Embed Size (px)
Citation preview

A
Aapihega©
K
C
P
0d
Journal of Steroid Biochemistry & Molecular Biology 105 (2007) 1–15
Review
Androgens and androgen receptors in breast cancer
B. Nicolas Dıaz-Chico a,b,∗, F. German Rodrıguez a,b, Ana Gonzalez a,c, Raquel Ramırez a,Cristina Bilbao a, A. Cabrera de Leon a,c, A. Aguirre Jaime a,c, Ricardo Chirino a,b,
Domingo Navarro a,b, Juan C. Dıaz-Chico a,b
a Instituto Canario de Investigacion del Cancer (ICIC), Canary Islands, Spainb Department of Biochemistry and Physiology, Faculty of Health Sciences, Universidad de Las Palmas de Gran Canaria,
P.O. Box 550, 35080 Las Palmas de Gran Canaria, Canary Islands, Spainc Research Unit, Hospital Universitario de La Candelaria, Servicio Canario de Salud, Santa Cruz de Tenerife, Spain
Received 19 June 2006; accepted 9 November 2006
bstract
Aromatase (CYP19) converts adrenal and ovarian androgens into estrogens, which supports the growth of estrogen-dependent breast cancers.nti-aromatase agents are displacing antiestrogens as the first-line treatment for estrogen receptor positive breast cancers. Androgens can act
s estrogen precursors, but besides this capability they can also directly act on breast cancer cells by binding to androgen receptors, which areresent in the majority of breast cancer specimens. Epidemiological and clinical evidences suggest that higher levels of circulating androgenncrease the risk of developing breast cancer. Androgen receptor gene polymorphisms which render the more transcriptionally active receptorsave been related to a lower risk of breast cancer. It is currently accepted that androgens act as antiproliferative agents in the presence ofstrogens in some breast cancer cell lines. However, emerging evidence suggests that direct androgenic activity might also stimulate cellrowth in a subset of estrogen-resistant breast tumors. Here we discuss the supporting evidence which proposes that androgens themselvesre actively involved in breast carcinogenesis and its clinical behaviour.
2007 Elsevier Ltd. All rights reserved.
eywords: Androgen; Androgen receptor; Gene polymorphism; Breast cancer; Carcinogenesis
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.1. Androgens and breast cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.2. Steroid hormones and breast cancer risk: epidemiological evidence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.3. Steroid hormones and breast carcinogenesis: current hypothesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2. Androgen receptors and breast cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62.1. Androgen receptor protein and breast cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72.2. The AR gene length polymorphisms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2.2.1. The GGC (polyglycine) repeat of the androgen receptor gene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2.2.2. The CAG (polyglutamine) repeat of the androgen receptor gene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82.2.3. Effects of expanded CAG repeats in the AR gene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82.2.4. The androgen receptor gene polymorphisms and plasma testosterone level . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93. AR polymorphisms and the risk of breast cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94. The AR polymorphisms and breast cancer prognosis and progression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
∗ Corresponding author at: Department of Biochemistry and Physiology, Faculty of Health Science, University of Las Palmas de Gran Canaria,.O. Box 550, 35080 Las Palmas de Gran Canaria, Canary Islands, Spain.Tel.: +34 928 451445; fax: +34 928 458653.
E-mail address: [email protected] (B. Nicolas Dıaz-Chico).
960-0760/$ – see front matter © 2007 Elsevier Ltd. All rights reserved.oi:10.1016/j.jsbmb.2006.11.019

2 B. Nicolas Dıaz-Chico et al. / Journal of Steroid Biochemistry & Molecular Biology 105 (2007) 1–15
5. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11. . . .. . .
1
1
wdogPtfatbshAfal
gti(
aibdeF
aeit[bccc
1e
d
Fdbp
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. Introduction
.1. Androgens and breast cancer
The ovarian production of steroid hormones throughout aoman’s lifetime is ample and varied, with important changesuring different life stages. Estrogens are the most prominentvarian hormones in terms of their morphogenetic and mito-enic capabilities, but they are secreted in low quantities.rogestagens (progesterone and 16�-OH progesterone) are
he steroid hormones most secreted by the ovaries during theertile period of a woman’s life. Androgens (androstenedionend testosterone) are necessary precursors for estrogen syn-hesis to occur in the ovary. Androgens are also secreted byoth ovaries and adrenals. They circulate in a concentrationimilar to estradiol during the preovulatory peak, and in aigher concentration during the rest of the menstrual cycle.fter menopause the secretion of progesterone and estrogens
alls dramatically, but androgens continue being secreted inn even higher proportion than during the fertile period ofife [1].
Since androgens are secreted by both ovaries and adrenal
lands, they constitute the dominant sex steroid hormoneshroughout a woman’s entire life. The main androgen secreteds dehidropeandrosterone, both sulphated and non-sulphatedDHEA-S or DHEA, respectively), but androstenedione isseam
ig. 1. Sex steroid hormone synthesis during menopause. Both adrenals and ovaehydroepiandrosterone-sulfate (DHEA-S). The enzymes 17�-hydroxysteroid dehrain, liver, adipose tissue and normal breast and pathological epithelium. The estroremenopausal tumors. Since breast cancer tissue also contains sulfatase activity, th
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
lso important and is a direct precursor of testosteronenvolved in the conversion of low-potency androgens secretedy both adrenals and ovaries into potent androgens and estra-iol are expressed in many cell types [1–3], including thepithelial and stromal breast cancer cells (summarized inig. 1).
Adaptation in order to survive in a low estrogenic milieufter menopause appears to be a crucial step in breast andndometrial epithelial cell biology. Such adaptation mightnvolve both increasing the capability of local estrogen syn-hesis and increasing the cell responsiveness to estrogen4–7]. This change could be brought about in epithelial cellsy increasing the levels of aromatase, estrogen receptors andoactivators. These adaptations would give the cells a higherapability of surviving when levels of estrogen are low, andould be at the basis of estrogen driven carcinogenesis [6,7].
.2. Steroid hormones and breast cancer risk:pidemiological evidence
Endogenous sex steroids have long been implicated in theevelopment of breast cancer [8]. Epidemiological evidence
upports that breast cancer risk is increased, relative to gen-ral population, among women who had an early menarchend is reduced among women who experience early naturalenopause or have a bilateral ovariectomy before the ageries produce low-potency androgens, predominantly androstenedione andydrogenase and aromatase are widespread in peripheral tissues, includinggen content of breast cancer cells during menopause is even higher than ine DHEA-S also behaves as a precursor of estrogens.
B.N
icolasD
ıaz-Chico
etal./JournalofSteroidB
iochemistry
&M
olecularB
iology105
(2007)1–15
3
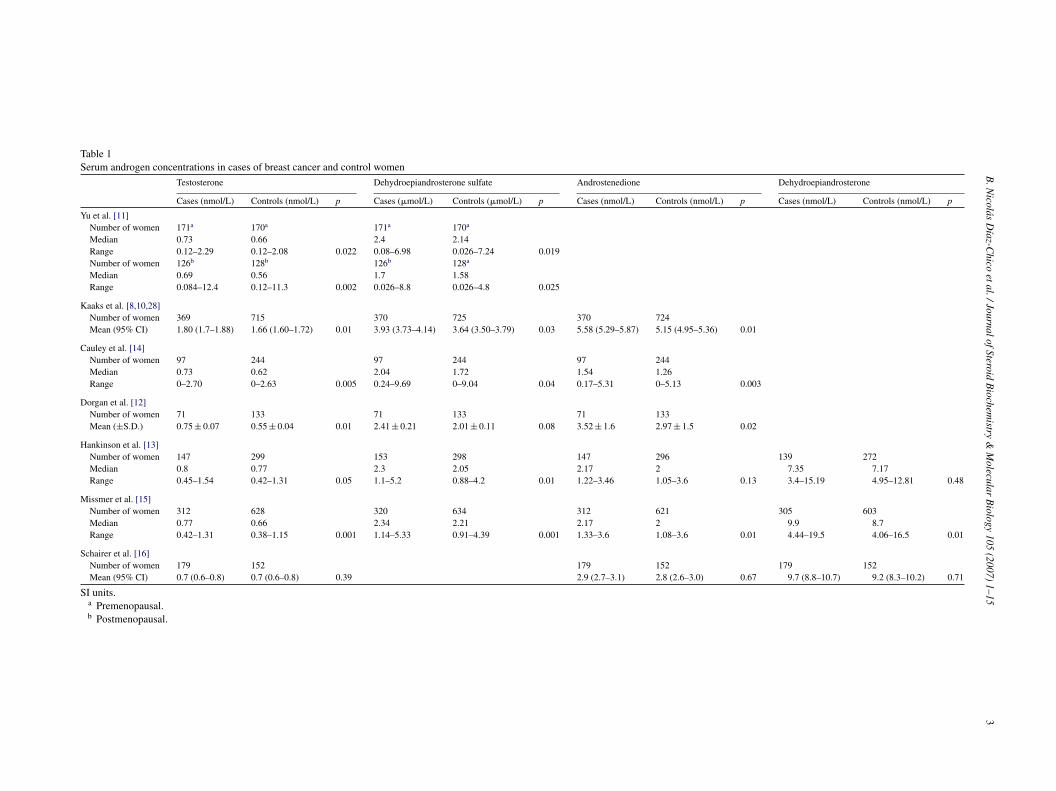
Table 1Serum androgen concentrations in cases of breast cancer and control women
Testosterone Dehydroepiandrosterone sulfate Androstenedione Dehydroepiandrosterone
Cases (nmol/L) Controls (nmol/L) p Cases (�mol/L) Controls (�mol/L) p Cases (nmol/L) Controls (nmol/L) p Cases (nmol/L) Controls (nmol/L) p
Yu et al. [11]Number of women 171a 170a 171a 170a
Median 0.73 0.66 2.4 2.14Range 0.12–2.29 0.12–2.08 0.022 0.08–6.98 0.026–7.24 0.019Number of women 126b 128b 126b 128a
Median 0.69 0.56 1.7 1.58Range 0.084–12.4 0.12–11.3 0.002 0.026–8.8 0.026–4.8 0.025
Kaaks et al. [8,10,28]Number of women 369 715 370 725 370 724Mean (95% CI) 1.80 (1.7–1.88) 1.66 (1.60–1.72) 0.01 3.93 (3.73–4.14) 3.64 (3.50–3.79) 0.03 5.58 (5.29–5.87) 5.15 (4.95–5.36) 0.01
Cauley et al. [14]Number of women 97 244 97 244 97 244Median 0.73 0.62 2.04 1.72 1.54 1.26Range 0–2.70 0–2.63 0.005 0.24–9.69 0–9.04 0.04 0.17–5.31 0–5.13 0.003
Dorgan et al. [12]Number of women 71 133 71 133 71 133Mean (±S.D.) 0.75 ± 0.07 0.55 ± 0.04 0.01 2.41 ± 0.21 2.01 ± 0.11 0.08 3.52 ± 1.6 2.97 ± 1.5 0.02
Hankinson et al. [13]Number of women 147 299 153 298 147 296 139 272Median 0.8 0.77 2.3 2.05 2.17 2 7.35 7.17Range 0.45–1.54 0.42–1.31 0.05 1.1–5.2 0.88–4.2 0.01 1.22–3.46 1.05–3.6 0.13 3.4–15.19 4.95–12.81 0.48
Missmer et al. [15]Number of women 312 628 320 634 312 621 305 603Median 0.77 0.66 2.34 2.21 2.17 2 9.9 8.7Range 0.42–1.31 0.38–1.15 0.001 1.14–5.33 0.91–4.39 0.001 1.33–3.6 1.08–3.6 0.01 4.44–19.5 4.06–16.5 0.01
Schairer et al. [16]Number of women 179 152 179 152 179 152Mean (95% CI) 0.7 (0.6–0.8) 0.7 (0.6–0.8) 0.39 2.9 (2.7–3.1) 2.8 (2.6–3.0) 0.67 9.7 (8.8–10.7) 9.2 (8.3–10.2) 0.71
SI units.a Premenopausal.b Postmenopausal.

4 d Bioch
ooa
eaacaadibdom
bDbcilgrm
Di[Nbo
csecbtobDil
cchtawcdpde
bmmhD
TS
R
S
S
B. Nicolas Dıaz-Chico et al. / Journal of Steroi
f 45 years. These observations implicate that both an earlynset and a longer lifetime duration of ovarian steroidogenicctivity influence breast cancer development.
Women who have elevated levels of androgens ofither adrenal and/or ovarian origin (i.e., DHEA, DHEAS,ndrostenedione, and testosterone) or estrogens (i.e., estronend estradiol) in the blood have an increased risk of breastancer [9,10]. Epidemiologic studies that have examinedssociations between breast cancer risk in a premenopausalnd postmenopausal plasma levels of sex steroids have pro-uced varying results [11–16] (Table 1). This type of studys particularly complicated for estradiol and progesteroneecause the circulating levels of these hormones vary greatlyuring the menstrual cycle, and a single blood sample takenn a given day of the cycle does not provide enough infor-ation about their levels.As regards to androgens, early data indicated a possi-
le inverse relationship between levels of adrenal androgenHEAS and its metabolites in the blood (and urine) andreast cancer risk in young women [17,18]. Recent carefullyontrolled studies [8,19] have found a statistically significantncrease in breast cancer risk for women who had elevatedevels of testosterone and comparatively low levels of pro-esterone. Even these studies do not find conclusive resultsegarding estrogen level and the risk of breast cancer in pre-enopausal women.In some studies, low serum or urine levels of DHEA,
HEAS, or their metabolites were associated with anncreased risk of breast cancer in premenopausal women
17,20,21]. Another recent prospective study within theurses Health Study cohort showed no clear associationetween breast cancer risk and premenopausal serum levelsf DHEA or DHEAS, irrespective of age at subsequent breastcptl
able 2erum androgen concentrations in blood and breast cancer tissue
Testosterone
Tumour tissue (pg/g) Blood (pg/mL) p
ecchione et al. [24]Number of cases 34 34Median 216 247Range 54–645 128–825 0
ecreto et al. [25]Number of cases 26 (<70 years) 26 (<70 years)Median 225 251Range 72–645 128–593 0Number of cases 24 (≥70 years) 24 (≥70 years)Median 196 294Range 54–644 110–825 0
Testosterone
Cysts fluid (ng/mL) Blood (ng/mL)
ecreto et al. [26]Number of cases 51 51Median 0.132 0.307Range 0.033–0.476 0.117–0.579
emistry & Molecular Biology 105 (2007) 1–15
ancer diagnosis [22]. In contrast, other studies [8] did nothow a reduction in breast cancer risk among women who hadlevated serum DHEAS, but did show a statistically signifi-ant direct association between serum levels of DHEAS andreast cancer risk. The risk was even more pronounced whenhe analysis was limited to women who were under the agef 49 years (the median age at diagnosis) when diagnosed ofreast cancer. By contrast, elevated blood levels of DHEA orHEAS have been consistently associated with an increase
n the risk of breast cancer in postmenopausal women in allarge prospective studies thus far conducted [8–10].
Epidemiologic findings have led to opposing hypothesisoncerning the relationship between the risk of breast can-er and DHEA or DHEAS (the levels of which are usuallyighly correlated with DHEA levels in blood). To reconcilehese contrasting observations, it has been proposed that, in
highly estrogenic milieu, such as that of premenopausalomen, DHEA might act as an estrogen antagonist through
ompetitive binding of its metabolite 5-androstene-3�,17�-iol (ADIOL) to the estrogen receptor �. By contrast, inostmenopausal women, who have much lower serum estra-iol levels, ADIOL would be more likely to act as a moderatestrogen agonist [23].
We must bear in mind that these case-control studies haveeen carried out measuring the serum level of steroid hor-ones, and there is increasing evidence to show that theyight not properly reflect the actual concentration of these
ormones inside both normal and neoplasic breast cells [1,2].ata on intracellular concentration of androgens in breast
ancer tissues are scarce [24–26] (Table 2), and as in hap-ens the case of estrogens [1], they cannot be inferred fromhe serum concetrations of androgens. In addition, intracel-ular dinamics of androgens and its capability for influencing
Dihydrotestosterone
Tumour tissue (pg/g) Blood (pg/mL) p
34 34249 86
.1513 110–698 29–222 0.0001
26 (<70 years) 26 (<70 years)259 88
.6099 116–698 40–200 0.000124 (≥70 years) 24 (≥70 years)
200 123.0001 78–467 20–290 0.0005
Dihydrotestosterone
p Cysts fluid (ng/mL) Blood (ng/mL) p
52 520.155 0.16
0.0001 0.023–1.57 0.074–0.331 0.013

B. Nicolas Dıaz-Chico et al. / Journal of Steroid Bioch
Table 3Blood concentration and relative binding affinities for the Androgen Recep-tor of endogenous androgens
Steroid hormone Relative bindingaffinitya
Blood concentration(nM)
5�-Dihydrotestosterone(DHT)
1.0 2
Testosterone 0.2 20Progesterone 0.02 1Estradiol 0.015 0.15Cortisol <0.0001 300Aldosterone <0.0001 4
Hipakka and Liao, 1995. Androgen receptors and action, in: De Groot (Ed.)Endrocinology, third ed. Saunders, New York.
Ta
astAtb
1c
rtaraa[cilschu
estaeCiaencb
p
csTitprsttrHrcds
rcpmoifiwcndh
oeohcmobsbcfbrcpa
aprt
a The Kd for DHT of the androgen receptor is in the range of 0.2–0.4 nM.he relative binding affinity values are normalized to DHT, which is givenvalue of 1.0.
ndrogen-dependent gene expression include other variables,uch as the Kd of each androgen for binding the AR (Table 3),he ability of each individual compound to form productiveR dimers able to bind coactivators, and the capability of
he androgen-AR-coactivaor(s) to attract other proteins andeing able to induce gene transcription [27].
.3. Steroid hormones and breast carcinogenesis:urrent hypothesis
Three hypothesis have been at the centre of the debateegarding steroid hormones and the risk of breast cancer. Inhe 1960s, Grattarola [17] put forward the “ovarian hyper-ndrogenism/luteal inadequacy” hypothesis, stating that theisk of breast cancer is increased among women who haven excess of ovarian androgens, chronic anovulation and anssociated reduction of luteal-phase progesterone production28]. This hypothesis was based on the observation that breastancer patients often show hyperplasia of the endometrium,ndicative of an excess of ovarian androgens, chronic anovu-ation and progesterone deficiency. The hypothesis receivedome initial confirmation from a series of traditional case-ontrol studies that showed that breast cancer patients haveigher plasma or urinary concentrations of testosterone or itsrinary metabolites [29,30] than cancer-free control subjects.
The ovarian hyperandrogenism/luteal inadequacy hypoth-sis has not been so readily acceptable for various reasonsuch as the fact that it describes androgens as being ableo induce proliferation or cell arrest “in vitro” and as beingble to increase, as well as to slow down the growth ofxperimentally induced mammary tumors in animals [31,32].onflictive results are due to the fact that, in addition to hav-
ng direct androgenic effects on breast tissue, elevated serumndrogen levels may lead indirectly to increased estrogenicxposures of breast tissue, because all steroidogenic enzymesecessary for the formation of estrogens from androgenic pre-
ursor molecules are present in normal mammary tissues andreast cancer specimens.A second major hypothesis is the “estrogen-plus-rogesterone” hypothesis, which states that the risk of breast
dioa
emistry & Molecular Biology 105 (2007) 1–15 5
ancer would be increased in women with an elevated expo-ure to a combination of both estrogens and progesterone.his hypothesis was largely motivated by observations of
ncreased proliferation rates of breast epithelium duringhe luteal phase of the menstrual cycle, when the ovariesroduce both estradiol and progesterone [33]. A moreecent observation that can be interpreted as providingupport for the ‘estrogen-plus-progesterone’ hypothesis ishat postmenopausal estrogen-plus-progestin replacementherapy increases breast cancer risk to a greater extent thaneplacement therapies that contain estrogen alone [34].owever, the interpretation of this second observation with
espect to the possible actions of natural progesterone isomplicated by the fact that many synthetic progestins areerived from androgenic precursor molecules and also haveome androgenic activity [34].
Experimental studies have also provided conflictingesults regarding the possible role of progesterone in breastancer development. Some animal studies have shown thatrogesterone is a critical determinant for the initiation ofammary tumors in response to carcinogens [35]; however,
ther studies have shown that progesterone reduces estrogen-nduced proliferation of breast epithelial cells [36,37]. Thending of an increased risk of breast cancer among womenith elevated premenopausal serum testosterone levels and
omparatively low serum levels of progesterone [28] doesot support the “estrogen-plus-progestin” hypothesis, butoes provide support for the ovarian hyperandrogenismypothesis.
A third hypothesis regarding the relationships betweenvarian sex steroids and breast cancer risk stipulates thatstrogens stimulate tumor development independently ofther hormones, i.e., the “estrogen-alone” hypothesis. Thisypothesis has gained considerable support because ofonsistent reports from prospective studies stating that post-enopausal women who have elevated serum concentrations
f estrone and estradiol have an increased risk of developingreast cancer [8]. Experimental studies have also providedtrong and consistent evidence that estrogens can promotereast tumor development and growth [38,39]. However,ase-control studies on premenopausal women [34] have notound a clear relationship between serum estrogen levels andreast cancer risk. It is conceivable that breast cancer risk iselated in a nonlinear fashion to circulating estrogen levels. Alear, direct relationship could exists only within the lower,ostmenopausal range of endogenous estrogen levels, but nott higher, premenopausal estrogen concentrations.
These studies [17,28] might be biased because theyssayed only one blood sample per woman, and it is alsoossible that the measured estradiol levels may not have accu-ately represented the average, long-term estradiol levels inhe women. Several studies that have addressed the repro-
ucibility of serum estradiol measurements made over timen premenopausal women found that a single measurementf estradiol does not accurately reflect a woman’s long-termverage blood concentration, unlike single measurements of
6 d Bioch
agicrcm
ecfibmp
2
ctsdbahdtrD
stfeo(w
cHdpiAphwa
shfthvg
Fcde(tT
B. Nicolas Dıaz-Chico et al. / Journal of Steroi
ndrogens [40]. In addition, the intracellular levels of estro-en and other steroid hormones are considered of relevancen breast carcinogenesis, and is of difficult assessment inase-control studies [1,2]. Although it is possible that theelationship between hormone levels and breast cancer riskould change abruptly after a woman’s transition throughenopause, such an abrupt change is unlikely.In conclusion, prospective cohort studies provide strong
vidence that the risk of breast cancer is directly related toirculating levels of testosterone and androstenedione. Thisnding appeared to be equally applicable to women bothefore and after the menopause. No clear statement can beade at this point on the association between premenopausal
rogesterone or estrogen levels and the risk of breast cancer.
. Androgen receptors and breast cancer
Androgens act on the target cells by binding to an intra-ellular protein, the androgen receptor (AR), a member ofhe nuclear receptors family, that are ligand-dependent tran-cription factors (Fig. 2). The AR protein possesses variousomains that mediate its different functions: (1) the DNA-inding domain (DBD), which contains two zinc fingersble to specifically interact with short DNA sequences calledormone response elements (HREs); (2) the ligand-binding
omain (LBD), where androgens and antiandrogens bind tohe AR by interacting with specific aminoacids; (3) the hingeegion, located between the ligand-binding domain and theNA-binding domain; (4) the nuclear localization function, aamr
ig. 2. The androgen receptor gene and protein. The AR gene is located at the Xontains two trinucleotide repeats that are polymorphic: the CAG repeat (polyglutaomains which confer different capabilities: the ligand-binding domain (LBD) whnables greater flexibility to the molecule; a nuclear localization domain (NLD), wDBD) which constitutes the part of AR that interacts with the hormone responsiveranscription activating functions (TAF), TAF-1 which is flanked and influenced byAF-2 which is fully operative only after the AR binds to an agonist ligand.
emistry & Molecular Biology 105 (2007) 1–15
tretch of basic aminoacids that when exposed help to locatehe AR in the cell nuclei; (5) two transcription-activationunctions (TAF), TAF-1 which is androgen-independent (itxists in the unliganded AR) and is located at the N-terminusf the AR protein, and TAF-2 which is androgen-dependentit is conformed after AR binds to an androgen) and co-locatesith the LBD [41].Unliganded AR exists in the cytoplasm as a complex
ontaining several molecular chaperones, including Hsp90,sp70 and Hsp56. The AR possesses a higher affinity forihidrotestosterone (DHT) than for testosterone, the two mostowerful natural androgens. The process of hormone bind-ng to the LBD results in a conformational change of theR molecule that promotes shedding of cytosolic heat shockroteins and translocation into the nucleus. Once bound to aormone, the AR is able to form homodimers that associateith nuclear chaperons and coactivators, and constitutes the
ctive form of the receptor.Coactivators (and co-repressors) bind to the two main tran-
cription activating functions of the AR. The final result oformone–receptor interaction is the conformation of an activeorm of AR able to recognize the HREs located at (or close to)he promoter region of androgen-dependent genes. Once theomodimers bind to HREs it recruits coactivators and acti-ate the transcription machinery, thus increasing the specificene transcription by one or two orders of magnitude.
An intact proteasome pathway is necessary for full ARctivity [42–44]. This can be explained by the cyclic recruit-ent of transcriptional activators (and of their cofactors) to
esponsive promoters followed by degradation, as an ulti-
chromosome, spans 90 kbases and is composed by 8 exons. The exon 1mine) and the GGN repeat (polyglycine). The AR protein contains severalich binds to androgens and antiandrogens; the hinge domain (HD) whichhich enables capability for nuclear localization; the DNA-binding domainelements at the promoter region of the androgen-regulated genes; and twothe two polymorphic tracts and is operative in the absence of ligand, and

Bioch
mmatsbdmrt
tTrtfbpow
toraTpg
2
heiptwbab((tbtr
erao[btm
bt[tp
tplniEstcticis
2
Xowt(a
e1pAab[r
2r
((aa[(C
G
B. Nicolas Dıaz-Chico et al. / Journal of Steroid
ate and necessary step of the transcription process. Thisodel suggests that new transcriptionally active complexes
re formed as long as newly formed AR homodimers bindo the HREs, and permits tight control of gene expres-ion. Accordingly, transcriptional activity has been found toe necessary for proteasome-dependent AR and coactivatoregradation, and an overlap between motifs which deter-ine protein turnover and transactivation domains has been
eported for many unstable transcription factors, especiallyhose involved in cell growth control [44–46].
There is increasing evidence supporting non-ranscriptional activity of the AR in target cells [47,48].his activity refers to a rapid (non-genomic and/or genomic)
esponse to androgens and is the result of an interaction ofhe AR with src and perhaps with other proteins downstreamrom the membrane receptors (the so-called cross-talketween membrane and nuclear receptor pathways). Thisathway might produce rapid changes in ionic channels,r even activation by androgen of the expression of genes,hich are indirectly regulated by the AR.Unliganded AR can also be activated by phosphoryla-
ion by several protein-kinases that are downstream targetsf membrane receptor signaling pathways [49,50]. Phospho-ylated AR might activate the expression of some of thendrogen-responsive genes, even in the absence of ligand.his might partly explain the presence of androgen-inducedroteins in prostate cancer patients treated with total andro-en suppression therapies.
.1. Androgen receptor protein and breast cancer
The presence of AR protein in breast cancer specimensas been known since the early studies with tritiated DHTxchange assays carried out by the McGuire’s laboratoryn the seventies [51]. However, most of the studies wereerformed using specific anti-AR antibodies in immunohis-ochemical (IHC) studies. Thus, positive AR immunostainingas described in a range of 31% (34) to 85% [52] ofreast cancers, but most authors cite that 60–70% of tumoursre AR-positive [53,54]. Since almost all the AR-positivereast tumours are also positive for prostate-specific antigenPSA) (98%) and for the gross cystic disease fluid protein-15GCDFP-15) (92%), two androgen-dependent proteins [52],here is no doubt that the AR is functionally active in humanreast cancer. Androgen itself has been used effectively inreating some breast cancer patients and the average responseate is about 20% [55].
A significant association has been found between thexpression of AR, estrogen receptors (ER) and progesteroneeceptors (PgR) as determined immunohistochemically ondjacent sections [53,54]. However, a significant percentagef tumours are positive for AR and negative for ER and PR
56]. This reveals the independent expression of AR in humanreast cancer. AR shows significant associations with impor-ant clinical and pathologic prognostic factors [54]. The vastajority of Grade 1 carcinomas are positive for AR (90%),daac
emistry & Molecular Biology 105 (2007) 1–15 7
ut a significant number of poorly differentiated carcinomashat are ER-negative and PR-negative, were also AR-positive53,54]. AR status has been related to age, menopausal sta-us and histopathological grade, and is accepted as a goodrognosis marker [57,58].
Whereas ER-positivity has been related to node negativeumors, the level of expression of both AR genes and ARroteins in breast cancer was found to be positively corre-ated with node invasiveness [57,58]. The AR category didot reveal any significant prognostic information concern-ng relapse free survival. However, in univariate analysis ofR-negative tumors, patients with AR-positive tumors hadignificantly better disease-free survival rate [54]. In a mul-ivariate analysis, the AR category (median value used as aut-off point) was shown to be an independent predictor ofhe likelihood of axillary metastases [59]. This associations suggestive of a specific action of the androgen on breastancer cells that gives them an advantage when coloniz-ng lymphatic nodes, an interesting finding devoid of furthertudies.
.2. The AR gene length polymorphisms
The AR is codified by the AR gene which is located on thechromosome (q11.2–q12). Thus, the AR-mediated effects
f androgen in males depend on a single AR allele. In femalesith two different AR alleles, random X-chromosome inac-
ivation leads to effects of different alleles in different cellsFig. 2). The AR gene spans 90 kbases, contains eight exonsnd encodes for a protein of around 917 amino acids [60,61].
The AR gene possesses two length polymorphisms at thexon 1: a 9–39 CAG repeat (polyglutamine, polyQ) and a4–27 GGN repeat (polyglycine, polyG). Together, theseolymorphisms make 90% of women heterocygotic for theR gene. The polyglutamine and polyglycine tracts flank the
ctivating function-1 (AF-1) of the AR protein and both haveeen reported to be modulators of AR transcriptional activity62]. Short alleles of both CAG and GGN repeats have beeneported to increase the risk of prostate cancer [63].
.2.1. The GGC (polyglycine) repeat of the androgeneceptor gene
The polyglycine tract in the AR protein is encoded byCGT)3GGG(GGT)2(GGC)n, an invariant six-glycine tractGGT/GGG) followed by a polymorphic GGC repeat. Mostuthors refer to the polyglycine tract as being encoded byGGN repeat, equivalent to the GGC repeat plus 6 triplets
45]. About 90% of normal AR contains 16–18 GGC repeats22–24 glycines). It is less polymorphic in length than theAG repeat [61].
Natural occurrence of an AR gene completely lacking theGN repeat has not been reported. Neither has any known
isease caused by abnormal GGN tracts (44). However, anssociation has been described between longer GGN lengthnd the risk of cryptorchidism and penile hypospadias, bothonditions considered consequences of low androgenicity
8 d Bioch
[ms4hcla
hnAAlirstanr
eD9hloofC
aDiot
2r
mCtiie
Vrtstrrs
gt
becdtCtotmt
laMliotrcdawl
2
o(T(mp[eAttbltwct
trf
B. Nicolas Dıaz-Chico et al. / Journal of Steroi
64,65]. Conversely, the shorter GGN repeats appear to beodestly associated with prostate cancer risk. A review of
tudies on GGC and prostate cancer, comprising a total of274 cases and 5275 controls, found that short GGN repeatsave an OR of 1.31 (1.06–1.61) for developing prostate can-er. These observations suggest that the polyglycine repeatength could modulate net AR protein activity, thus playingrelevant biological role.
The biological effects of changing the GGN repeat lengthave not been as widely studied as those of CAG. No sig-ificant effects of expanded GGN on androgen-dependentR transactivation activity have been described. However,R protein levels were inversely affected by the GGN repeat
ength and the net amount of AR activity per cell was highern cells expressing AR with a short polyglycine repeat. Theeason seems to be that the GGC repeat can form a hairpintructure in AR mRNA, whose stability may interfere withranslation. The ability of a short GGC repeat to enhancendrogen action provides a biologically plausible mecha-ism supporting reports which demonstrate that a short GGCepeat in the AR gene is a risk factor for prostate cancer [62].
The polymorphic polyglycine repeat is located near thend of the N-terminal transactivation domain, adjacent to theNA-binding domain. Deletion of the GGN repeat leads to0% reduction in AR transactivation activity. Authors thatave studied the effect of different GGN and CAG tractengths [62,66] have reported that the magnitude of the effectf physiological differences in repeat length on AR proteinr net AR activity could be greater for the GGC repeat (2.7-old difference between GGC13 versus GGC17) than for theAG repeat (40% difference, maximum) [66].
It has been proposed that the polyglycine repeat functionss a flexible hinge between the transactivation domain and theNA-binding domain [62]. Flexibility may be required for
nteractions between the N-terminal and C-terminal domainsr between the DNA-binding domains of dimerized receptorshat are required for AR transcriptional activity [67].
.2.2. The CAG (polyglutamine) repeat of the androgeneceptor gene
The polyglutamine tract of the AR is encoded by a poly-orphic CAG (glutamine) repeat followed by an invariantAA (also glutamine) codon located at the N-terminus of
he exon 1 [61]. Most authors refer to the CAG repeat lengthn the gene, but some refer to the glutamine repeat lengthn the protein. The equivalence is: the CAG repeat length isqual to the glutamine repeat length minus one.
The normal CAG repeat has between 6 and 39 repeats.ariation of the CAG-AR length polymorphism shows
emarkable ethnical differences. African populations havehe shortest CAG repeats, whereas Asian populations pos-ess the longest and Caucasians and America Indians are in
he middle [67,68]. African-American men have an increasedisk of developing prostate cancer when compared with otheracial and ethnic groups and they also have a greater propen-ity for developing aggressive prostate cancer. The CAG-ARlHlA
emistry & Molecular Biology 105 (2007) 1–15
ene polymorphisms are considered a potential risk factorhat may explain these racial differences in prostate cancer.
The availability of AR in target cells is regulated by a num-er of variables, including translational and posttranslationalvents, receptor cleavage and further processing. A signifi-ant effect of CAG repeat length on AR protein level has beenescribed [66] but the molecular mechanism involved has yeto be determined. As occurs in the case of GGC repeats, theAG repeats can form hairpin structures, which could affect
he AR mRNA degradation susceptibility or the efficiencyf movement along the ribosome which in turn could affectranslation efficiency. Thus, if CAG repeat length affects AR
RNA stability, this could provide an additional mechanismo help regulate AR protein levels.
The effect of different CAG tract lengths in the physio-ogical range (18–25 CAG repeats) on the AR transcriptionalctivity is modest [62,66] (40% difference, maximum) [66].ost of the experimental studies report on the effects of
engthening either the CAG or the GGN repeats, maintain-ng the other repeats as a constant. However, there are recentbservations on diverse human tumors [69,70] suggestinghat combinations of different repeat length in the normalange could increase the risk of developing cancer or causeancers with different clinical behaviour. It remains to beetermined whether the effects of CAG and GGC repeats onndrogen action are independent, since GGC repeat lengthas held constant when studying the effect of CAG repeat
ength on AR function and vice-versa.
.2.3. Effects of expanded CAG repeats in the AR genePolymorphisms of the androgen receptor were first rec-
gnized in patients of spinal and bulbar muscular atrophySBMA), all of whom had longer than normal AR genes [71].he relationship between an abnormally long CAG repeat
40–62 CAGs) in the exon 1 of the AR gene and the develop-ent of SBMA was the first observation to suggest that the
olyglutamine tract might play a critical role in AR function72,73] (Fig. 1). The biological evidence supports the hypoth-sis that an increased number of CAG repeats decreases theR-dependent responses, even when there is an excess of
estosterone. An important finding is that CAG repeats longerhan 40 provoke a disfunction in protein processing that mighte responsible for motor neuron death in SBMA. Mutant AR,ike other proteins with expanded glutamine tracts, may exertoxic affects by disrupting the ubiquitin-proteasome path-ay, by sequestering other factors such as transcriptional
o-regulators or by impairing synaptic function and axonalransport [74,75].
Deletion of the CAG repeat enhances AR activity [76] andhere is an inverse correlation between the number of CAGepeats (above 23) and AR transcriptional activity [77]. Dif-erences in normal AR-CAG repeat length, i.e., CAG repeat
engths in the range of 11–31, may also affect AR function.owever, recent data indicate that only certain CAG repeatengths within the normal range of 14–25 significantly affectR protein level and transactivation activity. Thus, an inverse

Bioch
etbcas[i(
CdahiA(amalmprcm
tottpgPiisAp
ltctoanptt
tTiup
dtdawt
wCbtiImde
2p
ofomFtChapiwsgp
3
kbgha
ri1abT
B. Nicolas Dıaz-Chico et al. / Journal of Steroid
ffect of 23, 43, and 65 CAG repeats on AR mRNA and pro-ein levels has been reported, whereas 14 and 23 CAG repeatsoth had the same effect. Rebbeck et al. [78] reported thatertain CAG repeat lengths were optimal for transactivationctivity and that the CAG repeat did not affect the AR tran-criptional activity in transfection assays. Finally, Ding et al.66] found that the main difference in transactivation activityn the normal range corresponded to 15 and 17 CAG repeatsa 40% increase with respect to 22 repeats).
Experimental data coincides with the observation that 17AG alleles are substantially more common in men whoevelop aggressive prostate cancer than in men who developless aggressive form of the disease and in the general
ealthy population [79,80]. In addition, case-control stud-es have also reported a higher than expected frequency ofR alleles with a short CAG repeat [64,65] in the germ line
non-tumor, inherited) DNA of men who developed a moreggressive form of prostate cancer. A biologically plausibleechanism to account for these data would be that an AR
llele with a shorter CAG repeat yields a higher AR proteinevel or a more transcriptionally active AR, thereby mediating
ore efficacious growth stimulation of androgen-dependentrostate epithelial cells. A short (but still within the normalange) CAG allele could thereby increase the risk of prostateells that undergo carcinogenic transformation developingore efficiently into a clinically significant cancer.Effects of the polyglutamine tract lengths on transactiva-
ion activity may be due to the effect of these repeat lengthsn the strength of interaction between the AR and otherranscription factors and co-regulators. The tertiary struc-ure of the AR polyglutamine repeat is unknown and atresent is not possible to predict how specific changes inlutamine repeat length might affect AR protein structure.olyglutamine tracts can form b-sheets and hydrogen bond
nteractions between b-sheets can mediate protein–proteinnteractions. An effect of glutamine repeat length on b-sheettability and protein–protein interactions could thereby affectR transactivation activity via an effect on transcription com-lex formation [81–84].
The ubiquitin proteasome pathway has only recently beeninked to nuclear hormone receptor-regulated gene transcrip-ion, although it had been previously well connected withell cycle regulation, signal transduction and cell differentia-ion of higher eukariotes [85]. Characterization of a numberf ubiquitin proteasome pathway enzymes as coactivatorsnd observations that several nuclear receptors are ubiquiti-ated and degraded in the course of their nuclear activitiesrovide evidence that ubiquitin proteasome-mediated pro-ein degradation plays an integral role in androgen-dependentranscription.
In addition to AR, studies have revealed that its coactiva-ors are also ubiquitinated and degraded via the proteasome.
he notion that the ubiquitin proteasome pathway is involvedn gene transcription is further strengthened by the fact thatbiquitin proteasome pathway enzymes are recruited at theromoters of target genes and that proteasome-dependent
r
ta
emistry & Molecular Biology 105 (2007) 1–15 9
egradation of nuclear receptors is required for efficientranscriptional activity. These findings suggest that proteinegradation is coupled with nuclear receptor coactivationctivity [85]. It is possible that the ubiquitin proteasome path-ay modulates transcription by promoting remodelling and
urnover of the nuclear receptor–transcription complex.The magnitude of differences in the transcriptional activity
hen CAG repeat length is within the normal range (14–25AG repeats) might be considered modest. However, it haseen suggested that small differences in androgen stimula-ion could have a large, cumulative effect over the life of anndividual which may well be physiologically relevant [71].n fact, one might expect modest effects to be normal in poly-orphisms that do not cause obvious disease whereas severe
iseases are more likely in polymorphic alleles that have largeffects on function [72].
.2.4. The androgen receptor gene polymorphisms andlasma testosterone level
In women, about 25% of testosterone is produced in thevaries, 25% in the adrenals and the remaining 50% is derivedrom peripheral conversion of proandrogens [20]. Since thevary expresses the AR, it is possible that the AR genotypeight affect the ovarian hormones, including Testosterone.our studies have investigated testosterone levels in relation
o the different AR CAG genotypes in women [86–89]. ShortAG repeat lengths have been reported to be associated withigher testosterone levels in young girls with ovarian hyper-ndrogenism [87], as well as in premenopausal [86] andostmenopausal women [88]. Hietala et al. found a decreasen the Testosterene levels in oral contraceptive user youngomen bearing long GGC repeats. These data suggest that
hort CAG and GGC repeats might increase either the andro-en sensitivity or a stimulatory effect of the AR on androgenroduction in women.
. AR polymorphisms and the risk of breast cancer
The presence of AR in BC tissue and cell lines has beennown for more than 30 years [90,91]. Several studies haveeen conducted to examine the effects of androgens on therowth of AR-positive breast cancer cell lines. These studiesave reported both inhibitory and stimulatory effects but thesere specific to the cell line under study [91].
A mutation in the DNA-binding domain of the AR gene,esulting in an inability to bind androgens, was first reportedn a pair of brothers with breast cancer [92]. In a study of3 male breast cancer patients one was observed to carrysimilar mutation [93]. In another small study of 11 male
reast cancer patients [67] this mutation was not observed.hese results first suggested that an AR mutation may play a
ole in the development of breast cancer in some males.The long AR-CAG repeat has been associated with a statis-
ically significant increase in BC risk in both Caucasians [94]nd Philippine women [95]. Similar results were described
10 B. Nicolas Dıaz-Chico et al. / Journal of Steroid Biochemistry & Molecular Biology 105 (2007) 1–15
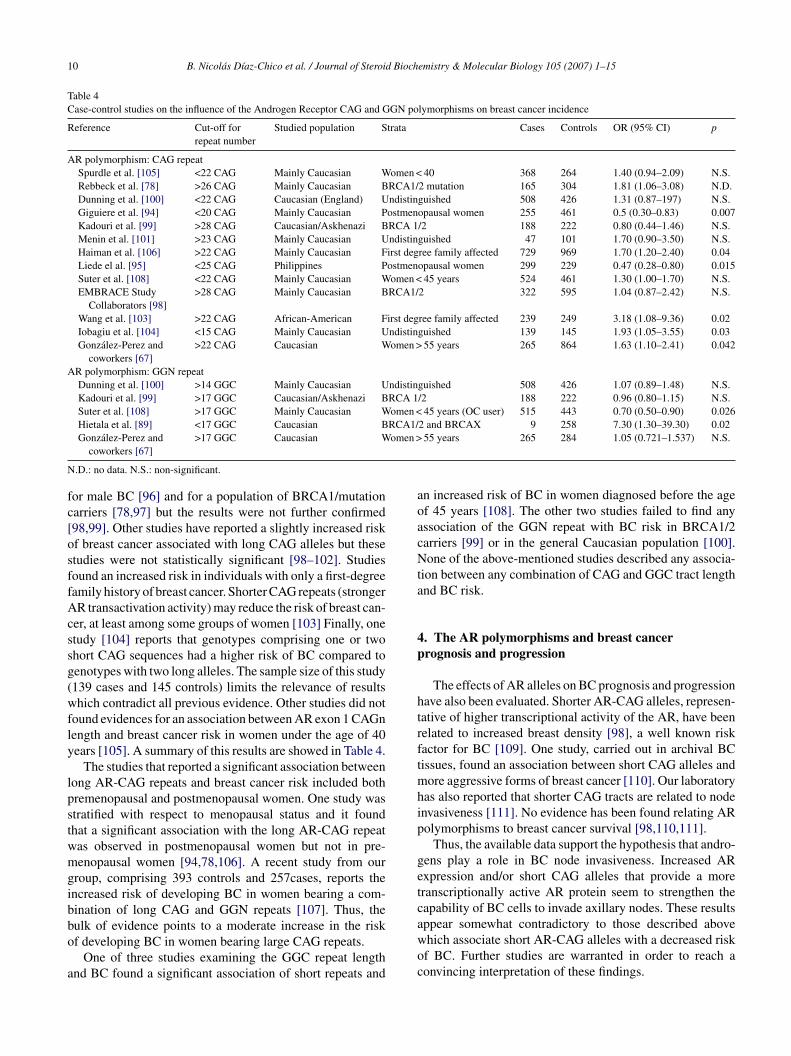
Table 4Case-control studies on the influence of the Androgen Receptor CAG and GGN polymorphisms on breast cancer incidence
Reference Cut-off forrepeat number
Studied population Strata Cases Controls OR (95% CI) p
AR polymorphism: CAG repeatSpurdle et al. [105] <22 CAG Mainly Caucasian Women < 40 368 264 1.40 (0.94–2.09) N.S.Rebbeck et al. [78] >26 CAG Mainly Caucasian BRCA1/2 mutation 165 304 1.81 (1.06–3.08) N.D.Dunning et al. [100] <22 CAG Caucasian (England) Undistinguished 508 426 1.31 (0.87–197) N.S.Giguiere et al. [94] <20 CAG Mainly Caucasian Postmenopausal women 255 461 0.5 (0.30–0.83) 0.007Kadouri et al. [99] >28 CAG Caucasian/Askhenazi BRCA 1/2 188 222 0.80 (0.44–1.46) N.S.Menin et al. [101] >23 CAG Mainly Caucasian Undistinguished 47 101 1.70 (0.90–3.50) N.S.Haiman et al. [106] >22 CAG Mainly Caucasian First degree family affected 729 969 1.70 (1.20–2.40) 0.04Liede el al. [95] <25 CAG Philippines Postmenopausal women 299 229 0.47 (0.28–0.80) 0.015Suter et al. [108] <22 CAG Mainly Caucasian Women < 45 years 524 461 1.30 (1.00–1.70) N.S.EMBRACE Study
Collaborators [98]>28 CAG Mainly Caucasian BRCA1/2 322 595 1.04 (0.87–2.42) N.S.
Wang et al. [103] >22 CAG African-American First degree family affected 239 249 3.18 (1.08–9.36) 0.02Iobagiu et al. [104] <15 CAG Mainly Caucasian Undistinguished 139 145 1.93 (1.05–3.55) 0.03Gonzalez-Perez and
coworkers [67]>22 CAG Caucasian Women > 55 years 265 864 1.63 (1.10–2.41) 0.042
AR polymorphism: GGN repeatDunning et al. [100] >14 GGC Mainly Caucasian Undistinguished 508 426 1.07 (0.89–1.48) N.S.Kadouri et al. [99] >17 GGC Caucasian/Askhenazi BRCA 1/2 188 222 0.96 (0.80–1.15) N.S.Suter et al. [108] >17 GGC Mainly Caucasian Women < 45 years (OC user) 515 443 0.70 (0.50–0.90) 0.026Hietala et al. [89] <17 GGC Caucasian BRCA1/2 and BRCAX 9 258 7.30 (1.30–39.30) 0.02
omen >
N
fc[osffAcssg(wfly
lpstwmgibbo
a
aoacNta
4p
htrftmhip
getcappear somewhat contradictory to those described above
Gonzalez-Perez andcoworkers [67]
>17 GGC Caucasian W
.D.: no data. N.S.: non-significant.
or male BC [96] and for a population of BRCA1/mutationarriers [78,97] but the results were not further confirmed98,99]. Other studies have reported a slightly increased riskf breast cancer associated with long CAG alleles but thesetudies were not statistically significant [98–102]. Studiesound an increased risk in individuals with only a first-degreeamily history of breast cancer. Shorter CAG repeats (strongerR transactivation activity) may reduce the risk of breast can-
er, at least among some groups of women [103] Finally, onetudy [104] reports that genotypes comprising one or twohort CAG sequences had a higher risk of BC compared toenotypes with two long alleles. The sample size of this study139 cases and 145 controls) limits the relevance of resultshich contradict all previous evidence. Other studies did not
ound evidences for an association between AR exon 1 CAGnength and breast cancer risk in women under the age of 40ears [105]. A summary of this results are showed in Table 4.
The studies that reported a significant association betweenong AR-CAG repeats and breast cancer risk included bothremenopausal and postmenopausal women. One study wastratified with respect to menopausal status and it foundhat a significant association with the long AR-CAG repeatas observed in postmenopausal women but not in pre-enopausal women [94,78,106]. A recent study from our
roup, comprising 393 controls and 257cases, reports thencreased risk of developing BC in women bearing a com-ination of long CAG and GGN repeats [107]. Thus, theulk of evidence points to a moderate increase in the risk
f developing BC in women bearing large CAG repeats.One of three studies examining the GGC repeat lengthnd BC found a significant association of short repeats and
woc
55 years 265 284 1.05 (0.721–1.537) N.S.
n increased risk of BC in women diagnosed before the agef 45 years [108]. The other two studies failed to find anyssociation of the GGN repeat with BC risk in BRCA1/2arriers [99] or in the general Caucasian population [100].one of the above-mentioned studies described any associa-
ion between any combination of CAG and GGC tract lengthnd BC risk.
. The AR polymorphisms and breast cancerrognosis and progression
The effects of AR alleles on BC prognosis and progressionave also been evaluated. Shorter AR-CAG alleles, represen-ative of higher transcriptional activity of the AR, have beenelated to increased breast density [98], a well known riskactor for BC [109]. One study, carried out in archival BCissues, found an association between short CAG alleles and
ore aggressive forms of breast cancer [110]. Our laboratoryas also reported that shorter CAG tracts are related to nodenvasiveness [111]. No evidence has been found relating ARolymorphisms to breast cancer survival [98,110,111].
Thus, the available data support the hypothesis that andro-ens play a role in BC node invasiveness. Increased ARxpression and/or short CAG alleles that provide a moreranscriptionally active AR protein seem to strengthen theapability of BC cells to invade axillary nodes. These results
hich associate short AR-CAG alleles with a decreased riskf BC. Further studies are warranted in order to reach aonvincing interpretation of these findings.

B. Nicolas Dıaz-Chico et al. / Journal of Steroid Biochemistry & Molecular Biology 105 (2007) 1–15 11
Fig. 3. Targeting estrogen action in breast cancer. There are three main ways of controlling the estrogen action caused by peripheral conversion of androgens.The use of LH-RH agonists is an effective procedure for controlling the ovarian activity of producing both androgens and estrogens. This treatment is seldomused in the menopausal status, but is very useful for reducing these hormones in premenopausal breast cancer patients. The aromatase antagonists block thee ine trea( he estroe
rlhdrTCict
sattaeavic
5
ip
HseTeaomw
acAelpsdafs(
gt
nzyme that converts androgens into estrogens and are emerging as the first-ltamoxifen) or total (fulvestrant) estrogen receptor antagonists to interrupt tstrogen-dependent breast cancer in postmenopausal patients.
As regards cancers in other organs, our laboratory haseported that short CAG and GGN tracts are associated withess aggressive endometrial cancer [52]. Longer CAG allelesave been related to increased colon cancer risk [112,113] andifferent combinations of CAG and GGN alleles have beenelated to prostate [114] and esophageal cancer risk [115].herefore, data from different types of cancer suggest thatAG and GGN repeats a have different capability of mod-
fying carcinogenesis in androgen responsive tissues. Thisapability may not be related to their possible influence onhe clinical behavior of the same cancer type.
Taking all of these factors into account, the available datauggest that a number of combinations in the CAG and GGNlleles might have an effect on the AR-mediated responseo androgens in different cell types and organs. The interac-ion of the AR protein is known to be dependent on tissuend promoter context [61]. However, the present knowl-dge regarding the influence of CAG and GGN repeats onndrogen-dependent gene transcription in cancer cells “inivo” does not allow us to put forward any solid hypothesisntegrating all the described observations on BC and otherancer types.
. Conclusions
Androgens are the most prominent sex-steroid hormonesn women after the menopause, an age at which a largeercentage of breast cancer (BC) patients are diagnosed.
avrd
tment for estrogen receptor positive breast cancers. The capability of partialgen action by blocking the receptor has long been used in order to control
ormone-related breast carcinogenesis after menopause pos-ibly is a consequence of subtle changes that allow thepithelial cells to survive in a low estrogenic environment.hese changes include androgen aromatization to producestradiol which is mitogenic in epithelial breast cells. Cellsdapted to a low estrogenic environment increase the numberf cell cycles per year, thus increasing the probability of accu-ulating uncorrected DNA replication errors and mutations,hich are at the basis of estrogen-related tumorigenesis.Both peripheral and local aromatization of androgens,
s well as increased estrogen responsiveness in epithelialells, could be critical for estrogen-related tumorigenesis.
number of epidemiological studies have unequivocallystablished a clear relationship between elevated circu-ating androgen levels and the risk of breast cancer inostmenopausal women. In premenopausal women, severaltudies have found a similar correlation although the evi-ence is not as strong as in postmenopausal women. Thenti-aromatase agents are emerging as the first-line treatmentor estrogen receptor (ER) positive breast cancers, which con-titutes an unequivocal proof of principle for this hypothesisFig. 3).
Besides being able to act as an estrogen precursor, andro-en can directly act on epithelial breast cells by activatinghe androgen receptor which modulates a number of genes
t the transcriptional level. Prospective cohort studies pro-ide strong evidence that the risk of breast cancer is directlyelated to circulating levels of testosterone and androstene-ione. This finding appears to be equally applicable to women
1 d Bioch
bapc
tolirgtaltrrbtbmip
rTimtcAphipasn
dit
A
ddPdJerRe
fw
R
2 B. Nicolas Dıaz-Chico et al. / Journal of Steroi
efore and after menopause. No clear statement can be madet this point about the association between premenopausalrogesterone or estrogen levels and the risk of breastancer.
Understanding which effects of androgens are direct fromhose exerted as androgen precursors in modulating the riskf breast cancer is a difficult task. The AR gene contains twoength polymorphisms in exon 1. The GGC repeat seems to benversely correlated with AR protein yield, whereas the CAGepeat seems to be inversely correlated with AR-dependentene transcription. The long AR-CAG repeat, that representshe less transcriptionally active AR variants has been associ-ted with an increase in breast cancer risk. Other studies haveimited the impact of the AR-CAG repeat on BC risk. Out ofhe three studies on the influence of the GGN repeat on BCisk, only one has found an association between short alleles,epresentative of a higher AR protein yield, and an increase inreast cancer risk in women under the age of 45 years. Sincehe direct effect of androgens on breast epithelium seems toe mainly antiproliferative, enlarged CAG and/or GGN tractsight represent a decreased androgenic potency, thus lead-
ng to a predominant estrogenic capability of increasing cellroliferation.
More than 60% of BC expresses AR and also androgen-egulated proteins, thus indicating androgen responsiveness.he dominant effect of androgens on estrogen-related tumors
s antiproliferative and a decrease in AR-mediated effectsight favour tumorigenesis. In that sense, a positive AR sta-
us is generally considered a good prognosis marker for breastancer progression. However, the expression level of bothR gene and AR protein in breast cancer was found to beositively correlated with node invasiveness. These findingsave raised the hypothesis that androgens might play a rolen breast cancer node invasiveness. This hypothesis is sup-orted by data from our laboratory showing that short CAGlleles, providing a more transcriptionally active AR protein,trengthen the capability of BC cells to invade the axillaryodes.
As a final statement, we feel that the accumulated evidenceemonstrating the direct role of androgen receptor signallingn modulating human breast carcinogenesis is too plausibleo be ignored any longer.
cknowledgements
This study has been sponsored by the Instituto Canarioe Investigacion del Cancer, Cabildo de Tenerife, Fondoe Investigaciones Sanitarias (FIS-ISCiii-RTICCC C03/10),royecto BIOPOLIS – Interroga IIIB, and Fundacion Canariae Investigacion y Salud (FUNCIS). Both B.N.D.-C and.C.D.-C received grants from the Canary Islands Gov-
rnment (Direccion General de Universidades). G.R. is aecipient of a fellowship from the University of Las Palmas;.R. is recipient of a fellowship from the Canary Islands Gov-rnment. C.B., A.G. and E.B. are recipients of fellowshipsemistry & Molecular Biology 105 (2007) 1–15
rom the ICIC. Authors gratefully acknowledge the editingork of Ms. Emer M. Pigott.
eferences
[1] N. Honma, K. Takubo, M. Sawabe, T. Arai, F. Akiyama, G. Sakamoto,T. Utsumi, N. Yoshimura, N. Harada, Estrogen-metabolizing enzymesin breast cancers from women over the age of 80 years, J. Clin.Endocrinol. Metab. 91 (2) (2006) 607–613.
[2] J.R. Pasqualini, G.S. Chetrite, Recent insight on the control ofenzymes involved in estrogen formation and transformation in humanbreast cancer, J. Steroid Biochem. Mol. Biol. 93 (2–5) (2005) 221–236(Review).
[3] H. Sasano, T. Suzuki, T. Nakata, T. Moriya, New development inintracrinology of breast carcinoma, Breast Cancer 13 (2) (2006)129–136 (Review).
[4] J.D. Yager, N.E. Davidson, Estrogen carcinogenesis in breast cancer,N. Engl. J. Med. 354 (3) (2006) 270–282 (Review).
[5] M. Clemons, P. Goss, Estrogen and the risk of breast cancer, N. Engl.J. Med. 344 (4) (2001) 276–285 (Review).
[6] J. Kurebayashi, Endocrine-resistant breast cancer: underlying mech-anisms and strategies for overcoming resistance, Breast Cancer 10 (2)(2003) 112–119 (Review).
[7] Y. Shang, Molecular mechanisms of oestrogen and SERMs inendometrial carcinogenesis, Nat. Rev. Cancer 6 (5) (2006) 360–368(Review).
[8] R. Kaaks, S. Rinaldi, T.J. Key, F. Berrino, P.H. Peeters, C. Biessy,L. Dossus, A. Lukanova, S. Bingham, K.T. Khaw, N.E. Allen, H.B.Bueno-de-Mesquita, C.H. van Gils, D. Grobbee, H. Boeing, P.H. Lah-mann, G. Nagel, J. Chang-Claude, F. Clavel-Chapelon, A. Fournier,A. Thiebaut, C.A. Gonzalez, J.R. Quiros, M.J. Tormo, E. Ardanaz,P. Amiano, V. Krogh, D. Palli, S. Panico, R. Tumino, P. Vineis, A.Trichopoulou, V. Kalapothaki, D. Trichopoulos, P. Ferrari, T. Norat,R. Saracci, E. Riboli, Postmenopausal serum androgens, oestrogensand breast cancer risk: the European prospective investigation intocancer and nutrition, Endocr. Relat. Cancer 12 (4) (2005) 1071–1082.
[9] D.Y. Wang, D.S. Allen, B.L. De Stavola, I.S. Fentiman, J. Brussen,R.D. Bulbrook, Urinary androgens and breast cancer risk: results froma long-term prospective study based in Guernsey, Br. J. Cancer 82(2000) 1577–1584.
[10] A. Zeleniuch-Jacquotte, Y. Gu, R.E. Shore, K.L. Koenig, A.A. Arslan,I. Kato, S. Rinaldi, R. Kaaks, P. Toniolo, Postmenopausal levels of sexhormones and risk of breast carcinoma in situ: results of a prospectivestudy, Int. J. Cancer 114 (2) (2005) 323–327.
[11] H. Yu, X.O. Shu, R. Shi, Q. Dai, F. Jin, Y.T. Gao, B.D. Li, W.Zheng, Plasma sex steroid hormones and breast cancer risk in Chinesewomen, Int. J. Cancer 105 (1) (2003) 92–97.
[12] J.F. Dorgan, C. Longcope, H.E. Stephenson Jr., R.T. Falk, R. Miller, C.Franz, L. Kahle, W.S. Campbell, J.A. Tangrea, A. Schatzkin, Serumsex hormone levels are related to breast cancer risk in postmenopausalwomen, Environ. Health Perspect. 105 (3) (1997) 583–585.
[13] S.E. Hankinson, W.C. Willett, J.E. Manson, G.A. Colditz, D.J. Hunter,D. Spiegelman, R.L. Barbieri, F.E. Speizer, Plasma sex steroid hor-mone levels and risk of breast cancer in postmenopausal women, J.Natl. Cancer Inst. 90 (17) (1998) 1292–1299.
[14] J.A. Cauley, F.L. Lucas, L.H. Kuller, K. Stone, W. Browner, S.R. Cum-mings, Elevated serum estradiol and testosterone concentrations areassociated with a high risk for breast cancer, study of osteoporoticfractures research group, Ann. Intern. Med. 130 (4) (1999) 270–
277.[15] S.A. Missmer, A.H. Eliassen, R.L. Barbieri, S.E. Hankinson, Endoge-nous estrogen, androgen, and progesterone concentrations and breastcancer risk among postmenopausal women, J. Natl. Cancer Inst. 96(24) (2004) 1856–1865.

Bioch
B. Nicolas Dıaz-Chico et al. / Journal of Steroid[16] C. Schairer, D. Hill, S.R. Sturgeon, T. Fears, C. Mies, R.G. Ziegler,R.N. Hoover, M.E. Sherman, Serum concentrations of estrogens, sexhormone binding globulin, and androgens and risk of breast hyperpla-sia in postmenopausal women, Cancer Epidemiol Biomarkers Prev.14 (7) (2005) 1660–1665.
[17] R. Grattarola, The premenstrual endometrial pattern of women withbreast cancer. A study of progestional activity, Cancer 17 (1964)1119–1122.
[18] R. Grattarola, Androgens in breast cancer. Atypical endometrialhyperplasia and breast cancer in married premenopausal women, Am.J. Obstet. Gynecol. 116 (1973) 423–428.
[19] G. Secreto, P. Toniolo, P. Pisani, C. Recchione, A. Cavalleri, G.Fariselli, Androgens and breast cancer in premenopausal women,Cancer Res. 49 (1989) 471–476.
[20] W. Somboonporn, S.R. Davis, Testosterone effects on the breast:implications for testosterone therapy for women, Endocr. Rev. 25 (3)(2004) 374–388.
[21] The Endogenous Hormones and Breast Cancer Collaborative Group,Endogenous steroid hormones and breast cancer risk in post-menopausal women: analysis of nine prospective studies, J. Natl.Cancer Inst. 94 (2002) 606–616.
[22] G. Secreto, B. Zumoff, Abnormal production of androgens in womenwith breast cancer, Anticancer Res. 14 (1994) 2113–2117.
[23] J.H. Page, G.A. Colditz, N. Rifai, R.L. Barbieri, W.C. Willett, S.E.Hankinson, Plasma adrenal androgens and risk of breast cancer in pre-menopausal women, Cancer Epidemiol. Biomarkers Prev. 13 (2004)1032–1036.
[24] C. Recchione, E. Venturelli, A. Manzari, A. Cavalleri, A. Martinetti,G. Secreto, Testosterone, dihydrotestosterone and oestradiol levels inpostmenopausal breast cancer tisues, J. Steroid Biochem. Mol. Biol.52 (6) (1995) 541–546.
[25] G. Secreto, E. Venturelli, A. Bucci, D. Piromalli, G. Fariselli, E.Galante, Intratumour amount of sex steroids in elderly breast cancerpatients, An approach to the biological characterization of mammarytumours in the elderly, J. Steroid Biochem. Mol. Biol. 58 (5–6) (1996)557–561.
[26] G. Secreto, C. Recchione, P. Ballerini, L. Callegari, A. Cavalleri, A.Attili, G. Fariselli, D. Moglia, I. Del Prato, Accumulation of activeandrogens in breast cyst fluids, Eur. J. Cancer 27 (1) (1991) 44–47.
[27] B.J. Feldman, D. Feldman, The development of androgen-independent prostate cancer, Nat. Rev. Cancer 1 (1) (2001) 34–45(Review).
[28] R. Kaaks, F. Berrino, T. Key, S. Rinaldi, L. Dossus, C. Biessy, G.Secreto, P. Amiano, S. Bingham, H. Boeing, H.B. Bueno de Mesquita,J. Chang-Claude, F. Clavel-Chapelon, A. Fournier, C.H. van Gils,C.A. Gonzalez, A.B. Gurrea, E. Critselis, K.T. Khaw, V. Krogh, P.H.Lahmann, G. Nagel, A. Olsen, N.C. Onland-Moret, K. Overvad, D.Palli, S. Panico, P. Peeters, J.R. Quiros, A. Roddam, A. Thiebaut, A.Tjonneland, M.D. Chirlaque, A. Trichopoulou, D. Trichopoulos, R.Tumino, P. Vineis, T. Norat, P. Ferrari, N. Slimani, E. Riboli, Serumsex steroids in premenopausal women and breast cancer risk within theEuropean Prospective Investigation into Cancer and Nutrition (EPIC),J. Natl. Cancer Inst. 97 (10) (2005) 755–765.
[29] C. Dimitrakakis, J. Zhou, C.A. Bondy, Androgens and mammarygrowth and neoplasia, Fertil. Steril. 77 (Suppl. 4) (2002) S26–S33.
[30] J.R. Pasqualini, G.S. Chetrite, Recent insight on the control ofenzymes involved in estrogen formation and transformation in humanbreast cancer, J. Steroid Biochem. Mol. Biol. 93 (2–5) (2005)221–236.
[31] B. Xie, S.W. Tsao, Y.C. Wong, Sex hormone-induced mammary car-
cinogenesis in female noble rats: the role of androgen, Carcinogenesis20 (1999) 1597–1606.[32] B. Xie, S.W. Tsao, Y.C. Wong, Induction of high incidence of mam-mary tumour in female Noble rats with a combination of 17-oestradioland testosterone, Carcinogenesis 20 (1999) 1069–1078.
emistry & Molecular Biology 105 (2007) 1–15 13
[33] M.C. Pike, D.V. Spicer, L. Dahmoush, M.F. Press, Estrogens, pro-gestogens, normal breast cell proliferation, and breast cancer risk,Epidemiol. Rev. 15 (1993) 17–35.
[34] C. Schairer, J. Lubin, R. Troisi, S. Sturgeon, L. Brinton, R. Hoover,Menopausal estrogen and estrogen-progestin replacement therapy andbreast cancer risk, JAMA 283 (2000) 485–491.
[35] C. Lanari, A.A. Molinolo, Progesterone receptors—animal modelsand cell signalling in breast cancer, diverse activation pathways forthe progesterone receptor: possible implications for breast biologyand cancer, Breast Cancer Res. 4 (2002) 240–243.
[36] O.M. Conneely, B.M. Jericevic, J.P. Lydon, Progesterone receptors inmam mary gland development and tumorigenesis, J. Mammary GlandBiol. Neoplasia 8 (2003) 205–214.
[37] Y.E. Shi, Y.E. Liu, M.E. Lippman, R.B. Dickson, Progestins andantiprogestins in mammary tumour growth and metastasis, Hum.Reprod. 9 (Suppl.) (1994).
[38] S.F. Doisneau-Sixou, C.M. Sergio, J.S. Carroll, R. Hui, E.A. Mus-grove, R.L. Sutherland, Estrogen and antiestrogen regulation of cellcycle progression in breast cancer cells, Endocr. Relat. Cancer 10(2003) 179–186.
[39] R.B. Dickson, G.M. Stancel, Estrogen receptor-mediated processesin normal and cancer cells, J. Natl. Cancer Inst. Monogr. (2000)135.
[40] F. Labrie, V. Luu-The, C. Labrie, A. Belanger, J. Simard, S.X. Lin,Endocrine and intracrine sources of androgens in women: inhibitionof breast cancer and other roles of androgens and their precursordehydroepiandrosterone, Endocr. Rev. 24 (2003) 152–182.
[41] A. Baniahmad, Nuclear hormone receptor co-repressors, J. SteroidBiochem. Mol. Biol. 93 (2–5) (2005) 89–97.
[42] Z. Kang, A. Pirskanen, O.A. Janne, J.J. Palvimo, Involvement ofproteasome in the dynamic assembly of the androgen receptor tran-scription complex, J. Biol. Chem. 277 (2002) 48366–48371.
[43] H.K. Lin, S. Altuwaijri, W.J. Lin, P.Y. Kan, L.L. Collins, C. Chang,Proteasome activity is required for androgen receptor transcriptionalactivity via regulation of androgen receptor nuclear translocation andinteraction with coregulators in prostate cancer cells, J. Biol. Chem.277 (2002) 36570–36576.
[44] E. Molinari, M. Gilman, S. Natesan, Proteasome-mediated degrada-tion of transcriptional activators correlates with activation domainpotency in vivo, EMBO J. 18 (1999) 6439–6447.
[45] S.E. Salghetti, A.A. Caudy, J.G. Chenoweth, W.P. Tansey, Regulationof transcriptional activation domain function by ubiquitin, Science293 (2001) 1651–1653.
[46] S.E. Salghetti, M. Muratani, H. Wijnen, B. Futcher, W.P. Tansey,Functional overlap of sequences that activate transcription and sig-nal ubiquitin-mediated proteolysis, Proc. Natl. Acad. Sci. U. S. A. 97(2000) 3118–3123.
[47] G.D. Lorenzo, R. Bianco, G. Tortora, F. Ciardiello, Involvement ofgrowth factor receptors of the epidermal growth factor receptor familyin prostate cancer development and progression to androgen indepen-dence, Clin. Prostate Cancer 2 (1) (2003) 50–57.
[48] Z. Culig, B. Comuzzi, H. Steiner, G. Bartsch, A. Hobisch, Expressionand function of androgen receptor coactivators in prostate cancer, J.Steroid Biochem. Mol. Biol. 92 (4) (2004) 265–271.
[49] Z. Culig, Androgen receptor cross-talk with cell signalling pathways,Growth Factors 22 (3) (2004) 179–184 (Review).
[50] S. Sengupta, B. Wasylyk, Physiological and pathological conse-quences of the interactions of the p53 tumor suppressor with theglucocorticoid, androgen, and estrogen receptors, Ann. N. Y. Acad.Sci. 1024 (2004) 54–71.
[51] J.C. Allegra, M.E. Lippman, E.B. Thompson, R. Simon, A. Barlock,L. Green, K.K. Huff, H.M. Do, S.C. Aitken, Distribution, frequency,
and quantitative analysis of estrogen, progesterone, androgen, andglucocorticoid receptors in human breast cancer, Cancer Res. 39(1979) 1447–1454.[52] R.E. Hall, J.A. Clements, S.N. Birrell, W.D. Tilley, Prostate-specificantigen and gross cystic disease fluid protein-15 are co-expressed in

1 d Bioch
4 B. Nicolas Dıaz-Chico et al. / Journal of Steroiandrogen receptor-positive breast tumours, Br. J. Cancer 78 (1998)360–365.
[53] V. Kuenen-Boumeester, T.H. Van der Kwast, W.L. van Putten, C.Claassen, B. van Ooijen, S.C. Henzen-Logmans, Immunohistochem-ical determination of androgen receptors in relation to oestrogen andprogesterone receptors in female breast cancer, Int. J. Cancer 52(1992) 581–584.
[54] S.N. Agoff, P.E. Swanson, H. Linden, S.E. Hawes, T.J. Lawton,Androgen receptor expression in estrogen receptor-negative breastcancer, Immunohistochemical, clinical, and prognostic associations,Am. J. Clin. Pathol. 120 (2003) 725–731.
[55] G. Secreto, P. Toniolo, F. Berrino, C. Recchione, S. Di Pietro, G.Fariselli, Increased androgenic activity and breast cancer risk in pre-menopausal women, Cancer Res. 44 (1984) 5902–5905.
[56] J.J. Isola, Immunohistochemical demonstration of androgen receptorin breast cancer and its relationship to other prognostic factors, J.Pathol. 170 (1993) 31–35.
[57] M. Brys, M. Wojcik, H. Romanowicz-Makowska, W.M. Krajewska,Androgen receptor status in female breast cancer: RT-PCR and West-ern blot studies, J. Cancer Res. Clin. Oncol. 128 (2002) 85–90.
[58] I. Bieche, B. Parfait, S. Tozlu, R. Lidereau, M. Vidaud, Quantita-tion of androgen receptor gene expression in sporadic breast tumorsby real-time RT-PCR: evidence that MYC is an AR-regulated gene,Carcinogenesis 22 (2001) 1521–1526.
[59] J.A. Soreide, O.A. Lea, J.E. Varhaug, A. Skarstein, S. Kvinnsland,Androgen receptors in operable breast cancer: relation to other steroidhormone receptors, correlations to prognostic factors and predictivevalue for effect of adjuvant tamoxifen treatment, Eur. J. Surg. Oncol.18 (1992) 112–118.
[60] H.C. Shen, G.A. Coetzee, The androgen receptor: unlocking thesecrets of its unique transactivation domain, Vitam. Horm. 71 (2005)301–319.
[61] D. Navarro, O.P. Luzardo, L. Fernandez, N. Chesa, B.N. Diaz-Chico,Transition to androgen-independence in prostate cancer, J. SteroidBiochem. Mol. Biol. 81 (3) (2002) 191–201.
[62] D. Ding, L. Xu, M. Menon, G.P. Reddy, E.R. Barrack, Effect of GGC(glycine) repeat length polymorphism in the human androgen receptoron androgen action, Prostate 62 (2) (2005) 133–139.
[63] A.W. Hsing, Y.T. Gao, G. Wu, X. Wang, J. Deng, Y.L. Chen, I.A.Sesterhenn, F.K. Mostofi, J. Benichou, C. Chang, Polymorphic CAGand GGN repeat lengths in the androgen receptor gene and prostatecancer risk: a population-based case-control study in China, CancerRes. 60 (18) (2000) 5111–5116.
[64] E.L. Aschim, A. Nordenskjold, A. Giwercman, K.B. Lundin, Y.Ruhayel, T.B. Haugen, T. Grotmol, Y.L. Giwercman, Linkagebetween cryptorchidism, hypospadias, and GGN repeat length in theandrogen receptor gene, J. Clin. Endocrinol. Metab. 89 (10) (2004)5105–5109.
[65] M. Iwakura, T. Nakamura, Effects of the length of a glycine linkerterminal interaction, J. Biol. Chem. 277 (2002) 25631–25639.
[66] D. Ding, L. Xu, M. Menon, G.P. Reddy, E.R. Barrack, Effect of ashort CAG (glutamine) repeat on human androgen receptor function,Prostate 58 (1) (2004) 23–32.
[67] E. Esteban, N. Rodon, M. Via, E. Gonzalez-Perez, J. Santamaria, J.M.Dugoujon, F.E. Chennawi, M. Melhaoui, M. Cherkaoui, G. Vona, N.Harich, P. Moral, Androgen receptor CAG and GGC polymorphismsin Mediterraneans: repeat dynamics and population relationships, J.Hum. Genet. 51 (2) (2006) 129–136.
[68] S.M. Gilbert, M.C. Benson, J.M. McKiernan, Linkage disequilibriumbetween the androgen receptor gene CAG and GGC repeats in theAfrican-American population, Curr. Urol. Rep. 3 (3) (2002) 189–193(Review).
[69] G. Rodriguez, C. Bilbao, R. Ramirez, O. Falcon, L. Leon, R. Chirino,O. Falcon Jr., B.P. Diaz, J.F. Rivero, M. Perucho, B.N. Diaz-Chico,J.C. Diaz-Chico, Alleles with short CAG and GGN repeats in theandrogen receptor gene are associated with benign endometrial can-cer, Int. J. Cancer 118 (2006) 1420–1425.
emistry & Molecular Biology 105 (2007) 1–15
[70] M.P. Zeegers, L.A. Kiemeney, A.M. Nieder, H.R. Ostrer, Howstrong is the association between CAG and GGN repeat lengthpolymorphisms in the androgen receptor gene and prostate cancerrisk? Cancer Epidemiol. Biomarkers Prev. 13 (11) (2004) 1765–1771.
[71] L.K. Beitel, T. Scanlon, B. Gottlieb, M.A. Trifiro, Progress in Spinob-ulbar muscular atrophy research: insights into neuronal dysfunctioncaused by the polyglutamine-expanded androgen receptor, NeurotoxRes. 7 (3) (2005) 219–230.
[72] A. Poletti, The polyglutamine tract of androgen receptor: from func-tions to dysfunctions in motor neurons, Front Neuroendocrinol. 25(1) (2004) 1–26 (Review).
[73] F. Piccioni, S. Simeoni, I. Andriola, E. Armatura, S. Bassanini, P.Pozzi, A. Poletti, Polyglutamine tract expansion of the androgenreceptor in a motoneuronal model of spinal and bulbar muscularatrophy, Brain Res. Bull. 1 (56) (2001) 215–220 (Review).
[74] A.R. La Spada, E.M. Wilson, D.B. Lubahn, A.E. Harding, K.H. Fis-chbeck, Androgen receptor gene mutations in X-linked spinal andbulbar muscular atrophy, Nature 4 (352) (1991) 77–79.
[75] J.L. Walcott, D.E. Merry, Trinucleotide repeat disease, the andro-gen receptor in spinal and bulbar muscular atrophy, Vitam. Horm. 65(2002) 127–147 (Review).
[76] J.M. Hakimi, M.P. Schoenberg, R.H. Rondinelli, S. Piantadosi, E.R.Barrack, Androgen receptor variants with short glutamine or glycinerepeats may identify unique subpopulations of men with prostatecancer, Clin. Cancer Res. 3 (1997) 1599–1608.
[77] N.L. Chamberlain, E.D. Driver, R.L. Miesfeld, The length and loca-tion of CAG trinucleotide repeats in the androgen receptor N-terminaldomain affect transactivation function, Nucl. Acids Res. 22 (1994)3181–3186.
[78] T.R. Rebbeck, P.W. Kantoff, K. Krithivas, S. Neuhausen, M.A. Black-wood, A.K. Godwin, M.B. Daly, S.A. Narod, J.E. Garber, H.T. Lynch,B.L. Weber, M. Brown, Modification of BRCA1-associated breastcancer risk by the polymorphic androgen-receptor CAG repeat, Am.J. Hum. Genet. 64 (1999) 1371–1377.
[79] P.E. Clark, R.A. Irvine, G.A. Coetzee, The androgen receptor CAGrepeat and prostate cancer risk, Methods Mol. Med. 81 (2003)255–266 (Review).
[80] M.P. Zeegers, L.A. Kiemeney, A.M. Nieder, H.R. Ostrer, How strongis the association between CAG and GGN repeat length polymor-phisms in the androgen receptor gene and prostate cancer risk? CancerEpidemiol. Biomarkers Prev. 13 (2004) 1765–1771.
[81] J. Beilin, E.M. Ball, J.M. Favaloro, J.D. Zajac, Effect of the andro-gen receptor CAG repeat polymorphism on transcriptional activity:specificity in prostate and non-prostate cell lines, J. Mol. Endocrinol.25 (2000) 85–96.
[82] P. Ferro, M.G. Catalano, R. Dell’Eva, N. Fortunati, U. Pfeffer, Theandrogen receptor CAG repeat: a modifier of carcinogenesis? Mol.Cell Endocrinol. 31 (193) (2002) 109–120 (Review).
[83] A.P. Lieberman, G. Harmison, A.D. Strand, J.M. Olson, K.H. Fis-chbeck, Altered transcriptional regulation in cells expressing theexpanded polyglutamine androgen receptor, Hum. Mol. Genet. 11(2002) 1967–1976.
[84] D.L. Stenoien, C.J. Cummings, H.P. Adams, M.G. Mancini, K.Patel, G.N. DeMartino, M. Marcelli, N.L. Weigel, M.A. Mancini,Polyglutamine-expanded androgen receptors form aggregates thatsequester heat shock proteins, proteasome components and SRC-1,and are suppressed by the HDJ-2 chaperone, Hum. Mol. Genet. 8(1999) 731–741.
[85] Z. Nawaz, B.W. O’Malley, Urban renewal in the nucleus: is proteinturnover by proteasomes absolutely required for nuclear receptor-regulated transcription? Mol. Endocrinol. 18 (2004) 493–499.
[86] L. Westberg, F. Baghaei, R. Rosmond, M. Hellstrand, M. Landen,M. Jansson, G. Holm, P. Bjorntorp, E. Eriksson, Polymorphisms ofthe androgen receptor gene and the estrogen receptor beta gene areassociated with androgen levels in women, J. Clin. Endocrinol. Metab.86 (2001) 2562–2568.

Bioch
B. Nicolas Dıaz-Chico et al. / Journal of Steroid[87] L. Ibanez, K.K. Ong, N. Mongan, J. Jaaskelainen, M.V. Marcos,I.A. Hughes, F. De Zegher, D.B. Dunger, Androgen receptor geneCAG repeat polymorphism in the Development of ovarian hyperan-drogenism, J. Clin. Endocrinol. Metab. 88 (2003) 3333–3338.
[88] I.S. Brum, P.M. Spritzer, F. Paris, M.A. Maturana, F. Audran, C.Sultan, Association between androgen receptor gene CAG repeatpolymorphism and plasma testosterone levels in postmenopausalwomen, J. Soc. Gynecol. Investig. 12 (2005) 135–141.
[89] M. Hietala, T. Sandberg, A. Borg, H. Olsson, H. Jernstrom, Testos-terone levels in relation to oral contraceptive use and the androgenreceptor CAG and GGC length polymorphisms in healthy youngwomen, Hum. Reprod. (2006) (Advance Access, August 18).
[90] D.T. Zava, W.L. McGuire, Estrogen receptors in androgen-inducedbreast tumor regression, Cancer Res. 37 (1977) 1608–1610.
[91] K.B. Horwitz, M.E. Costlow, W.L. McGuire, MCF-7; a human breastcancer cell line with estrogen, androgen, progesterone, and glucocor-ticoid receptors, Steroids 26 (1975) 785–795.
[92] R. Wooster, J. Mangion, R. Eeles, S. Smith, M. Dowsett, D. Averill,P. Barrett-Lee, D.F. Easton, B.A. Ponder, M.R. Stratton, A germlinemutation in the androgen receptor gene in two brothers with breastcancer and Reifenstein syndrome, Nat. Genet. 2 (1992) 132–134.
[93] H.E. MacLean, R.W. Brown, J. Beilin, G.L. Warne, J.D. Zajac,Increased frequency of long androgen receptor CAG repeats in malebreast cancers, Breast Cancer Res. Treat. 88 (3) (2004) 239–246.
[94] Y. Giguiere, E. Dewailly, J. Brisson, P. Ayotte, N. Laflamme, A.Demers, V.I. Forest, S. Dodin, J. Robert, F. Rousseau, Short polyg-lutamine tracts in the androgen receptor are protective against breastcancer in the general population, Cancer Res. 61 (2001) 5869–5874.
[95] A. Liede, W. Zhang, M.L. De Leon Matsuda, A. Tan, S.A. Narod,Androgen receptor gene polymorphism and breast cancer suscepti-bility in The Philippines, Cancer Epidemiol. Biomarkers Prev. 12 (9)(2003) 848–852.
[96] I.S. Brum, P.M. Spritzer, F. Paris, M.A. Maturana, F. Audran, C.Sultan, Association between androgen receptor gene CAG repeatpolymorphism and plasma testosterone levels in postmenopausalwomen, J. Soc. Gynecol. Investig. 12 (2) (2005) 135–141.
[97] E. Dagan, E. Friedman, T. Paperna, N. Carmi, R. Gershoni-Baruch,Androgen receptor CAG repeat length in Jewish Israeli women whoare BRCA1/2 mutation carriers: association with breast/ovarian can-cer phenotype, Eur. J. Hum. Genet. 10 (11) (2002) 724–728.
[98] EMBRACE Study Collaborators, The androgen receptor CAG repeatpolymorphism and modification of breast cancer risk in BRCA1 andBRCA2 mutation carriers, Breast Cancer Res. 7 (2) (2005) 176–183.
[99] L. Kadouri, D.F. Easton, S. Edwards, A. Hubert, Z. Kote-Jarai, B.Glaser, F. Durocher, D. Abeliovich, T. Peretz, R.A. Eeles, CAG andGGC repeat polymorphisms in the androgen receptor gene and breastcancer susceptibility in BRCA1/2 carriers and non-carriers, Br. J.Cancer 85 (2001) 36–40.
[100] A.M. Dunning, S. McBride, J. Gregory, F. Durocher, N.A. Foster,C.S. Healey, N. Smith, P.D. Pharoah, R.N. Luben, D.F. Easton, B.A.Ponder, No association between androgen or vitamin D receptor genepolymorphisms and risk of breast cancer, Carcinogenesis 20 (1999)2131–2135.
[101] C. Menin, G.L. Banna, G. De Salvo, V. Lazzarotto, A. De Nicolo,S. Agata, M. Montagna, G. Sordi, O. Nicoletto, L. Chieco-Bianchi,E. D’Andrea, Lack of association between androgen receptor CAGpolymorphism and familial breast/ovarian cancer, Cancer Lett. 168(2001) 31–36.
emistry & Molecular Biology 105 (2007) 1–15 15
[102] Y.A. Elhaji, B. Gottlieb, R. Lumbroso, L.K. Beitel, W.D. Foulkes, L.Pinsky, M.A. Trifiro, The polymorphic CAG repeat of the androgenreceptor gene: a potential role in breast cancer in women over 40,Breast Cancer Res. Treat. 70 (2001) 109–116.
[103] W. Wang, E.M. John, S.A. Ingles, Androgen receptor andprostate-specific antigen gene polymorphisms and breast cancer inAfrican-American women, Cancer Epidemiol. Biomarkers Prev. 14(12) (2005) 2990–2994.
[104] C. Iobagiu, C. Lambert, M. Normand, C. Genin, Microsatellite pro-file in hormonal receptor genes associated with breast cancer, BreastCancer Res. Treat. 95 (2) (2006) 153–159.
[105] A.B. Spurdle, G.S. Dite, X. Chen, C.J. Mayne, M.C. Southey, L.E.Batten, H. Chy, L. Trute, M.R. McCredie, G.G. Giles, J. Armes, D.J.Venter, J.L. Hopper, G. Chenevix-Trench, Androgen receptor exon 1CAG repeat length and breast cancer in women before age forty years,J. Natl. Cancer Inst. 91 (11) (1999) 961–966.
[106] C.A. Haiman, M. Brown, S.E. Hankinson, D. Spiegelman, G.A.Colditz, W.C. Willett, P.W. Kantoff, D.J. Hunter, The androgen recep-tor CAG repeat polymorphism and risk of breast cancer in the Nurses’Health Study, Cancer Res. 62 (2002) 1045–1049.
[107] A. Gonzalez, F.J. Dorta, F.G. Rodrıguez, B. Brito, M.C. Rodrıguez,A.Cabrera, J.C. Dıaz-Chico, R. Reyes, A. Aguirre-Jaime, B.N.Dıaz-Chico, Increased Risk of Breast Cancer in Women Bearing aCombination of Large CAG and GGN Repeats in the Exon 1 of theAndrogen Receptor Gene, Eur. J. Cancer (2007), in press.
[108] N.M. Suter, K.E. Malone, J.R. Daling, D.R. Doody, E.A. Ostrander,Androgen receptor (CAG)n and (GGC)n polymorphism and breastcancer risk in a population-based case-control study of young women,Cancer Epidemiol. Biomarkers Prev. 12 (2003) 127–135.
[109] G. Ursin, H. Ma, A.H. Wu, L. Bernstein, M. Salane, Y.R. Parisky,M. Astrahan, C.C. Siozon, M.C. Pike, Mammographic density andbreast cancer in three ethnic groups, Cancer Epidemiol. BiomarkersPrev. 12 (4) (2003) 332–338.
[110] H. Yu, B. Bharaj, E.J. Vassilikos, M. Giai, E.P. Diamandis, ShorterCAG repeat length in the androgen receptor gene is associated withmore aggressive forms of breast cancer, Breast Cancer Res. Treat. 59(2000) 153–161.
[111] E. Betancor, G. Rodriguez, A. Gonzalez, A. Cabrera, M. Sanchez,V. Vega, A. Murias, U. Bohn, M. Limeres, J. Pestano, B.N. Diaz-Chico, J.C. Diaz-Chico, The androgen receptor gene polymorphismsin human breast cancer: Short CAG repeats are associated with lymphnode involvement, in: 28th Annual San Antonio Breast Cancer Sym-posium, San Antonio, Texas, USA, 2005.
[112] M.L. Slattery, C. Sweeney, M. Murtaugh, K.N. Ma, B.J. Caan, J.D.Potter, R. Wolff, Associations between vitamin D, vitamin D receptorgene and the androgen receptor gene with colon and rectal cancer, Int.J. Cancer 118 (12) (2006) 3140–3146.
[113] M.L. Slattery, C. Sweeney, M. Murtaugh, K.N. Ma, R.K. Wolff,J.D. Potter, B.J. Caan, W. Samowitz, Associations between ERal-pha, ERbeta, and AR genotypes and colon and rectal cancer, CancerEpidemiol. Biomarkers Prev. 14 (12) (2005) 2936–2942.
[114] E.A. Platz, E. Giovannucci, D.M. Dahl, K. Krithivas, C.H. Hennekens,M. Brown, M.J. Stampfer, P.W. Kantoff, The androgen receptor
gene GGN microsatellite and prostate cancer risk, Cancer Epidemiol.Biomarkers Prev. 7 (1998) 379–384.[115] E. Dietzsch, R. Laubscher, M.I. Parker, Esophageal cancer risk inrelation to GGC and CAG trinucleotide repeat lengths in the androgenreceptor gene, Int. J. Cancer 20 (107) (2003) 38–45.











![Evaluation of androgen-induced effects on the uptake of [18F]FDG, [11C]choline and [11C]acetate in an androgen-sensitive and androgen-independent prostate cancer xenograft model](https://img.pdfslide.net/doc/110x75/6361861c2fd302ae64094544/evaluation-of-androgen-induced-effects-on-the-uptake-of-18ffdg-11ccholine-and.jpg)

















