Embed Size (px)
Citation preview

EUKARYOTIC CELL, Apr. 2006, p. 638–649 Vol. 5, No. 41535-9778/06/$08.00�0 doi:10.1128/EC.5.4.638–649.2006Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Conserved Fungal Genes as Potential Targets for Broad-SpectrumAntifungal Drug Discovery†
Mengping Liu,1 Matthew D. Healy,2 Brian A. Dougherty,2 Kim M. Esposito,3 Trina C. Maurice,1Charles E. Mazzucco,1 Robert E. Bruccoleri,2 Daniel B. Davison,2 Marybeth Frosco,1
John F. Barrett,1 and Ying-Kai Wang1*Departments of Infectious Diseases,1 Applied Genomics,2 and Lead Discovery,3 Bristol-Myers Squibb Company
Pharmaceutical Research Institute, 5 Research Parkway, Wallingford, Connecticut 06492
Received 2 December 2005/Accepted 26 January 2006
The discovery of novel classes of antifungal drugs depends to a certain extent on the identification of new,unexplored targets that are essential for growth of fungal pathogens. Likewise, the broad-spectrum capacity offuture antifungals requires the target gene(s) to be conserved among key fungal pathogens. Using a genomecomparison (or concordance) tool, we identified 240 conserved genes as candidates for potential antifungaltargets in 10 fungal genomes. To facilitate the identification of essential genes in Candida albicans, we developeda repressible C. albicans MET3 (CaMET3) promoter system capable of evaluating gene essentiality on agenome-wide scale. The CaMET3 promoter was found to be highly amenable to controlled gene expression, aprerequisite for use in target-based whole-cell screening. When the expression of the known antifungal targetC. albicans ERG1 was reduced via down-regulation of the CaMET3 promoter, the CaERG1 conditional mutantstrain became hypersensitive, specifically to its inhibitor, terbinafine. Furthermore, parallel screening againsta small compound library using the CaERG1 conditional mutant under normal and repressed conditionsuncovered several hypersensitive compound hits. This work therefore demonstrates a streamlined process forproceeding from selection and validation of candidate antifungal targets to screening for specific inhibitors.
Candidiasis, aspergillosis, and cryptococcosis are the leadinginvasive fungal infections that cause substantial morbidity andmortality in nosocomial settings and among immunocompro-mised patients (51). Candida species are considered the fourthmost common bloodstream isolates in the United States (15).For many years, amphotericin B and fluconazole have been thestandard of therapy for treatment of severe fungal infections(13). However, serious side effects such as nephrotoxicity areassociated with amphotericin B, while fluconazole suffers fromsuch drawbacks as frequent interactions with coadministereddrugs, limited spectrum (for example, it is not effective againstaspergillosis), and the increasing emergence of fluconazole-resistant Candida species (for example, Candida krusei andsome strains of Candida glabrata) (9, 13, 20). Encouragingly,several new drugs have recently been approved by the U.S.Food and Drug Administration, including voriconazole, caspo-fungin, and micafungin. Like the other two triazoles (posacon-azole and ravuconazole) that are in development, voriconazoledisplays a broader antifungal spectrum, including Aspergillusstrains and resistant Candida species (25, 30). Caspofungin andmicafungin represent the first new class (the echinocandinlipopeptides) of antifungal agents approved after several de-cades of intensive research (2, 28, 56).
In spite of these recent advances, the currently marketedantifungals and those in development represent only threemajor chemical classes (polyenes, azoles, and echinocandins)
and target only two cellular structures (the cell membrane andthe cell wall) (49). Therefore, there is a critical need for newclasses of broad-spectrum antifungals that bind to novel tar-gets. Microbial genomics offer an unprecedented opportunityto enlarge the repertoire of antimicrobial compounds with theidentification of genes that encode potential new targets forchemotherapy. To date, more than 10 fungal genomes havebeen sequenced (http://ncbi.nih.gov/RefSeq/ [accessed August2005]), including those of Saccharomyces cerevisiae, Candidaalbicans, C. glabrata, Aspergillus fumigatus, Cryptococcus neo-formans, Debaryomyces hansenii, Kluyveromyces lactis, Schizo-saccharomyces pombe, Yarrowia lipolitica, and Eremotheciumgossypii. It is expected that tremendous interest and effort willbe devoted to the identification of essential genes as potentialdrug targets from key fungal pathogens. While hundreds ofessential genes have been identified in S. cerevisiae (55) and C.albicans (11, 44, 55), target prioritization in terms of, for ex-ample, degree of essentiality, broad-spectrum potential, drugtarget potential, fungal specificity, availability of functionalassays, and amenability to high-throughput screening (HTS)would likely present a much greater challenge to the researchcommunity.
Recognizing the ever-increasing demands for broad-spec-trum antifungals, we sought to identify potential antifungaltargets that are conserved among the 10 fungal species men-tioned above and to develop a system to identify target-specificinhibitors. To this end, we identified 240 conserved genesamong the 10 fungi via the genome comparison tool, deter-mined essentiality of selected genes in C. albicans using arepressible C. albicans MET3 (CaMET3) promoter, and adaptedthe CaMET3 promoter system to a target-based whole-cell HTSplatform.
* Corresponding author. Mailing address: Bristol-Myers Squibb Com-pany Pharmaceutical Research Institute, 5 Research Parkway, Walling-ford, CT 06492. Phone: (203) 677-7015. Fax: (203) 677-6771. E-mail:[email protected].
† Supplemental material for this article may be found at http://ec.asm.org/.
638

MATERIALS AND METHODS
Bioinformatics. The completed genome sequences were acquired from theNCBI Reference Sequence database (http://ncbi.nih.gov/RefSeq/) for the follow-ing 10 fungal and human strains: S. cerevisiae S288c (5,866 open reading frames[ORFs]), C. albicans SC5314 (13,685 ORFs), C. glabrata CBS138 (5,192 ORFs),A. fumigatus Af293 (9,907 ORFs), Cryptococcus neoformans JEC21 (6,593ORFs), D. hansenii CBS767 (6,317 ORFs), K. lactis NRRL Y-1140 (5,336ORFs), Schizosaccharomyces pombe 972 h� (5,035 ORFs), Y. lipolitica CLIB99(6,544 ORFs), Eremothecium gossypii ATCC 10895 (4,718 ORFs), and Homosapiens (29,236 ORFs). Other sources of genome sequence and annotation datainclude Genome Therapeutics Corporation, Waltham, MA, The Institute forGenomic Research (http://www.tigr.org), the Yeast Proteome Database (YPD)(http://www.proteome.com/), and Incyte Pharmaceuticals, Palo Alto, CA. Whereincomplete genome sequences were encountered, coverage was increased byadding contiguous sequences for the same organism from multiple public and/orprivate sources and assembling the data using the PHRAP algorithm (http://www.phrap.org/). Likely genes that were not yet annotated in available yet incompletegenomes were identified using the program CRITICA (4), and MAGPIE (17)was used to visualize genes in context. Similar genes were identified using the“neighbors” function of the concordance system (7). To determine if the pre-dicted gene sequences were full length and of the proper reading frame forincomplete genomes, both BLAST searches and CLUSTAL multiple sequencealignments (1, 26) were conducted.
To identify conserved genes, whole-genome comparisons based on all-versus-all pairwise alignments with the FASTA algorithm (41) were conducted using theconcordance function essentially as described previously (7). Briefly, all ORFs(�100 amino acids) of the query genome were compared to ORFs from variousother genomes and stored. A list of ORFs and their associated amino acidsequences was then generated at a specified percent protein identity cutoff.When a gene of interest was not found in a genome, FRAMESEARCH (24) wasused to determine that it was not due to a failure of the gene-finding algorithm.
Strains and growth media. The C. albicans strains used were SC5314 (wildtype; Bristol-Myers Squibb Culture Collection) and its derivative, BWP17(ura3�::�imm434/ura3�::�imm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG),kindly provided by A. P. Mitchell of Columbia University. The S. cerevisiae strainused was ATCC 201390 (MATa/MAT� his3�1/his3�1 leu2�0/leu2�1 lys2�0/LYS2 met15�0/MET15 ura3�0/ura30). Escherichia coli strain DH5� was used forplasmid propagation. Yeast extract-Bacto peptone-dextrose medium (YEPD),synthetic complete (SC) medium, and synthetic dextrose (SD) were preparedaccording to standard procedures as previously described (46). Uridine (25 �gml�1) was added according to a method described previously by Fonzi and Irwin(16), and other supplements such as histidine and arginine (20 �g ml�1) wereadded as described previously (46). In general, 5 mM of methionine and 2.5 mMof cysteine were used in media unless otherwise indicated. Both S. cerevisiae andC. albicans strains were grown at 30°C, and the E. coli strain was grown at 37°C.
DNA manipulations. DNA manipulations were carried out according to stan-dard molecular methods (45) unless otherwise noted. Typical PCRs were carriedout in a volume of 100 �l with an initial 2-min jump start at 95°C, 30 cycles ofamplification (2 min at 95°C, 1 min at 52°C, and 5 min at 72°C), and a final10-min extension step at 72°C. Typical reaction mixtures contained 10 mMTris-HCl, pH 8.3, 1.5 mM MgCl2, 50 mM KCl, 0.2 mM deoxynucleoside triphos-phates, 10 pmol of primers, 100 ng of genomic template DNA, and 5 U of TaqDNA polymerase (Gibco-BRL) or Pfu Turbo DNA polymerase (Stratagene).Total genomic DNA from C. albicans was isolated by the glass bead lysis method(27). All PCR primers used in this study are listed in Table S1 in the supple-mental material.
Transformation of C. albicans was carried out as described previously (54).Typically, about 20 to 80 �l of the PCR mixture was used for transformation. Fordisrupting the first allele of a target gene, cells were plated onto selectivemedium (SC minus uridine or SC minus arginine), while the second round oftransformation to disrupt the second allele used SC minus uridine and argininemedium.
Construction of C. albicans MET3 promoter-swapping cassettes. Two plas-mids, pUMP and pAMP, which contain a CaMET3 promoter cassette, wereconstructed. Plasmid pUMP contains the CaURA3 gene, and pAMP harborsCaARG4 as the selective marker. The 1.4-kb C. albicans MET3 promoter regionwas amplified by PCR from genomic DNA of strain SC5314. To constructpUMP, the CaMET3 promoter PCR product was cut with restriction enzymesSphI and NcoI, gel purified, and ligated into pGEM-URA3 that was linearizedby SphI and NcoI. This placed the CaMET3 promoter sequence adjacent toCaURA3 but in the opposite orientation to avoid transcription read-through. Toconstruct pAMP, the C. albicans ARG4 gene was excised from pRS-ARG4�SpeI
after digestion with SacI and KpnI (blunt ended) and ligated into the SacII(blunt ended) and SacI sites of pGEM-URA3 to replace CaURA3. Theresulting plasmid was linearized with SphI and NcoI, gel purified, and ligatedwith the CaMET3 promoter PCR product treated with SphI and NcoI, yield-ing plasmid pAMP.
Construction of conditional mutants. Two steps were involved in constructingconditional mutants of C. albicans. The first step was the disruption of the firstallele of the gene of interest by the PCR-based gene disruption method (54) instrain BWP17, with subsequent verification by PCR analysis. Next, the promoterof the second copy of the gene was replaced by the CaMET3 promoter in theheterozygous strain. To do this, the CaMET3 promoter-swapping cassette wasamplified by PCR from plasmid pUMP or pAMP. The PCR primers usedcontained both template-specific common primer sequences and gene-specificflanking sequences of 50 to 60 bp in length. The common primer sequencesannealed to the plasmid template pUMP or pAMP for amplification of thepromoter-swapping cassette, whereas the flanking sequences directed the ampli-fied promoter cassette fragment to its specific location on the chromosomeduring recombination. The resulting PCR product of the CaMET3 promoter-swapping cassette was then introduced into the cells that were heterozygous forthe gene of the interest to replace the endogenous promoter of the remainingallele via homologous recombination. Typically, between 500- to 1,000-bp nucle-otides upstream of the ATG codon of the gene of interest were removed, andpromoter constructs were verified by PCR.
Cell-based high-throughput screening. C. albicans conditional mutants har-boring the gene of interest under the control of the CaMET3 promoter were usedfor screening of target-specific inhibitors in the absence and presence of methi-onine or cysteine. To carry out HTS, a culture grown overnight was diluted in SDplus His broth medium to 7.5 � 105 CFU/ml, 35 �l of which was dispensed intowells of 384-well plates containing 8 �l of compounds in each well. Ten micro-liters of SD plus His broth containing or lacking methionine or cysteine wasadded to these wells. The final compound concentration at screening was 14.2�M. The concentration of methionine or cysteine used was the 50% inhibitoryconcentration (IC50) value previously determined, such that cells would prolif-erate at half the growth rate compared to no-methionine or cysteine controlcultures. The plates were then incubated at 30°C for 18 h, and cell growth wasdetermined by reading the absorbance at 590 nm on a Perkin-Elmer 7000 platereader. Cell growth inhibition was calculated by using plain medium as a “blank”and cells grown in medium with dimethyl sulfoxide only as the “total.” Todetermine dose responses, the compound concentration that causes 50% growthinhibition (IC50) was calculated using the following two-parameter logistic equa-tion: Y 100/[1 � (IC50/I)H], where Y is the percent growth inhibition, I is theinhibitor concentration, and H is the hill slope, as described by GraphPadSoftware Inc. (San Diego, CA).
RESULTS
Identification of conserved fungal genes via concordanceanalysis. It is generally believed in the antimicrobial drug dis-covery community that conserved, essential genes have greatpotential to yield broad-spectrum antimicrobial agents. Usingthe 1,049 essential S. cerevisiae genes (http://www.yeastgenome.org) as a query, we conducted a pairwise genome comparisonor concordance analysis across the nine other complete fungalgenomes in an effort to identify genes that are conserved acrossthose fungi (see Materials and Methods for details). To deter-mine a legitimate cutoff for gene conservation, we profiled alist of known fungal “drug-able” targets against the 10 fungalgenomes. This allowed us to quantitatively correlate the broad-spectrum capacity of the currently marketed drugs to the per-cent protein identity of their corresponding drug targets. Vori-conazole (VFend) and caspofungin (Cancidas) are consideredto have broad-spectrum capacity against a number of medicallyimportant fungi, including Candida species and Aspergillus spe-cies (30, 34). Voriconazole targets lanosterol 14-�-demethyl-ase, encoded by the essential gene ERG11 in the yeast S.cerevisiae (50). The target of caspofungin is an essential cellwall enzyme, -1,3-glucan synthase, encoded by FKS1 andFKS2 and regulated by RHO1 in S. cerevisiae (35, 36). The
VOL. 5, 2006 IDENTIFICATION OF CONSERVED FUNGAL GENES 639

putative homologs of all four genes were identified in the 10fungal genomes with protein sequence identity of more than40% for Erg11p and more than 50% for each of the three geneproducts for glucan synthase (Table 1). In vitro, terbinafine(Lamisil) has an inhibitory activity against many strains ofCandida species, Aspergillus species, and Cryptococcus neofor-mans (although it is only marketed for the treatment of footfungus) (31), and its target, squalene epoxidase (Erg1p), wasfound to be conserved at �39% across the 10 fungi (Table 1).In addition, four other known fungal targets, CHS2, RAM2,NMT1, and EFT2, which are the subject of preclinical researchbut lack a marketed drug, were also compared side by sideacross the 10 fungal genomes and were found to be conservedat 30% or higher at the protein level (Table 1). Based on theabove-described data, we set the benchmark in our search forconserved genes across the fungal genomes to �40% identityat protein level.
A total of 240 genes, denoted conserved fungal genes, wereidentified with percent identity at �40% at the protein levelusing the 1,049 essential S. cerevisiae genes as a query (seeTable S2 in the supplemental material). To view the distribu-tion of those genes by function, we assigned them to the func-tional and cellular categories described by the YPD (http://www.proteome.com) for S. cerevisiae (Table 2). It was foundthat the 240 conserved fungal genes encode proteins involvedin all 39 cellular processes categorized by the YPD. As shownin Table 2, the top four cellular processes to which the con-served fungal genes were assigned are protein synthesis (13%),RNA processing modification (10%), protein degradation(7%), and polymerase II (Pol II) transcription (5%), the basic,evolutionarily conserved cellular processes. It is interesting thatthere are 3% of the conserved fungal genes whose biologicalfunctions were unknown in S. cerevisiae according to the Saccha-romyces Genome Database (SGD) (http://www.yeastgenome.org[accessed October 2005]).
The preferred profile for fungal targets are essential proteinsconserved among fungi yet absent from the human genome soas to minimize potential toxic side effects exerted by pharma-cological inhibition of the cellular targets. With this ideal inmind, the 240 conserved fungal genes were prioritized basedon their relatedness to human genes (Fig. 1A). Interestingly,more than half (59%) of the conserved fungal genes haveputative human homologs with percent identities ranging from41 to 60%, whereas each of those having the least (0 to 40%)and most (61 to 100%) identity with human genes constitutesaround 20% of the conserved fungal genes identified. Whenthe essential yeast gene set (1,049 genes) as well as the entireyeast gene set (5,866 genes) were compared with the humangenome, it was found that the majority of the yeast genes(�76% for the essential gene set and �88% for whole geneset) do not have significant human homologs (�40% proteinidentity) (Fig. 1B; data shown for human homologs of theentire yeast gene set only). Similar findings were also madewhen each of the remaining nine fungal genomes was com-pared with the human genome, in which between 85 to 93% ofthe fungal genes for any one organism were found to be uniqueto fungi (data not shown). This indicates that most (�80%) ofthe 240 conserved fungal genes identified here come from only7 to 15% of each fungal genome that is also conserved in the
TA
BL
E1.
Hom
olog
yco
mpa
riso
nof
know
nan
tifun
galt
arge
ts
Gen
eID
inS.
cere
visi
aeA
nnot
atio
nin
SGD
Kno
wn
drug
(s)
orin
hibi
tor(
s)
%Id
entit
yto
othe
rfu
ngia
tpr
otei
nle
vel
C.a
lbic
ans
C.g
labr
ata
A.f
umig
atus
C.n
eofo
rman
sD
.han
seni
iK
.lac
tisS.
pom
beY
.lip
oliti
caE
.gos
sypi
iH
.sap
iens
ER
G11
Lan
oste
rol-1
4�-d
emet
hyla
seA
zole
s64
8347
4364
7546
6071
36F
KS1
Subu
nit
of1,
3-
-glu
can
synt
hase
Cas
pofu
ngin
,m
icaf
ungi
n73
8663
5274
8256
6572
Non
e
FK
S2Su
buni
tof
1,3-
-g
luca
nsy
ntha
seC
aspo
fung
in,
mic
afun
gin
7386
6353
7482
5665
72N
one
RH
O1
GT
Pbi
ndin
gpr
otei
nre
gula
ting
gluc
ansy
ntha
se
Cas
pofu
ngin
,m
icaf
ungi
n77
9272
6876
8371
7384
64
CH
S2C
hitin
synt
hase
IIN
ikko
myc
ins
4076
3633
4165
3642
60N
one
ER
G1
Squa
lene
epox
idas
eT
erbi
nafin
e52
8339
3953
7437
4464
30R
AM
2Su
buni
tof
gera
nylg
eran
yltr
ansf
eras
eN
one
3955
3132
3851
3035
4820
NM
T1
N-M
yris
toyl
tran
sfer
ase
Non
e54
7246
3758
6447
4765
36E
FT
2T
rans
latio
nel
onga
tion
fact
or2
Sord
arin
8793
7974
8793
7985
9266
640 LIU ET AL. EUKARYOT. CELL

human genome, which at least partly explains the difficulty indeveloping safe and selective antifungal drugs.
Development of a CaMET3 promoter system for essentialgene identification in C. albicans. Although the aforemen-tioned conserved fungal genes are essential in S. cerevisiae, it is
necessary to verify their essentiality in the remaining fungi,especially in the pathogenic fungi, in order for them to beconsidered antifungal targets. Towards this goal, we developeda CaMET3 promoter system capable of large-scale identifica-tion of essential genes in the primary fungal pathogen C. albi-
TABLE 2. Functional distribution of the 240 conserved fungal genes identified
Function categorya Gene(s) of S. cerevisiae No. ofgenesb
% ofgenesc
Protein synthesis CDC95, GRS1, RIA1, RPS31, SUP35, YLL034C, RPL18B, SIS1, SUP45, DYS1, ILS1,MES1, NMD3, ALA1, CDC60, DED1, DED81, DPS1, EFB1, FRS1, FRS2, GLN4,HTS1, HYP2, KRS1, RLP24, RPL10, RPL15A, RPL17A, RPL3, RPL5, RPP0, RPS15,RPS2, RPS20, RPS3, RPS5, SES1, SUI2, SUI3, THS1, TIF11, TIF34, TIF5, VAS1,R341C, YEF3, YHR020W, YIF2, YNL247W
50 12.8
RNA processing modification CDC95, GRS1, HAS1, RIA1, RRP3, RVB1, RVB2, GLC7, SPB1, GSP1, ACC1, DBP5,MTR4, DIB1, DIS3, LSM2, PRP22, PRP43, SNU13, EMG1, BMS1, CBF5, DBP2,DBP9, DIM1, ERB1, FAL1, GAR1, HCA4, IMP3, IMP4, MAK16, NIP7, NOP1,NOP2, NOP5, SIK1
37 9.5
Protein degradation KAE1, RPS31, RPT4, RPT6, RPN11, RPN6, RPN8, RPT1, RPT2, RPT3, RPT5, SKP1,RSP5, UBA1, SRP1, CDC48, UBC9, PRE1, PRE10, PRE2, PRE3, PRE4, PRE5,PRE8, PUP1, PUP2, PUP3, SCL1
28 7.2
Pol II transcription RVB1, RVB2, RPT4, RPT6, RPN11, RPN6, RPN8, RPT1, RPT2, RPT3, RPT5, ACT1,SPT16, TOP2, SSL2, RPO26, RPB5, SPT15, RPO21
19 4.9
Cell structure ACT1, SSC1, CCT2, CCT3, CCT4, CCT5, CCT6, CCT7, CCT8, TCP1, TUB1, CDC42,ARC19, ARC35, TUB2
15 3.8
Protein folding SUP35, YLL034C, SIS1, SUP45, SSC1, CCT2, CCT3, CCT4, CCT5, CCT6, CCT7,CCT8, TCP1, HSP60, KAR2
15 3.8
Vesicular transport RPS31, ACT1, RHO3, ARP3, LST8, SEC13, YKT6, SEC23, COP1, GDI1, SAR1,SEC26, SEC27, SEC4, YPT1
15 3.8
Unknown BRX1, FRQ1, KRE30, KRE33, KRR1, NBP35, NSA2, RLI1, RPF2, R339C, YJR072C,YLR243W, YOR262W
13 3.3
Carbohrate metabolism GLC7, PCM1, SEC53, CDC19, TPI1, GFA1, ACS2, ENO2, FBA1, PGI1, PGK1, UGP1 12 3.1Chromatin chromosome
structureRVB1, RVB2, RPT4, RPT6, RSP5, SSL2, POL2, RFC4, RFC5, CDC2, CDC54, RAD3 12 3.1
DNA repair RVB1, RVB2, RPT4, RPT6, RSP5, SSL2, POL2, RFC4, RFC5, CDC2, CDC54, RAD3 12 3.1Protein modification DYS1, RSP5, UBA1, PCM1, SEC53, GPI8, PSA1, BET2, MAS1, MAS2, PMI40, STT3 12 3.1Other metabolism HEM1, NFS1, PMA1, GLN1, HEM15, HEM2, HEM12, HEM13, QNS1, RIB3, RIB5 11 2.8Cell cycle control GSP1, SKP1, SRP1, CDC48, UBC9, SPT16, TUB1, RHO1, CDC28, SDS22 10 2.6Cell polarity GLC7, ACT1, CDC42, ARC19, ARC35, RHO3, ARP3, RHO1, CDC12, PWP2 10 2.6DNA synthesis POL2, RFC4, RFC5, RFC2, RFC3, CDC2, CDC54, CDC47, MCM2, MCM6 10 2.6Lipid fatty acid and sterol
metabolismACC1, HEM1, ERG10, ERG11, ERG13, ERG20, ERG25, ERG7, IDI1, MVD1 10 2.6
Small-molecule transport RSP5, UBA1, LST8, SEC13, NFS1, PMA1, ATM1, PET9, NEO1 9 2.3Nuclear-cytoplasmic transport ILS1, MES1, GSP1, ACC1, DBP5, MTR4, SRP1, CRM1 8 2.0Amino acid metabolism SKP1, RSP5, UBA1, GLN1, ILV3, ILV5, SAH1 7 1.8Energy generation CDC19, TPI1, NFS1, HEM15, HEM2, ATM1, PET9 7 1.8Pol III transcription RPO26, RPB5, SPT15, RPC40, RET1, RPC11, RPO31 7 1.8Mitosis DIB1, DIS3, TOP2, TUB1, TUB2 6 1.5Pol I transcription RPO26, RPB5, SPT15, RPC40, RPA135, RPA190 6 1.5Protein translocation SUP35, SSC1, HSP60, KAR2, SEC61, SRP54 6 1.5RNA splicing DIB1, LSM2, PRP22, PRP43, SNU13, SUB2 6 1.5Nucleotide metabolism ADE13, GUK1, RNR1, RNR2, URA6 5 1.3Cell wall maintenance GFA1, GPI8, PSA1, RHO1 4 1.0Cytokinesis YLL034C, CDC48, CDC12, PWP2 4 1.0Signal transduction RSP5, RHO3, RHO1, NOG1 4 1.0Cell stress GLC7, HTB1, TRR1 3 0.8Mating response ACT1, CDC42, CDC12 3 0.8Meiosis GLC7, CDC28, NOP7 3 0.8Membrane fusion YLL034C, CDC48, YKT6 3 0.8Other ACT1, SEC23 2 0.5Protein complex assembly EMG1, RPO26 2 0.5RNA turnover NMD3, LSM2 2 0.5Recombination SUB2, RAD3 2 0.5Phosphate metabolism IPP1 1 0.3
Total 391
a The 39 function categories as described by the YPD (http://www.proteome.com).b Total counts were over 240 since certain genes were assigned to multiple functions by the YPD.c Percentage was calculated based on the number of genes in each category divided by the total number (391).
VOL. 5, 2006 IDENTIFICATION OF CONSERVED FUNGAL GENES 641

cans. In this system, a conditional mutant where one allele ofthe gene of interest was disrupted by the PCR-based genedisruption technique (54) and the second allele was placedunder the control of the CaMET3 promoter (see Fig. 2A for aschematic illustration of the promoter-swapping approach) wasconstructed. Homologous recombinations were verified byPCR confirmation analysis using various primer sets (Fig. 2B).Gene essentiality was determined by comparing phenotypes ofserially diluted conditional mutants grown on SD agar plates inthe absence and presence of methionine and cysteine (Fig.2C). Scores of 1 to 4 were used to assess the degree of essen-tiality, with 1 being absolutely required and 4 being not re-quired for cell growth. As expected, three known essential C.albicans genes (CaERG1, CaRAM2, and CaNMT1) were de-termined to be essential in this work, with scores of 1, 3, and 1,respectively (Fig. 2C). However, CaPFY1, whose essentiality isstrain dependent in S. cerevisiae, was found to be not essentialin C. albicans (Fig. 2C). It is important that no growth defectby methionine and/or cysteine was observed for any heterozy-
gous parental strains tested (Fig. 2C; data shown for a repre-sentative heterozygous strain).
We previously identified a list of conserved genes between S.cerevisiae and C. albicans using the S. cerevisiae essential geneset (52). A dozen genes with no known biological function(denoted conserved unknown reading frames [CURFs]) wereselected in this work to evaluate their essentiality in C. albicanswith the hope of identifying novel, broad-spectrum antifun-gals (Table 3). Each of the 12 CURFs was evaluated by theCaMET3 promoter system, and 10 were determined to beessential in C. albicans (Table 3 and Fig. 2C). To corroboratethe essentiality results obtained by the CaMET3 promotermethod, all genes tested (known or unknown) were indepen-dently evaluated for essentiality by the PCR-based gene dis-ruption method (54). Comparable results were obtained be-tween the two methodologies for all genes except CaYJR072c,which was deemed to be not essential by the CaMET3 pro-moter method, yet the disruption method failed to obtain thedisruption of both alleles (data not shown).
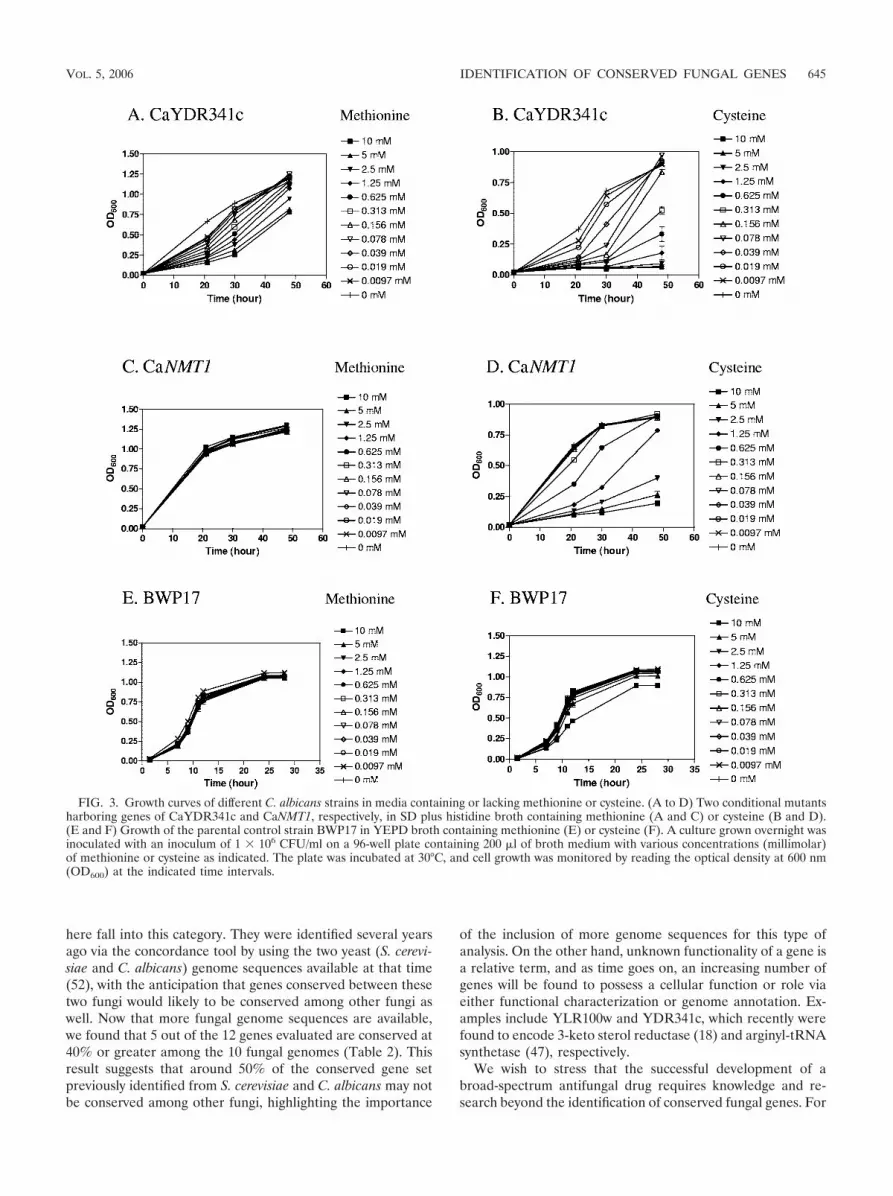
Characterization of the CaMET3 promoter for HTS amena-bility. It has previously been reported that both bacterial andfungal cells can become hypersensitive to specific inhibitorswhen the target molecule is rate limiting for cell growth byeither using heterozygous strains (11, 21) or using regulatable(either repressible/or inducible) promoters (12, 44). To assessthe utility of CaMET3 promoter in this regard, all 13 essentialgenes (3 known genes and 10 CURFs) identified in this workwere individually assessed for repression of growth via con-trolled expression of the CaMET3 promoter by the addition ofmethionine or cysteine. A variety of conditions such as inocu-lum size, methionine or cysteine concentration, and incubationtime were explored. Figure 3 shows growth curves of two rep-resentative conditional mutants and of the parental BWP17strain using various concentrations of either methionine orcysteine, demonstrating that the CaMET3 promoter could betitrated by either methionine (for example, CaYDR341c) orcysteine (for example, CaYDR341c and CaNMT1), while nei-ther methionine nor cysteine caused any significant growthdefect on the parental BWP17 strain at concentrations up to 5mM. To assist HTS, the concentration of either methionine orcysteine that inhibits 50% of the cell growth (IC50), was deter-mined for each of the 13 conditional mutants tested (Table 4).As discussed below, the IC50 was the concentration arbitrarilychosen and used to set cells that were rate limiting in growth inthe cell-based HTS to identify hypersensitive inhibitors. Wefound that cysteine alone was capable of repressing all 13essential genes tested, while methionine was only able to re-press 3 (CaYDR341c, CaYOL077c, and CaERG1) at up to 10mM (Fig. 3 and Table 4).
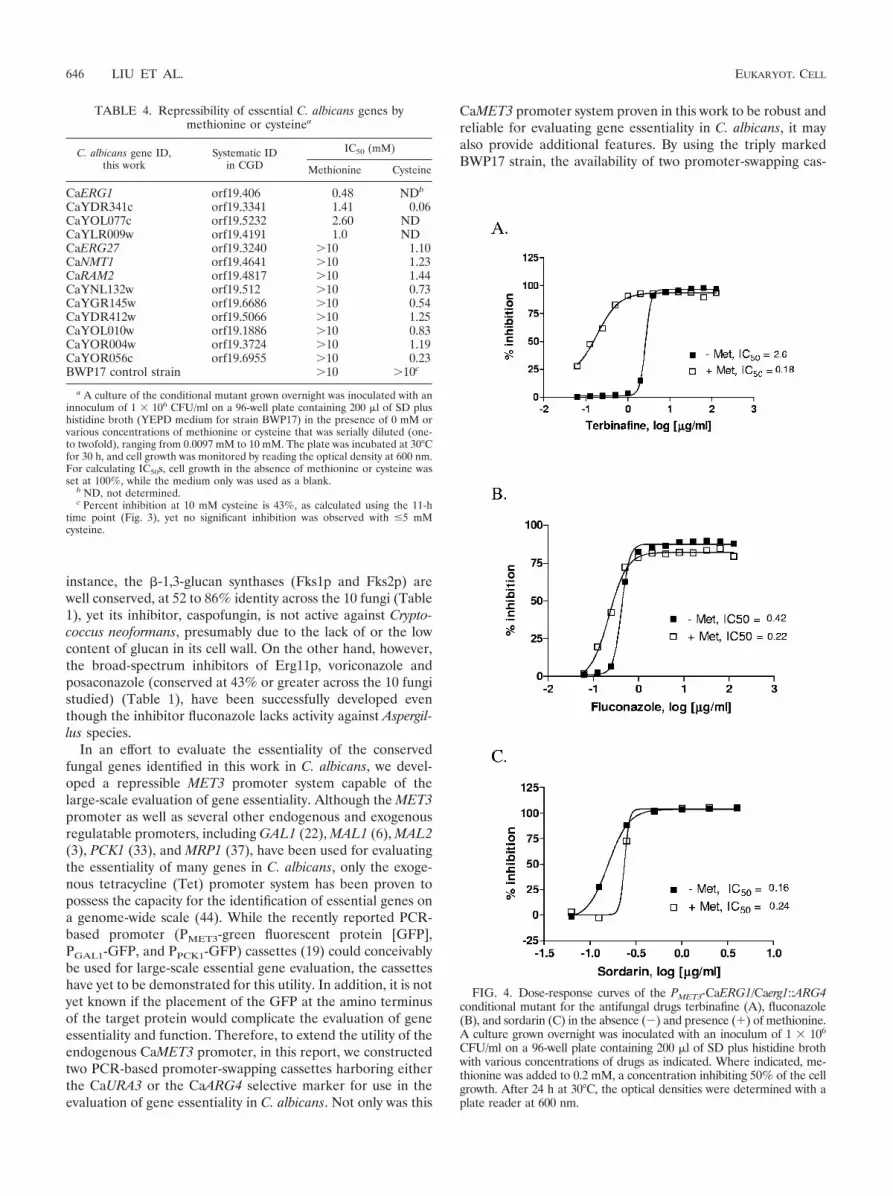
The C. albicans ERG1 gene, encoding a squalene epoxidase,is specifically inhibited by terbinafine (13, 20). To assess drughypersensitivity, growth inhibition by terbinafine was deter-mined for the PMET3-CaERG1 conditional mutant in the pres-ence and absence of methionine. As shown in Fig. 4A, a 15-fold increase in terbinafine potency (expressed as IC50) wasobserved in the presence of methionine compared to the no-methionine control. In addition, the growth inhibition in thepresence of methionine varied from about 25 to 90% over a logrange of inhibitor concentrations (0.06 to 1 �g/ml), whereasonly 0.5 to 3.5% growth inhibition was observed in the absence
FIG. 1. (A) Illustration of identifiable human homologs of the 240conserved fungal genes (FCGs) as percent protein identity, with thelower left corner showing the least homology and upper right cornershowing the most homology. (B) Human homologs of the genes(5,866) of S. cerevisiae plotted as percent protein identity.
642 LIU ET AL. EUKARYOT. CELL

of methionine over the same terbinafine concentration range(Fig. 4A). On the contrary, the experiments conducted withfluconazole (Fig. 4B) or sordarin (Fig. 4C), which are knowninhibitors of ERG11 and EFT2 (29), respectively, showed thatsensitivities of the PMET3-CaERG1 conditional mutant to thesedrugs were not altered by the presence of methionine. Similarresults were also obtained when a modified MIC test wasconducted for the three antifungals for comparison from thesame cultures (data not shown) (the MIC was defined as thedrug concentration that gave 100% of cell growth inhibition forterbinafine or sordarin and 80% of inhibition for fluconazole,respectively). Furthermore, the sensitivity of a PMET3-CaRAM2conditional mutant to these three drugs remained unchangedin either the absence or presence of 5 mM methionine and 1mM cysteine (data not shown). Taken together, these resultsestablished that reduced expression of CaERG1 resulted incells that were hypersensitive only to the target-specific inhib-itor terbinafine.
Parallel high-throughput screening. The utility of the CaMET3promoter for identifying hypersensitive inhibitors was furtherexplored by conducting a cell-based parallel HTS campaignusing the PMET3-CaERG1 conditional mutant. A library of�12,000 compounds in the Bristol-Myers Squibb Companycompound collection was screened in the absence and pres-ence of methionine (denoted Met minus and Met plus, respec-tively) (see Materials and Methods for details). Using �40%growth inhibition as a cutoff at the 14.2 �M compound con-centration, 37 hits were identified from the Met-plus screen, 18of which were also scored positive in the Met-minus screen(Fig. 5). By superimposing the 37 compounds from both theMet-plus and Met-minus screen with respect to percent inhi-bition of cell growth, we categorized them into three groups.Group I has six hits with percent growth inhibition rangingfrom 41 to 70% in the Met-plus screen and �11 to 10% in theMet-minus screen. Group II contains hits with �40% inhibi-tion in the Met-plus screen and 11 to 40% inhibition in the
FIG. 2. Construction of C. albicans conditional mutants by using the CaMET3 promoter-swapping cassette. (A) Scheme of promoter swappingin which the PCR-amplified CaMET3 promoter-swapping cassette is transformed to a heterozygous YFG1 strain, replacing the endogenouspromoter of the second allele via homologous recombination. Primer sets used for confirmation PCR are shown as arrows and are as follows: 5�promoter/3� PTURA, 5� PTURA/3� promoter, and 5� gene/3� gene. (B) Verification of CaMET3 promoter conditional mutants of the CaERG1gene, as an example, by PCR confirmation analysis. Lanes labeled “M” contain 1-kb DNA. Lanes 1 and 2 depict examples of wrong constructs,whereas lanes 3 and 4 are examples of correct conditional mutant constructs. The expected sizes of PCR products using the primer sets 5�PTURA/3� promoter, 5� promoter/3� PTURA, and 5� gene/3� gene are 1.6 kb, 1.2 kb, and 2.4 kb, respectively. (C) Growth phenotypes of variousCaMET3 promoter conditional mutants grown on SD plus histidine medium (SD plus histidine plus uridine for the CaERG1/Caerg1::ARG4 mutantused as a growth control) in the absence (�) or presence (�) of 5 mM of methionine and 2.5 mM of cysteine after incubation for 2 days at 30°C.Cells were serially diluted (1 to 10) starting at 2 � 106 CFU/ml for the first dilution, and 5 �l of the dilution was spotted from left to right. Anarbitrary score of 1, 2, 3, or 4 was assigned to a specific conditional mutant corresponding to its degree of growth defect in SD plus methionineand cysteine, with 1 being most essential and 4 being nonessential.
VOL. 5, 2006 IDENTIFICATION OF CONSERVED FUNGAL GENES 643

Met-minus screen. Group III consists of the 18 hits common toboth conditions (both �40%). By referring to the dose-re-sponse curves for terbinafine in the presence and absence ofmethionine (Fig. 4A), those six hits in group I therefore rep-resent the so-called “hypersensitive” hits that would otherwisenot be found if normal strains were used in HTS.
DISCUSSION
Identification of conserved fungal genes is a first step to-wards the discovery and development of future broad-spec-trum antifungal agents. Although the inclusion of medicallyimportant fungi should conceivably be adequate for the anal-ysis, we chose to include as many available completed fungalgenomes as possible for the following reasons. First, a humanpathogen is a relative term from a clinical perspective. Forexample, C. albicans and A. fumigatus are both opportunistichuman pathogens that did not produce deep-organ, potentiallyfatal diseases in humans until recently due to advances inmodern medicine (32, 38). Second, broad-spectrum antibioticsand antifungals do not discriminate between pathogenic andnonpathogenic microbes. In addition, we wish to emphasizethat the 240 conserved fungal genes identified in this workshould serve as a starting point for future fine tuning as morefungal genomes are sequenced and more essential genes ineach species are identified.
Although our concordance analysis is a powerful tool formultiple-genome analysis, certain genes would inevitably bemissed in our gene set. They include proteins that share con-served function due to structural and/or functional motif (forexample, active site) similarity but not primary sequence sim-ilarity (14). Other genes that can be missed are those that arenonessential when evaluated individually, yet the protein com-plex, for example, to which the gene product is assembled canbe vital for cell growth. The fungal -1,3-glucan synthase issuch an example, where neither S. cerevisiae FKS1 (ScFKS1)nor ScFKS2 is required individually for cell viability, yet thedisruption of both is lethal (35). Another example is transla-tional elongation factor 2, which is encoded by two nonessen-tial genes (EFT1 and EFT2) (42).
To qualify as potential antifungal targets, the conserved fun-gal genes have to be essential for fungal cell growth, especiallyfor medically important fungi. Since the verification of geneessentiality is a tremendous task even in the model yeast S.cerevisiae, we decided to initiate the conserved fungal geneidentification process by using the essential gene set of S.cerevisiae. We reasoned that fungal genes that are conservedwith the essential S. cerevisiae genes would more likely beessential in other fungi. In this regard, the conserved gene setidentified previously by Braun et al. (5) from six fungi (C.albicans, S. cerevisiae, Schizosaccharomyces pombe, A. niger,Magnaporthe grisea, and Neurospora crassa) would likely con-tain many genes that are not essential, even in S. cerevisiae (forexample, ARO4, DAL5, and AGP2, to name a few). Thosenonessential genes would therefore not be considered antifun-gal targets.
In addition to finding conserved fungal genes, the identifi-cation of conserved genes of unknown function would be an-other way of prioritizing targets in the hope of finding novelclasses of broad-spectrum inhibitors. The 12 CURFs evaluated
TA
BL
E3.
Prop
ertie
sof
fung
alge
nes
ofun
know
nfu
nctio
nid
entifi
edin
this
wor
k
Gen
eID
for
S.ce
revi
siae
Gen
eID
inC
GD
Des
crip
tiona
Nul
lmut
ant
phen
otyp
ein
CG
D
Ess
entia
lin
this
wor
k
%Pr
otei
nid
entit
yw
ithot
her
fung
i
C.a
lbic
ans
C.g
labr
ata
A.f
umig
atus
C.n
eofo
rman
sD
.han
seni
iK
.lac
tisS.
pom
beY
.lip
oliti
caE
.gos
sypi
iH
.sap
iens
YL
R10
0w/E
RG
27E
RG
27/o
rf19
.324
03-
Ket
ost
erol
redu
ctas
eV
iabl
e/in
viab
leY
es59
5725
2161
6835
4258
21Y
DR
341c
/RR
S1or
f19.
3341
Arg
inyl
-tR
NA
synt
heta
seN
otav
aila
ble
Yes
6363
5045
6271
5461
6235
YL
R02
2c/S
DO
1or
f19.
2708
Unk
now
npr
otei
nin
volv
edin
RN
Am
etab
olis
mN
otav
aila
ble
No
5654
2644
5474
4650
4645
YO
L07
7c/B
RX
1C
SI2/
orf1
9.52
325S
rRN
Abi
ndin
gin
S.ce
revi
siae
;In
volv
edin
chiti
nsy
nthe
sis
and
filam
ento
usgr
owth
inC
.al
bica
ns
Het
eroz
ygou
svi
able
Yes
7068
3644
6485
5063
6836
YN
L13
2w/K
RE
33or
f19.
512
Unk
now
nN
otav
aila
ble
Yes
7575
6654
7685
6368
7556
YJR
072c
/NP
A3
orf1
9.64
63Pr
otei
nbi
ndin
gIn
viab
leN
o65
6547
4260
7244
5562
40Y
GR
145w
/EN
P2
orf1
9.66
86U
nkno
wn
prot
ein;
rRN
Apr
oces
sing
Not
avai
labl
eY
es58
5835
3457
6539
4855
34
YD
R41
2wor
f19.
5066
Unk
now
nN
otav
aila
ble
Yes
4344
2522
4357
2137
4115
YO
L01
0w/R
CL
1or
f19.
1886
RN
Ate
rmin
alph
osph
ate
cycl
ase-
like
prot
ein
invo
lved
inrR
NA
proc
essi
ng
Not
avai
labl
eY
es69
6640
4368
7145
5468
41
YO
R00
4wor
f19.
3724
Unk
now
npr
otei
nin
volv
edin
rRN
Apr
oces
sing
Not
avai
labl
eY
es50
4633
3446
5842
3848
28
YO
R05
6c/N
OB
1or
f19.
6955
RN
A,p
rote
inbi
ndin
gN
otav
aila
ble
Yes
4946
3327
4958
3142
4824
YL
R00
9w/R
LP
24or
f19.
4191
Unk
now
npr
otei
nin
volv
edin
ribo
som
alla
rge-
subu
nit
biog
enes
is
Not
avai
labl
eY
es69
7142
4472
8043
6070
42
aC
ombi
ned
info
rmat
ion
take
nfr
ombo
thth
eSG
D(h
ttp:
//ww
w.y
east
geno
me.
org)
and
the
CG
D(h
ttp:
//ww
w.c
andi
dage
nom
e.or
g).
644 LIU ET AL. EUKARYOT. CELL
here fall into this category. They were identified several yearsago via the concordance tool by using the two yeast (S. cerevi-siae and C. albicans) genome sequences available at that time(52), with the anticipation that genes conserved between thesetwo fungi would likely to be conserved among other fungi aswell. Now that more fungal genome sequences are available,we found that 5 out of the 12 genes evaluated are conserved at40% or greater among the 10 fungal genomes (Table 2). Thisresult suggests that around 50% of the conserved gene setpreviously identified from S. cerevisiae and C. albicans may notbe conserved among other fungi, highlighting the importance
of the inclusion of more genome sequences for this type ofanalysis. On the other hand, unknown functionality of a gene isa relative term, and as time goes on, an increasing number ofgenes will be found to possess a cellular function or role viaeither functional characterization or genome annotation. Ex-amples include YLR100w and YDR341c, which recently werefound to encode 3-keto sterol reductase (18) and arginyl-tRNAsynthetase (47), respectively.
We wish to stress that the successful development of abroad-spectrum antifungal drug requires knowledge and re-search beyond the identification of conserved fungal genes. For
FIG. 3. Growth curves of different C. albicans strains in media containing or lacking methionine or cysteine. (A to D) Two conditional mutantsharboring genes of CaYDR341c and CaNMT1, respectively, in SD plus histidine broth containing methionine (A and C) or cysteine (B and D).(E and F) Growth of the parental control strain BWP17 in YEPD broth containing methionine (E) or cysteine (F). A culture grown overnight wasinoculated with an inoculum of 1 � 106 CFU/ml on a 96-well plate containing 200 �l of broth medium with various concentrations (millimolar)of methionine or cysteine as indicated. The plate was incubated at 30°C, and cell growth was monitored by reading the optical density at 600 nm(OD600) at the indicated time intervals.
VOL. 5, 2006 IDENTIFICATION OF CONSERVED FUNGAL GENES 645
instance, the -1,3-glucan synthases (Fks1p and Fks2p) arewell conserved, at 52 to 86% identity across the 10 fungi (Table1), yet its inhibitor, caspofungin, is not active against Crypto-coccus neoformans, presumably due to the lack of or the lowcontent of glucan in its cell wall. On the other hand, however,the broad-spectrum inhibitors of Erg11p, voriconazole andposaconazole (conserved at 43% or greater across the 10 fungistudied) (Table 1), have been successfully developed eventhough the inhibitor fluconazole lacks activity against Aspergil-lus species.
In an effort to evaluate the essentiality of the conservedfungal genes identified in this work in C. albicans, we devel-oped a repressible MET3 promoter system capable of thelarge-scale evaluation of gene essentiality. Although the MET3promoter as well as several other endogenous and exogenousregulatable promoters, including GAL1 (22), MAL1 (6), MAL2(3), PCK1 (33), and MRP1 (37), have been used for evaluatingthe essentiality of many genes in C. albicans, only the exoge-nous tetracycline (Tet) promoter system has been proven topossess the capacity for the identification of essential genes ona genome-wide scale (44). While the recently reported PCR-based promoter (PMET3-green fluorescent protein [GFP],PGAL1-GFP, and PPCK1-GFP) cassettes (19) could conceivablybe used for large-scale essential gene evaluation, the cassetteshave yet to be demonstrated for this utility. In addition, it is notyet known if the placement of the GFP at the amino terminusof the target protein would complicate the evaluation of geneessentiality and function. Therefore, to extend the utility of theendogenous CaMET3 promoter, in this report, we constructedtwo PCR-based promoter-swapping cassettes harboring eitherthe CaURA3 or the CaARG4 selective marker for use in theevaluation of gene essentiality in C. albicans. Not only was this
CaMET3 promoter system proven in this work to be robust andreliable for evaluating gene essentiality in C. albicans, it mayalso provide additional features. By using the triply markedBWP17 strain, the availability of two promoter-swapping cas-
FIG. 4. Dose-response curves of the PMET3-CaERG1/Caerg1::ARG4conditional mutant for the antifungal drugs terbinafine (A), fluconazole(B), and sordarin (C) in the absence (�) and presence (�) of methionine.A culture grown overnight was inoculated with an inoculum of 1 � 106
CFU/ml on a 96-well plate containing 200 �l of SD plus histidine brothwith various concentrations of drugs as indicated. Where indicated, me-thionine was added to 0.2 mM, a concentration inhibiting 50% of the cellgrowth. After 24 h at 30°C, the optical densities were determined with aplate reader at 600 nm.
TABLE 4. Repressibility of essential C. albicans genes bymethionine or cysteinea
C. albicans gene ID,this work
Systematic IDin CGD
IC50 (mM)
Methionine Cysteine
CaERG1 orf19.406 0.48 NDb
CaYDR341c orf19.3341 1.41 0.06CaYOL077c orf19.5232 2.60 NDCaYLR009w orf19.4191 1.0 NDCaERG27 orf19.3240 �10 1.10CaNMT1 orf19.4641 �10 1.23CaRAM2 orf19.4817 �10 1.44CaYNL132w orf19.512 �10 0.73CaYGR145w orf19.6686 �10 0.54CaYDR412w orf19.5066 �10 1.25CaYOL010w orf19.1886 �10 0.83CaYOR004w orf19.3724 �10 1.19CaYOR056c orf19.6955 �10 0.23BWP17 control strain �10 �10c
a A culture of the conditional mutant grown overnight was inoculated with aninnoculum of 1 � 106 CFU/ml on a 96-well plate containing 200 �l of SD plushistidine broth (YEPD medium for strain BWP17) in the presence of 0 mM orvarious concentrations of methionine or cysteine that was serially diluted (one-to twofold), ranging from 0.0097 mM to 10 mM. The plate was incubated at 30°Cfor 30 h, and cell growth was monitored by reading the optical density at 600 nm.For calculating IC50s, cell growth in the absence of methionine or cysteine wasset at 100%, while the medium only was used as a blank.
b ND, not determined.c Percent inhibition at 10 mM cysteine is 43%, as calculated using the 11-h
time point (Fig. 3), yet no significant inhibition was observed with �5 mMcysteine.
646 LIU ET AL. EUKARYOT. CELL

settes with two different selective markers permits the evalua-tion of a gene in the rare situation where three allelic copiesexist (23). In this case, the first allele is deleted using the HIS1selective marker, followed by sequential placement of the sec-ond and third alleles under the control of the CaMET pro-moter using either the URA3 or the ARG4 cassette. It is note-worthy that out of the 13 essential C. albicans genes identifiedhere, CaYJR072c, which appeared to be nonessential in thisstudy, was reported previously by Davis et al. (10) to be re-quired for cell growth. While the different methodologies used(Tn7-UAU1 insertional mutagenesis versus repressible pro-moter) may account for the discrepancy, other possibilitiescould exist, since our attempt to obtain double disruption ofthis gene also failed (data not shown).
The CaMET3 promoter was further characterized and foundto be highly amenable to controlled gene expression. By vary-ing the amount of methionine or cysteine added to the growthmedium and thus varying the cellular level of the essential geneproduct (8), cell growth was correspondingly delayed or inhib-ited (Fig. 3 and Table 4). Interestingly, we found that cysteinecould control expression of the CaMET3 promoter moretightly than methionine, because all 13 conditional mutantscould be repressed in growth by cysteine but not all could berepressed by methionine at concentrations up to 10 mM (Table4). It is conceivable that the CaMET3 promoter is leaky whenregulated by methionine alone, since the maximum inhibitionof cell growth by methionine never reached beyond 80% for allconditional mutants tested (data not shown). Although theCaMET3 promoter has been utilized for the evaluation of theessentiality of several genes in C. albicans (8, 40, 53), it is notknown whether methionine or cysteine was the more effectivemodulator, since methionine and cysteine were often usedsimultaneously. Contradictory results were also reported in theS. cerevisiae literature regarding whether methionine, cysteine,or both are the true repressors of the enzymes of sulfur me-tabolism (39, 48). Nevertheless, our data presented here sug-gest that in a C. albicans background, cysteine is a much tighter
effector of the CaMET3 promoter. It should be noted, though,that a high concentration of cysteine (10 mM) did appear toslightly slow the growth of the parental BWP17 strain (Fig. 3F),albeit with the IC50 of cysteine being �10 mM (Table 4).
We demonstrated that the CaERG1 conditional mutantstrain displayed, in the presence of a rate-limiting amount ofmethionine, hypersensitivity to the specific inhibitor terbin-afine but not to other antifungals such as fluconazole andsordarin (Fig. 3). It should be noted that hypersensitivity tofluconazole and other azoles was reported with a CaERG1conditional mutant placed under the control of the CaMET3promoter (40). While different strain backgrounds (CAI4 ver-sus BWP17) could account for the discrepancy, different assaymethods (a spot assay on solid medium [40] versus a modifiedCLSI macrodilution method used in this work) could also beresponsible for the observed differences in drug susceptibility.
Traditionally, antimicrobial agents were identified via whole-cell screening. This approach, however, does not identify thebiochemical target of a lead compound. On the other hand,enzyme- or target-based in vitro screening often yields potentenzyme inhibitors but with little or no whole-cell activity.Moreover, there has been little success with attempts to buildin “whole-cell activity” for these enzyme inhibitors (43). Usingthe down-regulatable PMET3-CaERG1 conditional mutantstrain, we demonstrated, as a proof of principle, that hyper-sensitive compound hits with whole-cell activity could be iden-tified in a single parallel HTS campaign. Several lines of evi-dence suggest that those hypersensitive hits are target specific.First, hypersensitivity of the PMET3-CaERG1 conditional mu-tant strain in the presence of methionine was observed onlywith terbinafine and those six hits in group I of Fig. 5. Second,none of the six hits were found to inhibit the growth of anefflux-deficient C. albicans strain as well as a strain of Staphy-lococcus aureus at the HTS screening concentration (14.2 �M)tested (data not shown). Third, the presence of methionine didnot alter the susceptibility of the PMET3-CaERG1 conditionalmutant to fluconazole and sordarin (Fig. 4B and C) as well asseveral other known antifungal inhibitors (control compoundsin the HTS), including papulacandin B, ascosteroside, pradi-mycin, cycloheximide, and benomyl, that bind to targets otherthan CaErg1p compared to the Met-minus controls (data notshown). On the other hand, while the hypersensitive hits arepotential specific inhibitors, the common hits identified fromthe Met-plus and Met-minus screens may contain specific hitsas well. One possible reason for this is that the active concen-trations of those compounds in the screening solution werehigh enough to inhibit cell growth under both Met-plus andMet-minus conditions. Therefore, individual dose-responsedeterminations are needed to identify which common hit(s) isspecific to the target under evaluation.
Finally, the construction of conditional mutants by using theCaMET3 promoter makes it possible to screen against anytarget placed under this promoter, whether or not its functionis known or if a functional in vitro assay is available. Althoughnew classes of inhibitors can be identified by rescreening ex-isting drug targets, the new targets identified in this workconceivably offer greater potential for the identification ofnovel classes of antifungal agents. Admittedly, the successfulidentification of novel targets as well as inhibitors does notnecessarily lead to a drug due to the complexity of the drug
FIG. 5. Superimposition of HTS hits from parallel screening of aPMET3-CaERG1/Caerg1::ARG4 conditional mutant in the presence (● )(0.4 mM) and absence (e) of methionine (Met-plus and Met-minusscreen, respectively). Based on the percent inhibition range in theMet-minus screen, the positive hits identified using a cutoff of �40%growth inhibition in the Met-plus screen were sorted into three groups(group I, �11 to 10%; group II, 11 to 40%; group III, �40% in theMet-minus screen), which are separated by the dashed vertical lines.
VOL. 5, 2006 IDENTIFICATION OF CONSERVED FUNGAL GENES 647

development process. Nevertheless, it is our hope that thestreamlined process we developed proceeding from selectionand validation of candidate antifungal targets to screening forspecific inhibitors can be one of many useful approaches forfuture antifungal drug discovery.
ACKNOWLEDGMENTS
We thank Aaron Mitchell for providing strain BWP17 and plasmidspGEM-URA3, pGEM-HIS1, and pRS-Arg4�SpeI. We are grateful toJoan C. Fung-Tomc for critical reading of the manuscript and toMahmound Ghannoum for constructive discussions.
This study was supported by the Bristol-Myers Squibb Company.We dedicate this paper to our coauthor, John F. Barrett, who died
24 January 2006. He was a strong advocate of genomics-based antimi-crobial drug discovery, and we will miss his leadership, enthusiasm, andboundless energy.
REFERENCES
1. Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller,and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generationof protein database search programs. Nucleic Acids Res. 25:3389–3402.
2. Arikan, S., and J. H. Rex. 2002. New agents for the treatment of systemicfungal infections—current status. Exp. Opin. Emerg. Drugs 7:3–32.
3. Backen, A. C., I. D. Broadbent, R. W. Fetherston, J. D. Rosamond, N. F.Schnell, and M. J. Stark. 2000. Evaluation of the CaMAL2 promoter forregulated expression of genes in Candida albicans. Yeast 16:1121–1129.
4. Badger, J. H., and G. J. Olsen. 1999. CRITICA: coding region identificationtool invoking comparative analysis. Mol. Biol. Evol. 16:512–524.
5. Braun, B. R., M. van het Hoog, C. d’Enfert, M. Martchenko, J. Dungan, A.Kuo, D. O. Inglis, M. A. Uhl, H. Hogues, M. Berriman, M. Lorenz, A.Levitin, U. Oberholzer, C. Bachewich, D. Harcus, A. Marcil, D. Dignard, T.Iouk, R. Zito, L. Frangeul, F. Tekaia, K. Rutherford, E. Wang, C. A. Munro,S. Bates, N. A. Gow, L. L. Hoyer, G. Kohler, J. Morschhauser, G. Newport,S. Znaidi, M. Raymond, B. Turcotte, G. Sherlock, M. Costanzo, J. Ihmels, J.Berman, D. Sanglard, N. Agabian, A. P. Mitchell, A. D. Johnson, M. White-way, and A. Nantel. 2005. A human-curated annotation of the Candidaalbicans genome. PLOS Genet. 1:e1.
6. Brown, D. H., Jr., I. V. Slobodkin, and C. A. Kumamato. 1996. Stabletransformation and regulated expression of an inducible reporter constructin Candida albicans using restriction enzyme-mediated integration. Mol.Gen. Genet. 251:75–80.
7. Bruccoleri, R., T. Dougherty, and D. Davison. 1998. Concordance analysis ofmicrobial genomes. Nucleic Acids Res. 26:4482–4486.
8. Care, R. S., J. Trevethick, K. M. Binley, and P. E. Sudbery. 1999. The MET3promoter: a new tool for Candida albicans molecular genetics. Mol. Micro-biol. 34:792–798.
9. Cha, R., and J. D. Sobel. 2004. Fluconazole for the treatment of candidiasis:15 years experience. Exp. Rev. Anti-Infect. Ther. 2:357–366.
10. Davis, D. A., V. M. Bruno, L. Loza, S. G. Filler, and A. P. Mitchell. 2002.Candida albicans Mds3p, a conserved regulator of pH responses and viru-lence identified through insertional mutagenesis. Genetics 162:1573–1581.
11. De Backer, M. D., B. Nelissen, M. Logghe, J. Viaene, I. Loonen, S. Vandon-inck, R. de Hoogt, S. Dewaele, F. A. Simons, P. Verhasselt, G. Vanhoof, F.Contreras, and W. H. M. L. Luyten. 2001. An antisense-based functionalgenomics approach for identification of genes critical for growth of Candidaalbicans. Nat. Biotechnol. 19:235–241.
12. DeVito, J. A., J. A. Mills, V. G. Liu, A. Agarwal, C. F. Sizemore, Z. Yao, D. M.Stoughton, M. G. Cappiello, M. D. Barbosa, L. A. Foster, and D. L. Pom-pliano. 2002. An array of target-specific screening strains for antibacterialdiscovery. Nat. Biotechnol. 20:478–483.
13. Dismukes, W. E. 2000. Introduction to antifungal drugs. Clin. Infect. Dis.30:653–657.
14. Drennan, D., and A. G. Ryazanov. 2004. Alpha-kinases: analysis of the familyand comparison with conventional protein kinases. Prog. Biophys. Mol. Biol.85:1–32.
15. Edmond, M. B., S. E. Wallace, D. K. McClish, M. A. Pfaller, R. N. Jones, andR. P. Wenzel. 1999. Nosocomial bloodstream infections in United Stateshospitals: a three-year analysis. Clin. Infect. Dis. 29:239–244.
16. Fonzi, W. A., and M. Y. Irwin. 1993. Isogenic strain construction and genemapping in Candida albicans. Genetics 134:717–728.
17. Gaasterland, T., and C. W. Sensen. 1996. MAGPIE: automated genomeinterpretation. Trends Genet. 12:76–78.
18. Gachotte, D., S. E. Sen, J. Eckstein, R. Barbuch, M. Krieger, B. D. Ray, andM. Bard. 1999. Characterization of the Saccharomyces cerevisiae ERG27gene encoding the 3-keto reductase involved in C-4 sterol demethylation.Proc. Natl. Acad. Sci. USA 96:12655–12660.
19. Gerami-Nejad, M., D. Hausauer, M. McClellan, J. Berman, and C. Gale.
2004. Cassettes for the PCR-mediated construction of regulatable alleles inCandida albicans. Yeast 21:429–436.
20. Ghannoum, M. A., and L. B. Rice. 1999. Antifungal agents: mode of action,mechanisms of resistance, and correlation of these mechanisms with bacte-rial resistance. Clin. Microbiol. Rev. 12:501–517.
21. Giaever, G., D. D. Shoemaker, T. W. Jones, H. Liang, E. A. Winzeler, A.Astromoff, and R. W. Davis. 1999. Genomic profiling of drug sensitivities viainduced haploinsufficiency. Nat. Biotechnol. 21:278–283.
22. Gorman, J. A., W. Chan, and J. W. Gorman. 1991. Repeated use of GAL1for gene disruption in Candida albicans. Genetics 129:19–24.
23. Gow, N., P. Robbins, J. Lester, A. Brown, W. Fonzi, T. Chapman, and O.Kinsman. 1994. A hyphal-specific chitin synthase gene (CHS2) is not essen-tial for growth, dimorphism, or virulence of Candida albicans. Proc. Natl.Acad. Sci. USA 91:6216–6220.
24. Healy, M. D. 2003. Finding homologs to nucleic acid or protein sequencesusing the FrameSearch program, p. 3.2.1–3.2.23. In A. D. Baxevanis, D. B.Davison, R. D. M. Page, G. A. Petsko, L. D. Stein, and G. D. Stormo (ed.),Current protocols in bioinformatics. John Wiley & Sons, New York, N.Y.
25. Herbrecht, R. 2004. Voriconazole: therapeutic review of a new azole anti-fungal. Exp. Rev. Anti-Infect. Ther. 2:485–497.
26. Higgins, D. G., J. D. Thompson, and T. J. Gibson. 1996. Using CLUSTALfor multiple sequence alignments. Methods Enzymol. 266:383–402.
27. Hoffman, C. S., and F. Winston. 1987. A ten-minute DNA preparation fromyeast efficiently releases autonomous plasmids for transformation of Esche-richia coli. Gene 57:267–272.
28. Hossain, M. A., and M. A. Ghannoum. 2000. New investigational antifungalagents for treating invasive fungal infections. Exp. Opin. Investig. Drugs9:1797–1813.
29. Justice, M. C., M.-J. Hsu, B. Tse, T. Ku, J. Balkovec, D. Schmatz, and J.Nielsen. 1998. Elongation factor 2 as a novel target for selective inhibition offungal protein synthesis. J. Biol. Chem. 273:3148–3151.
30. Kale, P., and L. B. Johnson. 2005. Second-generation azole antifungalagents. Drugs Today (Barcelona) 41:91–105.
31. Kontoyiannis, D. P., and G. P. Bodey. 2002. Invasive aspergillosis in 2002: anupdate. Eur. J. Clin. Microbiol. Infect. Dis. 21:161–172.
32. Latge, J.-P. 1999. Aspergillus fumigatus and aspergillosis. Clin. Microbiol.Rev. 12:310–350.
33. Leuker, C. E., A. Sonneborn, S. Delbruck, and J. F. Ernst. 1997. Sequenceand promoter regulation of the PCK1 gene encoding phosphoenolpyruvatecarboxykinase of the fungal pathogen Candida albicans. Gene 192:235–240.
34. Maschmeyer, G., and A. Glasmacher. 2005. Pharmacological properties andclinical efficacy of a recently licensed systemic antifungal, caspofungin. My-coses 48:227–234.
35. Mazur, P., N. Morin, W. Baginsky, M. El-Sherbeini, J. Clemas, J. Nielsen,and F. Floor. 1995. Differential expression and function of two homologoussubunits of yeast 1,3--D-glucan synthase. Mol. Cell. Biol. 15:5671–5681.
36. Mol, P., H. Park, J. Mullins, and E. Cabib. 1994. A GTP-binding proteinregulates the activity of (133)-beta-glucan synthase, an enzyme directlyinvolved in yeast cell wall morphogenesis. J. Biol. Chem. 269:31267–31274.
37. Munro, C. A., K. Winter, A. Buchan, K. Henry, J. M. Becker, A. J. Brown,C. E. Bulawa, and N. A. Gow. 2001. Chs1 of Candida albicans is an essentialchitin synthase required for synthesis of the septum and for cell integrity.Mol. Microbiol. 39:1414–1426.
38. Odds, F. C. 1988. Candida and candidosis. Bailliere Tindall, London, UnitedKingdom.
39. Ono, B. I., K. Kijma, N. Ishii, T. Kawato, and A. Matsuda. 1996. Regulationof sulphate assimilation in Saccharomyces cerevisiae. Yeast 12:1153–1162.
40. Pasrija, R., S. Krishnamurthy, T. Prasad, J. F. Ernst, and R. Prasad. 2005.Squalene epoxidase encoded by ERG1 affects morphogenesis and drug sus-ceptibilities of Candida albicans. J. Antimicrob. Chemother. 55:905–913.
41. Pearson, W. R., and D. J. Lipman. 1988. Improved tools for biologicalsequence comparison. Proc. Natl. Acad. Sci. USA 85:2444–2448.
42. Perentesis, J., L. Phan, W. Gleason, D. LaPorte, D. Livingston, and J.Bodley. 1992. Saccharomyces cerevisiae elongation factor 2. Genetic cloning,characterization of expression, and G-domain modeling. J. Biol. Chem. 267:1190–1197.
43. Pucci, M., T. J. Dougherty, and J. F. Barrett. 1998. Why are there no newantibiotics? Exp. Opin. Investig. Drugs 7:1233–1235.
44. Roemer, T., B. Jiang, J. Davison, T. Ketela, K. Veillette, A. Breton, F.Tandia, A. Linteau, S. Sillaots, C. Marta, N. Martel, S. Veronneau, S.Lemieux, S. Kauffman, J. Becker, R. Storms, C. Boone, and H. Bussey. 2003.Large-scale essential gene identification in Candida albicans and applicationsto antifungal drug discovery. Mol. Microbiol. 50:167–181.
45. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
46. Sherman, F. 1991. Getting started with yeast. Methods Enzymol. 194:3–21.47. Sissler, M., R. Giege, and C. Florentz. 1998. The RNA sequence context
defines the mechanistic routes by which yeast arginyl-tRNA synthetasecharges tRNA. RNA 4:647–657.
48. Thomas, D., H. Cherest, and Y. Surdin-Kerjan. 1989. Elements involved in
648 LIU ET AL. EUKARYOT. CELL

S-adenosylmethionine-mediated regulation of the Saccharomyces cerevisiaegene. Mol. Cell. Biol. 9:3292–3298.
49. Tkacz, J. S., and B. DiDomenico. 2001. Antifungals: what’s in the pipeline.Curr. Opin. Microbiol. 4:540–545.
50. Truan, G., J. C. Epinat, C. Rougeulle, C. Cullin, and D. Pompon. 1994.Cloning and characterization of a yeast cytochrome b5-encoding gene whichsuppresses ketoconazole hypersensitivity in a NADPH-P-450 reductase-de-ficient strain. Gene 142:123–127.
51. Walsh, T. J., and A. H. Groll. 1999. Emerging fungal pathogens: evolvingchallenges to immunocompromised patients for the twenty-first century.Transpl. Infect. Dis. 1:247–261.
52. Wang, Y.-K., M. Liu, B. A. Dougherty, M. D. Healy, D. B. Davison, C. E.Mazzucco, S. R. Krystek, and D. A. Bassolino. 6 November 2003. Novelessential fungal polynucleotides, polypeptides, and methods of use. Worldpatent WO2003091418 A2.
53. Warit, S., N. Zhang, A. Short, R. M. Walmsley, S. G. Oliver, and L. I. Stateva.2000. Glycosylation deficiency phenotypes resulting from depletion of GDP-mannose pyrophosphorylase in two yeast species. Mol. Microbiol. 36:1156–1166.
54. Wilson, R. B., D. Davis, and A. P. Mitchell. 1999. Rapid hypothesis testingwith Candida albicans through gene disruption with short homology regions.J. Bacteriol. 181:1868–1874.
55. Winzeler, E. A., D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson,B. Andre, R. Bangham, R. Benito, J. D. Boeke, H. Bussey, A. M. Chu, C.Connelly, K. Davis, F. Dietrich, S. W. Dow, M. El Bakkoury, F. Foury,S. H. Friend, E. Gentalen, G. Giaever, J. H. Hegemann, T. Jones, M.Laub, H. Liao, N. Liebundguth, D. J. Lockhart, A. Lucau-Danila, M.Lussier, N. M’Rabet, P. Menard, M. Mittmann, C. Pai, C. Rebischung,J. L. Revuelta, L. Riles, C. J. Roberts, P. Ross-MacDonald, B. Scherens,M. Snyder, S. Sookhai-Mahadeo, R. K. Storms, S. Veronneau, M. Voet,G. Volckaert, T. R. Ward, R. Wysocki, G. S. Yen, K. Yu, K. Zimmermann,P. Philippsen, M. Johnston, and R. W. Davis. 1999. Functional charac-terization of the Saccharomyces cerevisiae genome by gene deletion andparallel analysis. Science 285:901–906.
56. Zaas, A. K., and W. J. Steinbach. 2005. Micafungin: the US perspective. Exp.Rev. Anti-Infect. Ther. 3:183–190.
VOL. 5, 2006 IDENTIFICATION OF CONSERVED FUNGAL GENES 649























![Advances in the development of antifungal agents]](https://img.pdfslide.net/doc/110x75/635ea923095e4caf220652ff/advances-in-the-development-of-antifungal-agents.jpg)





