Embed Size (px)
Citation preview

www.elsevier.com/locate/theochem
Journal of Molecular Structure: THEOCHEM 850 (2008) 127–134
Ab initio study of luminescent substituted 8-hydroxyquinolinemetal complexes with application in organic light emitting diodes
Francisco Nunez-Zarur, Ricardo Vivas-Reyes *
Grupo de Quımica Cuantica y Teorica, Programa de Quımica, Universidad de Cartagena, Cartagena, Colombia
Received 2 May 2007; received in revised form 22 October 2007; accepted 23 October 2007Available online 6 November 2007
Abstract
The geometry, electronic structure and reactivity sites of a set of HQ–M (M = Be, Zn, Cd) with application in OLED’s were com-puted and compared through computational calculations of both electronic and DFT-based chemical reactivity descriptors. All the struc-tures were optimized at B3LYP level of theory and 6-31G(d) basis set for C, N, O, H and 3-21G(d) for Be, Zn and Cd. All the geometrieswere distorted tetrahedral, where both angles and bond length increase in the order Be+2 < Zn+2 < Cd+2. The molecular orbital anal-ysis reveals that the frontier orbitals are delocalized and preserve largely the electronic structure of the individual 8-hydroxyquinolineligand, being practically unaffected by both the amidopyridine group and the metal cation. The more reactive sites were found atomsbelonging to the lowest unoccupied molecular orbital, which are in para-position to the quinoline nitrogen.� 2007 Elsevier B.V. All rights reserved.
Keywords: 8-Hydroxyquinoline metal complexes; DFT Calculation; OLED; Chemical reactivity descriptors; Electronic descriptors; Reactivity descrip-tors; HOMO; LUMO; HF; DFT
1. Introduction
In recent years, the flat-panel display industry has growntremendously due mostly to the success of liquid-crystaldisplays (LCD). However during this period, the technol-ogy of organic light-emitting diodes (OLED’s) hasadvanced so that it now can compete directly with LCDfor high-information content display applications [1]. Thestructural arrangement of an OLED, first proposed byTang and Van Slyke [2], consists of three molecular films:a hole-transporting layer (HTL), an emissive layer (EL)and an electron-transport layer (ETL) which nature isessentially organic, sandwiched between two electrodesusually with ITO serving as a anode and a low work func-tion metal or alloy of them as a cathode [3]. In an OLED,electrons and holes are injected from each electrode intoETL and HTL, respectively. These charge carriers movestoward each other until they form h+–e� pairs in the EL,
0166-1280/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.theochem.2007.10.036
* Corresponding author. Tel.: +57 5 6698180; fax: +57 5 6698323.E-mail addresses: [email protected] (F. Nunez-Zarur), rvivasr
@unicartagena.edu.co (R. Vivas-Reyes).
called excitons, which radiate characteristic light in the vis-ible spectrum when they become relaxed [4].
The more important complex used in OLED has beentris-(8-hydroxyquinoline) aluminum (III), called Alq3 [2].This is an organometallic complex with one aluminum cat-ion in +3 formal oxidation state, and surrounded by threequinolate ligands, resulting in a pseudo-octahedral config-uration [5–7]. Alq3 can exist in two isomeric forms, whichare object of debate about film composition. An analysis ofthe electronic structure of the Alq3 was performed by Curi-oni et al. on the basis of ab initio calculations suggesting thecoexistence of the two isomers, called mer-Alq3 and fac-Alq3 [7]. However, experimental studies have not showedevidence of the facial isomer [8,9], although recently havebeen reported a new crystalline phase for this isomer [10]that is presents in a few amounts. Nevertheless this dis-agreement about the film composition, the most of boththeoretical and experimental treatments refer almost exclu-sively to mer-Alq3 [11].
Thin films of Alq3 are usually obtained by high-vacuumdeposition but, at times, isomerization occurs upon hightemperature sublimation. Thus, for instance, Alq3, which

128 F. Nunez-Zarur, R. Vivas-Reyes / Journal of Molecular Structure: THEOCHEM 850 (2008) 127–134
is synthesised in the mer form, has been shown to convert,in part, to the fac isomer by effect of sublimation process[12]. Since chemical degradation or crystallization are bothdetrimental for device stability and performance, Alq3films are insufficiently stable under long-term working con-ditions [3]. These factors limit the utility of OLED’s as analternative to LCD technology.
At present, research has been done in order to preparenew compounds with improved performances and stabilityand/or emitting in a particular spectral range. Exploringthese aspects, several complexes of metals such as Be, Znand Cd with substituted 8-hydroxyquinoline ligands havebeen reported. [3,12] At moment, these complexes havebeen characterized from an experimental point view,through photoluminescence studies, but it has not beenreported a theoretical study about their geometric and elec-tronic structures. In addition, for the best of our knowl-edge, this is the first contribution that makes use ofDFT-based chemical reactivity descriptors in order toexplore the interactions between OLED materials andreducing agents, such as metal-based cathodes.
2. Theory and computational details
The complexes objects of this study were taken from lit-erature [3,12]. These are a set of three bis-chelates com-plexes of Be, Zn and Cd, with 5-amidopyridine-8-hydroxyquinoline as ligands. Fig. 1 shows the general
N
O
N1
O1
M
N
H
Substituent 1
Liga
M Comp
Be H
Zn H
Cd H
Fig. 1. General structure for the studied compounds. It is denoted the twonitrogen atoms belonging to the inner shell are labelled too.
structure for the studied compounds, as also the keys toidentifying them.
All the geometries were optimized using the Gaussian 03suite of programs [13] in the ground (S0) state and in theanhydrous form by means of the Becke’s three parametershybrid method [14] with the Lee–Yang–Parr correlationfunctional [15], abbreviated B3-LYP and the split-valence6-31G(d) [16] basis set for C, N, H, O atoms and the 3-21G(d) [17] basis set for the metal atoms Be (II), Zn (II)and Cd (II). The corresponding electronic and DFT-basedchemical reactivity descriptors were computed at same levelof theory. The electronic descriptors calculated in thispaper were: energy of the highest occupied molecular orbi-tal (EHOMO), energy of the lowest unoccupied molecularorbital (ELUMO), the energy difference between HOMO–LUMO (Gap), contour maps of frontier molecular orbitalsand molecular orbital contributions (MOC’s) form differ-ent molecular moieties.
Also, there is a set of chemical reactivity descriptorswhich can be derived from Density Functional Theory[18]. These are the global hardness (g), global softness(S), Fukui function (FF), local softness (s) and the globaland local electrophilicity index (x). A detailed presentationand discussion about these descriptors can be found else-where [19–24] and only the relevant expressions will begiven here.
The global hardness and global softness are calculatedas follows:
N2
O2
N
O
NH
Substituent 2
nd 1
Ligand 2
ound key
Q-Be
Q-Zn
Q-Cd
ligands with a numeral and the respective substituents. The oxygen and

Table 1Bond lengths (A) and Angles (�) of the inner shell for the studiedcompounds optimized at B3LYP/6-31G(d)a level
Molecule M–O1 M–O2 M–N1 M–N2 O1–M–O2 N1–M–N2
HQ–Be 1.609 1.609 1.805 1.805 124.068 111.947HQ–Zn 1.873 1.873 1.955 1.955 125.571 123.147HQ–Cd 2.160 2.160 2.321 2.321 141.556 123.512
a The metal atoms Be, Zn and Cd were treated with the 3-21G(d) basisset.
F. Nunez-Zarur, R. Vivas-Reyes / Journal of Molecular Structure: THEOCHEM 850 (2008) 127–134 129
g � IE� EA
2ð1Þ
and
S ¼ 1
2gð2Þ
where IE and EA are the vertical ionization energy andelectron affinity, respectively, for the system. The globalhardness can be seen as the resistance of a chemical speciesto charge transfer or to the loss or gain electrons and theglobal softness is interpreted as the inverse of global hard-ness. These descriptors describe global properties for a sys-tem as a whole; however, in order to study selectivity andreactivity it is more advisable to use local reactivity descrip-tors. Within these we have the Fukui function [25]:
f ðrÞ ¼ oqðrÞoN
� �vðrÞ¼ dl
dvðrÞ
� �N
ð3Þ
The Fukui function is interpreted as the change of chemicalpotential given an external perturbation, or the variation ofthe electron density function when the electrons numberchange. In this paper, we will work exclusively with theequation for a nucleophilic attack, because the nature ofthe interactions between the OLED materials focused andreducing agents, such as metal-based cathodes. In this con-text, the Fukui function of interest is:
f þðrÞ ¼ oqðrÞoN
� �þvðrÞ
ð4Þ
Yang and Mortier [26] proposed a condensed version of theEq. (4), as follows:
f þA ¼ qAðN þ 1Þ � qAðNÞ ð5Þ
where qA (N + 1) and qA (N) are the atomic populations ofthe atom A for the anionic and neutral system, respectively.
In analogy of the condensed Fukui function, it is reason-able to introduce the local softness condensed in any atomfor a nucleophilic attack, sþx :
sþx ¼ Sf þx ð6Þ
where S is the global softness and fx the Fukui function asdefined above.
Another important DFT-based chemical reactivitydescriptor is the electrophilicity index, x [27], which mea-sures the second-order energy of an electrophile when it
Fig. 2. Optimized structure of the HQ–Zn complex at B3LYP/6-31G (d)
gets saturated with electrons, that is, the electrophilicpower of the system. This is one of the so called globalreactivity descriptors and it was calculated as follows:
x ¼ v2
2gð7Þ
where v is the electronegativity and g is the global hardnessfor the given system. Additionally, a local electrophilicityindex [28] can be computed from an expression analogueto Eq. (6) and it has been introduced to analyze the electro-phile–nucleophile reactions better:
xk ¼ xf þk ð8Þ
where f þk is the nucleophilic Fukui function and x is theglobal electrophilicity index.
3. Results and discussion
3.1. Geometries of the ground state (S0)
The three studied complexes showed the same geometryin the inner shell, i.e., distorted tetrahedral in which themetal center presents a +2 formal oxidation state. Asexample, Fig. 2 shows the optimized structure for the com-plex HQ–Zn. The main geometric parameters are onlyfunction of the type of central cation. Table 1 shows themain optimized parameters for the three complexes. Thebond lengths are equivalent for the two ligands and thisfact leads to a relationship between geometry of the groundstate and the electronic structure, as we will show below.
As can be seen, these parameters clearly increase withthe size of the metal center. For example, the two M–O dis-tances increase from HQ–Be to HQ–Zn and from HQ–Znto HQ–Cd, in nearly 0.3 A and the M–N distances increasefrom HQ–Be to HQ–Zn in 0.15 A and from HQ–Zn to
level of theory. The Zn atom was treated with the 3-21G(d) basis set.

130 F. Nunez-Zarur, R. Vivas-Reyes / Journal of Molecular Structure: THEOCHEM 850 (2008) 127–134
HQ–Cd in 0.37 A. The O–M–O bond angles have a smallincrease from HQ–Be to HQ–Zn, which is nearly to 1.5�and a great increase from HQ–Zn to HQ–Cd in around16�. Conversely, the N–M–N bond angles show a contrarytrend. These increase in a slight amount (0.37�) betweenHQ–Zn and HQ–Cd and a greater amount between HQ–Be and HQ–Zn (11�).
These general trends clearly indicate that the angles andbond lengths increase in the order HQ–Be < HQ–Zn < HQ–Cd and, consequently, the metal-ligand interac-tions decrease in the order HQ–Be > HQ–Zn > HQ–Cd.The result of this fact will be explored in next section.
3.2. Molecular orbital analysis
For the three complexes, the frontier molecular orbitalsdivide into ‘‘doublets’’ whose energy difference is slight.The electron distribution is similar for the set, and as exam-ple the Fig. 3 shows the contour surfaces of the HOMO
Fig. 3. Frontier molecular orbital surfaces of HQ–Zn calculated at B3LYP/6-30.03 e/au3).
and LUMO sets for HQ–Zn. These orbitals are delocalizedand preserve the nature of the individual 8-hydroxyquino-line (8-Hq) ligand. The delocalized nature is related to thesymmetry in the inner shell bond lengths, that is, the equiv-alency between the bond lengths of two ligands leads to astronger delocalization of the HOMO and LUMO orbitals.Thus, the grade of symmetry is an indicative of the orbitaldelocalization, which indicates that an asymmetrical innershell leads to strongly localized molecular orbitals. Thishas been proved for our group, for example, for tris-che-lates analogues compounds [29].
In order to confirm the similarity between the HOMOand LUMO sets of the studied complexes and those ofthe 8-Hq, it was carried out a molecular orbital calculationon the 8-Hq at B3LYP/6-31G(d) level of theory. The con-tour plots of HOMO and LUMO for 8-Hq is showed in theFig. 4 and their comparison with those in Fig. 3 results inalmost the same shape, independently of the type of metalcation and substituent and only a few amount of this elec-
1G(d) level. The Zn atom was treated with the 3-21G(d) basis set. (Isovalue
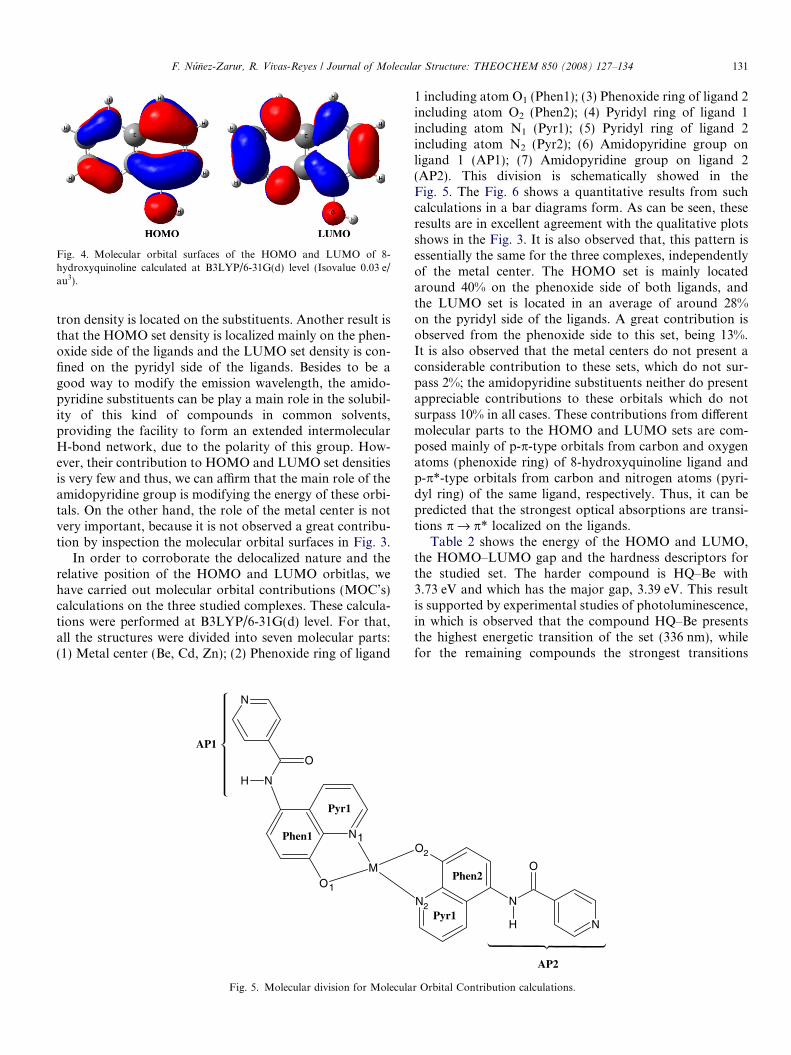
Fig. 4. Molecular orbital surfaces of the HOMO and LUMO of 8-hydroxyquinoline calculated at B3LYP/6-31G(d) level (Isovalue 0.03 e/au3).
F. Nunez-Zarur, R. Vivas-Reyes / Journal of Molecular Structure: THEOCHEM 850 (2008) 127–134 131
tron density is located on the substituents. Another result isthat the HOMO set density is localized mainly on the phen-oxide side of the ligands and the LUMO set density is con-fined on the pyridyl side of the ligands. Besides to be agood way to modify the emission wavelength, the amido-pyridine substituents can be play a main role in the solubil-ity of this kind of compounds in common solvents,providing the facility to form an extended intermolecularH-bond network, due to the polarity of this group. How-ever, their contribution to HOMO and LUMO set densitiesis very few and thus, we can affirm that the main role of theamidopyridine group is modifying the energy of these orbi-tals. On the other hand, the role of the metal center is notvery important, because it is not observed a great contribu-tion by inspection the molecular orbital surfaces in Fig. 3.
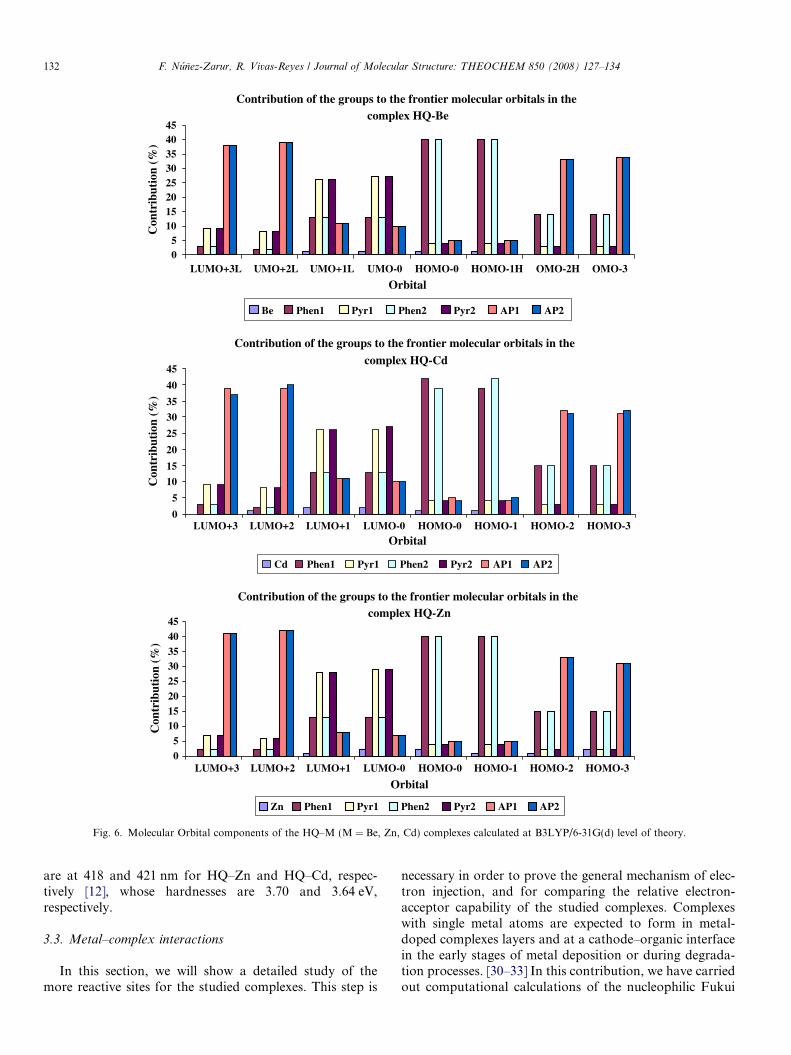
In order to corroborate the delocalized nature and therelative position of the HOMO and LUMO orbitlas, wehave carried out molecular orbital contributions (MOC’s)calculations on the three studied complexes. These calcula-tions were performed at B3LYP/6-31G(d) level. For that,all the structures were divided into seven molecular parts:(1) Metal center (Be, Cd, Zn); (2) Phenoxide ring of ligand
N
O
N1
O1
M
N
H
Phen1
Pyr1
AP1
Fig. 5. Molecular division for Molecula
1 including atom O1 (Phen1); (3) Phenoxide ring of ligand 2including atom O2 (Phen2); (4) Pyridyl ring of ligand 1including atom N1 (Pyr1); (5) Pyridyl ring of ligand 2including atom N2 (Pyr2); (6) Amidopyridine group onligand 1 (AP1); (7) Amidopyridine group on ligand 2(AP2). This division is schematically showed in theFig. 5. The Fig. 6 shows a quantitative results from suchcalculations in a bar diagrams form. As can be seen, theseresults are in excellent agreement with the qualitative plotsshows in the Fig. 3. It is also observed that, this pattern isessentially the same for the three complexes, independentlyof the metal center. The HOMO set is mainly locatedaround 40% on the phenoxide side of both ligands, andthe LUMO set is located in an average of around 28%on the pyridyl side of the ligands. A great contribution isobserved from the phenoxide side to this set, being 13%.It is also observed that the metal centers do not present aconsiderable contribution to these sets, which do not sur-pass 2%; the amidopyridine substituents neither do presentappreciable contributions to these orbitals which do notsurpass 10% in all cases. These contributions from differentmolecular parts to the HOMO and LUMO sets are com-posed mainly of p-p-type orbitals from carbon and oxygenatoms (phenoxide ring) of 8-hydroxyquinoline ligand andp-p*-type orbitals from carbon and nitrogen atoms (pyri-dyl ring) of the same ligand, respectively. Thus, it can bepredicted that the strongest optical absorptions are transi-tions p fi p* localized on the ligands.
Table 2 shows the energy of the HOMO and LUMO,the HOMO–LUMO gap and the hardness descriptors forthe studied set. The harder compound is HQ–Be with3.73 eV and which has the major gap, 3.39 eV. This resultis supported by experimental studies of photoluminescence,in which is observed that the compound HQ–Be presentsthe highest energetic transition of the set (336 nm), whilefor the remaining compounds the strongest transitions
N2
O2
N
O
NH
Phen2
Pyr1
AP2
r Orbital Contribution calculations.
Contribution of the groups to the frontier molecular orbitals in the complex HQ-Be
05
1015202530354045
LUMO+3L UMO+2L UMO+1L UMO-0 HOMO-0 HOMO-1H OMO-2H OMO-3
Orbital
Con
trib
utio
n (%
)
Be Phen1 Pyr1 Phen2 Pyr2 AP1 AP2
Contribution of the groups to the frontier molecular orbitals in the complex HQ-Cd
0
5
10
15
20
25
30
35
40
45
LUMO+3 LUMO+2 LUMO+1 LUMO-0 HOMO-0 HOMO-1 HOMO-2 HOMO-3Orbital
Con
trib
utio
n (%
)
Cd Phen1 Pyr1 Phen2 Pyr2 AP1 AP2
Contribution of the groups to the frontier molecular orbitals in the complex HQ-Zn
05
1015202530354045
LUMO+3 LUMO+2 LUMO+1 LUMO-0 HOMO-0 HOMO-1 HOMO-2 HOMO-3
Orbital
Con
trib
utio
n (%
)
Zn Phen1 Pyr1 Phen2 Pyr2 AP1 AP2
Fig. 6. Molecular Orbital components of the HQ–M (M = Be, Zn, Cd) complexes calculated at B3LYP/6-31G(d) level of theory.
132 F. Nunez-Zarur, R. Vivas-Reyes / Journal of Molecular Structure: THEOCHEM 850 (2008) 127–134
are at 418 and 421 nm for HQ–Zn and HQ–Cd, respec-tively [12], whose hardnesses are 3.70 and 3.64 eV,respectively.
3.3. Metal–complex interactions
In this section, we will show a detailed study of themore reactive sites for the studied complexes. This step is
necessary in order to prove the general mechanism of elec-tron injection, and for comparing the relative electron-acceptor capability of the studied complexes. Complexeswith single metal atoms are expected to form in metal-doped complexes layers and at a cathode–organic interfacein the early stages of metal deposition or during degrada-tion processes. [30–33] In this contribution, we have carriedout computational calculations of the nucleophilic Fukui

Table 2Energy of the HOMO, LUMO, gap (HOMO–LUMO) and globalhardness (eV) for the studied complexes calculated at B3LYP/6-31G(d)a
level of theory
Molecule EHOMO ELUMO Gap Hardness (g)
HQ–Be �5.336 �1.944 3.39 3.73HQ–Zn �5.264 �1.972 3.29 3.70HQ–Cd �5.210 �1.873 3.34 3.64
a The metal atoms Be, Zn and Cd were treated with the 3-21G(d) basisset.
F. Nunez-Zarur, R. Vivas-Reyes / Journal of Molecular Structure: THEOCHEM 850 (2008) 127–134 133
function (FF+), local softness (s+) and local electrophilicityindex (xk) on the three studied compounds. Fig. 7 showsthe atom numbering for identifying each one of the atomof the molecules.
Our results of the chemical reactivity descriptors FF+,s+ and xk show that the more reactive atoms for the threecomplexes are the C4 atoms located at para-position fromthe quinoline nitrogen on both ligands. The FF+ descriptorin this position presents values of 0.049, 0.057 and 0.051 eVfor HQ–Be, HQ–Zn and HQ–Cd, respectively. Thecorresponding values of s+ are 0.0066, 0.0077 and0.0070 eV�1, these values are in essence of the same orderof magnitude. In order to corroborate these results we alsohave computed the local electrophilicity index on eachatom of the ligands and substituent skeleton. The C4 atomspresent the greater local electrophilicity indexes, being0.1201, 0.1399 and 0.1216 eV for HQ–Be, HQ–Zn andHQ–Cd, respectively. This indicates that the reducingagents will attack the molecules at pyridyl side, being theC4-atom the prone to receive the electron from it. More-over, it has been demonstrated that when a low work func-tion metal acts as a cathode, this injects electrons to
C10
C9
C5
C8
C6
C7
C3
C2
C4
N1
O11
H12
H13
N17
H15
H14
H16
C19
C21
O20
C25
C26
N24C23
C22H27
H28 H29
H30
H18
M
Fig. 7. Atom numbering for the studied compounds. For simplicity it isshown a single ligand.
LUMO of electron transport layer. [34–37] Thus, the chem-ical reactivity descriptors FF+, s+ and xk are useful todescribe the electron injection mechanism in an OLED.This is a validity proof for the results obtained here.
The next atom prone to receive electrons from cathodeis N24 atoms (nitrogen atoms of the pyridine group ofthe substituents). For this atoms, the respective values ofFF+, s+ and xk are 0.044 eV, 0.0059 eV�1 and 0.1078 eV(HQ-Be), 0.040 eV, 0.0054 eV�1 and 0.0982 eV (HQ–Zn)and 0.044 eV, 0.0061 eV�1 and 0.1049 eV (HQ–Cd), respec-tively. It must be recall that these atoms also present someextent of LUMO density and this can be the reason forwhich they show the second values of the local reactivitydescriptors.
The following atoms prone to receive electrons for thecathode are the N1 and C2 atoms, which also belong tothe LUMO and display values of FF+ of 0.030 and0.026 eV (HQ–Be), 0.029 and 0.031 eV (HQ–Zn), 0.025and 0.027 eV (HQ–Cd), respectively.
By comparing s+ values, it can be predicted that the bet-ter electron-withdrawing complex will be HQ–Zn, due itslarger value of this descriptor compared to others ones.This is confirmed by the xk index, which show that theelectrophilicity power decrease in the order HQ–Zn > HQ–Cd > HQ–Be, in perfect agreement with FF+
and s+. Thus, all local descriptors are useful in describingthe relative electron-capture capability, displaying the pos-sibility of modifying/design some molecular regions inorder to improve the efficiency of the OLED’s devices.
4. Conclusions
We have computed and compared the geometry,electronic structure and the reactivity sites for a set ofbis-chelates of Be, Zn and Cd and 5-amidopyridine-8-hydroxyquinoline as ligands. Besides the use of electronicdescriptors, this is the first theoretical treatment whereare used DFT-based chemical reactivity descriptors suchas global hardness and softness, Fukui function, local soft-ness and local electrophilicity index. The geometriesaround the metal center are distorted tetrahedral, wherethe inner shell bond lengths and angles are only functionof the metal type. These parameters increase in the orderHQ–Be < HQ–Zn < HQ–Cd and, thus, the metal–ligandinteractions decrease in the order HQ–Be > HQ–Zn >HQ–Cd.
The molecular orbital analysis reveals some interestingfeatures. The HOMO and LUMO sets are delocalizedand divide into sets of two orbitals, which conserve the nat-ure of the individual ligand 8-Hq. The HOMO set islocated on phenoxide side of the ligands, while the LUMOset is located on the pyridyl side. Comparison of theelectronic structure of our compounds with those of the8-Hq ligand shows that the nodal patterns and relativeposition of the HOMO and LUMO are essentially the samefor both. This indicate that the electron distribution of8-Hq is practically unaffected upon chelating and substitu-

134 F. Nunez-Zarur, R. Vivas-Reyes / Journal of Molecular Structure: THEOCHEM 850 (2008) 127–134
tion and so the metal and amidopyridine substituent do notmodify the original nature of 8-Hq. The substitution of theligands, besides to be a good way to change the emissionwavelength, can be used for the establishment of anextended intermolecular H-bond network in solution.
The MOC’s analyses confirm and support the delocal-ized nature and relative positions of the HOMO andLUMO set, showing that the HOMO set is confined inaround 40% on the phenoxide side, and the LUMO set islocated on the pyridyl side in around 28% and on phenox-ide side in around 13%. We have attributed these contribu-tions to p-p-type orbitals from carbon and oxygen atoms(phenoxide ring) of 8-hydroxyquinoline ligand and p-p*-type orbitals from carbon and nitrogen atoms (pyridylring) of the same ligand, respectively. So, we can predictthat the strongest optical absorptions are transitionsp fi p* localized on the ligands.
The local reactivity descriptors FF+, s+ and xk revealedthat the atoms prone to receive electrons from reducingagents (cathode) are those related to the lowest unoccupiedmolecular orbital (LUMO-0). These atoms are opposed tothe pyridyl nitrogen (para) and showed the larger values ofFF+, s+ and xk of whole the molecules. This result is alsosupported by experimental studies of electron injectionmechanism carried out in an OLED, by which the presentstudy is validated from an experimental point view.
Acknowledgements
The authors are grateful to COLCIENCIAS and Uni-versidad de Cartagena, as well as the Program of YoungInvestigators sponsored by both institutions
References
[1] Meyers, Amy. The design and synthesis of metal-functionalized Poly(norbornenes) for potential use in light-emitting diodes. DoctoralThesis. Georgia Institute of Technology. December, 2004.
[2] C.W. Tang, S.A. Van Slyke, Appl. Phys. Lett. 51 (1987) 913–915.[3] M. Ghedini, I. Aiello, A. Grisolia, A. Crispini, M. La Deda,
Substituted 8-hydroxyquinolines metal complexes for application inorganic light emitting devices. N. Russo, D.R. Salahub, M. Witko,Metal–ligand interactions. Molecular, Nano-, Micro-, and Macrosystems in complex environment. NATO Science series II. Math.Phys. and Chem. 16 (2003).
[4] W. Zhao, W. Wei, J. Lozano, J.M. White, Chem. Mater. 16 (2004)750–756.
[5] M.D. Halls, H.B. Schlegel, Chem. Mater. 13 (2001) 2632.[6] M. Amati, F. Lelj, J. Phys. Chem. A. 107 (2003) 2560–2569.[7] A. Curioni, M. Boero, W. Andreoni, Chem. Phys. Lett. 294 (1998)
263.[8] G.P. Kushto, Y. Iizumi, J. Kido, Z.H. Kafafi, J. Phys. Chem. 104
(2000) 3670.[9] M. Brinkmann, G. Gadret, M. Muccini, C. Taliani, N. Masciocchi,
A. Sironi, J. Am. Chem. Soc. 122 (2000) 5147.[10] M. Braun, J. Gmeiner, M. Tzolov, M. Coelle, F.D. Meyer, W. Milius,
H. Hellebrecht, O. Wendland, J.U. von Schutz, W. Brutting, J. Chem.Phys. 114 (2001) 9625–9632.
[11] L.S. Sapochak, F.E. Benincasa, R.S. Schofield, J.L. Baker, K.K.C.Riccio, D. Fogarty, H. Kohlmann, K.F. Ferris, P.E. Burrows, J. Am.Chem. Soc 124 (2002) 6119–6125.
[12] M. Ghedini, M. LaDeda, I. Aiello, A. Grisolia, Synthetic Met. 138(2003) 189–192.
[13] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb,J.R. Cheeseman, J.A. Montgomery Jr., T. Vreven, K.N. Kudin, J.C.Burant, J.M. Millam, S.S. Iyengar, J. Tomasi, V. Barone, B.Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A. Petersson, H.Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa,M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X.Li, J.E. Knox, H.P. Hratchian, J.B. Cross, C. Adamo, J. Jaramillo, R.Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C.Pomelli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, G.A. Voth, P.Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich, A.D.Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K.Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S.Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P.Piskorz, I. Komaromi, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill,B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A. Pople,Gaussian 03, Revision B.01, Gaussian, Inc., Pittsburgh PA, 2003.
[14] A.D.J. Becke, Chem. Phys. 98 (1993) 5648.[15] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1998) 785.[16] (a) W.J. Hehre, R. Ditchfield, J.A. Pople, J. Chem. Phys. 56 (1972)
2257;(b) M.M. Francl, W.J. Petro, W.J. Hehre, J.S. Binkley, M.S. Gordon,D.J. DeFrees, J.A. Pople, J. Chem. Phys. 77 (1982) 3654.
[17] (a) M.J. Frisch, J.A. Pople, J.S. Binkley, J. Chem. Phys. 80 (1984)3265;(b) M.S. Gordon, J.S. Binkley, J.A. Pople, W.J. Pietro, W.J. Hehre, J.Am. Chem. Soc. 104 (1982) 2797.
[18] K. Hohenberg, W. Kohn, Phys. Rev. B 136 (1964) 864.[19] R.G. Pearson, J. Am. Chem. Soc. 85 (1963) 3533.[20] R.G. Parr, R.G. Pearson, J. Am. Chem. Soc. 105 (1983) 7512.[21] R.G. Parr, R.A. Donnelly, M. Levy, W.E. Palke, J. Chem. Phys. 68
(1978) 3801.[22] R.G. Pearson, J. Am. Chem. Soc. 107 (1985) 6801.[23] R.G. Parr, W. Yang, Density Functional Theory of Atoms and
Molecules, Oxford University Press, New York, 1989.[24] P. Geerlings, F. De Proft, W. Langenaekar, Adv. Quant. Chem. 33
(1999) 303.[25] R.G. Parr, W. Yang, J. Am. Chem. Soc. 106 (1984) 4049.[26] W. Yang, W. Mortier, J. Am. Chem. Soc. 108 (1986) 5708.[27] R.G. Parr, L.v. Szentpaly, S. Liu, J. Am. Chem. Soc. 121 (1999) 1922.[28] (a) P. Perez, A. Toro-Labbe, A. Aizman, R. Contreras, J. Org. Chem.
67 (2002) 4747;(b) E. Chamorro, P.K. Chattaraj, P. Fuentealba, J. Phys. Chem. A107 (2003) 7068.
[29] R. Vivas-Reyes, F. Nunez-Zarur, J. Chem. Theory Comput.,submitted for publication.
[30] A. Curioni, W. Andreoni, IBM J. Res. & Dev. 45 (1) (2001).[31] A. Rajagopal, A. Kahn, J. Appl. Phys. 84 (1998) 355.[32] J. Kido, T. Matsumoto, Appl. Phys. Lett. 73 (1998) 2866.[33] M.B. Huang et al., Appl. Phys. Lett. 72 (1998) 2914.[34] A. Greiner, Polym. Adv. Technol. 9 (1998) 371–389.[35] M. Stoßel, J. Staudigel, F. Steuber, J. Blassing, J. Simmerer, A.
Winnacker, H. Neuner, D. Metzdorf, H.-H. Johannes, W. Kowalsky,Synth. Met. 111-112 (2000) 19–24.
[36] T.A. Hopkins, K. Meerholz, S. Shaheen, M.L. Anderson, A. Schmidt,B. Kippelen, A.B. Padias, J.H.K. Hall, N. Peyghambarian, N.R.Armstrong, Chem. Mater. 8 (1996) 344–351.
[37] L.S. Sapochak, A. Padmaperuma, N. Washton, F. Endrino, G.T.Schmett, J. Marshall, D. Fogarty, P.E. Burrows, S.R. Forrest, J. Am.Chem. Soc. 123 (2001) 6300–6307.





























