSeq) and short read/assembly content
concluded that their short read assembly was not comprehensive.
“Low rate of read mapping (18-30%) is typically indicative of complex communities with a large
number of low abundance genomes or with
high degree of species and strain variations.”
Sharon et al., Banfield lab; PMID 25665577

The dirty not-so-secret (?) about sequence assembly:The assembler will simply discard two types of data.
1. Low coverage data - can’t be reconstructed with confidence; may be erroneous.
2. Highly polymorphic data – confuses the assembler.

So: why didn’t the Rifle data assemble?
There are no published approaches that will discriminate between low coverage and strain
variation.
But we’ve known about this problem for ages.
So we’ve been working with something called “assembly graphs”.

Assembly graphs.Assembly “graphs” are a way of representing
sequencing data that retains all variation; assemblers then crunch this down into a
single sequence.
Image from Iqbal et al., 2012. PMID 23172865

Our work on assembly graphs enables:
Evaluation of data set coverage profiles prior to assembly.
Variant calling and quantification on raw metagenomic data.
Analysis of strain variation.
Evaluation of “what’s in my reads but not in my assembly”.
(See http://ivory.idyll.org/blog/2015-wok-notes.html
for details.)
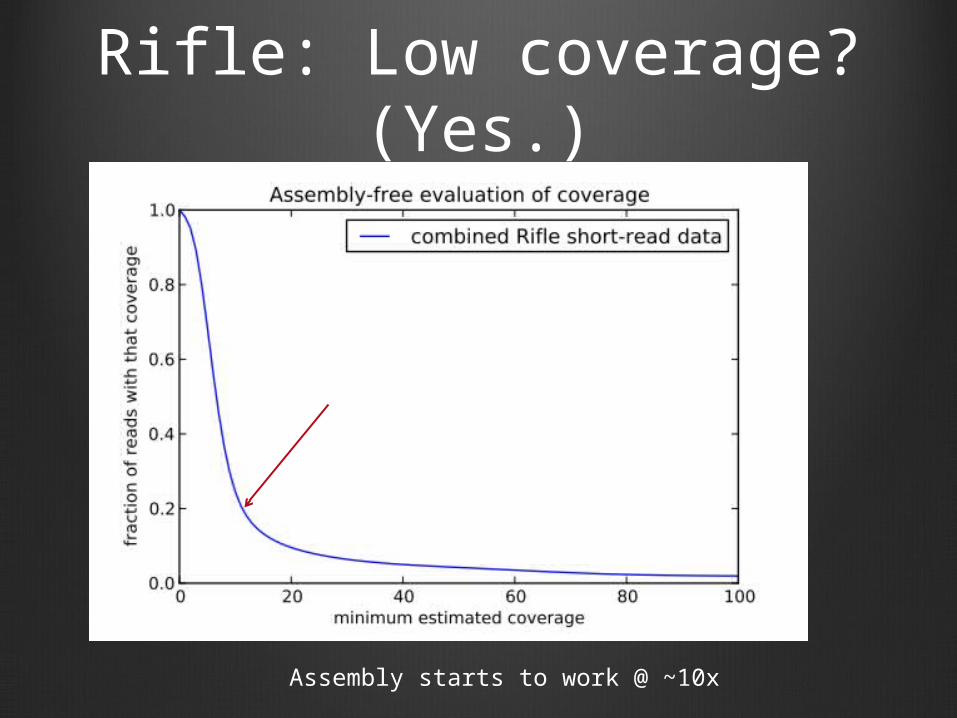
Rifle: Low coverage? (Yes.)
Assembly starts to work @ ~10x

Rifle: strain variation? (Maybe.)
ATTCGTCGATTGGCAAAAGTTCTTTCCAGAGCCTACGGGAGAAGTGTA|||||||||||||||||||||||||||||||| |||||||||||||||ATTCGTCGATTGGCAAAAGTTCTTTCCAGAGCTTACGGGAGAAGTGTA
GTCAAAATAAGGTGAGGTTGCTAATCCTCGAACTTTTCAC||||||||||||||| |||||||| ||||||| |||||||
GTCAAAATAAGGTGAAGTTGCTAACCCTCGAATTTTTCAC
A typical subalignment between all short reads & one long read:
If we saw many medium-high coverage alignments with this level of variation, => strain variation.

My thoughts on metagenome assembly & Rifle data:
The Rifle short-read data is low coverage, based on both indirect (in paper) and direct (our) observations. This is the first reason why it didn’t assemble well.
Strain variation is also present, within the limits of low coverage analysis. That will cause problems in future
=> Your methods limit and bias your results.

The problem:Assembly graphs are coming to all of genomics.
Because they are fundamentally different they require a completely new bioinformatics tool chain. (They don’t use FASTA…)
For better or for worse, us bioinformaticians are not going to write tools that are easy to use.
It’s hard;There’s little incentive;The tool/application needs are incredibly diverse;

Who ya gonna call??…to do your bioinformatics?

Choices
(1) Focus on biology and avoid computation as much as possible.
(2) Integrate large scale data analysis into your biology.
(3) Become purely computational
https://commons.wikimedia.org/wiki/File:Three_options_-_three_choices_scheme.png

Choices
(1) Focus on biology and avoid computation as much as possible.
(2) Integrate large scale data analysis into your biology.
(3) Become purely computational.

Towards a “bioinformatics middle class”
Most bioinformaticians are quite ignorant of the biology you’re doing; biologists are often more
aware of the bioinformatics they’re using.
There is amazing opportunity at the intersection of biology and computing.
I think of it as a “bioinformatics middle class” – biologists who are comfortable with computing,
and deploy large scale data analysis in the service of their biological work.

Towards a “bioinformatics middle class”
We need many more biologists who have an intuitive & deep understanding of the
computing.
Such people are rare, and there is no defined “pipeline” for them. Training
must be self-motivated.
(And higher ed has really abdicated its responsibilities in this area.)

My top four suggestions(more at end)
1. Don’t avoid computing; embrace it.
2. Invest in the Internet and social media (blogs, Twitter) – seqanswers, biostars, etc.
3. Be patient and aware of the time it takes time to effectively cross-train.
4. Seek out formal training opportunities.

If you’re a senior scientist, or know any:
Ask them to lobby for funding at this intersection.
Ask them to lobby for good (nay, excellent) funding for training opportunities.
Make sure they respect the challenges and opportunities of large scale data analysis and modeling (along with those who do it).

Career benefits of doing large-scale data analysis.
Alternative career paths (i.e. “jobs actually exist in this area.”)
Flexibility in work hours & location.
Work with an even broader diversity of people and projects.

Dangers:
It’s easy to get caught up in the computing and ignore the biology!
…but right now training & culture are tilted too much towards experimental and field research,
which presents its own problems in a data-intensive era of research.
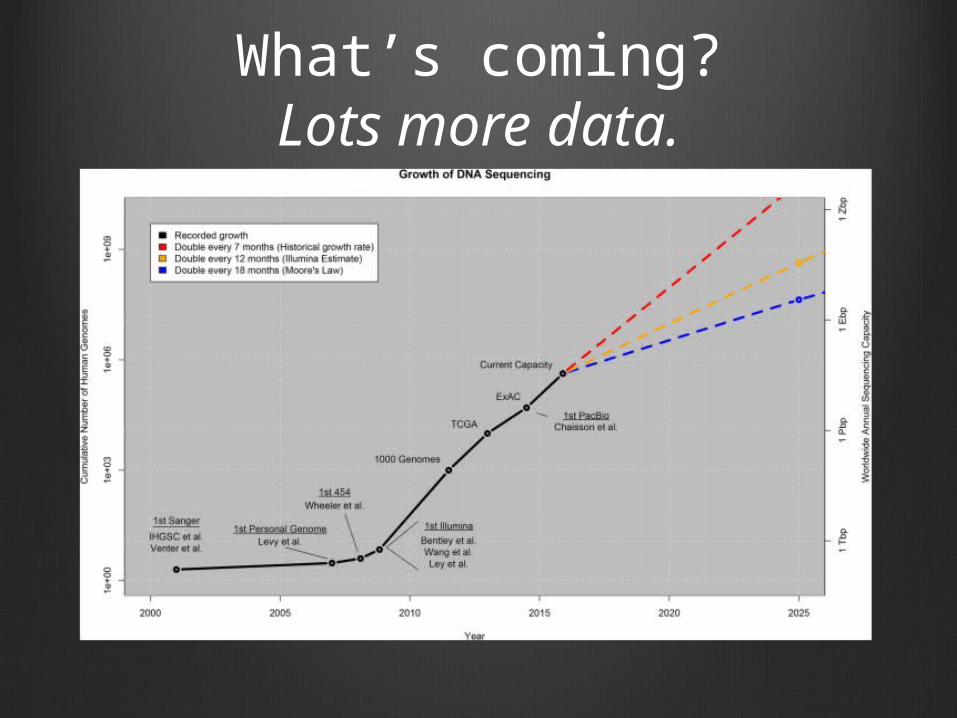
What’s coming?Lots more data.
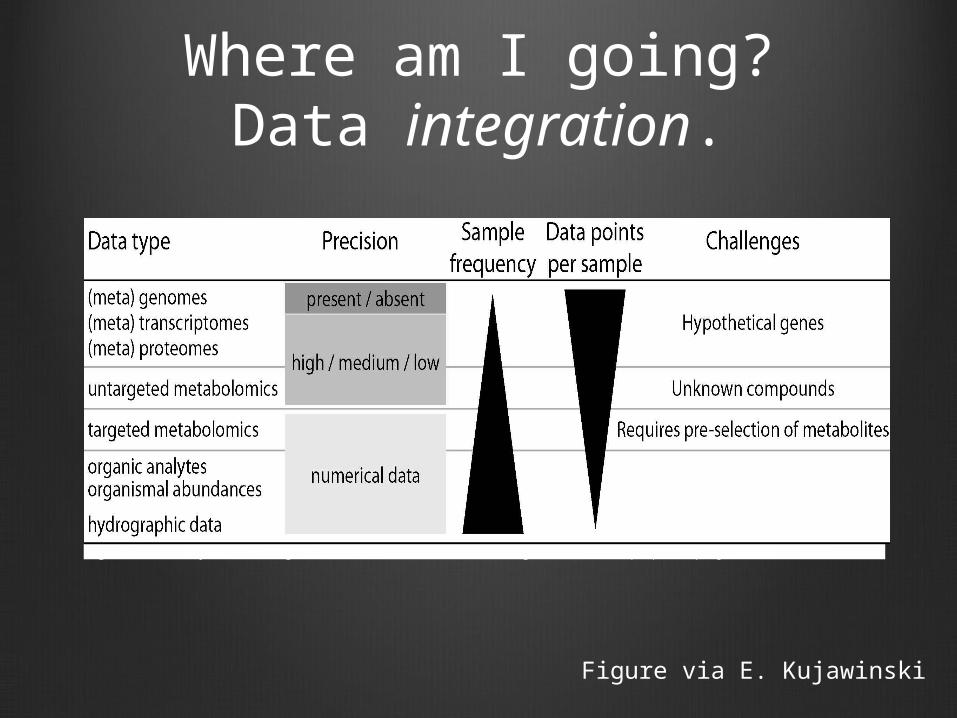
Where am I going?Data integration.
Figure 2. Summary of challenges associated with the data integration in the proposed project.
Figure via E. Kujawinski

An optimistic message
This is a great time to be alive and doing research!
We can look at & try to understand environmental microbes with many new
tools and new approaches!
The skills you need to do this extend across disciplines, across the public and
private sectors, and cannot be automated or outsourced!

Thank you for listening!
I’ll be here all week; really
looking forward to it!

More advice.
don’t avoid computing
teach and train what you do know; put together classes and workshops;
host and run software and data carpentry workshops, then put together more advanced workshops;
do hackathons or compute-focused events where you just sit down in groups and work on data analysis.
(push admin to support all this, or just do it without your admin);
invest in the internet and social media – blogs, twitter, biostars…
take a CS prof to lunch, seek joint funding, do a sabbatical in a purely compute lab, etc.
support open source bioinformatics software
invest in reproducibility
be aware that compute people’s time is as or more oversubscribed as yours & prospectively value it.





























