Embed Size (px)
Citation preview

A Systematic Characterization of MitochondrialProteome from Human T Leukemia Cells*□S
Karim Rezaul, Linfeng Wu, Viveka Mayya, Sun-Il Hwang, and David Han‡
Global understanding of tissue-specific differences in mi-tochondrial signal transduction requires comprehensivemitochondrial protein identification from multiple cell andtissue types. Here, we explore the feasibility and effi-ciency of protein identification using the one-dimensionalgel electrophoresis in combination with the nano liquid-chromatography tandem mass spectrometry (GeLC-MS/MS). The use of only 40 �g of purified mitochondrial pro-teins and data analysis using stringent scoring criteriaand the molecular mass validation of the gel slices en-ables the identification of 227 known mitochondrial pro-teins (membrane and soluble) and 453 additional proteinslikely to be associated with mitochondria. Replicate anal-yses of 60 �g of mitochondrial proteins on the fasterscanning LTQ mass spectrometer validate all the previ-ously identified proteins and most of the single hit pro-teins except the 81 single hit proteins. Among the identi-fied proteins, 466 proteins are known to functionallyparticipate in various processes such as respiration, tri-carboxylic acid cycle (TCA cycle), amino acid and nucle-otide metabolism, glycolysis, protection against oxidativestress, mitochondrial assembly, molecular transport, pro-tein biosynthesis, cell cycle control, and many knowncellular processes. The distribution of identified proteinsin terms of size, pI, and hydrophobicity reveal that thepresent analytical strategy is largely unbiased and veryefficient. Thus, we conclude that this approach is suitablefor characterizing subcellular proteomes form multiplecells and tissues. Molecular & Cellular Proteomics 4:169–181, 2005.
Mitochondria are one of the most complex and importantorganelles found in eukaryotic cells. In addition to their centralrole in energy metabolism, mitochondria are involved in manycellular processes and mitochondrial dysfunctions have beenassociated with apoptosis, aging, and a number of patholog-ical conditions, including Parkinson’s, diabetes mellitus, Alz-heimer’s, and cardiovascular diseases (1, 2). The fundamentalrole of mitochondria in cell life and death has driven experi-mental efforts to define mitochondrial proteome and to dis-cover new molecular target for drug development and thera-peutic intervention. In mammals, the mitochondrial genome is
approximately 16,500 nucleotides long and encodes the 12and 16S rRNA, 22 tRNAs, and 13 polypeptides, all of whichencode essential components of the respiratory chain. Thelow complexity of the mitochondrial genome indicates thatvast majority of the mitochondrial proteins (estimated to be1,500) are encoded by nuclear genome (1–3). So far, thelargest proteomic study of purified human heart mitochondriawas performed by Taylor et al., leading to the identification of615 mitochondrial and mitochondria-associated proteins witha coverage of �45% of the predicted human mitochondrialproteins (4, 5). However, to achieve this number of proteinidentifications, 701 LC-MS/MS runs were carried out with astarting material of 40 mg of mitochondrial proteins. MitoPro-teome database, a recently developed publicly accessibledatabase, lists 784 mitochondrial genes and proteins for hu-man, well short of the predicted human mitochondrial proteins(6). Traditionally, two-dimensional (2D)1-PAGE has been themethod of choice for the characterization of a complex pro-tein mixture prior to MS analysis followed by enzymatic di-gestion of the separated protein spots (7, 8). In 2D-PAGE, dueto the separation of protein into multiple gel spots, subse-quent digestion, extraction, and LC-MS/MS analysis of eachspot are tedious and time consuming. Moreover, despite therecent advances, this approach is biased against membrane-associated proteins, low-abundance proteins, or proteins withextremes in isoelectric point or molecular mass (9, 10). Toovercome the limitations of 2D-PAGE, approaches whichcombine one or several chromatographic separations withtandem mass spectrometry have been developed (11–13).One promising method, termed multidimensional capillary-scale LC-ESI-MS/MS protein identification technology (Mud-PIT) and pioneered by Yates and colleagues, permits shotgunsequencing of large numbers of proteins present in complexmixtures (11–13). MudPIT has been applied successfully toseveral model organisms, leading to identification of 1,484proteins in yeast, 2,363 proteins in rice, and most recently2,415 proteins in plasmodium (12, 14, 15), thus suggestingthat MudPIT is much more powerful for the global character-ization of proteins from cells and tissues compared with the
From the Department of Cell Biology, Center for Vascular Biology,University of Connecticut Health Center, Farmington, CT 06030
Received, August 31, 2004, and in revised form, December 9, 2004Published, MCP Papers in Press, December 14, 2004, DOI
10.1074/mcp.M400115-MCP200
1 The abbreviations used are: 2D, two dimensional; 1D, one dimen-sional; GeLC-MS/MS, 1D PAGE protein separation followed by nano-capillary LC-MS/MS; TMD, transmembrane domain; hnRNP, hetero-geneous nuclear ribonucleoprotein; MudPIT, multidimensionalcapillary-scale LC-ESI-MS/MS protein identification technology;LDH, lactate dehydrogenase; HFBA, heptaflourobutyric acid; PCNA,proliferating cell nuclear antigen; TCA, tricarboxylic acid.
Research
© 2005 by The American Society for Biochemistry and Molecular Biology, Inc. Molecular & Cellular Proteomics 4.2 169This paper is available on line at http://www.mcponline.org

2D-PAGE. However, successful analysis using the MudPITstrategy requires proficiency and sophistication in multidi-mensional chromatography to prevent sample loss associ-ated with each of the chromatography steps.
An alternative approach that is simple and easily performedby many in the field of proteomics with very little expertise isone-dimensional (1D)-SDS-PAGE protein separation andnano-LC-MS/MS. Recently, this method has been success-fully employed for profiling proteome of mitochondria andhuman cell lines (4, 16, 17). In this study, we have used asimilar approach to create a reference proteome database ofmitochondria from human Jurkat T leukemia cells. This cellline has been extensively utilized to study apoptosis, and all ofthese cells synchronously undergo apoptosis when the Fasreceptor is engaged (1–3, 18). Furthermore, identification ofthe mitochondrial protein makeup of this cell line may provideadditional biological insights of leukemic transformation.From the sucrose gradient-purified mitochondria, we haveidentified 680 mitochondrial and mitochondria-associatedproteins, of which 227 are known mitochondrial proteins.Thus, this study provides the first large-scale analysis of hu-man leukemia cells and allows further comparative analysis oftissue-specific protein expression in the mitochondria.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents—Anti-cytochrome c antibody (7H8.2c12, 6H2.B4; BD Pharmigen, San Diego, CA); cytosolic marker anti-lactate dehydrogenase (LDH; Sigma, St. Louis, MO); nuclear markeranti-PCNA (clone PC10; Oncogene Research Products, San Diego,CA); anti-F1� (Molecular Probes, Eugene, OR). All other reagentswere from Sigma.
Cell Culture—The human T leukemia cells (Jurkat A3) were ob-tained from the American Type Culture Collection (Bethesda, MD).Cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 25 mM HEPES, and antibiotics ina humidified incubator with 5% CO2 in air at 37 °C. The cells weregrown to a maximum density of 0.5–0.8 � 106/ml and split at a ratioof 1:10.
Subcellular Fractionation and Western Blotting—Mitochondriawere isolated as described previously with minor modifications asoutlined below (19). Jurkat A3 cells were collected by centrifugation at400 � g for 10 min at 4 °C. The cell pellets were washed twice withice-cold PBS (pH 7.4) and resuspended with 10 volumes of isolationbuffer (20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM
EDTA, 1 mM EGTA, 1 mM DTT, 0.25 M sucrose, and a mixture ofprotease inhibitors). After 10-min incubation on ice, the cells werehomogenized in a glass Dounce homogenizer until �75% of the cellsbecame trypan blue-positive. The homogenates were centrifugedtwice at 650 � g for 10 min at 4 °C to remove nuclei and unbrokencells. The postnuclear supernatants were centrifuged at 12,500 � gfor 25 min at 4 °C, and the pellets were saved as the heavy membranefraction (designated HM). The supernatants of the 12,500 � g spinwere further centrifuged at 100,000 � g for 1 h at 4 °C, and theresulting supernatants (designated cytosolic; S-100) and pellet (des-ignated light membrane; LM) were frozen as aliquots at �80 °C forsubsequent experiments. The heavy membrane fraction was resus-pended carefully in the isolation buffer and centrifuged again at12,500 � g for 25 min. The heavy membrane fraction was thenresuspended in isotonic sucrose buffer (0.25 M sucrose, 1 mM EDTA,and 10 mM Tris-HCl, pH 7.4), layered on a 1.0/1.5 M discontinuous
sucrose gradient, and centrifuged at 60,000 � g for 20 min at 4 °C.The mitochondria were collected from the phase between the 1.0 and1.5 M sucrose, diluted in the isolation buffer, and centrifuged again at15,000 � g for 20 min to pellet mitochondria. Purified mitochondrialpellets were washed with isolation buffer and then preserved at�80 °C until further analysis.
Purified mitochondrial fraction and HM fraction were solubilized inlysis buffer (1% n-dodecyl-�-D-maltoside, 50 mM Tris-HCl, pH 8.0,150 mM NaCl, 1 mM EDTA, and a mixture of protease inhibitors) for 1 hon ice and centrifuged at 15,000 � g for 5 min. The supernatant wascollected, and protein concentration was determined by a Micro-BCA protein concentration determination kit (Pierce, Rockford, IL).For Western blotting, equal amount of various subcellular fractionswere loaded in each lane of a 10% NuPAGE gel (Invitrogen, SanDiego, CA). After gel electrophoresis and protein transfer, the mem-branes were probed with various primary and corresponding second-ary antibodies against marker proteins from different cellular com-partments. Immunoreactivity was detected with an ECL method(PerkinElmer, Boston, MA).
In-gel Digestion with Trypsin—A total mitochondrial protein from0.75 � 107 cells (40 �g) was loaded on 10% NuPAGE gel (Invitrogen)and run at 20 mA for 30 min, then 30 mA for 2 h. The gel was stainedwith Coomassie blue R-250 (50% methanol, 10% acetic acid, 0.1%R-250) for 30 min and destained overnight in a solution containing 5%methanol, 7% acetic acid. After imaging, the area from the top to thebottom of each lane of the Coomassie-stained gel was cut at 2-mmintervals (some slices were wider because of the absence of anyprominent band at those positions). Each gel slice was cut into smallpieces (�1-mm cubes) and transferred to 500-�l microcentrifugetubes. The gel pieces were washed with 200 �l of 50 mM NH4HCO3
for 45 min at room temperature. The supernatant was removed andthen 200 �l of neutralization buffer (50 mM NH4HCO3 in 50% CH3CN)was added for 20 min at room temperature. The gel pieces werecompletely neutralized and destained by repeated washes withNH4HCO3 and NH4HCO3/CH3CN if necessary. The destained gelpieces were dehydrated with 100% CH3CN and dried in a vacuumconcentrator (CentriVap, Labconco Corporation, Kansas City, MO).Gel pieces were rehydrated with 15–20 �l of trypsin solution (25 ng/�lin 100 mM NH4HCO3) on ice for 45 min. In-gel digestion was per-formed at 37 °C for 18–20 h. The resulting peptides were extractedaccording to the protocol of Shevchenko et al. (20). Extracted pep-tides were dried in Centrivap, redissolved in solvent A [5% ACN, 0.4%acetic acid, and 0.005% heptaflourobutyric acid (HFBA)] and storedat �20 °C until mass spectrometric analysis was performed. For thetwo additional replicate analyses, 60 �g of mitochondrial proteinswere analyzed on a faster scanning Ion trap mass spectrometer(Finnigan LTQ; Thermo Finnigan, Palo Alto, CA).
Nano-LC-MS/MS Analysis—The digested mitochondrial proteinswere sequenced using a high-throughput tandem mass spectrometer(LCQ-DECA ion trap; Thermo Finnigan) equipped with an in-housebuilt nano-electrospray device. Samples were directly loaded onto a10-cm � 75-�m capillary reverse-phase column packed in-house(Magic C18; Michrom BioResources, Auburn, CA) by means of ahelium pressure cell. The column was previously equilibrated withsolvent A (5% ACN, 0.4% acetic acid, and 0.005% HFBA). Peptideswere eluted with a linear gradient from 100% solvent A to 80%solvent B (95% ACN, 0.4% acetic acid, and 0.005% HFBA) for 85 minat a flow rate of �240 nl/min. Peptides were eluted directly into theLCQ-DECA ESI ion trap mass spectrometer capable of data-depend-ent acquisition. Each full MS scan was followed by two MS/MS scansof the two most intense peaks in the full MS spectrum with dynamicexclusion enabled to allow detection of less-abundant peptide ions.Mass spectrometric scan events and HPLC solvent gradients werecontrolled by the Xcalibur software (Thermo Finnigan). For two addi-
Mitochondrial Proteome from Human Leukemia Cells
170 Molecular & Cellular Proteomics 4.2
tional replicate analyses, 60 �g of mitochondrial proteins were ana-lyzed by the LTQ using the same methodology as described above.
Analysis of MS/MS Data—A total of 32,298 MS/MS spectra werecollected from 22 nano-LC-MS/MS runs and were searched against ahuman nonredundant protein database from the National CancerInstitute (human.nci) protein database using the SEQUEST algorithmrunning on a 32 node Linux cluster (21). Search parameters usedincluded provision for both unmodified and oxidized (�16 Da) methi-onine at a mass tolerance �1.5 Da with proteolytic enzyme trypsinspecified. SEQUEST output files were automatically submitted toPeptideProphet for statistical data modeling and generation of com-puted probability (Pcomp) scores for each peptide assigned by SE-QUEST (22). The PeptideProphet-generated output files were filteredwith Pcomp � 0.85 using INTERACT (51). In addition to the Pcompscores, SEQUEST Xcorr, �Cn scores, and the spectra of each of thepeptides were carefully examined. Furthermore, the molecularmasses of identified proteins from each of the gel slices were sys-tematically compared with each other, with gel slices that are adja-cent, and with the molecular mass standards. Protein identificationsthat are outside of the acceptable molecular mass range or peptidesthat match to were excluded. After these filtering steps, we attemptedto validate all of the identified proteins including the single-hit proteinsby two independent purifications of mitochondria from Jurkat cellsfollowed by the GeLC-MS/MS procedure using the LTQ Ion trap massspectrometer. This filtering steps and validation experiments allowedthe identification of 3,490 peptides among which 2,618 were uniquepeptides.
Bioinformatics—For the predictions of subcellular locations of pro-teins, the programs PSORT II (psort.nibb.ac.jp/form2.html) (23) andTargetP (www.cbs.dtu.dk/services/TargetP/) (24) were used. GRAVYindex scores and transmembrane domains (TMD) were predicted withSOSUI (sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html) (25).
RESULTS AND DISCUSSION
Characterization of Isolated Jurkat Mitochondria—The qual-ity of a subcellular proteome, such as that of mitochondria, islargely dependent on its purity. Thus, we optimized the puri-fication procedure that allowed the isolation of high-puritymitochondria from Jurkat T leukemia cells. This procedureinvolved three differential centrifugations followed by the su-crose density gradient centrifugation using three sucrose den-
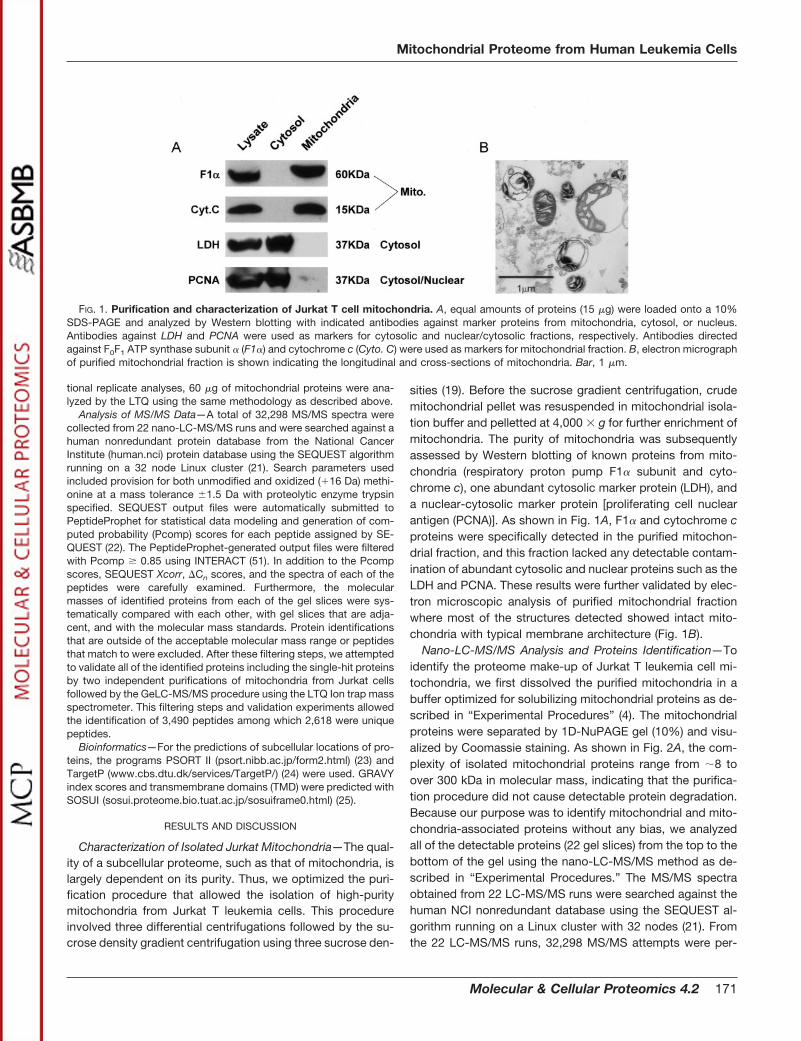
sities (19). Before the sucrose gradient centrifugation, crudemitochondrial pellet was resuspended in mitochondrial isola-tion buffer and pelletted at 4,000 � g for further enrichment ofmitochondria. The purity of mitochondria was subsequentlyassessed by Western blotting of known proteins from mito-chondria (respiratory proton pump F1� subunit and cyto-chrome c), one abundant cytosolic marker protein (LDH), anda nuclear-cytosolic marker protein [proliferating cell nuclearantigen (PCNA)]. As shown in Fig. 1A, F1� and cytochrome cproteins were specifically detected in the purified mitochon-drial fraction, and this fraction lacked any detectable contam-ination of abundant cytosolic and nuclear proteins such as theLDH and PCNA. These results were further validated by elec-tron microscopic analysis of purified mitochondrial fractionwhere most of the structures detected showed intact mito-chondria with typical membrane architecture (Fig. 1B).
Nano-LC-MS/MS Analysis and Proteins Identification—Toidentify the proteome make-up of Jurkat T leukemia cell mi-tochondria, we first dissolved the purified mitochondria in abuffer optimized for solubilizing mitochondrial proteins as de-scribed in “Experimental Procedures” (4). The mitochondrialproteins were separated by 1D-NuPAGE gel (10%) and visu-alized by Coomassie staining. As shown in Fig. 2A, the com-plexity of isolated mitochondrial proteins range from �8 toover 300 kDa in molecular mass, indicating that the purifica-tion procedure did not cause detectable protein degradation.Because our purpose was to identify mitochondrial and mito-chondria-associated proteins without any bias, we analyzedall of the detectable proteins (22 gel slices) from the top to thebottom of the gel using the nano-LC-MS/MS method as de-scribed in “Experimental Procedures.” The MS/MS spectraobtained from 22 LC-MS/MS runs were searched against thehuman NCI nonredundant database using the SEQUEST al-gorithm running on a Linux cluster with 32 nodes (21). Fromthe 22 LC-MS/MS runs, 32,298 MS/MS attempts were per-
FIG. 1. Purification and characterization of Jurkat T cell mitochondria. A, equal amounts of proteins (15 �g) were loaded onto a 10%SDS-PAGE and analyzed by Western blotting with indicated antibodies against marker proteins from mitochondria, cytosol, or nucleus.Antibodies against LDH and PCNA were used as markers for cytosolic and nuclear/cytosolic fractions, respectively. Antibodies directedagainst F0F1 ATP synthase subunit � (F1�) and cytochrome c (Cyto. C) were used as markers for mitochondrial fraction. B, electron micrographof purified mitochondrial fraction is shown indicating the longitudinal and cross-sections of mitochondria. Bar, 1 �m.
Mitochondrial Proteome from Human Leukemia Cells
Molecular & Cellular Proteomics 4.2 171
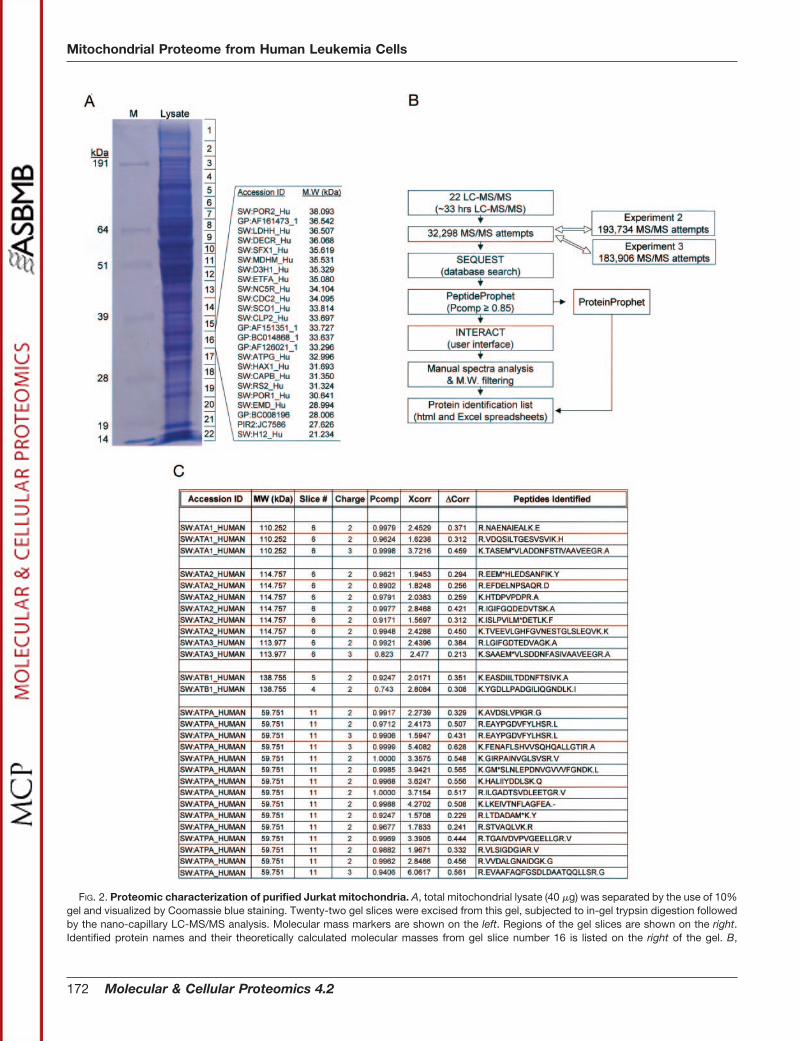
FIG. 2. Proteomic characterization of purified Jurkat mitochondria. A, total mitochondrial lysate (40 �g) was separated by the use of 10%gel and visualized by Coomassie blue staining. Twenty-two gel slices were excised from this gel, subjected to in-gel trypsin digestion followedby the nano-capillary LC-MS/MS analysis. Molecular mass markers are shown on the left. Regions of the gel slices are shown on the right.Identified protein names and their theoretically calculated molecular masses from gel slice number 16 is listed on the right of the gel. B,
Mitochondrial Proteome from Human Leukemia Cells
172 Molecular & Cellular Proteomics 4.2
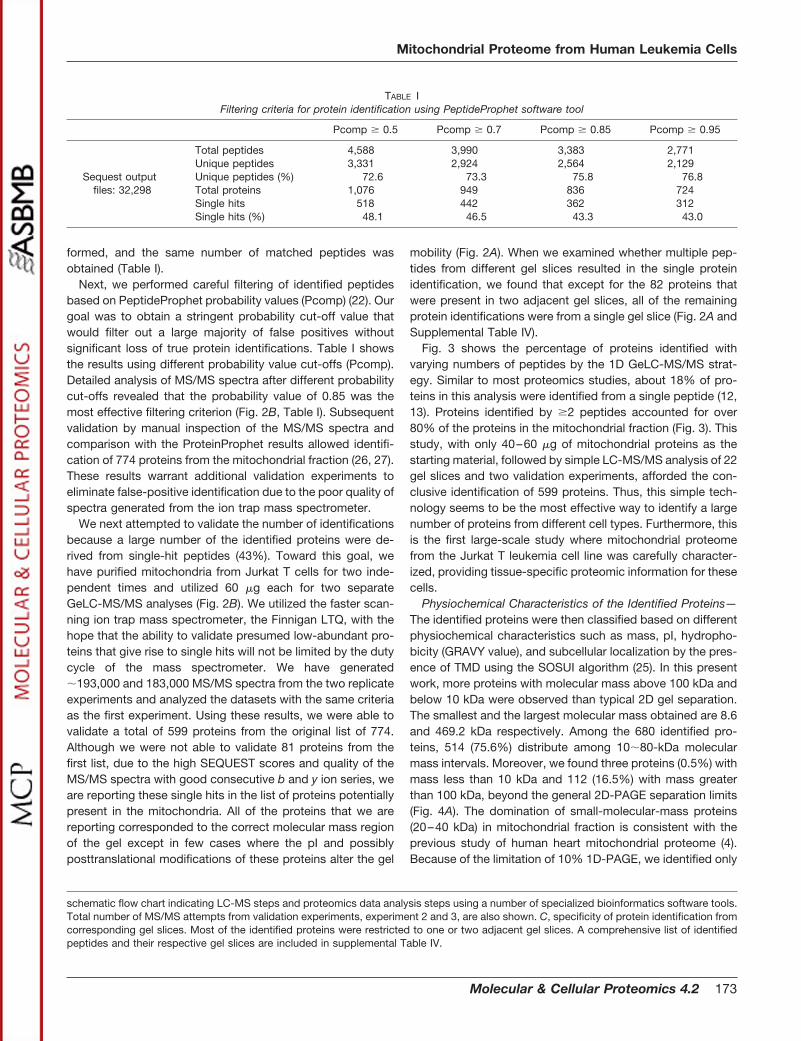
formed, and the same number of matched peptides wasobtained (Table I).
Next, we performed careful filtering of identified peptidesbased on PeptideProphet probability values (Pcomp) (22). Ourgoal was to obtain a stringent probability cut-off value thatwould filter out a large majority of false positives withoutsignificant loss of true protein identifications. Table I showsthe results using different probability value cut-offs (Pcomp).Detailed analysis of MS/MS spectra after different probabilitycut-offs revealed that the probability value of 0.85 was themost effective filtering criterion (Fig. 2B, Table I). Subsequentvalidation by manual inspection of the MS/MS spectra andcomparison with the ProteinProphet results allowed identifi-cation of 774 proteins from the mitochondrial fraction (26, 27).These results warrant additional validation experiments toeliminate false-positive identification due to the poor quality ofspectra generated from the ion trap mass spectrometer.
We next attempted to validate the number of identificationsbecause a large number of the identified proteins were de-rived from single-hit peptides (43%). Toward this goal, wehave purified mitochondria from Jurkat T cells for two inde-pendent times and utilized 60 �g each for two separateGeLC-MS/MS analyses (Fig. 2B). We utilized the faster scan-ning ion trap mass spectrometer, the Finnigan LTQ, with thehope that the ability to validate presumed low-abundant pro-teins that give rise to single hits will not be limited by the dutycycle of the mass spectrometer. We have generated�193,000 and 183,000 MS/MS spectra from the two replicateexperiments and analyzed the datasets with the same criteriaas the first experiment. Using these results, we were able tovalidate a total of 599 proteins from the original list of 774.Although we were not able to validate 81 proteins from thefirst list, due to the high SEQUEST scores and quality of theMS/MS spectra with good consecutive b and y ion series, weare reporting these single hits in the list of proteins potentiallypresent in the mitochondria. All of the proteins that we arereporting corresponded to the correct molecular mass regionof the gel except in few cases where the pI and possiblyposttranslational modifications of these proteins alter the gel
mobility (Fig. 2A). When we examined whether multiple pep-tides from different gel slices resulted in the single proteinidentification, we found that except for the 82 proteins thatwere present in two adjacent gel slices, all of the remainingprotein identifications were from a single gel slice (Fig. 2A andSupplemental Table IV).
Fig. 3 shows the percentage of proteins identified withvarying numbers of peptides by the 1D GeLC-MS/MS strat-egy. Similar to most proteomics studies, about 18% of pro-teins in this analysis were identified from a single peptide (12,13). Proteins identified by �2 peptides accounted for over80% of the proteins in the mitochondrial fraction (Fig. 3). Thisstudy, with only 40–60 �g of mitochondrial proteins as thestarting material, followed by simple LC-MS/MS analysis of 22gel slices and two validation experiments, afforded the con-clusive identification of 599 proteins. Thus, this simple tech-nology seems to be the most effective way to identify a largenumber of proteins from different cell types. Furthermore, thisis the first large-scale study where mitochondrial proteomefrom the Jurkat T leukemia cell line was carefully character-ized, providing tissue-specific proteomic information for thesecells.
Physiochemical Characteristics of the Identified Proteins—The identified proteins were then classified based on differentphysiochemical characteristics such as mass, pI, hydropho-bicity (GRAVY value), and subcellular localization by the pres-ence of TMD using the SOSUI algorithm (25). In this presentwork, more proteins with molecular mass above 100 kDa andbelow 10 kDa were observed than typical 2D gel separation.The smallest and the largest molecular mass obtained are 8.6and 469.2 kDa respectively. Among the 680 identified pro-teins, 514 (75.6%) distribute among 10�80-kDa molecularmass intervals. Moreover, we found three proteins (0.5%) withmass less than 10 kDa and 112 (16.5%) with mass greaterthan 100 kDa, beyond the general 2D-PAGE separation limits(Fig. 4A). The domination of small-molecular-mass proteins(20–40 kDa) in mitochondrial fraction is consistent with theprevious study of human heart mitochondrial proteome (4).Because of the limitation of 10% 1D-PAGE, we identified only
schematic flow chart indicating LC-MS steps and proteomics data analysis steps using a number of specialized bioinformatics software tools.Total number of MS/MS attempts from validation experiments, experiment 2 and 3, are also shown. C, specificity of protein identification fromcorresponding gel slices. Most of the identified proteins were restricted to one or two adjacent gel slices. A comprehensive list of identifiedpeptides and their respective gel slices are included in supplemental Table IV.
TABLE IFiltering criteria for protein identification using PeptideProphet software tool
Pcomp � 0.5 Pcomp � 0.7 Pcomp � 0.85 Pcomp � 0.95
Sequest outputfiles: 32,298
Total peptides 4,588 3,990 3,383 2,771Unique peptides 3,331 2,924 2,564 2,129Unique peptides (%) 72.6 73.3 75.8 76.8Total proteins 1,076 949 836 724Single hits 518 442 362 312Single hits (%) 48.1 46.5 43.3 43.0
Mitochondrial Proteome from Human Leukemia Cells
Molecular & Cellular Proteomics 4.2 173
three (0.5%) small-molecular-mass proteins (�10 kDa). In thiscase, the use of longer gradient gel and increasing the sampleamount will likely increase the chance of small-molecular-mass protein identification.
The 680 proteins distribute across a wide pI range (4.34–12.3) with a roughly trimodal distribution, typical characteristicof pI histograms for eukaryotic proteins (28). A total of 653proteins (96%) distribute among pI 4–10 intervals but 27 (4%)proteins have pI greater than 10 and may be not resolvable bythe 2D-PAGE separation (Fig. 4B). Interestingly, more thanhalf of the identified proteins in the mitochondrial fractiondistribute among pI 8–11 intervals (51.2%), suggesting thatsubcellular fractionation enriched alkaline proteins (pI 8–12).About 50% of annotated mitochondrial proteins are theoreti-cally in pI 8�10, consistent with the pI distribution pattern ofthe currently identified proteins in the mitochondrial fraction.The values of molecular mass and pI reported here do notreflect the mature forms of proteins where a significant num-ber of these are known to be posttranslationally processed.For example, a number of mitochondrial proteins are proteo-
lytically processed during translocation into the mitochondria,and therefore the mature forms differ significantly in theirrespective molecular mass and pI from gene sequencepredictions.
The proteins detected in 2D-PAGE gels are generally hy-drophilic with the negative GRAVY values (28–30). For the 680proteins we identified, their hydrophobicity values vary in therange of –1.6��0.74 (data not shown), indicating that recov-ery of membrane-bound proteins was not biased as in thecase of 2D-PAGE. Additionally, theoretical TMD predicted bySOSUI shows that 120 (17.6%) proteins of the total 680proteins have one or more predicted TMDs (data not shown),of which 30 proteins have three or more TMDs (Table II). Inparticular, 14 of the 30 proteins with three or more TMDs areall annotated as integral membrane proteins.
Functional Classification of Identified Proteins—Next, weassigned the function of 680 identified proteins based oninformation available in published literature as well as from thetwo most comprehensive mitochondrial protein resources:the MitoProteome database (www.mitoproteome.org) and the
FIG. 3. Distribution of identified pep-tide number per mitochondrial proteinfrom Jurkat T leukemia cells. Distribu-tion of the 680 identified mitochondrialproteins from Jurkat T cells in relation tothe number of the matching peptidesused for identity assignment is shown.The bars indicate the percentages of theproteins identified with the correspond-ing number of matching peptides.
FIG. 4. Physiochemical characteristics of mitochondrial proteins form Jurkat T leukemia cells. Distribution of the 680 identifiedmitochondrial proteins from Jurkat T cells in relation to their pI (A) and molecular mass (B) are shown. The 680 identified proteins were sortedbased on their theoretical pI and molecular mass ranges as indicated.
Mitochondrial Proteome from Human Leukemia Cells
174 Molecular & Cellular Proteomics 4.2
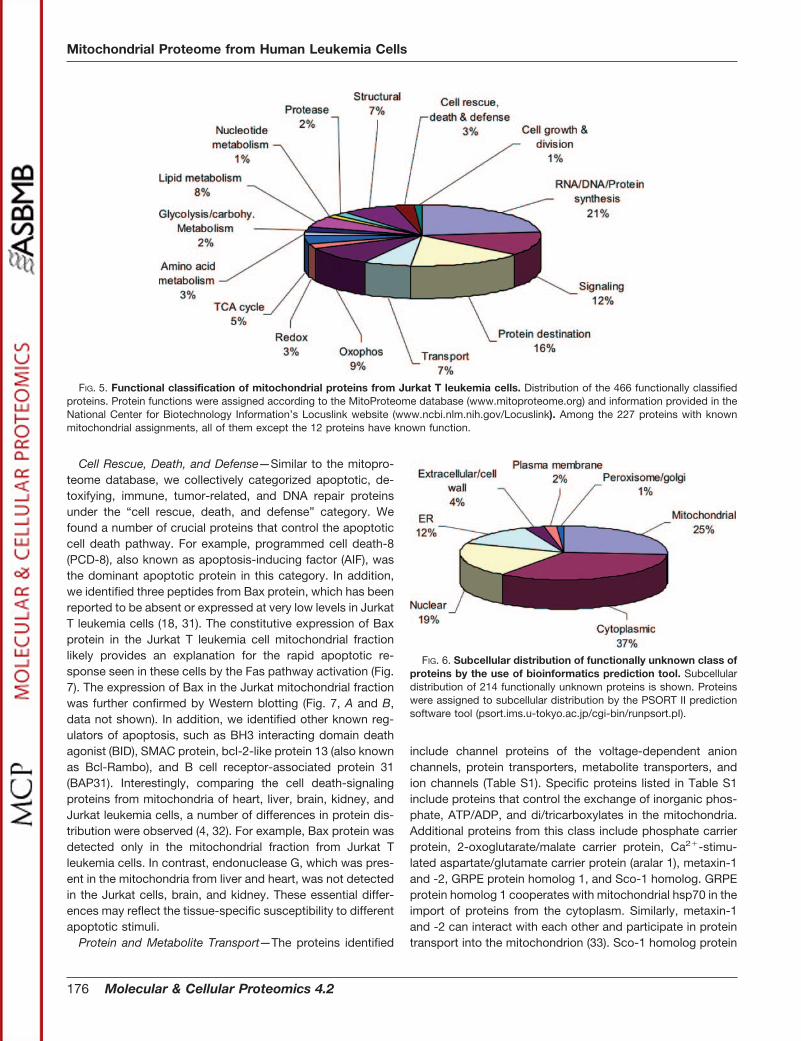
NCBI’s locus link database (www.ncbi.nlm.nih.gov/Lo-cusLink/). The identified proteins were functionally subdividedinto two classes: 466 proteins that can be assigned to knownfunctional classes (69%) and 214 proteins for which no func-tional information is available (31%). Similar to our findings, arecent study by Taylor et al. revealed 498 functionally knownproteins from human heart mitochondria (4). The distributionof the 466 functionally classified proteins is shown in Fig. 5and is listed in supplemental Table S1. To our knowledge, thisadditional information of mitochondrial proteins from Jurkat Tleukemia cells represent the most comprehensive proteomicdata deposited for this cell line.
The Oxidative Phosphorylation Machinery and Metabo-lism—The oxidative phosphorylation machinery is composedof five electron transport chain complexes, representing amajor group of functionally classified mitochondrial proteins.Among the 466 functionally classified proteins, 40 (9%) arecomponents of the five electron transport chain protein com-plexes from Jurkat T leukemia cells. We also identified thehuman homolog of one of the three newly discovered com-
plex I subunits in human heart mitochondria: NADH-ubiqui-none oxidoreductase ESSS subunit (neuronal protein p17.3).
The bulk of ATP used by many cells to maintain homeosta-sis is produced by the oxidation of pyruvate in the tricarbox-ylic acid (TCA) cycle. We identified 25 proteins involved in theTCA cycle (5%). At least one component of each of the sevenTCA cycle enzymes were identified along with components ofthe associated pyruvate dehydrogenase complex and NAD-malic enzymes (Tables S1 and S3). We also identified oneenzyme involved in carbohydrate metabolism, methylmalonyl-CoA epimerase, and 10 enzymes of glycolytic pathways. Fur-thermore, 28 proteins involved in lipid metabolism (6%) wereidentified, and these include � and � subunits of mitochon-drial trifunctional enzymes. We also identified multiple en-zymes involved in amino acid and nucleic acid metabolisms:serine hydroxymethyltransferase (both cytosolic and mito-chondrial isoform), glutaminase (kidney isoform), ornithineaminotransferase, �-1-pyrroline-5-carboxylate synthetase,�-1-pyrroline-5-carboxylate dehydrogenase, pyrroline-5-car-boxylate, and three adenylate kinases.
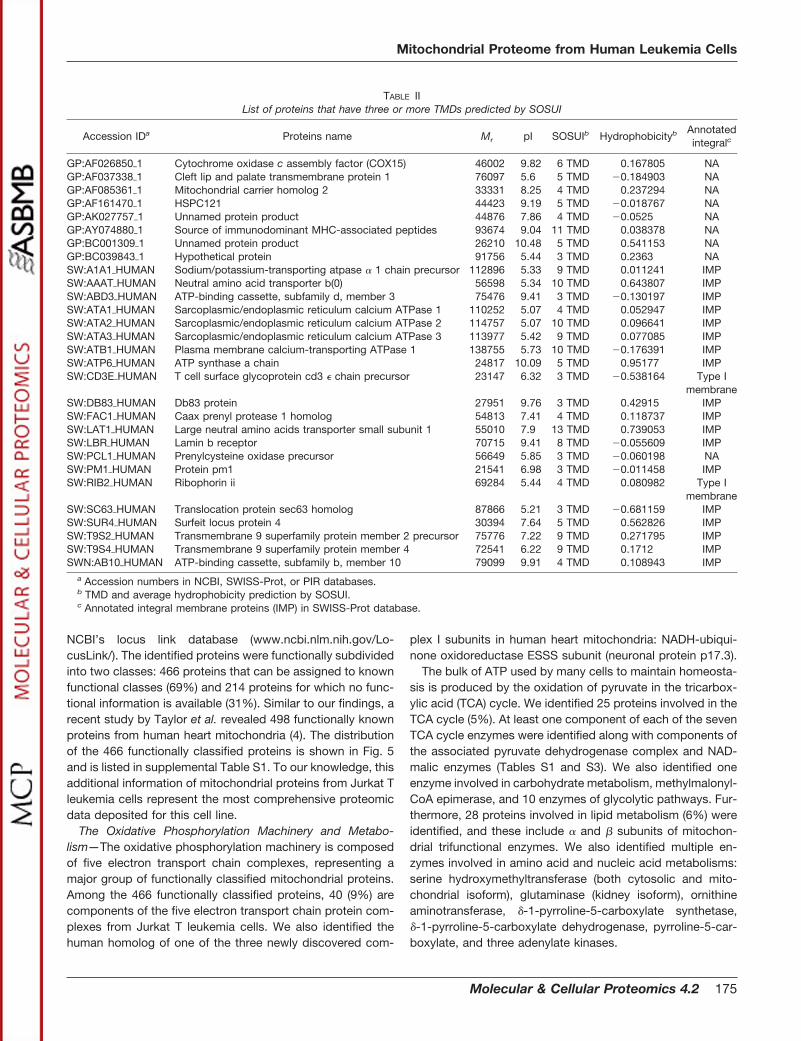
TABLE IIList of proteins that have three or more TMDs predicted by SOSUI
Accession IDa Proteins name Mr pI SOSUIb Hydrophobicityb Annotatedintegralc
GP:AF026850�1 Cytochrome oxidase c assembly factor (COX15) 46002 9.82 6 TMD 0.167805 NAGP:AF037338�1 Cleft lip and palate transmembrane protein 1 76097 5.6 5 TMD �0.184903 NAGP:AF085361�1 Mitochondrial carrier homolog 2 33331 8.25 4 TMD 0.237294 NAGP:AF161470�1 HSPC121 44423 9.19 5 TMD �0.018767 NAGP:AK027757�1 Unnamed protein product 44876 7.86 4 TMD �0.0525 NAGP:AY074880�1 Source of immunodominant MHC-associated peptides 93674 9.04 11 TMD 0.038378 NAGP:BC001309�1 Unnamed protein product 26210 10.48 5 TMD 0.541153 NAGP:BC039843�1 Hypothetical protein 91756 5.44 3 TMD 0.2363 NASW:A1A1�HUMAN Sodium/potassium-transporting atpase � 1 chain precursor 112896 5.33 9 TMD 0.011241 IMPSW:AAAT�HUMAN Neutral amino acid transporter b(0) 56598 5.34 10 TMD 0.643807 IMPSW:ABD3�HUMAN ATP-binding cassette, subfamily d, member 3 75476 9.41 3 TMD �0.130197 IMPSW:ATA1�HUMAN Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 110252 5.07 4 TMD 0.052947 IMPSW:ATA2�HUMAN Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 114757 5.07 10 TMD 0.096641 IMPSW:ATA3�HUMAN Sarcoplasmic/endoplasmic reticulum calcium ATPase 3 113977 5.42 9 TMD 0.077085 IMPSW:ATB1�HUMAN Plasma membrane calcium-transporting ATPase 1 138755 5.73 10 TMD �0.176391 IMPSW:ATP6�HUMAN ATP synthase a chain 24817 10.09 5 TMD 0.95177 IMPSW:CD3E�HUMAN T cell surface glycoprotein cd3 � chain precursor 23147 6.32 3 TMD �0.538164 Type I
membraneSW:DB83�HUMAN Db83 protein 27951 9.76 3 TMD 0.42915 IMPSW:FAC1�HUMAN Caax prenyl protease 1 homolog 54813 7.41 4 TMD 0.118737 IMPSW:LAT1�HUMAN Large neutral amino acids transporter small subunit 1 55010 7.9 13 TMD 0.739053 IMPSW:LBR�HUMAN Lamin b receptor 70715 9.41 8 TMD �0.055609 IMPSW:PCL1�HUMAN Prenylcysteine oxidase precursor 56649 5.85 3 TMD �0.060198 NASW:PM1�HUMAN Protein pm1 21541 6.98 3 TMD �0.011458 IMPSW:RIB2�HUMAN Ribophorin ii 69284 5.44 4 TMD 0.080982 Type I
membraneSW:SC63�HUMAN Translocation protein sec63 homolog 87866 5.21 3 TMD �0.681159 IMPSW:SUR4�HUMAN Surfeit locus protein 4 30394 7.64 5 TMD 0.562826 IMPSW:T9S2�HUMAN Transmembrane 9 superfamily protein member 2 precursor 75776 7.22 9 TMD 0.271795 IMPSW:T9S4�HUMAN Transmembrane 9 superfamily protein member 4 72541 6.22 9 TMD 0.1712 IMPSWN:AB10�HUMAN ATP-binding cassette, subfamily b, member 10 79099 9.91 4 TMD 0.108943 IMP
a Accession numbers in NCBI, SWISS-Prot, or PIR databases.b TMD and average hydrophobicity prediction by SOSUI.c Annotated integral membrane proteins (IMP) in SWISS-Prot database.
Mitochondrial Proteome from Human Leukemia Cells
Molecular & Cellular Proteomics 4.2 175
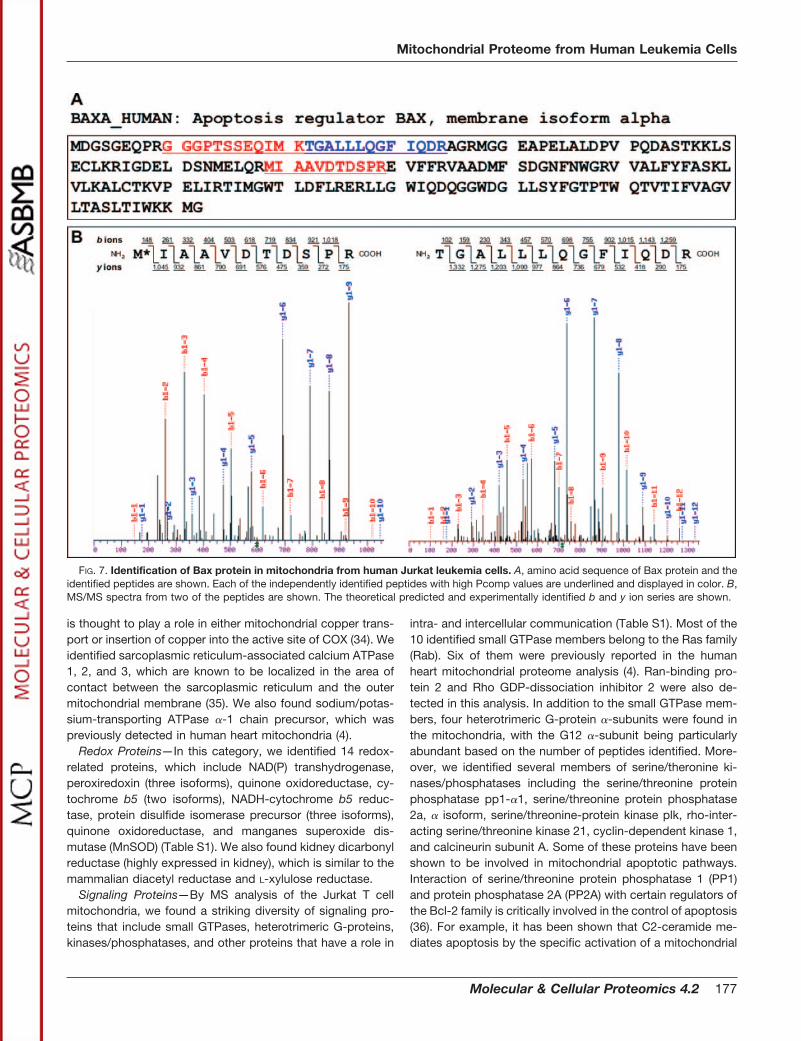
Cell Rescue, Death, and Defense—Similar to the mitopro-teome database, we collectively categorized apoptotic, de-toxifying, immune, tumor-related, and DNA repair proteinsunder the “cell rescue, death, and defense” category. Wefound a number of crucial proteins that control the apoptoticcell death pathway. For example, programmed cell death-8(PCD-8), also known as apoptosis-inducing factor (AIF), wasthe dominant apoptotic protein in this category. In addition,we identified three peptides from Bax protein, which has beenreported to be absent or expressed at very low levels in JurkatT leukemia cells (18, 31). The constitutive expression of Baxprotein in the Jurkat T leukemia cell mitochondrial fractionlikely provides an explanation for the rapid apoptotic re-sponse seen in these cells by the Fas pathway activation (Fig.7). The expression of Bax in the Jurkat mitochondrial fractionwas further confirmed by Western blotting (Fig. 7, A and B,data not shown). In addition, we identified other known reg-ulators of apoptosis, such as BH3 interacting domain deathagonist (BID), SMAC protein, bcl-2-like protein 13 (also knownas Bcl-Rambo), and B cell receptor-associated protein 31(BAP31). Interestingly, comparing the cell death-signalingproteins from mitochondria of heart, liver, brain, kidney, andJurkat leukemia cells, a number of differences in protein dis-tribution were observed (4, 32). For example, Bax protein wasdetected only in the mitochondrial fraction from Jurkat Tleukemia cells. In contrast, endonuclease G, which was pres-ent in the mitochondria from liver and heart, was not detectedin the Jurkat cells, brain, and kidney. These essential differ-ences may reflect the tissue-specific susceptibility to differentapoptotic stimuli.
Protein and Metabolite Transport—The proteins identified
include channel proteins of the voltage-dependent anionchannels, protein transporters, metabolite transporters, andion channels (Table S1). Specific proteins listed in Table S1include proteins that control the exchange of inorganic phos-phate, ATP/ADP, and di/tricarboxylates in the mitochondria.Additional proteins from this class include phosphate carrierprotein, 2-oxoglutarate/malate carrier protein, Ca2�-stimu-lated aspartate/glutamate carrier protein (aralar 1), metaxin-1and -2, GRPE protein homolog 1, and Sco-1 homolog. GRPEprotein homolog 1 cooperates with mitochondrial hsp70 in theimport of proteins from the cytoplasm. Similarly, metaxin-1and -2 can interact with each other and participate in proteintransport into the mitochondrion (33). Sco-1 homolog protein
FIG. 6. Subcellular distribution of functionally unknown class ofproteins by the use of bioinformatics prediction tool. Subcellulardistribution of 214 functionally unknown proteins is shown. Proteinswere assigned to subcellular distribution by the PSORT II predictionsoftware tool (psort.ims.u-tokyo.ac.jp/cgi-bin/runpsort.pI).
FIG. 5. Functional classification of mitochondrial proteins from Jurkat T leukemia cells. Distribution of the 466 functionally classifiedproteins. Protein functions were assigned according to the MitoProteome database (www.mitoproteome.org) and information provided in theNational Center for Biotechnology Information’s Locuslink website (www.ncbi.nlm.nih.gov/Locuslink). Among the 227 proteins with knownmitochondrial assignments, all of them except the 12 proteins have known function.
Mitochondrial Proteome from Human Leukemia Cells
176 Molecular & Cellular Proteomics 4.2
is thought to play a role in either mitochondrial copper trans-port or insertion of copper into the active site of COX (34). Weidentified sarcoplasmic reticulum-associated calcium ATPase1, 2, and 3, which are known to be localized in the area ofcontact between the sarcoplasmic reticulum and the outermitochondrial membrane (35). We also found sodium/potas-sium-transporting ATPase �-1 chain precursor, which waspreviously detected in human heart mitochondria (4).
Redox Proteins—In this category, we identified 14 redox-related proteins, which include NAD(P) transhydrogenase,peroxiredoxin (three isoforms), quinone oxidoreductase, cy-tochrome b5 (two isoforms), NADH-cytochrome b5 reduc-tase, protein disulfide isomerase precursor (three isoforms),quinone oxidoreductase, and manganes superoxide dis-mutase (MnSOD) (Table S1). We also found kidney dicarbonylreductase (highly expressed in kidney), which is similar to themammalian diacetyl reductase and L-xylulose reductase.
Signaling Proteins—By MS analysis of the Jurkat T cellmitochondria, we found a striking diversity of signaling pro-teins that include small GTPases, heterotrimeric G-proteins,kinases/phosphatases, and other proteins that have a role in
intra- and intercellular communication (Table S1). Most of the10 identified small GTPase members belong to the Ras family(Rab). Six of them were previously reported in the humanheart mitochondrial proteome analysis (4). Ran-binding pro-tein 2 and Rho GDP-dissociation inhibitor 2 were also de-tected in this analysis. In addition to the small GTPase mem-bers, four heterotrimeric G-protein �-subunits were found inthe mitochondria, with the G12 �-subunit being particularlyabundant based on the number of peptides identified. More-over, we identified several members of serine/theronine ki-nases/phosphatases including the serine/threonine proteinphosphatase pp1-�1, serine/threonine protein phosphatase2a, � isoform, serine/threonine-protein kinase plk, rho-inter-acting serine/threonine kinase 21, cyclin-dependent kinase 1,and calcineurin subunit A. Some of these proteins have beenshown to be involved in mitochondrial apoptotic pathways.Interaction of serine/threonine protein phosphatase 1 (PP1)and protein phosphatase 2A (PP2A) with certain regulators ofthe Bcl-2 family is critically involved in the control of apoptosis(36). For example, it has been shown that C2-ceramide me-diates apoptosis by the specific activation of a mitochondrial
FIG. 7. Identification of Bax protein in mitochondria from human Jurkat leukemia cells. A, amino acid sequence of Bax protein and theidentified peptides are shown. Each of the independently identified peptides with high Pcomp values are underlined and displayed in color. B,MS/MS spectra from two of the peptides are shown. The theoretical predicted and experimentally identified b and y ion series are shown.
Mitochondrial Proteome from Human Leukemia Cells
Molecular & Cellular Proteomics 4.2 177

PP2A, which dephosphorylates Bcl2 and inhibits its anti-ap-optotic function (37). In addition, cyclin-dependent kinase 1(Cdc2) has been shown to phosphorylate BAD and causeapoptosis (38). Thus, it is likely that the presence of thesekinases and phosphatases in the mitochondria is biologicallyrelevant.
Protein Destination and Proteases—Proteins identified inthe “protein destination” category are involved in proteincomplex assembly, protein modification, protein targeting,and protein stabilization. We found cytochrome oxidase as-sembly factor (COX15), which might be involved in electrontransport chain assembly in mitochondria (18). We identifiedsix subunits of the inner and outer mitochondrial membranetranslocases, TIM and TOM, which function in the proteinimport into the mitochondria. The proteins involved in proteinstabilization, such as the heat-shock proteins and three cy-clophilins A, B, and F, were found. We also identified protein-modifying enzymes, ubiquitin-like protein SUMO-1-conjugat-ing enzyme (Ubc9), and ubiquitin-protein ligase e3a.Ubiquitin-like protein SUMO-1-conjugating enzyme catalyzesthe covalent attachment of ubiquitin-like protein SUMO-1 toother proteins (39). Consistent with our findings, it has beenrecently shown that in addition to the nucleus, Ubc9 andSUMO-1 are localized in mitochondria in COS7 cells.SUMO-1 has been shown to conjugate to mitochondrial sub-strate(s) and participate in mitochondrial fission (39). In addi-tion, we also found a number of proteases such as the en-dopeptidase La homolog precursor, the lon proteasehomolog, the caax prenyl protease 1 homolog, and sentrin-specific protease 5. Caax prenyl protease 1 homolog proteo-lytically removes the C-terminal three residues of farnesylatedand geranylated proteins (40). On the other hand, sentrin-specific protease 5 releases SUMO-1 from its precursorsequence (41).
Structural Proteins—Mitochondrial fraction was found tocontain a large number of cytoskeleton proteins, particularlythose of actin family members. In addition to � and � actin, wealso found some actin binding or regulatory proteins, includ-ing � adducin, emerin, ezrin (p81), and coffilin. We found 26and 53 peptides for the nonerythrocytes � and � spectrins,respectively, in addition to six peptides for erythrocyte spec-trin (Table S1). Numerous proteins of microtubule cytoskele-ton and intermediate filaments were identified by MS analysisof mitochondria (Table S1). In our study, we found tubulin �-1chain, tubulin �-2 chain, tubulin �-6 chain, and vimentin. Wealso identified three septin family proteins in purified mito-chondria, including septin 1, septin 2, and septin 7. It is likelythat these proteins tether mitochondria to cell cytoskeleton asdescribed previously (32). This is also supported by the factthat tubulin is an inherent component of mitochondrial mem-branes, and it could play a role in apoptosis via interactionwith the permeability transition pore (42). However, we cannotconclusively rule out the possibility that there is a traceamount of contaminant cytoskeletal proteins in our prepara-
tion even though our preparation is devoid of the abundantcytosolic protein LDH.
RNA, DNA, and Protein Synthesis—In this major category,we found 31 proteins from the mitochondrial ribosomal pro-teins of the 39S and 29S subunits. Furthermore, we identifiedDNA-directed RNA polymerase, mitochondrial elongation fac-tor G1, tryptophanyl-tRNA synthetase, elongation factor ts,and elongation factor tu (Table S1). In addition to mitochon-drial protein synthesis machinery, LC-MS/MS analysis identi-fied multiple cytoplasmic ribosomal proteins (19 proteins) andfactors that regulate translation (Table S1). A number of RNAbinding proteins and heterogenous nuclear ribonucleopro-teins (hnRNPs) were also detected. It is widely known thatmost mitochondrial protein are encoded in the nucleus, syn-thesized by free cytosolic ribosomes, and translocated intomitochondria posttranslationally. However, evidence sug-gests that some proteins may be imported cotranslationally(43). Because outer mitochondrial membranes contain recep-tors specific for ribisomes, direct interaction between mito-chondria and cytosolic ribosomes support cotranlational im-port (43). In support of the association of hnRNP proteins withmitochondria, it has been shown that hnRNP K protein trans-locates into the mitochondria and interacts with multiple mi-tochondrial transcripts within this organelle (44). Proteomeanalysis of Jurkat T leukemia cells identified the hnRNP Kprotein during apoptosis (45). Involvement of several otherhnRNP family proteins in apoptosis has been reported (45).
Nonmitochondrial Proteins—A large number of proteins de-tected in our mitochondrial preparation have been describedso far for other subcellular compartments. A majority of thenonmitochondrial proteins identified here in this study arecytoplasmic proteins according to database information andprediction by bioinformatics tools. A similar result was alsoobtained from MS analysis of human heart and mouse livermitochondria (4, 46). We propose that the association ofcytoskeleton proteins, endoplasmic reticulum and its associ-ated proteins, cytoplasmic signaling proteins, and ribosomalproteins with mitochondria is likely to be physiologically rele-vant. Consistent with this notion, in our mitochondrial prepa-rations, we identified several glycolytic enzymes including thehexokinase I. This enzyme has been previously shown to belocalized in outer mitochondrial membrane and possibly tomodulate glycolysis (47). Among the other nonmitochondrialproteins, we also found four lysosomal proteins, two peroxy-somal proteins, some Golgi proteins, and some vesicle-asso-ciated membrane proteins, a large majority of which havebeen reported in other mitochondrial preparations (32, 46).This may indicate functional association of mitochondria withother cellular compartments rather than contaminations. Theintimate association between mitochondria and the nucleus,coupled with electrostatic effects, may explain the presenceof nuclear proteins in our preparation and also shown in otherpreparations. In addition to histones, we also found a numberof nuclear pore complex proteins and helicase enzymes. A
Mitochondrial Proteome from Human Leukemia Cells
178 Molecular & Cellular Proteomics 4.2

recent proteomic survey of highly purified yeast mitochondriaby Sickmann et al. (5) identified 750 proteins of which 436 hadbeen previously shown to be mitochondrial proteins. Similarto what is reported here, 314 proteins identified in the yeastmitochondrial study are proteins from multiple subcellularcompartments.
Prediction of Subcellular Localization—We classified all ofthe 680 identified proteins in our mitochondrial preparationinto two categories: “known functional group” and “unknownfunctional group” (Tables S1 and S2). About 31% of the 680proteins identified here have not been assigned to a functionalgroup. For these proteins with no data available, we utilizedtwo bioinformatic tools, PSORT II and TargetP, to predict theirsubculture localization (23, 24). We found that the newerversion of PSORT II can predict proteins into 17 differentsubcellular locations. The TargetP predictor has a more lim-ited prediction scope than PSORT II and therefore we areshowing the data from the PSORT II prediction only. To esti-mate the confidence of these predictions, we first processedsequence of known mitochondrial proteins. We found thehighest agreement with 62.0% for PSORT II (146 of 240proteins). As shown in Fig. 6, PSORT II predicted a putativemitochondrial localization for the additional 25% of 214 pro-teins. Thus, 58 additional proteins were predicted, and thisallows the total number of mitochondrial signal-containingproteins to be 285. Furthermore, 77 proteins were predictedfor cytoplasmic localization (37%), 41 proteins were predictedfor nuclear localization (19%), 26 proteins for endoplasmicreticulum (12%), and 15 proteins for other cellular compart-ments (7%). While several bioinformatics tools are currentlyavailable for detecting mitochondrial targeting sequence,such predictions still suffer from poor sensitivity and specific-ity. Clearly, additional studies are needed to fully validate theprediction of subcellular localization of all these unknownproteins.
CONCLUSION
In this study, with the combination of differential centrifu-gation, sucrose density gradient, GeLC/MS/MS, and multiplevalidation experiments, we identified 599 mitochondria andmitochondria-associated proteins. In addition, we are includ-ing the list of 81 proteins identified from the first experiment inthe list due to their quality of the spectra, fitting the gel bandmolecular mass constraint, as well as the SEQUEST scores.Similar to the recent replicate analysis of human heart mito-chondrial proteome, we found that each of the replicate sam-ples identified 135 common proteins with a number of uniqueproteins identified from each experiment (48). We believe thatthis incomplete coverage in replicate analyses is due in part tothe overall complexity of proteins and wide dynamic range ofthe mitochondrial proteome. Thus, it is conceivable that eventhe faster scanning mass spectrometers cannot completelyidentify all the available peptides from the replicate samples.We have included the list of 81 proteins identified only with a
single peptide hit in our final list as this information may becrucial for additional investigation and further validationexperiments.
This approach clearly utilizes significantly less starting ma-terial and thus reduced the protein separation and MS timewhen compared with a number of other proteomic ap-proaches (9–13). Using this simple strategy, we demonstratethat efficient identification mitochondrial proteins with a widerange of biochemical characteristics can be achieved. How-ever, the very-low-abundant proteins in mitochondria mayescape identification by current strategy. Assuming that cur-rent detection limit of LCQ-DECA is at 10 fmol, �6 � 109
molecules must be present in the samples for protein identi-fication. Thus, from our starting number of �0.75 � 107 cells,which provided 40 �g of mitochondrial proteins, we estimatethat we could detect proteins that are expressed at 800copies per cell. To identify a greater complement of the mi-tochondrial proteins including the proteins that are expressedat a much lower than 800 copies per cell, sample scale-up aswell as an additional increase in sensitivity of the mass spec-trometer are required.
Moreover, to comprehensively identify proteins that areexpressed in cells, many additional factors influence the suc-cess of the study; proteins solubility, type of chromatographiccolumn for peptide separation, and sensitivity and duty cycleof mass spectrometers. In terms of specific proteins withdifferent solubility, optimization of detergent and buffer con-ditions as well as the differential extraction of proteins with atleast two or more detergents may be required. Furthermore,for the identification of very hydrophobic proteins, both C-18and C-8 reverse-phase chromatographic columns could beused for the same sample. Finally, with the help of highlysensitive and faster scanning linear ion trap mass spectrom-eters such as the LTQ (Thermo Finnigan), approximately twotimes more proteins can be identified (unpublished results).Future studies and optimization steps are required to identifymitochondrial proteins comprehensively from different tissuesand cells.
Similar to the genomics community, we anticipate thatlarge-scale proteomic datasets in a standardized format willbe available to investigators in the future. We envision that theavailability of subcellular proteomes will allow numerous in-vestigators to perform comparative studies and further under-stand tissue-specific differences exhibited in human. Thereare many tissue-specific differences in response to hormonaland external stimuli exhibited in vivo and some of these arevery well characterized (49, 50). For example, it is known thatapoptotic-inducing anti-Fas IgM triggers apoptosis in the liverbut not in the heart in mouse, although both tissues expressFas receptors (50). Why these differences are exhibited in theliver and heart is not well understood. We anticipate thatcomprehensive characterization of proteomes from differentcell and tissue types will allow us to better understand howtissue-specific differences are exhibited. Thus, future pro-
Mitochondrial Proteome from Human Leukemia Cells
Molecular & Cellular Proteomics 4.2 179

teomic studies are expected to help understand essentialdifferences that are present in physiological and pathologicalconditions.
Acknowledgments—We thank Debbie Lundgren, Michael Fong,and members of the Han laboratory for helpful discussion and Mi-chael Fong for his help in graphics.
* This work was supported by R01 HL 67569, P01 HL70694, andRR 13186. The costs of publication of this article were defrayed inpart by the payment of page charges. This article must therefore behereby marked “advertisement” in accordance with 18 U.S.C. Section1734 solely to indicate this fact.
□S The on-line version of this manuscript (available at http://www.mcponline.org) contains supplemental material.
‡ To whom correspondence should be addressed: Center for Vas-cular Biology, Department of Cell Biology, University of ConnecticutHealth Center, 263 Farmington Avenue, Farmington, CT 06030. Tel.:860-679-2444; Fax: 860-679-1201; E-mail: [email protected].
REFERENCES
1. McDonald, T. G., and Van Eyk, J. E. (2003) Mitochondrial proteomics:Undercover in the lipid bilayer. Basic Res. Cardiol. 98, 219–227
2. Lopez, M. F., and Melov, S. (2002) Applied proteomics: Mitochondrialproteins and effect on functions. Circ. Res. 90, 380–389
3. Westermann, B., and Neupart, W. (2003) “Omimics of the mitochondrion”:Two complementary proteomics approaches promise to move us closerto definition of the complete complement of proteins that make up amitochondria. Nat. Biotechnol. 21, 239–240
4. Taylor, S. W., Fahy, E, Zhang, B., Glenn, G. M., Warnock, D. E., Willy, S.,Murphy, A. N., Gaucher, S. P., Capaldi, R. A., Gibson, B. W., and Ghos,S. S. (2003) Characterization of the human heart mitochondrial pro-teome. Nat. Biotechnol. 21, 281–286
5. Sickman, A., Reinder, J., Wagner, Y., Joppich, C., Zehedi, R., Meyer, H. M.,Schonfisch, B., Perschil, I., Chacinska, A., Guiard, B., Rehling, P., Pfan-ner, N., and Meisinger, C. (2003) The proteome of Saccharomyces cer-evissiae mitochondria. Proc. Natl. Acad. Sci. U. S. A. 100, 13207–13212
6. Cotter, D., Guda, P., and Subramanium, S. (2004) MitoProteome: Mito-chondrial protein sequence database and annotation system. NucleicAcids Res. 32, D463–D467
7. Hanash, S. M. (2000) Biomedical applications of two-dimensional electro-phoresis using immobilized pH gradient: Current status. Electrophoresis21, 1202–1209
8. Westbrook, J. A., Yan, J. X., Wait, R., Welson, S. Y., and Dunn, M. J. (2001)Zooming-in on the proteome: Very narrow-range immobilized pH gradi-ents reveal more protein species and isoforms. Electrophoresis 22,2865–2871
9. Corthals, G. L., Wasigner, V. C., Hochstrasser, D. F., and Sanchez, J. C.(2000) The dynamic range of protein expression: A challenge for pro-teomic research. Electrophoresis 21, 1104–1115
10. Gygi, S. P., Corthals, G. L., Zhang, Y., Rochon, Y., and Aebersold, R. (2000)Evaluation of two-dimensional gel electrophoresis-based proteome anal-ysis technology. Proc. Natl. Acad. Sci. U. S. A. 97, 9390–9395
11. Link, A. J., Eng, J., Scheiltz, D, M., Carmack, E., Mize, G. J., Morris, D. R.,Garvik, B. M., and Yates, J. R., III (1999) Direct analysis of proteincomplexes using mass spectrometry. Nat. Biotchnol. 17, 676–682
12. Washburn, M. P., Wolters, D., and Yates, J. R., III (2001) Large-scaleanalysis of the yeast proteome by multidimensional protein identificationtechnology. Nat. Bitechnol. 19, 242–247
13. Wolters, D., Washburn, M. P., and Yates, J. R., III (2001) An automatedmultidimensional protein identification technology for shotgun proteom-ics. Anal. Chem. 73, 5683–5690
14. Koller, A., Washburn, M. P., Lange, B. M., Andon, N. L., Deciu, C., Haynes,P.A., Hays, L., Scheiltz, D, M., Ulaszek, R., Wei, J., Wolters, D., andYates, J. R., III. (2002) Proteomic survey of metabolic pathways in rice.Proc. Natl. Acad. Sci. U. S. A. 99, 11969–11974
15. Florens, L., Washburn, M. P., Raine, J. D., Anthony, R. M., Grainer, M.,Haynes, J. D., Moch, J. K., Muster, N., Sacci, J. B., Tabb, D. L., Witney,A. A., Wolters, D., Wu, Y., Gardner, M. J., Holder, J. J., Siden, J. E.,
Yates, J. R., and Carucci, D. J. (2002) A proteomic view of the Plasmo-dium falciparum life cycle. Nature 419, 520–526
16. Pflieger, D., Le Caer, J. P., Lemaire, C., Bernard, B. A., and Dujardin, G.(2000) Systematic identification of mitochondrial proteins by LC-MS/MS.Anal. Chem. 74, 2400–2406
17. Schirle, M., Heurtier, M., A., and Kuster, B (2003) Profiling core proteomesof human cell lines by one-dimensional PAGE and liquid chromatogra-phy-tandem mass spectrometry. Mol. Cell. Proteomics 2, 1297–1305
18. Zipp, F., Martin, R., Lichtenfels, R., Roth, W., Dichgans, J., Krammer, P. H.,and Weller, M. (1997) Human autoreactive and foreign antigen-specific Tcells resist apoptosis induced by soluble recombinant CD95 ligand.J. Immunol. 159, 2108–2115
19. Desai, B. N., Myer, B. R., and Schreiber, S. L. (2002) FKBP12-rpamycin-associated protein associate with mitochondria and senses osmoticstress via mitochondrial dysfunction. Proc. Natl. Acad. Sci. U. S. A. 99,4319–4324
20. Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996) Mass spectro-metric sequencing of proteins from silver-stained polyacrylamide gels.Anal. Chem. 68, 850–858
21. Eng, J. K., McCormack, A. L., and Yates, J. R. III (1994) An approach tocorrelate tandem mass spectral data of peptides with amino acid se-quences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989
22. Keller, A., Nesvizhskii, A. I., Kolker, E., and Aebersold, R. (2002) Empiricalstatistical model to estimate the accuracy of peptide identifications madeby MS/MS and database search. Anal. Chem. 74, 5383–5392
23. Nakai, K. (2000) Protein sorting signals and prediction of subcellular local-ization. Adv. Protein. Chem. 54, 277–344
24. Emanuelsson, O., and Heijne, G. V. (2001) Prediction of organellar targetingsignals. Biochim. Biophys. Acta 1541, 114–119
25. Hirokawa, T., Boon-Chieng, S., and Mitaku, S. (1998) SOSUI: Classificationand secondary structure prediction systems for membrane proteins.Bioinformatics 14, 378–389
26. Nesvizhskii, A. I., Keller, A., Koller, E., and Aebersold, R. (2003) A statisticalmodel for identifying proteins by tandem mass spectrometry. Anal.Chem. 75, 4646–4658
27. Haller, P. D. V., Yi, E., Donohoe, S., Vaughn, K., Keller, A., Nesvizhskii, A. I.,Eng, J., Li, X., Goodlett, D. R., Aebersold, R., and Watts, J. D. (2003) Anapplication of new software tools to quantitative protein profiling viaisotope-coded affinity tag (ICAT) and tandem mass spectrometry. Mol.Cell. Proteomics 2, 426–442
28. Schartz, R., Ting, C. S., and King, J. (2001) Whole proteome pI valuescorrelate with subcellular localization of proteins for organisms with thethree domains of life. Genome Res. 11, 703–709
29. Fountoulakis, M., and Suter, L. (2000) Proteomic analysis of the rat livermitochondrial proteins. J. Chromatogr. B. 782, 197–218
30. Fountoulakis, M., and Takacs, B. (2001) Effect of strong detergents andchaotropes on the detection of proteins in two-dimensional gels. Elec-trophoresis 22, 1553–1602
31. Brimmell, M., Mendiola, R., Mangion, J., and Packham, G. (1998) BAXframeshift mutations in cell lines derived from human haemopoieticmalignancies are associated with resistance to apoptosis and microsat-ellite instability. Oncogene 16, 1803–1812
32. Ohlmeier, S., Kastaniotis, A. J., Hiltunen, J. K., and Bergmann, U. (2004)The yeast mitochondrial proteome, a study of fermentative and respira-tory growth. J. Biol. Chem. 279, 3956–3979
33. Armstrong, L. C., Saenz, A. J., and Bornstein, P. (1999) Metaxin 1 interactswith metaxin 2, a novel related protein associated with the mammalianmitochondrial outer membrane. J. Cell. Biochem. 74, 11–22
34. Petruzzella, V., Tiranti, V., Fernandez, P., Ianna, P., Carrozzo, R., andZeviani, M. (1998) Identification and characterization of human cDNAsspecific to BCS1, PET112, SCO1, COX15, and COX11, five genes in-volved in the formation and function of the mitochondrial respiratorychain. Genomics 54, 494–504
35. Simpson, R. B., and Russell, J. T. (1997) The role of sarcoplasmic/endo-plasmic reticulum Ca2�-ATPase in mediating Ca2� waves and localCa2�-release microdomains in culture glia. Biochem. J. 325, 239–247
36. Garcia, A., Cayla, X., Guergnon, J., Dessauge, F., Hospital, V., Rebollo,M. P., Fleischer, A., and Rebollo, A. (2003) Serine/threonine proteinphosphatases PP1 and PP2A are key players in apoptosis. Biochimie 85,721–726
37. Ruvolvo, P. P., Deng, X., Ito, T., Carr, B. K., and May, W. S. (1999) Ceramide
Mitochondrial Proteome from Human Leukemia Cells
180 Molecular & Cellular Proteomics 4.2

induces Bcl2 dephosphorylation via a mechanism involving mitochon-drial PP2A. J. Biol. Chem. 274, 20296–20300
38. Konoshi, Y., Lehtinen, M., Donovan, N., and Bonni, A (2002) Cdc2 phos-phorylation of BAD links the cell cycle to the cell death machinery. Mol.Cell 9, 1005–1016
39. Harder, Z., Zunino, R., and McBride, H. (2004) Sumo1 conjugates mito-chondrial substrates and participates in mitochondrial fission. Curr. Biol.14, 340–345
40. Freije, J. M. P., Blay, P., Pendas, A. M., Cadinanos, J., Crespo, P., andLopez-Otin, C. (1999) Identification and chromosomal location of twohuman genes encoding enzymes potentially involved in proteolytic mat-uration of farnesylated proteins. Genomics 58, 270–280
41. Yeh, E. T. H., Gong, L., Kamitani, T. (2000) Ubiquitin-like proteins: Newwines in new bottles. Gene 248, 1–14
42. Carre, M., Andre, N., Carles, G., Borrghi, H., Brichese, Briand, C., andBraguer, D. (2002) Tubulin is an inherent component of mitochondrialmembranes that interacts with the voltage-dependent anion channel.J. Biol. Chem. 277, 33664–33669
43. MacKenzie, J. A., and Payne, R. M. (2004) Ribosome specifically bind tomammalian mitochondria via protease-sensitive proteins on the outermembrane. J. Biol. Chem. 279, 9803–9810
44. Ostrowski, J., Wyrwicz, L., Rychlewski, L., and Bomstyk, K. (2002) Hertog-enous nuclear ribonucleoprotein K protein associates with multiple mi-tochondrial transcripts within the organelle. J. Biol. Chem. 277,6303–6310
45. Thiede, B., Siejak, F., Dimmler, C., and Rudel, T. (2002) Prediction oftranslocation and cleavage of heterogenous ribonuclear proteins andRho guanine nucleotide dissociation inhibitor 2 during apoptosis bysubcellualr proteome analysis. Proteomics 2, 996–1006
46. Mootha, V, K., Bunkenborg, J., Olsen, J. V., Hjerrild, M., Wisniewski, J. R.,Stahl, E., Bolouri, M. S., Ray, H. N., Sihag, S., Kamal, M., Patterson, N.,Lander, E. S., and Mann, M. (2003) Integrated analysis of protein com-position, tissue diversity, and gene regulation in mouse mitochondria.Cell 115, 629–640
47. Pastorino, J. G., Shugla, N., and Hoek, J. B. (2002) Mitochondrial binding ofhexokinase II inhibits Bax-induced cytochromr c release and apoptosis.J. Biol. Chem. 277, 7610–7618
48. Gaucher, S. P., Taylor, S. W., Fahy, E., Zhang, B., Warnock, D. E., Ghosh,S., and Gibson, B. W. (2004) Expanded coverage of the human heartmitochondrial proteome using multidimensional liquid chromatographycoupled with tandem mass spectrometry. J. Proteome Res. 3, 495–505
49. Natoli, G. (2004) Little things that count in transcriptional regulation. Cell118, 406–408
50. Ogasawara, J., Watanabe-Fukunaga, R., Adachi, M., Matsuzawa, A., Ka-sugai, T., Kitamura, Y., Itoh, N., Suda, T., Nagata, S. (1993) Lethal effectof the anti-Fas antibody in mice. Nature 364, 806–9
51. Han, D. K. M., Eng, J., Zhou, H., Aebersold, R. (2001) Quantitative profilingof differentiation induced microsomal proteins using isotope coded af-finity tags and mass spectrometry. Nat. Biotechnol. 19, 946–951
Mitochondrial Proteome from Human Leukemia Cells
Molecular & Cellular Proteomics 4.2 181





























