Embed Size (px)
Citation preview

IntroductionHypertrophic and dilated cardiomyopathies are impor-tant pathologies that increase myocardial mass, albeitwith distinct patterns of remodeling (reviewed in refs.1, 2). Hypertrophic cardiomyopathy produces ventric-ular wall thickening without increases in ventricularvolume, whereas both wall thickness and chamber vol-umes increase in dilated cardiomyopathy. Contractileparameters further discriminate between thesepathologies, with systolic function preserved orimproved in hypertrophic hearts but diminished indilated cardiomyopathy. Whereas the triggering eventsfor each of these pathologies usually appear to be dif-ferent, clinical observations indicate a potential over-lap in the molecular signals and/or cellular events that
remodel the heart, because some patients with hyper-trophic cardiomyopathy develop a dilated phenotype.
Mutations in cardiac myosin-binding protein-C(MyBP-C) account for approximately 20% of familialhypertrophic cardiomyopathy (FHC) (3–7). Whereasmutations of other sarcomere proteins that participatein force generation are expected to cause FHC by a dom-inant negative effect on contractile properties, themechanism(s) by which cardiac MyBP-C defects causeFHC are less certain (reviewed in refs. 8, 9). MyBP-C is alarge and abundant myofibrillar protein with bothstructural and regulatory functions (10). During cardiacdevelopment, MyBP-C expression corresponds to theonset of myofibrillogenesis, and 3 fibronectin-likemotifs are thought to be necessary for thick filament
The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9 1235
Dilated cardiomyopathy in homozygous myosin-bindingprotein-C mutant mice
Bradley K. McConnell,1 Karen A. Jones,1 Diane Fatkin,2 Luis H. Arroyo,3 Richard T. Lee,3
Orlando Aristizabal,4 Daniel H. Turnbull,4 Dimitrios Georgakopoulos,5 David Kass,5
Meredith Bond,6 Hideshi Niimura,1 Frederick J. Schoen,7 David Conner,1
Donald H. Fischman,8 Christine E. Seidman,2 and J.G. Seidman1
1Department of Genetics, Howard Hughes Medical Institute and Harvard Medical School, Boston, Massachusetts 02115, USA2Cardiovascular Division and Howard Hughes Medical Institute, Brigham and Women’s Hospital, Boston, Massachusetts 02115, USA
3Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts 02115, USA4Skirball Institute of Biomolecular Medicine, New York University School of Medicine, New York, New York 10016, USA5Division of Cardiology, The Johns Hopkins Medical Institute, Baltimore, Maryland 21287, USA6Department of Molecular Cardiology, Lerner Research Institute, The Cleveland Clinic Foundation, Cleveland, Ohio 44195, USA
7Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts 02115, USA8Department of Cell Biology, Weill Medical College of Cornell University, New York, New York 10021, USA
Address correspondence to: Jonathan Seidman, Department of Genetics, Harvard Medical School, 200 Longwood Avenue, Alpert Building, Boston, Massachusetts 02115, USA. Phone: (617) 432-7871; Fax: (617) 432-7832; E-mail: [email protected].
Bradley K. McConnell and Karen A. Jones contributed equally to this work.
Received for publication May 19, 1999, and accepted in revised form September 28, 1999.
To elucidate the role of cardiac myosin-binding protein-C (MyBP-C) in myocardial structure and func-tion, we have produced mice expressing altered forms of this sarcomere protein. The engineered muta-tions encode truncated forms of MyBP-C in which the cardiac myosin heavy chain-binding and titin-binding domain has been replaced with novel amino acid residues. Analogous heterozygous defects inhumans cause hypertrophic cardiomyopathy. Mice that are homozygous for the mutated MyBP-C alle-les express less than 10% of truncated protein in M-bands of otherwise normal sarcomeres. Homozygousmice bearing mutated MyBP-C alleles are viable but exhibit neonatal onset of a progressive dilated car-diomyopathy with prominent histopathology of myocyte hypertrophy, myofibrillar disarray, fibrosis,and dystrophic calcification. Echocardiography of homozygous mutant mice showed left ventriculardilation and reduced contractile function at birth; myocardial hypertrophy increased as the animalsmatured. Left-ventricular pressure-volume analyses in adult homozygous mutant mice demonstrateddepressed systolic contractility with diastolic dysfunction. These data revise our understanding of therole that MyBP-C plays in myofibrillogenesis during cardiac development and indicate the importanceof this protein for long-term sarcomere function and normal cardiac morphology. We also propose thatmice bearing homozygous familial hypertrophic cardiomyopathy–causing mutations may provide use-ful tools for predicting the severity of disease that these mutations will cause in humans.
J. Clin. Invest. 104:1235–1244 (1999).

assembly. In the mature heart, MyBP-C is found in 7–9transverse strips (spaced about 43 nm apart) located inthe C-zone of sarcomere A-bands (11–13). WhereasMyBP-C does not directly participate in force genera-tion, reversible phosphorylation by cAMP-dependentprotein kinase A (14) and calcium/calmodulin-depend-ent protein kinase II (15) may affect contractile functionby stimulating cardiac actomyosin ATPase (16) or influ-encing myofibril tension generation and contractilevelocity (17). Six FHC-causing MyBP-C mutations thatare predicted to encode truncated polypeptides (3–5)that lack carboxyl residues required to bind myosinheavy chain and titin and that have novel residues attheir carboxyl termini have been described. FHC is an
autosomal dominant trait, andaffected individuals are heterozygousfor these mutations. Individualsbearing these 6 FHC-causing muta-tions have clinically similar forms ofFHC. The mechanism by whichFHC-causing MyBP-C mutationsperturb cardiac structure and func-tion is unknown, because neithermutant peptides nor reduced levelsof MyBP-C has been found in cardiactissue from affected patients (18).
We have created mice bearing amutationally altered murine cardiacMyBP-C gene that encodes a trun-cated peptide, analogous to thatfound in human hypertrophic car-diomyopathy. Although FHCpatients are heterozygous, bearing 1mutant and 1 wild-type MyBP-Callele, we have bred the mice bearing
mutant MyBP-C alleles to homozygosity so that theycarry only mutated MyBP-C alleles. Normal MyBP-Cprotein cannot be expressed in cardiac tissues of thesemice, and mutant MyBP-C peptides are found atmarkedly reduced levels. We hypothesized thathomozygous mice bearing mutant MyBP-C allelesmight have a different cardiac phenotype than het-erozygous mice bearing a single mutant allele and a sin-gle wild-type allele. We have studied cardiac structureand function of the homozygous mutant mice to bet-ter define the mechanisms by which FHC-causingMyBP-C mutations alter cardiac physiology.
1236 The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9
Figure 1Schematic of (a) the genomic structure of wild-type MyBP-C, MyBP-C(LoxP), and MyBP-C(Neo) alleles, the RNAs produced by each allele,and (b) the structure of the MyBP-C proteins encoded by each allele.(a) Exons 29–32 of the murine cardiac MyBP-C gene are shown foreach allele (see Methods). The mutations alter only exon 30; all otherexons are identical. The black lines above each gene segment reflect thestructure of the encoded RNA indicated in the 5′→3′ orientation; thicklines are exons incorporated into the RNA, thin lines indicate skippedsegments of the gene not found in RNA. The genotypes of mice weredetermined with primers 1F, 1R, 2F, 2R, and 3R. Primers 4F and 4Rwere used to determine the structure of MyBP-C RNA; structures of thededuced RNAs are indicated above each allele. (b) The structure of car-boxyl ends of the MyBP-C polypeptides encoded by MyBP-C(Neo) andMyBP-C(Lox) alleles were deduced from the sequences of RNA foundin the left ventricles of homozygous mice bearing the indicated allele.The residues (between residues 1064 and 1111) of the wild-type pro-tein are encoded by exon 30. Novel amino acid residues at the carboxylend of the mutant proteins (bold) are encoded by altered reading ofexon 30 (Lox) or exon 31 (Neo). The wild-type MyBP-C protein is 1270amino acids whereas the MyBP-C(Lox) protein is 1240-amino acidslong; the complete sequences of these 2 carboxyl ends are not shown.The carboxyl end of the analogous human mutation found in familyNN (3) is shown for comparison. Differences between the family NNprotein and the MyBP-C(Neo) protein reflect sequence differencesbetween the mouse and human MyBP-C gene exon 31.
Figure 2MyBP-C mRNA and protein expression levels in hearts of 12-week-old homozygous MyBP-C(Neo) mice (designated t/t). (a) Northernblot analyses of RNAs from left ventricle of wild-type (+/+) andMyBP-C(Neo) mice hybridized with murine MyBP-C and GAPDHprobes. (b) Western blot analyses identified the 150-kDa MyBP-Cprotein in myofibrillar extracts of the LV of wild-type (+/+) andhomozygous MyBP-C(Neo) mice (t/t).

MethodsGeneration of homozygous mutant cardiac MyBP-C mice.Murine MyBP-C sequences were amplified from cardiacRNA by RT-PCR using oligonucleotide primers 3301Fand 3900R derived from the published human MYBPC3gene sequence (EMBL accession number X84075) andused to probe a 129SvJ sub-genomic library (unpub-lished). A bacteriophage clone, λMyBPc, containing themurine MyBP-C gene was isolated, from which a 7.4-kbSpeI fragment was subcloned into pBluescript and char-acterized by partial nucleotide sequence analysis.Intron-exon boundaries were deduced by comparisonwith the human gene (Figure 1a). A blunt-ended, 2-kbfragment encoding the neomycin gene flanked by LoxPsequences was excised from plasmid pPTloxPNeo (kind-ly provided by J. Rossant), and inserted into the EcoRVsite within exon 30.
The targeting construct was introduced into embryon-ic stem (ES) cells and selected (19). Colonies werescreened for homologous recombination by Southernblot analyses using an external probe (data not shown).Targeted ES cells were injected into mouse blastocysts asdescribed (20). Genotypes were ascertained from tailDNA in the offspring of chimeric animals by PCR analy-ses (25 µL assay) using primers (Figure 1a) to amplifyexon 30: 1F (CTAGGTACTAACAG GCTCCTGCTT), 1R(CCTACCATGCAGGAAACCAGAATA) and primers: 2F(GGTTCTTTTT GTCAAGACCGAC), 2R (GTAGCCGGAT-CAAGCGTATG) to amplify the inserted neomycin cassette.
In addition a male chimeric mouse was mated with anEIIa-Cre transgenic mouse (kindly provided by H. West-phal, National Institutes of Health, Bethesda, Maryland,USA; ref. 21) to produce progeny in which the LoxP-flanked Neo gene had been efficiently deleted (Figure1a). The MyBP-C (Lox) genotype was determined byPCR analyses of exon 30 and absence of Neo sequences.Mice homozygous for the MyBP-C(Neo) or MyBP-C(Lox)alleles (both designated MyBP-Ct/t) were produced bybreeding heterozygous animals.
RNA analyses. Total RNA was isolated from left ventri-cle (LV), right ventricle (RV), and atrium, using Trizol
(GIBCO BRL Life Technologies, Frederick, Maryland,USA) and analyzed by standard Northern blot proce-dures (22). MyBP-C RNA was detected using a 32P-labeled insert from a mouse cDNA clone designatedpcMyBPC, which encodes MyBP-C amino acid residues582–1110. Other RNAs were detected using 5′-32P-labeled transcript-specific oligonucleotide probes withstandard hybridization conditions (23) as follows: α-skeletal actin: 5′-TGGCTTTAATGCTTCAAGTTTT CATTTC-CTTTCCACAGGG; brain natriuretic peptide (BNP): 5′-CAGCTTGAGATATGTGTCACCTTGGAATTTTGAGGTCTCTGCTGGACC; sarcoplasmic reticulum Ca2+-ATPase(SERCA): 5′-AACAACGCACATGCACGCACCCGAACAC CTT-ATATTTCTGCAAATGG; GAPDH: 5′-GGAA-CATGTAGA-CCATGTAGTTGAGGTCAATGAAG. Hybridization signalswere quantified using ImageQuant software (MolecularDynamics, Sunnyvale, California, USA), and normalizedto the signal intensity observed with an oligonucleotidespecific for GAPDH RNA. Student’s t tests were per-formed to determine whether the data were significant-ly different between the groups of mice.
The structure of MyBP-C RNA was assessed bynucleotide sequence analysis of RT-PCR–amplifiedDNA fragments using conditions described previously(3) with primers 4F (TCAGGTGACCTGGACCAAAGAG)and 4R (ATGTTATGGCTGAAGACCCGG).
Protein analyses. Cardiac tissues were isolated,washed in Dulbecco’s PBS, blotted on filter paper, andhomogenized in ice-cold inhibiting buffer (24) con-taining 50 mM potassium dihydrogen phosphate(KH2PO4), 70 mM sodium fluoride (NaF), and 5 mMEDTA, with protease inhibitors (5 µg/mL antipain, 10µg/mL leupeptin, 5 µg/mL pepstatin A, 43 µg/mLPMSF, 5 mM EGTA, and 0.1 µM sodium orthovana-date). Myofibrillar fractions were isolated from totalprotein homogenates by centrifugation at 15,000 g for5 minutes at 4oC. The pellet was resuspended ininhibiting buffer plus 1% Triton X-100. Detergent-extracted myofibrils were again centrifuged at 5000 gfor 5 minutes, and the pellet was resuspended ininhibiting buffer.
The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9 1237
Table 1Left ventricular dimensions of wild-type and homozygous mutant MyBP-C mice
GenotypeA Body wt (g)C LVAW (mm) LVPW (mm) LVDD (mm) LVSD (mm) LVFS (%) HR (bpm)
<3 days+/+ 1.73 ± 0.35 0.21 ± 0.01 0.21 ± 0.02 1.58 ± 0.07 1.12 ± 0.10 29 ± 4 346 ± 17t/t 1.24 ± 0.15 0.14 ± 0.02 0.14 ± 0.02 1.67 ± 0.28 1.41 ± 0.31 16 ± 5 381 ± 43PB 0.002 NS NS <0.001 <0.001 0.001 NS
3 weeks+/+ 16.03 ± 1.45 0.86 ± 0.04 0.81 ± 0.07 2.76 ± 0.03 1.45 ± 0.15 47 ± 6 460 ± 86t/t 14.60 ± 1.00 1.02 ± 0.13 1.00 ± 0.10 3.42 ± 0.37 2.57 ± 0.48 25 ± 6 458 ± 18P NS NS 0.05 0.04 0.02 0.009 NS
8 weeks+/+ 22.10 ± 2.21 0.84 ± 0.03 0.82 ± 0.03 2.74 ± 0.43 1.54 ± 0.35 44 ± 6 470 ± 95t/t 22.99 ± 2.59 1.12 ± 0.13 1.10 ± 0.13 3.56 ± 0.53 2.64 ± 0.46 26 ± 5 414 ± 40P NS <0.001 <0.001 0.006 <0.001 <0.001 NS
ANumber of animals and age at study: 4 +/+ and 11 t/t (<3 days); 3 +/+ and 3 t/t (3 weeks); 6 +/+ and 10 t/t (8 weeks). Homozygous MyBP-C(Neo) and MyBP-C(LoxP) mice were indistinguishable, and these data (indicated as t/t) reflect measurements made on both types of mutant mice. BP-value (wild-type vs. mutantMyBP-C mice) derived by Student’s unpaired t test after raw data were corrected for body weight. CAverage body weight of mice.

Protein fractions were separated by standard methodson 6% polyacrylamide mini gels (Western blot analyses),6% polyacrylamide slab gels (cardiac α- and β-myosinheavy chain [MHC] isoforms; ref. 25), or 12% polyacry-lamide slab gels (total proteins). Protein concentrationwas determined using the Bradford protein assay. Theproportion of cardiac α- and β-MHC was quantified bydensitometry and NIH-Image software (National Insti-tutes of Health, Bethesda, Maryland, USA).
MyBP-C was identified by Western blot analyses (Amer-sham Pharmacia Biotech UK, Little Chalfont, Bucking-hamshire, England; ref. 26) of total protein or myofibril-lar homogenates separated by SDS-PAGE gels (125 V for2 hours), transferred to a nitrocellulose membranes (35V for 2.5 hours), and incubated with rabbit anti-chickenskeletal muscle MyBP-C γ-serum polyclonal antibody(diluted 1:10,000; ref. 27). Antibody-labeled proteins weredetected using an enhanced chemiluminescence plus kit(ECL+Plus; Amersham Life Sciences Inc.). The relativeamounts of antibody-labeled protein were quantified bydensitometry and NIH-Image software.
Morphology and microscopy. Animals were sacrificed bycervical dislocation and hearts were excised, rinsed inPBS (GIBCO), and weighed. Cardiac tissues were iso-lated as described (28) and pathologic evaluations wereperformed by an experienced cardiac pathologist.
Excised tissues were mounted in paraffin blocks asdescribed (25) and 3–7-µM sections were stained withhematoxylin and eosin (H&E) or Masson trichrome(American HistoLabs Inc. Gaithersburg, Maryland,USA). High-magnification images were obtained usinga Zeiss Axiophot light microscope (Carl Zeiss Inc.,Thornwood, New York, USA) equipped with a ×2.5, ×20,and ×40 oil immersion objective and a Sony digitalphoto camera (DKC-5000; Sony Corp., Tokyo, Japan).Images were viewed using Adobe Illustrator software(Adobe Systems Inc., Mountain View, California, USA).
LV and RV sections were prepared for transmissionelectron microscopy as described previously (29). Inbrief, sections were fixed with 2.5% glutaraldehyde + 2%
paraformaldehyde in 0.1 Mcacodylate buffer, pH 7.4, thenrinsed 3 times in 0.1 M cacody-late buffer, pH 7.4, and post-fixed in 1% osmium tetroxide(OsO4) for 2 hours, rinsed indH20, and then 1% uranylacetate for 1 hour (en-blockstaining). The fixed pellets weredehydrated in alcohol, rinsedtwice in propylene oxide for 20minutes, and embeddedovernight with a 1:1 ratio ofpropylene oxide to Spurr’s resinsolution. After 12 hours, thissolution was replaced with100% Spurr’s solution and thenpolymerized in a dry oven at70oC for 48 hours. Ultrathin
sections (100-nm thick) from the stained and embed-ded samples were observed in transmission mode usinga Phillips CM12 electron microscope (FEI Company,http://www.feic.com).
Assessment of LV function. Transthoracic echocardiog-raphy was performed in 0 to 3 day-old mice using ahigh-frequency (45 MHz) echocardiographic technique(25). Echocardiographic imaging was performed with-out anesthesia and with mice lightly restrained in asupine position using a Humphrey Ultrasound Biomi-croscope (model 840; Humphrey Instruments, SanLeandro, California, USA). LV parameters wereobtained from the 2-dimensional echocardiographicimages in a short-axis view. Neonatal mouse heart rateswere determined from continuous-wave Doppler trac-ing using a high-frequency Doppler system (25, 30).
Transthoracic echocardiography was performed inadult mice (> 3 weeks old) using a Sonos 5500 ultra-sonograph with a 12-MHz transducer (Hewlett-Packard, Andover, Massachusetts, USA). Mice wereanesthetized with 2.5% Avertin (0.010 mL/g) andwarmed with a heating pad during echocardiographicexamination. Two-dimensional echocardiographicimages were obtained with mice orientated in a left lat-eral decubitus or supine position. LV parameters wereobtained from M-mode interrogation in a short-axisview. Orthogonal left atrial diameter (LAD) wasobtained from 2-dimensional echocardiographicimages in a long-axis view. Echocardiographic meas-urements, for both neonatal and adult mouse studies,were averaged from at least 3 separate cardiac cycles: LVanterior wall (LVAW) thickness, LV posterior wall(LVPW) thickness, maximal LV diastolic diameter(LVDD), minimum LV systolic diameter (LVSD), LVfractional shortening (LVFS), maximal LAD, and heartrate. The statistical significance of differences inechocardiographic parameters between wild-type andMyBP-C mice was determined by unpaired Student’s t-test. Data are expressed as mean ± SD. A P value lessthan 0.05 was considered significant.
1238 The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9
Table 2Cardiac function of 12-week-old mice assessed by in vivo cardiac catheterization
Cardiac function Wild-type (+/+)A MyBP-Ct/t P-valueB
Stroke work(end diastolic volume) (SW(EDV)) (mmHg) 104.99 ± 7.23 56.30 ± 8.87 0.004End systolic pressure (mmHg) 100.11 ± 3.49 90.94 ± 7.43 NSdP/dtmax (mmHg/s) 11536.96 ± 852.22 10841.40 ± 794.17 NSNormalized end-systolic elastance
(mmHg/µl*100mg) 13.14 ± 3.38 3.53 ± 0.42 0.02dP/dtmin (mmHg/s) –9615.07 ± 488.19 –4841.86 ± 223.91 0.0001dP/dtmax / dP/dtmin 1.21 ± 0.10 2.25 ± 0.15 0.004End diastolic pressure (mmHg) 3.28 ± 0.72 3.73 ± 0.58 NSTime to peak filling (ms) 33.25 ± 1.46 42.70 ± 2.99 0.03tau (ms) 9.03 ± 0.62 14.56 ± 1.28 0.008Normalized beta (mmHg/µL–1/100 mg) 0.07 ± 0.01 0.03 ± 0.01 0.01Heart rate (min–1) 601.70 ± 19.65 580.20 ± 21.55 NS
A10 wild-type and 5 homozygous mutant MyBP-C(Neo) or MyBP-C(LoxP) mice (designated MyBP-Ct/t).BP-value (+/+ vs. t/t) derived by Student’s unpaired t test.

Analysis of LV-chamber systolic and diastolic prop-erties was performed in vivo as described (31). In brief,a miniaturized impedance/micromanometer catheterwas used to derive real-time LV pressure-volume rela-tionships. Animals were anesthetized with etomidate(10–20 mg/kg body weight), morphine (1–2 mg/kgbody weight), and urethane (750 mg/kg body weight),intubated, and artificially ventilated with a custom-designed murine ventilator, with 100% inspired oxygenat a tidal volume of 200 µL and frequency of 120breaths per minute. Heart rates were near normal(>500 beats per minute). The LV was catheterizedusing an apical stab exposed by a limited thoracotomy.Signals were recorded at steady state and during tran-sient load reduction produced by transiently occlud-ing the inferior vena cava. Data were sampled at 2 kHzand analyzed with custom software. Following initialdata collection, an ultrasound perivascular flow probe(1RB; Transonics, Ithaca, New York, USA) was placedaround the thoracic aorta to measure cardiac output.The stroke volume was used to calibrate relative vol-umes measured simultaneously by the volume-catheter signal, matching its value to the mean widthof the pressure-volume loop. Relative changes and themajority of derived hemodynamic parameters were cal-ibrated, although absolute volume was not. Heartweights and gross anatomic examinations were madeafter completion of each study. Comparisons betweenwild-type and mutant MyBP-C mice were performedby Student t tests. All data are reported as means ±SEM and P values less than 0.05 are numericallyreported; higher values are indicated as nonsignificant.
ResultsConstruction MyBP-C(Neo) and MyBP-C(LoxP) alleles. A seg-ment of the murine MyBP-C gene was isolated from abacteriophage λDASH genomic library and its nucleotidesequence determined (unpublished). Exons 29–31 were
identified on a 7.4-kb SpeI fragment that was subcloned;the neomycin resistance gene (PGK-Neo-polyA) flanked byLoxP sequences was inserted into exon 30 (Figure 1a andMethods). This construct was used to disrupt theendogenous MyBP-C gene in ES cells by homologousrecombination and the targeted allele was designatedMyBP-C(Neo). Blastocysts injected with ES cells bearingthe MyBP-C(Neo) allele produced chimeric animals. Theamount of mutant MyBP-C mRNA in cardiac tissuesfrom mice bearing this allele was significantly less thanthe amount of MyBP-C mRNA found in wild-type mice(see below). We hypothesized that the amount of mutantMyBP-C mRNA was reduced because the neomycin-resistant gene sequences in the mutant mRNA caused theRNA to be degraded rapidly or because these sequencesaltered the processing of MyBp-C RNA precursor. To testthis hypothesis, a second allele, designated MyBP-C(LoxP), was created by mating chimeric MyBP-C(Neo)mice with a transgenic mouse expressing CRE. TheMyBP-C(LoxP) allele resulted from excision of theneomycin resistance gene by CRE recombinase and con-tains a residual 97-bp duplicated LoxP sequence (Figure1a and data not shown). Heterozygous offspring ofchimeric animals were bred to produce mice homozy-gous for either the MyBP-C(Neo) or MyBP-C(LoxP) allele.Mice homozygous for each allele were characterized bySouthern blot analyses and by PCR amplification of theMyBP-C(LoxP) allele with primers 1F,1R (Figure 1a) andthe MyBP-C(Neo) allele with primers 1F,3R and 2F,2R(Figure 1a; data not shown). Homozygous MyBP-C(Neo)and MyBP-C(LoxP) mice were fertile, produced normallitter sizes, and survived more than 1 year.
Cardiac MyBP-C mRNA and protein expression frommutant MyBP-C alleles. The amounts of cardiac MyBP-CmRNA in MyBP-C(LoxP) and MyBP-C(Neo) mousehearts were assessed by Northern blot analyses. Theabundant cardiac MyBP-C transcripts (4.5 kb) found inwild-type mice were markedly reduced in the LV (14.5
The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9 1239
Figure 3Comparison of hearts from 8–12-week-old wild-type and homozygous mutant mice. (a) Gross morphology of homozygous MyBP-C(Neo) (des-ignated t/t) and wild-type (+/+) hearts and (b) heart-to-body weight or LV-to-body weight ratios demonstrate significant increases (P < 0.001)in cardiac size of homozygous mutant mice (t/t) versus wild-type mice (+/+).

± 4%; n = 6), RV (9.5 ± 1%; n = 3), and atria (18.4 ± 5%; n= 3) of MyBP-C(Neo) mice (Figure 2a and data notshown). Cardiac tissues from MyBP-C(Neo) mice andMyBP-C(LoxP) mice contained approximately the sameamount of cardiac MyBP-C mRNA (data not shown).
The structure of the cardiac MyBP-C RNA producedfrom each allele was characterized by nucleotidesequence analyses of RT-PCR–amplified productsderived from homozygous mutant mouse heart totalRNA (see Methods). The structures of the cardiacMyBP-C sequences found in the hearts of the mutantmice were compared with wild-type RNA sequencesand were consistent with the model that the differentmutant RNAs resulted from alternate splice patterns(Figure 1, a and b). That is, RNA derived from theMyBP-C(Neo) allele spliced out exon 30 whereas RNAderived from the MyBP-C(LoxP) allele lacked the 5′part of exon 30 because the RNA was spliced from the3′ end of exon 29 to the LoxP sequence (Figure 1). Thepredicted mutant MyBP-C polypeptides encoded bythe altered MyBP-C alleles differ at their carboxyl ter-mini (Figure 1b). The MyBP-C(LoxP) allele produces aprotein that has 166 novel amino acid residues and is30 residues shorter than the wild-type protein, where-as the MyBP-C(Neo) protein is 174 amino acidresidues shorter than the wild-type protein and has 32novel amino acid residues (Figure 1b).
The amount of mutant peptide found in the ventriclesof homozygous mutant MyBP-C mice was assessed byWestern blot analyses. Polyclonal MyBP-C antibodiesidentified a 150-kDa protein in LV total protein and
myofibrillar-enriched protein preparations that comi-grated with chicken MyBP-C (Figure 2b and data notshown). Although the mutant protein from MyBP-C(Neo) mice was theoretically 15 kDa smaller than wild-type MyBP-C protein, the mutant and wild-type proteinsmigrated at approximately the same position on a 6%SDS-PAGE gel. (Why the smaller mutant peptide comi-grated with the large wild-type protein is uncertain.).Mutant cardiac MyBP-C protein in total proteinhomogenates of the LV from MyBP-C(Neo) mice was 2.3± 1% (n = 6), the amount found in analogous wild-typeextracts (data not shown). Myofibrillar extracts containedsignificantly more mutant peptide than correspondingamounts of total protein extract. Mutant cardiac MyBP-C protein levels in myofibrillar extracts were 9.5 ± 3 % (n= 8) the amount of MyBP-C in wild-type myofibrillarextracts (Figure 2b). Similar analyses demonstrated thatthe MyBP-C(LoxP) mutant protein was also enriched inthe myofibrillar fraction compared with the amount inthe total cytoplasmic fraction (data not shown).
Cardiac morphology of homozygous mutant MyBP-C mice.Cardiac structure was characterized in mutant MyBP-Cmice at 8–12 weeks of age. Hearts from both homozy-gous MyBP-C(Neo) and homozygous MyBP-C(LoxP)mutant mice were visibly enlarged compared with thosefrom wild-type mice, and calcified plaques were presenton the LV chamber walls of homozygous mutant MyBP-C mouse hearts but not wild-type mice (Figure 3 anddata not shown). Total heart weights in mutant versuswild-type animals (177.0 ± 5.7 mg, n = 26 vs. 113.8 ± 3.8mg, n = 25) and LV weights (136.0 ± 7.5 mg, n = 12, vs.
1240 The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9
Figure 4Histology of 8–12-week-old wild-type (a) and MyBP-C(Neo) (b–d) mouse hearts. Sections were stained with H&E (a and c) or Massontrichrome (b and d).

80.0 ± 4.8 mg, n = 11) were significantly increased (P <0.001). Heart-to-body weights and LV-to-body weightratios were also greater in mutant MyBP-C mice than inwild-type mice (Figure 3b). In contrast, RV left and rightatria were of comparable size and weights in mutantand wild-type mice (Figure 3 and data not shown).
Both homozygous MyBP-C(Neo) and MyBP-C(LoxP)hearts demonstrated prominent histologic abnormal-ities, including myocyte hypertrophy, myofibrillar dis-array, and fibrosis. In addition 3 of 5 mutant hearts haddystrophic calcification (Figure 4, b and d), which var-ied in extent and location. Focal LV and RV calcifica-tions and multifocal intramural calcification werefound in 2 of 5 hearts, as well as subendocardial calci-fication with multifocal interstitial fibrosis (1 of 5hearts), and subendocardial papillary muscle withslight perivascular fibrosis (1 of 5 hearts).
Sarcomeres from homozygous mutant MyBP-C(Neo)mouse hearts were examined by electron microscopy(Figure 5) The sarcomeres of these mouse hearts werewell-organized with regularly aligned A-bands, I-bands,and Z-lines; the spacing of these bands was similar tothat observed in sarcomeres from wild-type mice. TheM-line was frequently absent in sarcomeres from theLV of homozygous MyBP-C mice (Figure 5), however insarcomeres derived from the RV, well-defined M-lineswere found (data not shown).
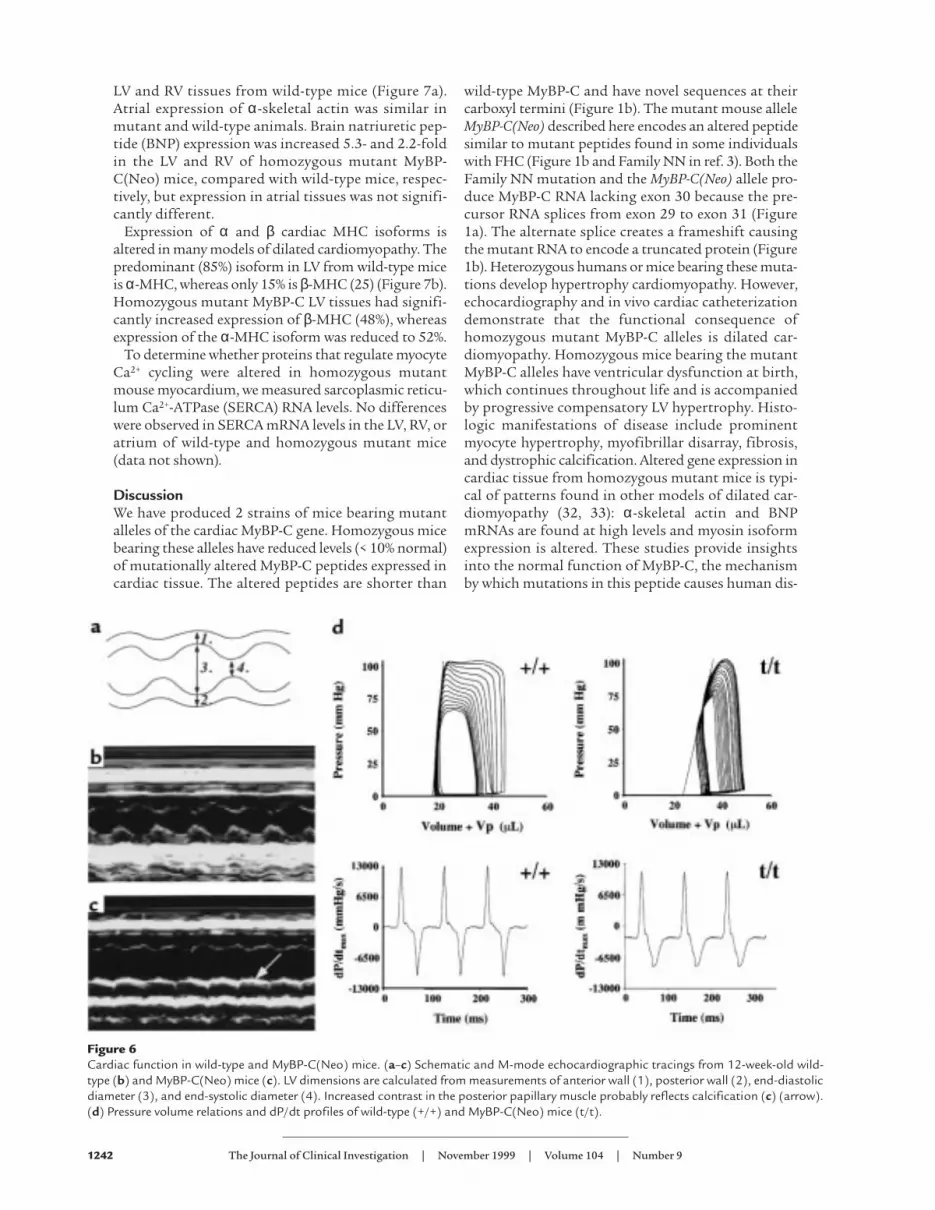
Cardiac function of homozygous mice bearing mutantMyBP-C alleles. Cardiac function was assessed in adultand neonatal homozygous MyBP-C(Neo) mice usingtransthoracic echocardiography (Figure 6, a–c, anddata not shown; see Methods). At birth (0–3 days) andthroughout adulthood (from 3 weeks after birth),mutant MyBP-C mouse hearts demonstratedincreased LV diastolic and systolic diameter as well asreduced LV fractional shortening (Table 1). LV wallthickness was used to assess whether ventricularhypertrophy was present. In the neonatal period, ante-rior and posterior LV wall thickness in homozygousmutant MyBP-C mice was not significantly differentfrom the wall thickness of wild-type mice after cor-rection for body weight differences (Table 1). As wild-type and mutant mice aged, LV wall thicknessincreased; however, this increase was greater inhomozygous mutant MyBP-C mice, and by 8 weeks ofage mutant mice had significantly thicker LV wallsthan did wild-type mice (Table 1). LAD of homozy-gous mutant MyBP-C mice were also significantlyincreased in comparison with wild-type mice (1.77 ±0.13 mm vs. 1.53 ± 0.08 mm; P = 0.01) at 8–12 weeksof age. Echo-dense material, consistent with calcifica-tion of the papillary muscles and focal lesions withinthe LV, was observed in all (10 of 10) homozygousmutant MyBP-C mice at 8 weeks; in 6 animals thesechanges were moderate to severe.
Cardiac function of homozygous MyBP-C(Neo) micewas further assessed in anesthetized mice by in vivocatheterization (31). Pressure-volume loops obtainedfrom homozygous MyBP-C(Neo) mice and wild-type
mice demonstrated significant differences between thecardiac function of these 2 strains (Figure 6d). Althoughthe isovolumic phase of systole was normal, abnormali-ties were evident with the onset of contraction, consis-tent with abnormal fractional shortening observed inechocardiographic studies. The abnormal profile of sys-tolic contraction was reflected in several quantitativeparameters (Table 2). Diastolic properties (dP/dTmin,dP/dTratio, end-diastolic volume (SW(EDV)), time to peakfilling, relaxation time (tau), and chamber compliance(normalized beta) were abnormal. Heart rates, dP/dTmax,end-diastolic pressure, and end-systolic pressure werenot different between mutant and wild-type mice.
RNA and protein expression associated with cardiomyopa-thy. Altered gene expression, a recognized feature ofdilated cardiomyopathy (reviewed in refs. 32, 33), wasexamined in homozygous mutant MyBP-C(Neo) andMyBP-C(LoxP) mice. Mutant mice expressed α-skele-tal actin at levels 16- and 8-fold above that found in
The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9 1241
Figure 5Transmission electron micrographs of sarcomeres from 12-week-oldwild-type (a) and MyBP-C(Neo) mouse hearts (t/t). Note the unusu-al appearance of the M-line (asterisk) in MyBP-C(Neo) mouse-derived sarcomeres. Bar, 0.5 µm.
LV and RV tissues from wild-type mice (Figure 7a).Atrial expression of α-skeletal actin was similar inmutant and wild-type animals. Brain natriuretic pep-tide (BNP) expression was increased 5.3- and 2.2-foldin the LV and RV of homozygous mutant MyBP-C(Neo) mice, compared with wild-type mice, respec-tively, but expression in atrial tissues was not signifi-cantly different.
Expression of α and β cardiac MHC isoforms isaltered in many models of dilated cardiomyopathy. Thepredominant (85%) isoform in LV from wild-type miceis α-MHC, whereas only 15% is β-MHC (25) (Figure 7b).Homozygous mutant MyBP-C LV tissues had signifi-cantly increased expression of β-MHC (48%), whereasexpression of the α-MHC isoform was reduced to 52%.
To determine whether proteins that regulate myocyteCa2+ cycling were altered in homozygous mutantmouse myocardium, we measured sarcoplasmic reticu-lum Ca2+-ATPase (SERCA) RNA levels. No differenceswere observed in SERCA mRNA levels in the LV, RV, oratrium of wild-type and homozygous mutant mice(data not shown).
DiscussionWe have produced 2 strains of mice bearing mutantalleles of the cardiac MyBP-C gene. Homozygous micebearing these alleles have reduced levels (< 10% normal)of mutationally altered MyBP-C peptides expressed incardiac tissue. The altered peptides are shorter than
wild-type MyBP-C and have novel sequences at theircarboxyl termini (Figure 1b). The mutant mouse alleleMyBP-C(Neo) described here encodes an altered peptidesimilar to mutant peptides found in some individualswith FHC (Figure 1b and Family NN in ref. 3). Both theFamily NN mutation and the MyBP-C(Neo) allele pro-duce MyBP-C RNA lacking exon 30 because the pre-cursor RNA splices from exon 29 to exon 31 (Figure1a). The alternate splice creates a frameshift causingthe mutant RNA to encode a truncated protein (Figure1b). Heterozygous humans or mice bearing these muta-tions develop hypertrophy cardiomyopathy. However,echocardiography and in vivo cardiac catheterizationdemonstrate that the functional consequence ofhomozygous mutant MyBP-C alleles is dilated car-diomyopathy. Homozygous mice bearing the mutantMyBP-C alleles have ventricular dysfunction at birth,which continues throughout life and is accompaniedby progressive compensatory LV hypertrophy. Histo-logic manifestations of disease include prominentmyocyte hypertrophy, myofibrillar disarray, fibrosis,and dystrophic calcification. Altered gene expression incardiac tissue from homozygous mutant mice is typi-cal of patterns found in other models of dilated car-diomyopathy (32, 33): α-skeletal actin and BNPmRNAs are found at high levels and myosin isoformexpression is altered. These studies provide insightsinto the normal function of MyBP-C, the mechanismby which mutations in this peptide causes human dis-
1242 The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9
Figure 6Cardiac function in wild-type and MyBP-C(Neo) mice. (a–c) Schematic and M-mode echocardiographic tracings from 12-week-old wild-type (b) and MyBP-C(Neo) mice (c). LV dimensions are calculated from measurements of anterior wall (1), posterior wall (2), end-diastolicdiameter (3), and end-systolic diameter (4). Increased contrast in the posterior papillary muscle probably reflects calcification (c) (arrow).(d) Pressure volume relations and dP/dt profiles of wild-type (+/+) and MyBP-C(Neo) mice (t/t).

ease, and further indicate a molecular relatedness ofhypertrophic and dilated cardiomyopathy.
During embryogenesis, appearance of MyBP-C cor-relates with the appearance of cross striations (13),implying a developmental role for this molecule inalignment of thick filaments within the sarcomeres.Because the mutated MyBP-C alleles described hereencode truncated peptide sequences that participate inbinding myosin and the level of foreshortened peptideis markedly less than normal MyBP-C, we were partic-ularly surprised to find near-normal sarcomeres inmutant hearts. Neither the banding patterns nor thelength of sarcomeres were adversely affected by MyBP-C deficits. These data either imply a nonessential rolefor MyBP-C in forming and maintaining sarcomereultrastructure or, alternatively, indicate that other mol-ecules (possibly MyBP-H) can substitute for theseMyBP-C functions. In any event, MyBP-C is notrequired in stoichiometric quantities to produce nor-mal looking sarcomeres.
Transmission electron microscopy of LV tissues fromhomozygous mice bearing mutant MyBP-C allelesshowed that the M-band was often less distinct andnonhomogenous compared with that found in wild-type specimens. M-bands are primarily composed ofthe thick filament–associated components, MyBP-C,M-protein, skelemin, and myomesin. MyBP-C normal-ly forms 7 to 9 stripes in the C-zone of the A-band, oneither side of the M-band (11–13, 34). Whereas thepoorly defined M-bands in LV from homozygousmutant mouse hearts could reflect altered compositionof these MyBP-C stripes, several findings indicate thisabnormal pattern is a consequence of the cardiac dys-function evident in mutant mice. First, equivalent lev-els of mutant peptide were found in both ventricularchambers; however, all M-bands in the RV (and someLV M-bands) of mutant animals appeared normal,implying that hemodynamic forces influence thisbanding pattern. Second, indistinct LV M-bands havealso been observed in mice overexpressing tropomod-ulin, a thin filament component (35). Since both tropo-modulin transgenics and homozygous mutant MyBP-C mice develop dilated cardiomyopathy, we suggestthat M-band loss within the LV is a consequence ratherthan cause of sarcomere dysfunction.
Heterozygous humans bearing cardiac MyBP-C genemutations that encode truncated polypeptides develophypertrophic cardiomyopathy. The mechanism bywhich these cause disease is unclear because neithermutant MyBP-C peptides nor reduced peptide levelswere found in myofibrillar fractions prepared from car-diac tissues of an affected individual (18). Transgenicmice have recently been reported to express mutant car-diac MyBP-C under the α-MHC promoter (36), but lev-els were so robust as to diminish endogenous wild-typeMyBP-C gene expression. Homozygous mice bearingmutant MyBP-C alleles provide evidence that mutatedMyBP-C genes that are regulated by the endogenouspromoter produce mutant peptides that appear to be
incorporated into sarcomeres. That is, 4-fold moremutant peptide was found in myofibrillar extracts thantotal cell extracts of LV tissue (see Figure 2b andResults). Extension of these findings to humans carry-ing analogous cardiac MyBP-C gene defects suggeststhat hypertrophic cardiomyopathy is caused by thedominant negative action of mutant MyBP-C peptideon sarcomere function, although the possibility thatreduction in the amount of MyBP-C has a role in thedisease process has not been excluded.
Mice expressing homozygous sarcomere genedefects may help to address another aspect of humanhypertrophic cardiomyopathy. Analyses of phenotypeand genotype in this disorder have demonstrated theinfluence that genetic etiology has on survival inaffected individuals (for reviews see ref. 8, 9).Although genetically engineered mice that are het-erozygous for a human gene mutation have providedimportant reagents for dissecting the biology of thisdisease, the natural history of disease in these modelshas not been useful in defining mutations that causepremature death in humans. For example, individualswith the myosin mutation Arg403Gln have markedlyshortened life expectancies, whereas mice with thecomparable mutation (designated α-MHC403/+) havenormal survival. In contrast, survival in 3 lines of micethat are homozygous for human hypertrophic muta-tions more precisely mimicked the premature deathobserved in humans with the corresponding muta-tions: survival is markedly reduced in α-MHC403/403
mice but near-normal in homozygous mice bearingmutant MyBP-C alleles. Whereas study of otherhypertrophic cardiomyopathy mutations is needed tovalidate these observations, we suggest that homozy-
The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9 1243
Figure 7Altered gene expression in hearts from MyBP-C(Neo) mice. North-ern blots (a) show increased expression of α-skeletal actin and BNPmRNAs in comparison with GAPDH transcripts. Silver-stained SDS-page gels (b) demonstrate altered expression of cardiac MHC iso-forms in total ventricular extracts from MyBP-C(Neo) mouse hearts(from 8–12-week-old animals; designated t/t) compared with pro-tein from wild-type mice (+/+).

gous murine models may be valuable reagents for pro-filing survival, particularly because relevant clinicalinformation from humans who share a mutation isoften unavailable.
An important question resulting from these studiesis how a 2-fold difference in the amount of mutant sar-comere proteins causes markedly different phenotypes:hypertrophy versus dilation. That is, heterozygous micebearing sarcomere protein gene mutations develophypertrophic cardiomyopathy whereas homozygousmutant mice bearing the same mutations developdilated cardiomyopathy. We hypothesize that parame-ters of sarcomere function, such as force generation, aremonitored and serve as the central signaling mecha-nism that triggers different pathways to remodel theheart. When force produced by an admixture of mutantand normal sarcomere proteins is sufficiently impaired,compensatory myocyte growth occurs, resulting in car-diac hypertrophy. However, when sarcomere functionremains inadequate despite growth (as in α-MHC403/403
and homozygous mutant MyBP-C hearts), other path-ways are activated, as evidenced by altered gene expres-sion (e.g., α-skeletal actin, BNP, and β-MHC), myocytedeath, and fibrosis. If severity of sarcomere dysfunctionis the central signal relating these pathways, then anextension of this model predicts that external forces onthe myocyte, such as hemodynamic load or extracellu-lar matrix remodeling, could exacerbate intrinsic sar-comere dysfunction and tip the balance from compen-sated hypertrophy toward uncompensated failure. Thismechanism could account for progression of hyper-trophic to dilated cardiomyopathy
AcknowledgmentsWe would like to thank Michael Giewat and JohnGabrovsek for technical assistance. This research wassupported by the Howard Hughes Medical Institute.
1. Chien, K.R., Grace, A.A., and Hunter, J.J. 1999. Molecular and cellularbiology of cardiac hypertrophy and failure. In Molecular basis of cardiovas-cular disease. K.R. Chien, editor. W.B. Saunders Co. Philadelphia, PA.251–264.
2. Rodkey, S.M., Ratliff, N.B., and Young, J.B. 1998. Cardiomyopathy andmyocardial failure. In Comprehensive cardiovascular medicine. E.J. Topal, edi-tor. Lippincott-Raven Publishers. Philadelphia, PA. 2589–2620.
3. Watkins, H., et al. 1995. Mutations in the cardiac myosin binding proteinC gene on chromosome 11 cause familial hypertrophic cardiomyopathy.Nat. Genet. 11: 434–437.
4. Bonne, G., et al. 1995. Cardiac myosin binding protein-C gene spliceacceptor site mutation is associated with familial hypertrophic car-diomyopathy. Nat. Genet. 11:438–440.
5. Niimura, H., et al. 1998. Mutations in the gene for cardiac myosin-bind-ing protein C and late-onset familial hypertrophic cardiomyopathy. N.Engl. J. Med. 338:1248–1257.
6. Bonne, G., Carrier, L., Richard, P., Hainque, B., and Schwartz, K. 1998.Familial hypertrophic cardiomyopathy: from mutations of functionaldefects. Circ. Res. 83:580–593.
7. Carrier, L., et al. 1997. Organization and sequence of human cardiacmyosin binding protein C gene (MYBPC3) and identification of muta-tions predicted to produce truncated proteins in familial hypertrophiccardiomyopathy. Circ. Res. 80:427–434.
8. Seidman, C.E., and Seidman, J.G. 1997. Gene defects that cause inherit-ed cardiomyopathy. In Molecular basis of heart disease. K.R. Chien, J.L. Bres-low, J.M. Leiden, R.D. Rosenberg, and C.E. Seidman, editors. W.B. Saun-ders. Philadelphia, PA. pp. 251–263.
9. Seidman, C.E., and Seidman, J.G. 1998. Molecular genetic studies of famil-
ial hypertrophic cardiomyopathy. Basic Res. Cardiol. 93(Suppl.):13–16.10. Gruen, M., and Gautel, M. 1999. Mutations in beta-myosin S2 that cause
familial hypertrophic cardiomyopathy (FHC) abolish the interactionwith the regulatory domain of myosin binding protein-C. J. Mol. Biol.286:933–949.
11. Dennis, J., Shimizu, E.T., Reinach, F.C., and Fischman, D.A. 1984. Local-ization of C-protein isoforms in chicken skeletal muscle: ultrastructur-al detection using monoclonal antibodies. J. Cell Biol. 98:1514–1522.
12. Bennett, P., Craig, R., Starr, R., and Offer, G. 1986. The ultrastructurallocation of C-protein, X-protein and H-protein in rabbit muscle. J. Mus-cle Res. Cell. Motil. 7:550–567.
13. Seiler, S.H., Fischman, D.A., and Leinwand, L.A. 1995. Modulation ofmyosin filament organization by C-protein family members. Mol. Biol.Cell. 7:113–127.
14. Gautel, M., Suffardi, O., Freiburg, A., and Labeit, S. 1995. Phosphoryla-tion switches specific for the cardiac isoform of myosin binding proteinC: a modulator of cardiac contraction? EMBO J. 14:1952–1960.
15. Schlender, K., and Bean, L.J. 1991. Phosphorylation of chick cardiac C-protein by calcium/calmodulin-dependent protein kinase II. J. Biol. Chem.266:2811–2817.
16. Hartzell, H.C. 1985. Effects of phosphorylated and unphosphorylatedC-protein on cardiac actomyosin ATPase. J. Mol. Biol. 186:185–195.
17. Hofmann, P.A., Hartzell, H.C., and Moss, R.L. 1991. Alterations in Ca2+
sensitive tension due to partial extraction of C-protein from rat skinnedcardiac myocytes and rabbit skeletal muscle fibers Ca2+ activation. J. Gen.Physiol. 97:1141–1163.
18. Rottbauer, W., et al. 1997. Novel splice donor site mutations in the car-diac myosin-binding protein-C gene in familial hypertrophic cardiomy-opathy: characterization of cardiac transcript and protein. J. Clin. Invest.100:475–482.
19. Hendrickson, B.A., et al. 1995. Altered hepatic transport of immunoglob-ulin A in mice lacking the J chain. J. Exp. Med. 182:1905–1911.
20. Ho, C., et al. 1995. A mouse model of human familial hypocalciurichypercalcemia and neonatal severe hyperparathyroidism. Nat. Genet.11:389–394.
21. Lakso, M., et al. 1996. Efficient in vivo manipulation of mouse genomicsequences at the zygote stage. Proc. Natl. Acad. Sci. USA. 93:5860–5865.
22. Ogawa, T., et al. 1996. Evidence for load-dependent and load-independ-ent determinants of cardiac natriuretic peptide production. Circulation.93:2059–2067.
23. Jones, W.K., et al. 1996. Ablation of the murine α myosin heavy chainleads to dosage effects and functional deficits in the heart. J. Clin. Invest.98:1906–1917.
24. McConnell, B.K., Moravec, C.S., Morano, I., and Bond, M. 1997. Tro-ponin I phosphorylation in spontaneously hypertensive rat hearts: effectof α-adrenergic stimulation. Am. J. Physiol. 273:H1440–H1451.
25. Fatkin, D., et al. 1999. Neonatal cardiomyopathy in mice homozygousfor the Arg403Gln mutant in the α-cardiac myosin heavy chain gene. J.Clin. Invest. 103:147–153.
26. McConnell B.K., Moravec, C.S., and Bond, M. 1998. Troponin I phos-phorylation and myofilament calcium sensitivity during decompensat-ed cardiac hypertrophy. Am. J. Physiol. 274:H385–H396.
27. Einheber, S., and Fischman, D.A. 1990. Isolation and characterization ofa cDNA clone encoding avian skeletal muscle C-protein: an intracellu-lar member of the immunoglobulin superfamily. Proc. Natl. Acad. Sci.USA. 87:2157–2161.
28. Geisterfer-Lowrance, et al. 1996. A mouse model of familial hypertrophiccardiomyopathy. Science. 272:731–734.
29. Di Francesco, A., et al., 1998. Changes in Mg content and subcellular dis-tribution during retinoic acid induced differentiation of HL60 cells.Arch. Biochem. Biophys. 360:149–157.
30. Aristizabal, O., Christopher, D.A., Foster, F.S., and Turnbull, D.H. 1998.40-MHz echocardiographic scanner for cardiovascular assessment ofmouse embryos. Ultrasound Med. Biol. 24:1407–1417.
31. Georgakopoulos, D., et al. 1999. Dissecting the pathogenesis of familialhypertrophic cardiomyopathy: primary and secondary effects of an α-cardiac myosin heavy chain missense mutation. Nat. Med. 5:327–330.
32. Vikstrom, K.L., Bohlmeyer, T., Factor, S.M., and Leinwand, L.A. 1998.Hypertrophy, pathology, and molecular markers of cardiac pathogene-sis. Circ. Res. 82:773–778.
33. Kubota, T., et al. 1997. Dilated cardiomyopathy in transgenic mice withcardiac-specific overexpression of tumor necrosis factor-alpha. Circ. Res.81:627–635.
34. Schiaffino, S., and Reggiani, C. 1996. Molecular diversity of myofibril-lar proteins: gene regulation and functional significance. Physiol. Rev.76:371–423.
35. Sussman, M.A., et al. 1998. Myofibril degeneration caused by tropo-modulin overexpression leads to dilated cardiomyopathy in juvenilemice. J. Clin. Invest. 101:51–61.
36. Yang, Q., et al. 1998. A mouse model of myosin binding protein C humanfamilial hypertrophic cardiomyopathy. J. Clin. Invest. 102:1292–1300.
1244 The Journal of Clinical Investigation | November 1999 | Volume 104 | Number 9





























