Embed Size (px)
Citation preview

(This is a sample cover image for this issue. The actual cover is not yet available at this time.)
This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Adenosine monophosphate-activated protein kinase regulates platelet-derivedgrowth factor-BB-induced vascular smooth muscle cell migration
Miki Iida a, Kumiko Tanabe a,⇑, Rie Matsushima-Nishiwaki b, Osamu Kozawa b, Hiroki Iida a
a Department of Anesthesiology and Pain Medicine, Gifu University Graduate School of Medicine, Yanagido 1-1, Gifu 501-1194, Japanb Department of Pharmacology, Gifu University Graduate School of Medicine, Yanagido 1-1, Gifu 501-1194, Japan
a r t i c l e i n f o
Article history:Received 27 June 2012and in revised form 26 November 2012Available online 5 January 2013
Keywords:AMPKMigrationPDGF-BBVascular smooth muscle
a b s t r a c t
Migration of vascular smooth muscle cells (VSMCs) is essential for repair of vascular injury, developmentof atherosclerotic lesions and restenosis after angioplasty or by-pass graft surgery. It has been reportedthat platelet-derived growth factor (PDGF)-BB induces VSMC migration via the p44/p42 mitogen-acti-vated protein (MAP) kinase pathway and the phosphatidylinositol 3 (PI3)-kinase/Akt pathway. Adenosinemonophosphate-activated protein kinase (AMPK) is generally known to regulate multiple metabolicpathway. In the present study, we investigated the involvement of AMPK in PDGF-BB-induced migrationof VSMCs using, a VSMC line, A10 cells. PDGF-BB induced phosphorylation of AMPK-a at Thr-172 residue.Treatment of A10 cells with compound C, an AMPK inhibitor, suppressed PDGF-BB-induced migration in aconcentration-dependent manner (0.01–1 lM). Compound C truly attenuated PDGF-BB induced phos-phorylation of acetyl-CoA carboxylase, a downstream substance of AMPK. Downregulation of AMPK-aexpression by the siRNA appeared an anti-migratory effect on PDGF-BB-induced migration. PDGF-BB-induced phosphorylation of c-Raf, MEK1/2 or p44/p42 MAP kinase, and phosphorylation of PI3-kinaseor Akt were markedly suppressed by compound C. In conclusion, our results strongly suggest thatPDGF-BB induces activation of AMPK in VSMCs, and subsequently regulates the migration via both thep44/p42 MAP kinase pathway and the PI3-kinase/Akt pathway.
� 2013 Elsevier Inc. All rights reserved.
Introduction
It is generally recognized that vascular smooth muscle cells(VSMCs)1 contribute the regulation of blood flow due to contractionof blood vessels [1]. VSMCs play important roles also in vascular in-jury repair, angiogenesis and atherosclerosis [2]. Among VSMC func-tions, VSMC migration is essential for vascular development,vascular injury repair, development of atherosclerotic lesions andrestenosis after angioplasty or by-pass graft surgery [3]. VSMC pro-trudes leading edge to contact with an extracellular substance andbinding the formation of focal adhesion complexes [4]. A cascadeof intracellular signal transduction, including GTP-binding proteinsand tyrosine kinases, results in actin filament alignment and myosincontraction within the leading edge, dissolution of adhesion com-plexes, so that movement can take place [4]. It has been shown thatVSMC migration is regulated by a variety of factors, including
platelet-derived growth factor (PDGF)-BB, basic fibroblast growthfactor (bFGF), transforming growth factor-b (TGF-b), epidermalgrowth factor and insulin-like growth factor [3]. However, the de-tails of VSMC migration remain to be clarified.
PDGF was first identified as a growth-promoting factor releasedby human platelets [5]. At the present, it is generally known thatPDGF is produced by not only platelets, but also monocyte/macro-phages, vascular endothelial cells and VSMCs [5]. PDGF is secretedat the site of wound and contributes to the healing [6]. It has beenreported that PDGF is implicated in fibrotic diseases of several dif-ferent organs, atherosclerosis and restenosis after intervention [7].In the cardiovascular system, PDGF is the strongest chemoattrac-tant for VSMC migration [8]. VSMC migration following vascularinjury is dependent on PDGF release by monocyte/macrophages,vascular endothelial cells and VSMCs [9]. PDGF expression is dy-namic and responsive to variety of stimuli, including hypoxia,thrombin, cytokines, and growth factors [7]. The PDGF family con-sists of four dimeric growth factors: PDGF-AA, -AB, -BB, and -DD[10]. Among them, PDGF-BB plays an important role in atheroscle-rosis, since PDGF-BB is expressed in macrophages and VSMCs with-in atherosclerotic lesions [10]. Basic expression level of PDGFreceptors is low in VSMCs, but increased dramatically by severalfactors including TGF-b, estrogen, interleukin-1a, bFGF, tumornecrosis factor-a, and lipopolysaccharide [7].
0003-9861/$ - see front matter � 2013 Elsevier Inc. All rights reserved.http://dx.doi.org/10.1016/j.abb.2012.12.019
⇑ Corresponding author. Fax: +81 58 6405.E-mail addresses: [email protected] (M. Iida), [email protected] (K.
Tanabe), [email protected] (R. Matsushima-Nishiwaki), [email protected] (O.Kozawa), [email protected] (H. Iida).
1 Abbreviations used: VSMCs, vascular smooth muscle cells; PDGF, platelet-derivedgrowth factor; bFGF, basic fibroblast growth factor; TGF-b, transforming growthfactor-b; MAP, mitogen-activated protein; AMPK, Adenosine monophosphate-acti-vated protein kinase.
Archives of Biochemistry and Biophysics 530 (2013) 83–92
Contents lists available at SciVerse ScienceDirect
Archives of Biochemistry and Biophysics
journal homepage: www.elsevier .com/ locate/yabbi

Author's personal copy
Regarding the intracellular signaling system, PDGF-BB signalingis engaged in several well-characterized cellular signaling path-ways such as the p44/p42 mitogen-activated protein (MAP) kinasepathway, the phosphatidylinositol 3 (PI3)-kinase/Akt pathway andthe phospholipase C (PLC)-protein kinase C (PKC) pathway, ulti-mately leading to cellular proliferation, differentiation, survivaland migration [7]. As for PDGF-BB-induced VSMC migration, ithas been reported that p44/p42 MAP kinase, stress-activated pro-tein kinase/c-Jun N-terminal kinase (SAPK/JNK), PI3-kinase andPKC are involved in PDGF-BB-induced rat aortic VSMC migration[11,12]. In a rat VSMC line, A10 cells, it has been reported thatPDGF-BB induces the migration through p44/p42 MAP kinase andPLCc activation [13]. However, the exact mechanism of PDGF-in-duced VSMC migration has not yet been fully elucidated.
Adenosine monophosphate-activated protein kinase (AMPK)plays a key role in the regulation of energy homeostasis and mon-itors of cellular energy charge [14]. AMPK consists of three sub-units such as a, b and c [14]. Among the subunits, a-subunit isrecognized as a catalytic site, whereas b and c-subunits are consid-ered as regulatory sites [14]. The activation of AMPK is mainly reg-ulated by phosphorylation of AMPK-a at Thr-172 residue [15].With regard to the role of AMPK in VSMCs, it has been reportedthat serum-induced VSMC migration is suppressed by 5-amino-imidazole-4-carboxamide-1-b-D-ribofuranosyl 50-monophosphate(AICAR), an activator of AMPK [16], and activation of AMPKstrongly suppresses the proliferation in VSMCs [17]. However,the role of AMPK on PDGF-BB-induced VSMC migration has notyet been clarified. Therefore, in the present study, we investigatedwhether AMPK is involved in PDGF-BB-induced A10 cell migrationand the detailed mechanism.
Materials and methods
Materials
PDGF-BB was obtained from R&D Systems, Inc (Minneapolis,MN). AICAR, was purchased from Sigma Chemical Co. (St. Louis,MO). Compound C, PD98059 and LY294002 were obtained fromCalbiochem-Novabiochem (La Jolla, CA). Antibodies against phos-pho-specific AMPK-a (Thr-172), AMPK-a, phospho-specific acet-yl-CoA carboxylase, phospho-specific c-Raf, phospho-specificMAP/extracellular signal-regulated kinase kinase (MEK) 1/2,MEK1/2, phospho-specific p44/p42 MAP kinase, p44/p42 MAP ki-nase, phosphor-specific Akt (Thr-308 or Ser-473) and Akt were ob-tained from Cell Signaling Technology, Inc (Beverly, MA).Antibodies against GAPDH and phospho-specific PI3-kinase p85were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz,CA). ECL Western blotting detection system was purchased fromGE Healthcare (Buckinghamshire, UK). Other materials and chem-icals were obtained from commercial sources. Compound C or AI-CAR was dissolved in dimethyl sulfoxide. The maximumconcentration of dimethyl sulfoxide was 0.1%, which did not affectthe cell migration assay or Western blot analysis.
Cell culture and treatments
Fetal rat aortic smooth muscle derived A10 cells were obtainedfrom American Type Culture Collection (Rockville, MD). The cellswere seeded into 35-mm (8 � 104 cells/dish) or 90-mm (4 � 105 -cells/dish) diameter dishes and maintained in Dulbecco’s modifiedEagle’s medium (DMEM) containing 10% fetal bovine serum (FBS)at 37 �C under a humidified atmosphere of 5% carbon dioxideand 95% air. After 6 days, the medium was exchanged for serum-free DMEM. The cells (90-mm diameter dishes) were then usedfor Western blot analysis after 24 h. The cells were pretreated with
compound C for 60 min before PDGF-BB or AICAR stimulationwhen indicated.
Cell migration assay
Cell migration was assessed in using Boyden chamber (polycar-bonate membrane with 8-lm pores, Transwell�, Coring CostarCorp, Cambridge, MA). The cells were trypsinized, and seeded(3 � 104 cells/well) onto the upper chamber in serum-free DMEM.The cells were pretreated with compound C in lower chamber for60 min at 37 �C. Then PDGF-BB or AICAR was added to lower cham-ber and incubated for 9 h at 37 �C. The cells on the upper surface ofthe membrane were mechanically removed. The migrated cellsadherent to the underside of the membrane were fixed with 4%paraformaldehyde and stained with 40,6-diamidino-2-phenylin-dole (DAPI) solution. The migrated cells were photographed andcounted using fluorescent microscopy at a magnification of 20�by counting the stained cells from three randomly chosen highpower fields.
Western blot analysis
The cultured cells (90-mm diameter dishes) were stimulated by30 ng/ml PDGF-BB or 3 mM AICAR in serum-free DMEM for theindicated periods. The cells were washed twice with phosphate-buffered saline, and then lysed and sonicated in a lysis buffer con-taining 62.5 mM Tris/HCl (pH 6.8), 2% sodium dodecyl sulfate(SDS), 50 mM dithiothreitol and 10% glycerol. The sample was sub-jected to SDS–polyacrylamide gel electrophoresis (PAGE) that wasperformed using by the method of Laemmli [18]. Western blotanalysis was performed using phospho-specific AMPK-a antibod-ies, AMPK-a antibodies, phospho-specific acetyl-CoA carboxylaseantibodies, phospho-specific c-Raf antibodies, phospho-specificMEK1/2 antibodies, MEK1/2 antibodies, phospho-specific p44/p42 MAP kinase antibodies, p44/p42 MAP kinase antibodies, phos-pho-specific PI3-kinase antibodies, phospho-specific Akt antibod-ies, Akt antibodies or GAPDH antibodies with peroxidase-labeledanti-rabbit IgG antibodies used as secondary antibodies. Peroxi-dase activity on polyvinylidene difluoride membrane was visual-ized on X-ray film by means of the ECL Western blottingdetection system.
Small interfering RNA transfection
The cells (35-mm diameter dishes) were seeded in DMEM con-taining 10% FBS and sub-cultured for 72 h. Predesigned small inter-fering RNAs (siRNAs) targeting rat AMPK-a (Prkaa1–5), and thenegative control-siRNA (Silencer� Negative Control No.1) werepurchased from Qiagen (Venlo, Netherlands), and Life technologiesCo. (Carlsbad, CA), respectively. Transfection was performedaccording to the manufacturer’s protocol (Bio-Rad, Tokyo, Japan).In brief, 5 ll of siLentFect (Bio-Rad, Tokyo, Japan) and finally30 nM of AMPK-a-siRNA or negative control-siRNA were dilutedwith serum-free DMEM, preincubated at room temperature for20 min. Cells were incubated at 37 �C for 72 h with siRNA–siLent-Fect complexes and subsequently harvested for cell migration as-say or exchanged to serum-free DMEM for Western blot analysis.
Statistical analysis
The data were analyzed by ANOVA followed by Bonferroni’smethod for multiple comparisons between pairs. P < 0.05 was con-sidered to be significant. All data are presented as the mean ± SD oftriplicate determinations from three independent cell prepara-tions. Each experiment was repeated three times with similarresults.
84 M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92
Author's personal copy
Results
Effect of PDGF-BB on AMPK-a phosphorylation in A10 cells
It has been reported that PDGF-BB induces migration of vascu-lar smooth muscle A10 cells [13,19]. We confirmed that when thecells were exposed to PDGF-BB, A10 cells significantly migrated,compared with unstimulated A10 cells in a time-dependent man-ner. The stimulatory effect of PDGF-BB was observed at 6 h, in-creased up to 12 h, and decreased thereafter (date not shown).To investigate whether AMPK-a is activated by PDGF-BB in vascu-lar smooth muscle A10 cells, we first examined the effect of PDGF-BB on phosphorylation of AMPK-a in A10 cells. We found thatPDGF-BB markedly induced phosphorylation of AMPK-a at Thr-172 residue in time-dependent manner. Phosphorylation ofAMPK-a was markedly increased at 3 min after the stimulation un-til 5 min and downregulated thereafter (Fig. 1).
Effect of compound C on PDGF-BB-induced A10 cell migration
Next, we investigated the involvement of AMPK on PDGF-BB-in-duced migration of A10 cells. Compound C, an inhibitor of AMPK
[20,21], which alone did not affect the cell migration, significantlysuppressed PDGF-BB-induced A10 cell migration (Fig. 2A, panel 4).The numbers of migrated cells were significantly decreased bycompound C in concentration-dependent manner (Fig. 2B). Theinhibitory effect of compound C was observed between 0.01 and1 lM.
Effect of compound C on PDGF-BB-induced acetyl-CoA carboxylasephosphorylation in A10 cells
It is generally recognized that acetyl-CoA carboxylase, whichregulates lipid synthesis, is a direct downstream substance ofAMPK and phosphorylated [22]. To investigate whether compoundC truly inhibits AMPK activity in A10 cells, we examined the effectof compound C on phosphorylation of PDGF-BB-induced acetyl-CoA carboxylase in A10 cells. PDGF-BB stimulated phosphorylationof acetyl-CoA carboxylase in a time-dependent manner (Fig. 3A).The phosphorylation was remarkably observed at 3 min, and con-tinued up to 5 min and decreased thereafter (Fig. 3A). CompoundC markedly suppressed acetyl-CoA carboxylase phosphorylationinduced by PDGF-BB (Fig. 3B). Therefore, it seems that compound
Fig. 1. Effect of PDGF-BB on phosphorylation of AMPK-a in A10 cells. The cultured cells were stimulated by 30 ng/ml PDGF-BB for the indicated period. The extracts of cellswere subjected to SDS–PAGE with subsequent Western blot analysis with antibodies against phospho-specific AMPK-a (Thr-172) or GAPDH. Similar results were obtainedwith two additional and different cell preparations. The histogram shows quantitative representations of the levels of PDGF-BB-induced phosphorylation of AMPK-a obtainedfrom laser densitometric analysis.
A B
Fig. 2. Effect of compound C on PDGF-BB induced migration of A10 cells. (A) Fluorescence microscopy of the migrated cells. The cells were pretreated with 0.1 lM compoundC (panels 3 and 4) or vehicle (panels 1 and 2) for 1 h and stimulated by 30 ng/ml PDGF-BB (panels 2 and 4) or vehicle (panels 1 and 3) for 9 h. The migrated cells were fixedwith paraformaldehyde, and stained with DAPI for nucleus (blue signal). The cells were photographed by fluorescent microscopy at a magnification of 20�. (B) The cells werepretreated with various concentrations of compound C for 1 h and then stimulated by 30 ng/ml PDGF-BB (d) or vehicle (s) for 9 h. Each value represents the mean ± SD oftriplicate independent determinations of a representative experiment carried out three times. Similar results were obtained with two additional and different cellpreparations. ⁄P < 0.05, compared to the value of PDGF-BB alone. (For interpretation of the references to color in this figure legend, the reader is referred to the web version ofthis article.)
M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92 85
Author's personal copy
C truly reduces PDGF-BB-stimulated AMPK activation in aorticsmooth muscle A10 cells.
Effect of AMPK-a-siRNA on PDGF-BB-induced A10 cell migration
Furthermore, to verify the role of AMPK in PDGF-BB-inducedA10 cell migration, we investigated the effect of AMPK-a downreg-ulation by AMPK-a-siRNA on PDGF-BB-induced cell migration. Wefound that the transfection with 30 nM AMPK-a-siRNA reducedAMPK-a protein level (Fig. 4A). The PDGF-induced migration inAMPK-a downregulated A10 cells was significantly suppressed in
comparison with that in negative control-siRNA-transfected cells(Fig. 4B).
Effects of compound C on PDGF-BB-induced phosphorylation of thep44/p42 MAP kinase pathway or the PI3-kinase/Akt pathway in A10cells
The MAP kinase superfamily, such as p38 MAP kinase, p44/p42MAP kinase and SAPK/JNK, plays an important role in transducingextracellular signaling into a variety of cellular responses [23]. Ithas been reported that PDGF-BB stimulates VSMC migration via
A
B
Fig. 3. Effect of PDGF-BB on phosphorylation of acetyl-CoA carboxylase and effect of compound C on PDGF-BB-induced phosphorylation of acetyl-CoA carboxylase in A10cells. (A) The cultured cells were stimulated by 30 ng/ml PDGF-BB for the indicated period. (B) The cultured cells were pretreated with various concentrations of compound Cfor 1 h and then stimulated by 30 ng/ml PDGF-BB or vehicle for 3 min. The extracts of cells were subjected to SDS–PAGE with subsequent Western blot analysis withantibodies against phospho-specific acetyl-CoA carboxylase or GAPDH. Similar results were obtained with two additional and different cell preparations. The histogramshows quantitative representations of the levels of PDGF-BB-induced phosphorylation of acetyl-CoA carboxylase obtained from laser densitometric analysis.
A B
Fig. 4. Effect of AMPK-a-siRNA on PDGF-BB-induced migration of A10 cells. The cells were transfected with 30 nM of AMPK-a-siRNA or negative control-siRNA at 37 �C for72 h in serum-free DMEM. (A) The extracts of cells were subjected to SDS–PAGE with subsequent Western blot analysis with antibodies against AMPK-a or GAPDH. Thehistogram shows quantitative representations of the levels of AMPK-a obtained from laser densitometric analysis. (B) The cells were harvested, seeded and stimulated by30 ng/ml PDGF-BB or vehicle for 9 h. Cell migration assay was performed. Each value represents the mean ± SD of triplicate independent determinations of a representativeexperiment carried out three times. Similar results were obtained with two additional and different cell preparations. ⁄P < 0.05, compared to the value of the unstimulatedcells. ⁄⁄P < 0.05, compared to the value of the stimulated cells in negative control-siRNA transfected cells.
86 M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92
Author's personal copy
p44/p42 MAP kinase activation [12]. It is well known that p44/p42MAP kinase is phosphorylated by MEK1/2, direct proximal kinaseof p44/p42 MAP kinase as a MAP kinase kinase, and then activated[24]. MEK1/2 is in turn regulated by c-Raf, as a MAP kinase [24,25].We confirmed that PDGF-BB induced phosphorylation of c-Raf,MEK1/2 and p44/p42 MAP kinase in A10 cells in a time dependentmanner. The phosphorylation of these kinases by PDGF-BB reachedthe peak at 10 min after stimulation (Fig. 5). In order to investigatethe relationship of AMPK and the p44/p42 MAP kinase pathway inPDGF-BB-induced A10 cell migration, we examined the effect of
compound C on PDGF-BB-induced phosphorylation of c-Raf,MEK1/2 or p44/p42 MAP kinase. Compound C markedly sup-pressed PDGF-BB-induced phosphorylation of c-Raf, MEK1/2 andp44/p42 MAP kinase (Fig. 6A–C).
In addition to the p44/p42 MAP kinase pathway, it has beenshown that the PI3-kinase/Akt pathway plays a positive role inVSMC migration [26,27]. PI3-kinase induces the translocation ofAkt to plasma membrane thorough generation of PI-3,4,5-triphos-phate, where Akt is phosphorylated at Thr-308 and Ser-473 resi-dues, and then activated [28]. It has been reported that PDGF-BB
Fig. 5. Effect of PDGF-BB on phosphorylation of the p44/p42 MAP kinase pathway. The cultured cells were stimulated by 30 ng/ml PDGF-BB for the indicated period. Theextracts of cells were subjected to SDS–PAGE with subsequent Western blot analysis with antibodies against phospho-specific c-Raf, phospho-specific MEK1/2, phospho-specific p44/p42 MAP kinase or GAPDH. Similar results were obtained with two additional and different cell preparations. The histogram shows quantitative representationsof the levels of PDGF-BB-induced phosphorylation of c-Raf, MEK1/2 and p44/p42 MAP kinase obtained from laser densitometric analysis.
A
B
C
Fig. 6. Effects of compound C on PDGF-BB-induced phosphorylation of (A) c-Raf, (B) MEK1/2, (C) p44/p42 MAP kinase in A10 cells. The cultured cells were pretreated withvarious concentrations of compound C for 1 h and then stimulated by 30 ng/ml PDGF-BB for 10 min. The extracts of cells were subjected to SDS–PAGE with subsequentWestern blot analysis with antibodies against phospho-specific c-Raf, GAPDH, phospho-specific MEK1/2, MEK1/2, phospho-specific p44/p42 MAP kinase or p44/p42 MAPkinase. Similar results were obtained with two additional and different cell preparations. The histogram shows quantitative representations of the levels of PDGF-BB-inducedphosphorylation of c-Raf, MEK1/2 and p44/p42 MAP kinase obtained from laser densitometric analysis.
M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92 87
Author's personal copy
stimulates human pulmonary VSMC or human aortic SMC migra-tion through PI3-kinase activation [26,27]. In A10 cells, it has beenreported that PDGF-BB stimulates the activation of PI3-kinase andAkt [29]. We confirmed that PDGF-BB induced phosphorylation ofPI3-kinase and Akt at Thr-308 and Ser-473 residues in A10 cells ina time dependent manner. PDGF-BB-induced phosphorylation ofPI3-kinase or Akt reached the peak at 10 min (Fig. 7). In order toinvestigate the relationship of AMPK and the PI3-kinase/Aktpathway in PDGF-BB-induced A10 cell migration, we examined
the effect of compound C on phosphorylation of PI3-kinase orAkt. Compound C remarkably reduced PDGF-BB-induced PI3-ki-nase phosphorylation level (Fig. 8A) and attenuated phosphoryla-tion level of Akt at Thr-308 and Ser-473 residues (Fig. 8B).
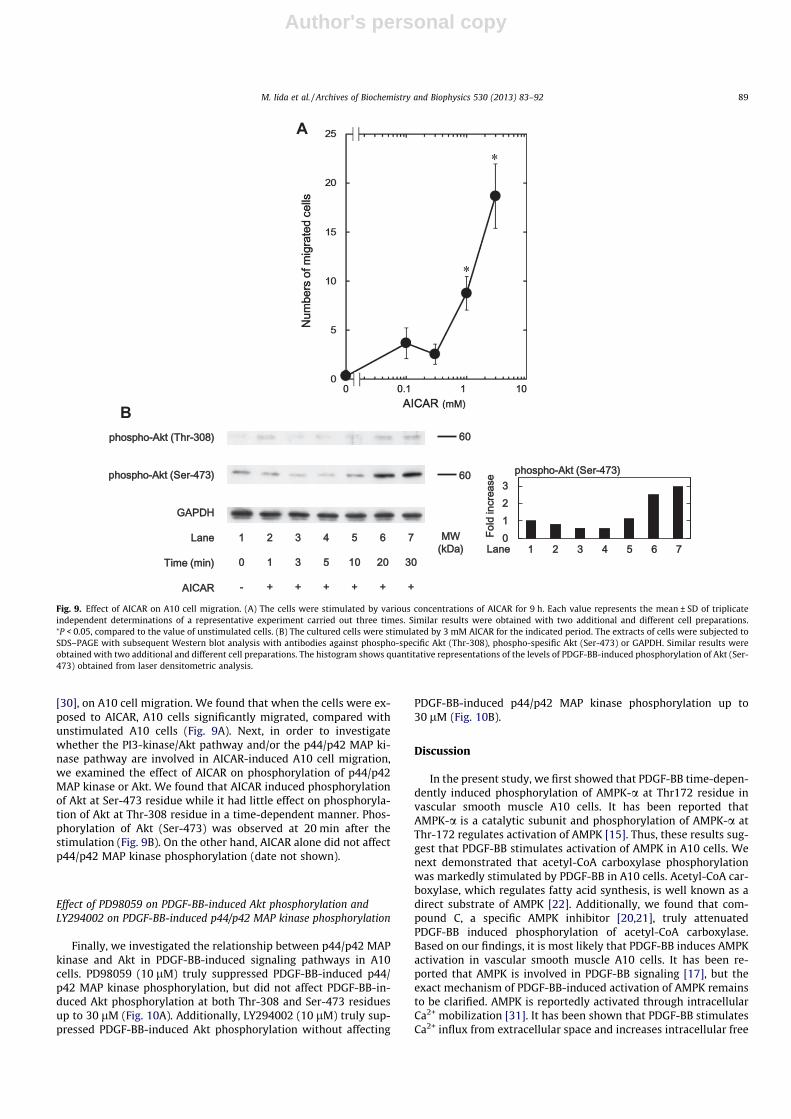
Effect of AICAR on A10 cell migration
To investigate whether AMPK activation induces A10 cellmigration, we examined the effect of AICAR, an activator of AMPK
Fig. 7. Effect of PDGF-BB on phosphorylation of the PI3-kinase/Akt pathway. The cultured cells were stimulated by 30 ng/ml PDGF-BB for the indicated period. The extracts ofcells were subjected to SDS–PAGE with subsequent Western blot analysis with antibodies against phospho-specific PI3-kinase, phospho-specific Akt (Thr-308), phospho-spesific Akt (Ser-473) or GAPDH. Similar results were obtained with two additional and different cell preparations. The histogram shows quantitative representations of thelevels of PDGF-BB-induced phosphorylation of PI3-kinase, Akt (Thr-308) and Akt (Ser-473) obtained from laser densitometric analysis.
A
B
Fig. 8. Effects of compound C on PDGF-BB-induced phosphorylation of (A) PI3-kinase or (B) Akt in A10 cells. The cultured cells were pretreated with various concentrations ofcompound C for 1 h and then stimulated by 30 ng/ml PDGF-BB for 10 min. The extracts of cells were subjected to SDS–PAGE with subsequent Western blot analysis withantibodies against phospho-specific PI3-kinase, GAPDH, phospho-specific Akt (Thr-308), phospho-spesific Akt (Ser-473) or Akt. Similar results were obtained with twoadditional and different cell preparations. The histogram shows quantitative representations of the levels of PDGF-BB-induced phosphorylation of PI3-kinase, Akt (Thr-308)and Akt (Ser-473) obtained from laser densitometric analysis.
88 M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92
Author's personal copy
[30], on A10 cell migration. We found that when the cells were ex-posed to AICAR, A10 cells significantly migrated, compared withunstimulated A10 cells (Fig. 9A). Next, in order to investigatewhether the PI3-kinase/Akt pathway and/or the p44/p42 MAP ki-nase pathway are involved in AICAR-induced A10 cell migration,we examined the effect of AICAR on phosphorylation of p44/p42MAP kinase or Akt. We found that AICAR induced phosphorylationof Akt at Ser-473 residue while it had little effect on phosphoryla-tion of Akt at Thr-308 residue in a time-dependent manner. Phos-phorylation of Akt (Ser-473) was observed at 20 min after thestimulation (Fig. 9B). On the other hand, AICAR alone did not affectp44/p42 MAP kinase phosphorylation (date not shown).
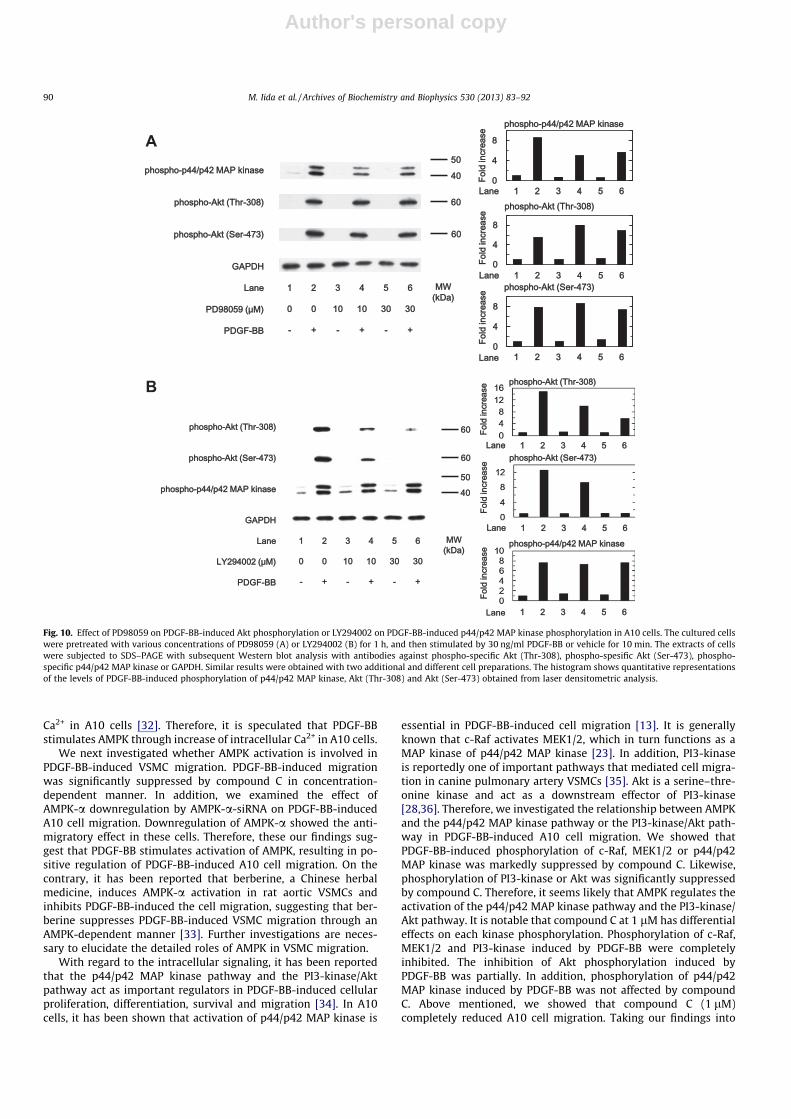
Effect of PD98059 on PDGF-BB-induced Akt phosphorylation andLY294002 on PDGF-BB-induced p44/p42 MAP kinase phosphorylation
Finally, we investigated the relationship between p44/p42 MAPkinase and Akt in PDGF-BB-induced signaling pathways in A10cells. PD98059 (10 lM) truly suppressed PDGF-BB-induced p44/p42 MAP kinase phosphorylation, but did not affect PDGF-BB-in-duced Akt phosphorylation at both Thr-308 and Ser-473 residuesup to 30 lM (Fig. 10A). Additionally, LY294002 (10 lM) truly sup-pressed PDGF-BB-induced Akt phosphorylation without affecting
PDGF-BB-induced p44/p42 MAP kinase phosphorylation up to30 lM (Fig. 10B).
Discussion
In the present study, we first showed that PDGF-BB time-depen-dently induced phosphorylation of AMPK-a at Thr172 residue invascular smooth muscle A10 cells. It has been reported thatAMPK-a is a catalytic subunit and phosphorylation of AMPK-a atThr-172 regulates activation of AMPK [15]. Thus, these results sug-gest that PDGF-BB stimulates activation of AMPK in A10 cells. Wenext demonstrated that acetyl-CoA carboxylase phosphorylationwas markedly stimulated by PDGF-BB in A10 cells. Acetyl-CoA car-boxylase, which regulates fatty acid synthesis, is well known as adirect substrate of AMPK [22]. Additionally, we found that com-pound C, a specific AMPK inhibitor [20,21], truly attenuatedPDGF-BB induced phosphorylation of acetyl-CoA carboxylase.Based on our findings, it is most likely that PDGF-BB induces AMPKactivation in vascular smooth muscle A10 cells. It has been re-ported that AMPK is involved in PDGF-BB signaling [17], but theexact mechanism of PDGF-BB-induced activation of AMPK remainsto be clarified. AMPK is reportedly activated through intracellularCa2+ mobilization [31]. It has been shown that PDGF-BB stimulatesCa2+ influx from extracellular space and increases intracellular free
A
B
Fig. 9. Effect of AICAR on A10 cell migration. (A) The cells were stimulated by various concentrations of AICAR for 9 h. Each value represents the mean ± SD of triplicateindependent determinations of a representative experiment carried out three times. Similar results were obtained with two additional and different cell preparations.⁄P < 0.05, compared to the value of unstimulated cells. (B) The cultured cells were stimulated by 3 mM AICAR for the indicated period. The extracts of cells were subjected toSDS–PAGE with subsequent Western blot analysis with antibodies against phospho-specific Akt (Thr-308), phospho-spesific Akt (Ser-473) or GAPDH. Similar results wereobtained with two additional and different cell preparations. The histogram shows quantitative representations of the levels of PDGF-BB-induced phosphorylation of Akt (Ser-473) obtained from laser densitometric analysis.
M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92 89
Author's personal copy
Ca2+ in A10 cells [32]. Therefore, it is speculated that PDGF-BBstimulates AMPK through increase of intracellular Ca2+ in A10 cells.
We next investigated whether AMPK activation is involved inPDGF-BB-induced VSMC migration. PDGF-BB-induced migrationwas significantly suppressed by compound C in concentration-dependent manner. In addition, we examined the effect ofAMPK-a downregulation by AMPK-a-siRNA on PDGF-BB-inducedA10 cell migration. Downregulation of AMPK-a showed the anti-migratory effect in these cells. Therefore, these our findings sug-gest that PDGF-BB stimulates activation of AMPK, resulting in po-sitive regulation of PDGF-BB-induced A10 cell migration. On thecontrary, it has been reported that berberine, a Chinese herbalmedicine, induces AMPK-a activation in rat aortic VSMCs andinhibits PDGF-BB-induced the cell migration, suggesting that ber-berine suppresses PDGF-BB-induced VSMC migration through anAMPK-dependent manner [33]. Further investigations are neces-sary to elucidate the detailed roles of AMPK in VSMC migration.
With regard to the intracellular signaling, it has been reportedthat the p44/p42 MAP kinase pathway and the PI3-kinase/Aktpathway act as important regulators in PDGF-BB-induced cellularproliferation, differentiation, survival and migration [34]. In A10cells, it has been shown that activation of p44/p42 MAP kinase is
essential in PDGF-BB-induced cell migration [13]. It is generallyknown that c-Raf activates MEK1/2, which in turn functions as aMAP kinase of p44/p42 MAP kinase [23]. In addition, PI3-kinaseis reportedly one of important pathways that mediated cell migra-tion in canine pulmonary artery VSMCs [35]. Akt is a serine–thre-onine kinase and act as a downstream effector of PI3-kinase[28,36]. Therefore, we investigated the relationship between AMPKand the p44/p42 MAP kinase pathway or the PI3-kinase/Akt path-way in PDGF-BB-induced A10 cell migration. We showed thatPDGF-BB-induced phosphorylation of c-Raf, MEK1/2 or p44/p42MAP kinase was markedly suppressed by compound C. Likewise,phosphorylation of PI3-kinase or Akt was significantly suppressedby compound C. Therefore, it seems likely that AMPK regulates theactivation of the p44/p42 MAP kinase pathway and the PI3-kinase/Akt pathway. It is notable that compound C at 1 lM has differentialeffects on each kinase phosphorylation. Phosphorylation of c-Raf,MEK1/2 and PI3-kinase induced by PDGF-BB were completelyinhibited. The inhibition of Akt phosphorylation induced byPDGF-BB was partially. In addition, phosphorylation of p44/p42MAP kinase induced by PDGF-BB was not affected by compoundC. Above mentioned, we showed that compound C (1 lM)completely reduced A10 cell migration. Taking our findings into
A
B
Fig. 10. Effect of PD98059 on PDGF-BB-induced Akt phosphorylation or LY294002 on PDGF-BB-induced p44/p42 MAP kinase phosphorylation in A10 cells. The cultured cellswere pretreated with various concentrations of PD98059 (A) or LY294002 (B) for 1 h, and then stimulated by 30 ng/ml PDGF-BB or vehicle for 10 min. The extracts of cellswere subjected to SDS–PAGE with subsequent Western blot analysis with antibodies against phospho-specific Akt (Thr-308), phospho-spesific Akt (Ser-473), phospho-specific p44/p42 MAP kinase or GAPDH. Similar results were obtained with two additional and different cell preparations. The histogram shows quantitative representationsof the levels of PDGF-BB-induced phosphorylation of p44/p42 MAP kinase, Akt (Thr-308) and Akt (Ser-473) obtained from laser densitometric analysis.
90 M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92

Author's personal copy
account, it is possible that activation of Akt and p44/p42 MAP ki-nase are regulated also by other signal pathway in addition toAMPK in A10 cells.
As for the time dependent effect of PDGF in the present study,we showed that maximum effects of PDGF-BB on phosphorylationof AMPK-a (Thr-172) and acetyl-CoA carboxylase reached its peakat 3 min or 5 min after the stimulation. Moreover, we found thatPDGF-BB induced phosphorylation of c-Raf, MEK1/2, p44/p42MAP kinase, PI3-kinase and Akt reached the peak at 10 min afterthe stimulation. The time course of PDGF-BB-induced phosphory-lation of AMPK seems to be faster than those of c-Raf, MEK1/2,p44/p42 MAP kinase, PI3-kinase and Akt. Therefore, it seems rea-sonable that PDGF-BB-induced activation of the p44/p42 MAP ki-nase pathway and the PI3-kinase/Akt pathway subsequentlyoccurs after activation of AMPK in vascular smooth muscle A10cells. Based on our findings, it is most likely that AMPK functionsat a point upstream from c-Raf and PI3-kinase, and positively reg-ulates PDGF-BB-induced A10 cell migration. It has been reportedthat treatment with AICAR, an activator of AMPK [30], inhibits ser-um-induced VSMC migration [16]. Thus, we investigated how AI-CAR affects A10 cell migration, and phosphorylation of Akt orp44/p42 MAP kinase. AICAR alone significantly induced A10 cellmigration. Furthermore, AICAR induced phosphorylation of Akt(Ser-473) but not p44/p42 MAP kinase. Based on our findings, itis probable that the activation of AMPK acts as a positive regulatorin A10 cell migration. We additionally demonstrated that PD98059did not affect PDGF-BB-induced Akt phosphorylation, andLY294002 had no effect on PDGF-BB-induced p44/p42 MAP kinasephosphorylation. Therefore, it is most likely that the p44/p42 MAPkinase pathway and the PI3-kinase/Akt pathway regulate PDGF-BB-induced migration of A10 cells independently of one another.The potential mechanism of AMPK activation in PDGF-BB-inducedA10 cell migration shown here is summarized in Fig. 11. Furtherinvestigations are necessary to clarify the exact mechanisms howAMPK activation and the phosphorylation of p44/p42 MAP kinaseand PI3-kinase/Akt pathway related behind PDGF-BB stimulation.And elucidation of the mechanism by which PDGF-BB activatesAMPK needs further exploration.
It is recognized that PDGF-BB is expressed at very low levels innormal vessels [8]. However, vascular injury occurs clinically afterangioplasty, vascular stent implantation, by-pass graft surgery or
organ transplantation [3,4]. In response to vascular injury, VSMCsdramatically increase its rate of cell proliferation, migration, andsynthetic capacity and play a pivotal role in vascular repair [2].PDGF-BB is a potent growth factor released from various cells afterinjury and induces VSMC migration [8]. PDGF-BB-induced VSMCmigration is an essential element during in the neovascular re-sponse [3]. Accordingly, PDGF-BB-induced VSMC migration playsimportant role in the repair of vessel injury with tissue damage[2]. Under certain pathological conditions, PDGF-BB-induced VSMCmigration might lead to cardiovascular disease, such as atheroscle-rosis and restenosis after angioplasty, stent implantation and by-pass graft surgery [8]. Thus, the blockade of PDGF-BB action inVSMC migration could prevent atherosclerosis and restenosis aftersurgical or catheter intervention depends on the condition. It isgenerally recognized that AMPK is a major regulator of cellularand whole-body energy homeostasis that coordinates metabolicpathway in order to balance nutrient supply with energy demand[14]. In vascular system, it has been reported that serum-inducedmigration of rat aortic VSMC, which are pretreated with AICAR,an AMPK activator, is suppressed [16]. In the present study, wedemonstrated that AMPK acts as positive regulator in the PDGF-BB-induced VSMC migration. Therefore, AMPK might be consid-ered as a therapeutic target for patient with atherosclerosis andafter surgical or catheter intervention. Based on our findings, it ispossible that AMPK plays an essential role in proper vascularremodeling. That is to say, there is possibility that inhibition ofAMPK activation is preventing to atherosclerosis and restenosisafter intervention depends on the condition.
In conclusion, our results strongly suggest that PDGF-BB in-duces activation of AMPK in VSMCs and subsequently regulatesVSMC migration via the p44/42 MAP kinase pathway and thePI3-kinase/Akt pathway, independently.
Acknowledgments
We are very grateful to Emiko Fuseya and Yumiko Kurokawa fortheir skillfull technical assistance. This work was supported in partby a Grant-in-Aid for Scientific Research (No. 23592247) from theMinistry of Education, Science, Sports and Culture of Japan.
References
[1] S.A. Fisher, Physiol. Genomics 42A (2010) 169–187.[2] G.K. Owens, M.S. Kumar, B.R. Wamhoff, Physiol. Rev. 84 (2004) 767–801.[3] A.I. Willis, D. Pierre-Paul, B.E. Sumpio, V. Gahtan, Vasc. Endovascular Surg. 38
(2004) 11–23.[4] W.T. Gerthoffer, Circ. Res. 100 (2007) 607–621.[5] K. Pierras, T. Sjöblom, K. Rubin, C.H. Heldin, A. Östman, Cancer Cell 3 (2003)
439–443.[6] D.F. Bowen-Pope, E.W. Raines, Arterioscler. Thromb. Vasc. Biol. 31 (2011)
2397–2401.[7] J. Andrae, R. Gallini, C. Betsholtz, Genes Dev. 22 (2008) 1276–1312.[8] E.W. Raines, Cytokine Growth Factor Rev. 15 (2004) 237–254.[9] A.C. Newby, Toxicol. Lett. 112–113 (2000) 519–529.
[10] N.S.M. Azahri, B.A.D. Bartolo, L.M. Khachigian, M.M. Kavurma, J. Cell. Biochem.113 (2012) 2597–2606.
[11] L. Pukac, J. Huangpu, M.J. Karnovsky, Exp. Cell Res. 242 (1998) 548–560.[12] Y. Zhan, S. Kim, Y. Izumi, Y. Izumiya, T. Nakao, H. Miyazaki, H. Iwao,
Arterioscler. Thromb. Vasc. Biol. 23 (2003) 795–801.[13] H.M. Lo, C.F. Hung, Y.L. Tseng, B.H. Chen, J.S. Jian, W.B. Wu, Biochem.
Pharmacol. 74 (2007) 54–63.[14] S. Fogarty, D.G. Hardie, Biochim. Biophys. Acta 1804 (2010) 581–591.[15] S.A. Hawley, M. Davison, A. Woods, S.P. Davies, R.K. Beri, D. Carling, D.G.
Hardie, J. Biol. Chem. 271 (1996) 27879–27887.[16] K.J. Peyton, Y. Yu, B. Yates, A.R. Shebib, X.M. Liu, H. Wang, W. Durante, J.
Pharmacol. Exp. Ther. 338 (2011) 476–484.[17] M. Igata, H. Motoshima, K. Tsuruzoe, K. Kojima, T. Matsumura, T. Kondo, T.
Taguchi, K. Nakamaru, M. Yano, D. Kukidome, K. Matsumoto, T. Toyonaga, T.Asano, T. Nishikawa, E. Araki, Circ. Res. 97 (2005) 837–844.
[18] U.K. Laemmli, Nature 5259 (1970) 680–685.[19] D.E. Stec, K.P. Gannon, J.S. Beaird, H.A. Drummond, Cell. Physiol. Biochem. 19
(2007) 121–128.
Fig. 11. Schematic representation of the involvement of adenosine monophos-phate-activated protein kinase (AMPK) in the mechanism of platelet-derivedgrowth factor-BB (PDGF-BB)-induced migration in vascular smooth muscle cells(VSMCs). MAP kinase, mitogen-activated protein kinase; MEK, MAP/extracellularsignal-regulated kinase kinase; PI3-kinase, phosphatidylinositol 3-kinase.
M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92 91

Author's personal copy
[20] G. Zhou, R. Myers, Y. Li, Y. Chen, X. Shen, J. Fleyk-Melody, M. Wu, J. Ventre, T.Doebber, N. Fujii, N. Musi, M.F. Hirshman, L.J. Goodyear, D.E. Moller, J. Clin.Invest. 108 (2001) 1167–1174.
[21] J. Bain, L. Plater, M. Elliott, N. Shpiro, C.J. Hastie, H. Mclauchalan, I. Klevernic,J.S. Arthur, D.R. Allessi, P. Cohen, Biochem. J. 408 (2007) 297–315.
[22] D.G. Hardie, D. Carling, A.T.R. Sim, Trends Biochem. Sci. 14 (1989) 20–23.[23] J.M. Kyriakis, J. Avruch, Physiol. Rev. 81 (2001) 807–869.[24] J.M. Kyriakis, H. App, X.F. Zhang, P. Banerjee, D.L. Brautigan, U.R. Rapp, J.
Avruch, Nature 358 (1992) 417–421.[25] F. Chang, L.S. Steelman, J.T. Lee, J.G. Shelton, P.M. Navolanic, W.L. Blalock, R.A.
Franklin, J.A. McCubrey, Leukemia 17 (2007) 1263–1293.[26] E.A. Goncharova, A.J. Ammit, C. Irani, R.G. Carroll, A.J. Eszterhas, R.A. Panettieri,
V.P. Krymskaya, Am. J. Physiol. Lung Cell. Mol. Physiol. 283 (2002) L354–L363.[27] K.H. Choi, J.E. Kim, N.R. Song, J.E. Son, M.K. Hwang, S. Byun, J.H. Kim, K.W. Lee,
H.J. Lee, Cardiovasc. Res. 85 (2010) 836–844.
[28] B.M. Burgering, P.J. Coffer, Nature 376 (1995) 599–602.[29] H. Yamada, T. Tsushima, H. Murakami, Y. Uchigata, Y. Iwamoto, Int. J. Exp.
Diabetes Res. 3 (2002) 131–144.[30] J.M. Corton, J.G. Gillespie, S.A. Hawley, D.G. Hardie, Eur. J. Biochem. 229 (1995)
558–565.[31] G.A. Rutter, I. Leclerc, Mol. Cell. Endocrinol. 297 (2009) 41–49.[32] H.A. Pershadsingh, J. Szollosi, S. Benson, W.C. Hyun, B.G. Feuerstein, T.W. Kurtz,
Hypertension 21 (1993) 1020–1023.[33] K.W. Liang, S.C. Yin, C.T. Ting, S.J. Lin, C.M. Hsueh, C.Y. Chen, S.L. Hsu, Eur. J.
Pharmacol. 590 (2008) 343–354.[34] Y. Dai, Expert Opin. Ther. Patents 20 (2010) 885–907.[35] I.A. Yamboliev, J. Chen, W.T. Gerthoffer, Am. J. Physiol. Cell. Physiol. 281 (2001)
C709–C718.[36] B.M. Marte, J. Downward, Trends Biochem. Sci. 22 (1997) 355–358.
92 M. Iida et al. / Archives of Biochemistry and Biophysics 530 (2013) 83–92





























