Embed Size (px)
Citation preview

PAPER www.rsc.org/dalton | Dalton Transactions
Chemoselective and biomimetic hydroxylation of hydrocarbons by non-hemel-oxo-bridged diiron(III) catalysts using m-CPBA as oxidant†
Ramasamy Mayilmurugan,a Helen Stoeckli-Evans,b Eringathodi Sureshc and Mallayan Palaniandavar*a
Received 20th November 2008, Accepted 31st March 2009First published as an Advance Article on the web 15th May 2009DOI: 10.1039/b820771b
Three novel non-heme m-oxo-bridged diiron(III) complexes [Fe2(m-O)(L1)2] 2, where H2(L1) isN,N¢-o-phenylenebis(salicylideneimine), [Fe2(m-O)(L2)2]·2H2O 4, where H2(L2) isN,N¢-o-phenylenebis(3,5-di-tert-butylsalicylideneimine), and [Fe2(m-O)(L3)2] 6, where H2(L3) =1,4-bis(2-hydroxybenzyl)-1,4-diazepane, have been isolated and studied as catalysts for the selectiveoxidative transformation of alkanes into alcohols using m-choloroperbenzoic acid (m-CPBA) asco-oxidant. The mononuclear iron(III) complexes [Fe(L1)Cl] 1 and [Fe(L4)Cl] 7, where H2(L4) =1,4-bis(2-hydroxy-3,5-di-tert-butylbenzyl)-1,4-diazepane, have been also isolated and thosecorresponding to the dimeric complexes 4 and 6 have been generated in CH3CN solution andcharacterized as [Fe(L2)Cl] 3 and [Fe(L3)Cl] 5 by using ESI-MS, absorption and EPR spectral andelectrochemical methods. The molecular structures of 4 and 6 have been successfully determined bysingle crystal X-ray diffraction. Both 4 and 6 possess the Fe–O–Fe structural motif with each iron atompossessing a distorted square pyramidal coordination geometry. The steric constraint at the iron(III)center in 6 is higher than that in 4 as understood from the values of the trigonality structural index(t : 4, 0.226, 0.273; 6, 0.449) due to the higher steric congestion built by the diazapane back bone. Them-oxo-to-Fe(III) LMCT band for 4 is observed around 622 nm (e, 1830 M-1 cm-1) in methanol but is notobserved in CH3CN solution and it is blue-shifted to around 485 nm (e, 5760 M-1 cm-1) in 6, possiblydue to the higher Fe–O–Fe bond angle in the latter (4, 177.4; 6, 180◦). The FeIII/FeII redox potentials ofthe dinuclear complexes (E1/2: 2, -0.606; 4, -0.329; 6, -0.889 V) are more negative than those for theircorresponding mononuclear complexes (E1/2: 1, -0.300 V; 3, -0.269; 5, -0.289 V) due to O2-
coordination. Interestingly, upon addition of peroxides (H2O2, t-BuOOH) and the peracid m-CPBA,the intensity of the phenolate-to-Fe(III) LMCT band for 2 and 6 decreases but does not exhibit anyappreciable change for 4. In the presence of m-CPBA cyclohexane is selectively (A/K, 12.2) oxidized bythe dimeric complex 4 to cyclohexanol (A, CyOH) and a small amount of the further oxidized productcyclohexanone (K, CyO). However, interestingly, the corresponding monomeric complex 3 affordsenhanced yields of both CyOH and CyO but with a lower selectivity (A/K = 1.7) and also1-chlorocyclohexane via oxidative ligand transfer (OLT). The oxidation of adamantane by 4 affordsexclusively 1-adamantanol (50.5%) and 2-adamantanol (9.5%) with enhanced yields over 12 h. Incontrast, 3 provides 1-adamantanol (32.4%) and 2-adamantanol (14.8%) and adamantanone (14.6%) inaddition to 1-chloroadamantane (14.1%) as the OLT product. The secondary C–H bond ofethylbenzene is randomly activated by both 3 and 4 to give 1-phenylethanol and acetophenone. Also,oxidation of cumene with tertiary C–H bonds to give 2-phenyl-2-propanol and the further oxidizedproduct acetophenone is illustrated by invoking the iron-phenoxyl radical species as invoked formetalloporphyrin-catalyzed systems. The strong chemoselectivity in C–H bond activation of alkanes by4 has been illustrated by invoking the involvement of a high-valent iron-oxo intermediate generated byusing m-CPBA rather than the conventional oxidants H2O2 and t-BuOOH. In contrast to 4, thecomplexes 2 and 6 fail to effect the oxidation of hydrocarbons in the presence of H2O2, t-BuOOH andm-CPBA as the co-oxidant.
aSchool of Chemistry, Bharathidasan University, Tiruchirappalli, 620 024,India. E-mail: [email protected], [email protected] of Chemistry, University of Neuchatel, Neuchatel, SwitzerlandcAnalytical Science Discipline, Central Salt and Marine Chemicals ResearchInstitute, Bhavnagar, 364 002, India† Electronic supplementary information (ESI) available: Electronic andEPR spectra, cyclic and differential pulse voltammograms, ESI-Masstable. CCDC reference numbers 710272 and 710273 for 4 and 6. For ESIand crystallographic data in CIF or other electronic format see DOI:10.1039/b820771b
Introduction
Selective oxidation and functionalization of hydrocarbons undermild conditions is an exciting and challenging scientific goal.1
Non-heme iron oxygenase enzymes like methane monooxygenase(MMO)2 have received much attention in recent years owing totheir ability to carry out a wide range of oxidative oxygen transferand oxidation reactions.3–5 Methane monooxygenases (MMOs)are widely investigated metalloenzymes that catalyze the oxidationof methane to methanol using dioxygen. Also, a wide variety
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 5101–5114 | 5101
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online / Journal Homepage / Table of Contents for this issue

of coordination environments around the iron center in non-heme enzymes generate distinct oxidizing intermediates, which aresupposed to result in intrinsic catalytic transformations.2,6 In theoxidizing intermediates the activated form of oxygen is distributedaround the iron center and then transferred to substrates. In thecase of heme enzymes the electronic structure of the oxidizingintermediate is well documented to be the oxoiron(IV) porphyrinp-cation radical.7 On the other hand, the active site observedfor the soluble diiron enzyme methane monooxygenase8 is Fe2O2
diiron center, which retains two oxidizing equivalents divided ontwo iron centers and generates the FeIV
2O2 diamond core.9 Thesynthetic diiron compounds have mainly served as benchmarksfor illustrating the oxidizing intermediates and catalytic pathwayof enzymes.
In earlier studies significant efforts have been made to reproducethe unique functional aspects of the non-heme diiron enzymes.Model complexes for these enzymes are attractive as they catalyzethe unique selective chemical transformations such as methaneoxidation using dioxygen or peroxide.10,11 Several bioinspired di-iron(III) catalysts for hydroxylation, epoxidation and sulfoxidationreactions have been reported previously.2,6,12,13 Further, severalm-oxo-diiron(III) complexes also have received much attention asthey represent structural mimics of the active centers of sMMOand related enzymes.14,15 The diiron(III) complexes of tris(2-pyridylmethyl)amine (TPA) and related ligands are well known aseffective sMMO models16 but the ligands in them do not stabilizethe m-oxo-diiron core in solution17 and the resulting complexesdisplay varied reactivity, depending on whether the complexes arepresent as mono- or diiron species in solution.18,19 The iron(III)complexes of TPA derivatives catalyze the alkene oxidation withH2O2 to give the 1,2-cis-diol and epoxide products, and Rieskedioxygenase type monoiron active species have been proposed.18,20
However, predominant epoxidation has been observed with theiron(III) complex of the ligand N,N¢-dimethyl-N,N¢-bis(2-pyridyl-methyl)ethane-1,2-diamine (BPEN),19 which is similar to TPA,and an sMMO-type diiron species has been proposed as theintermediate. Also, the sMMO model derived from a TPA-containing dinucleating ligand has been stabilized as a m-oxo-diiron core in solution and acts as an effective catalyst for epoxi-dation and alkane functionalization.21 The simple dimeric com-plex [Fe2O(phen)4(H2O)2](ClO4)4 (phen = 1,10-phenanthroline)catalyzes the epoxidation of a wide range of alkenes, includingterminal alkenes and combining low catalyst loading22 and knownas a good catalyst for selective cyclohexane oxidation with H2O2.23
Very recently, the chiral diiron complex [Fe2O(PB)4(H2O)2](ClO4)4
(PB = 4,5-pinene-2,2¢-bipyridine) has been explored as an enan-tioselective epoxidation catalyst.24 Also, surprisingly, to date thereare only a few reports concerning the oxidation of organicsubstrates using diiron(III) complexes of salen-based ligands.12
Tabushi et al. have used [Fe(salen)]2O, where salen is N,N¢-ethylenebis(salicylideneimine), as the w-hydoxylation catalyst forregioselective (secondary carbon) oxidation of adamantane inthe presence of molecular oxygen to produce 1-adamantanol(67%, based on the catalyst), 2-adamantanol (162%) and 2-adamantanone (51%).25 Collins and co-workers have shown thatthe non-heme m-oxo-bridged diiron(IV) complexes of a macrocylictetraamidate ligand (TAML) brings about the selective oxidationof benzylic alcohols into the corresponding aldehydes.26 In avery recent effort Sorokin et al. found that a m-nitrido-bridged
diiron(IV,III) phthalocyanine complex catalyzes the oxidation ofCH4 to mostly HCOOH via Fe(IV)=N–Fe(V)=O species in wateras solvent and H2O2 as co-oxidant.27 So, the design and studyof thermodynamically stable m-oxo-bridged non-heme diiron(III)complexes, which mimic the functions of enzymes with diironcore, has become important in understanding their functionsand ultimately the complexes may prove to be excellent catalystsfor the oxidation of organic substrates, particularly for alkanefunctionalisation and alkene epoxidation.
We report here the first m-oxo-bridged non-heme bis-phenolate-diiron(III) complex [Fe2(m-O)(L2)2]·2H2O 4, where H2-(L2) = N,N¢-o-phenylenebis(3,5-di-tert-butylsalicylidene)diimine(Scheme 1), as a catalyst capable of converting adamantaneinto its alcohol exclusively rather than a mixture of alcohol(A) and ketone (K) with m-chloroperbenzoic acid (m-CPBA)as the oxidant. Also, the selective hydroxylation of cyclohexaneto cyclohexanol (A/K, 12) has been observed; however, themononuclear complex [Fe(L2)Cl] 3 yields both the alcoholand ketone as a mixture (A/K, cyclohexane, 1.7; adamantane,2.8). This is interesting because the diiron(III) complexes ofpyridyl-based ligands, namely [Fe2O(OAc)2(bpy)2]Cl2 (bpy =2,2¢-bipyridine),28 [Fe2O(OAc)2(tmima)2](ClO4)3 (tmima =tris(1-methylimidazol-2-ylmethyl)amine),29 [Fe(pma)](ClO4)2
30
(pma = 2-(2¢,5¢-diazapentyl)-5-bromopyrimidine-6-carboxylicacid N-[2-(4¢-imidazolyl)ethyl]amine) and [Fe2O(bpy)4(H2O)2]-(ClO4)4
29 have been reported to produce both the alcohol andketone from cyclohexane in equimolar amounts (A/K, ~1).Also, the mononuclear systems such as [Fe(bpmen)(CH3CN)2]-(ClO4)2 (bpmen = N,N¢-dimethyl-N,N¢-bis(2-pyridylmethyl)-ethylenediamine (A/K = 6.3),6a [Fe(tpa)(CH3CN)2](ClO4)2
(tpa = tris(2-pyridylmethyl)amine) (A/K = 4.3)32 and also[Fe(N4Py)(CH3CN)](ClO4)2 (N4Py = N,N-bis(pyridylmethyl)-N-bis(2-pyridylmethyl)amine) (A/K = 7.9) elicited large A/Kratios.33 Also, the m-oxo-bridged complexes [Fe2(m-O)(L1)2] 2,where H2(L1) is N,N-o-phenylene-bis(salicylideneimine), and[Fe2(m-O)(L3)2] 6, where H2(L3) is 1,4-bis(2-hydroxybenzyl)-1,4-diazepane (Scheme 1) have been also isolated, and, interestingly,they are found to be ineffective towards alkane oxidation whenperacids and peroxides are used as the oxidants.
Scheme 1 Structures of the bis-phenolate ligands used in the study.
5102 | Dalton Trans., 2009, 5101–5114 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online

Experimental section
Materials
3,5-Di-tert-butyl-2-hydroxybenzaldehyde, 2,4-di-tert-butylphe-nol, homopiperazine, ethylbenzene, cumene, sodium cyanotri-hydroborate (Aldrich), 1,2-phenylenediamine, benzaldehyde(Loba, India), iron(III) chloride (anhydrous) (Merck, India) andadamantane (Acros Organics) were used as received and the purityof the other substrates were checked by gas chromatographybefore use. The supporting electrolyte tetra-n-butylammoniumperchlorate (NBu4ClO4, G. F. Smith, USA) was recrystallizedtwice from aqueous ethanol.
Physical measurements
Elemental analyses were performed on a Perkin Elmer Series IICHNS/O Analyzer 2400. 1H NMR spectra were recorded on aBruker 200 MHz NMR spectrometer. The electronic spectra wererecorded on a Varian Cary 300 double beam UV-VIS spectropho-tometer and Agilent 8453 diode array UV-Visible spectropho-tometer. The EPR spectra were recorded on a JEOL JES-TE 100X-band spectrometer. ESI-Mass spectrometry was performed ona Thermo Finnigan LCQ 6000 Advantage Max instrument. Cyclicvoltammetry (CV) and differential pulse voltammetry (DPV) wereperformed using a three electrode cell configuration. A platinumsphere, a platinum plate and a Ag(s)/Ag+ system were used asworking, auxiliary and reference electrodes respectively. The sup-porting electrolyte used was NBu4ClO4 (TBAP). The temperatureof the electrochemical cell was maintained at 25 ± 0.2 ◦C bya cryocirculator (HAAKE D8 G). By bubbling research gradenitrogen the solutions were deoxygenated and an atmosphere ofnitrogen was maintained over the solutions during measurements.The E1/2 values were observed under identical conditions forvarious scan rates. The instruments utilized included an EG &G PAR 273 Potentiostat/Galvanostat and Pentium-IV computeralong with EG & G M270 software to carry out the experimentsand to acquire the data. The product analyses were performedusing a HP 6890 GC series Gas Chromatograph equipped witha FID detector and a HP-5 capillary column (30 m ¥ 0.32 mm ¥2.5 mm) and GC-MS analysis was performed on a Perkin ElmerClarus 500 GC-MS instrument using a PE-5 column.
Synthesis of ligands
The ligand N,N¢-o-phenylenebis(salicylideneimine), H2(L1) wassynthesized as reported already.34
N ,N ¢-o-Phenylenebis(3,5-di-tert-butylsalicylideneimine) H2(L2).To 1,2-phenylenediamine (0.32 g, 3.0 mmol) in methanol(10 mL) was added 3,5-di-tert-butyl-2-hydroxybenzaldehyde(1.41 g, 6.0 mmol) and the solution was stirred for 12 h. Thebright yellow solid formed was filtered off and used as such withoutfurther purification for complex preparation. The yield was 60.0%(0.96 g). mp: 128–130 ◦C. 1H NMR (200 MHz, CDCl3): d 8.56(s, 2H, OH), 7.41 (s, 2H), 7.21 (s, 2H), 1.55 (s, 2H), 1.43 (s, 18H),1.30 (s, 18H).
1,4-Bis(2-hydroxybenzyl)-1,4-diazepane H2(L3). To a solutionof 2-hydroxybenzaldehyde (2.44 g, 20.0 mmol) in methanol(100 mL) were added homopiperazine (1.0 g, 10.0 mmol) and a
small amount of acetic acid. Sodium cyanotrihydroborate (1.26 g,20.0 mmol) in methanol (5 mL) was added dropwise to theresulting solution with stirring. After the solution was stirred for3 days at 25 ◦C, it was acidified by adding conc. HCl and thenevaporated almost to dryness under reduced pressure. The residuewas dissolved in saturated aqueous solution (50 mL) of Na2CO3
and extracted with CHCl3 (3 ¥ 50 mL). The combined extracts weredried over anhydrous Na2SO4 and filtered. On slow evaporationof the filtrate a white crystalline solid was obtained. Yield: 1.36 g(44.0%). It was purified by recrystallization from CH2Cl2. mp:108–110 ◦C. 1H NMR (200 MHz, CDCl3): d 7.14–7.26 (m, 2H),6.74–6.97 (m, 8H), 3.79 (s, 4H), 2.83 (t, 4H), 2.79 (s, 4H), 1.93(pentet, 1H).
1,4-Bis(2-hydroxy-3,5-di-tert-butylbenzyl)-1,4-diazepane H2(L4).A solution of 2,4-di-tert-butylphenol (5.0 g, 24.2 mmol), ho-mopiperazine (1.21 g, 12.1 mmol) and 37% aqueous formaldehyde(2.5 mL, 33.6 mmol) in methanol (10 mL) was stirred and refluxedfor 24 h. The product obtained upon cooling the solution wasfiltered and washed with ice-cold methanol to give a colorlesssolid. Yield: 4.71 (72.0%). The product was further purifiedby recrystallization from CH2Cl2. mp: 173–175 ◦C. 1H NMR(200 MHz, CDCl3): d 7.21 (s, 2H), 6.82 (s, 2H), 3.77 (s, 4H),2.82 (t, 4H), 2.77 (s, 4H), 1.94 (pentet, 1H), 1.42 (s, 18H), 1.27(s, 18H).
Synthesis of complexes
The complex [Fe(L1)Cl] (1) was prepared as reported34 already.The complexes [Fe(L2)Cl] (3) and [Fe(L3)Cl] (5) were preparedin situ by treating FeCl3 with an equivalent amount of the corre-sponding bisphenolate ligands in acetonitrile in an atmosphereof N2.
[Fe2(l-O)(L1)2] (2). This was prepared using a previouslyreported34 procedure by the reaction of FeCl3 with N,N-o-phenylenebis(salicylidene)diimine H2(L1) in the presence ofEt3N. Anal. Calcd. for C40H28N4O5Fe2: C, 63.52; H, 3.73; N, 7.41.Found C, 63.64; H, 3.75; N, 7.47%.
[Fe2(l-O)(L2)2]·2H2O (4). The complex 4 was prepared byadding a solution of anhydrous ferric chloride (0.16 g, 1.0 mmol) inmethanol (10 mL) with stirring to an acetonitrile (10 mL) solutionof an equivalent amount of the ligand H2(L2) (0.54 g, 1.0 mmol)in the presence of two equivalents of triethylamine (0.20 g, 280 mL,2.0 mmol) to abstract the phenolic hydrogens of the ligand. Thesolution was then refluxed for 2 h to obtain a green solution, whichwas cooled to room temperature to give dark red X-ray qualitycrystals. Yield: 0.98 g (79.0%). Anal. Calcd. for C72H96N4O7Fe2:C, 69.67; H, 7.80; N, 4.51. Found C, 69.64; H, 7.75; N, 4.54%.
[Fe2(l-O)(L3)2] (6). A methanolic slurry (4 mL) of the ligandH2(L3) (0.31 g, 1.0 mmol) was treated with FeCl3 (0.16 g, 1.0 mmol)in methanol (4 mL) and then triethylamine (0.20 g, 280 mL,2.0 mmol) in methanol (2 mL) was added with stirring. Thesolution was refluxed for 1 h under N2 atmosphere and the darkblue-green solution was cooled at room temperature to obtain redX-ray quality crystals, which were filtered off. Yield: 0.55 g (73%).Anal. Calcd. for C38H44N4O5Fe2: C, 60.98; H, 5.93; N, 7.49. FoundC, 60.97; H, 5.94; N, 7.52%.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 5101–5114 | 5103
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online

[Fe(L4)Cl] (7). The complex 7 was prepared by the reaction ofa methanolic slurry (4 mL) of the ligand H2(L4) (0.54 g, 1.0 mmol)with FeCl3 (0.16 g, 1.0 mmol) in methanol (4 mL) in presence ofEt3N (0.20 g, 280 mL, 2.0 mmol) in methanol (2 mL). The solutionwas stirred for 30 min and the dark blue crystalline precipitateobtained was filtered off, washed with small amounts of coldmethanol and dried over P4O10. Yield: 0.49 g (78.7%). Anal. Calcd.for C35H54N2O2FeCl: C, 67.14; H, 8.69; N, 4.47. Found C, 67.16;H, 8.62; N, 4.50.
Crystallographic data collection and structure refinement
Suitable X-ray quality crystals of 4 were obtained as dark redblocks. The intensity data were collected at 173 K (-100 ◦C) ona Stoe Mark II-Image Plate Diffraction System35 equipped witha two-circle goniometer and using Mo-Ka graphite monochro-mated radiation. Image plate distance was 135 mm, w rotationscans 0–180◦ at f 0◦, and 0–113◦ at f 90◦, step Dw = 1.5◦, 2qrange 1.70–51.55◦, dmin–dmax = 23.995–0.817 A. The structure wassolved by direct methods using the SHELXS-97.36 The refinementand all further calculations were carried out using SHELXL-97.37
The H-atoms were included in calculated positions and treatedas riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically using weighted full-matrixleast-squares on F 2. An empirical absorption correction wasapplied using the DELrefABS routine in PLATON;38 transmissionfactors: Tmin/Tmax = 0.682/0.909. It was not possible to locatethe hydrogen atoms on the water molecules that were disorderedover 4 sites, each with an occupancy of 0.5. The t-Bu group,involving the atom C29, undergoes considerable thermal motioncompared to other similar groups. Intensity data for the crystal6 were collected using Mo-Ka (l = 0.71073 A) radiation ona Bruker SMART APEX diffractometer equipped with CCDarea detector at 293 K. Absorption correction was applied bySADABS program.39 The structure was solved by direct methodsand refined on F 2 by full-matrix least-squares methods using theprogram SHELXTL version 5.1.40 All of the non-hydrogen atomswere refined anisotropically. H atoms were placed in calculatedpositions and constrained to ride on their parent atoms. The detailsof the crystallographic data for both the complexes 4 and 6 aresummarized in Table 1.
Catalytic oxidations
To an acetonitrile solution (2 mL) of alkanes (0.2 M), iron(III)complex (1 ¥ 10-3 M) and m-choloroperbenzoic acid (m-CPBA,0.05 M) was added a known amount of bromobenzene in N2 atm.After stirring for 12 h at room temperature an aliquot (1.0 ml) wastaken from the reaction mixture, filtered over a silica column andthe silica washed with diethyl ether to remove the iron complex.The GC analysis of the solution provides the amount of substrateconverted into the products and the amount of product yield wasexpressed relative to the internal standard. The products wereidentified by a comparison of the GC retention times of theauthentic sample and the reaction sample. In case the authenticsamples were not readily available the products were identified byGC-MS. The Electrospray Ionisation Mass Spectra (ESI-MS) forthe complexes and those of the reaction intermediates obtained
Table 1 Crystal data and structure refinement for [Fe2(m-O)(L2)2]·2H2O4 and [Fe2(m-O)(L3)2] 6
[Fe2(m-O)(L2)2]·2H2O [Fe2(m-O)(L3)2]
Empirical formula C72H96Fe2N4O7 C38H44Fe2N4O5
Fw 1241.23 748.47Crystal system Triclinic MonoclinicSpace group P1 P21/ca/A 14.1982(12) 11.852(4b/A 15.1411(13) 11.495(4)c/A 17.9586(16) 13.525(4)a/◦ 101.532(7) 90b/◦ 102.963(7) 106.298(7)g /◦ 100.443(7) 90V/A3 3581.5(5) 1768.7(10)T/K 173(2) 293(2)Mo-Ka l/A 0.71073 0.71073Density/Mg m-3 1.151 1.405Z 2 2m/mm-1 0.456 0.869F(000) 1328 784No. of reflections collected 34448 8608Goodness-of-fit on F 2 0.826 1.069Final R indicesR1
a [I > 2s(I)] 0.0645 0.0629wR2
b [I > 2s(I)] 0.1442 0.1187R1
a [R indices (all data)] 0.1370 0.1129wR2
b [R indices (all data)] 0.1766 0.1348
a R1 = ∑‖F o| - |F c‖/∑
|F o|. b wR2 = ∑w[(F o
2 - F c2)2/
∑w[(F o
2)2]}1/2.
immediately after mixing the complexes with peroxides/peracidwere recorded at room temperature.
Results and discussion
The ligands H2(L1) and H2(L2) (Scheme 1) were synthesized bySchiff base condensation respectively of 2-hydroxybenzaldehydeand 3,5-di-tert-butyl-2-hydroxybenzaldehyde with 1,2-phenylenediamine. The ligand H2(L3) was synthesized byreductive amination of 2-hydroxybenzaldehyde with the cyclicdiamine homopiperazine using sodium cyanoborohydride as thereducing agent. The ligand H2(L4) was obtained in good yieldby the reaction of the Mannich base formed in situ by treatingexcess of 37% aqueous formaldehyde with 2,4-di-tert-butylphenolin methanol using the reported method.41 The reaction of FeCl3
with H2(L2) and H2(L3) results in the immediate formationof the mononuclear iron(III) complexes [Fe(L2)Cl] 3 [ESI-MS,m/z, 631] and [Fe(L3)Cl] 5 [ESI-MS, m/z, 401] respectively insolution. These complexes generated in situ are extremely sensitiveto dioxygen42 and lead to the formation of red crystals of thedinuclear iron(III) complexes [Fe2(m-O)(L2)2]·2H2O 4 and [Fe2(m-O)(L3)2] 6 upon oxygenation or under atmospheric conditions.Exposure of the air sensitive complex 4 to the atmosphere yieldsinsoluble red products. The dinuclear iron(III) complexes 4 and 6were also prepared in good yields by the reaction of FeCl3 withan equivalent amount of the bisphenolate ligands in the presenceof two equivalents of Et3N in methanol. The complex 4 maybe regarded as the derivative of the previously reported m-oxodiiron complex [Fe2(m-O)(L1)2] 2.34 Interestingly, the reaction ofFeCl3 with the ligand H2(L4) results in immediate formation ofthe crystalline blue mononuclear iron(III) complex [Fe(L4)Cl] 7(ESI-MS, m/z, 630) and the sterically bulky tert-butyl substituents
5104 | Dalton Trans., 2009, 5101–5114 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online

Table 2 EPR spectral data for mononuclear iron(III) complexes inCH3CN–acetone solution at 77 K
Compound g values E/D
[Fe(L1)Cl] 1 8.36, 4.82, 2.12 0.0744.30,a 2.06, 1.94, 1.89
[Fe(L2)Cl] 3 8.28, 5.14, 2.18 0.0654.09,a 2.10, 1.97, 1.86
[Fe(L3)Cl] 5 8.48, 4.95, 2.10 0.0744.14,a 2.01, 1.93, 1.87
[Fe(L4)Cl] 7 8.49, 6.73, 4.99, 2.10 0.0734.10,a 2.01, 1.99, 1.88
a Minor component.
on the ligand H2(L4) appear to prevent the formation of thecorresponding (m-oxo)diiron(III) complex.2b,43 Also, the ESI-massspectra of 2, 4 and 6 in acetonitrile show a prominent peak clusterat m/z values of 754, 1241 and 749 respectively correspondingto [Fe2(m-O)(L)2] core illustrating the existence of the Fe–O–Festructural motif in solution. The asymmetric Fe–O stretchingvibration observed around 835 cm-1 in the IR spectra (KBr) of4 and 6 are consistent with the presence of the Fe–O–Fe coreobserved in their X-ray structures. The frozen solution EPRspectrum of the mononuclear iron(III) complex 1 (Table 2; ESI,Fig. S1†) display high-spin (S = 5/2) rhombic ferric signals43,44
around g = 8.4, 4.8 and 2.1 (E/D, 0.074) associated with the |5/2,-1/2> → |5/2, +1/2> transition and also signals correspondingto rhombic low-spin45 (S = 1/2) iron(III) species (g, 2.06, 1.94,1.89). On introducing sterically hindered tert-butyl groups in 1to obtain 3 similar high-spin ferric signals are observed but withshifts in g values and changes in E/D values (8.3, 5.1, 2.2, E/D,0.065) (Table 2; ESI, Fig. S1†) suggesting changes in rhombicityof the iron(III) coordination sphere. Also, the iron(III) complexes5 and 7 derived from the diazapane-based ligands also exhibitsimilar rhombic ferric signals but with smaller shifts in g (5: 8.5,5.0, 2.1; 7: 8.5, 6.7, 2.1) and E/D values (5, 0.074; 7, 0.073). Asexpected, the (m-oxo)diiron(III) complexes 2, 4 and 6 are found tobe EPR-silent due to large spin–spin coupling.24,26
Description of X-ray crystal structures of [Fe2(l-O)(L2)2]·2H2O 4and [Fe2(l-O)(L3)2] 6
The molecular structure of [Fe2(m-O)(L2)2]·2H2O 4 with the atomnumbering scheme is shown in Fig. 1 and the selected bond lengthsand bond angles are collected in Table 3. The molecule containsa Fe–O–Fe core and the coordination geometry around each ironatom is described as distorted square pyramidal as understoodfrom the trigonality index46,47 t of 0.226 and 0.273 [t = (b -a)/60◦; for perfect square pyramidal and trigonal bipyramidalgeometries the t values are zero and unity respectively] with thetwo amine nitrogen and two phenolate oxygen atoms of the ligandconstituting the corners of the square plane and the m-oxo oxygenatom occupying the axial position. Also, the FeN2O2 coordinationplane in each iron(III) is oriented trans to the other relative to theoxo bridge in order to avoid interligand steric repulsions. TheFe–(m-O) bond lengths observed (Fe1–O1, 1.779; Fe2–O1, 1.763A) are in the range found for other (m-oxo)diiron(III) complexes(1.75–1.80 A)20,28,29,48 and are very close to those of methemerythrin(~1.78 A).49 Each iron atom is asymmetrically bound to a bis-
Fig. 1 Molecular structure of complex [Fe2(m-O)(L2)2] 4 (40% probabilityfactor for the thermal ellipsoid). The t-Bu groups at C4, C6, C61 and C63and all hydrogen atoms have been omitted for clarity.
phenolate ligand (Fe1–O2, 1.931; Fe1–O3, 1.910; Fe2–O4, 1.915;Fe2–O5, 1.927 A) and the Fe–Ophenolate bond lengths are longerthan those observed in mononuclear iron(III) complexes44,46,50 ofbisphenolate ligands (~1.85 A) indicating that the pp orbital ofphenolate oxygen atom interacts46,51 less strongly with a half-filleddp* orbital of iron(III). The Fe–Nimine bonds (Fe1–N1, 2.115; Fe1–N2, 2.110; Fe2–N3, 2.105; Fe2–N4, 2.123 A) are also asymmetricbut fall in the range observed34,52 for other (m-oxo)-diiron(III)complexes. The Fe ◊ ◊ ◊ Fe distance in 4 (3.541 A) is in the samerange as those already reported for complexes with Fe–O–Fecore (3.35–3.55 A).16a ,20,28,29,48,53 It is interesting to compare thestructural parameters of 4 with those of its parent complex 2having a similar molecular structure. The Fe–(m-O) bond lengthsin 4 are almost the same as those in 2 (Fe1–O1, 1.78; Fe2–O1,1.78 A).34 However, the Fe–Nimine bond lengths observed for 4are much shorter, and the Fe–Ophenolate bond lengths shorter, thanthose for 2 (Fe–Nimine, 2.16, 2.17, 2.17, 2.18; Fe–Ophenolate, 1.92,1.94, 1.95, 1.95 A).32 This illustrates that incorporation of theelectron-releasing t-Bu groups on the phenolate moiety in 2 toobtain 4 leads to strong coordination of the H2(L2) ligand. Also,the complex 4 has a Fe–O–Fe bond angle (177.4◦) significantlyhigher than that in 2 (141◦). It is obvious that incorporation ofthe sterically hindering t-Bu groups in 2 to obtain 4 results in theexpansion of the Fe–O–Fe bond angle in 2 so as to make the Fe–O–Fe structural motif almost linear.54 A similar increase in theFe–O–Fe bond angle from 142.2◦ in [Fe(salen)]2O to 173.45◦ in[Fe(3-t-Busalen)]2O, where 3-t-Busalen is 2,3-dimethyl-2,3-bis[(3-tert-butylsalicylidene)amino]butane, upon introducing bulky t-Bugroups on the salen ligand without any significant increase in theFe–(m-O) bond distance, has been observed previously.52 Hence,for the m-oxo-bridged five-coordinate iron(III) complexes the stericrepulsion energies appear to outweigh any electronic preferencesfor a particular Fe–O–Fe angle.29–31
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 5101–5114 | 5105
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online

Table 3 Selected bond lengthsa [A] and bond anglesa [◦] for [Fe2(L2)2O]4 and Fe2(L3)2O] 6
[Fe2(m-O)(L2)2]·2H2O [Fe2(m-O)(L3)2]
Fe(1) ◊ ◊ ◊ Fe(2) 3.541 Fe(1) ◊ ◊ ◊ Fe(1a) 3.538Fe(1)–O(1) 1.779(4) Fe(1)–O(1) 1.869(4)Fe(1)–O(2) 1.931(3) Fe(1)–O(2) 1.902(3)Fe(1)–O(3) 1.910(3) Fe(1)–O(3) 1.7689(7)Fe(1)–N(1) 2.115(4) Fe(1)–N(1) 2.197(4)Fe(1)–N(2) 2.110(4) Fe(1)–N(2) 2.173(4)Fe(2)–O(1) 1.763(4)Fe(2)–O(4) 1.915(3)Fe(2)–O(5) 1.927(3)Fe(2)–N(3) 2.105(4)Fe(2)–N(4) 2.123(4)
O(1)–Fe(1)–O(3) 106.28(16) O(3)–Fe(1)–O(1) 112.70(13)O(1)–Fe(1)–O(2) 108.07(15) O(3)–Fe(1)–O(2) 108.67(11)O(3)–Fe(1)–O(2) 94.35(14) O(1)–Fe(1)–O(2) 94.02(16)O(1)–Fe(1)–N(2) 111.21(16) O(3)–Fe(1)–N(2) 94.90(9)O(3)–Fe(1)–N(2) 87.03(15) O(1)–Fe(1)–N(2) 88.53(17)O(2)–Fe(1)–N(2) 138.57(15) O(2)–Fe(1)–N(2) 153.06(14)O(1)–Fe(1)–N(1) 99.15(17) O(3)–Fe(1)–N(1) 101.38(9)O(3)–Fe(1)–N(1) 153.34(16) O(1)–Fe(1)–N(1) 142.61(16)O(2)–Fe(1)–N(1) 85.09(15) O(2)–Fe(1)–N(1) 89.07(15)N(2)–Fe(1)–N(1) 76.30(15) N(2)–Fe(1)–N(1) 73.22(16)O(1)–Fe(2)–O(4) 106.19(16) Fe(1a)–O(3)–Fe(1) 180.0O(1)–Fe(2)–O(5) 108.66(16) C(10)–N(1)–Fe(1) 113.2(3)O(4)–Fe(2)–O(5) 94.55(14) C(17)–O(2)–Fe(1) 131.8(3)O(1)–Fe(2)–N(3) 111.64(17)O(4)–Fe(2)–N(3) 86.88(14)O(5)–Fe(2)–N(3) 137.50(15)O(1)–Fe(2)–N(4) 98.68(16)O(4)–Fe(2)–N(4) 153.86(16)O(5)–Fe(2)–N(4) 84.84(15)N(3)–Fe(2)–N(4) 76.42(15)Fe(2)–O(1)–Fe(1) 177.4(2)C(1)–O(2)–Fe(1) 125.9(3)C(28)–O(3)–Fe(1) 133.8(3)C(37)–O(4)–Fe(2) 132.7(3)C(64)–O(5)–Fe(2) 125.2(3)
a Standard deviations in parenthesis.
A perspective view of the molecular structure of the complex[Fe2(m-O)(L3)2] 6 with atom numbering scheme is shown in Fig. 2and the selected bond lengths and bond angles are collectedin Table 3. The complex contains a Fe–O–Fe core and thecoordination geometry around each iron atom is a distorted squarepyramidal (t , 0.449; b, Fe1a–O3–Fe, 180.00◦; a, O2–Fe1–N2,153.06◦] with the two amine nitrogen and two phenolate oxygenatoms of the ligand constituting the corners of the square planeand the m-oxo oxygen atom (O3) occupying the axial position.Also, the FeN2O2 coordination plane in each iron(III) is orientedtrans to the other relative to the oxo bridge in order to avoidinterligand steric repulsions. The (m-oxo)diiron(III) unit is linear(Fe–O–Fe, 180◦), conferring a center of symmetry at the bridgingoxygen atom and the Fe-(m-O) bond distance is 1.7689 A, which isshorter than those reported for other m-oxo complexes.20,53 The Fe–Namine bonds (2.197, 2.173 A) are asymmetric and fall in the samerange (2.145–2.245 A) as that observed for the previously reportediron(III) complexes.54 The Fe–Ophenolate bonds (1.869, 1.902 A) arealso asymmetric and are shorter than the average octahedral Fe–Ophenolate bond distance42,44,46,50 of 1.92 A illustrating that the pporbital of the phenolate oxygen atom overlaps more strongly46,51
with a dp* orbital of iron(III). The Fe ◊ ◊ ◊ Fe distance (3.538 A) in 6
Fig. 2 Molecular structure of complex [Fe2(m-O)(L3)2] 6 (50% probabilityfactor for the thermal ellipsoid). Hydrogen atoms have been omitted forclarity.
falls in the range found for other mono(m-oxo)-bridged diiron(III)complexes (3.35–3.55 A)20,28,29,48,53 and is almost the same as thatin 4 (3.541 A). However, the Fe–(m-O) bond in 6 is slightlyshorter than that in 4 and the Fe–Ophenolate bonds in 6 (1.869,1.902 A) are much shorter than those in 4 illustrating that thediazapane ligand system L3 is coordinated more strongly thanthe L2 ligand. However, the Fe–Namine bond distances in 6 arelonger than the Fe–Nimine bond lengths in 4 obviously due to sp3
and sp2 hybridizations55 of the amine and imine nitrogen atomsrespectively in 6 and 4.
Electronic absorption spectral and redox properties
In acetonitrile solution all the mono- and dimeric complexesexhibit intense ligand-based bands in the range 198–288 nm.The complexes 3 and 5 were generated in situ for spectraland electrochemical studies. The less intense bands observedin the ranges 312–390 and 360–540 nm (Fig. 3, Table 4) areassignable44,46,50,56 respectively to phenolate (pp) → Fe(III) (ds*)and phenolate (pp) → Fe(III) (dp*) ligand-to-metal chargetransfer (LMCT) transitions. The complex 3 exhibits the LMCTbands (312 nm, e, 8410; 360 nm, e, 7400 M-1 cm-1) at energieshigher than those for the parent complex 1. The incorporation ofthe electron-releasing tert-butyl groups in 1 to obtain 3 enhancesthe electron density on iron(III) destabilizing the dp* orbitals,leading to an increase in energy gap between the ligand andmetal orbitals and hence the increase in energy of the observedLMCT bands.57 However, a similar blue shift is not observed ongoing from 5 to 7 indicating that the steric effect of the t-butylgroups in the diazapane ligand weakens the iron(III)-phenolateinteraction. The replacement of the imine nitrogens in 1 bythe strongly coordinating amine nitrogens as in 5 would beexpected to enhance the electron density on iron(III) destabilizingthe dp* orbitals, leading to an increase in energy gap betweenthe ligand and metal orbitals and hence an increase in energy
5106 | Dalton Trans., 2009, 5101–5114 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online
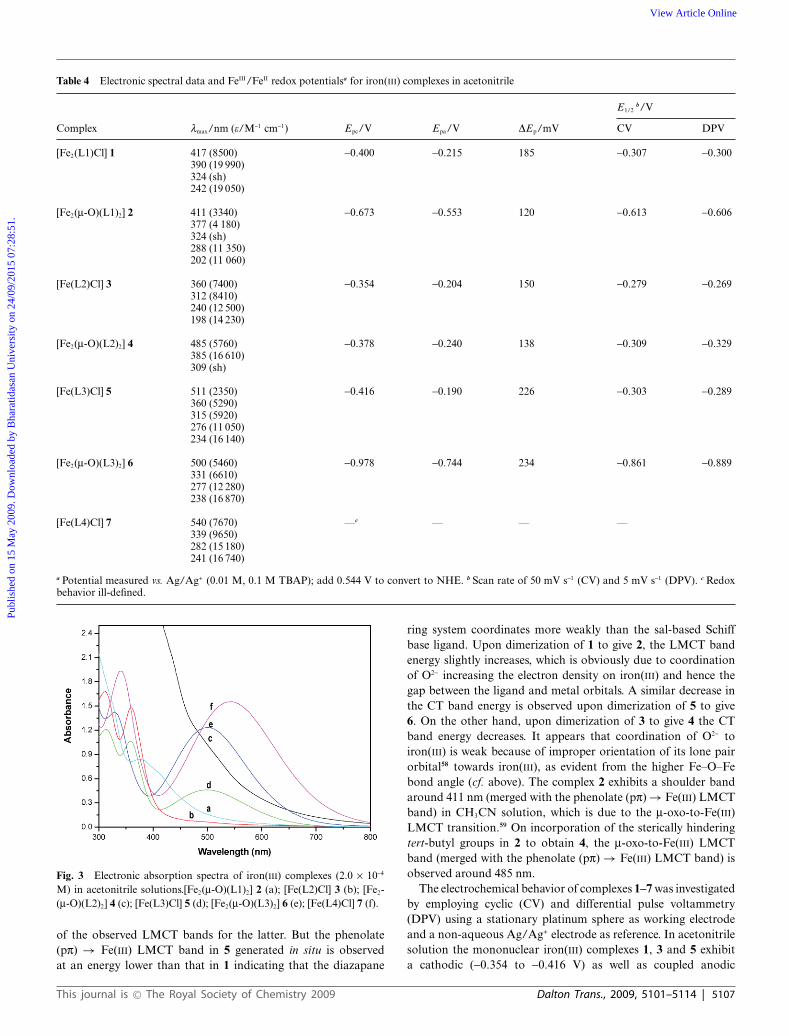
Table 4 Electronic spectral data and FeIII/FeII redox potentialsa for iron(III) complexes in acetonitrile
E1/2b/V
Complex lmax/nm (e/M-1 cm-1) Epc/V Epa/V DEp/mV CV DPV
[Fe2(L1)Cl] 1 417 (8500) -0.400 -0.215 185 -0.307 -0.300390 (19 990)324 (sh)242 (19 050)
[Fe2(m-O)(L1)2] 2 411 (3340) -0.673 -0.553 120 -0.613 -0.606377 (4 180)324 (sh)288 (11 350)202 (11 060)
[Fe(L2)Cl] 3 360 (7400) -0.354 -0.204 150 -0.279 -0.269312 (8410)240 (12 500)198 (14 230)
[Fe2(m-O)(L2)2] 4 485 (5760) -0.378 -0.240 138 -0.309 -0.329385 (16 610)309 (sh)
[Fe(L3)Cl] 5 511 (2350) -0.416 -0.190 226 -0.303 -0.289360 (5290)315 (5920)276 (11 050)234 (16 140)
[Fe2(m-O)(L3)2] 6 500 (5460) -0.978 -0.744 234 -0.861 -0.889331 (6610)277 (12 280)238 (16 870)
[Fe(L4)Cl] 7 540 (7670) —c — — —339 (9650)282 (15 180)241 (16 740)
a Potential measured vs. Ag/Ag+ (0.01 M, 0.1 M TBAP); add 0.544 V to convert to NHE. b Scan rate of 50 mV s-1 (CV) and 5 mV s-1 (DPV). c Redoxbehavior ill-defined.
Fig. 3 Electronic absorption spectra of iron(III) complexes (2.0 ¥ 10-4
M) in acetonitrile solutions.[Fe2(m-O)(L1)2] 2 (a); [Fe(L2)Cl] 3 (b); [Fe2-(m-O)(L2)2] 4 (c); [Fe(L3)Cl] 5 (d); [Fe2(m-O)(L3)2] 6 (e); [Fe(L4)Cl] 7 (f).
of the observed LMCT bands for the latter. But the phenolate(pp) → Fe(III) LMCT band in 5 generated in situ is observedat an energy lower than that in 1 indicating that the diazapane
ring system coordinates more weakly than the sal-based Schiffbase ligand. Upon dimerization of 1 to give 2, the LMCT bandenergy slightly increases, which is obviously due to coordinationof O2- increasing the electron density on iron(III) and hence thegap between the ligand and metal orbitals. A similar decrease inthe CT band energy is observed upon dimerization of 5 to give6. On the other hand, upon dimerization of 3 to give 4 the CTband energy decreases. It appears that coordination of O2- toiron(III) is weak because of improper orientation of its lone pairorbital58 towards iron(III), as evident from the higher Fe–O–Febond angle (cf. above). The complex 2 exhibits a shoulder bandaround 411 nm (merged with the phenolate (pp) → Fe(III) LMCTband) in CH3CN solution, which is due to the m-oxo-to-Fe(III)LMCT transition.59 On incorporation of the sterically hinderingtert-butyl groups in 2 to obtain 4, the m-oxo-to-Fe(III) LMCTband (merged with the phenolate (pp) → Fe(III) LMCT band) isobserved around 485 nm.
The electrochemical behavior of complexes 1–7 was investigatedby employing cyclic (CV) and differential pulse voltammetry(DPV) using a stationary platinum sphere as working electrodeand a non-aqueous Ag/Ag+ electrode as reference. In acetonitrilesolution the mononuclear iron(III) complexes 1, 3 and 5 exhibita cathodic (-0.354 to -0.416 V) as well as coupled anodic
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 5101–5114 | 5107
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online
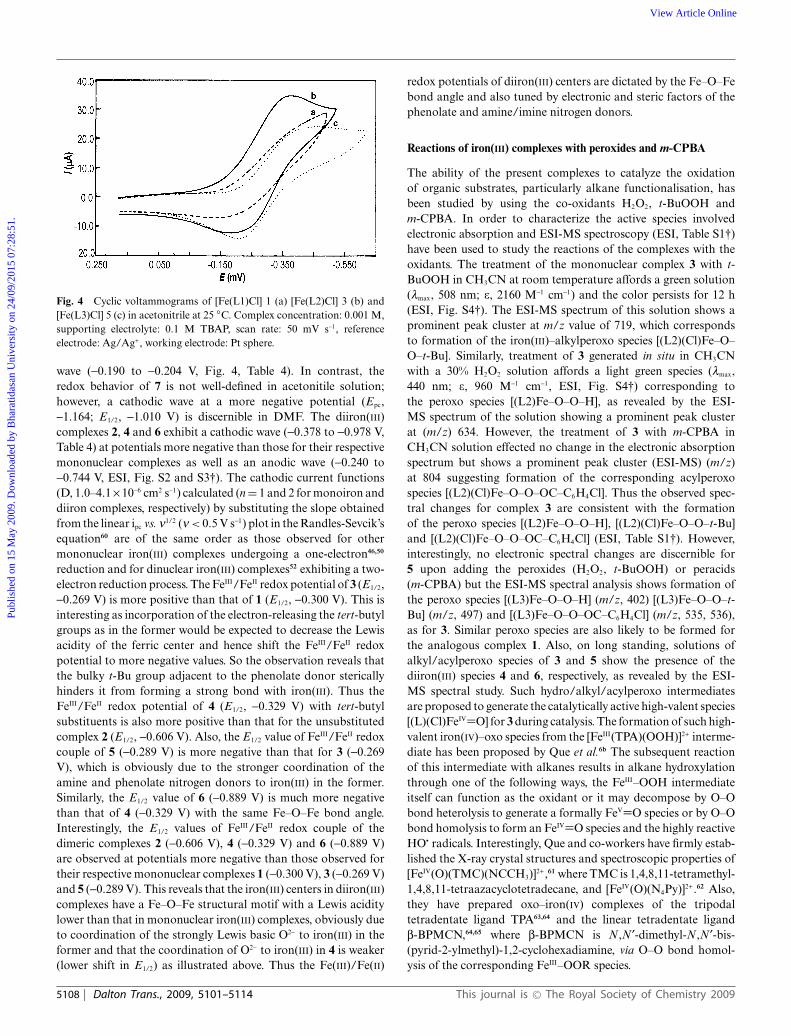
Fig. 4 Cyclic voltammograms of [Fe(L1)Cl] 1 (a) [Fe(L2)Cl] 3 (b) and[Fe(L3)Cl] 5 (c) in acetonitrile at 25 ◦C. Complex concentration: 0.001 M,supporting electrolyte: 0.1 M TBAP, scan rate: 50 mV s-1, referenceelectrode: Ag/Ag+, working electrode: Pt sphere.
wave (-0.190 to -0.204 V, Fig. 4, Table 4). In contrast, theredox behavior of 7 is not well-defined in acetonitile solution;however, a cathodic wave at a more negative potential (Epc,-1.164; E1/2, -1.010 V) is discernible in DMF. The diiron(III)complexes 2, 4 and 6 exhibit a cathodic wave (-0.378 to -0.978 V,Table 4) at potentials more negative than those for their respectivemononuclear complexes as well as an anodic wave (-0.240 to-0.744 V, ESI, Fig. S2 and S3†). The cathodic current functions(D, 1.0–4.1 ¥ 10-6 cm2 s-1) calculated (n = 1 and 2 for monoiron anddiiron complexes, respectively) by substituting the slope obtainedfrom the linear ipc vs. n1/2 (n < 0.5 V s-1) plot in the Randles-Sevcik’sequation60 are of the same order as those observed for othermononuclear iron(III) complexes undergoing a one-electron46,50
reduction and for dinuclear iron(III) complexes52 exhibiting a two-electron reduction process. The FeIII/FeII redox potential of 3 (E1/2,-0.269 V) is more positive than that of 1 (E1/2, -0.300 V). This isinteresting as incorporation of the electron-releasing the tert-butylgroups as in the former would be expected to decrease the Lewisacidity of the ferric center and hence shift the FeIII/FeII redoxpotential to more negative values. So the observation reveals thatthe bulky t-Bu group adjacent to the phenolate donor stericallyhinders it from forming a strong bond with iron(III). Thus theFeIII/FeII redox potential of 4 (E1/2, -0.329 V) with tert-butylsubstituents is also more positive than that for the unsubstitutedcomplex 2 (E1/2, -0.606 V). Also, the E1/2 value of FeIII/FeII redoxcouple of 5 (-0.289 V) is more negative than that for 3 (-0.269V), which is obviously due to the stronger coordination of theamine and phenolate nitrogen donors to iron(III) in the former.Similarly, the E1/2 value of 6 (-0.889 V) is much more negativethan that of 4 (-0.329 V) with the same Fe–O–Fe bond angle.Interestingly, the E1/2 values of FeIII/FeII redox couple of thedimeric complexes 2 (-0.606 V), 4 (-0.329 V) and 6 (-0.889 V)are observed at potentials more negative than those observed fortheir respective mononuclear complexes 1 (-0.300 V), 3 (-0.269 V)and 5 (-0.289 V). This reveals that the iron(III) centers in diiron(III)complexes have a Fe–O–Fe structural motif with a Lewis aciditylower than that in mononuclear iron(III) complexes, obviously dueto coordination of the strongly Lewis basic O2- to iron(III) in theformer and that the coordination of O2- to iron(III) in 4 is weaker(lower shift in E1/2) as illustrated above. Thus the Fe(III)/Fe(II)
redox potentials of diiron(III) centers are dictated by the Fe–O–Febond angle and also tuned by electronic and steric factors of thephenolate and amine/imine nitrogen donors.
Reactions of iron(III) complexes with peroxides and m-CPBA
The ability of the present complexes to catalyze the oxidationof organic substrates, particularly alkane functionalisation, hasbeen studied by using the co-oxidants H2O2, t-BuOOH andm-CPBA. In order to characterize the active species involvedelectronic absorption and ESI-MS spectroscopy (ESI, Table S1†)have been used to study the reactions of the complexes with theoxidants. The treatment of the mononuclear complex 3 with t-BuOOH in CH3CN at room temperature affords a green solution(lmax, 508 nm; e, 2160 M-1 cm-1) and the color persists for 12 h(ESI, Fig. S4†). The ESI-MS spectrum of this solution shows aprominent peak cluster at m/z value of 719, which correspondsto formation of the iron(III)–alkylperoxo species [(L2)(Cl)Fe–O–O–t-Bu]. Similarly, treatment of 3 generated in situ in CH3CNwith a 30% H2O2 solution affords a light green species (lmax,440 nm; e, 960 M-1 cm-1, ESI, Fig. S4†) corresponding tothe peroxo species [(L2)Fe–O–O–H], as revealed by the ESI-MS spectrum of the solution showing a prominent peak clusterat (m/z) 634. However, the treatment of 3 with m-CPBA inCH3CN solution effected no change in the electronic absorptionspectrum but shows a prominent peak cluster (ESI-MS) (m/z)at 804 suggesting formation of the corresponding acylperoxospecies [(L2)(Cl)Fe–O–O–OC–C6H4Cl]. Thus the observed spec-tral changes for complex 3 are consistent with the formationof the peroxo species [(L2)Fe–O–O–H], [(L2)(Cl)Fe–O–O–t-Bu]and [(L2)(Cl)Fe–O–O–OC–C6H4Cl] (ESI, Table S1†). However,interestingly, no electronic spectral changes are discernible for5 upon adding the peroxides (H2O2, t-BuOOH) or peracids(m-CPBA) but the ESI-MS spectral analysis shows formation ofthe peroxo species [(L3)Fe–O–O–H] (m/z, 402) [(L3)Fe–O–O–t-Bu] (m/z, 497) and [(L3)Fe–O–O–OC–C6H4Cl] (m/z, 535, 536),as for 3. Similar peroxo species are also likely to be formed forthe analogous complex 1. Also, on long standing, solutions ofalkyl/acylperoxo species of 3 and 5 show the presence of thediiron(III) species 4 and 6, respectively, as revealed by the ESI-MS spectral study. Such hydro/alkyl/acylperoxo intermediatesare proposed to generate the catalytically active high-valent species[(L)(Cl)FeIV=O] for 3 during catalysis. The formation of such high-valent iron(IV)–oxo species from the [FeIII(TPA)(OOH)]2+ interme-diate has been proposed by Que et al.6b The subsequent reactionof this intermediate with alkanes results in alkane hydroxylationthrough one of the following ways, the FeIII–OOH intermediateitself can function as the oxidant or it may decompose by O–Obond heterolysis to generate a formally FeV=O species or by O–Obond homolysis to form an FeIV=O species and the highly reactiveHO∑ radicals. Interestingly, Que and co-workers have firmly estab-lished the X-ray crystal structures and spectroscopic properties of[FeIV(O)(TMC)(NCCH3)]2+,61 where TMC is 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane, and [FeIV(O)(N4Py)]2+.62 Also,they have prepared oxo–iron(IV) complexes of the tripodaltetradentate ligand TPA63,64 and the linear tetradentate ligandb-BPMCN,64,65 where b-BPMCN is N,N¢-dimethyl-N,N¢-bis-(pyrid-2-ylmethyl)-1,2-cyclohexadiamine, via O–O bond homol-ysis of the corresponding FeIII–OOR species.
5108 | Dalton Trans., 2009, 5101–5114 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online
Upon adding H2O2 and m-CPBA to the dimeric complex 4in acetonitrile solution the hydroperoxo species [(L2)2OFe2(O–O–H)2] (m/z, 1308, 1275) and [(L2)2OFe2(O–O–OC–C6H4Cl)2](m/z, 1586, 1575) with the alkyl/hydroperoxide groups coor-dinated possibly to both the iron(III) centers are discernible.However, addition of t-BuOOH to 4 generates only themonoperoxo species [(L2)2OFe2(O–O–t-Bu)] (m/z, 1332) withonly one alkylperoxo group bound to only one of the twoiron(III) centers. These hydro/alkyl/acylperoxo intermediatesare proposed to generate the catalytically active high-valentspecies [(L2)(O)FeIV(m-O)FeIV(O)(L2)] for 4 during catalysis.Kodera and co-workers have proposed21 the involvement ofvery similar active species [(6-HPA)(O)FeIV(m-O)FeIV(O)(6-HPA)],where 6-HPA is 1,2-bis[2-{bis(2-pyridylmethyl)aminomethyl}-6-pyridyl]ethane), in the epoxidation of olefins with H2O2 asoxidant. In fact, Collins and co-workers have reported the firstcrystallographically characterized example of a non-heme oxo-bridged diiron(IV) complex using a tetraamidate macrocylic ligand(TAML).26 Que and co-workers have prepared the first exampleof a synthetic precedent complex with an [FeIV
2(m-O)2(L)2] coreby bulk electrolysis of [FeIIIFeIV(m-O)2(L)2] in CH3CN at -40 ◦C,which is the intermediate that was proposed to insert one oxygenatom of O2 into the C–H bond of various hydrocarbons includingCH4.
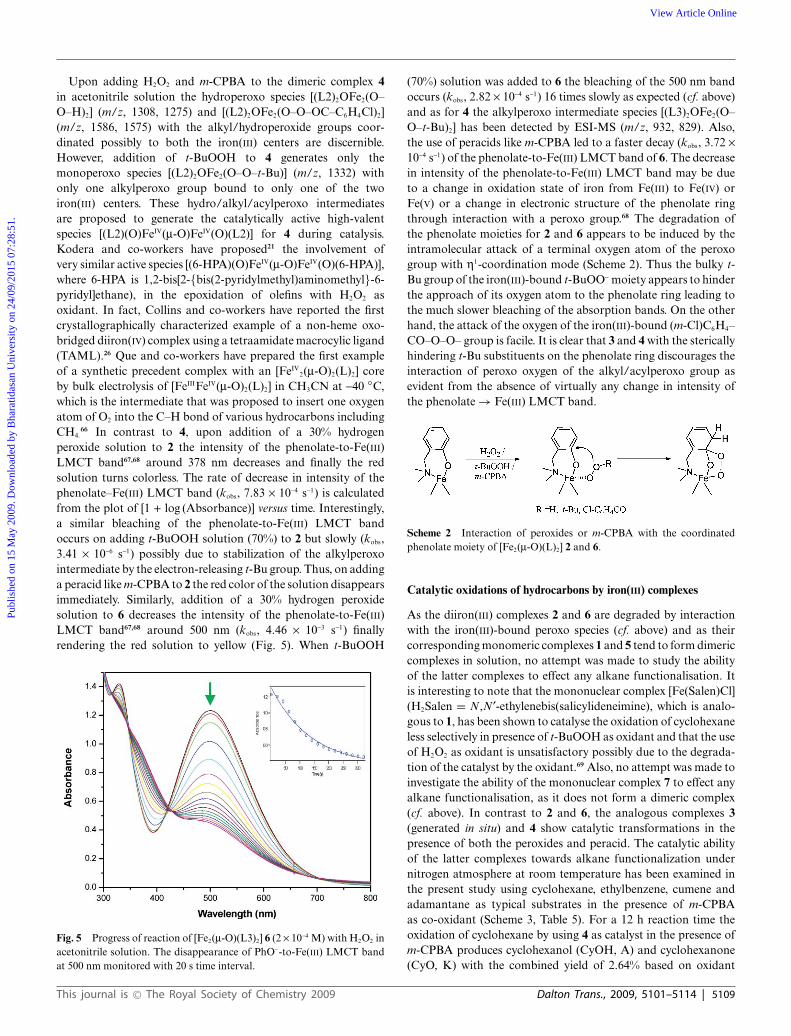
66 In contrast to 4, upon addition of a 30% hydrogenperoxide solution to 2 the intensity of the phenolate-to-Fe(III)LMCT band67,68 around 378 nm decreases and finally the redsolution turns colorless. The rate of decrease in intensity of thephenolate–Fe(III) LMCT band (kobs, 7.83 ¥ 10-4 s-1) is calculatedfrom the plot of [1 + log (Absorbance)] versus time. Interestingly,a similar bleaching of the phenolate-to-Fe(III) LMCT bandoccurs on adding t-BuOOH solution (70%) to 2 but slowly (kobs,3.41 ¥ 10-6 s-1) possibly due to stabilization of the alkylperoxointermediate by the electron-releasing t-Bu group. Thus, on addinga peracid like m-CPBA to 2 the red color of the solution disappearsimmediately. Similarly, addition of a 30% hydrogen peroxidesolution to 6 decreases the intensity of the phenolate-to-Fe(III)LMCT band67,68 around 500 nm (kobs, 4.46 ¥ 10-3 s-1) finallyrendering the red solution to yellow (Fig. 5). When t-BuOOH
Fig. 5 Progress of reaction of [Fe2(m-O)(L3)2] 6 (2 ¥ 10-4 M) with H2O2 inacetonitrile solution. The disappearance of PhO--to-Fe(III) LMCT bandat 500 nm monitored with 20 s time interval.
(70%) solution was added to 6 the bleaching of the 500 nm bandoccurs (kobs, 2.82 ¥ 10-4 s-1) 16 times slowly as expected (cf. above)and as for 4 the alkylperoxo intermediate species [(L3)2OFe2(O–O–t-Bu)2] has been detected by ESI-MS (m/z, 932, 829). Also,the use of peracids like m-CPBA led to a faster decay (kobs, 3.72 ¥10-4 s-1) of the phenolate-to-Fe(III) LMCT band of 6. The decreasein intensity of the phenolate-to-Fe(III) LMCT band may be dueto a change in oxidation state of iron from Fe(III) to Fe(IV) orFe(V) or a change in electronic structure of the phenolate ringthrough interaction with a peroxo group.68 The degradation ofthe phenolate moieties for 2 and 6 appears to be induced by theintramolecular attack of a terminal oxygen atom of the peroxogroup with h1-coordination mode (Scheme 2). Thus the bulky t-Bu group of the iron(III)-bound t-BuOO- moiety appears to hinderthe approach of its oxygen atom to the phenolate ring leading tothe much slower bleaching of the absorption bands. On the otherhand, the attack of the oxygen of the iron(III)-bound (m-Cl)C6H4–CO–O–O– group is facile. It is clear that 3 and 4 with the stericallyhindering t-Bu substituents on the phenolate ring discourages theinteraction of peroxo oxygen of the alkyl/acylperoxo group asevident from the absence of virtually any change in intensity ofthe phenolate → Fe(III) LMCT band.
Scheme 2 Interaction of peroxides or m-CPBA with the coordinatedphenolate moiety of [Fe2(m-O)(L)2] 2 and 6.
Catalytic oxidations of hydrocarbons by iron(III) complexes
As the diiron(III) complexes 2 and 6 are degraded by interactionwith the iron(III)-bound peroxo species (cf. above) and as theircorresponding monomeric complexes 1 and 5 tend to form dimericcomplexes in solution, no attempt was made to study the abilityof the latter complexes to effect any alkane functionalisation. Itis interesting to note that the mononuclear complex [Fe(Salen)Cl](H2Salen = N,N¢-ethylenebis(salicylideneimine), which is analo-gous to 1, has been shown to catalyse the oxidation of cyclohexaneless selectively in presence of t-BuOOH as oxidant and that the useof H2O2 as oxidant is unsatisfactory possibly due to the degrada-tion of the catalyst by the oxidant.69 Also, no attempt was made toinvestigate the ability of the mononuclear complex 7 to effect anyalkane functionalisation, as it does not form a dimeric complex(cf. above). In contrast to 2 and 6, the analogous complexes 3(generated in situ) and 4 show catalytic transformations in thepresence of both the peroxides and peracid. The catalytic abilityof the latter complexes towards alkane functionalization undernitrogen atmosphere at room temperature has been examined inthe present study using cyclohexane, ethylbenzene, cumene andadamantane as typical substrates in the presence of m-CPBAas co-oxidant (Scheme 3, Table 5). For a 12 h reaction time theoxidation of cyclohexane by using 4 as catalyst in the presence ofm-CPBA produces cyclohexanol (CyOH, A) and cyclohexanone(CyO, K) with the combined yield of 2.64% based on oxidant
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 5101–5114 | 5109
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online
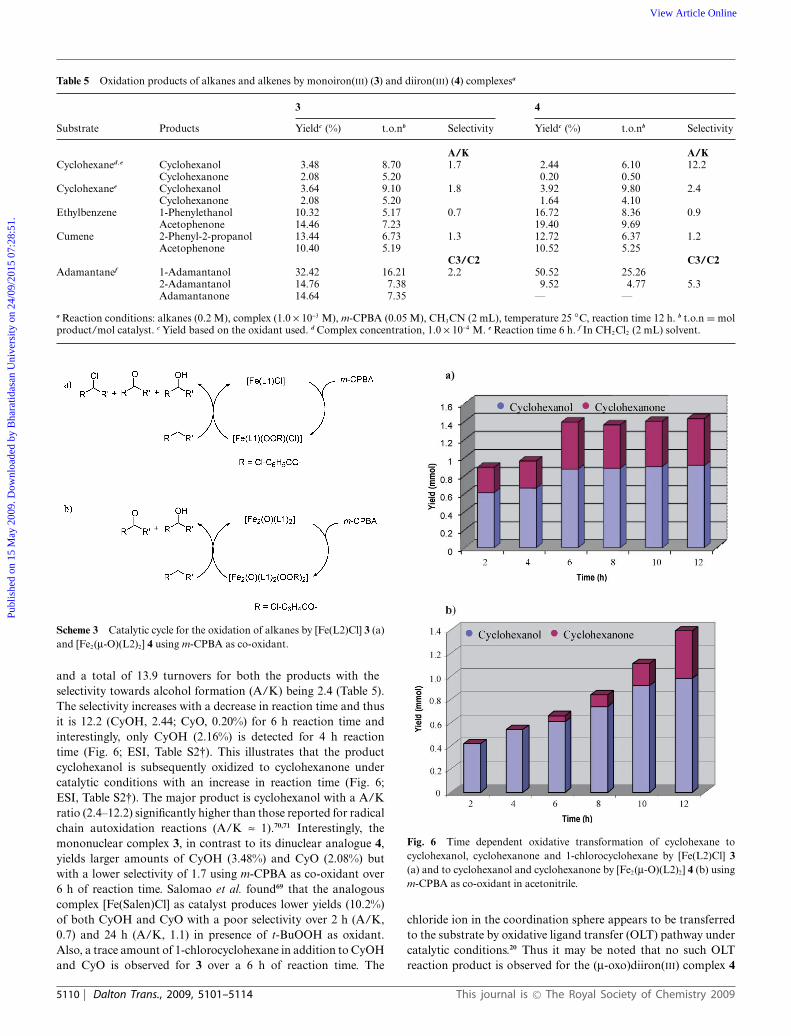
Table 5 Oxidation products of alkanes and alkenes by monoiron(III) (3) and diiron(III) (4) complexesa
3 4
Substrate Products Yieldc (%) t.o.nb Selectivity Yieldc (%) t.o.nb Selectivity
A/K A/KCyclohexaned ,e Cyclohexanol 3.48 8.70 1.7 2.44 6.10 12.2
Cyclohexanone 2.08 5.20 0.20 0.50Cyclohexanee Cyclohexanol 3.64 9.10 1.8 3.92 9.80 2.4
Cyclohexanone 2.08 5.20 1.64 4.10Ethylbenzene 1-Phenylethanol 10.32 5.17 0.7 16.72 8.36 0.9
Acetophenone 14.46 7.23 19.40 9.69Cumene 2-Phenyl-2-propanol 13.44 6.73 1.3 12.72 6.37 1.2
Acetophenone 10.40 5.19 10.52 5.25C3/C2 C3/C2
Adamantanef 1-Adamantanol 32.42 16.21 2.2 50.52 25.262-Adamantanol 14.76 7.38 9.52 4.77 5.3Adamantanone 14.64 7.35 — —
a Reaction conditions: alkanes (0.2 M), complex (1.0 ¥ 10-3 M), m-CPBA (0.05 M), CH3CN (2 mL), temperature 25 ◦C, reaction time 12 h. b t.o.n = molproduct/mol catalyst. c Yield based on the oxidant used. d Complex concentration, 1.0 ¥ 10-4 M. e Reaction time 6 h. f In CH2Cl2 (2 mL) solvent.
Scheme 3 Catalytic cycle for the oxidation of alkanes by [Fe(L2)Cl] 3 (a)and [Fe2(m-O)(L2)2] 4 using m-CPBA as co-oxidant.
and a total of 13.9 turnovers for both the products with theselectivity towards alcohol formation (A/K) being 2.4 (Table 5).The selectivity increases with a decrease in reaction time and thusit is 12.2 (CyOH, 2.44; CyO, 0.20%) for 6 h reaction time andinterestingly, only CyOH (2.16%) is detected for 4 h reactiontime (Fig. 6; ESI, Table S2†). This illustrates that the productcyclohexanol is subsequently oxidized to cyclohexanone undercatalytic conditions with an increase in reaction time (Fig. 6;ESI, Table S2†). The major product is cyclohexanol with a A/Kratio (2.4–12.2) significantly higher than those reported for radicalchain autoxidation reactions (A/K ª 1).70,71 Interestingly, themononuclear complex 3, in contrast to its dinuclear analogue 4,yields larger amounts of CyOH (3.48%) and CyO (2.08%) butwith a lower selectivity of 1.7 using m-CPBA as co-oxidant over6 h of reaction time. Salomao et al. found69 that the analogouscomplex [Fe(Salen)Cl] as catalyst produces lower yields (10.2%)of both CyOH and CyO with a poor selectivity over 2 h (A/K,0.7) and 24 h (A/K, 1.1) in presence of t-BuOOH as oxidant.Also, a trace amount of 1-chlorocyclohexane in addition to CyOHand CyO is observed for 3 over a 6 h of reaction time. The
Fig. 6 Time dependent oxidative transformation of cyclohexane tocyclohexanol, cyclohexanone and 1-chlorocyclohexane by [Fe(L2)Cl] 3(a) and to cyclohexanol and cyclohexanone by [Fe2(m-O)(L2)2] 4 (b) usingm-CPBA as co-oxidant in acetonitrile.
chloride ion in the coordination sphere appears to be transferredto the substrate by oxidative ligand transfer (OLT) pathway undercatalytic conditions.20 Thus it may be noted that no such OLTreaction product is observed for the (m-oxo)diiron(III) complex 4
5110 | Dalton Trans., 2009, 5101–5114 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online

most probably due to the absence of Cl- ion in the coordinationsphere. A reactivity similar to that in CH3CN is observed whenthe reactions are performed in both CH2Cl2 and acetone.
It is interesting to compare the catalytic behavior of 4 to-wards functionalization of cyclohexane with those of severalnon-heme iron catalysts. While the Gif family of catalysts af-fords mainly ketone products,72 catalysts such as [Fe2O(OAc)2-(bpy)2]Cl2,28 [Fe2O(OAc)2(tmima)2](ClO4)3,29 [Fe(pma)](ClO4)2
29
and [Fe2O(bpy)4(H2O)2](ClO4)431 exhibit A/K values of ª1.
Also, catalysts such as [Fe(bpmen)(CH3CN)2](ClO4)2 (A/K =6.3),6a [Fe(tpa)(CH3CN)2](ClO4)2 (A/K = 4.3)32 and [Fe(N4Py)-(CH3CN)](ClO4)2 (A/K = 7.2)33 with m-CPBA as oxidant showA/K values lower than that of 4 (A/K = 12.2). The use of theperoxides H2O2 and t-BuOOH leads to an enhanced yield, butwith a selectivity lower than that of m-CPBA. Thus, for 12 hreaction time, total turnovers of 12.2 (4.88%) and 51.8 (20.72%)with H2O2 and t-BuOOH, respectively, are observed. The catalyticoxidation of cyclohexane under aerobic conditions using m-CPBAleads to almost no change in selectivity (A/K, 12.0) and yield ofoxidation products (CyOH, 2.40; CyO, 0.20%) for 4 over 6 h.In contrast to 4, a decrease in selectivity (A/K, 1.1) and yieldof CyOH (2.84%) but increased yield of CyO (2.54%) over 6 hhave been observed for 3. Control reactions without 4 (or 3) asthe catalyst fail to effect any oxidative transformation of alkanesin the presence of both m-CPBA and TBHP revealing that thereactions are metal-catalyzed. The observation that the oxidationof cyclohexane by 4 using peroxides as co-oxidants affords amixture of alcohol and ketone (A/K, H2O2, 0.96; t-BuOOH, 0.68)clearly indicates the involvement of long-lived free radicals in thereaction leading to random oxidation.73 Thus the use of the peracid(m-CPBA) instead of the peroxides H2O2 and t-BuOOH leadsto high enhancement in chemoselectivity (CyOH formation) inthe oxidation of alkanes to alcohols. Obviously short-lived alkylradicals5,70,71 are formed with m-CPBA and not with H2O2 andt-BuOOH. The high value of the A/K ratio observed for the m-CPBA oxidation provides the direct evidence for involvement ofFeIV=O species as the reactive intermediate in the catalytic events,which determines the selectivity.5,33 Thus the lower A/K ratioobserved for 3 illustrates the formation of long-lived alkyl radicals5
by the high-valent radical species [L(Cl)FeIV=O]+∑. On the otherhand, the mechanistic pathway for the oxidation of alkanes by 4has been proposed to involve a two-electron process. The initialoxidation of the diferric complex 4 by m-CPBA generates an activeFeIV(O)–O–FeIV(O) intermediate33,74–76 (eqn (1)), which then reactswith alkanes to yield the hydroxylated products:
[(L2)Fe ( -O)Fe (L2)] [(L2)(O)Fe ( -O)FeIII III -CPBA IV Im mmæ Ææææ VV (O)(L2)]
(1)
The oxidation of adamantane, which has stronger C–Hbonds, by the diiron(III) complex 4 in the presence of m-CPBAexclusively affords the hydroxylation products 1-adamantanol(50.52%) and 2-adamantanol (9.52%) with TONs of 25.26 and4.77, respectively, but not the subsequent oxidation productadamantanone even for 12 h of reaction time. In contrast,the mononuclear analogue 3 in the presence of m-CPBA pro-vides 1-adamantanol (32.42%), 2-adamantanol (14.76%) andalso adamantanone (14.64%) in addition to 1-chloroadamantane.Obviously, the transfer of chloride ion from the coordination
sphere of the intermediate [L(Cl)FeIV=O]+∑ to adamantane undercatalytic conditions occurs through the OLT pathway as proposedabove for cyclohexane oxidation. Adamantane oxidation occursboth at secondary and tertiary carbon centers, the ratio oftertiary adamantanol to secondary adamantanol (C3/C2) being2.2 for complex 3; however, this ratio is high (5.3) for complex4. It is interesting to note that the C3/C2 ratio of 2.7 onan average has been found for Gif-type oxidations,77 about 2.0for the oxidation of alkanes by hydroxyl radicals,77b 3.5 foroxidation with [Fe2O(OAc)2(bpy)2]Cl2,28 9.5–10.0 for oxidationswith [Fe2O(bpy)4(H2O)2](ClO4)4 or [Fe(tpa)Cl2]ClO4,78,79 3.1 for[Fe(N4Py)(CH3CN)](ClO4)2
33 and 11–48 for oxidation with PhIOcatalyzed by P450 mimics.80 Hence the relatively low C3/C2 ratioobserved for the present complexes (3, 2.2; 4, 5.3) implies thatthe present catalytic reactions are distinct from a Fenton-typefree-radical oxidation reaction for which the typical C3/C2 ratiosare higher than 20.81 Also, the monomeric complex 3 is a bettercatalyst for secondary carbon oxidation than 4, as evident fromthe lower C3/C2 ratio observed for it.
In CH3CN solution the complexes 3 and 4 oxidize the substratesethylbenzene and cumene with active C–H bonds in presence ofm-CPBA to afford 1-phenylethanol (3, 10.32%; 4, 16.72%) and2-phenyl-2-propanol (3, 13.40%; 4, 12.72%), respectively, as thealcohol products.
Also, the subsequent oxidation product acetophenone is formedin major amounts for both the complexes (3, 14.46%; 4, 19.40%)when ethylbenzene is used as substrate and a smaller amount ofacetophenone (3, 10.40%; 4, 10.52%) is obtained when cumeneis used as the substrate. Small amounts (<1%) of the olefinproduct a-methylstyrene and styrene are observed for cumeneand ethylbenezene, respectively. The complex 3 affords an amountof alcohol product (10.32%) lower than the diiron(III) complex4 (16.72%) for the oxidation of secondary C–H bond of ethyl-benzene; however, the same amounts of alcohol products areobserved for cumene when 3 (13.44%) and 4 (12.72%) are usedas the catalyst. The mechanism for ethylbenzene and cumeneoxidation using iron(III) complexes is proposed in Scheme 4. Inthe first step iron(III) complex and m-CPBA initiate the abstractionof hydrogen atom from the substrate generating an intermediatealkyl radical.43b,82 They react with the alkyl radicals to producealcohol (3) and olefin (4) (path a). The alcohol would result eitherfrom the capture of the alkyl radical by the reactive iron(III)–alkylperoxo intermediate, which is similar to the oxygen reboundstep proposed for the heme-catalyzed hydroxylation or the electrontransfer between the alkyl radical and the iron(III)–alkylperoxointermediate. On the other hand, the olefin would be derived fromthe abstraction of a b-hydrogen of the alkyl radical by the ferrylor iron(III)–alkylperoxo complexes.83 However, only trace amountof olefins are detected demonstrating the involvement of anotherdistinct reaction path rather than path a. Further, the A/K ratio(3, 0.7; 4, 0.9) for ethylbenzene oxidation clearly illustrates theparticipation of a free-radical in the reaction (path b) but theA/K ratio (3, 1.2; 4, 1.3) for cumene oxidation indicates theinvolvement of both the reaction pathways (path a and b). The ox-idation of ethylbenzene and cumene to alcohol and acetophenonerespectively via the radical path b is proposed to proceed throughthe iron-phenoxyl radical species [(L2)Fe(m-ClC6H4CO3)]+∑ 8and [(L2)2Fe2(m-O)(m-ClC6H4CO3)2]2-∑ 9, which are generatedin situ from the iron(III)–arylperoxo species (discerned in the
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 5101–5114 | 5111
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online

Scheme 4 Proposed reaction pathways for ethylbenzene and cumene oxidations by 3 and 4 using m-CPBA as co-oxidant.
ESI-MS study as discussed above) obtained by the reaction of3 and 4 with m-CPBA. The species 8 and 9 are proposed togenerate the oxidizing intermediate oxo–iron(IV) phenoxyl radicalspecies in situ, similar to the oxoiron(IV)–porphyrin p-cationradical proposed in heme chemistry.71,83 Also, similar reactivespecies such as [FeIV=O(salen)]+∑12a and [FeIII(OIPh)(salen)]12b havebeen proposed for sulfide oxidation and similar iron-phenoxylradicals generated using m-CPBA have been proposed as oxidizingintermediates.43b
Conclusions
In the perspective of selective oxidation catalysis of alkanes, twonew m-oxo-bridged non-heme diiron(III) complexes of stericallyhindered bisphenolate ligands have been isolated and character-ized. Each iron(III) center of the linear Fe–O–Fe structural motif inthem adopts a distorted square pyramidal coordination geometry.Both the complexes are found to interact with H2O2, t-BuOOHand m-CPBA but only the complex of t-Bu-substituted sal-basedligand, but not that of diazapane-derived ligand, is effective incatalysing the oxidation of alkanes in the presence of m-CPBA asco-oxidant. Cyclohexane is selectively oxidized into cyclohexanolvia the formation of a high-valent diferryl intermediate generatedin situ by the co-oxidant m-CPBA. However, the selectivity(alcohol/ketone ratio) towards alcohol formation decreases withincrease in reaction time. Interestingly, adamantane is oxidized tothe hydroxylation products 1-adamantanol and 2-adamantanolexclusively with enhanced product yield over 12 h. On theother hand, the mononuclear analogue of the sal-based complexindiscriminately oxidizes cyclohexane and adamantane usingm-CPBA as co-oxidant to provide a mixture of alcohol and ketonesrevealing a poor selectivity towards alcohol formation; also,1-chlorocyclohexane and 1-chloroadamantane are formedthrough the unusual oxidative ligand transfer (OLT) reactionpathway. In the oxidation of secondary C–H bond of ethylbenzenethe diiron(III) complex affords the alcohol product higher inamount than the monomeric complex as expected; however, incumene oxidation both the complexes afford equal amounts of
alcohol and ketone products. The difference in product selectivityis explicable on the basis of formation of the high-valent FeIV(O)–FeIV(O) intermediate, which affords selective oxidation. Both themono- and diiron(III) complexes are active not only towardssubstrates containing activated C–H bonds (ethylbenzene andcumene) but also alkanes with stronger C–H bonds (cyclohexane,adamantane). It is remarkable that only the alcohols cyclohex-anol and adamantanols rather than the ketones are selectivelyobtained when the dimeric complex is used as the catalyst.Thus the diiron(III) complex of the sterically hindered N,N¢-o-phenylenebis(salicylideneimine) ligand has the potential to bedeveloped into a chemoselective catalyst for oxidation of alkanesto alcohols.
Acknowledgements
We sincerely thank the Council of Scientific and IndustrialResearch, New Delhi, for a Senior Research Fellowship to R. M.and the Department of Science and Technology, New Delhi,for supporting this research (Scheme No. SR/S5/BC-05/2006).Professor M. Palaniandavar is a recipient of DST RamannaFellowship. We thank Dr Balachandran Unni Nair, CentralLeather Research Institute, Chennai, for providing the ESI-MSfacility and Professor P. Sambasiva Rao, Pondicherry University,for providing EPR facility.
References
1 A. L. Feig and S. J. Lippard, Chem. Rev., 1994, 94, 759–805.2 M. Costas, K. K. Chen, L. Que and L. Jr., Coord. Chem. Rev., 2000,
200–202, 517–544.3 (a) L. Que, Jr. and R. Y. N. Ho, Chem. Rev., 1996, 96, 2607–2624;
(b) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Jr., Chem. Rev.,2004, 104, 939–986.
4 M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105,2227–2252.
5 S. V. Kryatov, E. V. Rybak-Akimova and S. Schindler, Chem. Rev.,2005, 105, 2175–2226.
6 (a) K. Chen and L. Que, Jr., Chem. Commun., 1999, 1375–1376;(b) K. Chen, L. Que and L. Jr., J. Am. Chem. Soc., 2001, 123, 6327–6337; (c) Y. Mekmouche, S. Menage, C. Toia-Duboc, M. Fontecave,
5112 | Dalton Trans., 2009, 5101–5114 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online

J.-B. Galey, C. Lebrun and J. Pecaut, Angew. Chem., Int. Ed., 2001, 40,949–952; (d) K. Chen and L. Que Jr., Angew. Chem., Int. Ed., 1999, 38,2227–2229; (e) W. Nam, R. Ho and J. S. Valentine, J. Am. Chem. Soc.,1991, 113, 7052–7054; (f) K. Chen, M. Costas and L. J. Que, J. Chem.Soc., Dalton Trans., 2002, 672–679.
7 M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev.,1996, 96, 2841–2887.
8 A. C. Rosenzweig, C. A. Frederick, S. J. Lippard and P. Nordlund,Nature, 1993, 366, 537–543.
9 (a) S.-K. Lee, J. C. Nesheim and J. D. Lipscomb, J. Biol. Chem.,1993, 268, 21569–21577; (b) S.-K. Lee, B. G. Fox, W. A. Froland,J. D. Lipscomb and E. Munck, J. Am. Chem. Soc., 1993, 115, 6450–6451; (c) K. E. Liu, A. M. Valentine, D. Wang, B. H. Huynh, D. E.Edmondson, A. Salifoglou and S. J. Lippard, J. Am. Chem. Soc.,1995, 117, 10174–10185; (d) L. Shu, J. C. Nesheim, K. Kauffmann,E. Munck, J. D. Lipscomb and L. Que, Jr., Science, 1997, 275,515–518.
10 Y. Jin and J. D. Lipscomb, J. Biol. Inorg. Chem., 2001, 6, 717–725.11 J. H. Green and H. Dalton, J. Biol. Chem., 1989, 264, 17698–17703.12 (a) V. K. Sivasubramanian, M. Ganesan, S. Rajagopal and R. Ramaraj,
J. Org. Chem., 2002, 67, 1506–1514; (b) K. P. Bryliakov and E. P. Talsi,Angew. Chem., Int. Ed., 2004, 43, 5228–5230.
13 E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012.14 M. H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Chem.
Rev., 2003, 103, 2385–2420.15 (a) B. J. Wallar and J. D. Lipscomb, Chem. Rev., 1996, 96, 2625; (b) D. M.
Kurtz, Jr., Chem. Rev., 1990, 90, 585–606.16 (a) Y. Dong, H. Fujii, M. P. Hendrich, R. A. Leising, G. Pan, C. R.
Randall, E. C. Wilkinson, Y. Zang and L. Que Jr., J. Am. Chem.Soc., 1995, 117, 2778–2792; (b) C. Kim, Y. Dong and Que Jr., J. Am.Chem. Soc., 1997, 119, 3635–3636; (c) Y. Dong, Y. Zang, L. Shu, E. C.Wilkinson and L. Que, Jr., J. Am. Chem. Soc., 1997, 119, 12683–12684;(d) H.-F. Hsu, Y. Dong, L. Shu, V. G. Young, Jr. and L. Que Jr., J. Am.Chem. Soc., 1999, 121, 5230–5237.
17 J. Kim, Y. Dong, E. Larka and L. Que Jr., Inorg. Chem., 1996, 35,2369–2372.
18 (a) K. Chen, M. Costas, J. Kim, A. K. Tipton and L. Que, Jr., J. Am.Chem. Soc., 2002, 124, 3026–3035; (b) M. Costas and L. Que, Jr., Angew.Chem., Int. Ed., 2002, 41, 2179–2181; (c) M. Fujita, M. Costas and QueJr., J. Am. Chem. Soc., 2003, 125, 9912–9913.
19 (a) M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc.,2001, 123, 7194; (b) J. Y. Ryu, J. Kim, M. Costas, K. Chen, W. Namand L. Que, Jr., Chem. Commun., 2002, 1288–1289.
20 T. Kojima, R. A. Leising, S. Yan and L. Que, Jr., J. Am. Chem. Soc.,1993, 115, 11328–11335.
21 M. Kodera, M. Itoh, K. Kano, T. Funabiki and M. Reglier, Angew.Chem., Int. Ed., 2005, 44, 7104–7106.
22 G. Dubois, A. Murphy and T. D. P. Stack, Org. Lett., 2003, 5, 2469–2472.
23 S. Menage, J. M. Vincent, C. Lambeaux, G. Chottard, A. Grand andM. Fontecave, Inorg. Chem., 1993, 32, 4766–4773.
24 C. Marchi-Delapierre, A. Jorge-Robin, A. Thibon and S. Menage,Chem. Commun., 2007, 1166–1168.
25 I. Tabushi, T. Nakajima and K. Seto, Tetrahedron Lett., 1980, 21, 2565–2568.
26 A. Ghosh, F. Tiago, de Oliveira, T. Yano, T. Nishioka, E. S. Beach, I.Kinoshita, E. Munck, A. D. Ryabov, C. P. Horwitz and T. J. Collins,J. Am. Chem. Soc., 2005, 127, 2505–2513.
27 A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008,2562–2564.
28 R. H. Fish, M. S. Konnings, K. J. Oberhausen, R. H. Fong, W. M. Yu,G. Christou, J. B. Vincent, D. K. Coggin and R. M. Buchanan, Inorg.Chem., 1991, 30, 3002–3006.
29 R. M. Buchanan, S. Chen, J. F. Richardson, M. Bressan, L. Forti, A.Morvillo and R. H. Fish, Inorg. Chem., 1994, 33, 3208–3209.
30 C. Nguyen, R. J. Guajardo and P. K. Mascharak, Inorg. Chem., 1996,35, 6273–6281.
31 S. Menage, J. M. Vincent, C. Lambeaux and M. Fontecave, J. Mol.Catal. A: Chem., 1996, 113, 61–75.
32 C. Kim, K. Chen, J. Kim and L. Que, Jr., J. Am. Chem. Soc., 1997, 119,5964–5965.
33 T. A. van den Berg, J. W. de Boer, W. R. Brownne, G. Roefles and B. N.Feringa, Chem. Commun., 2004, 2550–2551.
34 K. Oyaizu, E. L. Dewi and E. Tsuchida, Inorg. Chim. Acta, 2001, 321,205–208.
35 X-Area V1.17 & X-RED32 V1.04 Software, Stoe & Cie GmbH,Darmstadt, Germany. 2002.
36 G. M. Sheldrick, SHELXS-97 Program for Crystal Structure Determi-nation, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473.
37 G. M. Sheldrick, SHELXL-97, Program for refinement of crystalstructures, University of Gottingen, Germany, 1997.
38 A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13.39 G. M. Sheldrick, SADABS, Program for area detector adsorption
correction, Institute for Inorganic Chemistry, University of Gottingen,Germany, 1996.
40 G. M. Sheldrick, SHELXTL V5.1 Software Reference Manual, BrukerAXS, Inc., Madison, Wisconsin, USA, 1998.
41 E. Y. Tshuva, I. Goldberg, M. Kol and Z. Goldschmidt,Organometallics, 2001, 20, 3017–3028.
42 B. Carre, J.-P. Costes, J.-B. Tommasino, D. de. Montauzon, F. Souletand P.-L. Fabre, Polyhedron, 1993, 12, 641–649, and references therein.
43 (a) H. Fujii and Y. Funabashi, Angew. Chem., Int. Ed., 2002,41, 3638–3641; (b) T. Kurahashi, Y. Kobayashi, S. Nagatomo, T.Tosha, T. Kitagawa and H. Fujii, Inorg. Chem., 2005, 44, 8156–8166;(c) T. Kurahashi, K. Oda, M. Sugimoto, T. Ogura and H. Fujii, Inorg.Chem., 2006, 45, 7709–7721.
44 R. Mayilmurugan, E. Suresh and M. Palaniandavar, Inorg. Chem.,2007, 46, 6038–6049.
45 M. R. Bukowshi, P. Comba, C. Limberg, M. Merz, L. Que Jr. and T.Wistuba, Angew. Chem., Int. Ed., 2004, 43, 1283–1287.
46 M. Velusamy, M. Palaniandavar, R. Srinivasa, Gopalan and G. U.Kulkarni, Inorg. Chem., 2003, 42, 8283–8293.
47 A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Vershcoor,J. Chem. Soc., Dalton Trans., 1984, 1349–1356.
48 X. Wang, S. Wang, L. Li, E. B. Sundberg and G. P. Gacho, Inorg.Chem., 2003, 42, 7799–7808.
49 L. Que, Jr and A. E. True, Prog. Inorg. Chem., 1990, 38, 97.50 M. Velusamy, R. Mayilmurugan and M. Palaniandavar, Inorg. Chem.,
2004, 43, 6284–6293.51 M. R. McDevitt, A. W. Addision, E. Sinn and L. K. Thompson, Inorg.
Chem., 1990, 29, 3425–3433.52 R. N. Mukherjee, T. D. P. Stack and R. H. Holm, J. Am. Chem. Soc.,
1988, 110, 1850–1861.53 E. Wilkinson, Y. Dong and L. Que, Jr., J. Am. Chem. Soc., 1994, 116,
8394–8395.54 (a) A. Hazell, K. B. Jensen, C. J. Mckenzie and H. Toftlund, J. Chem.
Soc., Dalton Trans., 1993, 3249–3258; (b) E. C. Wilkinson, Y. Dong andL. Que, Jr., J. Am. Chem. Soc., 1994, 116, 8394–8395.
55 R. Mayilmurugan, H. S. Evans and M. Palaniandavar, Inorg. Chem.,2008, 47, 6645–6658.
56 (a) L. Casella, M. Gullotti, A. Pintar, L. Messouri, A. Rockenbauer andM. Gyor, Inorg. Chem., 1987, 26, 1031–1038; (b) S. Wang, L. Wang, X.Wang and Q. Luo, Inorg. Chim. Acta, 1997, 254, 71–77; (c) B. Krebs,K. Schepers, B. Bremer, G. Henkel, E. Althus, W. Muller-Warmuth,K. Griesar and W. Haase, Inorg. Chem., 1996, 35, 2360–2368; (d) B. P.Gaber, V. Miskowski and T. G. Spiro, J. Am. Chem. Soc., 1974, 96,6868–6873; (e) M. S. Shongwe, C. H. Kaschula, M. S. Adsetts, E. W.Ainscough, A. M. Brodie and M. J. Morris, Inorg. Chem., 2005, 44,3070–3079; (f) C. Imbert, H. P. Hratchian, M. Lanznaster, M. J. Heeg,L. M. Hryhorczuk, B. R. McGarvey, H. B. Schlegel and C. Verani,Inorg. Chem., 2005, 44, 7414–7422.
57 D. D. Cox, S. J. Benkovic, L. M. Bloom, F. C. Bradley, M. J. Nelson,L. Que, Jr. and D. E. Wallick, J. Am. Chem. Soc., 1988, 110, 2026–2032.
58 (a) K. Visvaganesan, R. Mayilmurugan, E. Suresh and M.Palaniandavar, Inorg. Chem., 2007, 46, 10294–10306; (b) D. D. Coxand L. Que, Jr., J. Am. Chem. Soc., 1988, 110, 8085–8092.
59 R. E. Norman, R. C. Holz, S. Menage, C. J. O’Connor, J. H. Zhangand L. Que, Jr., Inorg. Chem., 1990, 29, 4629–4637.
60 A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentaland Applications, John Wiley & Sons, New York, 1980, pp. 218.
61 J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski,A. Stubna, E. Munck, W. Nam and L. Que, Jr, Science, 2003, 299,1037–1039.
62 E. J. Klinker, J. Kaizer, W. W. Brennessel, N. L. Woodrum, C. J. Cramerand L. Que Jr., Angew. Chem., Int. Ed., 2005, 44, 3690–3694.
63 M. H. Lim, J.-U. Rohde, A. Stubna, M. R. Bukowski, M. Costas,R. Y. N. Ho, E. Munck, W. Nam and L. Que, Jr., Proc. Natl. Acad. Sci.U. S. A., 2003, 100, 3665–3670.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 5101–5114 | 5113
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online

64 J. Kaizer, M. Costas and L. Que, Jr., Angew. Chem., Int. Ed., 2003, 42,3671–3673.
65 M. P. Jensen, M. Costas, R. Y. N. Ho, J. Kaizer, A. Mairata i Payeras,E. Munck, L. Que Jr., J.-U. Rohde and A. Stubna, J. Am. Chem. Soc.,2005, 127, 10512–10525.
66 G. Xue, D. Wang, R. De Hont, A. T. Fiedler, X. Shan, E. Munck andL. Que, Jr., Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20713–20718.
67 S. Ito, M. Suzuki, T. Kobayashi, H. Itoh, A. Harada, S. Ohba and Y.Nishida, J. Chem. Soc., Dalton Trans., 1996, 2579–2580.
68 S. Ito, Y. Ishikawa, S. Nishino, T. Kobayashi, S. Ohba and Y. Nishida,Polyhedron, 1998, 17, 4379–4391.
69 G. C. Salomao, M. H. N. Olsen, V. Drago, C. Fernandes, L. C. Filhoand O. A. C. Antunes, Catal. Commun., 2007, 8, 69–72.
70 P. A. MacFaul, K. U. Ingold, D. D. M. Wayner and L. Que, Jr., J. Am.Chem. Soc., 1997, 119, 10594–10598.
71 (a) P. A. MacFaul, I. W. C. E. Arends, K. U. Ingold and D. D. M.Wayner, J. Chem. Soc., Perkin Trans. 2, 1997, 135–145; (b) S. Evan andJ. R. L. Smith, J. Chem. Soc., Perkin Trans. 2, 2000, 1541–1551.
72 D. H. R. Barton and M. Doller, Acc. Chem. Res., 1992, 25, 504–512.73 J. Kim, R. G. Harrison, C. Kim and L. Que Jr., J. Am. Chem. Soc.,
1996, 118, 4373–4379.
74 F. Avenier, L. Dubois and J. M. Latour, New J. Chem., 2004, 28, 782–784.
75 F. Avenier, L. Dubois, P. Dubourdeaux and J. M. Latour, Chem.Commun., 2005, 480–482.
76 A. M. Valentine, S. S. Stahl and S. J. Lippard, J. Am. Chem. Soc., 1999,121, 3876–3887.
77 (a) D. H. R. Barton, J. Boivin, W. B. Motherwell, N. Ozabalik, K. M.Schwarzentruber and K. Jankowski, New J. Chem., 1986, 10, 387–398;(b) B. Singh, J. R. Long, F. F. de Biani, D. Gatteschi and P. Stavropoulos,J. Am. Chem. Soc., 1997, 119, 7030–7047.
78 T. Kojima, R. A. Leising, S. Yan and L. Que Jr., J. Am. Chem. Soc.,1993, 115, 11328–11335.
79 J. T. Groves and T. E. Nemo, J. Am. Chem. Soc., 1983, 105, 6243–624.80 N. Kitajima, H. Fukui and Y. Moro-oka, J. Chem. Soc., Chem.
Commun., 1988, 485–486.81 A. J. Appleton, S. Evans and J. R. Lindsay Smith, J. Chem. Soc., Perkin
Trans. 2, 1996, 281–285.82 C. Kim, Y. Dong and L. Que, Jr., J. Am. Chem. Soc., 1997, 119, 3635–
3636.83 J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J.
Evans, J. Am. Chem. Soc., 1981, 103, 2884–2886.
5114 | Dalton Trans., 2009, 5101–5114 This journal is © The Royal Society of Chemistry 2009
Publ
ishe
d on
15
May
200
9. D
ownl
oade
d by
Bha
ratid
asan
Uni
vers
ity o
n 24
/09/
2015
07:
28:5
1.
View Article Online





























