Embed Size (px)
Citation preview

The 2.2-Å Crystal Structure of Human Pro-granzyme K Reveals aRigid Zymogen with Unusual Features*
Received for publication, August 5, 2002, and in revised form, October 8, 2002Published, JBC Papers in Press, October 15, 2002, DOI 10.1074/jbc.M207962200
Clara Hink-Schauer‡§, Eva Estebanez-Perpina§¶, Elke Wilharm‡, Pablo Fuentes-Prior¶,Wolfgang Klinkert‡, Wolfram Bode¶, and Dieter E. Jenne‡�
From the ‡Department of Neuroimmunology, Max-Planck-Institute of Neurobiology and the ¶Department of StructuralResearch, Max-Planck-Institute of Biochemistry, Am Klopferspitz 18a, Planegg-Martinsried D-82152, Germany
Granzyme K (GzmK) belongs to a family of trypsin-likeserine proteases localized in electron dense cytoplasmicgranules of activated natural killer and cytotoxicT-cells. Like the related granzymes A and B, GzmK cantrigger DNA fragmentation and is involved in apoptosis.We expressed the Ser195 3 Ala variant of human pro-GzmK in Escherichia coli, crystallized it, and deter-mined its 2.2-Å x-ray crystal structure. Pro-GzmK pos-sesses a surprisingly rigid structure, which is mostsimilar to activated serine proteases, in particular com-plement factor D, and not their proforms. The N-termi-nal peptide Met14-Ile17 projects freely into solution andcan be readily approached by cathepsin C, the naturalconvertase of pro-granzymes. The pre-shaped S1 pocketis occupied by the ion paired residues Lys188B-Asp194
and is hence not available for proper substrate binding.The Ser214-Cys220 segment, which normally provides atemplate for substrate binding, bulges out of the activesite and is distorted. With analogy to complement factorD, we suggest that this strand will maintain its non-productive conformation in mature GzmK, mainly dueto the unusual residues Gly215, Glu219, and Val94. Wehypothesize that GzmK is proteolytically active only to-ward specific, as yet unidentified substrates, whichupon approach transiently induce a functional active-site conformation.
Granzymes are chymotrypsin-like serine proteases of cyto-toxic T-lymphocytes and natural killer cells that are stored inspecialized secretory granules together with the membrano-philic perforin and proteoglycans (1). Upon recognition of anti-genetically altered target cells or transformed or virally in-fected host cells, granzymes and other granule components arereleased as high molecular weight complexes and are taken upby the target cell via membrane receptors. Perforin enhancesthis uptake and appears to contribute to the release of gran-zymes from endosomal vesicles (2–4).
Granzyme K (GzmK)1 was discovered in granules of humanlymphokine-stimulated killer cells together with granzyme A(GzmA) as a trypsin-like non-glycosylated serine protease (mo-lecular mass, �28 kDa) that cleaves N�-benzyloxycarbonyl-L-lysine thiobenzyl ester (BLT) bonds (5). GzmA is 10-fold moreabundant in human lymphokine-stimulated killer cell granulesthan GzmK (5), whereas granules from the RNK-16 rat tumorcell line contain significantly higher amounts of GzmK thanGzmA (6). The rat enzyme (also known as fragmentin-3) wasoriginally identified as the third proteolytic inducer of DNAfragmentation in perforin-treated YAC-1 target cells apartfrom GzmA and GzmB. DNA fragmentation initiated by GzmKor GzmA in the presence of perforin shows similar time kineticsand was observed after 20 h, whereas GzmB/perforin-mediatedapoptosis occurred already within 2 h (6). In evolutionaryterms, GzmK and GzmA are the most closely related gran-zymes. They both are encoded by single copy genes in humanand rodents and map to orthologous chromosomal segments onhuman chromosome 5q11.2 and mouse chromosome 13, bandD2.2, respectively (7–9). Transcripts of the murine GzmK genehave been detected in certain regions of the mouse brain, in-cluding the cerebral cortex, hippocampus, diencephalon, andthe pineal and pituitary glands (10).
GzmK and GzmA differ from GzmB by their substrate spec-ificity. Although the latter cleaves after Asp residues, bothGzmK and GzmA preferentially cut after basic residues. Todate only the thioesters Z-Arg-SBzl and BLT, and a highlybasic 13-amino acid oligopeptide (Cys-Gly-Tyr-Gly-Pro-Lys-Lys-Lys-Arg-Lys-Val-Gly-Gly) have been reported to be cleavedby GzmK at neutral pH. GzmK cleaves this oligopeptide be-tween basic residues, most rapidly after position 6 and 9 andmore slowly after position 7 and 8 (11). The best syntheticinhibitors of GzmK known are 3,3-diphenylpropanoyl-Pro-4-amidinophenylglycyldiphenylphosphonate esters (12) andD-Phe-Pro-Arg-chloromethyl ketone (13). The most efficientnatural inhibitor is inter-�-trypsin inhibitor, a heterotrimericprotein complex containing bikunin. Inhibition by this plasmaprotein complex is mediated by the second Kunitz-type domainof its bikunin subunit, the Ki value being 22 nM (13).
GzmK, like GzmA and GzmB, is synthesized as a zymogenprecursor, containing a signal and a short pro-peptide preced-ing the mature polypeptide chain (14). The cleavage site of thesignal peptidase in the GzmK precursor has not been experi-mentally determined, and thus the exact length of the propep-tide is uncertain. The N-terminal propeptides of GzmA, GzmB,and mast cell chymase precursors, however, have been identi-
* This work was supported by the Sonderforschungsbereich 469(projects A1 and A5) of the German Research Council, the Training andMobility program (ERBFMRXCT98) and the Biotech program (BIO-CT98-0418) of the European Community, the Human Frontier ScienceProgram (RG-203/98), and the Fonds der Chemischen Industrie. Thecosts of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked “adver-tisement” in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
The atomic coordinates and structure factors (code 1MZA and 1MZD)have been deposited in the Protein Data Bank, Research Collaboratoryfor Structural Bioinformatics, Rutgers University, New Brunswick, NJ(http://www.rcsb.org/).
§ Both authors contributed equally to this work.� To whom correspondence should be addressed. Tel.: 49-89-8578-
3588; Fax: 49-89-8578-3541; E-mail: [email protected].
1 The abbreviations used are: Gzm, granzyme; BLT, N�-benzyloxy-carbonyl-L-lysine thiobenzyl ester; BPTI, bovine pancreatic trypsin in-hibitor; DF, complement factor D (adipsin); pro-DF, pro-factor D; FIXa,coagulation factor IXa; r.m.s., root mean square.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 277, No. 52, Issue of December 27, pp. 50923–50933, 2002© 2002 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 50923

fied in cultured cells as Glu-Arg, Gly-Glu, and Gly-Glu, respec-tively (15–17). Removal of these dipeptides is ensured by thelysosomal cysteine protease cathepsin C (dipeptidyl peptidase1) (18), which is deficient in the Papillon-Lefevre syndrome(19). Conclusive confirmation of the essential role of cathepsinC in the conversion process was obtained from cathepsin C-deficient knockout mice, whose lymphocytes contain only cat-alytically inactive GzmA and GzmB precursors at normal lev-els (15). Our recent studies indicate that an N-terminal Met-Glu dipeptide is also efficiently removed by cathepsin C in vitrofrom the human granzyme K zymogen (13). Thus, the Met-Gluprecursor of GzmK most likely represents the natural proformor at least a functional intermediate during biosynthesis ofcatalytically active GzmK in vivo. Transport of zymogensthrough the endoplasmic reticulum and Golgi network beforerecognition by cathepsin C requires a certain degree of confor-mational stability, which prevents both premature proteolyticactivity and degradation of precursors intracellularly.
The natural Met-Glu precursor of human GzmK lacks N-linked carbohydrates and can be produced by recombinantexpression in Escherichia coli and refolding from inclusionbody proteins at high quantities, which is a prerequisite forfurther structural analyses (13). To understand the specificconversion of pro-GzmK by cathepsin C as well as the weakproteolytic activity of mature GzmK on a structural basis, weproduced the more stable but otherwise identical Ser1953 Alavariant of human pro-GzmK, crystallized this variant, anddetermined its crystal structure at 100 and 298 K. Our struc-tural work reveals an unanticipated mechanism of zymogenstabilization and suggests a distorted conformation for acti-vated GzmK, which is incompatible with the binding and proc-essing of protein substrates.
EXPERIMENTAL PROCEDURES
Construction of the Human S195A Pro-GzmK Expression Plasmid—The cDNA segment between the NdeI and ApaI site of the wild typeplasmid pET24c� (Novagen) was removed by NdeI and ApaI cleavage(New England BioLabs) and replaced by a mutated cDNA sequence.This cDNA sequence was generated from the wild type construct (13)using the oligonucleotides DJ601 (upper primer, 5�-TGTGTTTCCATA-TGGAAATTAT-3�) and DJ582 (lower primer, 5�-TCAAGGGGCCCCC-TGCGTCAC-3�). Oligonucleotide DJ582 introduces a codon for Ala atposition 195 in the wild type cDNA sequence of GzmK. The resultingPCR product was digested with NdeI and ApaI and ligated into thecleaved pET24c� vector carrying the 3� coding portion of the cDNA. Therecombinant plasmid was amplified in DH5� cells (Invitrogen) andtransformed into B834(DE3) cells (Novagen) for protein expression.
Expression and Purification of the Ser195 3 Ala Zymogen—The ex-pression of the Ser195 3 Ala mutant of pro-GzmK in B834(DE3) cells,grown in Luria-Bertani broth (LB) with 50 �g/ml kanamycin at 37 °C,was induced by addition of isopropyl-1-thio-�-D-galactopyranoside to afinal concentration of 1 mM as described previously (13). Bacteria wereharvested after 5-h induction and lysed in 50 mM Tris-HCl, 2 mM MgCl2,10 �g/ml DNase I, and 0.25 mg/ml lysozyme, pH 7.2. Inclusion bodieswere collected by centrifugation; washed twice in 50 mM Tris-HCl, 60mM EDTA, 1.5 M NaCl, 6% Triton X-100, pH 7.2, and three times in 50mM Tris-HCl, 60 mM EDTA, pH 7.2; and dissolved in 6 M guanidiniumchloride, 100 mM Tris-HCl, 20 mM EDTA, 15 mM GSH, 150 mM GSSG,pH 8.0, by stirring at room temperature over night. Solubilized proteinswere then dialyzed against 6 M guanidinium hydrochloride, pH 5.0 at4 °C. Refolding was initiated by diluting the solubilized proteins (typi-cally 10–15 mg/ml) 1:100 (v/v) in 50 mM Tris-HCl, 0.5 M L-arginine, 20mM CaCl2, 1 mM EDTA, 0.1 M NaCl, 0.5 mM L-cysteine, pH 8.5 (refoldingbuffer). Two further aliquots were added to this solution after 8 and16 h. Diluted proteins were kept at 4 °C in refolding buffer for anadditional period of 96 h. The protein solution was dialyzed againstphosphate-buffered saline, pH 7.3, concentrated by ultrafiltration, andloaded onto a Mono S-Sepharose column (Amersham Biosciences). Pro-teins were recovered from the column using a linear salt gradient (from0.137 to 2 M NaCl).
Crystallization, Data Collection, and Processing—Crystals suitablefor diffraction analysis were grown by the sitting drop vapor-diffusion
method. One microliter of the protein solution (10 mg/ml) was mixedwith 1 �l of the reservoir solution (3.2 M sodium formate, pH 8.4) andconcentrated against 300 �l of the reservoir. After 2 days of incubation,100 �l of 6 M sodium formate solution, pH 8.4, were added to thereservoir. Within 3 months at 20 °C, crystals grew to maximal dimen-sions of 0.3 mm � 0.2 mm � 0.5 mm. A complete data set to 2.9-Åresolution was recorded at room temperature from a single crystalmounted in a thin-walled glass capillary. A second complete data set to2.23 Å was collected at 100 K from a flash-cooled crystal that hadpreviously been equilibrated in a cryo-solution (3.9 M sodium formate,pH 8.4, 15% (v/v) glycerol). Both data sets were recorded “in house” ona 300-mm MAR Research image plate detector, using monochromatizedCuK� radiation from a RIGAKU rotating anode x-ray generator. Thecrystals of pro-GzmK Ser195 3 Ala did not experience appreciableradiation decay over the 2 days that they were exposed to the x-raybeam at room temperature. This feature seems to be related to therelatively low solvent content of the crystals (39%, corresponding to aMatthews coefficient of 2.02). The crystals belong to space groupP212121 and contain one molecule per asymmetric unit.
Diffraction data sets were evaluated with MOSFLM (20), reduced,and scaled without applying a sigma cut-off, using programs supportedby the Collaborative Computational Project No. 4 (21). Crystal struc-tures were solved by molecular replacement with AMoRe (available atwww.ccp4.ac.uk/dist/html/amore.html) using 15.0- to 3.5-Å data and amodified GzmB (1FQ3) search model (22) in which all residues differingbetween the two proteases had been truncated to alanine. Model build-ing was done on a Silicon Graphics Indigo 2 workstation using MAIN(23, 24). Calculation of the electron density maps and crystallographicrefinement were performed with X-PLOR (25) and CNS (26) using thetarget parameters of Engh and Huber (27). In the final steps of refine-ment, highly restrained atomic B-values were also refined. Models werebuilt in agreement with the human GzmK sequence as deposited in theSwiss-Prot data base (accession code P49863). They start with theN-terminal Met14, which was confirmed by sequence analysis of twocrystals, and end with Asn248 (chymotrypsinogen A numbering). In bothcases, there is continuous electron density from Gly19 to Ser92 and fromSer100 to Pro244. For the N-terminal segment Met14-Gly18, segmentArg93-Gln99, and the C-terminal residues from Pro245 to Asn248, lackinginterpretable electron density, the occupancy of all atoms was set tozero. The final model comprises 221 defined residues and 131 solvent(water) molecules. 99% of all residues fall into the most favored oradditionally favored regions (PROCHECK version 3.5.4 (28)). The finalR-factors are 24.07% (Rfree, 28.45%) and 22.36% (31.67%) for data setsmeasured at 100 and 298 K, respectively (see Table I for statistics of thedata collection, processing, and refinement).
Modeling of Active GzmK—Active GzmK was manually modeledusing MAIN (24). The S1 pocket and surrounding loops were modeledfollowing bovine trypsin in complex with bovine pancreatic trypsininhibitor (BPTI) (2PTC and 1TPA) (29) and human leukocyte elastasein complex with the third domain of turkey ovomucoid inhibitor (1PPF)
TABLE IStatistics for data collection and refinement
Pro-GzmK S195A Crystal 1a Crystal 2b
Molecules/asymmetric unit 1 1Space group P212121 P212121
Cell constant, a/b/c (Å) 31.46/78.16/83.62 31.75/78.86/85.28Resolution (Å) 2.23 2.90Reflections measured 65,669 39,313Rmerge (%)c 12.5 14.7Unique reflections 23,808 10,537Completeness (%), overall 97.1 94.5Completeness (%), outermost shell 95.1 92.5Reflections used for refinement 22,617 9,771Resolution range (Å) 22–2.23 22–2.90Rfactor (%) finald 24.07 22.36Rfree (%) finale 28.45 31.67r.m.s standard deviations:
Bond length (Å) 0.0074 0.0067Bond angles (°) 1.435 1.28Average B value/S.D. (Å) 23.6 26.8a Data set measured at 100 K.b Data set measured at room temperature.c Rmerge � �hkl��I� I�/�hkl�I�.d Rfactor � �hkl�Fobs� �Fcalc�/�hkl�Fobs�.e Rfree is the R value calculated with 500 reflections that were not
used for the refinement.
Crystal Structure of Human Pro-granzyme K50924
(30). Residues Gly89 to Phe94 comprising the reactive-site loop of thesecond bikunin domain were taken from the reported crystal structure(1BIK) (31) and adjusted to the model of active GzmK according to themain-chain trace in the above-mentioned complexes. The side chains ofLys192 of GzmK and Phe94 of bikunin D2 were rotated to avoid clashesbetween the two moieties (inter atom distances below 3 Å). The struc-ture of the complex was finally minimized with CNS, and the quality ofthe model was controlled with PROCHECK version 3.5.4 (28). Addi-tional atomic coordinates for serine proteases used in this work wereobtained from the Structural Bioinformatics Protein Data Bank (PDB)with the accession codes 2PTC, 1TPA, 1PPF, 1BIK, 1FDP, 1DIC, 1HFD,1BIO, 1DFP, 1DSU, 1DST, 1PJP, 1KLT, 1CGH, 1FQ3, 2PKA, 2CGA,1FDP, 1FAX, and 1TGB.
RESULTS AND DISCUSSION
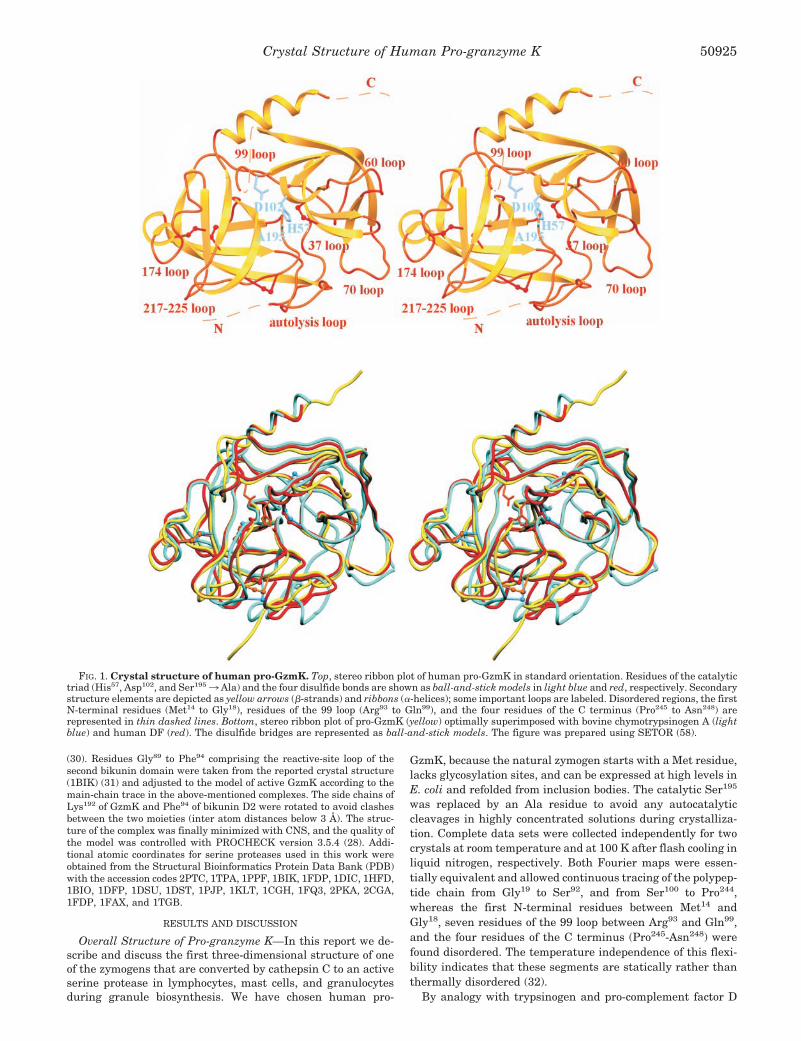
Overall Structure of Pro-granzyme K—In this report we de-scribe and discuss the first three-dimensional structure of oneof the zymogens that are converted by cathepsin C to an activeserine protease in lymphocytes, mast cells, and granulocytesduring granule biosynthesis. We have chosen human pro-
GzmK, because the natural zymogen starts with a Met residue,lacks glycosylation sites, and can be expressed at high levels inE. coli and refolded from inclusion bodies. The catalytic Ser195
was replaced by an Ala residue to avoid any autocatalyticcleavages in highly concentrated solutions during crystalliza-tion. Complete data sets were collected independently for twocrystals at room temperature and at 100 K after flash cooling inliquid nitrogen, respectively. Both Fourier maps were essen-tially equivalent and allowed continuous tracing of the polypep-tide chain from Gly19 to Ser92, and from Ser100 to Pro244,whereas the first N-terminal residues between Met14 andGly18, seven residues of the 99 loop between Arg93 and Gln99,and the four residues of the C terminus (Pro245-Asn248) werefound disordered. The temperature independence of this flexi-bility indicates that these segments are statically rather thanthermally disordered (32).
By analogy with trypsinogen and pro-complement factor D
FIG. 1. Crystal structure of human pro-GzmK. Top, stereo ribbon plot of human pro-GzmK in standard orientation. Residues of the catalytictriad (His57, Asp102, and Ser1953 Ala) and the four disulfide bonds are shown as ball-and-stick models in light blue and red, respectively. Secondarystructure elements are depicted as yellow arrows (�-strands) and ribbons (�-helices); some important loops are labeled. Disordered regions, the firstN-terminal residues (Met14 to Gly18), residues of the 99 loop (Arg93 to Gln99), and the four residues of the C terminus (Pro245 to Asn248) arerepresented in thin dashed lines. Bottom, stereo ribbon plot of pro-GzmK (yellow) optimally superimposed with bovine chymotrypsinogen A (lightblue) and human DF (red). The disulfide bridges are represented as ball-and-stick models. The figure was prepared using SETOR (58).
Crystal Structure of Human Pro-granzyme K 50925

(pro-DF) and because of the weak proteolytic activity of acti-vated GzmK, the pro-GzmK structure was expected to be quiteflexible as in trypsinogen (33) and DF (34). To our surprise, thecrystal structure analysis, however, revealed a rigid zymogenwith a preformed active-site cleft (see below). Especially theactivation domain and the S1 specificity pocket are well-or-dered. The pro-GzmK molecule has the shape of an oblateellipsoid with principal axes of about 35 and 50 Å (Fig. 1). As inother chymotrypsin-like serine proteases, the single GzmKpolypeptide chain folds into two �-barrels each comprising sixantiparallel strands labeled �1 to �6, and �7 to �12 (Fig. 2),which are strapped together by the domain-linking segmentsVal22-Pro28, Leu121-Ser129, and Thr229-Lys232A (Fig. 2). Theresidues of the catalytic triad, His57, Asp102, and Ser1953 Ala,are located at the junction of the two �-barrels. The preformedactive-site cleft (see below) runs perpendicular to this junction
along the surfaces of both barrels. Three helical elements arefound on the surface of the molecule, a single 310 turn (Ala55 toGln59, �1), an “intermediate” �-helix (Ser164 to Asn169, �2), anda long C-terminal �-helix extending from Lys232A to Leu243
(�3). The optimal alignment to bovine chymotrypsinogen Arequired insertions of one tetrapeptide (60A to 60D), threedipeptides (170A and 170B; 173A and 173B; 188A and 188B),and two single amino acid residues (223A and 232A), anddeletions of three single residues (36, 127, 218) and of thetetrapeptide segment between 203 and 206 (see Fig. 2).
A direct topological comparison between pro-GzmK and theavailable enzyme-zymogen pairs deposited with the ProteinData Bank (35) revealed a better superposition with the activeenzymes than with their respective zymogens. The best fit wasobtained with the six PDB entries for active human comple-ment factor D (DF), 1DIC, 1HFD, 1BIO, 1DFP, 1DSU, and
FIG. 2. Topological sequence alignment of GzmK and related proteases. Initial multiple sequence alignment was done using “pileup”(Genetics Computer Group, GCG, Madison, WI). Final alignments were adjusted according to the structural alignment of each serine protease withbovine chymotrypsinogen A (btCtrA). Homologous regions were highlighted using “prettybox” (GCG program package). The vote weights formmGzmk and rnGzmK were reduced to 0.6; no vote weight was specified for all other sequences. Helices and �-strands of pro-GzmK arerepresented by cylinders and arrows. The residues of the active site triad are emphasized in boldface numbers and the unique residues 188B, 215,and 219 are shown by italic numbers. Residues, which are identical in more than two of the aligned sequences, are emphasized by black shading.The species designations Hs (Homo sapiens), Rn (Rattus norvegicus), Mm (Mus musculus), Bt (Bos taurus) are used as prefixes in combination withaccepted gene symbols (Ctra, chymotrypsinogen A; DF, complement factor D; Gzm, granzyme). The numbering of pro-GzmK residues is based ontopological similarity to chymotrypsinogen A. Inserted residues are identified with the number of the last topologically equivalent chymotrypsino-gen residue, and capital letters as a suffix in alphabetical order.
Crystal Structure of Human Pro-granzyme K50926

1DST. Considering only � carbon atom pairs having C�-C�
distances of 1.5 Å or less, 181 residues turned out to be topo-logically equivalent between pro-GzmK and DF, showing anaverage root mean square (r.m.s.) deviation of 0.72 Å (Fig. 1B).This topological similarity extends into many structural details(see below). By contrast, pro-GzmK and the four independentpro-DF molecules in the asymmetric unit of 1FDP possess only157 topologically equivalent residues, with a slightly higherr.m.s. deviation of 0.81 Å. Second and third on the score rank-ing list are two groups of active serine proteases, comprisinghuman mast cell chymase (1PJP and 1KLT) (36, 37) and hu-man cathepsin G (1CGH) (38), followed by GzmB (1FQ3) (22)and porcine kallikrein (2PKA) (39). From the two latter pairs162 and 161 �-carbon atoms, respectively, can be superimposedon pro-GzmK with r.m.s. deviations of 0.74 and 0.76 Å. Thetopological fit with chymotrypsin and trypsin was worse, butstill better than for their zymogens, chymotrypsinogen andtrypsinogen.
Mast cell chymase, cathepsin G, and GzmB are slightly moredistant in agreement with their evolution and chromosomalclustering. These three proteases and GzmH lack the disulfidebond Cys191-Cys220 and are clustered together on chromosome14q11.2 within 130 kb (4), whereas granzyme-related memberswith four disulfide bonds are distributed over two separate locion chromosome 5q11.2 (GzmA and GzmK) and chromosome19p13.3 (GzmM, azurocidin 1, proteinase 3, neutrophil elas-tase, and complement factor D).
Heparin Binding Regions of Human Pro-GzmK—Comparedwith other serine proteases, the percentage of polar residues inGzmK is not particularly high. The 6 glutamate and 11 aspar-tate residues, however, do not compensate for the positivecharges of the 8 arginine and 23 lysine residues, renderingpro-GzmK very basic in accordance with a calculated isoelectricpoint of 10.2. The basic amino acid residues are unevenlydistributed on the surface and form several clusters of posi-tively charged surface patches (Fig. 3). A particularly largebasic region extends from the intermediate helix toward theC-terminal helix running along the upper rim (in the standardrepresentation, Fig. 3A). This patch comprises residues Lys126,Arg131, Arg165, Lys166, Lys178, Lys232A, Lys233, and Lys239 andshows clear topological similarities with the heparin bindingsite (also called anion binding exosite II) of �-thrombin (40).Other positively charged surface patches comprise residuesLys86, Lys87, Lys107, and Lys113 on the front side (Fig. 3A) and
Lys20, Arg27, Lys135, Lys137, and Lys202 on the back side of themolecule (Fig. 3B). In and around the S1-pocket, the lysines(Lys188B, Lys192, and Lys224) and arginines (Arg60A, Arg93, andArg150) and the catalytic His57 are charge compensated byaspartates (Asp97, Asp102, Asp145, and Asp194) and glutamates(Glu173B and Glu219) (Fig. 3A).
In line with these structural features, we were able to dem-onstrate strong interactions of both pro-GzmK and matureGzmK with a heparin matrix, from which they were eluted athigh salt concentrations (0.8 M NaCl). Using the same buffersystem, pro-GzmK also bound to a cation-exchange column(S15 Sepharose; Amersham Biosciences) but eluted at 0.6 M
NaCl, suggesting that heparin-like ligands specifically interactwith pro-GzmK with high affinity in vivo. This latter notion isalso supported by our observations that heparin affects proteo-lytic activity of both murine and human GzmK. In activityassays using 0.5 unit/ml heparin, the activities of human andmouse GzmK were elevated by 50 and 30%, respectively, ascompared with measurements in the absence of heparin (datanot shown).
The fact that esterolytic activity is not reduced by heparinindicates that heparin and small substrates bind to separateregions on the surface of the molecule. This heparin binding(exo)site could thus contribute to the recognition of macromo-lecular substrates, as shown e.g. for �-thrombin, resulting in ahighly selective function in vivo. Alternatively, the heparin-binding region of GzmK may play a role in the binding anduptake of GzmK by the target cells during killer cell attack. Inthis regard, all three mammalian homologs of GzmK are devoidof carbohydrates and thus cannot be transported into endoso-mal vesicles via the mannose 6-phosphate receptor during tar-get cell killing, as recently shown for GzmB (41).
Pro-GzmK, a Rigid Zymogen with a Preformed Active-siteCleft—Fig. 1 illustrates the preformed active-site cleft of pro-GzmK, which runs across the front side in standard view. Thefloor of this cleft is made by segments Ser214-Cys220, Gly193-Asp194 and the inner part of the 145 loop. The “upper” rim(addressed from right to left) is mainly formed by the 37 (�1�2)loop, the 60 (�3�4) loop, the 99 (�5�6) loop, and the 175 (�2�9)loop (loops are named after the central residue or the strands/helices flanking the loop, respectively). The 37 loop projects outof the molecule in a DF-like manner, with the side chains ofSer32 and His40 connected by a hydrogen bond. The 60 loop ofpro-GzmK, four residues longer than in chymotrypsinogen but
FIG. 3. Solid surface representations of pro-GzmK. A, the molecule is rotated downward with respect to the standard orientation as shownin Fig. 1. B, pro-GzmK is further rotated by 180° around the x-axis. The colors indicate positive (blue) and negative (red) electrostatic potential atthe molecular surface, contoured at �10 kT/e to 10 kT/e. Basic and acidic residues are highlighted by yellow labels consisting of single-lettersymbols for amino acid residues and sequence numbers; the N and C termini of pro-GzmK are marked with yellow labels. The figure was made withGRASP (59).
Crystal Structure of Human Pro-granzyme K 50927

still compact, bulges out of the main molecular body away fromthe active site. The 99 loop, normally forming the “roof” of theactive-site cleft, is fully disordered between Ser92 and Ser100.GzmK is special in that its residue 94 is not a tyrosine, as inalmost all other chymotrypsin-like serine proteases, or (morerarely) a phenylalanine, but a smaller valine residue. In con-trast to the bulkier side chains of Tyr94 or Phe94 residues, theVal94 side chain does not cover the catalytic Asp102 side chainand is hence unable to shield the “catalytic hydrogen bond”between His57 N�1 and Asp102 O�2 from bulk solvent mole-cules. With respect to position 94, GzmK resembles DF, whichalso carries a small residue, a serine, at this position (42).Adjacent to the flexible 99 loop of pro-GzmK lies the well-ordered 175 loop. This loop is two residues longer than inchymotrypsinogen and intrudes, in particular with its Pro174
pyrrolidine ring, deeply into the active-site cleft (Fig. 1).The “lower” boundary of the pro-GzmK active-site cleft (de-
scribed from left to right) is mainly formed by the 222 (�11�12)loop, the Cys191-Gly193 S1 base, the well-defined compact 145(“autolysis” or �7�8) loop, and the 70–80 (�4�5) loop (Fig. 1).The latter possesses a conformation similar to the “calciumbinding loops” of the calcium-stabilized pancreatic and coagu-lation serine proteases, but differs e.g. from pro-DF. In contrastto calcium-dependent serine proteases, pro-GzmK lacks thenegatively charged residues at positions 70 and 80, which con-tribute to the calcium coordination sphere (43). Instead, thedistal ammonium group of Lys80 of pro-GzmK occupies theposition of the calcium ion and is within hydrogen bond dis-tance to the carbonyl oxygens of Ser72 and Lys75 and to a bulksolvent molecule. Thus, GzmK should be stable and functionindependently of calcium, in agreement with our experimentalfindings (data not shown).
The catalytic triad residues Ser195, His57, and Asp102 arelocated in the center of the active-site cleft as in functionalactive proteases (Figs. 4 and 5A). The Asp102 side chain isplaced almost “normally,” without being shielded by residue 94from bulk solvent (see above). The His57 imidazoyl group, how-ever, is rotated away from its normal gauche� (�1 about 64°) toa trans conformation (�1 about 165.5°) and is thus unable toform the mechanistically required hydrogen bonds between itsN�1 atom and Asp102 O�2, and between its N�2 atom andSer195 O�. This inactive His57 conformation apparently resultsfrom the unusual conformation of the spatially adjacent resi-due Gly215, which would collide with a functionally arrangedHis57 side chain, rather than from the absence of the Ser195
hydroxyl group in the crystallized Ser195 3 Ala variant ofpro-GzmK.
The flanking Ser214-His217 segment, which represents theC-terminal part of strand �11 and forms the kinked “entranceframe” of the S1 pocket in functional trypsin-like proteases,
considerably curves outwards beyond the molecular body ofpro-GzmK (Figs. 1, 4, and 5A). This unusual conformationseems to be stabilized by a strong hydrogen bond connectingthe Gly215 nitrogen with the Asp102 carboxylate instead of theusually observed Ser214-Asp102 hydrogen bond, with the Ser214
side chain rotated into the molecule. In addition, the imidazolgroup of His217 slips into a pocket on the left side between the214–217 segment and the main molecular surface. Gly216,which in active trypsin-like proteases typically anchors the P3substrate residue (Fig. 6), exhibits a conformation incompatiblewith the donation (via N-H) or acceptation (via C-O) of sub-strate hydrogen bonds. Also with respect to the 214–217 loop,the pro-GzmK structure most closely resembles active DF (Fig.5A). In the latter, a serine residue at position 215 similarlypushes the His57 side chain away from its functional position.In the overwhelming majority of serine proteases with trypsin-like specificity, the carbonyl group of Gly219 points into the S1pocket. Pro-GzmK, DF, and coagulation factor IX representnotable exceptions, because position 219 is occupied by a glu-tamate residue. In the pro-GzmK structure, the main chaincarbonyl group of Glu219 points away from the S1 pocket andforms a hydrogen bond with the Lys224 N� atom. The followingCys220-Pro225 loop of pro-GzmK is loosely arranged on top ofthe Gln187-Asp189 segment similarly to trypsin. SegmentPro225-Tyr228, which follows the �12 strand, is placed as inactivated proteases, where it forms the back side of the S1pocket.
The main chain segment Asp189-Ala195, like in functionallyactive serine proteases, forms the lower base of a pre-shaped S1pocket in pro-GzmK. Due to the inversion of the Lys188B-Asp189
segment, however, the Lys188B side chain intrudes into the S1pocket toward Ala195, whereas the side chain of the immedi-ately following Asp189 residue points away from the pocket andinto the solvent (Fig. 4). The segment from Ser190 onward, inparticular the Gly193-Ala195 loop of pro-GzmK follows approx-imately the usual main chain course observed in active en-zymes (Fig. 5A). The N-H groups of Gly193 and Ser195 provide afunctional “oxyanion hole,” important for the stabilization ofthe negatively charged transition state intermediate. The con-formation of the oxyanion hole seems to be stabilized by theLys188B-Asp194 ion pair, which is held together by a very short(2.5 Å) charged hydrogen bond (Fig. 4). The N� atom of Lys188B
is further hydrogen-bonded to the main chain oxygen of Ser190.The Asp194 side chain of pro-GzmK is arranged as in matureenzymes with regard to its �-carbon atom, but differs withregard to its carboxylate group, which is directed toward theLys188B ammonium group (�1 � 75°) rather than toward theIle16 pocket (requiring a large �1 rotation of about 150° to adoptthe active conformation, see Fig. 6). Noteworthy, a similarconformation of Asp194 has been found in the zymogen of coag-
FIG. 4. The S1 specificity pocket ispreformed in pro-GzmK. Stereo view ofthe final electron density map around thepreformed S1 pocket of pro-GzmK. Theactive site region of pro-GzmK is shown instandard orientation, with residues dis-played as a ball-and-stick model. The oxy-gens are highlighted in red, the nitrogensin blue, and the Cys191-Cys220 disulfidebridge in yellow color. The 214–220 seg-ment, the basement of the preformed S1pocket, the residues of the unusual zymo-gen triad of pro-GzmK, Lys188B, Asp194,and Ser190, and His57 are completely de-fined. The figure was generated with Bob-script version 2.5 (60, 61).
Crystal Structure of Human Pro-granzyme K50928
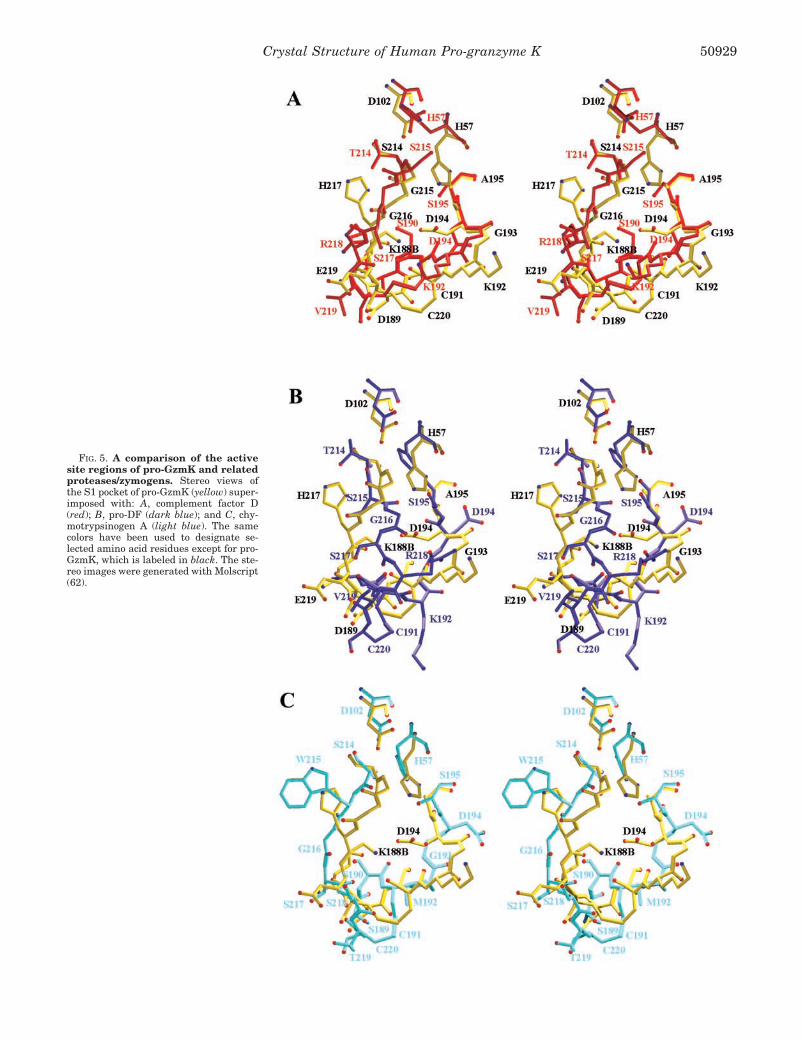
FIG. 5. A comparison of the activesite regions of pro-GzmK and relatedproteases/zymogens. Stereo views ofthe S1 pocket of pro-GzmK (yellow) super-imposed with: A, complement factor D(red); B, pro-DF (dark blue); and C, chy-motrypsinogen A (light blue). The samecolors have been used to designate se-lected amino acid residues except for pro-GzmK, which is labeled in black. The ste-reo images were generated with Molscript(62).
Crystal Structure of Human Pro-granzyme K 50929

ulation factor VII (44), but the adjacent main chain segmentsare more flexibly arranged than in pro-GzmK.
Stability of the Inactive Zymogen—Stabilization of catalyti-cally inactive conformations in multidomain serine proteases isachieved in several ways, whereas serine protease precursorswith short propeptides can be stabilized by a hydrogen bondnetwork between Ser32, His40, and Asp194, known as the “zy-mogen triad,” because its first description in trypsinogen andchymotrypsinogen (45). These three residues are conserved inhuman, rat, and mouse GzmK but do not form a trypsinogen-like triad in pro-GzmK.
By contrast, the inactivity of pro-GzmK is ensured by anumber of other structural features. The pre-shaped S1 pocketis occupied by the Lys188B-Asp194 ion pair, which althoughleaving space for substrates with small P1 residues, is notsuited to bind a substrate correctly. In all pro-GzmK homologssequenced so far (Fig. 2), both residues Lys188B and Asp194 areconserved. This suggests that the zymogen-stabilizing saltbridge Asp194-Lys188B is a common feature among all GzmKhomologs, and in turn points to a higher stability of this ionicinteraction compared with the zymogen triad. More important,the template segment Ser214-His217 is far from allowing pep-tide substrates to correctly align to the active site. Finally, theHis57 imidazoyl side chain is pushed out of its functional site bythe uncommon Gly215 (Figs. 4 and 5).
Conversion of Granzyme Precursors by Cathepsin C—Ca-thepsin C, a papain-like dipeptidyl-aminopeptidase, seems tobe the genuine activator of several granule-targeted serineprotease precursors produced by mast cells, natural killer cells,and activated lymphocytes (17), including pro-GzmK (13). Ourcrystallographic study of pro-GzmK was also aimed at under-standing zymogen recognition by cathepsin C. In particular, we
explored the possibility that exosites on the cathepsin C sur-face, distinct from its catalytic center, contribute to this highrecognition specificity and cleavage efficacy via docking exper-iments between human cathepsin C (46) and pro-GzmK. Wefound that all four substrate binding regions of the cathepsin Ctetramer are freely accessible. Any monomer of the tetramericcathepsin C molecule can productively bind to the exposedN-terminal Met14-Gly18 segment of pro-GzmK through subsitesS2 to S3� without the imminence of clashes between the twoapproaching molecules. In particular, the deep S2 specificitypocket of cathepsin C appears to be well-suited to accommodatethe side chain of the N-terminal Met14 of pro-GzmK. Subse-quent cleavage can occur between Glu15 (P1) and Ile16 (P1�). Wecould not discern any additional complementary surfaces invicinity to the N terminus of pro-GzmK and the substratebinding region of cathepsin C.
Pro-chymase, one of the known cathepsin C substrates, hasbeen reported to be resistant to cathepsin C in the absence ofheparin. To explain this finding, favorable intramolecular elec-trostatic interactions between the Glu15 carboxylate group anda basic patch on the surface of pro-chymase and their disrup-tion by heparin have been postulated by some investigators(47) but not proven (47, 48). The crystal structure of pro-GzmKnow disapproves this postulated role for Glu15. Consistent withour structural findings are recent investigations showing thatheparin and other sulfated polysaccharides with high negativecharge density do not stimulate the activation of pro-chymase,but rather inhibit its cleavage by cathepsin C at physiologicalsalt concentrations. Increasing ionic strength counteracts theinhibitory effect of heparin, but also leads to some inhibition ofthe conversion reaction, suggesting that the N terminus ofgranzyme precursors may be less accessible in heparin/pro-
FIG. 6. Putative structure of active GzmK bound to a substrate/inhibitor. The crystal structure of pro-GzmK (blue) is shownsuperimposed with a model of active GzmK (red). In addition, the reactive site loop of the second Kunitz-type domain of bikunin (1BIK,residues Gly89-Phe94, yellow) has been modeled into the active site region using the known complex between BPTI and trypsin as a template.The figure was prepared with WEBLABVIEWER (available at www.msi.com).
Crystal Structure of Human Pro-granzyme K50930

granzyme complexes and at high salt concentrations (48). Al-ternatively, binding of heparin-like proteoglycans to zymogensmight induce allosteric changes that impair cathepsin C bind-ing. Such mechanisms could explain the inhibitory role of hep-arin in the pro-chymase conversion process and would not be atvariance with our finding of a highly flexible N terminus inpro-GzmK.
A Structural Model for Mature GzmK—The conformationalrearrangements that occur after cathepsin C conversion can be
predicted with some confidence (Fig. 6) on the basis of struc-tures available for several zymogen/enzyme pairs and in par-ticular by analogy with pro-DF (1FDP) and DF (1DFP) (34, 49).After removal of the N-terminal dipeptide Met-Glu, Ile16-Gly19
rotates around Gly18-Gly19, dives into the pre-shaped Ile16
pocket between Asp189 and Ala143, and forms a salt bridge withthe Asp194 side chain. This change must be preceded or accom-panied by disruption of the salt bridge between Lys188B andAsp194 and rotation of the Asp194 side chain around its C�-C�
bond (from �1 � 75° to about 70°). Competition of the newlyformed N-terminal segment with the Lys188B side chain forforming a charged hydrogen bond with Asp194 would possiblyrequire some support by a fitting substrate.
The N-terminal Ile16 ammonium group, however, can easilyform a second hydrogen bond with the carbonyl oxygen ofAla143, which by contrast to most other zymogens is already inan “active” conformation in pro-GzmK (Fig. 6). The followingresidue, Ile17, most likely interacts with Asp189 via two intermain chain hydrogen bonds, which necessitates a C–CO bondrotation of Asp189 and a complete rearrangement of the preced-ing segment Asp185-Asp189. The conformational changes asso-ciated with an intruding N terminus would thus affect thestructure of pro-GzmK at multiple sites. The mutual flipping ofLys188B and Asp189 would result in the exposure of the Lys188B
side chain to the bulk solvent, and in the “correct” positioningof the Asp189 side chain at the bottom of the S1 pocket. Theseconformational rearrangements would also affect segmentSer190-Gly193 and the Ser214-Pro225 loop, including the connect-ing Cys191-Cys220 disulfide bridge (Fig. 6).
Not exactly predictable is, of course, the shape of the Ser214-His217 segment, whose extended conformation is critical forproductive substrate binding, and the conformation of the seg-ment His217-Cys220, which is important for the correct forma-tion of a trypsin-like S1 pocket. In analogy with observations inseveral independent structures for DF (49), which also pos-sesses a non-aromatic residue at position 215, we assume first,that segment Ser214-His217 and in particular Gly216 maintaintheir projecting pro-GzmK conformations and remain exposedafter activation cleavage, and second, that the His57 imidazoylside chain cannot move to its mechanistically correct position.Third, and in analogy to coagulation factor FIXa (1FAX) (50),Glu219 does not adopt the “high energy” trypsin-like conforma-tion. Fourth, a trypsin-like S1 entrance frame is not opened up.Finally, formation of a functional catalytic triad is disfavoredby the lack of an aromatic side chain at position 94 as in DF.
The non-productive resting-state conformation, which wepostulate for mature GzmK most likely originates from threeatypical residues in human GzmK, namely Gly215, Glu219, andVal94 for the following reasons. In trypsin and related typicalactive serine proteases, the side chain of the aromatic residue215 fixes the exposed �11 template segment to the underlyingstrand �12. Simultaneously, this residue provides the basis forsubsites S2 and S4. Certainly, and as supported by the DFstructures, Gly215 of GzmK cannot substitute for an aromaticresidue and, rather, might push His57 out of its catalyticposition.
Gly219 in functional serine proteases with trypsin-like struc-ture and specificity, i.e. with a kinked entrance frame, adopts amain-chain conformation that would be of high energy whenreplaced by any other amino acid. Upon substrate occupation ofthe S1 pocket, the carbonyl group of Gly219 forms a hydrogenbond with the distal ammonium or guanidyl group of a P1-lysine or arginine residue, respectively. A non-glycine residueat position 219 would have to adopt a high energy main chainconformation to fulfill an equivalent function, but Glu219 ofpro-GzmK (Lys219 in the mouse and rat GzmK, see Fig. 2)
FIG. 7. Cartoon illustrating the proposed structural transfor-mation of pro-GzmK (A) to mature, yet inactive GzmK (B) andsubstrate-activated GzmK (C). The critical structural elements (yel-low ribbons) contributing to this transformation consist of the N-termi-nal residues 16–17 (not shown in A), 188B–195, and 214–220. Approx-imate positions of C� atoms for these residues are marked by black dots.The N-terminal �-ammonium group of Ile16 and the side chains ofLys188B, Asp189, and Asp194 that swap places are highlighted as blue(basic) and red (acidic) bullets. A tripeptide substrate (P1 to P3, C�atoms in white) inducing the active state of GzmK (C) is represented bythe uppermost red ribbon.
Crystal Structure of Human Pro-granzyme K 50931

adopts a relaxed conformation ( � 100°; � 126°) instead.The extremely low affinity of GzmK for benzamidine (in themillimolar range (13)) seems to suggest such a cryptic, i.e.suboptimally widened S1 pocket. We had previously advanceda similar explanation for the weak activity of coagulation factorIXa toward chromogenic substrates (49), one of the very rareexamples for a non-glycine residue at position 219 in trypsin-like enzymes. Another example is the mannan-binding lectin(mannose-binding protein)-associated serine protease 3, whichcan interact with mannan-binding lectin complexes (51). Withregard to the factor IX-related, but much more active humancoagulation factor Xa, we have shown that the substitution ofGly219 by Glu in factor Xa (Gly219 3 Glu mutation) lowers itsactivity about 1000-fold (52), thus proving the detrimentaleffect of a Glu residue in this position.
Finally, the typical Tyr/Phe residue 94 is replaced by theshorter aliphatic Val in GzmK. Normally, an aromatic sidechain covers the reactive site Asp102 and shields the importanthydrogen bond to His57 N�2 from bulk solvent. Obviously, andsimilar to the even smaller Ser94 in DF (42), Val94 of humanGzmK cannot protect this Asp102-His57 pair, but leaves someextra space around the His57 imidazoyl side chain to adopt aninactive tautomer conformation, as seen in some DF structures(42). This interpretation is consistent with experimental datashowing that a triple mutant of DF (S94Y,T214S,S215W) ex-hibits a more than 16-fold higher esterolytic activity towardBLT than wild type DF (53).
Substrate-dependent Activation of Mature GzmK, a Hypoth-esis—In view of all these considerations and previous findings,we hypothesize that mature GzmK is activated only transientlyby a well fitting protein or extended peptide substrate that isable to induce a functional conformation of the nonspecificsubstrate-binding template strand 214–217 (Fig. 7). In fact,GzmK can interact with the peptide mimetic inhibitor D-Phe-Pro-Arg-chlorometyl ketone (13), although quite slowly com-pared with the specific reaction of thrombin, for instance. Mac-romolecular substrates that are efficiently cleaved mightexclude solvent on top of the Asp102-His57 pair and/or packtheir P2, P3, and/or P4 side chains in the encounter complexwith GzmK to promote formation of a functional catalyticmachinery.
To illustrate the anticipated conformational changes ofGzmK after binding to a macromolecular substrate, we mod-eled residues Gly89 to Phe94 of the reactive-site loop of thebikunin subunit D2 into the hypothetical S4 to S2� subsites ofmature GzmK (Fig. 6). In this model, the bikunin Arg92 sidechain occupies the S1 pocket, as observed in the BPTI-trypsincomplex (54), which was taken as the template (see “Experi-mental Procedures”). In addition, the Ser214-His217 stretch hadto be switched from a “resting” to an “active state” conformationto achieve an anti-parallel juxtaposition of this segment withthe main chain of the substrate (Figs. 6 and 7). Concomitantly,the main chain conformation of Glu219 had to be changed toallow for hydrogen bond formation between its carbonyl groupand the terminal guanidyl group of the P1 Arg residue. His217
has been shifted away by �8 Å with its side chain now beingexposed to the solvent. In this way the characteristic kink ofthe trypsin-like “entrance frame” to the S1 pocket can begenerated.
GzmK evidently belongs to the growing subclass of trypsin-like serine proteases with low proteolytic activity such as FIXa(with a cryptic S1 pocket and a distorted 99 loop) (50), DF (42),degP (with a blocked and simultaneously distorted 214–217template) (55), and �-tryptase (with a kinked 214–217 tem-plate and a blocked S1 pocket) (56). All these proteases arecharacterized by a non-functional substrate-interacting tem-
plate, incompatible with productive substrate binding andprocessing. Interestingly, these proteases appear to developtheir full proteolytic potential by diverse means. Binding ofcoagulation FIXa to coagulation cofactor VIIIa generates acomplex (intrinsic X-ase) with proteolytic activity toward coag-ulation FX, its specific macromolecular substrate. Similarly,DF efficiently cleaves and activates factor B upon binding tothe C3b-factor B complex (57). On the other hand, degP seemsto become proteolytically active toward partially unfolded pro-teins upon large conformational changes at elevated tempera-tures. Advantages for such a mechanism would be high speci-ficity for macromolecular substrates, inactivity toward shortpeptides, and dispensability of high affinity inhibitors.
Acknowledgments—We thank D. Turk, H. Pfister, K. Dornmaier, K.Maskos, M. Braun, and Rainer Friedrich for helpful discussions andProf. H. Wekerle for continuous interest and support of the project.
REFERENCES
1. Blott, E. J., and Griffiths, G. M. (2002) Nat. Rev. Mol. Cell. Biol. 3, 122–1312. Barry, M., and Bleackley, R. C. (2002) Nat. Rev. Immunol. 2, 401–4093. Russell, J. H., and Ley, T. J. (2002) Annu. Rev. Immunol. 20, 323–3704. Trapani, J. A. (2001) Genome Biol. 2, 3014.1–3014.75. Hameed, A., Lowrey, D. M., Lichtenheld, M., and Podack, E. R. (1988) J. Im-
munol. 141, 3142–31476. Shi, L., Kam, C. M., Powers, J. C., Aebersold, R., and Greenberg, A. H. (1992)
J. Exp. Med. 176, 1521–15297. Denizot, F., Brunet, J. F., Roustan, P., Harper, K., Suzan, M., Luciani, M. F.,
Mattei, M. G., and Golstein, P. (1989) Eur. J. Immunol. 19, 631–6358. Fink, T. M., Lichter, P., Wekerle, H., Zimmer, M., and Jenne, D. E. (1993)
Genomics 18, 401–4039. Baker, E., Sayers, T. J., Sutherland, G. R., and Smyth, M. J. (1994) Immuno-
genetics 40, 235–23710. Suemoto, T., Taniguchi, M., Shiosaka, S., and Yoshida, S. (1999) Brain Res.
Mol. Brain Res. 70, 273–28111. Babe, L. M., Yoast, S., Dreyer, M., and Schmidt, B. F. (1998) Biotechnol. Appl.
Biochem. 27, 117–12412. Jackson, D. S., Fraser, S. A., Ni, L. M., Kam, C. M., Winkler, U., Johnson,
D. A., Froelich, C. J., Hudig, D., and Powers, J. C. (1998) J. Med. Chem. 41,2289–2301
13. Wilharm, E., Parry, M. A., Friebel, R., Tschesche, H., Matschiner, G.,Sommerhoff, C. P., and Jenne, D. E. (1999) J. Biol. Chem. 274, 27331–27337
14. Sayers, T. J., Lloyd, A. R., McVicar, D. W., O’Connor, M. D., Kelly, J. M.,Carter, C. R., Wiltrout, T. A., Wiltrout, R. H., and Smyth, M. J. (1996)J. Leukoc. Biol. 59, 763–768
15. Pham, C. T., and Ley T. J. (1999) Proc. Natl. Acad. Sci. U. S. A. 96, 8627–863216. Smyth, M. J., McGuire, M. J., and Thia, K. Y. (1995) J. Immunol. 154,
6299–630517. Urata, H., Karnik, S. S., Graham, R. M., and Husain, A. (1993) J. Biol. Chem.
268, 24318–2432218. McGuire, M. J., Lipsky, P. E., and Thiele, D. L. (1993) J. Biol. Chem. 268,
2458–246719. Toomes, C., James, J., Wood, A. J., Wu, C. L., McCormick, D., Lench, N.,
Hewitt, C., Moynihan, L., Roberts, E., Woods, C. G., Markham, A., Wong,M., Widmer, R., Ghaffar, K. A., Pemberton, M., Hussein, I. R., Temtamy,S. A., Davies, R., Read, A. P., Sloan, P., Dixon, M. J., and Thakker, N. S.(1999) Nat. Genet. 23, 421–424
20. Leslie, A. (1991) in Crystallographic Computing V (Moras, D., Podjarny, A. D.,and Thierry, J. C., eds) pp. 50–61, Oxford University Press, Oxford, UK
21. Collaborative Computational Project, Number 4 (1994) Acta Crystallogr. Sect.D Biol. Crystallogr. 50, 760–763
22. Estebanez-Perpina, E., Fuentes-Prior, P., Belorgey, D., Braun, M.,Kiefersauer, R., Maskos, K., Huber, R., Rubin, H., and Bode, W. (2000) Biol.Chem. 381, 1203–1214
23. Turk D (1991) NATO Sci. Ser. I 325, 148–15524. Turk, D. (1992) Weiterentwicklung eines Programms fur Molekulgraphik und
Elektronendichte-Manipulation und seine Anwendung auf verschiedeneProtein-Strukturaufklarungen, Technische Universitat, Munchen,Germany
25. Brunger, A. T. (1992) X-PLOR, version 3.1. A System for X-ray Crystallographyand NMR, Yale University Press, New Haven, CT
26. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, N., Pannu, N. S., Read,R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr.Sect. D Biol. Crystallogr. 54, 905–921
27. Engh, R. A., and Huber, R. (1991) Acta Crystallogr. Sect. A 47, 392–40028. Laskowski, R. A., MacArthur, M. W., and Thornton, J. M. (1993) J. Appl.
Crystallogr. 26, 283–29129. Huber, R., Kukla, D., Bode, W., Schwager, P., Bartels, K., Deisenhofer, J., and
Steigemann, W. (1974) J. Mol. Biol. 89, 73–10130. Bode, W., Wei, A. Z., Huber, R., Meyer, E., Travis, J., and Neumann, S. (1986)
EMBO J. 5, 2453–245831. Xu, Y., Carr, P. D., Guss, J. M., and Ollis, D. L. (1998) J. Mol. Biol. 276,
955–96632. Fehlhammer, H., Bode, W., and Huber, R. (1977) J. Mol. Biol. 111, 415–43833. Singh, T. P., Bode, W., and Huber, R. (1980) Acta Crystallogr. Sect. B Struct.
Sci. 36, 621–627
Crystal Structure of Human Pro-granzyme K50932

34. Jing, H., Macon, K. J., Moore, D., DeLucas, L. J., Volanakis, J. E., andNarayana, S. V. (1999) EMBO J. 18, 804–814
35. Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H.,Shindyalov, I. N., and Bourne, P. E. (2000) Nucleic Acids Res. 28, 235–242
36. McGrath, M. E., Mirzadegan, T., and Schmidt, B. F. (1997) Biochemistry 36,14318–14324
37. Pereira, P. J., Wang, Z. M., Rubin, H., Huber, R., Bode, W., Schechter, N. M.,and Strobl, S. (1999) J. Mol. Biol. 286, 163–173
38. Hof, P., Mayr, I., Huber, R., Korzus, E., Potempa, J., Travis, J., Powers, J. C.,and Bode, W. (1996) EMBO J. 15, 5481–5491
39. Bode, W., Chen, Z., Bartels, K., Kutzbach, C., Schmidt-Kastner, G., andBartunik, H. (1983) J. Mol. Biol. 164, 237–282
40. Bode, W., Turk, D., and Karshikov, A. (1992) Protein Sci. 1, 426–47141. Motyka, B., Korbutt, G., Pinkoski, M. J., Heibein, J. A., Caputo, A., Hobman,
M., Barry, M., Shostak, I., Sawchuk, T., Holmes, C. F., Gauldie, J., andBleackley, R. C. (2000) Cell 103, 491–500
42. Jing, H., Babu, Y. S., Moore, D., Kilpatrick, J. M., Liu, X. Y., Volanakis, J. E.,and Narayana, S. V. (1998) J. Mol. Biol. 282, 1061–1081
43. Bode, W., and Schwager, P. (1975) FEBS Lett. 56, 139–14344. Eigenbrot, C., Kirchhofer, D., Dennis, M. S., Santell, L., Lazarus, R. A.,
Stamos, J., and Ultsch, M. H. (2001) Structure 9, 627–63645. Bode, W. (1979) J. Mol. Biol. 127, 357–37446. Turk, D., Janjic, V., Stern, I., Podobnik, M., Lamba, D., Dahl, S. W., Lauritzen,
C., Pedersen, J., Turk, V., and Turk, B. (2001) EMBO J. 20, 6570–658247. Murakami, M., Karnik, S. S., and Husain, A. (1995) J. Biol. Chem. 270,
2218–2223
48. McEuen, A. R., Ashworth, D. M., and Walls, A. F. (1998) Eur. J. Biochem. 253,300–308
49. Cole, L. B., Chu, N. M., Kilpatrick, J. M., Volanakis, J. E., Volanakis, J. E., andBabu, Y. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 143–150
50. Brandstetter, H., Bauer, M., Huber, R., Lollar, P., and Bode, W. (1995) Proc.Natl. Acad. Sci. U. S. A. 92, 9796–9800
51. Dahl, M. R., Thiel, S., Matsushita, M., Fujita, T., Willis, A. C., Christensen, T.,Vorup-Jensen, T., and Jensenius, J. C. (2001) Immunity 15, 127–135
52. Hopfner, K. P., Brandstetter, H., Karcher, A., Kopetzki, E., Huber, R., Engh,R. A., and Bode, W. (1997) EMBO J. 16, 6626–6635
53. Kim, S., Narayana, S. V., and Volanakis, J. E. (1995) J. Biol. Chem. 270,24399–24405
54. Grzesiak, A., Helland, R., Smalas, A. O., Krowarsch, D., Dadlez, M., andOtlewski, J. (2000) J. Mol. Biol. 301, 205–217
55. Krojer, T., Garrido-Franco, M., Huber, R., Ehrmann, M., and Clausen, T.(2002) Nature 416, 455–459
56. Marquardt, U., Zettl, F., Huber, R., Bode, W., and Sommerhoff, C. P. (2002) J.Mol. Biol. 321, 491–502
57. Xu, Y. Y., Narayana, S. V. L., and Volanakis, J. E. (2001) Immunol. Rev. 180,123–135
58. Evans, S. V. (1993) J. Mol. Graphics 11, 134–13859. Nicholls, A., Bharadwaj, R., and Honig, B. (1993) Biophys. J. 64, A16660. Merritt, E. A., and Bacon, D. J. (1997) Methods Enzymol. 277, 505–52461. Esnouf, R. M. (1999) Acta Crystallogr. Sect. D Biol. Crystallogr. 55, 938–94062. Kraulis, J. (1991) J. Appl. Crystallogr. 24, 946–950
Crystal Structure of Human Pro-granzyme K 50933





























