Embed Size (px)
Citation preview

J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r . com/ loca te / j p ro t
Review
Advances in top-down proteomics for diseasebiomarker discovery
David Calligaris, Claude Villard, Daniel Lafitte⁎
INSERM UMR 911, Centre de Recherche en Oncologie biologique et en Oncopharmacologie, Aix-Marseille Université, Plateforme Protéomique etInnovation Technologique Timone, Faculté de Pharmacie, 27 Boulevard Jean Moulin, 13385 Marseille Cedex 5, France
A R T I C L E I N F O
⁎ Corresponding author. Tel.: +33 49 183 5680E-mail address: [email protected]
1874-3919/$ – see front matter © 2011 Elsevidoi:10.1016/j.jprot.2011.03.030
A B S T R A C T
Article history:Received 19 January 2011Accepted 29 March 2011Available online 6 April 2011
Top-down mass spectrometry strategies allow identification and characterization ofproteins and protein networks by direct fragmentation. These analytical processesinvolve a panel of fragmentation mechanisms, some of which preserve protein post-translational modifications. Thus top-down is of special interest in clinical biochemistry toprobe modified proteins as potential disease biomarkers. This review describes separatingmethods, mass spectrometry instrumentation, bioinformatics, and theoretical aspects offragmentation mechanisms used for top-down analysis. The biological interest of thisstrategy is extensively reported regarding the characterization of post-translationalmodifications in biochemical pathways and the discovery of biomarkers. One has to bearin mind that quantitative aspects that are beyond the focus of this review are also of criticalimportant for biomarker discovery. The constant evolution of technologiesmakes top-downstrategies crucial players in clinical and basic proteomics.
© 2011 Elsevier B.V. All rights reserved.
Keywords:Top-downFragmentationProteomicsBiomarkersPost-translational modifications
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9212. Top-down workflow. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 921
2.1. Separation and sample handling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9212.2. Mass spectrometry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9232.3. Dedicated search engines for top-down analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 923
3. Fragmentation mechanisms for primary sequence analysis observed during top-down analysis . . . . . . . . . . . . 9253.1. Collision-induced dissociation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9253.2. IRMPD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9253.3. Electron capture dissociation and electron transfer dissociation . . . . . . . . . . . . . . . . . . . . . . . . . 9253.4. Laser-induced dissociation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9283.5. MALDI in-source decay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 928
.(D. Lafitte).
er B.V. All rights reserved.

921J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
4. Contribution of top-down approaches for characterization of biomarkers and biochemical pathways . . . . . . . . 9285. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 930References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 930
Protein separation
Electrophoretic methods
2D gel electrophoresisContinuous-elution electrophoresis, GELFrEEMicrofluidic-based gel protein recovery system
Chromatographic methodsRPLC, HILIC, UHPLC
Ion exchange chromatographyChromatofocusing
Mass spectrometryMALDI-TOF
ISD
Q-TOF LTQ-Orbitrap FTICR
CID HCD IRMPDETD/ECD
Data analyses
ProSight BIG Mascot Spectra alignment approach
Fig. 1 – Overall view of top-down analysis.
1. Introduction
Proteomics approaches embeddingmass spectrometry (MS) arewidely used in biomarker research and discovery to findsignatures for disease diagnosis or prognosis [1,2]. Some ofthese biomarkers have been identified [3]. For example, Bon etal. showed that levels of decarboxyprothrombin and alpha-fetoprotein were increased in hepatocellular carcinoma andcould serve in diagnosing the disease [4]. Another study showedthat CYFRA 21-1 is a predictive factor of survival of non-smallcell lung cancer patients treated with gefitinib [5]. The mostwidely used ionization techniques for analyzing biomoleculesare electrospray ionization (ESI) and matrix-assisted laserdesorption ionization (MALDI) [for a review, see 6]. These softionization modes enable ionization of large biopolymers, suchas proteins, lipids, and nucleic acids. Two complementarystrategies based on ESI or MALDI ionization are used for proteinidentification. These approaches are named "bottom-up" [forreviews, see 7–12] and "top-down" [for reviews, see 13–19]mainly because of the initial digestion or not of the analyte. Inthe case of bottom-up approaches, proteins of interest undergoenzymatic digestion before MS analysis. This digestion can beperformed directly in solution, after separation by 1D- or 2D-gelelectrophoresis, but also by direct digestion on tissues. Peptidesobtained by enzymatic digestion, generally with a mass below3500 Da, are then directly analyzed by MALDI MS or ESI. Proteinidentity is then determined by the comparison, in this case, ofpeptide masses from MS analyses (peptide mass fingerprint byMALDI-TOF [for a review, see 20]) or MS/MS (tandem massspectrometry [for a review, see 21]), with peptide theoreticalmasses derived from proteomic or genomic databases. Thisapproach, the most widely used serves well for analyzingcomplex samples. However, separation by mono or bidimen-sional HPLC is needed to concentrate and separate peptidesissued from protein digestion. This strategy has two maindisadvantages. It is time-consumingbecauseproteinsmust firstbe digested, and it is limited because the protein sequence israrely exhaustively covered [9,22]. Moreover, ion signals for N-and C-termini fragments are not oftenmeasured, and fragmen-tation processes commonly used during bottom-up approachesinduce loss of labile post-translational modifications (PTMs)(phosphorylation, glycosylation…), which are potential bio-markers in clinical proteomics.
Top-down approaches seem more appropriate to directlyobtain, without preliminary digestion, protein N and C-termini.This strategy is often compared to Edman sequencing. Molec-ular ions from intact proteins are obtained by MALDI or ESIionization and fragmented by MS/MS. When ESI-MS is theionization method, Electron Capture Dissociation (ECD) andElectronTransferDissociation (ETD) [for a review, see 23] are themost suitable fragmentation techniques for top-down analysis.They are used for gas phase fragmentation involving electroncapture by an analyte to form a free radical with high energy-inducing peptides/proteins N–Cα bond cleavage and preserving
labile PTMs upon electron transfer. The fragmentation can alsobe achieved by collision of analyte molecules with an inert gassuch as heliumor argon. This process, which promotes a higherenergy pathway than ECD and ETD, is called Collision-InducedDissociation (CID) and often leads to the loss of labile PTMs [24].This dissociation can be achieved by another process such asphoto-fragmentation, which is induced by absorption of mul-tiple infrared photons (IRMPD for Infrared Multiphoton Disso-ciation). MALDI top-down is also possible by a relatively oldstrategy called In-Source Decay (ISD) updated for protein by theuseofMALDI-TOF/TOFmass spectrometers [for a review, see 25]with pseudo MS3 strategies.
In this review, we investigate the various top-down strate-gies available for protein biochemistry and their importance inthe clinical field (Fig. 1). We focus on the workflow, whichbasically includes instrumentation and bioinformatics, andtheoretical aspects of fragmentation processes (Table 1). Then,we examine the importance of top-down for the characteriza-tion of biomarkers and discuss how to improve top-downanalysis for in situ characterization of biopolymers.
2. Top-down workflow
2.1. Separation and sample handling
For top-down analysis, samples must often be made lesscomplex. Electrophoretic or chromatographic methods serveto separate proteins prior to top-down analysis [26,27] (Table 1).

Table 1 – Top-down proteomics workflow.
Proteins separation Electrophoretic methods 2D gel electrophoresis [30]Continuous-elution electrophoresis [30]Microfluidic-based gel protein recoverysystem
[31]
Gel-eluted liquid fraction entrapmentelectrophoresis (GELFrEE)
[40]
Chromatographic methods Reverse phase liquid chromatography(RPLC)
[30,32,33,35,104,106–108,110,111,114]
Hydrophilic interactionchromatographic (HILIC)
[34,37,111]
Ion exchange chromatography [36]Chromatofocusing [38,39]Ultra-high performance liquidchromatography (UHPLC)
[34]
Mass spectrometry Instruments ESI-triple quadrupole [42,43]Hybrid quadrupole TOF [44,45,106,114]LTQ-Orbitrap [33,48,61,62,104,106,119]FTICR [35,49,105,109]Hybrid quadrupole-FTICR [30,33,110,111]LTQ-FTICR [107,108]MALDI-TOF [50–54,87,89–105,112–120,124]
Fragmentation Collision-induced dissociation (CID) [63–75,105–107,119]Higher energy collisionally activateddissociation (HCD)
[61,62]
Infrared multiphoton dissociation (IRMPD) [76–78,105,109,111]Electron capture dissociation (ECD) [79–85,105,108,109,111],Electron transfer dissociation (ETD) [86,117]In-source decay (ISD) [50–54,87,89–100,112,114,120,124]
Data analyses Bioinformatics ProSight [56,57,104,107]BIG mascot [59]Spectra alignment approach [60]
922 J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
In the case of mono or bidimensional electrophoresis, whichseparate proteins on the basis of their molecular weight andisoelectric point, the recovery of the proteins from gel spots isdifficult and often leads to protein fragmentation [28]. Thisrecovery performed via passive elution [29] or gel electroelutionis not readily compatible with ESI-MS because of the chemicalsused for protein extraction. Despite these technical limitations,Meng et al. characterized more than 400 proteins by massspectrometry after separation by 2D gel electrophoresis and off-line recovery into pools of 5–20 proteins. Proteins wereseparated in the first dimension by continuous-elution electro-phoresis, replacing SDS by an acid-labile surfactant (ALS "Acid-Labile Surfactant"), and then by Reverse Phase Liquid Chroma-tography (RPLC) in the second dimension. Characterization ofintact protein ions is done by on-line analysis with a 9.4Tquadrupole-Fourier Transform MS hybrid instrument [30]. Toovercome this problem, Hydrophilic Interaction Chromatogra-phy (HILIC) can also be performed after sample elution prior toMS. A new microfluidic-based gel protein recovery system,which uses a proprietary plastic microfluidic chip in a highvoltage environment, allows extraction from 10% “ALS-PAGE”gels of proteins with molecular weights between 6 and 65 kDaand is compatible with MALDI and ESI mass spectrometry [31].Chromatography is an alternative to electrophoreticmigrationsfor fractionation of intact proteins and top-down analysis. Thisseparation strategy can be done on-line with tandem massspectrometry using High Performance Liquid Chromatography(HPLC) or off-line with infusion of separated fractions of intactproteins. RPLC is themost often used stationary phase inmono
dimensional HPLC. Wang et al. explored parameters like alkylchain length, capillary temperature, or ion-pairing agent toenhance chromatographic resolution in intact protein separa-tion with reverse phase column [32]. They found that the bestconditions involved the use of C18 column heated to 60 °C andaddition of trifluoroacetic acid as ion-pairing agent. Theseoptimized parameters allow the same separation performanceof yeast cell lysates in their native or denatured form. Mono-dimensional separation does not provide enough peak capacityto resolve complex protein mixtures. However, Pesavento et al.used RPLC andmetabolic labeling coupled with high resolutionelectrospray mass spectrometry to separate protein isoforms,revealing progressive methylation of lysine20 in Histone-4during cell cycle [33]. A fast, automated, on-line methodinvolving Ultra-High Performance Liquid Chromatography(UHPLC) andmass spectrometrywas reported to profile histonemodifications and variants from human fibroblast [34]. Intactproteins separatedby reversephase capillary columncoupled toa Fourier Transform Ion Cyclotron Resonance (FTICR) massspectrometer revealed the presence of over 20 casein isoformsarising from genetic variants with different numbers ofphosphorylation sites [35]. To increase peak capacity and toresolve more complex mixtures of intact proteins, 2D chro-matographic strategies are coupled to high resolution massspectrometer detection. Reverse phase, highly compatible withelectrospray, is the second dimension of choice whether ionexchange [36], hydrophilic interaction chromatography (HILIC)[34,37], or chromatofocusing. [38,39] is used in first dimension.Another attractive and recent techniquecalledGELFrEE for "Gel-

923J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
Eluted Liquid Fraction Entrapment Electrophoresis" is used fortop-down analysis. The equipment includes a precision-castgel column surrounded by fraction collection chambers.Charged molecules are separated according to their molecu-lar weight. Proteins are then eluted into the fractionationcollection chamber where they are trapped in a defined liquidvolume. This technique allows rapid fractionation of intactproteins from 3.5 to 250 kDa [40]. Ionmobility devices are newseparating devices with the advantage of direct coupling tomass spectrometers [for a review, see 41]. This techniqueallows separation of ions in the gas phase not only by theirmass but also their shape. Isobaric proteins or peptides cantherefore be analyzed separately, which is not possible usingconventional mass spectrometry. Although there are noreports on coupling of ion mobility separation and top-down, this new technology could rapidly emerge as a powerfultool in the field.
2.2. Mass spectrometry
Early top-down analyses have been done on mass spectrom-eters equipped with quadrupole analyzers. Loo et al. studied a14-kDa protein such as ribonuclease A by using ESI-triplequadrupolemass spectrometers [42]. Feng et al. used the sametype of instrument for the analysis of larger proteins, such asan antibody (150 kDa) [43]. Nemeth-Cawley et al. used a hybridquadrupole TOF instrument to characterize a recombinant,immunoglobulin gamma-1 (IgG-1) fusion protein [44]. Al-though this study allowed isotopic mass assignment offragment ions with mass above 10 kDa, low resolution(between 8000 and 12,000) and accuracy (between 5 and20 ppm) limits the study of intact proteins to a mass of35 kDa. Instrument optimization is necessary to characterizeproteins of higher molecular weight. As an example, modifi-cation of the ion guide voltage (1–100 V) of an ESI-Q-TOFallowed the top-down study of N-terminal heavy (50-kDa) andlight (25-kDa) chains of a solution of gamma globulin [45].Beyond 35–40 kDa, however, Fourier Transform mass spec-trometers seem to be the solution [46,47]. These spectrome-
Fig. 2 – MALDI-reISD spectrum of HeLa cell tubulin. Fragment ionMALDI in-source decay.Monoisotopical specieswere assigned usy28-ion (m/z 3107.27 Da) and y28-ion (m/z 3253.24 Da) of α1B- an
ters, with resolution above 105 and accuracy below ppm levelare potentially the best instruments to obtain protein se-quences from top-down fragmentations patterns. Two typesof technologies encompass the term FT, orbital trap and ioncyclotron resonance. These mass spectrometers transformthe frequency of ion oscillation in a cell into molecularmasses. Orbitraps are used for analyses, by infusion, ofproteins with molecular mass not exceeding 50 kDa or, byRPLC-MS/MS, of proteins with molecular mass below 25 kDa[48]. Instruments such as FTICR can characterize proteins withnominal mass exceeding 100 kDa [49]. Several top-downstudies were also undertaken on MALDI mass spectrometers.Loo et al., for example, studied proteins from Escherichia colithrough a virtual 2 D strategy [50]. The proteins were firstseparated according to their Pi and then analyzed withoutelution or digestion directly in the gel by MALDI-TOF MS. Byusing this strategy, 2.5 times more proteins were detectedthan in classical 2D gel electrophoresis coupled to silverstaining. In addition, some proteins such as MetE protein, amethionine synthase of 84.6 kDa, were identified by MALDItop-down, also called MALDI in-source decay. Some studiesreported on MALDI in-source decay (ISD) for protein identifi-cation [51–54]. As an example, tubulin isotypes of HeLa cellswere characterized using ISD, which shows this technology isof interest for characterizing protein isoforms [54] (Fig. 2). Theemergence of MALDI-TOF–TOF mass spectrometers hasallowed ISD fragment sequencing by a pseudo MS3 techniquenamed T3-sequencing [51–54].
2.3. Dedicated search engines for top-down analysis
There are several search engines for identifying proteins forbottom-up approaches: Mascot™, X!tandem™, Sequest™,Phenyx™, and ProteinProspector™ are the most popular. Forpeptidemass fingerprint experiments, protein identification isdone by comparing experimental m/z values of enzymaticdigests with theoretical m/z values from in silico proteindigestion. Identification is also done by database searchingwith experimental MS/MS data sets of selected ions in the low
s corresponding to C-termini tubulin isotypes obtained bying BioTools 3.1 software. Insets show isotopic patterns of thed βI-tubulin isotypes, respectively.

B
CA
Fig. 3 – Mechanisms of fragmentation used in top-down proteomics. (A) Model of mobile proton explaining protonated peptidedissociation during CID fragmentation. Relocation of the N-terminus proton promotes the formation of an activatedintermediate, which leads to charge-directed amide bond cleavage. (B) Formation of cn- and zn-series ions via cleavage of atransient hypervalent radical produced by intermolecular hydrogen transfer under matrix-assisted laser desorption/ionizationconditions. (C) ETD fragmentation scheme of amultiply protonated peptide after reaction with a low energy electron to producecn- and zn-type ions.
924 J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
mass range. These search engines are constantly improved todecrease the number of false positives (typical for “database-based” search engine) [55]. It has been proposed to combinesearches from various engines but such an approach has juststarted and is still at the stage of experimentation. Softwaresolutions for the analysis of intact protein fragmentationand top-down analysis are not largely developed. There isan increasing demand for software that allows web-basedidentification and characterization of proteins by directcomparison of mass list of observed parent and fragmentions against annotated proteomic databases. ProSight was thefirst web application for top-down identification of proteins[56,57]. This software corresponds to the combination ofsearch engines and a browser environment for the analysisof fragments above 10 kDa. ProSight is centered on organism-specific databases of protein- and site-specific informa-tion. Fragment ion masses of high mass parent ions are usedto performsearches against the constantly updated databases,which include post-translational modifications and “single
nucleotide polymorphisms” (SNPs). It is also possible todetect and localize not annotated PTMs by using a “Δmsearching mode” that considers an error tolerance (Δm)between protein experimental mass and potential candidates’theoretical masses. To take into account the possibility thatseveral PTMs, splice variants, or a variation in amino acidsequence due to polymorphisms of a single genome base pairor SNPs arepresent simultaneously, the softwarehas amethodthat is commonly referred to as “shotgun annotation” [58].Besides ProSight, new bioinformatic tools have been devel-oped. Mascot, the most used web-based database searchengine for bottom-up experiment due to its probability-basedscoring method and its fast operating algorithm, is limited to16 kDa for precursor ion selection. BIG Mascot has beendesigned for top-down analysis and is therefore used toidentify larger proteins fragmented by CID or ECD [59]. Anotherapproach is to align spectra in order to assign the differentisomers of a protein [60]. This algorithm can find optimalalignments between an experimental mass list issued from

925J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
spectrum and a theoretical mass list of a protein sequenceby using a determined number of mass shifts that correspondto post-translational modifications, mutations, or insertions/deletions.
3. Fragmentation mechanisms for primarysequence analysis observed duringtop-down analysis
Fragmentation of biopolymers can occur inside or post themass spectrometer source through different mechanismscompatible with top-down analysis.
3.1. Collision-induced dissociation
The soft ionization sources currently used in biological massspectrometry generate ions in the gas phase by low energyprocesses. Collision of ionized molecules with an inert gas(helium, argon, nitrogen, or xenon) in the cell of the massspectrometer leads molecules to an excited state. Part of theions’ kinetic energy is converted into internal energy. Theseactivated ions undergo unimolecular decomposition thatleads to fragmentation. Fragmentation by CID can occur viahigh or low energy collisions. High-energy collisions can takeplace only in mass spectrometers with magnetic or TOF/TOFanalyzers where the energy involved is a few keV. In this case,ion fragmentation usually occurs in one step. Low energymass spectra are obtained by temporal or spatial tandemmassspectrometry, in which ion fragmentation occurs with acollision energy of a few eV. The collision cell is usually aquadrupole or a linear or 3D ion trap working in radiofrequency mode. Because ions’ residence time is longer andpressure in the collision cell is higher, fragments obtained bylow energy collision are the result of successive dissociations.Contrary to triple quadrupoles that provide a widemass rangefor fragment detection, ion traps suffers from lowmass cut off.Makarov and his group therefore upgraded the Curved LinearTrap (C-trap), a curved quadrupole where ions are stored byRF, coupled to the orbitrap with a higher energy collisionallyactivated dissociation (HCD) [61]. Top-down analysis of bovinecarbonic anhydrase II was obtained through fragmentation byHCD [62]. The CID process can also occur in the source; it iscalled in-source CID. Studies have shown that by varyingparameters such as voltage (source sampling cone, skimmer),temperature, pressure, drying gas, or at last the sourcegeometry, it is possible to fragment multiple charged ions ofhighmassmolecules [63–67]. This occurs because ions leavingthe capillary collide with the drying gas in the source. Thus,the increase in internal energy leads to ion fragmentation.This in-source fragmentation phenomenon is similar to thelow energy collisions obtained by tandem mass spectrometryin triple quadrupole analyzer. The advantage of this approachis the possibility to perform pseudo MS3 experiments frominstruments configured to perform tandem mass spectrome-try (MS/MS) [68,69].
The mechanism of CID fragmentation is usually explainedby the theory of the mobile proton [70] (Fig. 3A). In this model,it is the amino acids’ location and nature that influencepeptide or protein fragmentation. The addition of energy by
collisional activation leads to the relocation of the proton onexcitedmolecular ions. This relocation induces fragmentationat peptide backbones [70–74] (Fig. 3A). For most peptides orproteins, the fragmentation occurs at the amide bonds,leading preferentially to bn- and yn-series ions. High-energyCID induces additional fragmentation of residue side chains,generating vn- and wn-series ions. CID may favor certainschemes of fragmentation, such as amide bond rupture at N-terminal proline [71,74] and Gln–Gly bond cleavage [75].
3.2. IRMPD
During IRMPD dissociation, precursor ions undergo a series ofinfrared radiations by infrared photons, usually in the cell ofan FTICR mass spectrometer. This excitation enables ionheating and leads to the dissociation of peptide bonds. The ionfragments produced are of bn- and yn-series [76]. Thistechnique has the advantage over CID of preserving peptidephosphorylation [77]. IRMPD has also been used on otherinstruments. Raspopov et al. used IRMPD on a triple quadru-pole TOF to study top-down post-translational modificationsof a cancer biomarker, chaperonin-10 [78].
3.3. Electron capture dissociation and electron transferdissociation
In 1998 Zubarev et al. discovered ECD, a new process of peptideandprotein fragmentation [79]. Precursor ions are irradiatedwiththermal electrons (kinetic energy <0.2 eV) in the cell of FTICRmass spectrometers. Such fragmentation is exothermic (6 eV)and non-ergodic, which mean that the energy provided by therecombination necessary for fragmentation is not redistributedthroughout the molecule. Several devices produce the electronflow of electrons in ECD. The most widely used device iscomposed of a non-directly heated cathode irradiating for a fewms the precursor ions. Themain products of irradiation are [M+nH](n−1)+•. Another product of ECD is the [M+(n−1) H](n−1)+ ion.ECD induces the cleavage of parent ion N–Cα bonds, producing c′and z• daughter ions (Fig. 3B) or less frequently c• and z′ ions. Asecond fragmentation mode, leading to the formation of a• andy-type ions, can also be observed [79]. ECD fragmentation occursrandomly andprovides extensive coverage of peptides or proteinsequence [80]. However, there are some specific cleavagesinherent to this technique. Peptide cleavage at N-terminalproline is rarely observed [81] and disulfide bond disruption isfavored at the protein level [82]. Moreover, Fagerquist et al.showed that ECD is able to break disulfide bonds [83]. Electroncapture dissociation of peptides and proteins can also inducecleavage of certain amino acid side chains such as Lys, Arg, His,Asp/Glu, and Met [84]. Finally, ECD keeps PTMs (phosphoryla-tions, ubiquitination…) labile upon CID fragmentation [85].Although this phenomenon is poorly understood, it is probablydue to a lower intramolecular oscillation energy distribution,whichusually leads to the loss of thesemodificationsduringCID.
However, only 5 to 30% of precursor ions are converted intofragment ions and, because of the low kinetic energy ofelectrons for ECD, this approach is difficult to implement inmass spectrometers other than FTICR instruments [79]. So, anew technique emerged a few years ago to overcome theselimits while maintaining ECD's advantages. Electron transfer
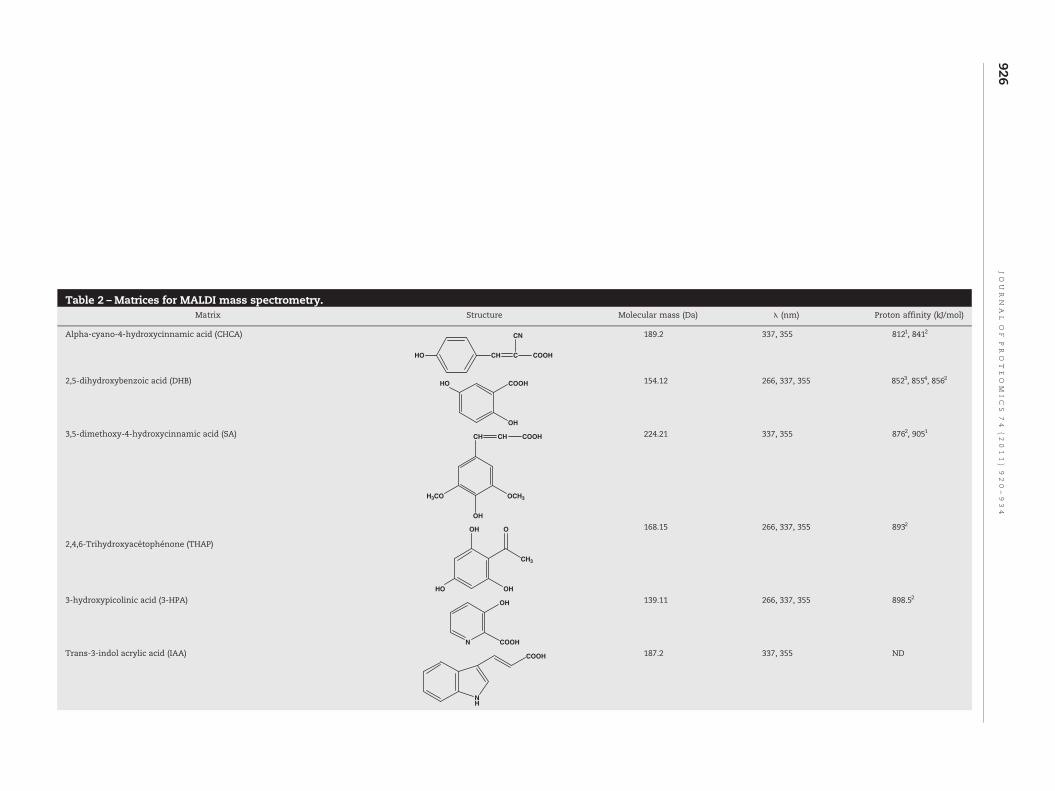
Table 2 –Matrices for MALDI mass spectrometry.Matrix Structure Molecular mass (Da) λ (nm) Proton affinity (kJ/mol)
Alpha-cyano-4-hydroxycinnamic acid (CHCA)
HO CH C
CN
COOH
189.2 337, 355 8121, 8412
2,5-dihydroxybenzoic acid (DHB) HO COOH
OH
154.12 266, 337, 355 8523, 8554, 8562
3,5-dimethoxy-4-hydroxycinnamic acid (SA)
H3CO OCH3
OH
CH CH COOH 224.21 337, 355 8762, 9051
2,4,6-Trihydroxyacétophénone (THAP)
OH
HO
O
CH3
OH
168.15 266, 337, 355 8932
3-hydroxypicolinic acid (3-HPA)
N
OH
COOH
139.11 266, 337, 355 898.52
Trans-3-indol acrylic acid (IAA)
NH
COOH 187.2 337, 355 ND
926JO
UR
NA
LO
FPR
OT
EO
MIC
S74
(2011)
920–934

Dithranol (DIT) O OHOH 226.23 266, 337, 355 885.52
T-2-[3-(4-t-butyl-phényl)-2-méthyl-2-propénylidène]malononitrile (DCTB)
H3C
NC CN
t-Bu
250.34 266, 337, 355 ND
1,5-diaminonaphthalene (1,5-DAN) NH2
NH2
158.2 266, 337, 355 9425
5-aminosalicylic acid (5-ASA) NH2
OHO
HO
153.135 337, 355 8775
1, 2, 3, 4 and 5 Data from [120–124] respectively.
927JO
UR
NA
LO
FPR
OT
EO
MIC
S74
(2011)
920–934

928 J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
dissociation developed by Hunt et al. in 2004 is mechanisticallyrelated toECDbutwasdesigned for instrumentswithan ion trapanalyzer, more widespread and less expensive [86]. Thistechnique is adaptable to mass spectrometers with 2D typelinear, spherical geometry or Orbitrap ion trap analyzers. ETDinvolves an electron transfer onto protonated proteins orpeptides (multicharged cations) from a reactive electron carrier(anion) formed outside the analyzer in a chemical ionizationsource. This source can be positioned opposite to the ESI sourceas in the case of LTQ-Orbitrap ETD or fitted between ESI sourceand the analyzer. Reactive species transfer to protonatedprotein/peptide occurs in gas phase by confinement of the twospecies in the analyzer. The fragmentation, which leads to thesame species as those in the ECDprocess, is a two-step reaction.First the electron transfer produces radical anion formation thatwill capture a proton. The result is the formation of an amino-ketal radical site that will trigger the dissociation of thecompound in cn- and zn-series ions (Fig. 3B).
3.4. Laser-induced dissociation
DuringMALDI ionization, ionized peptides/proteins can undergounimolecular reactions leading to the cleavage of the peptidebackbone. These dissociations are called metastable fragmenta-tions. Laser-Induced Dissociation (LID) corresponds to a frag-mentationof acceleratedmolecular ionsduring their travel in theflight tube (TOF) due to excess energy acquired in the sourceduring MALDI. The fragment ions, principally of yn-, an- and bn-series, have the same velocity as their precursor ion and reachtheselectiongateat thesametime. InMS/MSmode, ionswith thesame velocity are then selected according to their time of flight.Reflectron then allows the separation of the different ionsaccording to theirkinetic energy [87]. Reflectron,however, cannotrefocus in one step the ions with kinetic energy differenceshigher than 30%. For this, mass spectrometer manufacturershave designed TOF–TOF instruments equipped with “LIFT”technique [52]. Through an increase in kinetic energy of all ions(+19 keV), the LIFT cell reduces the kinetic energy differencebetween parent and fragment ions under the 30% threshold.Thus, a gridless two-stage reflectroncan focus, inonestep,wholeions accelerated after the LIFT cell. This technical progress is ofinterest because it decreases analysis time and enhances themass resolutionofparent and fragment ions. Inaddition, gridlesstwo-stage reflectron focuses the ion beam for better detection ofthe different ionic species, increasing sensitivity to the sub-femtomole level [52]. The curved-field reflectron can alsoperform MS/MS analysis in one step, but the absence ofreacceleration decreases mass resolution and sensitivity [52,88].
3.5. MALDI in-source decay
ISD fragmentation occurs in the hot MALDI plume afterion desorption/ionization steps. Indeed, the laser beam withfluence 5–20% above analyte ionization threshold leads tointermolecular transfer of hydrogen atoms between excitedmatrix molecules and carbonyl oxygen on the peptidebackbone [89–91]. The transfer depends on the hydrogenbonding network between matrix molecules and analytes.This fragmentation process preferentially gives cn- and (zn+2)-series ions [89] (Fig. 3C). Other ions of yn-, an-, xn-, and bn-series
can also be observed [90]. Numerous studies have beenperformed to determine the influence of the matrix and thematrix to analyte ratio in the ISD process. Thus matrices canbe ordered in function of their ability to donate protons. Themost widely used matrices for ISD have the followingclassification: picolinic acid (PA)>1,5 diaminonaphthalene(1,5-DAN)>5-aminosalicylic acid (5-ASA)>2,5-DHB>sinapinicacid (SA)>α-cyano-4-hydroxycinnamic acid (CHCA) [92,93]. Astudy has shown that the optimal matrix for inducing ISDfragmentation is a mixture of PA (20 mg/mL) and 1,5-DAN(saturated) with a ratio of 25/75 (v:v). Such a mix generatesalbumin (66 kDa) ion fragments ofmasses around 1000 Da [93].In addition, 1,5-DAN is a matrix with reducing properties,which makes it possible to study peptide/protein moleculeswith disulfide bridges [92,94,95]. This chemical, however, istoxic and can easily be replaced by 5-ASA [92]. These twomatrices produce mainly cn-, zn-, and yn-series ions, whereas2,5-DHB matrix, the most commonly used, and SA favor ionfragments of cn- and yn-series [96] (Table 2). In the case of 2,5-DHB, a study has indicated that the 5-hydroxy group plays animportant role in the ISD process [91]. Finally, the CHCAmatrix is the least able to induce ISD fragmentation andproduces yn-, bn-, and an-series ions [96]. This CID-likefragmentation is due to collisions between analytes in theearly step of ablation in the fragmentation process. Thisphenomenon is associated with analyte initial velocity duringdesorption. The lower the initial velocity, the higher thecollision rate and therefore the fragmentation. Matrix proper-ties take an important place in this process and a study hasproposed a classification of initial velocity of certain matrices:CHCA>THAP>SA>2,5-DHB [97]. To obtain proton transferbetween the matrix molecules and analytes, the matrix toanalyte ratio is set higher than in conventional MALDI analysis.Thus, Takayama et al. indicated that this ratio should bebetween 5000/1 and 10,000/1 [91]. Protein/peptide ISD fragmen-tation during top-down analysis can also be influenced byprimary and secondary structure elements. The gas phasebasicity (GB) of eachamino acid and therefore the proton affinity(PA) associatedwith it can influence biomolecule fragmentation[98]. Studies showed that the presence at polypeptide N- and C-termini of high gas basicity amino acids such as lysine, arginine,or glutamic acid is relatively important because they favorfragmentation [99,100]. Moreover, fragmentation depends onprotein sequence since in-source decay is almost impossible atthe N-terminus side of the proline due to its cyclic nature but isfavored when Glycine and Valine are present [91]. Finally, thesecondary structure, such as a-helix and β-sheet, inhibitsbackbone cleavage in ISD. Indeed, Takayama et al. showed thatMALDI-ISD of equine-apomyoglobin, a protein composed ofthree kinds of secondary structures, revealed that helix struc-tures can inhibit cleavage at amine bonds on the backbone [89].
4. Contribution of top-down approaches forcharacterization of biomarkers andbiochemical pathways
Top-down proteomic analysis is a major asset for discoveringbiomarkers because, by measuring the intact protein molecular
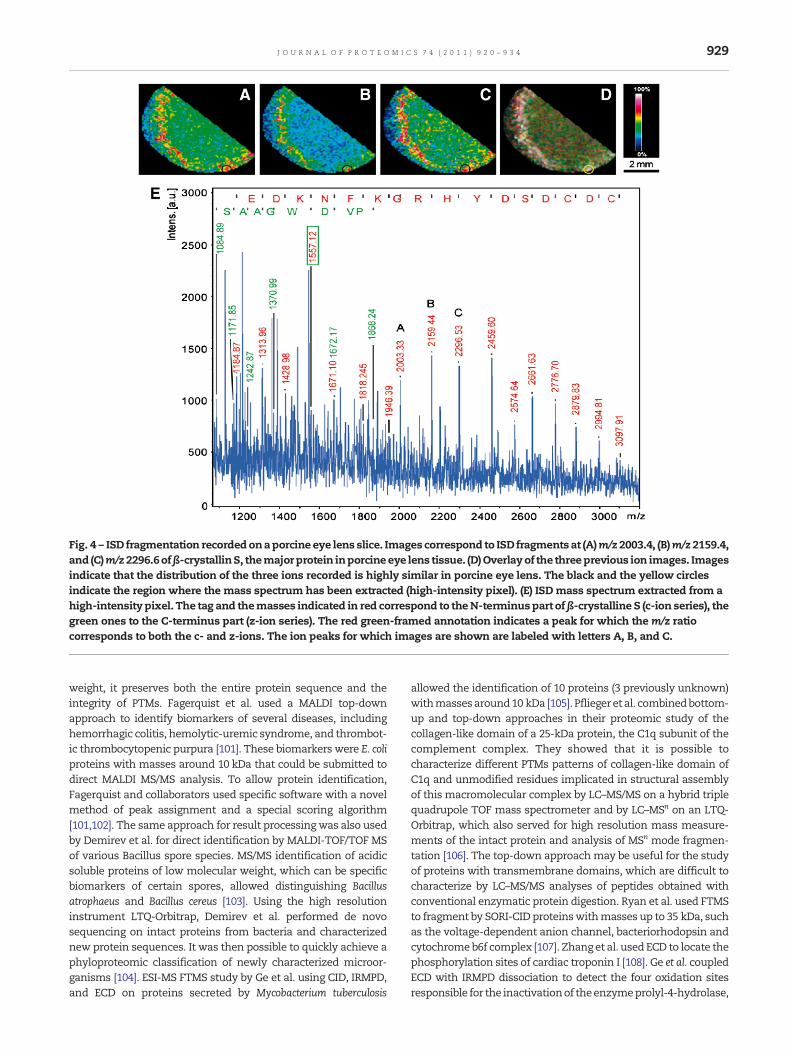
Fig. 4 – ISD fragmentation recordedonaporcine eye lens slice. Images correspond to ISD fragmentsat (A)m/z2003.4, (B)m/z2159.4,and (C)m/z2296.6ofβ-crystallinS, themajorprotein inporcineeye lens tissue. (D)Overlayof the threeprevious ion images. Imagesindicate that the distribution of the three ions recorded is highly similar in porcine eye lens. The black and the yellow circlesindicate the region where the mass spectrum has been extracted (high-intensity pixel). (E) ISD mass spectrum extracted from ahigh-intensity pixel. The tag and themasses indicated in red correspond to theN-terminuspart ofβ-crystalline S (c-ion series), thegreen ones to the C-terminus part (z-ion series). The red green-framed annotation indicates a peak for which the m/z ratiocorresponds to both the c- and z-ions. The ion peaks for which images are shown are labeled with letters A, B, and C.
929J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
weight, it preserves both the entire protein sequence and theintegrity of PTMs. Fagerquist et al. used a MALDI top-downapproach to identify biomarkers of several diseases, includinghemorrhagic colitis, hemolytic-uremic syndrome, and thrombot-ic thrombocytopenic purpura [101]. These biomarkers were E. coliproteins with masses around 10 kDa that could be submitted todirect MALDI MS/MS analysis. To allow protein identification,Fagerquist and collaborators used specific software with a novelmethod of peak assignment and a special scoring algorithm[101,102]. The same approach for result processingwas also usedby Demirev et al. for direct identification by MALDI-TOF/TOF MSof various Bacillus spore species. MS/MS identification of acidicsoluble proteins of low molecular weight, which can be specificbiomarkers of certain spores, allowed distinguishing Bacillusatrophaeus and Bacillus cereus [103]. Using the high resolutioninstrument LTQ-Orbitrap, Demirev et al. performed de novosequencing on intact proteins from bacteria and characterizednew protein sequences. It was then possible to quickly achieve aphyloproteomic classification of newly characterized microor-ganisms [104]. ESI-MS FTMS study by Ge et al. using CID, IRMPD,and ECD on proteins secreted by Mycobacterium tuberculosis
allowed the identification of 10 proteins (3 previously unknown)withmassesaround10 kDa [105]. Pfliegeret al. combinedbottom-up and top-down approaches in their proteomic study of thecollagen-like domain of a 25-kDa protein, the C1q subunit of thecomplement complex. They showed that it is possible tocharacterize different PTMs patterns of collagen-like domain ofC1q and unmodified residues implicated in structural assemblyof this macromolecular complex by LC–MS/MS on a hybrid triplequadrupole TOF mass spectrometer and by LC–MSn on an LTQ-Orbitrap, which also served for high resolution mass measure-ments of the intact protein and analysis of MSn mode fragmen-tation [106]. The top-down approachmay be useful for the studyof proteins with transmembrane domains, which are difficult tocharacterize by LC–MS/MS analyses of peptides obtained withconventional enzymatic protein digestion. Ryan et al. used FTMSto fragment by SORI-CID proteinswithmasses up to 35 kDa, suchas the voltage-dependent anion channel, bacteriorhodopsin andcytochromeb6f complex [107]. Zhang et al.used ECD to locate thephosphorylation sites of cardiac troponin I [108]. Ge et al. coupledECD with IRMPD dissociation to detect the four oxidation sitesresponsible for the inactivationof theenzymeprolyl-4-hydrolase,

930 J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
which catalyses the hydroxylation of proline in procollagen [109].Contrary to the bottom-up approach, the top-down approachdetects K216 oxidation directly from a single spectrum. Thisinteresting work by top-down MS/MS focused on oxidationkinetics data of prolyl-4-hydrolase. In a top-down kinetics studyby FTMS, Zhai et al. reported on this-thiocarboxylate (this-SH), aprotein involved in the synthesis of the thiazole subunit ofthiamine phosphate. They pointed out that it was possible tocharacterize aminoacids involved in the formationof imineThiGfrom its substrate, deoxy-D-xylulose 5-phosphate [110]. Top-down analysis can be an asset to label-free quantify proteins.Pesavento et al. showed that ECD not only identifies acetylationsites, but also quantifies samples composed of more than 15isomers of histone H4 presenting one or more acetylations bymeasuring fragment ion abundance [111]. MALDI top-down is anefficient approach for protein isoform characterization. A recentstudycharacterized theβIII-tubulin isotype, apotential biomarkerof poor prognosis cancers [54]. This isotype was identified in asolution of HeLa cell tubulin by ISD sequencing of its C-terminus.ISD fragmentation is of interest in biomarker discovery since ithas made it possible to sequence the N- and C-termini ofubiquitin (8.6 kDa), myoglobin (17 kDa), and bovine serumalbumin (66 kDa) [93] and to analyze PTMs like glycosylation inpermethylated glycans prepared frommouse IgG1 [112]. Thus byupdating bioinformatics tools, ISD fragmentation appears apromising tool for direct protein profiling in cell lysate but alsofor thedirect studyof intact proteinson tissue slices as in thecaseof imaging MALDI mass spectrometry (IMSS). This techniquereveals the relative abundance of proteins and their localizationin tissue sections with high spatial resolution neighboring 50 μm[113]. A recent study reported that ISD fragmentation can beapplied to in situ identify proteins located by IMSS to avoidmaker delocalization due to tryptic digestion Althoughpromising in situ ISD is up to now limited to identification ofmajor proteins [114] (Fig. 4). Therefore microextraction andtandem mass spectrometry using bottom-up or top-downapproaches are still complementary to IMSS for proteinidentification. Using a combination of IMSS and RPLC-ESI-MS/MS, Lemaire et al. evidenced a potential ovary cancermarker corresponding to a fragment of the 11S proteasomeactivator complex named Reg-alpha [115]. Meistermann et al.used IMSS to localize biomarkers of toxicity induced bygentamicin in rat kidney sections [116]. The identification oftransthyretin, which accumulates in the renal cortex inresponse to treatment, was performed using tissue micro-extraction followed by RPLC-ESI-MS/MS analysis. Rauser et al.focused on the specific expression of proteins based onoverexpression of HER2, a membrane glycoprotein on thesurface of breast cancer cells [117]. ESI coupled to ETDfragmentation identified a cystein-rich protein 1, CRIP1, abiomarker that serves to accurately define HER2-positivefrom HER2-negative tissues by IMSS. Another study hasshown the relevance of a fragment of mitogen-activatedprotein kinase/extracellular signal-regulated kinase kinasekinase 2 in prostate cancer (PCa) from uninvolved prostatetissue (MEKK2) [118]. IMSS indicated that this fragment has ahigh expression profile in the PCa area and MALDI MS/MSanalysis allowed its identification. These studies showed thatmicroextraction can produce enough material for ms/msanalysis so that top-down can be envisioned. This was nicely
demonstrated by Lagarrigue et al. who combined MALDIimaging at 20 μMofmammalian testis and top-down analysisby CID MS/MS [119]. This allowed identification of a proteinmarker of development of germ cells within the seminiferousepithelium.
5. Conclusion
Top-down strategies are constantly being improved ininstrumentation, bioinformatics, and data processing. Theincreased sensitivity and resolution of MS instruments andthe adaptation of techniques now allow direct analysis ofproteins with molecular masses exceeding 100 kDa. Furtheroptimization of fragmentation methods such as ISD, ECD, orETD coupled to ESI and MALDI ionization now allowsextensive coverage of protein sequences, especially N- andC-termini. These regions are highly variablewithin a family ofproteins and are the sites of many labile PTMs. These PTMsare usually lost during conventional CID but not by ISD, EDC,and ETD. This feature combined with the possibility ofdirectly analyzing intact protein suggests that the top-downapproach is particularly suitable for locating and character-izing PTMs. However, to study complex mixtures for theidentification of disease biomarkers, several parameters needto be optimized, for example, the separation of proteins priorto mass spectrometry. The use of MALDI mass spectrometerscan be a solution to directly study tissue sections andconsequently locate and characterize biomarkers, but fist animprovement of in situ fragmentation capabilities andsecond a better data treatment are needed for such a complexanalysis.
R E F E R E N C E S
[1] Draisma G, Etzioni R, Tsodikov A, Mariotto A, Wever E, GulatiR, et al. Lead time and overdiagnosis in prostate-specificantigen screening: importance of methods and context.J Natl Cancer Inst 2009;101:374–83.
[2] Ross JS, Hatzis C, Symmans WF, Pusztai L, Hortobagyi GN.Commercializedmultigene predictors of clinical outcome forbreast cancer. Oncologist 2008;13:477–93.
[3] Solassol J, Boulle N, Maudelonde T, Mange A. Clinicalproteomics: towards early detection of cancers. Med Sci(Paris) 2005;21:722–9.
[4] Bon C, Brillard B, Gelineau MC, Mailliavin A, Trépo C, Pichot J.Decarboxyprothrombin: importance in the diagnosis ofhepatocellular carcinoma. AnnBiol Clin (Paris) 1998;56:175–81.
[5] Barlesi F, Tchouhadjian C, Doddoli C, Torre JP, Astoul P,Kleisbauer JP. CYFRA 21-1 level predicts survival innon-small-cell lung cancer patients receiving gefitinib asthird-line therapy. Br J Cancer 2005;92:13–4.
[6] Lane CS. Mass spectrometry-based proteomics in the lifesciences. Cell Mol Life Sci 2005;62:848–69.
[7] Chait BT. Chemistry. Mass spectrometry: bottom-up ortop-down? Science 2006;314:65–6.
[8] Aebersold R, Mann M. Mass spectrometry-based proteomics.Nature 2003;422:198–207.
[9] Henzel WJ, Watanabe C, Stults JT. Protein identification: theorigins of peptide mass fingerprinting. J Am Soc MassSpectrom 2003;14:931–42.

931J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
[10] McDonald WH, Yates III JR. Shotgun proteomics: integratingtechnologies to answer biological questions. Curr Opin MolTher 2003;5:302–9.
[11] Meng Z, Limbach PA. Mass spectrometry of RNA: linking thegenome to the proteome. Brief Funct Genomic Proteomic2006;5:87–95.
[12] Hossain M, Limbach PA. Mass spectrometry-based detectionof transfer RNAs by their signature endonuclease digestionproducts. RNA (New York, NY) 2007;13:295–303.
[13] Kellie JF, Tran JC, Lee JE, Ahlf DR, Thomas HM, Ntai I, et al.The emerging process of Top Down mass spectrometry forprotein analysis: biomarkers, protein-therapeutics, andachieving high throughput. Mol Biosyst 2010;6:1532–9.
[14] Reid GE, McLuckey SA. 'Top down' protein characterizationvia tandem mass spectrometry. J Mass Spectrom 2002;37:663–75.
[15] Bogdanov B, Smith RD. Proteomics by FTICR massspectrometry: top down and bottom up. Mass Spectrom Rev2005;24:168–200.
[16] Han X, Jin M, Breuker K, McLafferty FW. Extending top-downmass spectrometry to proteins with masses greater than 200kilodaltons. Science 2006;314:109–12.
[17] Armirotti A, Damonte G. Achievements and perspectives oftop-down proteomics. Proteomics 2010;10:3566–76.
[18] Casado-Vela J, Cebrian A, Gomez Del Pulgar MT,Sanchez-Lopez E, Vilaseca M, Menchen L, et al. Lights andshadows of proteomic technologies for the study of proteinspecies including isoforms, splicing variants and proteinpost-translational modifications. Proteomics 2011;11:590–603.
[19] McLafferty FW, Breuker K, Jin M, Han X, Infusini G, Jiang H.Top-downMS, a powerful complement to the high capabilitiesof proteolysis proteomics. FEBS J 2007;274:6256–68.
[20] Cottrell JS. Protein identification by peptide massfingerprinting. Pept Res 1994;7:115–24.
[21] Issaq HJ, Blonder J. Electrophoresis and liquidchromatography/tandem mass spectrometry in diseasebiomarker discovery. J Chromatogr B Analyt Technol BiomedLife Sci 2009;877:1222–8.
[22] McDonald WH, Yates III JR. Shotgun proteomics andbiomarker discovery. Dis Markers 2002;18:99–105.
[23] Bakhtiar R, Guan Z. Electron capture dissociation massspectrometry in characterization of peptides and proteins.Biotechnol Lett 2006;28:1047–59.
[24] Sobott F, Watt SJ, Smith J, Edelmann MJ, Kramer HB, KesslerBM. Comparison of CID versus ETD based MS/MSfragmentation for the analysis of protein ubiquitination.J Am Soc Mass Spectrom 2009;20:1652–9.
[25] Hardouin J. Protein sequence information bymatrix-assistedlaser desorption/ionization in-source decay massspectrometry. Mass Spectrom Rev 2007;26:672–82.
[26] Wehr T. Separation technology in proteomics. LCGC NorthAmerica 2001;19:702–11.
[27] Wehr T. Multidimensional liquid chromatography inproteomic studies. LCGC North America 2002;20:954–62.
[28] Wehr T. Affinity selection techniques for proteomic studies.LCGC North America 2003;21:274–84.
[29] Claverol S, Burlet-Schiltz O, Gairin JE, Monsarrat B.Characterization of protein variants and post-translationalmodifications: ESI-MSn analyses of intact proteins elutedfrompolyacrylamide gels. Mol Cell Proteomics 2003;2:483–93.
[30] Meng F, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM,Kelleher NL. Processing complex mixtures of intact proteinsfor direct analysis by mass spectrometry. Anal Chem2002;74:2923–9.
[31] Powell MJ, Razunguzwa TT, Biddle AD, Asbury GR. A novelchip-based electroelution system for rapid and efficientrecovery of intact proteins from polyacylamide gels.BioTechniques Spec Issue 2009;46:373–4.
[32] Wang Y, Balgley BM, Rudnick PA, Lee CS. Effects ofchromatography conditions on intact protein separations fortop-down proteomics. J Chromatogr 2005;1073:35–41.
[33] Pesavento JJ, Yang H, Kelleher NL, Mizzen CA. Certain andprogressivemethylation of histone H4 at lysine 20 during thecell cycle. Mol Cell Biol 2008;28:468–86.
[34] Contrepois K, Ezan E, Mann C, Fenaille F. Ultra-highperformance liquid chromatography–mass spectrometry forthe fast profiling of histone post-translational modifications.J Proteome Res 2010;9:5501–9.
[35] Wu S, Yang F, Zhao R, Tolic N, Robinson EW, Camp II DG,et al. Integrated workflow for characterizing intactphosphoproteins from complex mixtures. Anal Chem2009;81:4210–9.
[36] Roth MJ, Parks BA, Ferguson JT, Boyne II MT, Kelleher NL."Proteotyping": population proteomics of human leukocytesusing top down mass spectrometry. Anal Chem 2008;80:2857–66.
[37] Pesavento JJ, Yang H, Kelleher NL, Mizzen CA. Certain andprogressivemethylation of histone H4 at lysine 20 during thecell cycle. Mol Cell Biol 2008;28:468–86.
[38] Zhao J, Zhu K, Lubman DM, Miller FR, Shekhar MP, Gerard B,et al. Proteomic analysis of estrogen response ofpremalignant human breast cells using a 2-D liquidseparation/mass mapping technique. Proteomics 2006;6:3847–61.
[39] Roth MJ, Forbes AJ, Boyne II MT, Kim YB, Robinson DE,Kelleher NL. Precise and parallel characterization of codingpolymorphisms, alternative splicing, and modifications inhuman proteins by mass spectrometry. Mol Cell Proteomics2005;4:1002–8.
[40] Tran JC, Doucette AA. Gel-eluted liquid fraction entrapmentelectrophoresis: an electrophoretic method for broadmolecular weight range proteome separation. Anal Chem2008;80:1568–73.
[41] Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJ. Ionmobility mass spectrometry of proteins and proteinassemblies. Chem Soc Rev 2010;39:1633–55.
[42] Loo JA, EdmondsCG, SmithRD. Primary sequence informationfrom intact proteins by electrospray ionization tandemmass spectrometry. Science 1990;248:201–4.
[43] Feng R, Konishi Y. Collisionally activated dissociation ofmultiply charged 150 kDa antibody ions. Anal Chem 1993;65:645–9.
[44] Nemeth-Cawley JF, Tangarone BS, Rouse JC. "Top Down"characterization is a complementary technique to peptidesequencing for identifying protein species in complexmixtures. J Proteome Res 2003;2:495–505.
[45] Ren D, Pipes GD, Hambly D, Bondarenko PV, Treuheit MJ,Gadgil HS. Top-down N-terminal sequencing ofImmunoglobulin subunits with electrospray ionization timeof flight mass spectrometry. Anal Biochem 2009;384:42–8.
[46] Loo JA, Quinn JP, Ryu SI, Henry KD, Senko MW, McLaffertyFW. High-resolution tandem mass spectrometry of largebiomolecules. Proc Natl Acad Sci U S A 1992;89:286–9.
[47] Senko MW, Beu SC, McLafferty FW. High-resolution tandemmass spectrometry of carbonic anhydrase. Anal Chem1994;66:415–8.
[48] Ren D, Pipes GD, Hambly D, Bondarenko PV, Treuheit MJ,Gadgil HS. Top-down N-terminal sequencing ofimmunoglobulin subunits with electrospray ionization timeof flight mass spectrometry. Anal Biochem 2009;384:42–8.
[49] Kelleher NL, Senko MW, Siegel MM, McLafferty FW. Unitresolution mass spectra of 112 kDa molecules with 3 Daaccuracy. J Am Soc Mass Spectrom 1997;8:380–3.
[50] Loo RR, Cavalcoli JD, VanBogelen RA, Mitchell C, Loo JA,Moldover B, et al. Virtual 2-D gel electrophoresis:visualization and analysis of the E. coli proteome by massspectrometry. Anal Chem 2001;73:4063–70.

932 J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
[51] Suckau D, Resemann A. T3-sequencing: targetedcharacterization of the N- and C-termini of undigestedproteins by mass spectrometry. Anal Chem 2003;75:5817–24.
[52] Suckau D, Resemann A, Schuerenberg M, Hufnagel P,Franzen J, Holle A. A novel MALDI LIFT-TOF/TOF massspectrometer for proteomics. Anal Bioanal Chem 2003;376:952–65.
[53] Suckau D, Resemann A. MALDI Top-Down sequencing:calling N- and C-terminal protein sequences with highconfidence and speed. J Biomol Tech 2009;20:258–62.
[54] Calligaris D, Villard C, Terras L, Braguer D, Verdier-Pinard P,Lafitte D. MALDI in-source decay of high mass proteinisoforms: application to alpha- and beta-tubulin variants.Anal Chem 2010;82:6176–84.
[55] Nesvizhskii AI. Protein identification by tandem massspectrometry and sequence database searching. MethodsMol Biol 2007;367:87–119.
[56] Taylor GK, Kim YB, Forbes AJ, Meng F, McCarthy R, KelleherNL. Web and database software for identification of intactproteins using "top down" mass spectrometry. Anal Chem2003;75:4081–6.
[57] Leduc RD, Kelleher NL. Using ProSight PTM and related toolsfor targeted protein identification and characterization withhigh mass accuracy tandem MS data. Current protocols inbioinformatics / editoral board, Andreas D Baxevanis et al.2007;Chapter 13:Unit 13 6.
[58] Pesavento JJ, Kim YB, Taylor GK, Kelleher NL. Shotgunannotation of histone modifications: a new approach forstreamlined characterization of proteins by top down massspectrometry. J Am Chem Soc 2004;126:3386–7.
[59] Karabacak NM, Li L, Tiwari A, Hayward LJ, Hong P, EasterlingML, et al. Sensitive and specific identification of wild typeand variant proteins from 8 to 669 kDa using top-downmassspectrometry. Mol Cell Proteomics 2009;8:846–56.
[60] Frank AM, Pesavento JJ, Mizzen CA, Kelleher NL, Pevzner PA.Interpreting top-down mass spectra using spectralalignment. Anal Chem 2008;80:2499–505.
[61] Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M.Higher-energy C-trap dissociation for peptide modificationanalysis. Nat Methods 2007;4:709–12.
[62] Olsen JV, Schwartz JC, Griep-Raming J, Nielsen ML, Damoc E,Denisov E, et al. A dual pressure linear ion trap Orbitrapinstrument with very high sequencing speed. Mol CellProteomics 2009;8:2759–69.
[63] Meng CK, McEwen CN, Larsen BS. Peptide sequencing withelectrospray ionization on a magnetic sector massspectrometer. Rapid Commun Mass Spectrom 1990;4:151–155.
[64] Loo JA, Udseth HR, Smith RD. Collision effects on the chargedistribution of ions from large molecules, formed byelectrospray ionization-mass spectrometry. Rapid CommunMass Spectrom 1988;2:207–10.
[65] Collette C, Drahos L, De Pauw E, Vékey K. Comparison of theinternal energy distributions of ions produced by differentelectrospray sources. Rapid Commun Mass Spectrom1998;12:1673–8.
[66] Weinmann W, Stoertzel M, Vogt S, Wendt J. Tunecompounds for electrospray ionisation/in-sourcecollision-induced dissociation with mass spectral librarysearching. J Chromatogr 2001;926:199–209.
[67] Schneider BB, Douglas DJ, Chen DD. Ion fragmentation in anelectrospray ionization mass spectrometer interface withdifferent gases. Rapid Commun Mass Spectrom 2001;15:249–57.
[68] Chen H, Tabei K, Siegel MM. Biopolymer sequencing using atriple quadrupole mass spectrometer in the ESInozzle-skimmer/precursor ion MS/MS mode. J Am Soc MassSpectrom 2001;12:846–52.
[69] Ginter JM, Zhou F, Johnston MV. Generating proteinsequence tags by combining cone and conventional collisioninduced dissociation in a quadrupole time-of-flight massspectrometer. J Am Soc Mass Spectrom 2004;15:1478–86.
[70] Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Mobile andlocalized protons: a framework for understanding peptidedissociation. J Mass Spectrom 2000;35:1399–406.
[71] Tang XJ, Thibault P, Boyd RK. Fragmentation reactions ofmultiply-protonated peptides and implications forsequencing by tandem mass spectrometry with low-energycollision-induced dissociation. Anal Chem 1993;65:2824–34.
[72] Dongré AR, Jones JL, Somogyi A, Wysocki VH. Influence ofpeptide composition, gasphase basicity, and chemicalmodification on fragmentation efficiency: evidence for themobile proton model. J Am Chem Soc 1996;118:8365–74.
[73] Johnson RS, Krylov D, Walsh KA. Proton mobility withinelectrosprayed ions. J Mass Spectrom 1995;30:386–7.
[74] Adamczyk M, Gebler JC, Wu J. Charge derivatization ofpeptides to simplify their sequencing with an ion trap massspectrometer. Rapid Commun Mass Spectrom 1999;13:1413–22.
[75] Griffiths WJ, Jonsson AP. Gas phase conformation can havean influence on peptide fragmentation. Proteomics 2001;1:934–45.
[76] Ge Y, Horn DM, McLafferty FW. Blackbody infrared radiativedissociation of larger (42 kDa) multiply charged proteins. IntJ Mass Spectrom 2001;210/211:203–14.
[77] Crowe MC, Brodbelt JS. Infrared multiphoton dissociation(IRMPD) and collisionally activated dissociation of peptidesin a quadrupole ion trap with selective IRMPD ofphosphopeptides. J Am Soc Mass Spectrom 2004;15:1581–92.
[78] Raspopov SA, El-Faramawy A, Thomson BA, Siu KW. Infraredmultiphoton dissociation in quadrupole time-of-flight massspectrometry: top-down characterization of proteins. AnalChem 2006;78:4572–7.
[79] Zubarev RA, Kelleher NL, McLafferty FW. Electron capturedissociation of multiply charged protein cations.A nonergodic process. J Am Chem Soc 1998;120:3265–6.
[80] Zubarev RA. Electron-capture dissociation tandem massspectrometry. Curr Opin Biotechnol 2004;15:12–6.
[81] Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, KrugerNA, Lewis MA, et al. Electron capture dissociation forstructural characterization of multiply charged proteincations. Anal Chem 2000;72:563–73.
[82] Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM,Carpenter BK, et al. Electron capture dissociation of gaseousmultiply-charged proteins is favored at disulfide bonds andother sites of high hydrogen atom affinity. J Am Chem Soc1999;121:2857–62.
[83] Fagerquist CK, Hudgins RR, Emmett MR, Hakansson K,Marshall AG. An antibiotic linked to peptides and proteins isreleased by electron capture dissociation Fourier transformion cyclotron resonance mass spectrometry. J Am Soc MassSpectrom 2003;14:302–10.
[84] Cooper HJ, Hudgins RR, Hakansson K, Marshall AG.Characterization of amino acid side chain losses in electroncapture dissociation. J Am Soc Mass Spectrom 2002;13:241–249.
[85] Kelleher NL, Zubarev RA, Bush K, Furie B, Furie BC,McLafferty FW, et al. Localization of labile posttranslationalmodifications by electron capture dissociation: the case ofgamma-carboxyglutamic acid. Anal Chem 1999;71:4250–3.
[86] Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF.Peptide and protein sequence analysis by electron transferdissociation mass spectrometry. Proc Natl Acad Sci U S A2004;101:9528–33.
[87] Mamyrin B. Time-of-flight mass spectrometry (concepts,achievements, and prospects). Int J Mass Spectrom 2001;206:251–66.

933J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
[88] Giannakopulos AE, Thomas B, Colburn AW, Reynolds DJ,Raptakis EN, Makarov AA, et al. Tandem time-of-flight massspectrometer (TOF–TOF) with a quadratic-field ion mirror.Rev Sci Instrum 2002;73:2115–23.
[89] Takayama M. Susceptible region of myoglobins to in-sourcedecay using matrix-assisted laser desorption/ionizationcoupled with reflectron time-of-flight mass spectrometer.J Mass Spectrom Soc Jpn 2002;50:304–10.
[90] Brown RS, Feng J, Reiber DC. Further studies of in-sourcefragmentation of peptides in matrix-assisted laserdesorption–ionization. Int J Mass Spectrom Ion Process1997;169/170:1–18.
[91] Takayama M. N–Ca bond cleavages of the peptide backbonevia hydrogen abstraction. J Am Soc Mass Spectrom 2001;12:1044–9.
[92] Sakakura M, Takayama M. In-source decay andfragmentation characteristics of peptides using5-aminosalicylic acid as a matrix in matrix-assisted laserdesorption/ionization mass spectrometry. J Am Soc MassSpectrom 2010;21:979–88.
[93] Demeure K, Quinton L, Gabelica V, De Pauw E. Rationalselection of the optimum MALDI matrix for top-downproteomics by in-source decay. Anal Chem 2007;79:8678–85.
[94] Fukuyama Y, Iwamoto S, Tanaka K. Rapid sequencing anddisulfide mapping of peptides containing disulfide bonds byusing 1,5-diaminonaphthalene as a reductive matrix. J MassSpectrom 2006;41:191–201.
[95] Quinton L, Demeure K, Dobson R, Gilles N, Gabelica V, DePauw E. New method for characterizing highlydisulfide-bridged peptides in complex mixtures: applicationto toxin identification from crude venoms. J Proteome Res2007;6:3216–23.
[96] Takayama M. In-source decay characteristics of peptides inmatrix-assisted laser desorption/ionization time-of-flightmass spectrometry. J Am Soc Mass Spectrom 2001;12:420–7.
[97] Sachon E, Clodic G, Blasco T, Jacquot Y, Bolbach G. In-sourcefragmentation of very labile peptides in matrix-assistedlaser desorption/ionization time-of-flight massspectrometry. Anal Chem 2009;81:8986–92.
[98] HarrisonAG. The gas-phase basicities and proton affinities ofamino acids and peptides. Mass Spectrom Rev 1997;16:201–17.
[99] Takayama M, Tsugita A. Sequence information of peptidesand proteins with in-source decay in matrix assisted laserdesorption/ionization-time of flight-mass spectrometry.Electrophoresis 2000;21:1670–7.
[100] Takayama M, Tsugita A. Does in-source decay occurindependant of the ionization process in matrix-assistedlaser desorption? Int J Mass Spectrom 1998;181:L1–6.
[101] Fagerquist CK, Garbus BR, Miller WG, Williams KE, Yee E,Bates AH, et al. Rapid identification of protein biomarkers ofEscherichia coli O157:H7 by matrix-assisted laser desorptionionization-time-of-flight-time-of-flight massspectrometry and top-down proteomics. Anal Chem2010;82:2717–25.
[102] Fagerquist CK, Garbus BR, Williams KE, Bates AH, Boyle S,Harden LA. Web-based software for rapid top-downproteomic identification of protein biomarkers, withimplications for bacterial identification. Appl EnvironMicrobiol 2009;75:4341–53.
[103] Demirev PA, Feldman AB, Kowalski P, Lin JS. Top-downproteomics for rapid identification of intactmicroorganisms. Anal Chem 2005;77:7455–61.
[104] Wynne C, Fenselau C, Demirev PA, Edwards N. Top-downidentification of protein biomarkers in bacteria withunsequenced genomes. Anal Chem 2009;81:9633–42.
[105] Ge Y, El-Naggar M, Sze SK, Oh HB, Begley TP, McLafferty FW,et al. Top down characterization of secreted proteins from
Mycobacterium tuberculosis by electron capture dissociationmass spectrometry. J Am Soc Mass Spectrom 2003;14:253–61.
[106] Pflieger D, Przybylski C, Gonnet F, Le Caer JP, Lunardi T,Arlaud GJ, et al. Analysis of human C1q by combinedbottom-up and top-down mass spectrometry: detailedmapping of post-translational modifications and insightsinto the C1r/C1s binding sites. Mol Cell Proteomics 2010;9:593–610.
[107] Ryan CM, Souda P, Bassilian S, Ujwal R, Zhang J, Abramson J,et al. Post-translational modifications of integral membraneproteins resolved by top-down Fourier transform massspectrometry with collisionally activated dissociation. MolCell Proteomics 2010;9:791–803.
[108] Zhang J, Dong X, Hacker TA, Ge Y. Decipheringmodificationsin swine cardiac troponin I by top-down high-resolutiontandem mass spectrometry. J Am Soc Mass Spectrom2010;21:940–8.
[109] Ge Y, Lawhorn BG, ElNaggar M, Sze SK, Begley TP, McLaffertyFW. Detection of four oxidation sites in viralprolyl-4-hydroxylase by top-down mass spectrometry.Protein Sci 2003;12:2320–6.
[110] Zhai H, Dorrestein PC, Chatterjee A, Begley TP, McLaffertyFW. Simultaneous kinetic characterization of multipleprotein forms by top down mass spectrometry. J Am SocMass Spectrom 2005;16:1052–9.
[111] Pesavento JJ, Mizzen CA, Kelleher NL. Quantitative analysisof modified proteins and their positional isomers by tandemmass spectrometry: human histone H4. Anal Chem 2006;78:4271–80.
[112] Smargiasso N, De Pauw E. Optimization of matrix conditionsfor the control of MALDI in-source decay of permethylatedglycans. Anal Chem 2010;82:9248–53.
[113] Chaurand P, Schwartz SA, Caprioli RM. Imaging massspectrometry: a new tool to investigate the spatialorganization of peptides and proteins in mammalian tissuesections. Curr Opin Chem Biol 2002;6:676–81.
[114] Debois D, Bertrand V, Quinton L, De Pauw-Gillet MC, DePauw E. MALDI-in source decay applied to massspectrometry imaging: a new tool for protein identification.Anal Chem 2010;82:4036–45.
[115] Lemaire R, Menguellet SA, Stauber J, Marchaudon V, Lucot JP,Collinet P, et al. Specific MALDI imaging and profiling forbiomarker hunting and validation: fragment of the 11Sproteasome activator complex, Reg alpha fragment, is a newpotential ovary cancer biomarker. J Proteome Res 2007;6:4127–34.
[116] Meistermann H, Norris JL, Aerni HR, Cornett DS, Friedlein A,Erskine AR, et al. Biomarker discovery by imaging massspectrometry: transthyretin is a biomarker forgentamicin-induced nephrotoxicity in rat.Mol Cell Proteomics 2006;5:1876–86.
[117] Rauser S, Marquardt C, Balluff B, Deininger SO, Albers C,Belau E, et al. Classification of HER2 receptor status in breastcancer tissues by MALDI imaging mass spectrometry.J Proteome Res 2010;9:1854–63.
[118] Cazares LH, Troyer D, Mendrinos S, Lance RA, Nyalwidhe JO,Beydoun HA, et al. Imaging mass spectrometry of a specificfragment of mitogen-activated protein kinase/extracellularsignal-regulated kinase kinase kinase 2 discriminates cancerfrom uninvolved prostate tissue. Clin Cancer Res 2009;15:5541–51.
[119] Lagarrigue M, Becker M, Lavigne R, Deininger SO, Walch A,Aubry F, et al. Revisiting rat spermatogenesis with MALDIimaging at 20–microm resolution. Mol Cell Proteomics2010;10 M110 005991.
[120] Schulz E, Karas M, Rosu F, Gabelica V. Influence of thematrixon analyte fragmentation in atmospheric pressure MALDI.J Am Soc Mass Spectrom 2006;17:1005–13.

934 J O U R N A L O F P R O T E O M I C S 7 4 ( 2 0 1 1 ) 9 2 0 – 9 3 4
[121] Mirza SP, Raju NP, Vairamani M. Estimation of the protonaffinity values of fifteen matrix-assisted laser desorption/ionization matrices under electrospray ionizationconditions using the kinetic method. J Am Soc MassSpectrom 2004;15:431–5.
[122] Bourcier S, Hoppilliard Y. B3LYP DFTmolecular orbitalapproach, an efficient method to evaluate the thermochemicalproperties of MALDI matrices. Int J Mass Spectrom 2002;217:231–4.
[123] Mormann M, Bashir S, Derrick PJ, Kuck D. Gas-phasebasicities of the isomeric dihydroxybenzoic acids andgas-phase acidities of their radical cations. J Am Soc MassSpectrom 2000;11:544–52.
[124] Demeure K, Gabelica V, De Pauw EA. New advances in theunderstanding of the in-source decay fragmentation ofpeptides in MALDI-TOF-MS. J Am Soc Mass Spectrom2010;21:1906–17.




























