Embed Size (px)
Citation preview

Current Pharmaceutical Design, 2011, 17, 2113-2129 2113
1381-6128/11 $58.00+.00 © 2011 Bentham Science Publishers
Drug-induced Cardiac Mitochondrial Toxicity and Protection: From Doxorubicin to Carvedilol
Gonçalo C. Pereira1, Ana M. Silva1, Cátia V. Diogo1, Filipa S. Carvalho1, Pedro Monteiro2 and Paulo J. Oliveira1,*
1Center for Neuroscience and Cell Biology, Department of Life Sciences, University of Coimbra, Portugal,
2Department of Cardiol-
ogy of the Coimbra University Hospital and Medical School, Coimbra, Portugal
Abstract: Mitochondria have long been involved in several cellular processes beyond its role in energy production. The importance of this organelle for cardiac tissue homeostasis has been greatly investigated and its impairment can lead to cell death and consequent organ failure. Several compounds have been described in the literature as having direct effects on cardiac mitochondria which can provide a mechanistic explanation for their toxicological or pharmacological effects. The present review describes one classic example of drug-induced cardiac mitochondrial toxicity and another case of drug-induced mitochondrial protection. For the former, we present the case of doxorubicin, an anticancer agent whose treatment is associated with a cumulative and dose-dependent cardiomyopathy with a mitochon-drial etiology. Following this, we present the case of carvedilol, a -blocker with intrinsic antioxidant activity, which has been described to protect cardiac mitochondria from oxidative injury. The final part of the review integrates information from the previous chapters, demonstrating how carvedilol can contribute to reduce doxorubicin toxicity on cardiac mitochondria. The two referred examples result in important take-home messages: a) drug-induced cardiac mitochondrial dysfunction is an important contributor for drug-associated organ failure, b) protection of mitochondrial function is involved in the beneficial impact of some clinically-used drugs and c) a more accurate prediction of toxic vs. beneficial effects should be an important component of drug development by the pharmaceutical industry.
Keywords: Cardioprotection, carvedilol, doxorubicin, heart, mitochondria, toxicity.
1. MITOCHONDRIA AS A DRUG TARGET: PROTECTION VS. TOXICITY
Mitochondria are usually considered the power plants of the cells, supplying most of the energy needed. Isolated mitochondrial fractions from different tissues, more frequently from liver, proved to be good in vitro systems to predict drug toxicity before more complex and expensive in vivo experiments [1]. Isolated mitochon-drial fractions were used to obtain toxicological parameters such as IC50 in order to establish a ranking of compound toxicity, however, one obvious disadvantage is the lack of complexity of the biological system. Nevertheless, isolated mitochondrial fractions can be used as a good indicator to predict drug safety, being in fact in use by several major pharmaceutical companies [2, 3].
But why is mitochondrial toxicity screening that important? For example, after extensive research mitochondria began to be linked with several pathologies such as Huntington’s and Parkinson’s disease [4], diabetes [5] and several cardiovascular disorders, in-cluding atherosclerosis, cardiac failure and ischemic heart disease [6] which demonstrates that mitochondrial function is critical for tissue homeostasis. Organs such as cardiac and skeletal muscles and the brain are metabolically more active, implying a higher depend-ence of mitochondrial-produced energy [7], with a consequent in-crease in tissue damage resulting from drug-induced mitochondrial failure.
At a first glance, mitochondria may appear an organelle mostly dedicated to the production of cell energy. In fact, not only mito-chondria constitute a dynamic filamentous network that can un-dergo remodeling by fusion and fission (as opposed to the classic “peanut” representation in the text books), but also mitochondria are much more than simple batteries. Mitochondria play an impor-tant role in the regulation of cell survival, proliferation [8] and death [9], calcium signaling [10], as well as redox homeostasis [11].
*Address correspondence to this author at the Center for Neuroscience and Cell Biology, Largo Marquês de Pombal, 3004-517 Coimbra - Portugal; Tel: +351 239855760; Fax: +351 239853409; E-mail: [email protected]
This initial section will briefly describe the physiology and function of mitochondria in the context of cardiomyocyte homeo-stasis, serving as framework to justify the need to preserve cardiac mitochondrial performance during drug-induced toxicity. Excellent reviews are available focusing in detail alterations of cardiac mito-chondrial energetic during stress [12, 13].
1.1. Mitochondrial Energy Production by Oxidative Phosphory-
lation
Mitochondria is one of the endomembrane systems found in cells, being constituted by two lipid bilayer membranes: an outer membrane containing cholesterol and highly permeable to ions due to the presence of non-specific high conducting channels and an inner membrane, rich in cardiolipin and mostly impermeable, which allows for selective ion and metabolite transport as well as to for-mation of ion gradients. Two other spaces are generally considered in mitochondria, the intermembrane space and the matrix, the gel-like organelle interior moiety [14, 15].
The degree of internal mitochondrial structure varies from tis-sue to tissue, depending on their metabolic states [16]. Even within the same tissue, mitochondrial heterogeneity can occur. One exam-ple occurs in the cardiomyocyte, where two distinct mitochondrial populations exist, one close to the T-tubules and sarcoplasmic re-ticulum and another one closely associated with contractile fila-ments. In adult cardiomyocytes, both populations are largely asso-ciated with cytoskeleton and, therefore, the dynamic properties of the organelle are limited [17]. However, the highly energy demand-ing character of the heart requires a bioenergetic state of the highest level which can only be achieved if refined quality control is veri-fied. In this case, damaged or non-functional mitochondria are re-moved by autophagy and new mitochondria are formed by biogene-sis. Both situations depend on fission/fusion events but the spatial constrains and crystal-like pattern of those mitochondria, which are found packed between the myofibrils, has generated discussion about the possibility of such events to occur [16, 17]. Supporting the notion of mitochondrial fusion and fission in the myocardium is the fact that cardiomyocytes have all the necessary machinery for the two events and, in certain pathological situations very small

2114 Current Pharmaceutical Design, 2011, Vol. 17, No. 20 Pereira et al.
mitochondria (e.g., dilated cardiomyopathy, cardiac rhadbomyoma and ventricular-associated congenital heart diseases) or abnormally large and defective mitochondria (e.g., senescent/aged cardiomyo-cytes) can be found [16, 17]. Studies not only in cardiac-like cell lines (e.g., HL-1 and H9C2) but also in neonatal cardiomyocytes have demonstrated a high degree of motility and morphological changes [17, 18]. However, although this type of cell lines shares some characteristics of differentiated cardiac cells, they lack the well organized and dense packing that is typical of adult cardio-myocytes. Likewise, studies where mRNA or protein levels are quantified in whole heart content should be interpreted cautiously since the organ consists in several types of cells besides cardiomyo-cytes [16]. The ultimate answer would be live imaging of adult cardiomyocytes but this carries some technical difficulties: myo-cytes are difficult to transfect and the spatial resolution of confocal microscopy may not be optimal [16, 17]. Nevertheless, an interest-ing work compared the dynamics of mitochondria between cell lines and adult cardiomyocytes showing, that under normal condi-tions, although fusion and fission do not occur, mitochondrial movements were observed which were attributed to transitions between the orthodox and condensed form and not to the formation of a continuous network [18].
Depending on the tissue, several energy substrates are primarily used to produce adenine nucleotide triphosphate (ATP). In muscle tissue, preferential substrates are fatty acids which are metabolized by -oxidation in mitochondria and peroxisomes [19]. Fatty acid oxidation generates substrates for the mitochondrial electron trans-port chain (ETC). The transference of electrons among several multi-subunit proteins (Complexes I-IV) is driven by the decrease in their redox potential, until reaching oxygen as the final acceptor. Electron transfer is coupled to proton ejection by specific sites of the ETC, generating a proton gradient along the two sides of the inner mitochondrial membrane. The proton gradient, or proton mo-tive force, is comprised by two main components: an electric factor, which is the major component in the energy transduction system, and a pH-dependent element. The proton gradient allows the phos-phorylation of adenine nucleotide diphosphate (ADP) by the en-zyme ATP synthase; ATP is then translocated back to the cytosol via the inner mitochondrial membrane adenine nucleotide transloca-tor (ANT) and the outer membrane voltage-dependent anion chan-nel (VDAC) [11]. The overall process of ATP generation due to substrate oxidation coupled to ADP phosphorylation is called oxi-dative phosphorylation (OXPHOS) [20].
Since mitochondria are responsible for the production of about 90% of the energy used by the cardiomyocyte [7], impairment of mitochondrial function will likely affect normal organ homeostasis. Therefore, it is not surprising that cardiac mitochondria, and spe-cifically OXPHOS, are a preferential target for several clinically used drugs and other xenobiotics including local anesthetics [21, 22], anti-cancer agents [23, 24] and non-reverse transcriptase in-hibitors [25], among others [26].
1.2. Calcium Homeostasis and the Mitochondrial Permeability Transition
Mitochondrial calcium concentration is one of the key factors regulating mitochondrial function and ATP synthesis [27]. Calcium enters mitochondria through a specific non-ATP dependent unipor-ter and exits by exchanging either with Na+ through the Na+/Ca2+ carrier (e.g. heart) or with the Ca+/H+ antiporter (e.g. liver) to create a continuous cycle of calcium across the inner membrane [11]. Matrix calcium accumulation is eletrophoretically driven by the proton gradient generated by the respiratory chain [28].
In excitable cells such as cardiomyocytes, the close proximity between the sarcoplasmic reticulum and the mitochondria allows the latter to act as a cytoplasmic calcium buffer when the ion is released from intracellular stores during contraction, being able to spatially and temporally modulate calcium signaling [29]. Because
such events are intrinsically regulated, disturbances on calcium homeostasis can lead to organ failure, as for example during cardiac ischemia and reperfusion [27].
During stressful events, including those resulting from drug-induced toxicity, excessive mitochondrial calcium accumulation can result in the induction of the mitochondrial permeability transi-tion (MPT). This phenomenon is characterized by a sudden increase in the permeability of the inner mitochondrial membrane. It is ac-cepted that the MPT originates due to the opening of non-selective pores in the inner membrane which allows the free passage of any molecule smaller than 1.5 kDa [30]. When the permeability barrier is disrupted, low molecular weight solutes move freely through the membrane while larger proteins do not, resulting in a colloidal os-motic pressure that leads to mitochondrial swelling [30]. If the MPT pores remain open indefinitely, as usually occurs during pathologi-cal events, mitochondrial structural alterations will cause the expan-sion of the inner membrane and consequent mechanical disruption of the outer membrane, which has a smaller area. Other conse-quences for the mitochondrion during the MPT, include mitochon-drial membrane depolarization and OXPHOS uncoupling, inhibi-tion of respiration due to loss of co-factors and/or cytochrome c and increase in oxidative stress. Also, mitochondrial membrane depo-larization leads to reversal of the ATP synthase activity, which starts hydrolyzing ATP instead of synthesizing it, in an attempt to re-establish the lost transmembrane potential [30].
As described above, mitochondrial dysfunction induced by MPT is a consequence of mitochondrial calcium overload [28], particularly when accompanied by oxidative stress [31], mitochon-drial depolarization, elevated phosphate concentrations and adenine nucleotide depletion [30]. In opposition, inhibition of the MPT pore can be achieved with cyclosporin A (CsA), Mg2+, ADP and anti-oxidants [32, 33]. A number of compounds and deleterious condi-tions increase the sensitivity to cardiac MPT pore opening in vitro as well as in vivo and that peculiarity is often used to explain their deleterious effects in the organ. Examples include the anti-cancer doxorubicin (DOX, [34-36] and below) and ischemia and reperfu-sion [37, 38]. Several strategies to prevent such actions are usually applied in the form of antioxidants, calcium chelators or specific inhibitors of the MPT pore which sometimes yield positive results [27].
1.3. Mitochondrial Oxidative Stress and Role in Cell Death
Mitochondrial respiration is generally identified as the prime source of reactive oxygen species (ROS) [39]. ROS generation by Complex I and III [31] is increased under pathological conditions such as ischemia and reperfusion [39]. Increased mitochondrial oxidative stress results in mitochondrial proteins damage and lipid peroxidation [31] which consequently promotes loss of mitochon-drial functional integrity [39]. Another consequence of increased mitochondrial oxidative stress is damage of the genetic apparatus of the organelle. In fact, accumulation of oxidized mtDNA has been reported in several human pathologies and lead ultimately to a de-crease in mtDNA copy number and progressive loss of proteins involved in energy production [40]. During normal mitochondrial activity, about 1% of oxygen is converted in superoxide radical anion (O2
-) [41] which is further converted into hydrogen peroxide (H2O2) [42], a relatively inert molecule but which is able to cross membranes and react with free metal ions originating a more dan-gerous ROS, the hydroxyl radical (HO ) [43]. Cardiac mitochondria are equipped with a wide range of enzymatic defenses, including manganese-superoxide dismutase (Mn-SOD), glutathione reduc-tase, glutathione peroxidase and peroxiredoxin, among others, which are modulated to answer to alterations in the redox balance [44]. A chronic increase of mitochondrial free radical production in the heart, which is not counteracted by either enzymatic or non-enzymatic defenses, can have further consequences besides oxida-tive damage to mitochondrial proteins, lipids and DNA. Myocardial

Doxorubicin, Carvedilol and Cardiac Mitochondria Current Pharmaceutical Design, 2011, Vol. 17, No. 20 2115
remodeling can also constitute a maladaptive consequence of in-creased ROS generation through activation of specific proteins including metalloproteinases [44, 45].
If mitochondrial ROS generation is too extensive and overload the antioxidant network, one consequence would be the removal of damaged mitochondria by autophagy [46], or a more drastic solu-tion, the triggering of cell death [42]. If the imbalance in oxidative stress is present concomitantly with alterations in cytosolic and mitochondrial calcium, the MPT may occur, with the rupture of the outer membrane leading to the release of several apoptotic initiators usually enclosed in the inter-membrane space [2], e.g., cytochrome c, the apoptosis-inducing factor (AIF) and the second mitochondria-derived activator of caspase/direct inhibitor of apoptosis (IAP)-binding protein with low isoelectric point (SMAC/DIABLO) pro-tein. Alternatively to the MPT pore pathway, pro-apoptotic factor release from mitochondria can also occur through outer membrane permeabilization by channels formed by specific pro-apoptotic proteins including the Bcl-2-associated protein (BAX), Bcl-2 ho-mologous antagonist killer (BAK) and the truncated form of BH3 interacting domain death agonist (tBid) [47]. After its release, cyto-chrome c interacts with several proteins in the cytosol, forming the apoptosome complex and consequently activating the caspases cascade [33], which leads to cleavage of a number of targets and apoptosis. Several other excellent reviews are available concerning the role of mitochondria in cell death in different tissues [9, 48, 49].
Various links between mitochondrial oxidative stress and pro-tein-mediated pro-apoptotic permeabilization of the mitochondrial outer membrane have already been described [50, 51]. Furthermore, oxidative stress can lead to the oxidation of cardiolipin residues, which decreases the affinity of this phospholipid for cytochrome c, contributing for its release to the cytosol after outer mitochondrial permeabilization due to a pro-apoptotic stimuli [52, 53].
As consequence of the post-mitotic nature of cardiomyocytes, loss of these cells by apoptosis may have deleterious effects in the organ. For example, it has been described that apoptosis or necrosis can follow ischemia and reperfusion damage in the heart, which can seriously limit the recovery of the myocardium [54]. Apoptosis in the heart has been detected in different conditions including diabe-tes [55] and heart failure [56].
The above sections describe how the disruption of cardiac mi-tochondrial balance can result in a grave danger to the cell, ulti-mately ending in organ failure. The next two chapters will provide one clear example of drug-induced mitochondrial toxicity (DOX) and drug-induced mitochondrial protection (carvedilol). Ultimately, we will focus on the work available of how one of the compounds can prevent the damage caused by the other. Further reviews on drug-induced cardiac mitochondrial dysfunction can be found in the literature [57].
2. DRUG-INDUCED CARDIAC MITOCHONDRIAL TOXIC-ITY: THE CASE FOR DOXORUBICIN
DOX (Fig. 1), {(7S, 9S)-7-[(2R, 4S, 5S, 6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6, 9, 11-trihydroxy-9-(2-hydroxy-acetyl)-4-methoxy-8, 10-dihydro-7H-tetracene-5, 12-dione}, was discovered in the late 1960s by the pharmaceutical company “Far-mitalia Research Laboratories” and initially named Adriamycin, due to the site of discovery (the Adriatic Sea). DOX is an antibiotic belonging to the anthracycline family and was isolated from a vari-ant culture of the Streptomyces peucetius (Streptomyces peucetius caesius) by aerobic fermentation after mutagenic treatment [58, 59]. The new molecule was chemically characterized and found to pos-sess an anthraquinone moiety connected to a glycoside group [59, 60]. The specific chemical structure is responsible not only for the antineoplastic activity but also for its toxicity, and will be discussed further on in this review.
Initial studies have shown that DOX is able to stop the progres-sion of tumors in rats [61] and its use in clinical trials achieved great success [62, 63]. However, it was later described that patients under DOX treatment developed signs of cardiac toxicity and car-diomyopathy [63, 64], hence a cardio-selective toxicity was soon considered one effect of DOX-chemotherapy. Nevertheless, the clinical use of DOX continued and still nowadays is probably one of the most potent anticancer agents in the clinical practice [65]
2.1. Clinical Use and Side Effects
DOX is commonly used to treat several types of human and non-human tumors, from leukemia and lymphomas to soft-tissue sarcomas and solid tumors, including: breast carcinoma, osteosar-coma, Kaposi’s sarcoma, Hodgkin’s and non-Hodgkin’s lympho-mas [62, 63]. Patients subjected to DOX-chemotherapy usually exhibit symptoms such as nausea, vomiting, alopecia, myelosup-pression, stomatitis and gastrointestinal disturbances [66, 67], which are all clinically manageable side effects. Nevertheless, car-diotoxicity is a completely different story, being probably the most hazardous side-effect associated with DOX treatment, sometimes reaching 50% mortality for the highest cumulative dosages. Acute toxicity accounts for 11% of all cases [68] with symptoms such as myopericardits, sinus tachycardia, reversible arrhythmias, pro-longed QT interval and flattening of the T wave [68], occurring during or right after the last administered dose, and disappearing right after discontinuation of the treatment. Alternatively, chronic DOX-induced cardiotoxicity can be observed within one month or even years later after the treatment ceases [64]. Although the inci-dence of this type of delayed toxicity is much lower (around 1.7%) it is rather dose-dependent, i.e., the probability of developing car-diomyopathy increases with the total cumulative dose [69]. Total dosages between 500-550 mg/m2 have a 5% probability of inducing cardiac heart failure [69] and, therefore, this threshold was estab-lished as the maximum dosage of DOX tolerable in chemotherapy. Naturally, the downside of such approach is a decrease in the per-formance of the drug as an anti-cancer agent. In addition to the cumulative dose, other risk factors can lower the toxicity threshold, including co-treatment with other drugs, mediastinal radiation ther-apy, age, gender, as well as a previous history of cardiovascular problems, hypertension and liver disease [66, 68]. Still, not all pa-tients show signs of cardiomyopathy even when high doses of DOX are applied suggesting intrinsic differences between subjects, at the genetic background. The idea of genetic polymorphisms in DOX-induced toxicity has been previously suggested [70] with other clinical situations using this type of approach to deliver a more selective therapeutically dosage to patients [71]. Although so far there are no experiments performed regarding polymorphism asso-ciated with DOX toxicity, isolated experiments with knock-out or genetically engineered animals give new understandings about the topic of polymorphism or SNPs and its relationship with drug-induced toxicity. In a relatively recent report [72], authors reviewed these aspects and pointed out genes related to drug efflux transport-ers, antioxidant and detoxification enzymes as well as some en-zymes capable of reducing DOX as possible targets for this type of analysis. Nevertheless, the authors mention that further work is needed to better address this important question.
2.2. Monitoring DOX-induced Cardiotoxicity
Presently, there is no effective approach that fully prevents or even treats DOX-associated toxicity and, therefore, the best strategy is the early detection of cardiomyopathy end-points. However, the idea may not be so straightforward because clinical manifestation of DOX-induced toxicity share symptoms with other cardio and non-cardiovascular illnesses and the current methods used to follow heart function lacks sensitivity or specificity [66]. Currently, from all the techniques available [66, 70] only two of them constitute the

2116 Current Pharmaceutical Design, 2011, Vol. 17, No. 20 Pereira et al.
best methodology to follow-up the progression of cardiomyopathy during and/or after DOX treatment: serial echocardiography and endomyocardial biopsy. Although the latter is the most sensitive and specific method it is also the more invasive and less available for the clinician. Echocardiography has its advantages over other methods because is widely available, noninvasive and highly reli-able being the study of ejection fraction to detect signs of early cardiomyopathy one of the main advantages of the technique [66, 68], the sensitivity of which can be increased when associated with some particular protocols (e.g., exercise) or Doppler imaging [73, 74]. Measurement of cardiac enzymes or humoral factors, such as troponin I and T and B-type natriuretic peptide, can be an additional strategy to detect early signs of DOX-induced cardiotoxicity, how-ever these markers represent general cardiac tissue damage and are not specific of DOX-induced damage [75]. Nevertheless, the com-bined use of specific cardiac damage markers together with imaging techniques can increase the sensitivity of the detection and demon-strate that marker levels are directly proportional to cardiomyopa-thy score.
Unfortunately, there is no effective treatment available for pa-tients with established cardiomyopathy even though the mortality rate is incredibly high once the prognosis is revealed [69]. The rea-son for that situation is probably related to the fact that standard therapy for cardiac heart failure fails to relieve the symptoms of DOX-treated patients [70, 76]. The only solution is cardiac trans-plantation but besides its radical character, it has also specific dis-advantages in addition to the drawbacks of general cardiac trans-plantation [76]. First, the immunosuppressive condition after trans-plantation may increase the risk of cancer recurrence. Second, pa-tients qualified as able to receive a new heart must provide evi-dences that are cancer-free for at least a period of 5 years. However, because the incidence of cancer-related death is so high in patients receiving DOX therapy, the majority of them die before the time
frame is complete. Still, the use of ventricular-assist devices may be a temporary alternative for this group of patients until the require-ments for cardiac transplantation are met [76].
2.3. Mechanisms for DOX Antineoplastic Activity
Despite the existing controversy, it is being more widely ac-cepted that DOX-induced cardiotoxicity is completely independent from its anticancer activity. The idea gets further ground if one agrees that cardiomyocytes, as terminal differentiated cells, should not be as sensitive to the primary antineoplastic activity, which is related with cell cycle blockage. Nevertheless, the inhibition of transcription of certain proteins can contribute to the cardiomyopa-thy, although a causal relationship between both still needs to be addressed.
The planar structure of DOX anthraquinone moiety allows the intercalation of the drug into the DNA double strand, which will not only unwind nucleic acids, as it will also cause a stereochemical disorder and consequent block transcription and replication proc-esses [77]. Meanwhile, topoisomerase II is also inhibited and down-stream effects are triggered due to the formation of DNA double-strand breaks. All these effects result in cell cycle arrest that culmi-nates in the activation of the apoptosis machinery leading to the death of cancer cells and tumor growth arrest. Obviously, one could not ignore the hypothesis that DNA interference may also occur in cardiomyocytes. However, other DNA intercalating compounds such as cisplatin or actinomycin D do not cause cardiotoxicity [78] and therefore the mechanism described is not likely responsible for degeneration of cardiac function.
2.4. Mitochondria and DOX-induced Cardiotoxicity
The cardiac tissue has a high energy demand and consequently any interference in the energy production machinery will clearly affect the whole physiology and heart contraction. Cardiomyocytes
Fig. (1). Chemical structure of DOX and carvedilol. Panel A represents the anticancer agent DOX with its planar anthraquinone tetracyclic ring attached to the deoxy amino sugar daunosamine. DOX results from a 14-hydroxylation of daunorubicin, here represented by the triangle. In I, the molecular part of DOX responsible for the redox cycle is shown, whereas in II, the portion of the molecule that allows the formation of DOX-iron complexes (see Fig. 2 for further details) is highlighted. Panel B represents the cardioprotective and -blocker compound carvedilol, also emphasizing the different parts of the molecule that accounts for its pharmacological properties. The asterisk denotes the hydroxylated carbon in the carbazole group of BM-910228, a carvedilol metabolite. The arrow represents the protonation/deprotonation moiety of carvedilol (see text for details). Both structures were withdrawn from the public website PubChem (www.pubchem.ncbi.nlm.nih.gov/). Panel B is an adaptation from [192].

Doxorubicin, Carvedilol and Cardiac Mitochondria Current Pharmaceutical Design, 2011, Vol. 17, No. 20 2117
depend approximately 90% on ATP production by the mitochon-drial OXPHOS, thus mitochondria appear as one attractive target for DOX toxicity. In fact, it is widely recognized that DOX-induced cardiotoxicity has a strong mitochondrial component [79, 80]. Mi-tochondria from DOX-treated animals show traces of the drug [81], which could be due to its affinity for particular biomolecules within the organelle. As with nuclear DNA, DOX also forms adducts with mitochondrial DNA (mtDNA) [82], while complexes between DOX and cardiolipin, one of the most abundant phospholipid in the inner mitochondrial membrane, have also been described [83]. Once accumulated in the mitochondrion, DOX can initiate its direct and deleterious effects which include: a) stimulation of ROS pro-duction, b) induction of MTP and c) inhibition of OXPHOS.
As pointed out earlier, the chemical structure of DOX, namely the anthraquinone group, also accounts for the toxic response of the drug. The quinone at the central ring (Fig. 1) is prone to reduction with a redox potential of approximately -320 mV, similar to re-duced nicotinamide adenine dinucleotide (NADH) [84]. Therefore, DOX can divert electrons from Complex I as well as from several other cellular dehydrogenases, namely, NADH transdehydrogenase, xanthine oxidase and cytochrome P450, being converted into a semiquinone radical along the process (Fig. 2) [84]. Afterwards, in the presence of molecular oxygen, DOX univalent reduced form, can donate its unpaired electron resulting in the formation of O2
-, regenerating the parent drug in the process [85]. The fact that in vitro studies with Complex I-specific inhibitor rotenone exacer-bated ROS formation suggests that the site of DOX activation in the enzyme is upstream to the binding site of the inhibitor [86]. Since, theoretically, no net consumption of DOX occurs, the redox-cycle can take place endlessly.
In addition to the possible action of iron as a catalyst for production of HO through Fenton reaction with DOX semiquinone participat-ing by recycling ferric anion, the metal ion may also establish com-plexes with DOX (Fig. 2), stimulating the production of H2O2 [82, 87]. However, significant levels of free iron are unlikely to be ob-served in cells during physiological conditions [82, 88] and there-fore, it is possible that DOX may induce first a dysregulation of iron homeostasis. Aconitase is known to be very sensitive to varia-tions in oxidative stress and it is possible that when damaged, iron from aconitase iron-sulfur clusters can be displaced thus increasing free iron in mitochondria. The most important aspect is the fact that this enzyme has a double role, since in its iron-free form, it can act as an iron-regulating protein, facilitating iron uptake over iron se-questration hence increasing the pool of free iron available to react with DOX [82, 87].
In the context of reactive radical species, nitric oxide (NO) plays a particular important role in the cardiovascular system, being responsible for the increase in contraction, myocardial relaxation and diastolic function, Frank-Starling response, heart rate, -adrenergic response, myocardial energetics and substrate metabo-lism [89, 90]. For this basal action to occur, cardiomyocytes and endothelial cells constitutively express two different isoforms, neu-ronal NO synthase (nNOS) and endothelial NO synthase (eNOS). Still, a third isoform with higher output, the inducible NO synthase (iNOS), whose abundance increases upon several stimuli, was pro-posed to be linked to different cardiac pathologies [91]. During DOX administration, transcription of eNOS in endothelial cells and iNOS in cardiomyocytes was shown to be increased, resulting in unbalanced amounts of NO and consequent deleterious effects [68, 90]. Moreover, DOX can be directly metabolized by all NOS iso-forms similarly to the mentioned reductases, uncoupling NO pro-duction and increasing O2
- [68, 90]. In fact, the turnover of DOX cycle for iNOS is ~10 fold higher than for nNOS and eNOS, which alone have a Km about ~100 fold lower than NADH-dehydrogenase [68, 90]. During the deregulated production of NO and O2
- both molecules can react decreasing their cellular amount. Although this may seem like a protective mechanism, in fact it leads to the pro-
duction of peroxynitrite (ONOO-), a much more reactive molecule when compared to NO which is relatively inert [90]. The impor-tance of NO in the cardiac toxicity induced by DOX was confirmed when in vivo studies performed by inhibiting iNOS, or by using ONOO- scavengers, delayed or prevented DOX-induced cardiomy-opathy [90, 92]. Nevertheless, some contradictory information is reported in the literature regarding the impact of NO production inhibition during DOX treatment [89, 93]. One view suggested that the protection afforded by NO was mediated by O2
- since both radical species can react together, which synergizes with superoxide dismutase (SOD) in the removal of O2
- [89]. In fact, overexpres-sion of Mn-SOD in NO null mice prevented the deleterious effects of DOX [89]. A second study demonstrated that iNOS overexpres-sion or delivered as a bolus increases the toxicity of DOX on hu-man breast cancer cells in culture but not on H9c2 cardiomyoblasts, suggesting that NO can act to prevent DOX toxicity in cardiac cells but can act to improve the clinical efficacy of DOX as an anti-tumor agent in cancer cells [93].
DOX-increased oxidative stress mediated by oxygen and nitro-gen species results in damage to several biomolecules, including DNA, lipids and proteins. Biomolecule oxidation can directly affect mitochondrial function, or alternatively lead to secondary re-sponses. For instance, oxidation of mtDNA can result in a defective respiratory chain, which will not only be unable to respond to high energy demands but will also increase electron leak to molecular oxygen, increasing ROS production [94]. ROS can directly inhibit the respiratory complexes of OXPHOS, e.g., cytochrome c oxidase (COX) [95], but alternatively the oxidation of certain residues or functional groups can induce conformational changes inhibiting protein function. One particular example is thiol groups, which are very vulnerable to oxidation. Several mitochondrial proteins show oxidation of this group after DOX treatment which forms disulfide bonds upon oxidation [94, 96]. One of such proteins is the ANT, in which some authors suggest that oxidation of vicinal cysteines al-lows the formation of dimers which are crucial for the formation and opening of MPT pores [97]. In fact, mitochondria treated in vitro with DOX show increased susceptibility to the MPT [35, 36] when treated with inhibitors and modulators of the MPT pore, e.g., ATP, dithiothreitol (DTT), butylated hydroxytoluene (BHT) and CsA, this phenomenon was prevented. The same authors showed that secondary metabolites, such as DOX aglycones, are even more powerful in the induction of the MPT [36]. The in vitro MPT-inducing effects may actually be due to increased oxidative stress, oxidation of pyridine nucleotides, depletion of high energy adenine nucleotides and decrease in membrane potential, while increased sensitivity to MPT observed in cardiac mitochondria from DOX-treated animals [34] is probably due to the synergistic action of such effects with calcium homeostasis dysregulation. In fact, DOX has been described to interfere with the flux of calcium from the sarcoplasmic reticulum by increasing the opening state probability of calcium release channels [98]. Additionally, DOX also interferes with the Na+/Ca2+ and Na+/K+ pumps which can contribute for en-hanced cytoplasmic calcium accumulation and, therefore contribut-ing to augment mitochondrial calcium accumulation [98]. Similarly to in vitro experiments, cardiac mitochondria from DOX-treated animals have a lower capacity to accumulate and retain calcium [34, 79, 96, 99-101]. The effect is more pronounced if the cumula-tive dose of DOX is increased and persists for long periods after the end of the treatment [34]. Therefore, the loss of mitochondrial cal-cium loading capacity can be classified as a sensitive marker for DOX-induced cardiotoxicity [102]. The co-administration of CsA in vivo was capable to prevent DOX-induced cardiomyopathy dem-onstrating the importance of calcium homeostasis and induction of MPT in the toxicity mechanism of the drug. Nonetheless, caution should be taken when inferring the results, since FK-506 also inhib-ited DOX cardiotoxicity and this compound is not a MPT inhibitor, being in fact an immunosuppressant similarly as CsA [101]. An improved knowledge on this issue could be accomplished if specific
2118 Current Pharmaceutical Design, 2011, Vol. 17, No. 20 Pereira et al.
MPT inhibitors were used instead [103, 104]. Another intriguing aspect is the ex vivo use of CsA on cardiac mitochondria from DOX-treated animals resulted in restoration of respiration to control values, which suggests that the MPT pore or cyclophilin D, the target of CsA in mitochondria, are involved in DOX-induced inhi-bition of mitochondrial respiration [96]. The authors suggest that binding of CsA to cyclophilin D may cause a disassembling of pre-formed pores allowing crucial components such as the ANT to become available again to participate in OXPHOS. Although there is currently some controversy about the structural characteristic of the MPT pore [105], it is likely that the ANT may have at least a regulatory effect. Agreeing with the possible role of the ANT on
DOX-induced toxicity, a decrease in functional ANT was detected after DOX treatment, suggesting a genetic or biochemical interfer-ence of DOX with this protein which results in a reduction of bio-energetic capacity and decrease mitochondrial calcium loading capacity [100]. Nevertheless, an evident direct interaction between drug-enzyme is still to be described in the literature for almost all OXPHOS enzymes, as pointed before [83], and the described in-hibitory effects are due to increased oxidative stress or to the dis-placement of lipid-enzyme environment, as is in the case of DOX-induced inhibition of COX [83].
A decrease on state 3 respiration also seems to be another mito-chondrial marker of DOX-induced cardiomyopathy, as it was al-
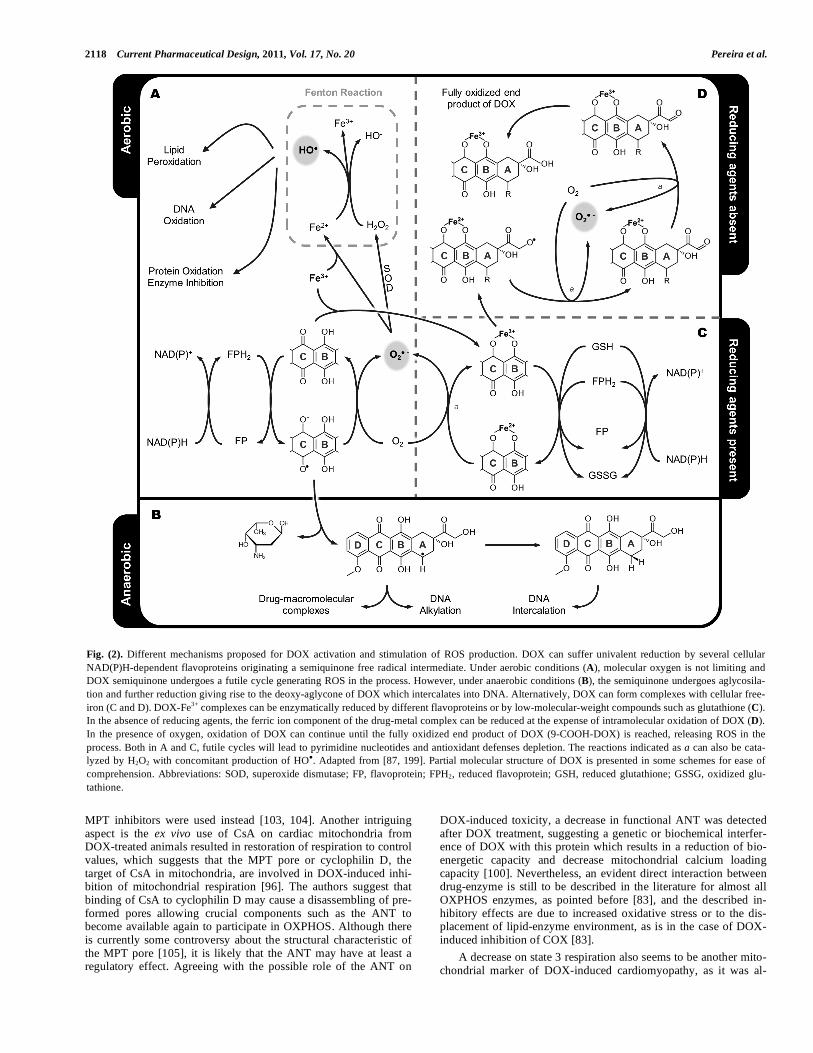
Fig. (2). Different mechanisms proposed for DOX activation and stimulation of ROS production. DOX can suffer univalent reduction by several cellular NAD(P)H-dependent flavoproteins originating a semiquinone free radical intermediate. Under aerobic conditions (A), molecular oxygen is not limiting and DOX semiquinone undergoes a futile cycle generating ROS in the process. However, under anaerobic conditions (B), the semiquinone undergoes aglycosila-tion and further reduction giving rise to the deoxy-aglycone of DOX which intercalates into DNA. Alternatively, DOX can form complexes with cellular free-iron (C and D). DOX-Fe3+ complexes can be enzymatically reduced by different flavoproteins or by low-molecular-weight compounds such as glutathione (C). In the absence of reducing agents, the ferric ion component of the drug-metal complex can be reduced at the expense of intramolecular oxidation of DOX (D). In the presence of oxygen, oxidation of DOX can continue until the fully oxidized end product of DOX (9-COOH-DOX) is reached, releasing ROS in the process. Both in A and C, futile cycles will lead to pyrimidine nucleotides and antioxidant defenses depletion. The reactions indicated as a can also be cata-lyzed by H2O2 with concomitant production of HO . Adapted from [87, 199]. Partial molecular structure of DOX is presented in some schemes for ease of comprehension. Abbreviations: SOD, superoxide dismutase; FP, flavoprotein; FPH2, reduced flavoprotein; GSH, reduced glutathione; GSSG, oxidized glu-tathione.

Doxorubicin, Carvedilol and Cardiac Mitochondria Current Pharmaceutical Design, 2011, Vol. 17, No. 20 2119
ready been described in several models of chronic exposure to the drug. The same effect was observed in vitro together with a slight stimulation of state 4 [106], usually attributed to the diversion of electrons from the respiratory chain to DOX during its redox cycle with concomitant increase of oxidation. However, is the ex vivo effect of DTT on mitochondrial respiration from DOX-treated rats that allows to draw major conclusions since it reverses respiratory state 3 inhibition, supporting the idea that DOX oxidation of thiol groups causes several of the mitochondrial alterations observed [96]. Nevertheless, as has been already pointed out [79], one must have some moderation in mind when analysis and conclusions are inferred from two distinct models of toxicity. In vitro effects are mainly due to the direct interaction of the drug with mitochondria and its membranes and may result from direct production of ROS by drug redox cycling. The changes in bioenergetics observed after the in vivo treatment may reflect alterations that are persistent, even after the drug is washed out from the system (memory effect). This may also result, among other possibilities, from genetic alterations.
Exposure to DOX has been described to inhibit several OX-PHOS enzymes, including NADH dehydrogenase, Rieske iron-sulfur protein, succinate dehydrogenase, COX, creatine kinase (CK), carnitine palmitoyl tranferase, fatty acid -oxidation-related enzymes as well as the translocation of phosphate and pyruvate to the mitochondrial matrix ([79] and references therein). A proteomic profile of sub-chronic anthracycline-induced cardiotoxicity was recently performed [107]. In order to perform the experiments, authors used the well establish rabbit model [108] and chose the parent compound of DOX, daunorubicin, to induce cardiac toxicity. Although interpretations should to be taken cautiously, since the degree of toxicity between the two compounds is slightly different [109], we can infer that the most drastic changes found in the study can be shared between both drugs. The authors showed that most of the observed alterations were in proteins related with the cytoskele-ton, OXPHOS and antioxidant detoxifying system [107], all of which are being covered in this review and which are considered typical markers of DOX-toxicity. One limitation of the work is related with the end-stage of the experiment and, therefore, one cannot discern cause from consequence. Nevertheless, daunorubicin caused a tremendous specific increase of desmin in cardiomyocytes which was suggested to act as rescue event for the loss of myofibril stability and for reposition of energy production and consumption sites since proteins related to energy production and channeling (see below) were decreased. Interestingly, desmin abundance was di-rectly proportional to the extension of DOX-induced cardiac dys-function and increased levels of this protein were detected even in animals without congestive heart failure and thus might be used as another early marker of DOX-toxicity progression. An additional aspect is the dichotomy between cytoplasmic and mitochondrial abundance of SOD, which is increased in the former and decreased in the latter, suggesting the possible compartmentalization of oxida-tive stress, as suggested by the authors. Supporting this study, an-other previous work investigated the variation of transcripts in the heart over time after a single treatment with DOX [110].
Contrarily to the increased expression and activity of COX observed in some works, the expression of at least some of its subunits as well as its activity appear diminished in other reports [95], which makes this respiratory complex another possible target during DOX toxicity. Interestingly, an analysis of different meta-bolic pathways (including OXPHOS) through the use of microar-rays and quantitative PCR showed that despite the variety of altered enzymes, the most affected biochemical networks are fatty acids -oxidation and glycolysis [111]. This result suggests alterations on substrate utilization, an event that is usually associated with myo-cardial remodeling and the development of cardiac pathologies. In fact, decrease in cardiac fatty acid utilization was already reported in DOX-treated animals, occurring before any functional change [112]. More recently, 13C-isotopomer analysis of isolated perfused
hearts from DOX-treated rats showed a metabolic shift from fatty acid oxidation towards glycolysis [113]. In this particular study, the use of the cardioprotector dexrazoxane proved to be capable of reversing the observed shift, shedding some light once again in the role of the imbalance of oxidative stress. Nevertheless, it remains to be elucidated if it is the impairment of the mitochondrial function that results in the metabolic remodeling. If indeed is this the case, the inability of the cardiac tissue to sustain energy production by glycolysis may be due to the fact that DOX treatment results in alterations of glucose intracellular transport and initial metabolism [114]. Following proteomic studies, treatment with anthracyclines induces a decrease in the fatty acid binding protein responsible for the import of such molecule fuels into cardiomyocytes [107]. Such decrease, in addition to the increased abundance of the rate limiting enzyme pyruvate dehydrogenase [107] might explain the metabolic shift observed during DOX-treatment. In addition, the same study attempts to link heart failure to metabolic remodeling since it was also found that anthracyclines decrease the triosephosphate isomerase enzyme important in the glucose metabolism and which was previous found to be sensitive to oxidative stress and associ-ated with cardiomyocytes and mitochondrial degeneration [107].
Another problematic issue identified in cardiomyocytes after exposure to DOX is the interference with the channeling of ATP and phosphocreatine (PCr) from mitochondria to the cytosol, i.e., the energy shuttle between production and consumption sites. The easily diffusible creatine (Cr) and its phosphorylated form PCr act like global or local buffers of ATP/ADP ratios, and are extremely important in heart physiology [115]. The term “channeling” is gen-erally used when the CK is associated with ATP-providing or -consuming transporters, pumps or enzymes. In the heart, different CK isoenzymes can be found free in the cytosol or bound to sarco-plasmic or mitochondrial membranes. The mitochondrial isoform (mtCK) has high affinity for cardiolipin forming complexes with it and establishing a strict environment around the ANT, allowing not only the regeneration of intramitochondrial ADP but also the gen-eration of the diffusible energy molecule PCr from ATP and Cr [115]. As it was mentioned before, DOX competes for cardiolipin binding sites and therefore, mtCK is a very likely target. In fact, in vitro assays showed that the drug was not only able to displace the complexes between the enzyme and phospholipids, but it was also capable to cause a direct oxidation of mtCK, namely in its cysteine residues [114]. In vivo, DOX also decreases the activity of mtCK and induces a shift of the expression of the isoenzymes towards an embryonic profile, an event that was already observed in several other cardiac pathologies [116]. All these effects are likely to be responsible for the decrease in the cellular PCr pool as assessed by nuclear magnetic resonance spectroscopy [117].
All adverse effects here stated, can drive the cardiomyocyte to undergo signaling resulting into cell death; being post-mitotic cells, cardiomyocytes cannot be regenerated leading to cardiac failure. There are several apoptotic markers present in individuals after DOX treatment which are covered elsewhere [118]. Several studies indicate that the transcription factor p53 is also involved in DOX cardiotoxicity. Targeted disruption of p53, either in knockout mice [119] or by using pifthrin- , a specific p53 inhibitor on cardio-myoblasts in culture [120, 121] prevents DOX cardiotoxicity. Mito-chondrial dysfunction has been attributed to induction of BAX ex-pression and mitochondrial translocation following initial nuclear DOX accumulation and DNA damage in cardiomyoblasts [121, 122]. Furthermore, DOX induces mitochondrial p53 translocation [121] but it is however unclear whether this translocation is in-volved in the apoptotic process or rather is part of a defense mecha-nism [123]. DOX-induced cell death also results in depletion of cardiac stem cell pool which is critical for cardioprotection and repair [124].
Lastly, the importance of autophagy in mitochondrial quality control in the heart should also been taken into account due to the

2120 Current Pharmaceutical Design, 2011, Vol. 17, No. 20 Pereira et al.
intensive work accomplished in this area in the last decades [46, 125-131]. Autophagy is the process by which the cell seeks to recy-cle damaged macromolecules or non-functional organelles and therefore promote cell survival. This is particular important in the cardiac tissue due to its high energy and terminal differentiated state. Nevertheless, autophagy is a two-edged sword because when disproportionate, it can lead to cell death [125], being in fact aug-mented under certain pathological conditions; however, the precise event responsible for that dichotomy remains to be elucidated. Autophagy might be triggered by starvation, i.e. energy depletion, ROS, accumulation of misfolded/oxidized proteins, mitochondrial depolarization and MPT induction [127-130], all of which are pre-sent after DOX treatment. Therefore, it is reasonable to consider that the treatment would activate the autophagic pathway in a pro-tective manner to remove damaged mitochondria but interestingly the few works performed so far show the opposite [132, 133]. Al-though at this time is difficult to discriminate if autophagy is di-rectly activated by DOX or it is induced as a consequence of mito-chondrial dysfunction it arises as another putative pathway for car-diomyocyte death, (in)dependent of apoptosis or necrosis. Kobaya-shi and co-workers stressed out the role of the heart-specific tran-scription factor GATA4 in the progression of DOX-toxicity and found that its abundance is inversely proportional to the autophagic flux, which is modulated by the expression of Bcl-2 related genes [132]. However, further investigations should be carried out to prove that the direct or indirect inhibition of the autophagic path-way is actually protective during DOX treatment.
2.5. DOX Toxicity in Other Organs – Why is the Heart More Affected?
Despite all the knowledge acquired over the years, one impor-tant doubt remains about DOX toxicity - why is the heart the most affected organ? Obviously, several authors have tried to explain the observed organ selectivity, however, most ideas still remain as sim-ple hypothesis with little practical support. Different drug accumu-lation in tissues was previously proposed [134]. Apparently, during its systemic circulation, DOX accumulates slowly in the tissues being the liver the organ showing the highest accumulation, perhaps because biliary secretion constitutes the major route of DOX elimi-nation [135]. However, after several DOX administrations, its lev-els in the cardiac tissue can increase up to 6-fold without changes in the rate of accumulation in other tissues [134]. It could be a specific cellular accumulation or differences in drug efflux as it was pointed earlier in this section. Another possible explanation is that the car-diac tissue apparently shows decreased levels of antioxidant de-fenses when compared with other organs, e.g. the liver [136]. This would make the heart less capable of dealing with the increased oxidative stress induced by DOX. One author also suggested the existence of a cardio-selective external NADH dehydrogenase, alternative to OXPHOS Complex I and similar to those found in plants [86]. The presence of this enzyme in the outer leaflet of the inner mitochondrial membrane would exacerbate the DOX redox cycle. Also, the fact that myocardial tissue possesses a higher mito-chondrial content [137] due to higher dependence on mitochondrial ATP could also contribute to enhanced DOX activation. Finally, one could point to the fact that different tissues possess different energy demands and therefore different dependence on mitochon-drial bioenergetics. In addition, some tissues can present higher reserves of certain important proteins for mitochondrial function, being consequently less sensitive to damage or decline in expres-sion of those same proteins or respiratory complex [138]. This idea is usually presented to explain the severity of certain mitochondrial hereditary disorders in specific tissues but it might also be applied to DOX toxicity.
Despite the fact that the heart is particularly affected by DOX, other organs do present signs of DOX toxicity as well. As has pre-viously reviewed [67], DOX also affects the liver, kidney and brain although it must be stressed that DOX itself cannot cross the blood-
brain barrier. Therefore, DOX-induced effects in the brain are probably indirect and mediated through cytokines, mainly tumor necrosis factor-alpha (TNF- ). The role of the cytokine on brain injury gains further ground when treatment is accompanied by an anti-TNF- , decreasing the high levels of the circulating cytokine usually observed in patients and preventing brain damage [67]. In the brain, DOX indirectly induces an increase in oxidative stress seen as increased levels of malondialdehyde, thiobarbituric acid reactive substances (TBARS) and protein carbonyl groups [67]. Once more, the mechanism underlying DOX-induced toxicity ap-pears to be related to the stimulation of ROS production. Mitochon-dria fractions isolated from this tissue also present increased sensi-tivity to the MPT, a phenomenon clearly modulated by the redox balance of the organelle, supporting the previous idea [139].
During DOX treatment, liver tissue accumulates high amounts of DOX and its metabolites, since it is one of the principal organs responsible for detoxification in the organism. The liver is also responsible for drug clearance since bile is the major route for drug elimination. Certainly is not a surprise that about 40% of patients present signs of liver failure after treatment [67]. Besides inducing changes in the expression of antioxidant enzymes and decreases the energy charge of hepatocytes, DOX also increases the basal produc-tion of ROS since one can find several other NADPH-oxireductases in the tissue [67]. As it was observed in the heart, mtDNA from hepatic mitochondria is oxidized after DOX treatment although in a reduced manner when compared to cardiac mitochondria [140]. Other effects on mitochondrial function include a slight decrease in the respiratory control ratio (RCR) and decrease calcium loading capacity which contributes to the observed mitochondrial vacuoli-zation and swelling pattern in histology [67, 141].
Recently, our lab has found that sub-chronic treatment with DOX does not induce alterations in the levels of apoptotic and mi-tochondrial membrane permeabilization proteins in the lung but exhibit markers of lipid peroxidation, however if it is a direct effect of the drug or rather an indirect effect as observed in the brain tis-sue remains to be elucidated [142]. Nephrotoxicity sometimes ac-companies the treatment and its cause is probably related to apopto-sis of glomerular podocytes [67]. If the extent of injury is mild to high, the kidneys filtration process is likely to be affected and pro-teinuria can be observed. One of the mechanisms proposed for renal toxicity is the formation of iron-DOX complexes, i.e., increased ROS production which will cause mtDNA damage and the produc-tion of a defective respiratory chain [67].
2.6. Prevention of DOX-Induced Cardiotoxicity
As the cure for DOX-induced cardiac heart failure is not pres-ently available, prevention remains the best therapeutic. The idea is not only to counteract DOX-induced toxicity but to increase the therapeutic efficacy of the drug either by allowing the use of higher dosage (>500 mg/m2) or to decrease the toxic response without interfering with the antineoplastic activity. However, the benefits from all preventive approaches currently available are limited. Nev-ertheless, a preventive approach can be achieved either by pharma-cologically or non- pharmacologically means. One obvious ap-proach is achieved by limiting the cumulative dose of DOX to less than 450 mg/m2 or by decreasing the drug peak plasma results in a reduction of the toxicity. A precise schedule for drug administration and/or choosing a continuous bolus infusion over a single infusion has proven to attain less damage to the heart and therefore decrease the probability of heart failure since both strategies can decrease drug concentration in the plasma [82, 98].
Alternatively, new anthracyclines analogs that have the same or better therapeutically effect but lack the toxic activity has been proposed. The rationale behind this idea goes back to late 1960s, since DOX is derived from its parent compound, daunorubicin, by the substitution of a hydrogen atom by a hydroxyl group, increasing the therapeutic efficacy (Fig. 1) [59]. Due to the tetracyclic ring and

Doxorubicin, Carvedilol and Cardiac Mitochondria Current Pharmaceutical Design, 2011, Vol. 17, No. 20 2121
amino-sugar moiety, several modifications can be made. However, from the almost 2000 analogues investigated in the last 4 decades, only a few have reached clinical approval [82, 98].
Use of liposome-encapsulated or tumor-targeted delivery of DOX is probably one of the most investigated strategies in the past decade [143]. The fact that vasculature around tumors is more per-meable, i. e., higher fenestration, allows for higher accumulation of liposomes at the tumor site, therefore decreasing systemic drug concentration and improving the targeting of the treatment. Lipos-somal formulations of DOX have proven improved efficacy over free DOX and are currently used in the clinic [70, 82, 98].
If no plausible replacement is obtained for DOX, one sounding strategy is to spare the non-target organ by co-administrating DOX with compounds capable of prevent side effects, e.g. by preventing DOX-induced cardiomyopathy. Along the years, strategies used included antioxidants, iron chelators, lipid-lowering drugs and -blockers. Several antioxidants either naturally occurring or syn-thetic have been co-administered with DOX [144, 145]. Whereas some in vitro studies demonstrated positive effects caused by direct antioxidant activity or indirectly, by stimulating the production of natural antioxidant defenses, in vivo results were disappointing [146]. The lack of positive outcome might be due to the poor bioavailability of the protective compounds after oral administra-tion or to a limited availability in the critical sites where DOX ex-erts its toxic effects, one example being mitochondria. Related to this latter point, one solution, suggested previously [98], would be the specific mitochondrial targeting of such antioxidants. Currently, two similar approaches are proposed to achieve such objective: linking antioxidants to the lipophilic cationic ion tetraphenilphos-phonium, e.g., coenzyme Q10 forming the molecule MitoQ [147] or plastoquinone in the case of Skulachev ions [148]. Due to the posi-tively charge nature of these compounds and higher negative-inside membrane potential of mitochondria, the compounds can accumu-late up to 200-fold in the matrix. Besides, the natural character of such molecules enables the recycling of the molecule by OXPHOS after scavenging the radical species and, theoretically, the process can continue indefinitely. Regarding DOX-induced oxidative stress, MitoQ was recently reported to confer protection against DOX toxicity [95]. This cationic molecule was capable to prevent altera-tions on the echocardiographic profile and the extension of fibrosis in cardiac tissue. Moreover, MitoQ restored the expression of COX subunits and therefore the activity of the enzyme [95], although no further end-points of mitochondrial dysfunction were investigated.
Dexrazoxane is currently the best therapeutic approach of choice, reaching significant efficacy in counteracting DOX-asso-ciated toxicity [82]. In fact, dexrazoxane is the only approved drug used in co-administration with DOX with the ultimate aim of pre-venting DOX-induced cardiac damage. The mechanism of action involves its ability to chelate free iron and to displace iron from DOX-iron complexes. Once accumulated into cardiomycocytes, dexrazoxane is hydrolyzed to form an open-ring which has strong chelating characteristics. However, some caution should be taken when using this compound since it can also deplete iron from intra-cellular stores and interfere with iron transport [149]. Moreover, it possesses myelosuppressive action which can aggravate the same suppressive action of DOX [150].
Another approach would be to decrease the rate of cardiac con-traction. The rationale behind this strategy is that by decreasing the energy demand of a myocardium with impaired mitochondrial ca-pacity, one can maintain heart function with a limited mitochondrial supply. -blockers are commonly used to decrease hypertension and can be used to control cardiomyopathies although some caution should be taken when interpreting results or long treatment periods [151]. Carvedilol is a -adrenergic receptor antagonist, which has demonstrated efficacy in preventing DOX-induced cardiac toxicity [102]. It has been described that the protective effects of carvedilol against DOX-induced cardiotoxicity go beyond hemodynamic
maintenance and directly involve maintenance of mitochondrial function, as we will see briefly.
Finally, reports exist that exercise can prevent DOX-induced cardiac damage [152-154] and specifically protect cardiac mito-chondria [99, 155] from the deleterious effects of that anti-neo-plastic agent. This subject is explored elsewhere [156].
3. DRUG-INDUCED MITOCHONDRIAL PROTECTION -
CARVEDILOL
Carvedilol (Fig. 1), {1-(9H-carbazol-4-yloxy)-3-[2-(2-methoxy-phenoxy)ethylamino]propan-2-ol} is a non-selective -blocker with both 1-, 2- and 1-adrenoreceptor blocking properties [157] which was initially used in hypertension patients [158]. In addition to vasodilator effects [159] which are probably due to its -blockade activity, carvedilol also presents antioxidant properties, including the ability to inhibit lipid peroxidation in myocardial cells and the preservation of antioxidant systems [160]. Carvedilol is currently used as an anti-hypertensive drug [157] to manage several cardiac pathologies, e.g. congestive heart failure or angina pectoris [161]. More recently, it has been shown in models of myocardial ische-mia-reperfusion that carvedilol can reduce infarct size and the num-ber of apoptotic myocytes in the ischemic area [160] demonstrating the cardioprotective action of the compound in myocardial infarc-tion.
Cardiac dysfunction is often correlated with changes in mito-chondrial bioenergetics [162] and since mitochondria has a critical role in myocyte biology several studies about the interaction carve-dilol-mitochondria were already performed [39] in order to explain why carvedilol presented a higher protective impact when com-pared with other similar compounds. Similar -blockers have the ability to decrease cardiac rhythm or to decrease systemic blood pressure contributing to improvements in after load and offering cardiac protection by the Frank-Starling law theory, nevertheless carvedilol always presented higher protective impact [102, 163, 164]. One rationale to explain the discrepancy lies in the fact that carvedilol is a very lipophilic compound with a high partition coef-ficient and therefore, can easily cross cell membranes reaching much higher concentration in cytoplasm and even in mitochondria, exerting intracellular responses unrelated with its hemodynamic effects [164].
3.1. Antioxidant Protection of Cardiac Mitochondrial Function by Carvedilol
In several in vitro systems, carvedilol was demonstrated to have intrinsic antioxidant activity. Several mechanisms have been pro-posed for carvedilol antioxidant activity. Yue et al. [164] demon-strated that carvedilol was able to inhibit iron-mediated lipid per-oxidation and -tocopherol depletion for concentrations much lower than other -adrenergic antagonists counterparts. The authors proposed a direct free radical scavenging mechanism that was mainly resident in the carbazole moiety and potentiated by the in-troduction of hydroxyl groups in specific positions of the molecule [164]. This was later confirmed by the same authors by providing evidence that SB 211475 (4-[2-hydroxy-3-[2-(2-methoxyphenoxy) ethylamino]propoxy]-9H-carbazol-3-ol), one of carvedilol hydroxy-lated derivatives, was more effective than carvedilol against iron and hydroxyl-mediated lipid peroxidation, macrophage-induced oxidation of low density lipoproteins and xanthine oxidase/xan-thine-related damage to endothelial cells [165]. Aruoma provided specific evidence that carvedilol react rapidly with peroxyl radicals, which provides support for its anti-lipid peroxidation effects [166]. In opposition, it was also proposed that carvedilol antioxidant effect on lipid peroxidation is not due to direct scavenging of free radicals but instead to a chelating effect on Fe3+, a species that is a catalyst in the process [167]. An apparent support for both theories was later published [168] and in fact, based on the entire set of data collected on carvedilol antioxidant activity, this seems to be a more reason-

2122 Current Pharmaceutical Design, 2011, Vol. 17, No. 20 Pereira et al.
able explanation for the superior antioxidant properties of this -adrenergic antagonist. The clinical relevance of carvedilol antioxi-dant effects was confirmed by demonstrating that carvedilol was able to decrease elevated oxidative stress in the failing human myo-cardium [169]. With this as background, antioxidant protection of mitochondrial function during deleterious events was investigated [170].
As described in the previous section, Nohl et al. suggested the existence of an organo-specific NADH dehydrogenase in heart mitochondria [171]. The oxidation of external NADH promoted by this very controversial and hypothetical [172] NADH dehydro-genase located in the external side of the inner membrane of mito-chondria is linked to a high respiratory rate even though no ADP phosphorylation occurs [173]. The enzyme has also been associated with pathological increased ROS production [173]. Oliveira et al. demonstrated that carvedilol inhibit the respiratory activity attrib-uted to the proposed exogenous NADH dehydrogenase in rat heart mitochondria, diminishing its activity and therefore contributing to protect cardiac cells from oxidative damage [173]. Also, protection against cardiac mitochondrial oxidative damage caused by O2
- production through the xanthine oxidase/hypoxanthine system was achieved by carvedilol [174]. Carvedilol improved mitochondrial respiratory activity in state 3 and was able to prevent the loss of mitochondrial respiratory coupling [174].
As with several other compounds, carvedilol is metabolized after entering the systemic circulation. Between all its metabolites, BM-910228 (Fig. 1) [1-3-hydroxycarbazolyl-(4)-oxy-3-(2-methoxyphenoxy-ethyl)amino-propanol-(2)], is one of the most important and results from the addition of a hydroxyl group at the position 3 of the carbazole group [175]. BM-910228 showed a bet-ter antioxidant activity, when compared with carvedilol since it is effective at concentrations 30 times lower than the parent com-pound [175]. In a work performed with hepatic mitochondrial frac-tions, both compounds inhibited lipid peroxidation and prevented the collapse of transmembranar electric potential [175]. Moreover, no toxic effect was observed on mitochondrial bioenergetics for all the concentrations effective as antioxidants [175]. One can infer therefore that some of carvedilol metabolites, namely BM-910228 further contribute to the antioxidant and therapeutic activity of the drug. Also, the antioxidant and scavenger properties of carvedilol are primarily attributed to the carbazole moiety [164] and as previ-ously mentioned hydroxylation at certain positions of the phenyl ring increases its antioxidant activity.
The antioxidant properties of carvedilol suggest that its hy-droxylated metabolites may be responsible for the cardioprotective action in vivo. After administration, carvedilol is distributed to the tissues and the concentration on plasma is low, but yet effective [176]. Keeping this in mind, it is reasonable to think that the amount of drug available is capable of protecting mitochondrial membranes from oxidative injuries, including lipid peroxidation and to prevent deleterious effects. Abreu et al. supported this idea by providing data where it was shown that the toxicity of carvedilol was verified for concentrations above 40 M [175]. However, such concentrations are not reached in vivo after an oral dose of 50mg since the absolute bioavailability only reaches 24% [176].
3.2. Carvedilol Effects on the Respiratory Chain – protono-phore vs. Complex I Inhibitor
Several studies investigated the mechanism by which carvedilol causes a mild decrease in mitochondrial transmembrane potential on isolated heart mitochondria [177]. Results suggest that carvedilol depresses mitochondrial membrane potential through a weak pro-tonophoretic mechanism, as demonstrated by the induction of a pH-dependent increased proton permeability without corresponding increasing potassium permeability across the mitochondrial mem-brane [177]. Accordingly, carvedilol increases state 4 respiration in succinate-energized cardiac, which is a hallmark shared by classical
protonophores. However, Monteiro et al. did not obtain such effect when cardiac mitochondria were isolated from hearts perfused with carvedilol and submitted to ischemia/reperfusion [178]. Neverthe-less, under these experimental conditions, carvedilol promoted mi-tochondrial protection against ischemia and reperfusion damage.
Since mitochondrial membrane potential is critical for mito-chondrial basic functions, such as being the driving force for the overall ionic fluxes and ATP synthesis, one should ask whether a mild mitochondrial depolarization is beneficial or hazardous. For example, a mild decrease in mitochondrial membrane potential can prevent calcium overload during pathological events, thus reducing the damaging effects of that cation in mitochondria. Also, it has been described that a small decrease in the membrane potential promotes a reduction of ROS produced by the mitochondrial respi-ratory chain [179], which can result in further antioxidant activity by carvedilol.
In an attempt to analyze the interaction of carvedilol with the components of the OXPHOS system, Cocco et al. performed spe-cific assays in mitochondria isolated from several rat organs [39]. The results obtained showed that carvedilol inhibited respiration supported by NAD-dependent substrates through decreasing the activity of respiratory chain Complex I, inducing toxic effects on mitochondrial function for high concentrations but without apparent tissue selectivity. In an apparent paradox, besides its intrinsic anti-oxidant activity, carvedilol appears to induce the production of ROS as a consequence of Complex I inhibition [39]. Similar effects were observed in another in vitro model, the cardiomyoblast cell line H9c2, where a consistent decline of the activity of mitochon-drial Complex I was observed concomitantly with an increase in mitochondrial H2O2 production and total glutathione and protein thiols content [157]. Nonetheless, the use of an oxidant insult such as H2O2 in cells pre-treated with carvedilol did not result in further cell damage; in fact, carvedilol prevented oxidative damage, sug-gesting that despite its effects on Complex I, carvedilol can provide some precondition mechanism that allows cells to be more resistant to oxidative damage [157].
3.3. Inhibitory Effect of Carvedilol on the Cardiac Mitochon-drial Permeability Transition
Carvedilol and BM-910228 were also tested for their in vitro protective effects on heart MPT, due to the role of this phenomenon on cardiac damage [27]. Several experimental protocols were used to induce the MPT in order to investigate the protective effect of carvedilol and its metabolite. Isolated rat heart mitochondria [180] pre-incubated with carvedilol had shown inhibition of mitochon-drial thiol groups oxidation induced by calcium/phosphate [180], the golden-standard for MPT induction. It was described that carvedilol had a dual action on mitochondrial calcium accumula-tion: a negligible effect when calcium concentrations were below the threshold for pore opening and a protective effect when calcium concentration reaches the pore activation threshold level. In this way, carvedilol is proposed to be an inhibitor of the high-conductance state of the MPT pore, acting on mitochondrial swel-ling and calcium efflux without preventing mitochondrial mem-brane depolarization or swelling in an ionic media [180]. Later [181, 182] it was described that carvedilol was effective only when MPT was triggered by a primary oxidative process which leads to protein thiol-groups oxidation and consequently induce MPT [181, 182]. To better understand how carvedilol manages to prevent MPT induction, it is important to consider that the drug does not prevent pore opening when the event relies on structural changes of some pore components. In contrast, when primary oxidation of mito-chondrial thiol groups occurs, carvedilol is able to exert is protec-tive role, as demonstrated when incubating mitochondria with the oxidative pair calcium/phosphate as opposed to results achieved with carboxyatractyloside, a specific ANT inhibitor and MPT in-ducer [183].This idea reinforces once more that the enhanced pro-

Doxorubicin, Carvedilol and Cardiac Mitochondria Current Pharmaceutical Design, 2011, Vol. 17, No. 20 2123
tective action of carvedilol is due to its antioxidant activity. Other studies confirm the same effects in events of cardiac stress, such as during cardiac ischemia/reperfusion, where heart mitochondria suffer from increased oxidative injury and accumulation of calcium and phosphate [182]. Moreover, the inhibitory effect of carvedilol decreases MPT-related cytochrome c release, which is relevant in the reduction of apoptosis in myocardium [177, 184]. One particu-lar work demonstrated that carvedilol ameliorates the overload of mitochondrial and cytosolic calcium, an effect that is completely dependent on the antioxidant properties of the compound rather than its adrenergic blocker activity since other -blockers lacking antioxidant activity, such as metroprolol, had no protective effect [184].
Interestingly, carvedilol, but not its metabolite, prevented mem-brane depolarization, osmotic swelling and release of matrix cal-cium when the MPT was induced by chenodeoxycholic acid (CDCA), which strengthens the role of carvedilol in the protection of mitochondrial function [185]. Nevertheless, inhibition of CDCA-induced MPT may not be due to its antioxidant activity, as the hy-droxylated metabolite does not offer such protection. Moreover, in this study, one cannot ignore the possible interaction between the bile acid and the drug or even the competition for potential binding sites and the ability of the carvedilol to insert into the membrane causing both protein-protein and lipid-protein disturbance which could ultimately interfere with the kinetic of the MPT pore, as was suggested by the authors [185].
3.4. Non-mitochondrial Targets for Carvedilol in the Heart
So far, several evidences that carvedilol also targets other struc-tures outside mitochondria have been reported. For example, carve-dilol and its metabolite attenuated H2O2-mediated decrease in sar-coplasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) mRNA and protein levels in rat primary cardiomyocytes. Furthermore, carve-dilol significantly enhanced SERCA2 gene transcription, suggesting that this compound specifically restores SERCA2 gene transcription being therefore suggested as a subjacent mechanism of the benefi-cial effects of carvedilol on cardiac function [186].
Yue et al. explored the cardioprotective effects of carvedilol in the ischemic myocardium and to understand its mechanism of ac-tion using rabbits subjected to coronary artery occlusion followed by reperfusion [187]. When carvedilol was administered before reperfusion, a decrease of over 77% in the number of apoptotic myocytes in the ischemic area was observed. Ischemic cardiomyo-cytes from rabbit hearts revealed an upregulation of Fas protein which carvedilol was able to reduce. The ischemia/reperfusion process induces fast activation of stress-activated protein kinase (SAPK) in the ischemic area and carvedilol was also capable to inhibit the activation of this protein by approximately 50%. In this case, the protective action of carvedilol against ischemia/reper-fusion injury is thought to be due to the downregulation of the SAPK signaling pathway, through the inhibition of Fas receptor expression, and by -adrenergic blockade instead of its antioxidant activity [187].
4. FIGHTING DRUG-INDUCED MITOCHONDRIAL TOX-
ICITY BY USING DRUG-INDUCED MITOCHONDRIAL PROTECTION: CARVEDILOL VS. DOXORUBICIN
The main goal of introducing a cardioprotective substance con-comitantly during DOX-chemotherapeutic program is to prevent the deleterious effects of the anti-cancer drug (Fig. 3), especially at the mitochondrial level, with few or no toxic side effects. Carvedilol, with its intrinsic antioxidant activity in addition to its -blocker properties, was a logical choice in an attempt to prevent DOX car-diac and mitochondrial damage by multiple mechanisms.
An early study by Fazio et al. [188] was the first to demonstrate that carvedilol was able to induce complete recovery in a patient undergoing DOX treatment and used to show signs of congestive
heart failure. Following this work, an animal model of DOX-induced cardiac toxicity showed a remarkable protective effect of carvedilol, attributed to its antioxidant and lipid-lowering properties [189]. Nevertheless, it remained to be known whether the protective effect of carvedilol on DOX-induced cardiac toxicity originated from the prevention of cardiac mitochondrial degeneration.
One work by Santos et al. demonstrated that carvedilol protects against cardiac and hepatic mitochondrial bioenergetic dysfunction associated with a sub-chronic DOX toxicity [190]. Co-adminis-tration of carvedilol decreased the extent of cellular vacuolization in cardiac myocytes and prevented the inhibitory effect of DOX on mitochondrial respiration in both heart and liver. DOX decreased mitochondrial calcium loading capacity in 50% and only 10% in liver. Interestingly, co-treatment with carvedilol protected cardiac mitochondria from DOX-induced decrease in that parameter but did not protect liver mitochondria [190].
One logical concern would be that carvedilol, besides prevent-ing DOX mitochondrionopathy, may also decrease the anti-neo-plastic activity of DOX. Nevertheless, it was observed that carve-dilol reduces P-glycoprotein activity increasing therefore the sensi-tivity to DOX cytotoxicity in cell cultures of two human breast cell sub-lines [191].
In cultured cardiomyoblast H9c2 cells, carvedilol inhibited free radical production, apoptotic pathways and the hallmarks associated with those events induced by DOX and consequently increased cell viability [163]. However, that effect was specific to carvedilol since atenolol, another -blocker, did not prevent DOX-induced increase in the BAX:Bcl-2 ratio. Confirming, once more that the cardiopro-tective effect of carvedilol is a consequence of its antioxidant activ-ity and not to its -blocking function. The possible toxic effect of carvedilol per se was not confirmed since carvedilol did not in-crease cell apoptosis.
Further in vivo studies with DOX-treated rats have confirmed that carvedilol is able to prevent cardiac mitochondrionopathy through an antioxidant mechanism [192]. Rats submitted to long-term treatment with DOX present several mitochondrial alterations, including decreased calcium loading capacity [102]. Although both compounds were able to preserve ATP levels in the tissue, only carvedilol was capable to preserve mitochondrial function by pre-venting the loss of mitochondrial calcium loading capacity and by inhibiting the appearance of protein carbonyl groups. The protective effect of carvedilol was not shared by atenolol, once more demon-strating the importance of carvedilol antioxidant activity [102]. Another common consequence of DOX-induced increase in oxida-tive stress is lipid peroxidation and once more, carvedilol was able to avoid such harmful occurrence in DOX-co-treated animals as assessed by the levels of TBARS [189].
Soon, clinical studies in humans have started in an attempt to confirm improvement of cardiac function in DOX-treated patients. Kalay et al. [193] performed the first clinical trial with 50 patients undergoing anthracyclines therapy, with half of them receiving carvedilol at the same time. The carvedilol group showed preserved left ventricular diastolic and systolic function, showing cardiac function enhancement. However, further clinical trials need to be performed with larger study groups.
One should apply the acquired knowledge regarding mitochon-drial studies to design superior approaches using carvedilol or simi-lar molecules to protect cardiac function without compromising the antitumor activity of DOX.
CONCLUDING REMARKS
The cardiac muscle is highly dependent on ATP production from mitochondria, which also makes this tissue highly susceptible to mitochondrial failure caused by different pathologies or drugs [194-197].
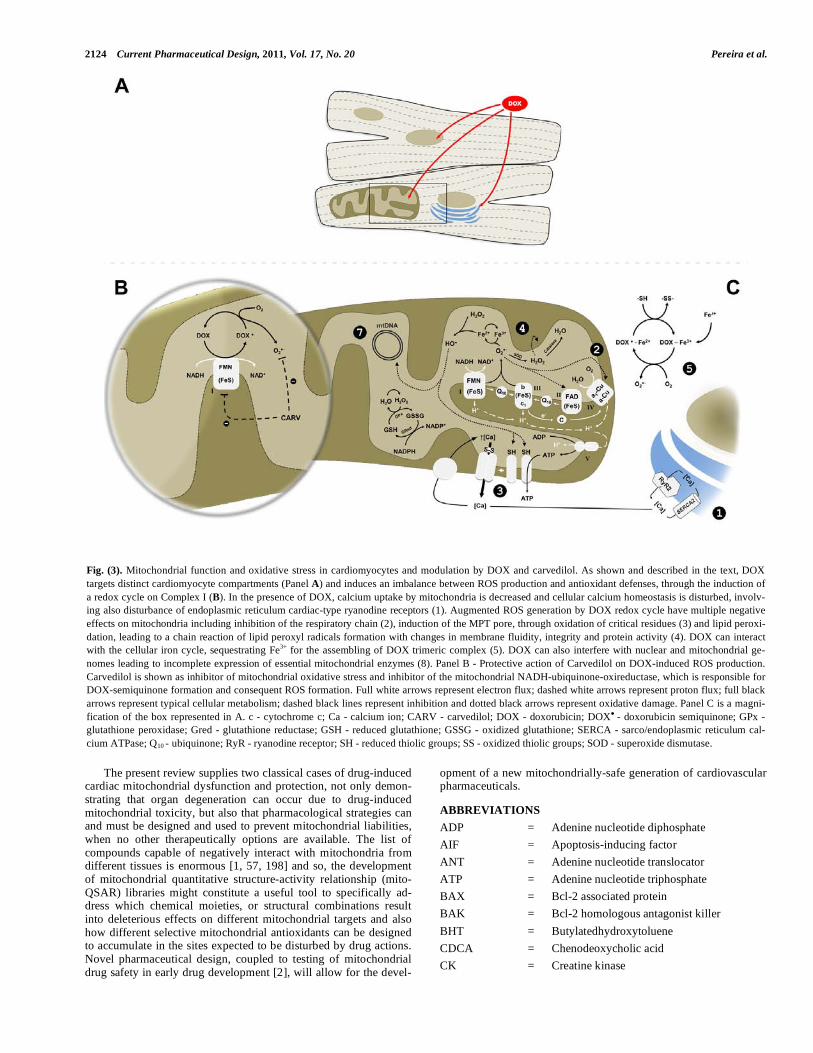
2124 Current Pharmaceutical Design, 2011, Vol. 17, No. 20 Pereira et al.
The present review supplies two classical cases of drug-induced cardiac mitochondrial dysfunction and protection, not only demon-strating that organ degeneration can occur due to drug-induced mitochondrial toxicity, but also that pharmacological strategies can and must be designed and used to prevent mitochondrial liabilities, when no other therapeutically options are available. The list of compounds capable of negatively interact with mitochondria from different tissues is enormous [1, 57, 198] and so, the development of mitochondrial quantitative structure-activity relationship (mito-QSAR) libraries might constitute a useful tool to specifically ad-dress which chemical moieties, or structural combinations result into deleterious effects on different mitochondrial targets and also how different selective mitochondrial antioxidants can be designed to accumulate in the sites expected to be disturbed by drug actions. Novel pharmaceutical design, coupled to testing of mitochondrial drug safety in early drug development [2], will allow for the devel-
opment of a new mitochondrially-safe generation of cardiovascular pharmaceuticals.
ABBREVIATIONS
ADP = Adenine nucleotide diphosphate
AIF = Apoptosis-inducing factor
ANT = Adenine nucleotide translocator
ATP = Adenine nucleotide triphosphate
BAX = Bcl-2 associated protein
BAK = Bcl-2 homologous antagonist killer
BHT = Butylatedhydroxytoluene
CDCA = Chenodeoxycholic acid
CK = Creatine kinase
Fig. (3). Mitochondrial function and oxidative stress in cardiomyocytes and modulation by DOX and carvedilol. As shown and described in the text, DOX targets distinct cardiomyocyte compartments (Panel A) and induces an imbalance between ROS production and antioxidant defenses, through the induction of a redox cycle on Complex I (B). In the presence of DOX, calcium uptake by mitochondria is decreased and cellular calcium homeostasis is disturbed, involv-ing also disturbance of endoplasmic reticulum cardiac-type ryanodine receptors (1). Augmented ROS generation by DOX redox cycle have multiple negative effects on mitochondria including inhibition of the respiratory chain (2), induction of the MPT pore, through oxidation of critical residues (3) and lipid peroxi-dation, leading to a chain reaction of lipid peroxyl radicals formation with changes in membrane fluidity, integrity and protein activity (4). DOX can interact with the cellular iron cycle, sequestrating Fe3+ for the assembling of DOX trimeric complex (5). DOX can also interfere with nuclear and mitochondrial ge-nomes leading to incomplete expression of essential mitochondrial enzymes (8). Panel B - Protective action of Carvedilol on DOX-induced ROS production. Carvedilol is shown as inhibitor of mitochondrial oxidative stress and inhibitor of the mitochondrial NADH-ubiquinone-oxireductase, which is responsible for DOX-semiquinone formation and consequent ROS formation. Full white arrows represent electron flux; dashed white arrows represent proton flux; full black arrows represent typical cellular metabolism; dashed black lines represent inhibition and dotted black arrows represent oxidative damage. Panel C is a magni-fication of the box represented in A. c - cytochrome c; Ca - calcium ion; CARV - carvedilol; DOX - doxorubicin; DOX - doxorubicin semiquinone; GPx - glutathione peroxidase; Gred - glutathione reductase; GSH - reduced glutathione; GSSG - oxidized glutathione; SERCA - sarco/endoplasmic reticulum cal-cium ATPase; Q10 - ubiquinone; RyR - ryanodine receptor; SH - reduced thiolic groups; SS - oxidized thiolic groups; SOD - superoxide dismutase.

Doxorubicin, Carvedilol and Cardiac Mitochondria Current Pharmaceutical Design, 2011, Vol. 17, No. 20 2125
CsA = Cyclosporin A
Cr = Creatine
COX = Cytochrome c oxidase
DOX = Doxorubicin
DTT = Dithiothreitol
eNOS = Endothelial nitric oxide synthase
ETC = Electron transport chain
H2O2 = Hydrogen peroxide
HO = Hydroxyl radical
IAP = Inhibitor of apoptosis
iNOS = Inducible nitric oxide synthase
mitoQSAR = Mitochondrial quantitative structure-activity relationship
Mn-SOD = Manganese-superoxide dismutase
MPT = Mitochondrial permeability transition
mtCK = Mitochondrial isoform of creatine kinase
mtDNA = Mitochondrial DNA
NADH = Nicotinamide adenine dinucleotide reduced form
nNOS = Neuronal nitric oxide synthase
NO = Nitric oxide
O2- = Superoxide radical anion
ONOO- = Peroxynitrite
OXPHOS = Oxidative phosphorylation
PCr = Phosphocreatine
RCR = Respiratory control ratio
ROS = Reactive oxygen species
SAPK = Stress-activated protein kinase
SERCA2 = Sarcoplasmic reticulum Ca2+-ATPase iso-form 2
SMAC/DIABLO = Second mitochondria-derived activator of capases/direct IAP-binding protein with low isoelectric point
TBARS = Thiobarbituric acid reactive substances
tBid = Truncated form of BH2 interacting domain death agonist
TNF- = Tumor necrosis factor-alpha
VDAC = Voltage-dependent anion channel
CONFLICT OF INTEREST
None to declare.
ACKNOWLEDGMENTS
Research in author’s lab is supported by research grants PTDC/SAU-OSM/104731/2008, PTDC/AGR-ALI/108326/2008 and PTDC/SAU-TOX/110952/2009 and by Ph.D. fellowships SFRH/BD/36938/2007, SFRH/BD/48133/2008 and SFRH/BD/64694/2009, all from the Portuguese Foundation for Science and Technology. The authors would also like to thank Inês Barbosa for proofreading the present manuscript.
REFERENCES
[1] Pereira SP, Pereira GC, Moreno AJ, Oliveira PJ. Can drug safety be predicted and animal experiments reduced by using isolated mi-tochondrial fractions? Altern Lab Anim 2009; 37: 355-65.
[2] Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov Today 2007; 12: 777-85.
[3] Nadanaciva S, Dillman K, Gebhard DF, Shrikhande A, Will Y. High-content screening for compounds that affect mtDNA-encoded
protein levels in eukaryotic cells. J Biomol Screen 2010; 15: 937-48.
[4] Witte ME, Geurts JJ, Vries HE de, Valk P van der, Horssen J van. Mitochondrial dysfunction: a potential link between neuroinflam-mation and neurodegeneration? Mitochondrion 2010; 10: 411-8.
[5] Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal 2010; 12: 537-77.
[6] Rosca MG, Vazquez EJ, Kerner J, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphoryla-tion. Cardiovasc Res 2008; 80: 30-9.
[7] Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev 2002; 54: 101-27.
[8] McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 2006; 16: R551-60.
[9] Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane per-meabilization in cell death. Physiol Rev 2007; 87: 99-163.
[10] Hajnóczky G, Csordás G, Das S, et al. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mito-chondrial Ca2+ uptake in apoptosis. Cell Calcium 2006; 40: 553-60.
[11] Nicholls DG, Ferguson SJ. Bioenergetics 3. London: Academic Press 2002.
[12] Halestrap AP. Mitochondria and reperfusion injury of the heart--a holey death but not beyond salvation. J Bioenerg Biomembr 2009; 41: 113-21.
[13] Baines CP. The cardiac mitochondrion: nexus of stress. Annu Rev Physiol, 2010; 72: 61-80.
[14] Scatena R, Bottoni P, Botta G, Martorana GE, Giardina B. The role of mitochondria in pharmacotoxicology: a reevaluation of an old, newly emerging topic. Am J Physiol Cell Physiol 2007; 293: C12-21.
[15] Montero J, Mari M, Colell A, Morales A, Basañez G, Garcia-Ruiz C, et al. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim Biophys Acta 2010; 1797: 1217-24.
[16] Hom J, Sheu SS. Morphological dynamics of mitochondria--a special emphasis on cardiac muscle cells. J Mol Cell Cardiol 2009; 46: 811-20.
[17] Kane LA, Youle RJ. Mitochondrial fission and fusion and their roles in the heart. J Mol Med 2010; 88: 971-9.
[18] Beraud N, Pelloux S, Usson Y, et al. Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J Bio-energ Biomembr 2009; 41: 195-214.
[19] Eaton S, Bartlett K, Pourfarzam M. Mammalian mitochondrial beta-oxidation. Biochem J 1996; 320(Pt 2): 345-57.
[20] Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961; 191144-8.
[21] Miró O, Barrientos A, Alonso JR, et al. Effects of general anaes-thetic procedures on mitochondrial function of human skeletal muscle. Eur J Clin Pharmacol 1999; 55: 35-41.
[22] Dabadie P, Bendriss P, Erny P, Mazat JP. Uncoupling effects of local anesthetics on rat liver mitochondria. FEBS Lett 1987; 226: 77-82.
[23] Biasutto L, Dong L-F, Zoratti M, Neuzil J. Mitochondrially tar-geted anti-cancer agents. Mitochondrion 2010; 10: 670-81.
[24] Grad JM, Cepero E, Boise LH. Mitochondria as targets for estab-lished and novel anti-cancer agents. Drug Resist Updat 2001; 4: 85-91.
[25] Lund KC, Wallace KB. Direct effects of nucleoside reverse tran-scriptase inhibitors on rat cardiac mitochondrial bioenergetics. Mi-tochondrion 2004; 4: 193-202.
[26] Finsterer J, Segall L. Drugs interfering with mitochondrial disor-ders. Drug Chem Toxicol 2010; 33: 138-51.
[27] Javadov SA, Karmazyn M, Escobales N. Mitochondrial permeabil-ity transition pore opening as a promising therapeutic target in car-diac diseases. J Pharmacol Exp Ther 2009; 330: 670-8.
[28] Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999; 341(Pt 2): 233-49.
[29] Robert V, Gurlini P, Tosello V, et al. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J 2001; 20: 4998-5007.
[30] Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res 2004; 61: 372-85.

2126 Current Pharmaceutical Design, 2011, Vol. 17, No. 20 Pereira et al.
[31] Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol 2009; 104: 181-8.
[32] Kass GE. Mitochondrial involvement in drug-induced hepatic injury. Chem Biol Interact 2006; 163: 145-59.
[33] Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria as targets for cancer chemotherapy. Semin Cancer Biol 2009; 19: 57-66.
[34] Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB. Cumula-tive and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res 2001; 61: 771-7.
[35] Solem LE, Wallace KB. Selective activation of the sodium-independent, cyclosporin A-sensitive calcium pore of cardiac mito-chondria by doxorubicin. Toxicol Appl Pharmacol 1993; 121: 50-7.
[36] Sokolove PM. Inhibition by cyclosporin A and butylated hydroxy-toluene of the inner mitochondrial membrane permeability transi-tion induced by adriamycin aglycones. Biochem Pharmacol 1990; 40: 2733-6.
[37] Machado NG, Alves MG, Carvalho RA, Oliveira PJ. Mitochondrial involvement in cardiac apoptosis during ischemia and reperfusion: can we close the box? Cardiovasc Toxicol 2009; 9: 211-27.
[38] Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol 2009; 104: 181-8.
[39] Cocco T, Cutecchia G, Montedoro G, Lorusso M. The antihyper-tensive drug carvedilol inhibits the activity of mitochondrial NADH-ubiquinone oxidoreductase. J Bioenerg Biomembr 2002; 34: 251-8.
[40] Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgak-ilas AG, Bonner WM. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat Res 2010; 704: 152-9.
[41] Bayir H, Kagan VE. Bench-to-bedside review: Mitochondrial in-jury, oxidative stress and apoptosis--there is nothing more practical than a good theory. Crit Care 2008; 12: 206.
[42] Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis 2007; 12: 913-22.
[43] Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci: 2009; 141197-218.
[44] Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res 2009; 81: 449-56.
[45] Hori M, Nishida K. Oxidative stress and left ventricular remodel-ling after myocardial infarction. Cardiovasc Res 2009; 81: 457-64.
[46] Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12: 9-14.
[47] Perkins G, Bossy-Wetzel E, Ellisman MH. New insights into mito-chondrial structure during cell death. Exp Neurol 2009; 218: 183-92.
[48] Tait SWG, Green DR. Mitochondria and cell death: outer mem-brane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11: 621-32.
[49] Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regula-tors of apoptosis and autophagy. Autophagy 2008; 4: 600-6.
[50] Szigeti A, Hocsak E, Rapolti E, et al. Facilitation of mitochondrial outer and inner membrane permeabilization and cell death in oxida-tive stress by a novel Bcl-2 homology 3 domain protein. J Biol Chem 2010; 285: 2140-51.
[51] Kim R, Emi M, Tanabe K, et al. Regulation and interplay of apop-totic and non-apoptotic cell death. J Pathol 2006; 208: 319-26.
[52] Petrosillo G, Ruggiero FM, Paradies G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mito-chondria. FASEB J 2003; 17: 2202-8.
[53] Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med 2009; 46: 1439-53.
[54] Machado NG, Alves MG, Carvalho RA, Oliveira PJ. Mitochondrial involvement in cardiac apoptosis during ischemia and reperfusion: can we close the box? Cardiovasc Toxicol 2009; 9: 211-27.
[55] Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am J Physiol Heart Circ Physiol 2011; 300: H118-24.
[56] Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogene-sis of heart disease: mechanisms and significance. Annu Rev Physiol, 2010; 72: 19-44.
[57] Sardão VA, Pereira SL, Oliveira PJ. Drug-induced mitochondrial dysfunction in cardiac and skeletal muscle injury. Expert Opin Drug Saf 2008; 7: 129-46.
[58] Arcamone F, Cassinelli G, Di Marco A, Gaetani M, inventors; Società Farmaceutici Italia, assignee. Adriamycin derivatives. United States patent US 3590028. 1971 Jun.
[59] Arcamone F, Cassinelli G, Fantini G, et al. Adriamycin, 14-hydroxydaimomycin, a new antitumor antibiotic from S. Peucetius var. caesius. Biotechnol Bioeng 1969; 11: 1101–1110.
[60] Arcamone F, Franceschi G, Penco S, Selva A. Adriamycin (14-hydroxydaunomycin), a novel antitumor antibiotic. Tetrahedron Lett 1969; 13: 1007.
[61] Di Marco A, Gaetani M, Scarpinato B. Adriamycin (NSC-123, 127): a new antibiotic with antitumor activity. Cancer Chemother Rep 1969; 53: 33-7.
[62] Bonadonna G, Monfardini S, De Lena M, Fossati-Bellani F. Clini-cal evaluation of adriamycin, a new antitumour antibiotic. Br Med J 1969; 3: 503.
[63] Bonadonna G, Monfardini S, De Lena M, Fossati-Bellani F, Beretta G. Phase I and preliminary phase II evaluation of adriamy-cin (NSC 123127). Cancer Res 1970; 30: 2572-82.
[64] Steinherz LJ, Steinherz PG, Tan CT, Heller G, Murphy ML. Car-diac toxicity 4 to 20 years after completing anthracycline therapy. JAMA 1991; 266: 1672-7.
[65] Weiss RB. The anthracyclines: will we ever find a better doxorubi-cin? Semin Oncol 1992; 19: 670-86.
[66] Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med 1998; 339: 900-5.
[67] Carvalho C, Santos RX, Cardoso S, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem 2009; 16: 3267-85.
[68] Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis 2007; 49: 330-52.
[69] Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxoru-bicin-induced congestive heart failure. Ann Intern Med 1979; 91(5): 710-7.
[70] Ludke AR, Al-Shudiefat AA, Dhingra S, Jassal DS, Singal PK. A concise description of cardioprotective strategies in doxorubicin-induced cardiotoxicity. Can J Physiol Pharmacol 2009; 87: 756-63.
[71] Robert J, Morvan VL, Smith D, Pourquier P, Bonnet J. Predicting drug response and toxicity based on gene polymorphisms. Crit Rev Oncol Hematol 2005; 54: 171-96.
[72] Deng S, Wojnowski L. Genotyping the risk of anthracycline-induced cardiotoxicity. Cardiovasc Toxicol 2007; 7: 129-34.
[73] Smibert E, Carlin JB, Vidmar S, Wilkinson LC, Newton M, Wein-traub RG. Exercise echocardiography reflects cumulative anthracy-cline exposure during childhood. Pediatr Blood Cancer 2004; 42: 556-62.
[74] Neilan TG, Jassal DS, Perez-Sanz TM, et al. Tissue Doppler imag-ing predicts left ventricular dysfunction and mortality in a murine model of cardiac injury. Eur Heart J 2006; 27: 1868-75.
[75] Germanakis I, Anagnostatou N, Kalmanti M. Troponins and natri-uretic peptides in the monitoring of anthracycline cardiotoxicity. Pediatr Blood Cancer 2008; 51: 327-33.
[76] Christiansen S. Surgical treatment of doxorubicin-induced heart failure. Thorac Cardiovasc Surg 2010; 58: 8-10.
[77] Bodley A, Liu LF, Israel M, et al. DNA topoisomerase II-mediated interaction of doxorubicin and daunorubicin congeners with DNA. Cancer Res 1989; 49: 5969-78.
[78] Canobbio L, Fassio T, Gasparini G, et al. Cardiac arrhythmia: possible complication from treatment with cisplatin. Tumori 1986; 72: 201-4.
[79] Wallace KB. Adriamycin-induced interference with cardiac mito-chondrial calcium homeostasis. Cardiovasc Toxicol 2007; 7: 101-7.
[80] Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mito-chondrial cardiotoxicity. Cell Biol Toxicol 2007; 23(1): 15-25.
[81] Anderson AB, Xiong G, Arriaga EA. Doxorubicin accumulation in individually electrophoresed organelles. J Am Chem Soc 2004; 126: 9168-9.
[82] Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracy-clines: molecular advances and pharmacologic developments in an-titumor activity and cardiotoxicity. Pharmacol Rev 2004; 56: 185-229.
[83] Jung K, Reszka R. Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev 2001; 49: 87-105.

Doxorubicin, Carvedilol and Cardiac Mitochondria Current Pharmaceutical Design, 2011, Vol. 17, No. 20 2127
[84] Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol 2003; 93: 105-15.
[85] Doroshow JH. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res 1983; 43: 460-72.
[86] Nohl H, Gille L, Staniek K. The exogenous NADH dehydrogenase of heart mitochondria is the key enzyme responsible for selective cardiotoxicity of anthracyclines. Z Naturforsch 1998; C53: 279-85.
[87] Sim nek T, Stérba M, Popelová O, Adamcová M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies ex-amining the roles of oxidative stress and free cellular iron. Pharma-col Rep 2009; 61: 154-71.
[88] Cairo G, Recalcati S, Pietrangelo A, Minotti G. The iron regulatory proteins: targets and modulators of free radical reactions and oxida-tive damage. Free Radic Biol Med 2002; 32: 1237-43.
[89] Cole MP, Chaiswing L, Oberley TD, et al. The protective roles of nitric oxide and superoxide dismutase in adriamycin-induced car-diotoxicity. Cardiovasc Res 2006; 69: 186-97.
[90] Fogli S, Nieri P, Breschi MC. The role of nitric oxide in anthracy-cline toxicity and prospects for pharmacologic prevention of car-diac damage. FASEB J 2004; 18: 664-75.
[91] Cotton JM, Kearney MT, Shah AM. Nitric oxide and myocardial function in heart failure: friend or foe? Heart 2002; 88: 564-6.
[92] Mukhopadhyay P, Rajesh M, Batkai S, et al. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol 2009; 296: H1466-83.
[93] Evig CB, Kelley EE, Weydert CJ, et al. Endogenous production and exogenous exposure to nitric oxide augment doxorubicin cyto-toxicity for breast cancer cells but not cardiac myoblasts. Nitric Oxide 2004; 10: 119-29.
[94] Zhou S, Palmeira CM, Wallace KB. Doxorubicin-induced persis-tent oxidative stress to cardiac myocytes. Toxicol Lett 2001; 121: 151-7.
[95] Chandran K, Aggarwal D, Migrino RQ, et al. Doxorubicin inacti-vates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys J 2009; 96: 1388-98.
[96] Oliveira PJ, Santos MS, Wallace KB. Doxorubicin-induced thiol-dependent alteration of cardiac mitochondrial permeability transi-tion and respiration. Biochemistry (Mosc) 2006; 71: 194-199.
[97] Costantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nu-cleotides and dithiol oxidation at two separate sites. J Biol Chem 1996; 271: 6746-51.
[98] Pereira GC, Oliveira PJ. Pharmacological strategies to counteract doxorubicin-induced cardiotoxicity: the role of mitochondria. J Pharmacol Exp Ther 2008; 1: 39-53.
[99] Ascensão A, Lumini-Oliveira J, Machado NG, et al. Acute exercise protects against calcium-induced cardiac mitochondrial permeabil-ity transition pore opening in doxorubicin-treated rats. Clin Sci (Lond) 2010; 120: 37-49.
[100] Oliveira PJ, Wallace KB. Depletion of adenine nucleotide translo-cator protein in heart mitochondria from doxorubicin-treated rats--relevance for mitochondrial dysfunction. Toxicology 2006; 220: 160-8.
[101] Al-Nasser IA. In vivo prevention of adriamycin cardiotoxicity by cyclosporin A or FK506. Toxicology 1998; 131: 175-81.
[102] Oliveira PJ, Bjork JA, Santos MS, et al. Carvedilol-mediated anti-oxidant protection against doxorubicin-induced cardiac mitochon-drial toxicity. Toxicol Appl Pharmacol 2004; 200: 159-68.
[103] Guo HX, Wang F, Yu KQ, et al. Novel cyclophilin D inhibitors derived from quinoxaline exhibit highly inhibitory activity against rat mitochondrial swelling and Ca2+ uptake/ release. Acta Pharma-col Sin 2005; 26: 1201-11.
[104] Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosup-pressive cyclosporin derivative NIM811. Mol Pharmacol 2002; 62: 22-9.
[105] Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res 2009; 83: 213-25.
[106] Sardão VA, Santos DL, Oliveira PJ. Doxorubicin-induced mito-chondrial dysfunction. In: Moreno AJ, Oliveira PJ, Palmeira CM, Ed. Mitochondrial Pharmacology and Toxicology. Trivandrum: Transworld Research Network 2006; pp. 85-112.
[107] Sterba M, Popelová O, Lenco J, et al. Proteomic insights into chronic anthracycline cardiotoxicity. J Mol Cell Cardiol 2011; 50: 849-862.
[108] Robert J. Long-term and short-term models for studying anthracy-cline cardiotoxicity and protectors. Cardiovasc Toxicol 2007; 7: 135-9.
[109] Le Bot MA, Begue JM, Kernaleguen D, et al. Different cytotoxic-ity and metabolism of doxorubicin, daunorubicin, epirubicin, esorubicin and idarubicin in cultured human and rat hepatocytes. Biochem Pharmacol 1988; 37: 3877-87.
[110] Pointon AV, Walker TM, Phillips KM, et al. Doxorubicin in vivo rapidly alters expression and translation of myocardial electron transport chain genes, leads to ATP loss and caspase 3 activation. PLoS One 2010; 5: e12733.
[111] Berthiaume JM, Wallace KB. Persistent alterations to the gene expression profile of the heart subsequent to chronic Doxorubicin treatment. Cardiovasc Toxicol 2007; 7: 178-91.
[112] Wakasugi S, Fischman AJ, Babich JW, et al. Myocardial substrate utilization and left ventricular function in adriamycin cardiomyopa-thy. Journal J Nucl Cardiol 1993; 34: 1529-35.
[113] Carvalho RA, Sousa RP, Cadete VJ, et al. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicol-ogy 2010; 270: 92-8.
[114] Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U. New insights into doxorubicin-induced cardiotoxic-ity: the critical role of cellular energetics. J Mol Cell Cardiol 2006; 41: 389-405.
[115] Saks V, Dzeja P, Schlattner U, Vendelin M, Terzic A, Wallimann T. Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J Physiol 2006; 571: 253-73.
[116] Younes A, Schneider JM, Bercovici J, Swynghedauw B. Redistri-bution of creatine kinase isoenzymes in chronically overloaded myocardium. Cardiovasc Res 1985; 19: 15-9.
[117] Bittner V, Reeves RC, Digerness SB, Caulfield JB, Pohost GM. 31P NMR spectroscopy in chronic adriamycin cardiotoxicity. Magn Reson Med Sci 1991; 17: 69-81.
[118] Zhang YW, Shi J, Li YJ, Wei L. Cardiomyocyte death in doxorubi-cin-induced cardiotoxicity. Arch Immunol Ther Exp (Warsz) 2009; 57: 435-45.
[119] Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM. Tar-geted disruption of p53 attenuates doxorubicin-induced cardiac tox-icity in mice. Mol Cell Biochem 2005; 273: 25-32.
[120] Chua CC, Liu X, Gao J, Hamdy RC, Chua BHL. Multiple actions of pifithrin-alpha on doxorubicin-induced apoptosis in rat myoblas-tic H9c2 cells. Am J Physiol Heart Circ Physiol 2006; 290: H2606-13.
[121] Sardão VA, Oliveira PJ, Holy J, Oliveira CR, Wallace KB. Doxorubicin-induced mitochondrial dysfunction is secondary to nuclear p53 activation in H9c2 cardiomyoblasts. Cancer Chemother Pharmacol 2009; 64: 811-27.
[122] L�Ecuyer T, Sanjeev S, Thomas R, et al. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am J Physiol Heart Circ Physiol 2006; 291: H1273-80.
[123] Nithipongvanitch R, Ittarat W, Velez JM, Zhao R, St Clair DK, Oberley TD. Evidence for p53 as guardian of the cardiomyocyte mitochondrial genome following acute adriamycin treatment. J His-tochem Cytochem 2007; 55: 629-39.
[124] De Angelis A, Piegari E, Cappetta D, et al. Anthracycline cardio-myopathy is mediated by depletion of the cardiac stem cell pool and is rescued by restoration of progenitor cell function. Circula-tion 2010; 121: 276-92.
[125] Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol 2008; 44: 654-61.
[126] Iglewski M, Hill JA, Lavandero S, Rothermel BA. Mitochondrial fission and autophagy in the normal and diseased heart. Curr Hy-pertens Rep 2010; 12: 418-25.
[127] Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 2010; 6.
[128] Gottlieb RA, Carreira RS. Autophagy in health and disease. 5. Mitophagy as a way of life. Am J Physiol Cell Physiol 2010; 299: C203-10.
[129] Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 2001; 15: 2286-7.

2128 Current Pharmaceutical Design, 2011, Vol. 17, No. 20 Pereira et al.
[130] Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36: 30-8.
[131] Gottlieb RA, Gustafsson AB. Mitochondrial turnover in the heart. Biochim Biophys Acta 2010.
[132] Kobayashi S, Volden P, Timm D, et al. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyo-cyte death. J Biol Chem 2010; 285: 793-804.
[133] Lu L, Wu W, Yan J, et al. Adriamycin-induced autophagic cardio-myocyte death plays a pathogenic role in a rat model of heart fail-ure. Int J Cardiol 2009; 134: 82-90.
[134] Peters JH, Gordon GR, Kashiwase D, Acton EM. Tissue distribu-tion of doxorubicin and doxorubicinol in rats receiving multiple doses of doxorubicin. Cancer Chemother Pharmacol 1981; 7: 65-9.
[135] Lal S, Mahajan A, Chen WN, Chowbay B. Pharmacogenetics of target genes across doxorubicin disposition pathway: a review. Curr Drug Metab 2010; 11: 115-28.
[136] Odom AL, Hatwig CA, Stanley JS, Benson AM. Biochemical determinants of Adriamycin toxicity in mouse liver, heart and in-testine. Biochem Pharmacol 1992; 43: 831-6.
[137] Rumsey WL, Wilson DF. Tissue Capacity for Mitochondrial Oxi-dative Phosphorylation and its Adaptation to Stress. John Wiley & Sons, Inc. 2011.
[138] Rossignol R, Malgat M, Mazat JP, Letellier T. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J Biol Chem 1999; 274: 33426-32.
[139] Cardoso S, Santos RX, Carvalho C, et al. Doxorubicin increases the susceptibility of brain mitochondria to Ca(2+)-induced perme-ability transition and oxidative damage. Free Radic Biol Med 2008; 45: 1395-402.
[140] Palmeira CM, Serrano J, Kuehl DW, Wallace KB. Preferential oxidation of cardiac mitochondrial DNA following acute intoxica-tion with doxorubicin. Biochim Biophys Acta 1997; 1321: 101-6.
[141] Santos DJ, Moreno AJ, Leino RL, Froberg MK, Wallace KB. Carvedilol protects against doxorubicin-induced mitochondrial car-diomyopathy. Toxicol Appl Pharmacol 2002; 185: 218-27.
[142] Machado NG, Baldeiras I, Pereira GC, Pereira SP, Oliveira PJ. Sub-chronic administration of doxorubicin to Wistar rats results in oxidative stress and unaltered apoptotic signaling in the lung. Chem Biol Interact 2010; 188: 478-86.
[143] Patil RR, Guhagarkar SA, Devarajan PV. Engineered nanocarriers of doxorubicin: a current update. Crit Rev Ther Drug Carrier Syst 2008; 25: 1-61.
[144] Hideg K, Kálai T. Novel antioxidants in anthracycline cardiotoxic-ity. Cardiovasc Toxicol 2007; 7: 160-4.
[145] Wattanapitayakul SK, Chularojmontri L, Herunsalee A, Cha-ruchongkolwongse S, Niumsakul S, Bauer JA. Screening of anti-oxidants from medicinal plants for cardioprotective effect against doxorubicin toxicity. Basic Clin Pharmacol Toxicol 2005; 96: 80-7.
[146] Berthiaume JM, Oliveira PJ, Fariss MW, Wallace KB. Dietary vitamin E decreases doxorubicin-induced oxidative stress without preventing mitochondrial dysfunction. Cardiovasc Toxic 2005 Jan; 5: 257-67.
[147] Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 2007; 47629-56.
[148] Skulachev VP, Anisimov VN, Antonenko YN, et al. An attempt to prevent senescence: a mitochondrial approach. Biochim Biophys Acta 2009; 1787: 437-61.
[149] Hasinoff BB, Kala SV. The removal of metal ions from transferrin, ferritin and ceruloplasmin by the cardioprotective agent ICRF-187 [(+)-1, 2-bis(3, 5-dioxopiperazinyl-1-yl)propane] and its hydrolysis product ADR-925. Agents Actions 1993; 39: 72-81.
[150] Hofland KF, Thougaard AV, Sehested M, Jensen PB. Dexrazoxane protects against myelosuppression from the DNA cleavage-enhancing drugs etoposide and daunorubicin but not doxorubicin. Clin Cancer Res 2005; 11: 3915-24.
[151] Hampton JR. Beta-blockers in heart failure--the evidence from clinical trials. Eur Heart J 1996; 17 Suppl B: 17-20.
[152] Hydock DS, Lien C-Y, Schneider CM, Hayward R. Exercise pre-conditioning protects against doxorubicin-induced cardiac dysfunc-tion Med Sci Sports Exerc 2008; 40: 808-17.
[153] Wonders KY, Hydock DS, Schneider CM, Hayward R. Acute exercise protects against doxorubicin cardiotoxicity. Integr Cancer Ther 2008; 7: 147-54.
[154] Hydock DS, Wonders KY, Schneider CM, Hayward R. Voluntary wheel running in rats receiving doxorubicin: effects on running ac-
tivity and cardiac myosin heavy chain. Anticancer Res 2009; 29: 4401-7.
[155] Kavazis AN, Smuder AJ, Min K, Tumer N, Powers SK. Short-term exercise training protects against doxorubicin induced cardiac mi-tochondrial damage independent of HSP72. Am J Physiol Heart Circ Physiol 2010; 299: H1515-24.
[156] Ascensão A, Ferreira R, Magalhães J. Exercise-induced cardiopro-tection--biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. Int J Cardiol 2007; 117: 16-30.
[157] Sgobbo P, Pacelli C, Grattagliano I, Villani G, Cocco T. Carvedilol inhibits mitochondrial complex I and induces resistance to H2O2 -mediated oxidative insult in H9C2 myocardial cells. Biochim Bio-phys Acta 2007; 1767: 222-32.
[158] Eggertsen R, Sivertsson R, Andrén L, Hansson L. Haemodynamic effects of carvedilol, a new beta-adrenoceptor blocker and precapil-lary vasodilator in essential hypertension. J Hypertens 1984; 2: 529-34.
[159] Ruffolo RR, Gellai M, Hieble JP, Willette RN, Nichols AJ. The pharmacology of carvedilol. Eur J Clin Pharmacol 1990; 38 Suppl 2: S82-8.
[160] Dulin B, Abraham WT. Pharmacology of carvedilol. Am J Cardiol 2004; 93: 3B-6B.
[161] McPhillips JJ, Schwemer GT, Scott DI, Zinny M, Patterson D. Effects of carvedilol on blood pressure in patients with mild to moderate hypertension. A dose response study. Drugs 1988; 36 Suppl 6: 82-91.
[162] Marques MP, Oliveira PJ, Moreno AJ, Batista de Carvalho LA. Study of carvedilol by combined Raman spectroscopy and ab initio MO calculations. J Raman Spectrosc 2002; 33: 778-783.
[163] Spallarossa P, Garibaldi S, Altieri P, et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardio-myocytes in vitro. J Mol Cell Cardiol 2004; 37: 837-46.
[164] Yue TL, Cheng HY, Lysko PG, et al. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J Pharmacol Exp Ther 1992; 263: 92-8.
[165] Yue TL, Mckenna PJ, Lysko PG, et al. SB 211475, a metabolite of carvedilol, a novel antihypertensive agent, is a potent antioxidant. Eur J Pharmacol 1994; 251: 237-43.
[166] Aruoma OI. Peroxyl radical scavenging activity of the antihyper-tensive drug carvedilol. Toxicol In Vitro 1996; 10: 625-9.
[167] Tadolini B, Franconi F. Carvedilol inhibition of lipid peroxidation. A new antioxidative mechanism. Free Radic Res 1998; 29: 377-87.
[168] Oettl K, Greilberger J, Zangger K, et al. Radical-scavenging and iron-chelating properties of carvedilol, an antihypertensive drug with antioxidative activity. Biochem Pharmacol 2001; 62: 241-8.
[169] Nakamura K, Kusano K, Nakamura Y, et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 2002; 105: 2867-71.
[170] Oliveira PJ, Gonçalves LM, Monteiro P, Providência LA, Moreno AJ. Are the antioxidant properties of carvedilol important for the protection of cardiac mitochondria? Curr Vasc Pharmacol 2005; 3: 147-58.
[171] Nohl H. Demonstration of the existence of an organo-specific NADH dehydrogenase in heart mitochondria. Eur J Biochem 1987; 169: 585-91.
[172] Fraisse L, Rey E, Rigoulet M. The organo-specific external NADH dehydrogenase of mammal heart mitochondria has an artefactual origin. Biochim Biophys Acta 1993; 1143: 190-8.
[173] Oliveira PJ, Santos DJ, Moreno AJ. Carvedilol inhibits the exoge-nous NADH dehydrogenase in rat heart mitochondria. Arch Bio-chem Biophys 2000; 374: 279-85.
[174] Oliveira PJ, Rolo AP, Palmeira CM, Moreno AJ. Carvedilol re-duces mitochondrial damage induced by hypoxanthine/xanthine oxidase: relevance to hypoxia/reoxygenation injury. Cardiovasc Toxic 2001; 1: 205-13.
[175] Abreu RM, Santos DJ, Moreno AJ. Effects of carvedilol and its analog BM-910228 on mitochondrial function and oxidative stress. J Pharmacol Exp Ther 2000; 295: 1022-30.
[176] Neugebauer G, Akpan W, Möllendorff E von, Neubert P, Reiff K. Pharmacokinetics and disposition of carvedilol in humans. J Car-diovasc Pharmacol 1987; 10 Suppl 11: S85-8.
[177] Oliveira PJ, Rolo AP, Sardão VA, Coxito PM, Palmeira CM, Moreno AJ. Carvedilol in heart mitochondria: protonophore or opener of the mitochondrial K(ATP) channels? Life Sci 2001; 69: 123-32.

Doxorubicin, Carvedilol and Cardiac Mitochondria Current Pharmaceutical Design, 2011, Vol. 17, No. 20 2129
[178] Monteiro P, Duarte AI, Moreno AJ, Gonçalves LM, Providência LA. Carvedilol improves energy production during acute global myocardial ischaemia. Eur J Pharmacol 2003; 482: 245-53.
[179] Korshunov SS, Skulachev VP, Starkov A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 1997; 416: 15-8.
[180] Oliveira PJ, Coxito PM, Rolo AP, Santos DJ, Palmeira CM, More-no AJ. Inhibitory effect of carvedilol in the high-conductance state of the mitochondrial permeability transition pore. Eur J Pharmacol 2001; 412: 231-7.
[181] Oliveira PJ, Esteves T, Rolo AP, et al. Carvedilol: relation between antioxidant activity and inhibition of the mitochondrial permeabil-ity transition. Rev Port Cardiol 2003; 22: 55-62.
[182] Oliveira PJ, Esteves T, Rolo AP, Palmeira CM, Moreno AJ. Carve-dilol inhibits the mitochondrial permeability transition by an anti-oxidant mechanism. Cardiovasc Toxic2004; 4: 11-20.
[183] Oliveira PJ, Marques MP, Batista de Carvalho LA, Moreno AJ. Effects of carvedilol on isolated heart mitochondria: evidence for a protonophoretic mechanism. Biochem Biophys Res Commun 2000; 276: 82-7.
[184] Wang R, Miura T, Harada N, et al. Pleiotropic effects of the beta-adrenoceptor blocker carvedilol on calcium regulation during oxi-dative stress-induced apoptosis in cardiomyocytes. J Pharmacol Exp Ther 2006; 318: 45-52.
[185] Rolo AP, Oliveira PJ, Moreno AJ, Palmeira CM. Protective effect of carvedilol on chenodeoxycholate induction of the permeability transition pore. Biochem Pharmacol 2001; 61: 1449-54.
[186] Koitabashi N, Arai M, Tomaru K, et al. Carvedilol effectively blocks oxidative stress-mediated downregulation of sarcoplasmic reticulum Ca2+-ATPase 2 gene transcription through modification of Sp1 binding. Biochem Biophys Res Commun 2005; 328: 116-24.
[187] Yue TL, Ma XL, Wang X, et al. Possible involvement of stress-activated protein kinase signaling pathway and Fas receptor expres-sion in prevention of ischemia/reperfusion-induced cardiomyocyte apoptosis by carvedilol. Circ Res 1998; 82: 166-74.
[188] Fazio S, Palmieri EA, Ferravante B, Bonè F, Biondi B, Saccà L. Doxorubicin-induced cardiomyopathy treated with carvedilol. Clin Cardiol 1998; 21: 777-9.
[189] Matsui H, Morishima I, Numaguchi Y, Toki Y, Okumura K, Haya-kawa T. Protective effects of carvedilol against doxorubicin-induced cardiomyopathy in rats. Life Sci 1999; 65: 1265-74.
[190] Santos DJ, Moreno AJ, Leino RL, Froberg MK, Wallace KB. Carvedilol protects against doxorubicin-induced mitochondrial car-diomyopathy. Toxicol Appl Pharmacol 2002; 185: 218-27.
[191] Jonsson O, Behnam-Motlagh P, Persson M, Henriksson R, Grankvist K. Increase in doxorubicin cytotoxicity by carvedilol in-hibition of P-glycoprotein activity. Biochem Pharmacol 1999; 58: 1801-6.
[192] Oliveira PJ, Moreno AJ. Carvedilol, An Hypertensive Drug, Pro-tects Heart Mitochondria From Oxidative Damage. In: Moreno AJ, Oliveira PJ, Palmeira CM, Ed. Mitochondrial Pharmacology and Toxicology. Trivandrum: Transworld Research Network 2006; pp. 23-53.
[193] Kalay N, Basar E, Ozdogru I, et al. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. Am J Cardiol 2006; 48: 2258-62.
[194] Balaban RS. The mitochondrial proteome: a dynamic functional program in tissues and disease states. Environ Mol Mutagen 2010; 51: 352-9.
[195] Bugger H, Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res 2010; 88: 229-40.
[196] Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res 2010; 88: 40-50.
[197] Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 2011; 18.
[198] Pessayre D, Mansouri A, Berson A, Fromenty B. Mitochondrial involvement in drug-induced liver injury. Handb Exp Pharmacol 2010; 196: 311-65.
[199] Hardin R, Gersl V, Klimtová I, Sim nek T, Machácková J, Adam-cová M. Anthracycline-induced cardiotoxicity. Acta Medica (Hradec Kralove) 2000; 43: 75-82.
Received: June 8, 2011 Accepted: June 24, 2011

Mitochondrion xxx (2011) xxx–xxx
MITOCH-00664; No of Pages 11
Contents lists available at ScienceDirect
Mitochondrion
j ourna l homepage: www.e lsev ie r.com/ locate /mi to
Review
Regulation and protection of mitochondrial physiology by sirtuins
Claudia V. Pereira a, Magda Lebiedzinska b, Mariusz R. Wieckowski b, Paulo J. Oliveira a,⁎a CNC-Center for Neuroscience and Cell Biology, University of Coimbra, Portugalb Nencki Institute of Experimental Biology, Warsaw, Poland
Abbreviations: Acetyl-CoA, acetyl-coenzyme A; AceCfactor; AMPK, 5′ adenosine monophosphate-activated prphosphate synthase; CR, calorie restriction; CREB, cAMPEstrogen-related receptor alpha; FAO, fatty acid oxidatioHIF1α, Hypoxia-inducible factor 1, alpha subunit; HMdehydrogenase; MEFs, mouse embryonic fibroblasts; MPdismutase; NAD+, nicotinamide adenine dinucleotide;ornithine transcarbamoylase; PPARγ, peroxisome proligamma coactivator 1-alpha; Pol1, polimerase1; ROS, reacTumor necrosis factor; UCP2, uncoupling protein 2.⁎ Corresponding author at: Center for Neuroscience an
+351 239 855789.E-mail address: [email protected] (P.J. Oliveira).
1567-7249/$ – see front matter © 2011 Elsevier B.V. andoi:10.1016/j.mito.2011.07.003
Please cite this article as: Pereira, C.V., etdoi:10.1016/j.mito.2011.07.003
a b s t r a c t
a r t i c l e i n f oArticle history:Received 3 November 2010Received in revised form 16 February 2011Accepted 7 July 2011Available online xxxx
Keywords:MitochondriaSirtuinsToxicologyOxidative stressMetabolism
The link between sirtuin activity and mitochondrial biology has recently emerged as an important field. Thisconserved family of NAD+-dependent deacetylase proteins has been described to be particularly involved inmetabolism and longevity. Recent studies on protein acetylation have uncovered a high number of acetylatedmitochondrial proteins indicating that acetylation/deacetylation processes may be important not only forthe regulation of mitochondrial homeostasis but also for metabolic dysfunction in the context of variousdiseases such as metabolic syndrome/diabetes and cancer. The functional involvement of sirtuins as sensorsof the redox/nutritional state of mitochondria and their role in mitochondrial protection against stressare hereby described, suggesting that pharmacological manipulation of sirtuins is a viable strategy againstseveral pathologies.
© 2011 Elsevier B.V. and Mitochondria Research Society. All rights reserved.
Contents
1. Sirtuins: function and families . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.2. Classification of sirtuins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.3. Sirtuin enzymatic activity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.4. Sirtuin protein targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.5. Tissue specificity and intracellular localization of mammalian sirtuins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.6. Cytosolic and nuclear sirtuins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.7. Mitochondrial sirtuins: characterization and activity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
1.7.1. Localization and processing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.7.2. Regulation of mitochondrial metabolism by sirtuins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01.7.3. Protection of mitochondrial function by sirtuins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
1.8. Future perspectives: are sirtuins good therapeutic targets?. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0Acknowledgments. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
S2, acetyl-coenzyme A synthethase 2; ACC, Acetyl-CoA carboxylase; ADP, adenosine diphosphate; AIF, apoptosis-inducingotein kinase; ANT, adenine nucleotide translocator; ATP, adenosine triphophate; BER, base excision repair; CPS1, Carbamoylresponse element-binding; CypD, cyclophilin D; DNA-PK, DNA-dependent protein kinase; DSB, double-strand break; ERR α,n; FOXO, Forkhead box O; GDH, glutamate dehydrogenase; H4, histone 4; HDL, high-density lipoprotein; HFD, high fat diet;GCS2, 3-hydroxy-3-methylglutaryl CoA synthase 2 Km, Michaelis-Menten constant; LCAD, long chain acyl coenzyme AP, mitochondrial processing peptidase; mPTP, mitochondrial permeability transition pore; MnSOD, manganese superoxideNDUFA9, NADH dehydrogenase [ubiquinone]-1 alpha subcomplex subunit; OSCC, oral squamous cell carcinoma; OTC,ferator-activated receptor; PARP-1, Poly [ADP-ribose] polymerase 1; PGC-1α, peroxisome proliferator-activated receptortive oxygen species; SIRT, sirtuin; Sir2, Silent Information Regulator Two (Sir2) protein; SOD2, superoxide dismutase 2; TNF,
d Cell Biology, Largo Marques de Pombal, University of Coimbra, 3004-517 Coimbra, Portugal. Tel.: +351 239 855760; fax:
d Mitochondria Research Society. All rights reserved.
al., Regulation and protection of mitochondrial physiology by sirtuins, Mitochondrion (2011),

2 C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
1. Sirtuins: function and families
1.1. Introduction
The involvement of mitochondria in multiple fundamental cellularprocesses places these organelles under the highlight of manyresearchers. In fact, mitochondria are now considered one majortarget for many therapeutic approaches. Oxidative stress has been aparticular fulcrum of interest since mitochondrial oxidative damagehas been recognized as being involved in many diseases and in theaging process itself. Also, disrupted mitochondrial metabolism maybe one of the critical elements leading to cancer, diabetes, age-associated and neurodegenerative disorders (Campisi and Yaswen,2009; Harman, 1956; Sultana and Butterfield, 2010; Weber andReichert, 2010; Zhu and Chu, 2010). In this fascinating area ofmitochondrial disease and protection, sirtuins are being speciallyfocused, since these proteins are able to regulate stress responsesand cell survival (Canto and Auwerx, 2009; Gan and Mucke, 2008).One of the most intensively investigated compound, resveratrol, is aknown sirtuin 1 (SIRT1) activator, shown to have a positive effect onmitochondrial metabolism and to delay the aging process (Alcain andVillalba, 2009; Finkel et al., 2009). The present review comprises ourrecent knowledge in the field of sirtuins with a special focus on themost recent and breakthrough studies related with the protectionand regulation of mitochondrial physiology by SIRT3, SIRT4 andSIRT5. Several other reviews on the subject are available, including thevery interesting work by Huang et al.(2010). The present reviewexpands the previous works by incorporating novel and excitingresults, which allows the reader to get a full picture of how sirtuins ingeneral, andmitochondrial sirtuins in particular, contribute to cellularand mitochondrial protection.
1.2. Classification of sirtuins
Sirtuins, or Silent information regulator proteins (Sir), and theirhomologs are present in a very wide range of organisms, from bacteriato humans, forming a conserved family of proteins (Denu, 2005). Upto date, seven sirtuin homologues have been identified in mammaliancells. These have different intracellular localization as well as variousroles in cell physiology (Blander and Guarente, 2004; Tanny et al.,1999). Based on the phylogenetic analysis of the core domain,mammalian sirtuins have been classified into four classes togetherwith other Sir2-related proteins widely distributed in eukaryotesand prokaryotes (Frye, 2000; Smith et al., 2000). Mammalian SIRT1(62.0 kDa), SIRT2 (41.5 kDa) and SIRT3 (43.6 kDa) are consideredClass I sirtuins, while SIRT4 (35.2 kDa) is a Class II sirtuin. Class IIISIRT5 (33.9 kDa) and Class IV SIRT6 (39.1 kDa) and SIRT7 (44.8 kDa)are other examples. All the described sirtuins contain a conserved 275amino acid catalytic core domain together with N-terminal and/or C-terminal domains. Additionally, a novel class (“U”) has been createdto include sirtuins with unique features, such as gram-positivebacteria and Termoga maritime sirtuins. An appropriate Classaffiliation of the individual Sir2-related proteins has been describedin the comprehensive review article by Michan and Sinclair(2007).A list of the different sirtuins, plus other details on their physiology/activity, can be seen in Table 1.
1.3. Sirtuin enzymatic activity
All sirtuins, with only one exception (SIRT4), catalyze proteindeacetylation in which the lysine acetyl group is transferred fromthe target protein to the ADP-ribose component of NAD+, which leadsto their full dependence on NAD+ availability and indicates thatsirtuins can be sensors of the cellular redox state. As a consequence,sirtuin activity leads to the generation of deacetylated proteins, 2′-O-acetyl-ADP ribose and nicotinamide (Sauve, 2010). Moreover, SIRT6
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
additionally demonstrates ADP-ribosyl transferase activity whileSIRT4 (as mentioned earlier) demonstrates ADP-ribosyl transferaseactivity only. As described above, sirtuin activity is regulated by NAD+
availability. On the other hand, nicotinamide noncompetitivelyinhibits sirtuins suggesting that the deacetylation reaction productcan also act as an endogenous regulator of sirtuin activity. Interest-ingly, isonicotinamide, which binds to the nicotinamide pocket ofyeast sirtuin (Sir2), only inhibits the base exchange, with thedeacetylation activity remaining unaffected or even being increased.This fact indicates that chemical compounds such as isonicotinamidecan act as potent sirtuins activators in mammalian cells. Additionally,it has been demonstrated that activity of yeast sirtuin Hst2 (a Class Imember) can be regulated by homo-oligomerization of the enzyme.Anotherwell knownand currently intensively studied sirtuin activatoris resveratrol, a polyphenol present in different sources, including forexample grapes skin and red wine (Lagouge et al., 2006). It has beendemonstrated that polyphenols and especially resveratrol, reduce Km
values for sirtuin substrates, resulting in different biological effectsas increased resistance to apoptosis and to stress stimuli. Apart fromnicotinamide, an inhibitory effect on sirtuins has also been describedfor several other compounds. The most well-known are sirtinol,splitomicin (specific for yeast sirtuins Hst1 and Sir2) and dehydros-plitomicin (specific mostly to sirtuin Hst1). Human sirtuins are notinhibited significantly by splitomicin aswell as by dehydrosplitomicin.More about the mechanism of protein deacetylation carried out bysirtuins as well as detail on their inhibitors and activators can be foundin the review of Sauve et al. (Sauve et al., 2006).
1.4. Sirtuin protein targets
Potential sirtuins substrates are various acetylated proteinsinvolved in cell metabolism, apoptosis and regulation of genetranscription. Sirtuins can thus determine the ability of cells to adaptto different conditions (Finkel et al., 2009; Haigis and Guarente,2006; Vaquero, 2009). Published data suggest a significant role ofdeacetylation inmetabolic responses to fasting or caloric restriction aswell as in the response to different stress stimuli, including forexample oxidative stress (Choudhary et al., 2009; Schwer et al., 2009).Among the several proteins which are deacetylated by sirtuins,histones and transcription factors such as p53 (Li et al., 2010), FOXO(Brunet et al., 2004), peroxisome proliferator activated receptor γ(PPARγ) (Picard et al., 2004), nuclear factor-κB (NFκB) (Kawaharaet al., 2009) and PGC-1α (Sugden et al., 2010) can be found. Sirtuinsare also able to deacetylate α-tubulin (Tang and Chua, 2008) andacetyl-CoA synthetase (Hallows et al., 2006). Another importantprotein for metabolism, glutamate dehydrogenase (GDH) is a well-known example of ADP-ribosylation substrates for SIRT4 (Haigiset al., 2006). The variety of sirtuin substrates, either being crucialenzymes or gene regulatory elements, indicates the possibility thatmultiple sirtuins may modulate cell physiology and metabolismthrough interacting with distinct targets regulated under a widerange of physiological conditions.
1.5. Tissue specificity and intracellular localization of mammalian sirtuins
Molecular analysis revealed that SIRT1, 2, 3, 5 and 6 areubiquitously expressed in different tissues and organs. The highestamount of SIRT6 was detected in muscles, brain and heart, whileSIRT4 is mostly present in muscle, kidney, testis and liver (Michishitaet al., 2005). By its turn, SIRT7 has been found in brain, kidney, liver,lung and adipose tissue (Michishita et al., 2005). Further detailedstudies determined subcellular localization of several sirtuins. Amongseven mammalian sirtuins, SIRT1 and SIRT2 show both cytoplasmicand nuclear localization, while SIRT6 and SIRT7 are only locatedin the nucleus and nucleoli, respectively (Michishita et al., 2005).SIRT3, SIRT4 and SIRT5 are mitochondrial proteins, although SIRT3
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),
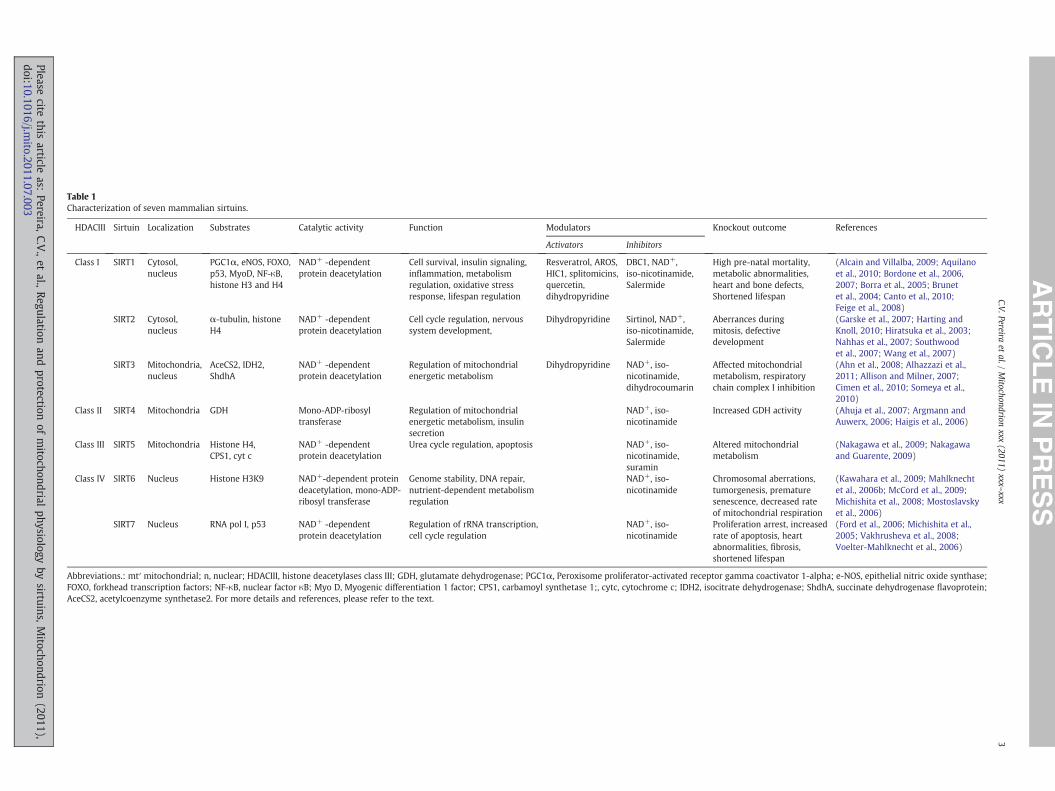
Table 1Characterization of seven mammalian sirtuins.
HDACIII Sirtuin Localization Substrates Catalytic activity Function Modulators Knockout outcome References
Activators Inhibitors
Class I SIRT1 Cytosol,nucleus
PGC1α, eNOS, FOXO,p53, MyoD, NF-κB,histone H3 and H4
NAD+ -dependentprotein deacetylation
Cell survival, insulin signaling,inflammation, metabolismregulation, oxidative stressresponse, lifespan regulation
Resveratrol, AROS,HIC1, splitomicins,quercetin,dihydropyridine
DBC1, NAD+,iso-nicotinamide,Salermide
High pre-natal mortality,metabolic abnormalities,heart and bone defects,Shortened lifespan
(Alcain and Villalba, 2009; Aquilanoet al., 2010; Bordone et al., 2006,2007; Borra et al., 2005; Brunetet al., 2004; Canto et al., 2010;Feige et al., 2008)
SIRT2 Cytosol,nucleus
α-tubulin, histoneH4
NAD+ -dependentprotein deacetylation
Cell cycle regulation, nervoussystem development,
Dihydropyridine Sirtinol, NAD+,iso-nicotinamide,Salermide
Aberrances duringmitosis, defectivedevelopment
(Garske et al., 2007; Harting andKnoll, 2010; Hiratsuka et al., 2003;Nahhas et al., 2007; Southwoodet al., 2007; Wang et al., 2007)
SIRT3 Mitochondria,nucleus
AceCS2, IDH2,ShdhA
NAD+ -dependentprotein deacetylation
Regulation of mitochondrialenergetic metabolism
Dihydropyridine NAD+, iso-nicotinamide,dihydrocoumarin
Affected mitochondrialmetabolism, respiratorychain complex I inhibition
(Ahn et al., 2008; Alhazzazi et al.,2011; Allison and Milner, 2007;Cimen et al., 2010; Someya et al.,2010)
Class II SIRT4 Mitochondria GDH Mono-ADP-ribosyltransferase
Regulation of mitochondrialenergetic metabolism, insulinsecretion
NAD+, iso-nicotinamide
Increased GDH activity (Ahuja et al., 2007; Argmann andAuwerx, 2006; Haigis et al., 2006)
Class III SIRT5 Mitochondria Histone H4,CPS1, cyt c
NAD+ -dependentprotein deacetylation
Urea cycle regulation, apoptosis NAD+, iso-nicotinamide,suramin
Altered mitochondrialmetabolism
(Nakagawa et al., 2009; Nakagawaand Guarente, 2009)
Class IV SIRT6 Nucleus Histone H3K9 NAD+-dependent proteindeacetylation, mono-ADP-ribosyl transferase
Genome stability, DNA repair,nutrient-dependent metabolismregulation
NAD+, iso-nicotinamide
Chromosomal aberrations,tumorgenesis, prematuresenescence, decreased rateof mitochondrial respiration
(Kawahara et al., 2009; Mahlknechtet al., 2006b; McCord et al., 2009;Michishita et al., 2008; Mostoslavskyet al., 2006)
SIRT7 Nucleus RNA pol I, p53 NAD+ -dependentprotein deacetylation
Regulation of rRNA transcription,cell cycle regulation
NAD+, iso-nicotinamide
Proliferation arrest, increasedrate of apoptosis, heartabnormalities, fibrosis,shortened lifespan
(Ford et al., 2006; Michishita et al.,2005; Vakhrusheva et al., 2008;Voelter-Mahlknecht et al., 2006)
Abbreviations.: mt′ mitochondrial; n, nuclear; HDACIII, histone deacetylases class III; GDH, glutamate dehydrogenase; PGC1α, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; e-NOS, epithelial nitric oxide synthase;FOXO, forkhead transcription factors; NF-κB, nuclear factor κB; Myo D, Myogenic differentiation 1 factor; CPS1, carbamoyl synthetase 1;, cytc, cytochrome c; IDH2, isocitrate dehydrogenase; ShdhA, succinate dehydrogenase flavoprotein;AceCS2, acetylcoenzyme synthetase2. For more details and references, please refer to the text.
3C.V
.Pereiraet
al./Mitochondrion
xxx(2011)
xxx–xxx
Pleasecite
thisarticle
as:Pereira,
C.V.,et
al.,Regulation
andprotection
ofmitochondrial
physiologyby
sirtuins,Mitochondrion
(2011),doi:10.1016/j.m
ito.2011.07.003

4 C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
can be also found in the nucleus.(Schwer and Verdin, 2008; Shobaet al., 2009).
1.6. Cytosolic and nuclear sirtuins
SIRT1, the closest ortholog of yeast Sirt2, is the most well studiedmammalian sirtuin, harboring both cytosolic and nuclear localization(although absent from nucleoli). SIRT1 has multiple functions andits role was confirmed in the regulation of energy metabolism (Yuand Auwerx, 2010), embryonic development (Mayanil et al., 2006;Saunders et al., 2010), myocyte differentiation (Fulco et al., 2003), cellsurvival and apoptosis (Guo et al., 2010; Yi and Luo, 2010) as well asin the regulation of gene transcription by histone deacetylation(Zhang and Kraus, 2010). SIRT1 is also known tomediate the effects ofcaloric restriction (Gillum et al., in press) and to play an importantrole in response to different stress stimuli (Leiser and Kaeberlein,2010). Additionally, SIRT1 may have an impact on the aging rate(Donmez and Guarente, 2010). The nuclear fraction of SIRT1 is alsolocalized to promyelocytic leukemia protein (PML) bodies, where itinteracts with the tumor suppressor p53 which carries severalacetylation sites. Deacetylation of lysine residues in the p53 proteinprocessed by SIRT1 decreases its transactivation activity, which inturn can suppress apoptosis initiated by DNA damage or oxidativestress (Kume et al., 2006; Sedding, 2008; Yi and Luo, 2010). The effectof SIRT1 on cell survival can be also mediated by FOXO transcriptionfactors that play an important role in cell adaptation to stressconditions including Foxo1, Foxo3a and Foxo4. Under oxidative stress,deacetylation of Foxo3a induces its translocation from cytosol to thenucleus where it forms a complex with SIRT1, regulating theexpression of some antioxidant enzymes, such as mitochondrialsuperoxide dismutase (SOD2) and catalase. At the same time, FOXO3aincreases the expression of cell-cycle checkpoint and DNA repairgenes, arresting the cell cycle (Brunet et al., 2004; Jacobs et al., 2008;Motta et al., 2004; Wang et al., 2007). FOXO1-mediated inhibition ofapoptosis occurs via FOXO4 activity regulation. In transformedepithelial cells, deacetylated FOXO4 suppresses two proapoptoticcaspases, caspase-3 and caspase-7, leading to an arrest of apoptosis(Ford et al., 2005; Frescas et al., 2005). In turn, deacetylation of FOXO1results in the expression of gluconeogenesis genes under control ofFOXO1 (Frescas et al., 2005).
Other SIRT1 targets such as PPARγ and PGC-1α are importantelements controlling energetic balance, especially fatty acid andglucose metabolism (Michan and Sinclair, 2007; Sugden et al., 2010).SIRT1 content and activity is increased under starvation and caloricrestriction (CR) (Chen et al., 2008; Nisoli et al., 2005). Deacetylationof PGC-1α by SIRT1 also stimulates mitochondrial biogenesis.Experiments by using transgenic animals showed that not all effectsof CR are mediated by SIRT1 but it is noteworthy that both SIRT1overexpression and caloric restriction are manifested by lower bodyweight, reduced cholesterol, glucose and insulin serum level and alsoby improved physical performance (Chen et al., 2005; Nisoli et al.,2005) Lower fat accumulation in SIRT1-overexpressing animals isassociated with PPARγ transcriptional repression which results inthe inhibition of adipogenesis (Picard et al., 2004). In the liver,SIRT1 regulates glycolysis, gluconeogenesis and fatty acid oxidationthrough PGC-1α deacetylation (Rodgers and Puigserver, 2007).Lipid metabolism (HDL biogenesis) in the liver is also stimulated bySIRT1-dependent Liver's X receptor deacetylation (Li et al., 2007).Deacetylation of PGC-1α by SIRT1 also stimulates mitochondrialbiogenesis and induces oxidative phosphorylation (Lagouge et al.,2006). Similarly, activation of SIRT1 by resveratrol is connected toenhanced mitochondrial biogenesis and to more efficient metabolismin the skeletal muscle (Aquilano et al., 2010; Chabi et al., 2009). SIRT1is also found to be involved in insulin secretion in pancreatic β-cells(Chen et al., 2010; Liang et al., 2009). The most convincing proofof concept comes from experiments on SIRT1-overexpressing mice
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
which present more efficient energy metabolism of pancreatic β-cellsthanwild-type individuals (Bordone et al., 2006). This is caused by therepression of uncoupling protein 2 (UCP2) gene expression by SIRT1,resulting into decreased content of mitochondrial UCP2 in pancreaticβ-cells, higher mitochondrial coupling and higher rate of ATPproduction, which loops to enhanced insulin secretion by pancreaticβ-cells (Bordone et al., 2006). SIRT1 knockout animals showed a lowerlevel of ATP in pancreatic β-cells as a result of increased UCP2 leveland they exhibited a decrease insulin secretion in the response toglucose. As indicated above, the feedback between mitochondrial ATPand insulin secretion is enhanced in SIRT1-overexpressing mice(Bordone et al., 2007; Chen et al., 2005).
SIRT2 is a mainly cytosolic protein, although a small amount wasalso detected in the nucleus,where it carries out histone deacetylation.In vitro studies demonstrated that SIRT2, similarly to SIRT1, preferen-tially deacetylates histone H4 (Vaquero et al., 2007). In the cytosol,SIRT2 was found to be associated with microtubules, where itdeacetylates α-tubulin (Harting and Knoll, 2010). Based on theobservations that SIRT2 increases during the mitotic phase and co-localizes with chromatin during transition from G2 to M phase, it isthought that this sirtuin can be involved in the regulation of the cellcycle (Dryden et al., 2003). More detailed studies on sirtuins showedthat SIRT2 plays an important role in glial cells development,microtubule dynamics in oligodendrocytes and maintenance ofaxonal integrity (Michan and Sinclair, 2007; Tang and Chua, 2008),which makes this isoform an attractive therapeutic target inneurodegenerative diseases (Wu et al., 2010). SIRT2 was also foundto be involved in Parkinson's and Alzheimer's disease and also ina Huntington's disease model induced in Drosophila (Pallos et al.,2008). Additionally, the development and progression of manybrain tumors are connected with a disturbance in SIRT2 deacetylaseactivity (Hiratsuka et al., 2003). Thus, many reports describe attemptsto modulate SIRT2 activity with the objective of treating severalmalignancies (Harting and Knoll, 2010).
Less published data exists for SIRT6 and SIRT7. Nonetheless, it hasbeen demonstrated that SIRT6 is similar to SIRT1 regarding themaintenance of genome integrity, since deletion of this protein leadsto different chromosomal aberrations, impairs cells proliferation andincreases the rate of DNA damage accumulation (Michishita et al.,2008). There is growing evidence indicating that SIRT6 is involvedin base excision repair (BER) of single-stranded DNA breaks.Moreover, SIRT6 is directly involved in DNA repair by forming amacromolecular complex with the DNA double-strand break (DSB)repair factor, DNA-PK (DNA-dependent protein kinase) promotingDSB repair. Cells lacking SIRT6 are more sensitive to DNA damagingagents (McCord et al., 2009). Moreover, SIRT6 can be considered alsoas a regulator of metabolism under variable nutrient availability(Kanfi et al., 2008). Although SIRT6 has weak deacetylase activity, itcan deacetylate histone H3K9, which makes SIRT6 involved in thestability of telomeric chromatin structure (Michishita et al., 2008).SIRT6 depletion is associated with premature senescence due toabnormal telomere structure. It was found that interaction of WRN,a factor mutated in human premature aging syndrome (Wernersyndrome), with histone H3K9 requires proper SIRT6 activity(Michishita et al., 2008). By deacetylating histone H3K9, SIRT6 mayadditionally regulate expressionof glycolytic genes, especially becauseit co-represses transcription of Hif1α, which is crucial for the responseto nutrient deprivation. In SIRT6 knockout cells, increased glucoseuptake and higher glycolysis rate is followed by a decrease inmitochondrial respiration. All these metabolic changes are associatedwith a higherHif1α activity. The regulatory role seems to be importantin several pathologic states associated with diabetes and obesity(Zhong et al., 2010). Moreover, SIRT6 has been associated with theproduction of pro-inflammatory cytokines such as TNF-α by immunecells (VanGool et al., 2009). Recently, it was described that SIRT6 playsa role in the control of somatic growth and in the prevention of obesity
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),

5C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
by modulating neural chromatin structure and gene activity (Schweret al., 2010).
SIRT7 was found to regulate transcription of ribosomal genes in thenucleus by interacting and regulating RNA polymerase (Pol1) activitywhichmeans that this sirtuin can have an important impact onmultiplecellular processes (Ford et al., 2006). In fact, it has been shown thattranscription of rRNA directly depends on SIRT7 content (Ford et al.,2006; Mostoslavsky et al., 2006), with the highest SIRT7 level found inblood and CD33+ myeloid bone marrow precursor cells. On the otherhand, lower SIRT7 levels were observed in ovaries and mammalianskeletal muscle (Voelter-Mahlknecht et al., 2006). In case of SIRT7ablation, cells do not proliferate and enter the apoptotic pathway (Fordet al., 2006), as opposed to an increased SIRT7 level, which has beenassociated with carcinogenesis. In fact SIRT7 overexpression was foundto occur in breast cancer cells (Ashraf et al., 2006). Therefore, the roleof SIRT7 in the regulation of cell cycle is important for cell adaptationprocesses under stress conditions. Interestingly, SIRT7 knockoutanimals suffer from heart hypertrophy, fibrosis and inflammatorycardiomyopathy and shortened lifespan (Vakhrusheva et al., 2008).
1.7. Mitochondrial sirtuins: characterization and activity
1.7.1. Localization and processingAmong the seven sirtuins already here described, three (SIRT3,
SIRT4 and SIRT5) are present inside mitochondria, regulating severalmetabolic pathways through protein deacetylation (Fig. 1). It hasbeen shown that about 1/5 of mitochondrial proteins undergo
Fig. 1. Mitochondrial sirtuins and their targets. SIRT3 is the major mitochondrial deacetyla(NDUFA9) and also with ShdhA, one of the flavoprotein subunits of complex II. SIRT3 plays ato regulate the Krebs cycle since it deacetylates Acetyl-CoA synthase 2, which catalyzesdehydrogenase 2 which catalyzes a key regulation point in the Krebs cycle. SIRT3 can also douter membrane. SIRT4 is a NAD-dependent mono-ADP-ribosyltransferase that is known to tand allows it to undergo subsequential steps in the Krebs cycle. GDH can also be deacetylatnegative regulator of oxidative metabolism in contrast with SIRT3. Finally, SIRT5 is knownphosphate synthetase (CPS1), the enzyme that catalyzes the first step of this cycle. Also, SIRTwas also pointed to SIRT3.
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
acetylation, being one of the highest pools in the cell. Proteomicstudies ofmice livermitochondria revealed almost 300 lysine residuesthat can be acetylated in 133 proteins involved in different metabolicpathways, including Krebs and urea cycle, fatty acid beta-oxidationand oxidative phosphorylation (Kim et al., 2006). Lysine acetylationprofile of mitochondrial proteins demonstrates some similaritieswith prokaryotic organisms, which appears to support the symbioticorigin of mitochondria hypothesis (Huang et al., 2010). All threemitochondrial sirtuins are located in themitochondrial matrix (Huanget al., 2010); however, SIRT5 was also found in the mitochondrialintermembrane space (Nakamura et al., 2008; Schlicker et al.,2008). Interestingly, SIRT5 overexpression does not increase themitochondrial intermembrane pool of this protein but resultsinstead into a preferential accumulation in the mitochondrial matrix(Nakagawa et al., 2009). There are some controversial data showingthat in COS7 cells co-expressing SIRT3 and SIRT5, the 44 kDa isoformof SIRT3 can be found in the nucleus, although its function stillwaits to be elucidated (Nakamura et al., 2008; Scher et al., 2007). Itwas speculated that under normal conditions, both a mitochondrialand nuclear localization of SIRT3 is possible, although under stressfulconditions, most of the nuclear pool moves to mitochondria (Scheret al., 2007). Mitochondrial sirtuins, especially SIRT3 and SIRT4,are responsible for sensing NAD+/NADH balance, which by beingdisrupted by several factors may affect cellular redox homeostasis(Yang et al., 2007; Yu and Auwerx, 2009).
All three mitochondrial sirtuins are encoded by nuclear DNAand contain a mitochondrial targeting sequence located on their
se which has been shown to physically interact with one of the subunits of complex Imajor role in fatty acid oxidation and in maintaining ATP levels. SIRT3 is also importantthe conversation of acetyl-CoA into acetate. Another target is the enzyme isocitrateeacetylate cyclophilin D and induce hexokinase II dissociation from the mitochondrialarget glutamate dehydrogenase (GDH), which converts glutamate into α-ketoglutarateed by SIRT3. SIRT4 also seems to interact with the ANT isoforms 2 and 3 and to act as ato play an important role in the urea cycle, deacetylating and activating carbamoyl
5 might deacetylate cytochrome c, pointing a pro-apoptotic function for this sirtuin as it
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),

6 C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
N′ terminal end, which is immediately cleaved after reaching themitochondrial matrix. Tissue expression profile of SIRT3, SIRT4 andSIRT5 is similar; however, relatively higher levels of SIRT5 have beenobserved in muscles (Hallows et al., 2008; Nakamura et al., 2008).
SIRT3 is a 28 kDa deacytelase, with, as mentioned before, a 44 kDaisoform found in nucleus, which requires proteolitic processing toachieve its maximal deacetylase activity. After reachingmitochondria,premature SIRT3 is cleaved at the N′-terminus targeting peptide bythe mitochondrial processing peptidase (MPP) (Schwer et al., 2002).
SIRT4 is a 35 kDa protein with a ADP-ribosylotransferase activitypresent in mitochondrial matrix. SIRT4 transfers ADP-ribose fromNAD+ to target proteins. In vitro studies showed no detectabledeacetylase activity of SIRT4 but it cannot be excluded that a veryspecific substrate would have to be present in order to observe somerelevant activity (Huang et al., 2010). Similarly to SIRT3, SIRT4 isalso processed in the mitochondrial matrix where a 28 amino acidsequence is cleaved when pro-SIRT4 reaches its destination (Schweret al., 2002).
The 33.0 kDa SIRT5 is present in the mitochondrial matrix, butwhen overexpressed, it can also be found in the mitochondrialintermembrane space. SIRT5 N-terminal 36 amino acids sequence isimmediately cleaved after import into the mitochondrial matrix(Mahlknecht et al., 2006a; Michishita et al., 2005).
1.7.2. Regulation of mitochondrial metabolism by sirtuinsAs described above, reversible protein acetylation allows cells to
adjust to a changing of the environment conditions. A key questionis why and where do mitochondrial proteins become acetylated?This question has not been answered in detail yet but it has beenconfirmed that protein acetylation does occur in mitochondria (Guanand Xiong, 2011) and, in fact, it is also been suggested that theacetylation mechanism in mitochondrial proteins is different fromcytosolic and nuclear proteins (Kim et al., 2006). Studies in knockoutSIRT4 and SIRT5 mice showed that there was no change in theacetylation level of mitochondrial proteins, but studies on SIRT3−/−
showed a significant increase in protein acetylation (Huang et al.,2010; Lombard et al., 2007). This finding suggests that SIRT3 seemsto play a central role in protein deacetylation in mitochondria. Itis known that sirtuins are directly or indirectly involved in manydifferent metabolic pathways including lipid metabolism (Lomb et al.,2010), calorie restriction conditions/insulin uptake (Ahuja et al.,2007; Guarente, 2008; Qiu et al., 2010b), urea cycle (Nakagawa et al.,2009; Nakagawa and Guarente, 2009), glycolysis, gluconeogenesisand Krebs cycle (Huang et al., 2010; Verdin et al., 2010).
Nowadays, major threats to human health are cancer, neurode-generative diseases, immune dysfunction and certainly, the metabolicsyndrome (Guarente, 2006). In the US, around 32% of adults and17% of children and adolescents are obese (Elliott and Jirousek, 2008).The human body is not adapted to process the excess of calorieintake per day. Also, in many regards, all of these diseases are alsoassociated with the aging process. When organisms are calorierestricted (30–40% decrease in food intake), it was shown to bepossible to increase the lifespan up to 50%, having sirtuins a veryimportant role in the adaptation process (Guarente, 2007; Qiuet al., 2010b). When food is scarce, mammals may have to adaptto the limited resources in order to maintain the basic functionssuch as growth, synthesis and reproduction, having to shut downunnecessary energy expenditures in order to survive. Thus, tissuesthat are crucial for survival such as the muscle, the heart andcertain parts of the brain are protected from starvation and gothrough mild calorie restriction. On the other hand, tissues thatare normally associated with endocrine and exocrine activity, suchas the liver, pancreas and the reproductive system, are very likelyto experience severe calorie restriction (Qiu et al., 2010b). Uponabundance of resources, energy is stored as fat. During calorierestriction, animals up-regulate fatty-acid oxidation and switch
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
fuel usage from glucose to fatty-acids. At a cellular level, increasedmitochondrial biogenesis in calorie-restricted tissues suggests atissue-specific increase in metabolic rate of the affected animals (Qiuet al., 2010b). Calorie-restricted animals have low levels of bloodinsulin in response to limited food intake. Low insulin levels suppressglycolysis and maintain blood glucose levels. As described above,SIRT1 suppresses UCP2 in pancreatic β-cells, which allows for a moreefficient ATP production (Bordone et al., 2006). Nevertheless, thefulcrum of calorie restriction response seems to be mitochondrialactivation which allows a metabolic adaptation to chronic energydeficit demand (Qiu et al., 2010b). Here is where sirtuins have nowthe spotlight. Fig. 1 shows a summary of the interplay betweenmitochondrial and in-house sirtuins.
1.7.2.1. SIRT3 and regulation of mitochondrial metabolism. SIRT3 is aunique mitochondrial sirtuin once it is the only one that was relatedwith extended lifespan in humans (Rose et al., 2003). SIRT3 may becritical in sensing NAD+ levels in the mitochondria since increasedNAD+ would trigger a regulatory pathway that would activate SIRT3leading to the deacetylation of specific targets. It has been demon-strated that mice deficient in SIRT3 present hyper protein acetylation(Lombard et al., 2007), including of the metabolic enzyme glutamatedehydrogenase (GDH) suggesting that this sirtuin may have animportant impact in metabolic control (Schlicker et al., 2008).Lombard et al. concluded that SIRT3 deficient mice are metabolicallyunremarkable under both fed and fasted conditions, including normalthermogenesis (Lombard et al., 2007). Another study demonstratedthat SIRT3 was able to deacetylate and activate the mitochondrialenzyme acetyl-CoA synthase 2 (Hallows et al., 2006), an enzyme thatcatalyzes the formation of Acetyl-CoA from acetate (Hallows et al.,2006; North and Sinclair, 2007). Under ketogenic conditions, such asduring calorie restriction period, the liver releases a large amount ofacetate into the blood. The heart and the muscles express acetyl-CoAsynthase 2 and use acetate in an efficient way as an energy source.Deacetylation of acetyl-CoA synthetase switches on its activity so it isrelevant that SIRT3 is indeed activated during CR (Hirschey et al.,2010). A study by Ahn et al. (2008) stressed the importance of NAD+-dependent deacetylation in the regulation of energy homeostasis andalso provided evidence for SIRT3-dependent regulation of globalmitochondrial function. The authors of this paper showed that SIRT3is an important regulator of basal ATP levels and observed that SIRT3can physically interact with at least one of the subunits of complex I,the 39-KDa protein NDUFA9, although in a reversible manner (Ahnet al., 2008). Other studies demonstrated that mitochondria fromSIRT3−/− animals display a selective inhibition of complex I activityand altered basal ATP content (Ahn et al., 2008). Another target ofSIRT3 is also the enzyme isocitrate dehydrogenase 2 (Schlicker et al.,2008), which promotes regeneration of antioxidants and catalyzes akey regulation point of the citric acid cycle. Schilicker C et al. reportedthat the N- and C-terminal regions of SIRT3 regulate its activity againstglutamate dehydrogenase and a peptide substrate, indicating rolesof these regions in substrate recognition and sirtuin regulation(Schlicker et al., 2008). Cimen et al. identified that the succinatedehydrogenase flavoprotein (ShdhA) subunit is also another SIRT3target in the mitochondrial respiratory chain (Cimen et al., 2010). Itwas demonstrated that SdhA is highly acetylated in SIRT3 knockoutmice and also that the activation of complex II was dependent onSIRT3 both in wild-type mice and in cells over-expressing SIRT3.The regulation of complex II by reversible acetylation is actually animportant point of control since is a crossroad between oxidativephosphorylation and the Krebs cycle, acting on the regulation ofmetabolism in mammalian mitochondria.
The skeletal muscle is a metabolically active organ and crucialfor insulin-mediated disposal and lipid catabolism. Palacios et al.demonstrated that exercise signals can regulate SIRT3 in skeletalmuscle, with a dynamic response of that sirtuin to coordinate
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),

7C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
downstream molecular responses (Palacios et al., 2009). The authorsshowed that exercise training increases SIRT3 level and CREBphosporylation and up-regulates PGC-1α. Interestingly, SIRT3 ismore highly expressed in slow oxidative type I soleus muscle incomparison with fast type II or gastrocnemius muscles (Palacios et al.,2009). In addition, it was found out that SIRT3 levels in skeletalmuscle are sensitive to diet, with SIRT3 decreasing by a high-fat diet,and increased by short-term fasting (24-hour) or long term nutrientdeprivation (12-month CR) and exercise training. Since caloricrestriction regiment leads to phospho-activation of AMPK in themuscle and that SIRT3−/− mice have low phosphorylation levels ofboth AMPK/CREB and low expression of PGC-1α, it was proposed thatSIRT3 levels may respond to various nutritional/energetic andphysiological challenges by regulating muscle energy homeostasisvia AMPK and PGC-1α (Palacios et al., 2009). Another important studyby Hirschey et al. strengths the role of SIRT3 in the regulation of fatty-acid oxidation (Hirschey et al., 2010). It was reported that SIRT3expression is up-regulated during fasting in liver and brown adiposetissues. Mass spectrometry results showed that the long chain acylcoenzyme A dehydrogenase (LCAD) is hyperacetylated at lysine 42 inthe absence of SIRT3, with LCAD hyperacetylation reducing itsenzymatic activity (Hirschey et al., 2010). Mice lacking SIRT3presented hallmarks of fatty-acid oxidation disorders during fasting,including reduced ATP levels and intolerance to cold exposure(Hirschey et al., 2010). This study identified acetylation as a novelregulatory mechanism for mitochondrial fatty acid oxidation anddemonstrates that SIRT3 modulates mitochondrial intermediarymetabolism and fatty-acid use during fasting. Interestingly, it wasalso reported that SIRT3 deacetylates cyclophilin D (Shulga et al.,2010), diminishing its activity and inducing its dissociation from theadenine nucleotide traslocator (ANT). In addition, SIRT3-inducedinteraction with cyclophilin D also induces the detachment ofhexokinase II from mitochondria. These findings might be importantfor the role of SIRT3 in the metabolism of cancer cells and theirsusceptibility to toxicity by foreign agents. Recently, Hafner et al.showed that SIRT3 can regulate the mPTP through the deacetylationof CypD at lysine 166, suppressing age-related cardiac hypertrophy(Hafner et al., 2010). In an elegant study, the authors reported thatSIRT3 deacetylates the regulatory component of the mPTP, cyclophilinD (CypD), at lysine 166, which is adjacent to the binding site ofcyclosporin A, a CypD inhibitor. Cardiac myocytes from SIRT3knockout mice showed increased mitochondrial swelling due to themPTP opening and accelerated signs of cardiac aging including cardiachypertrophy and fibrosis at 13 months of age (Hafner et al., 2010). Alltogether, the data shows that SIRT3 controls the mPTP and that a lossof SIRT3 promotes mitochondrial alterations resulting in enhancedROS production and cell dysfunction. The results are also a clearevidence that SIRT3 activity is necessary to prevent mitochondrialdysfunction and cardiac hypertrophy during aging (Hafner et al.,2010). Although it is not new that under certain conditions SIRT3mediates the reduction of oxidative damage, a study from Someya etal. relates age-related hearing loss and calorie restriction with thistopic (Someya et al., 2010). Under caloric restriction conditions, areduction of oxidative damage in multiple tissues and a decrease ofage-related hearing loss in WT mice were observed, as opposed tomice lacking SIRT3. The authors of this study identified SIRT3 as anessential player in enhancing the mitochondrial glutathione antiox-idant defense system during caloric restriction and concluded thatmitochondrial adaptations and aging retardation in mammals may bedependent on SIRT3 (Someya et al., 2010). Another paper showed thatthe protective effects of CR against oxidative stress and damage arediminished inmice lacking SIRT3, with the effects of this protein beingmediated by deacetylation of two critical lysine residues in SOD2,enhancing its antioxidant activity (Qiu et al., 2010a).
Several other SIRT3 targets involved in metabolism havebeen recently described. Shimazu et al. identified 3-hydroxy-3-
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
methylglutaryl CoA synthase 2 (HMGCS2) as another SIRT3 deacety-lated protein (Shimazu et al., 2010). Mice lacking SIRT3 present adecrease in β-hydroxybutyrate levels during fasting, which candemonstrate that SIRT3 regulates ketone body production duringfasting (Shimazu et al., 2010). Also, Hallows et al. identified theurea cycle enzyme ornithine transcarbomoylase (OTC) and otherenzymes involved in β-oxidation as likely SIRT3 targets. Fastedmice lacking SIRT3 revealed alterations in β-oxidation and in the ureacycle, demonstrating a direct role of SIRT3 in the regulation of thetwo important metabolic pathways during CR (Hallows et al., 2011).The results also suggested that under low energy input conditions,SIRT3 modulates mitochondria by promoting amino acid catabolismand β-oxidation (Hallows et al., 2011). Recently, it has been shownthat livers from mice maintained on a high fat diet (HFD) exhibitedreduced SIRT3 activity, a 3-fold decrease in hepatic NAD+ levels, andincreased mitochondrial protein oxidation. In contrast, neither SIRT1nor histone acetyltransferase activities were altered, suggesting SIRT3as a crucial factor contributing to the observed phenotype (Kendricket al., 2011). In SIRT3−/− mice, HFD further increased the acetylationstatus of liver proteins and reduced the activity of respiratorycomplexes III and IV. This study identified acetylation patterns inliver proteins from HFD-fed mice and the results suggest that SIRT3 isan integral regulator of mitochondrial function, with its depletionresulting in hyperacetylation of critical mitochondrial proteins thatprotect against liver lipotoxicity under conditions of nutrient excess(Kendrick et al., 2011).
1.7.2.2. SIRT4 and regulation of mitochondrial metabolism. The mito-chondrial isoform of SIRT4 does not show detectable deacetylaseactivity but possesses NAD-dependent mono-ADP-ribosyltransferaseactivity (Ahuja et al., 2007; Yamamoto et al., 2007). It is known thatglutamate dehydrogenase (GDH) is a substrate of SIRT4 (Haigis et al.,2006). By ADP-ribosylating GDH, SIRT4 suppresses its activity andprevents the usage of amino acids as an energy source. During calorierestriction, although NAD+ levels are increased in mitochondria,SIRT4 expression is decreased. The decrease in SIRT4 activity results inthe activation of GDH and it induces the usage of glutamate andglutamine in order to generate ATP (Haigis et al., 2006). Thismechanism is important in pancreatic β-cells of calorie-restrictedmice. Due to low blood glucose levels, glucose-stimulated insulinsecretion is suppressed. However, the activation of GDH allowsamino-acid stimulated insulin secretion and thus maintain basallevels of insulin (Argmann and Auwerx, 2006). GDH can also bedeacetylated by SIRT3, although the functional link between acety-lation and ADP-ribosylation of this important enzyme is not yet wellunderstood. Also, a new role for mitochondrial SIRT4 in the regulationof insulin secretion was identified, with SIRT4 considered as a proteinthat negatively regulates insulin secretion (Ahuja et al., 2007). Massspectrometry analysis of proteins that co-immunoprecipitate withSIRT4 identified insulin-degrading enzyme and the adenine nucleo-tide translocator isoforms ANT2 and ANT3 (Ahuja et al., 2007). It wasdemonstrated that depletion of SIRT4 from insulin-producing INS-1Ecells results in increased insulin secretion in response to glucose(Ahuja et al., 2007). Nasrin et al. investigatedwhether the depletion ofSIRT4 would enhance liver and muscle metabolism (Nasrin et al.,2010). An increase in gene expression of mitochondrial and fatty acidmetabolism enzymes was found in hepatocytes SIRT4 KO cells (Nasrinet al., 2010). Interestingly, it was also found out that SIRT1 mRNAlevels and protein expression were increased after SIRT4 knockdown(Nasrin et al., 2010). Interestingly, the loss of activity of SIRT4 inprimary hepatocytes and in whole liver led to an increased expressionof SIRT3. Fatty acid oxidation (FAO) was increased in SIRT4 KOprimary hepatocytes and this effect was dependent on SIRT1 (Nasrinet al., 2010). In SIRT4 KO primary myotubes, increased fatty acidoxidation, cellular respiration and pAMPK levels were detected. Thefindings demonstrate that SIRT4 inhibition increases fat oxidative
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),

8 C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
capacity in liver and mitochondrial function in skeletal muscle. Takentogether, it seems like SIRT4 is a negative regulator of oxidativemetabolism, in contrast with the functions of SIRT1 and SIRT3, whichenhance the oxidative capacity of tissues (Nasrin et al., 2010).
1.7.2.3. SIRT5 and mitochondrial metabolism. As compared with othersirtuins, there is a lot still to be discovered about SIRT5. For example, itis known that SIRT5 deacetylates and activates carbomoyl phospha-tase synthetase 1 (CPS1), an enzyme that catalyzes the first step ofthe urea cycle (Gertz and Steegborn, 2010; Michishita et al., 2005;Nakagawa et al., 2009). The expression levels of SIRT5 are unalteredbut NAD+ is increased by 50% in mitochondria from calorie restrictedliver (Nakagawa et al., 2009). This increase in the activity of SIRT5might regulate the urea cycle during caloric restriction conditions bydeacetylation and activation of CPS1. Schilicker et al. reported thatSIRT5 did not deacetylate any of the mitochondrial matrix proteinstested (Schlicker et al., 2008). The surprising result was that SIRT5can deacetylate cytochrome c (Schlicker et al., 2008). By using amitochondrial import assay, the authors determined that SIRT5 can betranslocated not only to the mitochondrial intermembrane space butalso to the matrix, indicating that localization might contribute toSIRT5 regulation and substrate selection (Schlicker et al., 2008). Invivo, SIRT5 appears to co-localize with cytochrome c in theintermembrane space. The reversible acetylation of cytochrome ccould either affect its function in respiration or in apoptosis, or both. Incerebellar granule neurons overexpressing SIRT5, a pro-apoptoticfunction for this sirtuin was pointed out (Gertz and Steegborn, 2010).Several other target proteins for SIRT5 are possible due to the inter-mitochondrial space localization of the sirtuin. One of them is theapoptosis inducing factor (AIF), which was already reported to beacetylated depending on the feeding status (Gertz and Steegborn,2010). Further and exciting studies are thus needed to understand thephysiological role of SIRT5 as a mitochondrial inter-membrane spacedeacetylase.
1.7.3. Protection of mitochondrial function by sirtuinsThe role of mitochondria in cell physiology and survival, as well as
in drug-induced toxicity is well established (Pereira et al., 2009a,b;Sardao et al., 2008). Besides control of mitochondrial metabolism,sirtuins are also involved in the lines of defense of that organelle.In fact, there is actually evidence that at least SIRT3 can protectmitochondria from exogenous and endogenous stresses (Kong et al.,2010; Pillai et al., 2010; Scher et al., 2007; Sundaresan et al., 2008).Mitochondria are an important site of reactive oxygen species (ROS)production in a cell, which is why a tight ROS production by thatorganelle must be exerted by different mechanisms in order toprevent structural damage and accelerated aging (Guarente, 2008).The role of mitochondrial sirtuins in the protection against mito-chondrial damage is now beginning to be clarified. One starting pointwas the finding that NAD+ levels dictate cell survival (Yang et al.,2007). It is well known that one of the major causes for cell deathdue to genotoxic stress is the hyperactivation of PARP-1 that depletesnuclear and cytosolic NAD+ causing the translocation of the AIFfrom themitochondrialmembrane to the nucleus. Yang et al. identifiedNampt as a stress- and nutrient-responsive gene that increasesmitochondrial NAD+ levels (Yang et al., 2007). It has been demon-strated that increased mitochondrial NAD+ levels improve cellsurvival during genotoxic stress and that this protection is dependenton SIRT3 and SIRT4,whichmeans thatNampt-mediated cell protectionrequires mitochondrial sirtuins (Yang et al., 2007). Nampt increasedSIRT3 activity since Nampt overexpression decreased the acetylationlevel of AceCS2. Allison et al. exploited apoptosis trough Bcl-2/p53regulation and verified a SIRT3 involvement in this process (Allisonand Milner, 2007). Bcl-2 and SIRT3 were silenced separately andin combination in human epithelial cancer and non-cancer cells. Itwas demonstrated that SIRT3 is required for apoptosis under basal
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
conditions, by selective silencing of Bcl-2 in HCT116 human epithelialcancer cells. SIRT3 is dispensable for stress-induced apoptosis inHCT116 human epithelial cancer cells but it is an essential pro-apoptotic mediator for both Bcl-2/p53-regulated apoptosis. Interest-ingly, SIRT3 functions in JNK2-regulated apoptosis but it is dispensablefor both SIRT-1 regulated and stress-induced apoptosis. It is thenconcluded that SIRT3 is a pro-apoptotic protein that participates indistinct basal apoptotic pathways (Allison and Milner, 2007).Nevertheless, this paper does not really point to a direct link betweenSIRT3 and protection against apoptosis under stress conditions incontrast to many other papers. For example, it has been shown thatSIRT3 protects the mouse heart by blocking the cardiac hypertrophicresponse (Sundaresan et al., 2009). In cardiomyocytes, SIRT3 pre-vented cardiac hypertrophy activation by activating the fork headbox O3a-dependent (Foxo3a-dependent), anti-oxidant superoxidedismutase and catalase genes and decreasing cellular levels of ROS(Sundaresan et al., 2009). The results demonstrate that SIRT3 is anendogenous negative regulator of cardiac hypertrophy by thesuppression of ROS levels. Another study showed that SIRT3 levelsare increased under stress not only in mitochondria but also in thenuclei of cardiomyocytes (Sundaresan et al., 2008). This particularpaper identified Ku70 as a new SIRT3 target, since SIRT3 physicallybinds to ku70 and deacetylates the latter, promoting its interactionwith Bax. Under stress, SIRT3 overexpression protects cardiomyocytesby partially preventing the translocation of Bax to mitochondria.This study points an essential role of SIRT3 in the survival ofcardiomyocytes under stress conditions. Recently, SIRT3 was pointedout as a tumor-suppressormitochondrial-localized protein (Kim et al.,2010). In this study, it has been shown that the expression of a singleoncogene (Myc or Ras) in SIRT3 knockout MEFs results in in vitrotransformation and altered intracellular metabolism. In addition,SIRT3 knockout mice developed ER/PR-positive mammary tumors(Kim et al., 2010). In this work, it is also shown that several humancancer tissues exhibit reduced SIRT3 levels, thus leading the authorsto propose SIRT3 as a mitochondrial fidelity protein. In this regard,loss of SIRT3 results in a decrease in the antioxidant defenses, resultingin an increase of ROS production. It is also speculated that a pro-oxidant environment may be permissive for in vivo carcinogenesis.It is known that PGC-1α is an important inducer of detoxifyingenzymes, even though the molecular mechanism is not clearlyunderstood. Kong et al. showed that PGC-1α activated the mouseSIRT3 promoter mediated by an estrogen-related receptor bindingelement (ERRE) (Kong et al., 2010). The knockdown of ERR α reducedthe induction of SIRT3 by PGC-1α in C2C12 myotubes. In the samework, it was found out that SIRT3 was crucial for PGC-1α-dependentinduction of ROS-detoxifying enzymes and other components of themitochondrial respiratory chain, such as glutathione peroxidase-1,superoxidase dismutase-2, ATP synthase 5c and cytochrome c (Konget al., 2010). Both overexpression of SIRT3 and PGC-1αdecreased basalROS levels in myotubes. SIRT3 stimulated mitochondrial biogenesisand might provide a novel target for treating ROS-related diseases.Another interesting study focused on SIRT3 capacity of reducing lipidaccumulation via AMKP activation in human hepatic cells (Shi et al.,2010). The knockdown of SIRT3 downregulated the phosphorylationof AMPK and acetyl coenzyme A carboxylase (ACC), promotingincreased lipid accumulation. It was concluded that the capacityof SIRT3 to activate AMPK is dependent on its deacetylase activity(Shi et al., 2010). A very interesting paper correlates p53, SIRT3 andprotection of in vitro-fertilized mouse against oxidative stress(Kawamura et al., 2010). During pre-implantation development,mitochondrial dysfunction or increased levels of oxidative stressadversely affect the developmental outcome. In this study, it wasshown that SIRT3 inactivation increases mitochondrial ROS produc-tion leading to p53 up-regulation and alterations in downstream geneexpression.The findings indicate that SIRT3 might play a protectiverole in pre-implantation of embryos under stress conditions during
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),

9C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
in vitro fertilization and culture, pointing out a new and interestingfuture clinical application related with manipulation of SIRT3 andpost-implantation success.
Recently, another study involved SIRT3 in oral cancer (Alhazzaziet al., 2011). The authors demonstrated for the first time that SIRT3is overexpressed in oral squamous cell carcinoma (OSCC) in vitro and invivo, when compared with other sirtuins. Down-regulation ofSIRT3 inhibited cell growth, increased cell sensitivity to chemotherapyand reduced tumor burden in vivo, demonstrating that SIRT3 can be anovel potential therapeutic target for oral cancer (Alhazzazi et al., 2011).Another paper showed that SIRT3-mediated deacetylation of evolu-tionary conserved lysine 122 inMnSOD results into increased activity inresponse to stress. SIRT3-knockout results in increased mitochondrialsuperoxide, formation of a tumor-permissive environment and finally,enhanced mammary carcinogenesis (Tao et al., 2010).
To the best of our knowledge, there is not much information aboutSIRT4 or even SIRT5 but it seems very likely that both sirtuins can alsobe important in mitochondrial protection against genotoxic stresssuggesting that they can contribute to apoptosis in tumor-supressiveor stress resistant conditions (Verdin et al., 2010). Interestingly, SIRT4is apparently involved in mitochondrial oxidative metabolism. SIRT4KO primary hepatocytes and myotubes were used in order to studyfatty acid oxidation and oxygen consumption under these conditions(Nasrin et al., 2010). As described above, the findings from this studysuggest that SIRT4 is a negative regulator of oxidative metabolism,in contrast with SIRT3 and SIRT1, which means that a functionalinterplay between different sirtuins must exist in order to coordinatethe flow of energy during a given metabolic state. It seems that SIRT4inhibition may improve hepatic insulin sensitivity via increased fatoxidative capacity and hence may be beneficial for the treatment oftype 2 diabetes.
Despite the limited knowledge on in vivo substrates for SIRT5, thecontribution of this sirtuin in disease has been suggested. Repetitiveelements in its gene structure suggested possible genomic instabilitiesand malignant diseases (Gertz and Steegborn, 2010). Furthermore,one particular study indicates that SIRT5 is decreased after alcoholexposure in rats, which increased hepatic mRNA expression of FoxO1and p53 (Lieber et al., 2008). Thus, alcohol consumption compromisesnuclear mitochondrial interactions by post-translational modifica-tions which contribute to alteration of mitochondrial biogenesisthrough the newly discovered decrease in SIRT5. It seems thatSIRT5 contributes to liver damage induced through chronic exposureto alcohol but would also be interesting to understand the role ofthis sirtuin under other stress conditions and understand its role inmitochondrial protection against various stressors.
1.8. Future perspectives: are sirtuins good therapeutic targets?
It is plausible that drugs aimed at modulating sirtuins activityand/or expression may have important consequences in cellularresponses to stress and life span. It is extremely important to fullyidentify and characterize targets of these exciting class of proteins andunderstand how the manipulation of their enzymatic activity orprotein expression can be involved in the protection, not only ofmitochondria but also of the entire cell. It seems that sirtuins arestrategically positioned along the different organs and cellularcompartments and that they may play distinct roles (Schwer andVerdin, 2008; Smith et al., 2002; Vaquero, 2009; Verdin et al., 2010;Yu and Auwerx, 2009). Furthermore, it would be very interestingto understand how sirtuins communicate within each other andexplore their communication network code that should in theory,contribute to protect the cell. What do these proteins have incommon? How do they interact with each other? Is the stoichiometryof the different sirtuins inmitochondria constant? How does it changethere and in other cellular locations upon different stimuli? Manyof these questions remain unanswered. Mitochondrial sirtuins might
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
contribute to the decrease of “unhealthy” mitochondria and some ofthem (e.g. SIRT3) might even be protective against drug-inducedtoxicity (Scher et al., 2007; Sundaresan et al., 2008, 2009), which iswhy it is fundamental to understand the mechanism behind sirtuin-mediated mitochondrial protection and how targeting sirtuins can bea valid future therapeutic approach for several different diseases.Although many recent reviews have provided new directions inthis field, there is still much to be elucidated. There is still a gap ofinformation regarding SIRT4 and SIRT5 functions that warrantsfurther studies in order to understand the roles of these importantplayers in the regulation and protection of mitochondrial function.This review provided an overall view of sirtuin function with a specialrelevance on the most recent findings on the function and relevanceof mitochondrial sirtuins. Furthermore, the new and exciting dataopens a completely new road for novel pharmaceutical applications ofinhibitors and inducers of this class of proteins.
Acknowledgments
Work in the authors' laboratory is funded by the PortugueseFoundation for Science and Technology (FCT) (research grantPTDC/SAU-TOX/110952/2009 to Paulo Oliveira). Claudia Pereira is therecipient of a Ph.D. fellowship from the FCT (SFRH/BD/48029/2008).The work was also partially supported by the Polish Ministry ofScience and Higher Education under grant NN407 075 137 for MagdaLebiedzinsk and Mariusz R. Wieckowski.
References
Ahn, B.H., Kim, H.S., Song, S., Lee, I.H., Liu, J., Vassilopoulos, A., Deng, C.X., Finkel, T., 2008.A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis.Proc. Natl. Acad. Sci. U. S. A. 105, 14447–14452.
Ahuja, N., Schwer, B., Carobbio, S., Waltregny, D., North, B.J., Castronovo, V., Maechler, P.,Verdin, E., 2007. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 282, 33583–33592.
Alcain, F.J., Villalba, J.M., 2009. Sirtuin activators. Expert Opin. Ther. Pat. 19, 403–414.Alhazzazi, T.Y., Kamarajan, P., Joo, N., Huang, J.Y., Verdin, E., D'Silva, N.J., Kapila, Y.L.,
2011. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer117, 1670–1687.
Allison, S.J., Milner, J., 2007. SIRT3 is pro-apoptotic and participates in distinct basalapoptotic pathways. Cell Cycle 6, 2669–2677.
Aquilano, K., Vigilanza, P., Baldelli, S., Pagliei, B., Rotilio, G., Ciriolo, M.R., 2010. Peroxisomeproliferator-activated receptor gammaco-activator1alpha (PGC-1alpha) and sirtuin 1(SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis.J. Biol. Chem. 285, 21590–21599.
Argmann, C., Auwerx, J., 2006. Insulin secretion: SIRT4 gets in on the act. Cell 126,837–839.
Ashraf, N., Zino, S., Macintyre, A., Kingsmore, D., Payne, A.P., George, W.D., Shiels, P.G.,2006. Altered sirtuin expression is associated with node-positive breast cancer. Br.J. Cancer 95, 1056–1061.
Blander, G., Guarente, L., 2004. The Sir2 family of protein deacetylases. Annu. Rev.Biochem. 73, 417–435.
Bordone, L., Motta, M.C., Picard, F., Robinson, A., Jhala, U.S., Apfeld, J., McDonagh, T.,Lemieux, M., McBurney, M., Szilvasi, A., Easlon, E.J., Lin, S.J., Guarente, L., 2006.Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells.PLoS Biol. 4, e31.
Bordone, L., Cohen, D., Robinson, A., Motta, M.C., van Veen, E., Czopik, A., Steele, A.D.,Crowe, H., Marmor, S., Luo, J., Gu, W., Guarente, L., 2007. SIRT1 transgenic miceshow phenotypes resembling calorie restriction. Aging Cell 6, 759–767.
Borra, M.T., Smith, B.C., Denu, J.M., 2005. Mechanism of human SIRT1 activation byresveratrol. J. Biol. Chem. 280, 17187–17195.
Brunet, A., Sweeney, L.B., Sturgill, J.F., Chua, K.F., Greer, P.L., Lin, Y., Tran, H., Ross, S.E.,Mostoslavsky, R., Cohen, H.Y., Hu, L.S., Cheng, H.L., Jedrychowski, M.P., Gygi, S.P.,Sinclair, D.A., Alt, F.W., Greenberg, M.E., 2004. Stress-dependent regulation ofFOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015.
Campisi, J., Yaswen, P., 2009. Aging and cancer cell biology, 2009. Aging Cell 8, 221–225.Canto, C., Auwerx, J., 2009. Caloric restriction, SIRT1 and longevity. Trends Endocrinol.
Metab. 20, 325–331.Canto, C., Jiang, L.Q., Deshmukh, A.S., Mataki, C., Coste, A., Lagouge, M., Zierath, J.R.,
Auwerx, J., 2010. Interdependence of AMPK and SIRT1 for metabolic adaptationto fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219.
Chabi, B., Adhihetty, P.J., O'Leary, M.F., Menzies, K.J., Hood, D.A., 2009. Relationshipbetween Sirt1 expression and mitochondrial proteins during conditions of chronicmuscle use and disuse. J. Appl. Physiol. 107, 1730–1735.
Chen, D., Steele, A.D., Lindquist, S., Guarente, L., 2005. Increase in activity during calorierestriction requires Sirt1. Science 310, 1641.
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),

10 C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
Chen, D., Bruno, J., Easlon, E., Lin, S.J., Cheng, H.L., Alt, F.W., Guarente, L., 2008. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757.
Chen, Y.R., Fang, S.R., Fu, Y.C., Zhou, X.H., Xu, M.Y., Xu, W.C., 2010. Calorie restrictionon insulin resistance and expression of SIRT1 and SIRT4 in rats. Biochem. Cell Biol.88, 715–722.
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M.L., Rehman, M., Walther, T.C., Olsen, J.V.,Mann, M., 2009. Lysine acetylation targets protein complexes and co-regulatesmajor cellular functions. Science 325, 834–840.
Cimen, H., Han, M.J., Yang, Y., Tong, Q., Koc, H., Koc, E.C., 2010. Regulation of succinatedehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 49,304–311.
Denu, J.M., 2005. The Sir 2 family of protein deacetylases. Curr. Opin. Chem. Biol. 9,431–440.
Donmez, G., Guarente, L., 2010. Aging and disease: connections to sirtuins. Aging Cell 9,285–290.
Dryden, S.C., Nahhas, F.A., Nowak, J.E., Goustin, A.S., Tainsky, M.A., 2003. Role for humanSIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cellcycle. Mol. Cell. Biol. 23, 3173–3185.
Elliott, P.J., Jirousek, M., 2008. Sirtuins: novel targets for metabolic disease. Curr. Opin.Investig. Drugs 9, 371–378.
Feige, J.N., Lagouge, M., Canto, C., Strehle, A., Houten, S.M., Milne, J.C., Lambert, P.D.,Mataki, C., Elliott, P.J., Auwerx, J., 2008. Specific SIRT1 activation mimics low energylevels and protects against diet-induced metabolic disorders by enhancing fatoxidation. Cell Metab. 8, 347–358.
Finkel, T., Deng, C.X.,Mostoslavsky, R., 2009. Recent progress in the biology and physiologyof sirtuins. Nature 460, 587–591.
Ford, J., Jiang, M., Milner, J., 2005. Cancer-specific functions of SIRT1 enable humanepithelial cancer cell growth and survival. Cancer Res. 65, 10457–10463.
Ford, E., Voit, R., Liszt, G., Magin, C., Grummt, I., Guarente, L., 2006. Mammalian Sir2homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 20,1075–1080.
Frescas, D., Valenti, L., Accili, D., 2005. Nuclear trapping of the forkhead transcriptionfactor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogeneticgenes. J. Biol. Chem. 280, 20589–20595.
Frye, R.A., 2000. Phylogenetic classification of prokaryotic and eukaryotic Sir2-likeproteins. Biochem. Biophys. Res. Commun. 273, 793–798.
Fulco, M., Schiltz, R.L., Iezzi, S., King, M.T., Zhao, P., Kashiwaya, Y., Hoffman, E., Veech,R.L., Sartorelli, V., 2003. Sir2 regulates skeletal muscle differentiation as a potentialsensor of the redox state. Mol. Cell 12, 51–62.
Gan, L., Mucke, L., 2008. Paths of convergence: sirtuins in aging and neurodegeneration.Neuron 58, 10–14.
Garske, A.L., Smith, B.C., Denu, J.M., 2007. Linking SIRT2 to Parkinson's disease. ACSChem. Biol. 2, 529–532.
Gertz, M., Steegborn, C., 2010. Function and regulation of the mitochondrial sirtuinisoform Sirt5 in Mammalia. Biochim. Biophys. Acta 1804, 1658–1665.
Gillum, M.P., Erion, D.M., Shulman, G.I., in press. Sirtuin-1 regulation of mammalianmetabolism. Trends Mol. Med.
Guan, K.L., Xiong, Y., 2011. Regulation of intermediary metabolism by proteinacetylation. Trends Biochem. Sci. 36, 108–116.
Guarente, L., 2006. Sirtuins as potential targets for metabolic syndrome. Nature 444,868–874.
Guarente, L., 2007. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol.72, 483–488.
Guarente, L., 2008. Mitochondria—a nexus for aging, calorie restriction, and sirtuins?Cell 132, 171–176.
Guo, X., Williams, J.G., Schug, T.T., Li, X., 2010. DYRK1A and DYRK3 promote cell survivalthrough phosphorylation and activation of SIRT1. J. Biol. Chem. 285, 13223–13232.
Hafner, A.V., Dai, J., Gomes, A.P., Xiao, C.Y., Palmeira, C.M., Rosenzweig, A., Sinclair, D.A.,2010. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923.
Haigis, M.C., Guarente, L.P., 2006. Mammalian sirtuins—emerging roles in physiology,aging, and calorie restriction. Genes Dev 20, 2913–2921.
Haigis, M.C., Mostoslavsky, R., Haigis, K.M., Fahie, K., Christodoulou, D.C., Murphy, A.J.,Valenzuela, D.M., Yancopoulos, G.D., Karow, M., Blander, G., Wolberger, C., Prolla,T.A., Weindruch, R., Alt, F.W., Guarente, L., 2006. SIRT4 inhibits glutamatedehydrogenase and opposes the effects of calorie restriction in pancreatic betacells. Cell 126, 941–954.
Hallows, W.C., Lee, S., Denu, J.M., 2006. Sirtuins deacetylate and activate mammalianacetyl-CoA synthetases. Proc. Natl. Acad. Sci. U. S. A. 103, 10230–10235.
Hallows, W.C., Albaugh, B.N., Denu, J.M., 2008. Where in the cell is SIRT3? Functionallocalization of an NAD+-dependent protein deacetylase. Biochem. J. 411, e11–e13.
Hallows,W.C., Yu,W., Smith, B.C., Devires, M.K., Ellinger, J.J., Someya, S., Shortreed, M.R.,Prolla, T., Markley, J.L., Smith, L.M., Zhao, S., Guan, K.L., Denu, J.M., 2011. Sirt3promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol.Cell 41, 139–149.
Harman, D., 1956. Aging: a theory based on free radical and radiation chemistry.J. Gerontol. 11, 298–300.
Harting, K., Knoll, B., 2010. SIRT2-mediated protein deacetylation: an emerging keyregulator in brain physiology and pathology. Eur. J. Cell Biol. 89, 262–269.
Hiratsuka, M., Inoue, T., Toda, T., Kimura, N., Shirayoshi, Y., Kamitani, H., Watanabe, T.,Ohama, E., Tahimic, C.G., Kurimasa, A., Oshimura, M., 2003. Proteomics-basedidentification of differentially expressed genes in human gliomas: down-regulationof SIRT2 gene. Biochem. Biophys. Res. Commun. 309, 558–566.
Hirschey, M.D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D.B., Grueter, C.A.,Harris, C., Biddinger, S., Ilkayeva, O.R., Stevens, R.D., Li, Y., Saha, A.K., Ruderman, N.B.,Bain, J.R., Newgard, C.B., Farese Jr., R.V., Alt, F.W., Kahn, C.R., Verdin, E., 2010. SIRT3
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation.Nature 464, 121–125.
Huang, J.Y., Hirschey, M.D., Shimazu, T., Ho, L., Verdin, E., 2010. Mitochondrial sirtuins.Biochim. Biophys. Acta 1804, 1645–1651.
Jacobs, K.M., Pennington, J.D., Bisht, K.S., Aykin-Burns, N., Kim, H.S., Mishra, M., Sun, L.,Nguyen, P., Ahn, B.H., Leclerc, J., Deng, C.X., Spitz, D.R., Gius, D., 2008. SIRT3 interactswith the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3adependent gene expression. Int. J. Biol. Sci. 4, 291–299.
Kanfi, Y., Shalman, R., Peshti, V., Pilosof, S.N., Gozlan, Y.M., Pearson, K.J., Lerrer, B.,Moazed, D., Marine, J.C., de Cabo, R., Cohen, H.Y., 2008. Regulation of SIRT6 proteinlevels by nutrient availability. FEBS Lett. 582, 543–548.
Kawahara, T.L., Michishita, E., Adler, A.S., Damian, M., Berber, E., Lin, M., McCord, R.A.,Ongaigui, K.C., Boxer, L.D., Chang, H.Y., Chua, K.F., 2009. SIRT6 links histone H3lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismallife span. Cell 136, 62–74.
Kawamura, Y., Uchijima, Y., Horike, N., Tonami, K., Nishiyama, K., Amano, T., Asano, T.,Kurihara, Y., Kurihara, H., 2010. Sirt3 protects in vitro-fertilized mouse preimplan-tation embryos against oxidative stress-induced p53-mediated developmentalarrest. J. Clin. Invest. 120, 2817–2828.
Kendrick, A.A., Choudhury, M., Rahman, S.M., McCurdy, C.E., Friederich, M., Van Hove,J.L., Watson, P.A., Birdsey, N., Bao, J., Gius, D., Sack, M.N., Jing, E., Kahn, C.R.,Friedman, J.E., Jonscher, K.R., 2011. Fatty liver is associated with reduced SIRT3activity and mitochondrial protein hyperacetylation. Biochem. J. 433, 505–514.
Kim, S.C., Sprung, R., Chen, Y., Xu, Y., Ball, H., Pei, J., Cheng, T., Kho, Y., Xiao, H., Xiao, L.,Grishin, N.V., White, M., Yang, X.J., Zhao, Y., 2006. Substrate and functional diversityof lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618.
Kim, H.S., Patel, K., Muldoon-Jacobs, K., Bisht, K.S., Aykin-Burns, N., Pennington, J.D., vander Meer, R., Nguyen, P., Savage, J., Owens, K.M., Vassilopoulos, A., Ozden, O., Park,S.H., Singh, K.K., Abdulkadir, S.A., Spitz, D.R., Deng, C.X., Gius, D., 2010. SIRT3 is amitochondria-localized tumor suppressor required for maintenance of mitochon-drial integrity and metabolism during stress. Cancer Cell 17, 41–52.
Kong, X., Wang, R., Xue, Y., Liu, X., Zhang, H., Chen, Y., Fang, F., Chang, Y., 2010. Sirtuin 3,a new target of PGC-1alpha, plays an important role in the suppression of ROS andmitochondrial biogenesis. PLoS One 5, e11707.
Kume, S., Haneda, M., Kanasaki, K., Sugimoto, T., Araki, S., Isono, M., Isshiki, K., Uzu, T.,Kashiwagi, A., Koya, D., 2006. Silent information regulator 2 (SIRT1) attenuatesoxidative stress-inducedmesangial cell apoptosis via p53 deacetylation. Free Radic.Biol. Med. 40, 2175–2182.
Lagouge, M., Argmann, C., Gerhart-Hines, Z., Meziane, H., Lerin, C., Daussin, F.,Messadeq, N., Milne, J., Lambert, P., Elliott, P., Geny, B., Laakso, M., Puigserver, P.,Auwerx, J., 2006. Resveratrol improves mitochondrial function and protects againstmetabolic disease by activating SIRT1 and PGC-1alpha. Cell 127, 1109–1122.
Leiser, S.F., Kaeberlein, M., 2010. A role for SIRT1 in the hypoxic response. Mol. Cell 38,779–780.
Li, X., Zhang, S., Blander, G., Tse, J.G., Krieger, M., Guarente, L., 2007. SIRT1 deacetylatesand positively regulates the nuclear receptor LXR. Mol. Cell 28, 91–106.
Li, S., Banck, M., Mujtaba, S., Zhou, M.M., Sugrue, M.M., Walsh, M.J., 2010. p53-inducedgrowtharrest is regulatedby themitochondrial SirT3deacetylase. PLoSOne5, e10486.
Liang, F., Kume, S., Koya, D., 2009. SIRT1 and insulin resistance. Nat. Rev. Endocrinol. 5,367–373.
Lieber, C.S., Leo, M.A., Wang, X., Decarli, L.M., 2008. Alcohol alters hepatic FoxO1, p53,and mitochondrial SIRT5 deacetylation function. Biochem. Biophys. Res. Commun.373, 246–252.
Lomb, D.J., Laurent, G., Haigis, M.C., 2010. Sirtuins regulate key aspects of lipidmetabolism. Biochim. Biophys. Acta 1804, 1652–1657.
Lombard, D.B., Alt, F.W., Cheng, H.L., Bunkenborg, J., Streeper, R.S., Mostoslavsky, R.,Kim, J., Yancopoulos, G., Valenzuela, D., Murphy, A., Yang, Y., Chen, Y., Hirschey,M.D., Bronson, R.T., Haigis, M., Guarente, L.P., Farese Jr., R.V., Weissman, S., Verdin,E., Schwer, B., 2007. Mammalian Sir2 homolog SIRT3 regulates global mitochon-drial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814.
Mahlknecht, U., Ho, A.D., Letzel, S., Voelter-Mahlknecht, S., 2006a. Assignment of theNAD-dependent deacetylase sirtuin 5 gene (SIRT5) to human chromosome band6p23 by in situ hybridization. Cytogenet. Genome Res. 112, 208–212.
Mahlknecht, U., Ho, A.D., Voelter-Mahlknecht, S., 2006b. Chromosomal organizationand fluorescence in situ hybridization of the human Sirtuin 6 gene. Int. J. Oncol. 28,447–456.
Mayanil, C.S., Pool, A., Nakazaki, H., Reddy, A.C., Mania-Farnell, B., Yun, B., George, D.,McLone, D.G., Bremer, E.G., 2006. Regulation of murine TGFbeta2 by Pax3 duringearly embryonic development. J. Biol. Chem. 281, 24544–24552.
McCord, R.A., Michishita, E., Hong, T., Berber, E., Boxer, L.D., Kusumoto, R., Guan, S., Shi,X., Gozani, O., Burlingame, A.L., Bohr, V.A., Chua, K.F., 2009. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging(Albany NY) 1, 109–121.
Michan, S., Sinclair, D., 2007. Sirtuins inmammals: insights into their biological function.Biochem. J. 404, 1–13.
Michishita, E., Park, J.Y., Burneskis, J.M., Barrett, J.C., Horikawa, I., 2005. Evolutionarilyconserved and nonconserved cellular localizations and functions of human SIRTproteins. Mol. Biol. Cell 16, 4623–4635.
Michishita, E., McCord, R.A., Berber, E., Kioi, M., Padilla-Nash, H., Damian, M., Cheung, P.,Kusumoto, R., Kawahara, T.L., Barrett, J.C., Chang, H.Y., Bohr, V.A., Ried, T., Gozani, O.,Chua, K.F., 2008. SIRT6 is a histone H3 lysine 9 deacetylase that modulatestelomeric chromatin. Nature 452, 492–496.
Mostoslavsky, R., Chua, K.F., Lombard, D.B., Pang, W.W., Fischer, M.R., Gellon, L., Liu, P.,Mostoslavsky, G., Franco, S., Murphy, M.M., Mills, K.D., Patel, P., Hsu, J.T., Hong, A.L.,Ford, E., Cheng, H.L., Kennedy, C., Nunez, N., Bronson, R., Frendewey, D., Auerbach,W., Valenzuela, D., Karow, M., Hottiger, M.O., Hursting, S., Barrett, J.C., Guarente, L.,
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),

11C.V. Pereira et al. / Mitochondrion xxx (2011) xxx–xxx
Mulligan, R., Demple, B., Yancopoulos, G.D., Alt, F.W., 2006. Genomic instability andaging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329.
Motta, M.C., Divecha, N., Lemieux, M., Kamel, C., Chen, D., Gu,W., Bultsma, Y., McBurney,M., Guarente, L., 2004. Mammalian SIRT1 represses forkhead transcription factors.Cell 116, 551–563.
Nahhas, F., Dryden, S.C., Abrams, J., Tainsky, M.A., 2007. Mutations in SIRT2 deacetylasewhich regulate enzymatic activity but not its interaction with HDAC6 and tubulin.Mol. Cell. Biochem. 303, 221–230.
Nakagawa, T., Guarente, L., 2009. Urea cycle regulation by mitochondrial sirtuin, SIRT5.Aging (Albany NY) 1, 578–581.
Nakagawa, T., Lomb, D.J., Haigis, M.C., Guarente, L., 2009. SIRT5 deacetylates carbamoylphosphate synthetase 1 and regulates the urea cycle. Cell 137, 560–570.
Nakamura, Y., Ogura, M., Tanaka, D., Inagaki, N., 2008. Localization of mousemitochondrial SIRT proteins: shift of SIRT3 to nucleus by co-expression withSIRT5. Biochem. Biophys. Res. Commun. 366, 174–179.
Nasrin, N., Wu, X., Fortier, E., Feng, Y., Bare, O.C., Chen, S., Ren, X., Wu, Z., Streeper, R.S.,Bordone, L., 2010. SIRT4 regulates fatty acid oxidation and mitochondrial geneexpression in liver and muscle cells. J. Biol. Chem. 285, 1995–2002.
Nisoli, E., Tonello, C., Cardile, A., Cozzi, V., Bracale, R., Tedesco, L., Falcone, S., Valerio, A.,Cantoni, O., Clementi, E., Moncada, S., Carruba, M.O., 2005. Calorie restrictionpromotes mitochondrial biogenesis by inducing the expression of eNOS. Science310, 314–317.
North, B.J., Sinclair, D.A., 2007. Sirtuins: a conserved key unlocking AceCS activity.Trends Biochem. Sci. 32, 1–4.
Palacios, O.M., Carmona, J.J., Michan, S., Chen, K.Y., Manabe, Y., Ward 3rd, J.L., Goodyear,L.J., Tong, Q., 2009. Diet and exercise signals regulate SIRT3 and activate AMPK andPGC-1alpha in skeletal muscle. Aging (Albany NY) 1, 771–783.
Pallos, J., Bodai, L., Lukacsovich, T., Purcell, J.M., Steffan, J.S., Thompson, L.M., Marsh, J.L.,2008. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in aDrosophila model of Huntington's disease. Hum. Mol. Genet. 17, 3767–3775.
Pereira, C.V., Moreira, A.C., Pereira, S.P., Machado, N.G., Carvalho, F.S., Sardao, V.A.,Oliveira, P.J., 2009a. Investigating drug-induced mitochondrial toxicity: a biosensorto increase drug safety? Curr. Drug Saf. 4, 34–54.
Pereira, S.P., Pereira, G.C., Moreno, A.J., Oliveira, P.J., 2009b. Can drug safety be predictedand animal experiments reduced by using isolated mitochondrial fractions? Altern.Lab. Anim. 37, 355–365.
Picard, F., Kurtev, M., Chung, N., Topark-Ngarm, A., Senawong, T., Machado De Oliveira,R., Leid, M., McBurney, M.W., Guarente, L., 2004. Sirt1 promotes fat mobilization inwhite adipocytes by repressing PPAR-gamma. Nature 429, 771–776.
Pillai, V.B., Sundaresan, N.R., Jeevanandam, V., Gupta, M.P., 2010. Mitochondrial SIRT3and heart disease. Cardiovasc. Res. 88, 250–256.
Qiu, X., Brown, K., Hirschey, M.D., Verdin, E., Chen, D., 2010a. Calorie restriction reducesoxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667.
Qiu, X., Brown, K.V., Moran, Y., Chen, D., 2010b. Sirtuin regulation in calorie restriction.Biochim. Biophys. Acta 1804, 1576–1583.
Rodgers, J.T., Puigserver, P., 2007. Fasting-dependent glucose and lipid metabolicresponse through hepatic sirtuin 1. Proc. Natl. Acad. Sci. U. S. A. 104, 12861–12866.
Rose, G., Dato, S., Altomare, K., Bellizzi, D., Garasto, S., Greco, V., Passarino, G., Feraco, E.,Mari, V., Barbi, C., BonaFe, M., Franceschi, C., Tan, Q., Boiko, S., Yashin, A.I., DeBenedictis, G., 2003. Variability of the SIRT3 gene, human silent information regulatorSir2 homologue, and survivorship in the elderly. Exp. Gerontol. 38, 1065–1070.
Sardao, V.A., Pereira, S.L., Oliveira, P.J., 2008. Drug-induced mitochondrial dysfunctionin cardiac and skeletal muscle injury. Expert Opin. Drug Saf. 7, 129–146.
Saunders, L.R., Sharma, A.D., Tawney, J., Nakagawa, M., Okita, K., Yamanaka, S.,Willenbring, H., Verdin, E., 2010. miRNAs regulate SIRT1 expression during mouseembryonic stem cell differentiation and in adult mouse tissues. Aging (Albany NY)2, 415–431.
Sauve, A.A., 2010. Sirtuin chemicalmechanisms. Biochim. Biophys. Acta 1804, 1591–1603.Sauve, A.A., Wolberger, C., Schramm, V.L., Boeke, J.D., 2006. The biochemistry of sirtuins.
Annu. Rev. Biochem. 75, 435–465.Scher, M.B., Vaquero, A., Reinberg, D., 2007. SirT3 is a nuclear NAD+-dependent histone
deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev.21, 920–928.
Schlicker, C., Gertz, M., Papatheodorou, P., Kachholz, B., Becker, C.F., Steegborn, C., 2008.Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3and Sirt5. J. Mol. Biol. 382, 790–801.
Schwer, B., Verdin, E., 2008. Conserved metabolic regulatory functions of sirtuins. CellMetab. 7, 104–112.
Schwer, B., North, B.J., Frye, R.A., Ott, M., Verdin, E., 2002. The human silent informationregulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adeninedinucleotide-dependent deacetylase. J. Cell Biol. 158, 647–657.
Schwer, B., Eckersdorff, M., Li, Y., Silva, J.C., Fermin, D., Kurtev, M.V., Giallourakis, C.,Comb, M.J., Alt, F.W., Lombard, D.B., 2009. Calorie restriction alters mitochondrialprotein acetylation. Aging Cell 8, 604–606.
Schwer, B., Schumacher, B., Lombard, D.B., Xiao, C., Kurtev, M.V., Gao, J., Schneider, J.I.,Chai, H., Bronson, R.T., Tsai, L.H., Deng, C.X., Alt, F.W., 2010. Neural sirtuin 6 (Sirt6)ablation attenuates somatic growth and causes obesity. Proc. Natl. Acad. Sci. U.S.A.107, 21790–21794.
Sedding, D.G., 2008. FoxO transcription factors in oxidative stress response andageing—a new fork on the way to longevity? Biol. Chem. 389, 279–283.
Shi, T., Fan, G.Q., Xiao, S.D., 2010. SIRT3 reduces lipid accumulation via AMPK activationin human hepatic cells. J. Dig. Dis. 11, 55–62.
Please cite this article as: Pereira, C.V., et al., Regulation and protectiodoi:10.1016/j.mito.2011.07.003
Shimazu, T., Hirschey, M.D., Hua, L., Dittenhafer-Reed, K.E., Schwer, B., Lombard, D.B.,Li, Y., Bunkenborg, J., Alt, F.W., Denu, J.M., Jacobson, M.P., Verdin, E., 2010. SIRT3deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 andregulates ketone body production. Cell Metab. 12, 654–661.
Shoba, B., Lwin, Z.M., Ling, L.S., Bay, B.H., Yip, G.W., Kumar, S.D., 2009. Function ofsirtuins in biological tissues. Anat. Rec. (Hoboken) 292, 536–543.
Shulga, N., Wilson-Smith, R., Pastorino, J.G., 2010l. Sirtuin-3 deacetylation of cyclophilinD induces dissociation of hexokinase II from the mitochondria. J. Cell. Sci. 123,894–902.
Smith, J.S., Brachmann, C.B., Celic, I., Kenna, M.A., Muhammad, S., Starai, V.J., Avalos, J.L.,Escalante-Semerena, J.C., Grubmeyer, C., Wolberger, C., Boeke, J.D., 2000. Aphylogenetically conserved NAD+−dependent protein deacetylase activity inthe Sir2 protein family. Proc. Natl. Acad. Sci. U. S. A. 97, 6658–6663.
Smith, J.S., Avalos, J., Celic, I., Muhammad, S., Wolberger, C., Boeke, J.D., 2002. SIR2family of NAD(+)-dependent protein deacetylases. Methods Enzymol. 353,282–300.
Someya, S., Yu, W., Hallows, W.C., Xu, J., Vann, J.M., Leeuwenburgh, C., Tanokura, M.,Denu, J.M., Prolla, T.A., 2010. Sirt3 mediates reduction of oxidative damage andprevention of age-related hearing loss under caloric restriction. Cell 143, 802–812.
Southwood, C.M., Peppi, M., Dryden, S., Tainsky, M.A., Gow, A., 2007. Microtubuledeacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 32, 187–195.
Sugden, M.C., Caton, P.W., Holness, M.J., 2010. PPAR control: it's SIRTainly as easy asPGC. J. Endocrinol. 204, 93–104.
Sultana, R., Butterfield, D.A., 2010. Role of oxidative stress in the progression ofAlzheimer's disease. J. Alzheimers Dis. 19, 341–353.
Sundaresan, N.R., Samant, S.A., Pillai, V.B., Rajamohan, S.B., Gupta, M.P., 2008. SIRT3 is astress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 28, 6384–6401.
Sundaresan, N.R., Gupta, M., Kim, G., Rajamohan, S.B., Isbatan, A., Gupta, M.P., 2009.Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependentantioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2771.
Tang, B.L., Chua, C.E., 2008. SIRT2, tubulin deacetylation, and oligodendrogliadifferentiation. Cell Motil. Cytoskeleton 65, 179–182.
Tanny, J.C., Dowd, G.J., Huang, J., Hilz, H., Moazed, D., 1999. An enzymatic activity in theyeast Sir2 protein that is essential for gene silencing. Cell 99, 735–745.
Tao, R., Coleman, M.C., Pennington, J.D., Ozden, O., Park, S.H., Jiang, H., Kim, H.S., Flynn,C.R., Hill, S., Hayes McDonald, W., Olivier, A.K., Spitz, D.R., Gius, D., 2010. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSODactivity in response to stress. Mol. cell 40, 893–904.
Vakhrusheva, O., Smolka, C., Gajawada, P., Kostin, S., Boettger, T., Kubin, T., Braun, T.,Bober, E., 2008. Sirt7 increases stress resistance of cardiomyocytes and preventsapoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 102, 703–710.
Van Gool, F., Galli, M., Gueydan, C., Kruys, V., Prevot, P.P., Bedalov, A., Mostoslavsky, R.,Alt, F.W., De Smedt, T., Leo, O., 2009. Intracellular NAD levels regulate tumor necrosisfactor protein synthesis in a sirtuin-dependent manner. Nat. Med. 15, 206–210.
Vaquero, A., 2009. The conserved role of sirtuins in chromatin regulation. Int. J. Dev.Biol. 53, 303–322.
Vaquero, A., Sternglanz, R., Reinberg, D., 2007. NAD+-dependent deacetylation of H4lysine 16 by class III HDACs. Oncogene 26, 5505–5520.
Verdin, E., Hirschey, M.D., Finley, L.W., Haigis, M.C., 2010. Sirtuin regulation ofmitochondria: energy production, apoptosis, and signaling. Trends Biochem. Sci.
Voelter-Mahlknecht, S., Letzel, S., Mahlknecht, U., 2006. Fluorescence in situhybridization and chromosomal organization of the human Sirtuin 7 gene. Int. J.Oncol. 28, 899–908.
Wang, F., Nguyen, M., Qin, F.X., Tong, Q., 2007. SIRT2 deacetylates FOXO3a in responseto oxidative stress and caloric restriction. Aging Cell 6, 505–514.
Weber, T.A., Reichert, A.S., 2010. Impaired quality control of mitochondria: aging from anew perspective. Exp. Gerontol. 45, 503–511.
Wu, F., Schweizer, C., Rudinskiy, N., Taylor, D.M., Kazantsev, A., Luthi-Carter, R.,Fraering, P.C., 2010. Novel gamma-secretase inhibitors uncover a commonnucleotide-binding site in JAK3, SIRT2, and PS1. FASEB J. 24, 2464–2474.
Yamamoto, H., Schoonjans, K., Auwerx, J., 2007. Sirtuin functions in health and disease.Mol. Endocrinol. 21, 1745–1755.
Yang, H., Yang, T., Baur, J.A., Perez, E., Matsui, T., Carmona, J.J., Lamming, D.W., Souza-Pinto,N.C., Bohr, V.A., Rosenzweig, A., de Cabo, R., Sauve, A.A., Sinclair, D.A., 2007. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107.
Yi, J., Luo, J., 2010. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim.Biophys. Acta 1804, 1684–1689.
Yu, J., Auwerx, J., 2009. The role of sirtuins in the control of metabolic homeostasis. Ann.N. Y. Acad. Sci. 1173 (Suppl. 1), E10–E19.
Yu, J., Auwerx, J., 2010. Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation. Pharmacol. Res. 62, 35–41.
Zhang, T., Kraus, W.L., 2010. SIRT1-dependent regulation of chromatin and transcrip-tion: linking NAD(+) metabolism and signaling to the control of cellular functions.Biochim. Biophys. Acta 1804, 1666–1675.
Zhong, L., D'Urso, A., Toiber, D., Sebastian, C., Henry, R.E., Vadysirisack, D.D., Guimaraes,A., Marinelli, B., Wikstrom, J.D., Nir, T., Clish, C.B., Vaitheesvaran, B., Iliopoulos, O.,Kurland, I., Dor, Y., Weissleder, R., Shirihai, O.S., Ellisen, L.W., Espinosa, J.M.,Mostoslavsky, R., 2010. The histone deacetylase Sirt6 regulates glucose homeosta-sis via Hif1alpha. Cell 140, 280–293.
Zhu, J., Chu, C.T., 2010. Mitochondrial dysfunction in Parkinson's disease. J. AlzheimersDis. 20 (Suppl. 2), S325–S334.
n of mitochondrial physiology by sirtuins, Mitochondrion (2011),

Tales of Broken Mitochondria: Drug‐induced Cardiac Mitochondrionopathy Paulo J. Oliveira, Ph.D. Center for Neuroscience and Cell Biology University of Coimbra, Portugal Email: [email protected] Drug‐induced cardiac mitochondrial dysfunction can progressively result into organ degeneration. Classical examples of drug‐induced cardiac mitochondrionopathy include nucleoside reverse transcriptase inhibitors, local anesthetics and anthracyclines. Doxorubicin (adriamycin, DOX) is a clear study case as a well known pharmaceutical can lead to progressive degeneration of cardiac mitochondrial function. DOX is a potent anthracycline antineoplastic agent, whose clinical use is limited by a dose‐dependent and cumulative cardiotoxicity, with a clear mitochondrial component. In this particular case, protection of cardiac mitochondrial function during DOX treatment appears to be critical for preventing the maintenance of the myocyte bioenergetics. Funding: Work funded by FCT research grants PTDC/SAU‐TOX/110952/2009, PTDC/SAU‐OSM/64084/2006 and PTDC/SAU‐OSM/104731/2008.

Register by March 4 and
Save up to $350!
Organized by Cambridge Healthtech Institute n 250 First Avenue Suite 300 n Needham, MA 02494 n 781.972.5400 n healthtech.com
Monitoring Cardiotoxicity
Predicting Hepatotoxicity
Detecting Nephrotoxicity
Early ADME/DMPK Predictions
June 7 - 9, 2011 Sheraton Philadelphia City CenterPhiladelphia, PA
WorldPharmaCongress.com
Corporate Sponsor
M e s o S c a l e D i s c o v e ry
®
SHORT COURSESMonday, June 6
n Use of Stem Cells for Safety Screening
n Advanced Topics in Drug Metabolism
n Translating Safety Biomarkers from the Lab to the Clinic
n Addressing Safety Concerns for Biological Drugs
Wednesday, June 8
n Mechanistic Insights into Hepatotoxicity
Cambridge Healthtech Institute’s 10th Annual
WPCWorld Pharma Congress
Promising Assays and Technologies forBetter Pre-Clinical Predictions
Thomas Force, M.D. Professor of Medicine and Clinical Director of the Center for Translational Medicine, Thomas Jefferson University
Stephen Furlong, Ph.D. Safety Science Lead, U.S., Patient Safety, AstraZeneca
Paul B. Watkins, M.D. Director, Hamner-UNC Institute for Drug Safety Sciences, Verne S. Caviness Distinguished Professor of Medicine, University of North Carolina at Chapel Hill
FEATURED SPEAKERS
Early, Efficient and Effective Tools for Predicting Drug-Induced Adverse Events
SCIENTIFIC PROGRAMS
2 | World Pharma Congress WorldPharmaCongress.com
STAY CONNECTED Join our group on Follow us on @chi_healthtech #WPC11
*Separate registration required, please see page 3 for details.
WORLD PHARMA CONGRESS CONFERENCE-AT-A-GLANCEDRUG SAFETY SUMMIT DRUG DISCOVERY SUMMIT SCREENING SUMMIT
Monday June 6
Pre-Conference Short Courses*
Tuesday June 7
Innovative Approaches for Monitoring Cardiotoxicity
Detecting Nephrotoxicity Using
Early Markers and Imaging Tools
Targeting Pain with Novel Therapeutics
Successful Targeting of Alzheimer’s Disease
Tools & Technologies for HTS
Wednesday June 8
Innovative Approaches for Monitoring Cardiotoxicity
Detecting Nephrotoxicity Using
Early Markers and Imaging Tools
Targeting Pain with Novel Therapeutics
Successful Targeting of Alzheimer’s Disease
Tools & Technologies for HTS
New Assays and Tools for Predicting
Hepatotoxicity
Early ADME and DMPK Predictions for Better
Lead Optimization
In vivo Molecular Imaging in Drug
Discovery & Development
Targeting Parkinson’s Disease
Evaluating Novel Technologies for Cell-Based Screening
Dinner Short Courses*
Thursday June 9
New Assays and Tools for Predicting
Hepatotoxicity
Early ADME and DMPK Predictions for Better
Lead Optimization
In vivo Molecular Imaging in Drug
Discovery & Development
Targeting Parkinson’s Disease
Evaluating Novel Technologies for Cell-Based Screening
CHI’s World Pharma Congress 2011 encompasses a broad spectrum of topics that are very important and relevant to scientists in academia as well as those in the pharmaceutical and biotechnology industry. Building on last year’s focus on the pre-clinical aspects of drug discovery and development, the congress has now expanded the coverage of each of its three summits by adding two new conferences to the program. The conferences all offer informative and pragmatic viewpoints for tackling issues relevant to chemists, biologists, pharmacologists, toxicologists and clinicians alike. Each conference features presentations, interactive panels and technology talks that cover the very latest on the topic, both on the scientific and the technical side. The World Pharma Congress continues to offer attendees and exhibitors ample opportunity to network, brain-storm and collaborate on various fronts.
SPONSORSHIP AND EXHIBITOR INFORMATIONCHI offers comprehensive sponsorship packages which include presentation opportunities, exhibit space, branding, as well as the use of the pre and post show delegate list.
Sponsorships allow you to achieve your objectives before, during, and long after the event. Any sponsorship can be customized to meet with your company’s needs and budget.
Opportunities Include:Sponsored PresentationsSpeak to a captive audience about your latest technology. Sponsorship includes a 15 minute podium presentation within the scientific agenda, exhibit space, onsite branding, and access to cooperative marketing efforts by CHI.
Breakfast & Luncheon PresentationsInvite session attendees to enjoy breakfast or lunch on your company’s behalf while you give your talk. Includes a 30-minute presentation and concludes with a 15-minute Q&A session.
Invitation-Only VIP Dinner/ Hospitality SuiteSponsor will select invitees from the conference pre-registration list for an evening of networking at the hotel or a top local venue. CHI will extend
invitations, conduct follow-up and monitor responses. Reminder cards will be placed in the badges of those delegates who will be attending.
Other Networking and Promotional Opportunities:
•BadgeLanyardsSponsor(exclusive)
•ExhibitHallReception•HotelRoomDoorHanger(1night
exclusive)•HotelRoomDrop•KeynoteChairDrop
•SpecificConferenceChairDrop•PadfolioSponsor•RefreshmentBreak•ToteBagSponsor•ToteBagInsert•Program&ExhibitGuideSponsor
(exclusive)
ExhibitsExhibitors at the World Pharmaceutical Congress will enjoy facilitated networking opportunities with over 500 high-level decision-makers. Speak face to face with prospective clients and showcase your latest product, service or solution.
For information, contact:Suzanne CarrollManager, Business Development781-972-5452 n [email protected]

WorldPharmaCongress.com World Pharma Congress | 3
WPC Short Courses*
MONDAY, JUNE 6 (9 AM - 12 pM)
ANIMAL MODELS OF PAIN: PrOgrESS AND CHALLENgESDue to frustration with translational progress, animal models of pain are currently being reconsidered. This course will cover:• Implementationofclassicalmodelsofacute,tonicand
chronic pain• Limitationsoftheseclassicalmodels• Refinementofclassicalmodelsviaaconsiderationof
modulatory factors (sex, genetics, testing environment, social modulation)
• Developmentofnewanimalmodels(e.g.,operantmethods, spontaneous behaviors)
Course Instructor:Jeffrey S. Mogil, Ph.D., E.P. Taylor Professor of Pain Studies, McGill University
USE OF STEM CELLS FOr SAFETy SCrEENINgThe course provides new insights into the use of embryonic and pluripotent stem cells for drug safety testing, especially cardiac safety.• Differentiationofhumanstemcellsintocardiacmyocytes• Comparisonofelectrophysiologyandpharmacology• Overcomingtechnicalchallengesrelatedtoworkingwith
stem cells• Methodologiestomaintainandusestemcellsfor
predictive safety testingCourse Instructor:Craig T. January, M.D., Ph.D., Professor, Medicine and Physiology, Division of Cardiovascular Medicine, University of Wisconsin, Madison
ADVANCED TOPICS IN DrUg METABOLISMThe purpose of this course is to cover advanced topics related to drug metabolism with a focus on newer developments in the field. • In vitro tools to study drug metabolism• Newbiotransformationpathwaysincludingsomethatlead
to reactive metabolites• Evidencelinkingreactivemetabolitesandidiosyncratic
drug toxicity• In silico tools to predict metabolism
Course Instructor:John C. Erve, Ph.D., Investigator III, Analytical Sciences, Novartis Institutes for Biomedical Research
MONDAY, JUNE 6 (2 pM - 5 pM)
TrANSLATINg SAFETy BIOMArkErS FrOM THE LAB TO THE CLINICThe course offers a unique and practical perspective for successfully translating the pre-clinical work done for testing and validating safety biomarkers to the clinic.• Designandimplementationofstudiestoidentifynewbiomarkers• Designingclinicalstudiestotestandvalidatebiomarkers• Clinicalmethodologiesforcost-effectiveandreliabledecision-making• Bridgingthegapbetweenpre-clinicalandclinicalfindings• Practicalconsiderationswhenusingbiomarkersintheclinic• PointstoconsiderforasuccessfultransferfromthelabtotheclinicCourse Instructors:Stephen Furlong, Ph.D., Safety Science Lead, U.S., Patient Safety, AstraZenecaWilliam B. Mattes, Ph.D., DABT, Independent Consultant, PharmPoint Consulting
ADDrESSINg SAFETy CONCErNS FOr BIOLOgICAL DrUgSThe course offers guidance from experts in the field on what is being used and looked at for early safety assessments for biological molecules, and how these early predictions are then being applied for clinical testing.• Overviewofchallengespertainingtothesafetyofbiologics• Tools,markersandassaysforearlysafetypredictions• Assessingimmunogenicityandoff-targeteffects• Regulatoryguidelinesandtheirinterpretations• CriteriafordeterminingwhatneedstobetestedandwhenCourse Instructors:Lisa M. Plitnick, Ph.D., Senior Investigator, Safety Assessment, Merck & Co., Inc.Noël Dybdal, Ph.D., D.V.M., Associate Director, Principal Scientist, Safety Assessment, Genentech, Inc. Vivek Kadambi, Ph.D., Senior Director, Drug Safety Evaluation, Millennium, The Takeda Oncology Company
PrACTICAL APPLICATION OF PLATE-BASED LABEL-FrEE BIOSENSOrS: gETTINg THE BASICS & DETAILS yOU NEED FOr DECISION MAkINgThis course will provide the attendee with all the necessary information to select the most appropriate label free method for their work. A wide variety of technologies are available, each with specific advantages depending on the scope of individual projects. This course will walk the attendee through evaluating instrumentation, comparing and selecting methodologies, as well as learning the basics of each technology. All current label free methods will be presented.• Basicunderstandingofcurrenttechnologies• Evaluatinginstruments• Latestapplicationsinbiochemicalmeasurementsofproteinsandsmallmolecules• Latestapplicationsincellbasedassaysfromtargetvalidationthroughinter/intracell
signaling and cardiac toxicity• Optimizationofyourlabel-freeassaysCourse Instructor:Lance Laing, Ph.D., Executive Director, Business Development, ACEA BiosciencesLisa K. Minor, Ph.D., President, In vitro Strategies, LLC.
WEDNESDAY, JUNE 8 (6 pM - 9 pM)
MOLECULAr IMAgINg IN DrUg DISCOVEry AND DEVELOPMENT: BACk TO BASICS. This course will provide knowledge needed to choose the appropriate imaging modality for a pre-clinical study and the basic requirements for generation of imaging agents for optical, MR, and nuclear imaging. It will consist of two parts:•Strengthsandlimitationsofimagingmodalities• Imagingagentdesignandsynthesis
Course Instructors:Thomas Krucker, Ph.D., Head, Molecular Imaging, Global Imaging Group, Novartis Institutes for Biomedical Research, Inc.Hisataka Kobayashi, M.D., Ph.D., Chief Scientist, Molecular Imaging Program, NCI/NIHVania Kenanova, Ph.D., Head , Pre-Clinical PET/SPECT/CT Imaging Laboratory, Novartis Institutes for Biomedical Research, Inc.
MECHANISTIC INSIgHTS INTO HEPATOTOxICITy The course is designed for both pre-clinical and clinical scientists looking to better understand the mechanisms underlying drug-induced liver injury or DILI, to help in the development of early predictive technologies for hepatotoxicity including mechanism-based assays. It provides an overview of cellular pathways involved in:• Mitochondrialdysfunctionandoxidativestress• Inflammation• Excessivegenerationofreactivemetabolites• Inhibitionofbilesalteffluxproteinandinvolvementofhepatictransportersindrug-
induced hepatotoxicity
Course Instructors:Dylan P. Hartley, Ph.D., Senior Scientist, Investigative Toxicology, Genentech, Inc.José E. Manautou, Ph.D., Associate Professor of Toxicology, Department of Pharmaceutical Sciences, University of ConnecticutRobert A. Roth, Ph.D., DABT, Professor, Pharmacology and Toxicology, Director, Graduate Program in Environmental and Integrative Toxicological Sciences, Michigan State UniversityYvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D
*Separate Registration Required

4 | World Pharma Congress WorldPharmaCongress.com
4th Annual
Innovative Approaches for Monitoring CardiotoxicityJuNE 7 - 8, 2011
TUESDAY, JUNE 7
7:45 am registration and Morning Coffee
EArLy In VItro MODELS AND MArkErS FOr CArDIAC SAFETy PrEDICTIONS
8:45 Chairperson’s Opening remarksPeter Hoffmann, M.D., Ph.D., Executive Director, Pre-Clinical Safety, Novartis Institutes for BioMedical Research
8:55 Expanding in vitro Biochemical and Cellular Models for Earlier Drug Safety Assessment Mary Ellen Cvijic, Ph.D., Principal Scientist, Lead Evaluation, Molecular Sciences and Candidate Optimization, Bristol-Myers Squibb Co. We have developed a high-throughput in vitro assay panel including target classessuchasGPCRs,kinases,transporters,ionchannels,andnuclearhormone receptors to determine which critical targets can be used to bestflagpotentialcardiacliabilityissuesbeforecompoundsadvancetoin vivo or late-stage drug safety evaluation. We have investigated which in vitro assays offer more sensitive and physiologically-relevant read-outs for assessing compound safety profiles and have identified current gaps in harnessing state-of-art technology platforms. We can now address how to establish a comprehensive pharmacologic liability tool kit for structure liability relationship studies and how to build connectivity between cause and effect.
9:25 Stem Cell Cardiomyocyte ScreeningCraig T. January, M.D., Ph.D., Professor, Medicine and Physiology, Division of Cardiovascular Medicine, University of Wisconsin, Madison
9:55 Networking Coffee Break
10:25 Development of Translatable Biomarkers for Cardiovascular Safety Jennifer Colangelo, Ph.D., Associate Director, Drug Safety R&D, Pfizer Global Research and Development Safety biomarkers are an integral part of the decision-making process for drug development at all stages, aiding in compound selection for early pre-clinical studies and ensuring patient safety in clinical trials. Mass spectrometry is one of the critical tools for safety biomarker discovery, development and deployment, providing assays that are often easily translatable between species. This presentation will provide examples of mass spectrometry applications to the development of biomarkers for drug-induced cardiovascular toxicity, including MS-based metabonomics for biomarker discovery and stable label approaches for the quantitation of novel proteins.
10:55 Pre-Clinical Strategies for De-risking the Potential of Cardiovascular Toxicity Peter Hoffmann, M.D., Ph.D., Executive Director, Pre-Clinical Safety and Co-Chair, Translational Cardiovascular Advisory Team, Novartis Institutes for BioMedical Research The introduction of in vitro and in vivo cardiovascular safety tests as suggested by guidelines S7A and S7B was successful in preventing acute and catastrophic effects in Phase 1 studies, primarily in healthy male volunteers. On the contrary, recent experiences show that the manifestation of human cardiovascular adverse effects during late stage clinical development or post-marketing is poorly predicted. The presentation summarizes current status and emerging trends of preclinical strategies for de-risking the potential of cardiovascular toxicity in the target population, e.g., diabetic patients.
11:25 Biologicals and Cardiac Toxicity risk: relating Toxicity to Mechanism of Action Noël Dybdal, Ph.D., D.V.M., D.A.C.V.P., Associate Director, Principal
Scientist, Safety Assessment, Genentech, Inc. High molecular weight biological therapeutics in general and monoclonal antibodies specifically are highly targeted in their activity and risk of off-target toxicity is low. Adverse cardiovascular effects associated with these drugs to-date result from on-target pharmacology and relate to their mechanism of action. Species specificity of biologics presents challenges that limit the extent to which cardiotoxicity risk can be assessed preclinically. However, novel approaches including in vitro strategies are increasingly offering better opportunities for focused safety assessments. This presentation will include case studies as examples of preclinical cardiac toxicity assessments for biologicals of various types.
11:55 Luncheon Presentation Sponsored bySpeaker to be Announced
M e s o S c a l e D i s c o v e ry
®
12:25 pm Luncheon Presentation II (Sponsorship Opportunitiy Available)
CHALLENgES COrrELATINg In VItro AND In VIVo DATA
1:30 Chairperson’s remarksVivek Kadambi, Ph.D., Senior Director, Drug Safety Evaluation, Millennium, The Takeda Oncology Company
1:35 Evolving Trends in Pre-Clinical Cardiac Safety: gazing into the Crystal Ball Gary Gintant, Ph.D., Senior Group Leader, Integrative Pharmacology, Global Pharmaceutical Research & Development, Abbott LaboratoriesIncreasing emphasis on the safety of novel drug candidates demands more efficient discovery and development efforts with a balanced focus onrisk/benefitconsiderations.Thisgoalwillberealizedonlywitha)early “frontloading” of appropriate assays to derisk compounds early in discovery, b) an understanding of the strengths and limitations of present (and evolving) assays, and c) an appreciation of present and emerging cardiac safety issues within the context of drug efficacy in development. This presentation will provide some instructive preclinical examples of where we have succeeded present-day gaps in our understanding, and future challenges for pre-clinical cardiac safety.
2:05 Does it Help or Hurt to know Cardiovascular Biology? Douglas B. Sawyer, M.D., Ph.D., Lisa M. Jacobson Professor of Medicine and Chief, Cardiovascular Division, Vanderbilt University Medical Center
2:35 you Don’t give Drugs to Normal People, so Why Search for Toxicity in Normal Animals? Robert Hamlin, Ph.D., Professor, Veterinary Biosciences, Ohio State University It is important to search for potential risks of drugs in subjects possessing the clinical substrates for which they are indicated. Heart failure, ventricular hypertrophy, and diabetes are present in many persons manifesting drug toxicity, therefore it appears reasonable to search for potential toxicity inanimalsafflictedwiththem.Furthermoreitisessentialtointerrogateall physiological parameters that if, affected by a drug may translate to morbidityand/ormortality.Inmodelsofrabbitsanddogspossessinghypertophy and heart failure, sensitivity to detect drug toxicity is improved dramtically over studies conducted in normals, and specificity does not appear to be reduced signficantly.
3:05 Sponsored Presentations (Opportunities Available)3:35 grand Opening refreshment Break in the Exhibit Hall

WorldPharmaCongress.com World Pharma Congress | 5
4th Annual
Innovative Approaches for Monitoring CardiotoxicityJuNE 7 - 8, 2011
»4:35 FEATURED pRESENTATION: pre-Clinical Strategies for predicting and preventing Cardiotoxicity
Thomas Force, M.D., Professor of Medicine and Clinical Director of the Center for Translational Medicine, Thomas Jefferson University
Despite intense interest in strategies to predict which tyrosine kinase inhibitor (TKI) cancer therapeutics may be
associated with cardiotoxicity, current approaches are inadequate. Herein I will explore a variety of pre-clinical models, starting with “best guess” approaches (virtual cardiotoxicity) based on what we do, and do not know about the role of protein kinases in maintaining homeostasis in the heart. I will also discuss various pre-clinical models, highlighting pros and cons of each. Finally, I will focus on the zebrafish as a pre-clinical model, and present some recent data suggesting it may be a viable tool for prediction and for defining mechanisms of cardiotoxicity.
5:05 PANEL DISCUSSION: Minimizing the Disconnect Between the In Vitro and In Vivo WorldsModerator: Vivek Kadambi, Ph.D., Senior Director, Drug Safety Evaluation, Millennium, The Takeda Oncology Company
5:35 - 6:30 Happy Hour in the Exhibit Hall
WEDNESDAY, JUNE 8
7:30 am Continental Breakfast Breakout Discussions Breakout Discussion Topics:l Trends in Safety Screening for Small Molecule Drugs Versus Biologicsl New Approaches and Insights for Early Pre-Clinical Safety Testing
MITOCHONDrIAL INVOLVEMENT IN CArDIAC, rENAL AND LIVEr TOxICITy
(Joint session for Cardiotoxicity and Nephrotoxicity tracks)
8:30 Chairperson’s remarksYvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D
8:40 Introduction to Mitochondrial Function and Drug-Induced DysfunctionJames Dykens, Ph.D., CEO, EyeCyte Therapeutics Mitochondria typically produce more than 90% of the ATP in aerobically poised cells, plus the majority of potentially injurious, free radicals. Inhibition of the electron transfer system, or the uncoupling of the membrane potential it generates from phosphorylation, reduce the bioenergetic capacity of the cell. Many drugs have direct and deleterious mitochondrial effects that contribute to the etiology of idiosyncratic drug-induced organ toxicity. These off-target effects are a function of compound chemistry, but also of genetic diversity in plasma membrane transporters that facilitate bio-accumulate of drugs into the mitochondria. Emerging animal models will be discussed with an emphasis on hepatotoxicity.
9:10 Tales of Broken Mitochondria: Drug-Induced Cardiac MitochondrionopathyPaulo Oliveira, Ph.D., Group Leader, Mitochondrial Toxicology and Disease, Center for Neuroscience and Cell Biology, University of Coimbra, Portugal and Visiting Research Associate, University of Minnesota Medical School Drug-induced cardiac mitochondrial dysfunction can progressively result in organ degeneration. Classical examples of drug-induced cardiac mitochondrionopathy include nucleoside reverse transcriptase inhibitors, local anesthetics and anthracyclines. Doxorubicin (adriamycin, DOX) is a clear case study of a well known pharmaceutical that can lead to progressive degeneration of cardiac mitochondrial function. DOX is a potent anthracycline anti-neoplastic agent, whose clinical use is limited by a dose-dependent and cumulative cardiotoxicity, with a clear mitochondrial component. In this case, protection of cardiac mitochondrial function during DOX treatment appears to be critical for preventing the maintenance of the myocyte bioenergetics.
9:40 Sponsored Presentations (Opportunities Available)
10:10 Networking Coffee Break in the Exhibit Hall
10:50 Mitochondrial Homeostasis in Acute kidney InjuryRick Schnellmann, Ph.D., Professor, Chair, Pharmaceutical and Biomedical Sciences, South Carolina College of Pharmacy Mitochondrial damage is a major contributor to the initiation of tubular cell injury and the progression of acute kidney injury (AKI) produced by drugs, toxicants, and ischemia. To understand the role of mitochondria in organ damage and repair, we think that mitochondria need to be examined holistically by measuring mitochondrial homeostasis. This includes changesinmitochondrialloss,fission/fusion,mitophagy,andbiogenesisover time. Using this approach, temporal differences in mitochondrial loss, dynamics and biogenesis were observed with mitochondrial lossoccurringearlyandchangesinmitochondrialfission/fushionandbiogenesis occurring later after AKI.
11:20 Mechanistic Insights into Mitochondrial-Based Organ Toxicity Yvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D Previous speakers have elucidated in detail on the contribution of mitochondrial impairment to different organ toxicities. It is apparent that in order to ovoid late stage attrition due to mitochondrial toxicity, early predictive screens need to be deployed early in the drug discovery process. Here I will show organelle and cell-based HTS applicable screens to detect such liabilities. I will describe the screens using examples from differentdrugclassessuchasantidiabetics/antilipidemics,antivirals,antibiotics, and NSAIDs.
11:50 PANEL DISCUSSION: Strategies for Assessing Mitochondrial Involvement in Drug-Induced Organ Toxicities Moderator: Yvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D
12:20 pm End of Conference
The Intro-Net offers you the opportunity to set up meetings with selected attendees before, during and after this conference, allowing you to connect to the key people you want to meet. This online system was designed with your privacy in mind and is available only
to registered session attendees of this event.
Registered conference attendees will receive more information on accessing the Intro-Net in the weeks leading up to the event!
Maximize your experience on-site at World Pharma Congress 2011!

6 | World Pharma Congress WorldPharmaCongress.com
3rd Annual
New Assays and Tools for Predicting HepatotoxicityWEDNESDAY, JUNE 8
12:30 pm registration
IDIOSyNCrATIC DILI: TrANSITIONINg FrOM PrE-CLINICAL TO CINICAL
1:55 Chairperson’s Opening remarksArie Regev, M.D., Hepatology Consultant and Chair, Liver and GI Safety Committee, Global Patient Safety, Eli Lilly and Company
2:00 How good are Currently Available Biomarkers for Idiosyncratic DILI, and Which New Biomarkers Should We Look for? Arie Regev, M.D., Chair, Liver and GI Safety Advisory Committee, Global Patient Safety, Eli Lilly and Company; Adjunct Associate Professor of Medicine, Indiana University School of Medicine Efforts to develop new biomarkers for IDILI have been largely unsuccessful due to limitations such as, incomplete understanding of the mechanisms, lack of effective in-vitro models, absence of universally accepted animal models, and often, lacking scientific exchange between clinicians and basic science researchers. One important prerequisite for the facilitation of new IDILI biomarkers is enhanced understanding on the types of questions that can and should be addressed. This presentation will outline pertinent clinical issues that could theoretically be addressed by specific diagnostic or predictive biomarkers.
» 2:30 FEATURED pRESENTATION: Genetic Basis of Susceptibility to DILI
Paul B. Watkins, M.D., Director, Hamner-UNC Institute for Drug Safety Sciences, Verne S. Caviness Distinguished Professor of Medicine, University of North Carolina at Chapel Hill
Over the last several years, some pharmaceutical companies have successfullyusedgenomewideassocation(GWA)tofindgeneticsusceptibility loci for hepatoxicity observed in clinical trials. In one case, this approach has lead to a proposal to reintroduce with routine genetic testing a drug that has been withdrawn from worldwide marketsduetoDILI.GenotypingdatafromthelargestDILIgenebanks(the International Severe Adverse Events Consortium and the U.S. based Drug Induced Liver Injury Network) have been recently pooled. GWAanalysisofthisdataset,whichreflectsDILIduetoover200different drugs, is providing fresh insight into mechanisms underlying DILI and should inform the hunt for epigenetic and environmental factors underlying DILI.
3:00 Sponsored Presentations (Opportunities Available)
3:30 Networking refreshment Break in the Exhibit Hall
4:30 Pediatric Drug Induced Liver Injury- Children Are Not Just Small Adults William Salminen, Ph.D., DABT, Director, Center for Hepatotoxicity, U.S. FDA, National Center for Toxicological Research Children are not simply small adults and it follows that children may exhibit differential sensitivity to drug-induced adverse events. This also applies to drug-induced liver injury (DILI). As an embryo develops, leading to the birth of a child, and eventually maturation into an adult, the human body goes through many different development phases. Various factors involved with the developmental phases may make the developing human more or less susceptible to DILI when compared to adults. This presentation will review the major developmental phases of the maturing liver with an emphasis on phases that may pose unique sensitivities to DILI.
5:00 PANEL DISCUSSION: Early Prediction of Idiosyncratic DILI: recent Progress and Interesting Trends Moderator: Arie Regev, M.D., Hepatology Consultant and Chair, Liver and GI Safety Committee, Global Patient Safety, Eli Lilly and Company
5:30 End of Day
ThURSDAY, JUNE 9
7:20 am Continental Breakfast Breakout Discussions Breakout Discussion Topics:l Utilization of Appropriate Models and Markers for Predicting Liver Injury l Promising in vitro Tools for Early Pre-Clinical Testingl Challenges with Predicting and Monitoring Liver Injury in the Clinic
EFFECTIVE USE OF MODELS AND BIOMArkErS FOr PrEDICTINg LIVEr INJUry
8:20 Chairperson’s remarksRobert A. Roth, Ph.D., DABT, Professor, Pharmacology and Toxicology, Director, Graduate Program in Environmental and Integrative Toxicological Sciences, Michigan State University
8:30 DILI-sim: An in silico Approach to Understanding and Predicting Drug-Induced Liver InjuryBrett A. Howell, Ph.D., Research Scientist, The Hamner-University of North Carolina Institute for Drug Safety SciencesDrug-induced liver injury (DILI) is the adverse drug event that most frequently leads to termination of clinical development programs and regulatory actions on drugs. A predictive model has been developed based on physiological processes involved in DILI. The model initially focuses on acetaminophenandincludesmultiplescales,spanningfromtheorgan/tissue level to the molecular and cellular levels. The model accurately reproduced acetaminophen pharmacokinetic and other measures for rats, mice, and humans. The use of N-acetyl-cysteine (NAC) as a treatment for acetaminophen overdose was analyzed to predict optimal use. Finally, the model successfully predicted the species differences in hepatotoxicity of methapyrilene using in vitro to in vivo extrapolation.
9:00 Virtual Liver: Integrating in vitro and in vivo Data to Predict Chemical-induced ToxicityImran Shah, Ph.D., Head, Computational Systems Biology, Natl .Ctr. for Computational Toxicology, U.S. Environmental Protection AgencyIt is difficult to assess the health impact of long-term exposure to low levels of contaminants from animal studies. Current methods for testing the toxicity of a single chemical can cost millions of dollars, take up to two years and sacrifice thousands of animals. In vitro models offer a more efficient and humane alternative, however, translating chemical-induced molecular changes in cell cultures to clinical outcomes remains an open problem. This talk will explain how the EPA Virtual Liver aims to bridge in vitro data from the EPA ToxCast Project to in vivo effects through a novel cellular systems model of hepatic tissues. ThisabstractdoesnotnecessarilyreflectUSEPApolicy.
9:30 Inflammatory Stress responses and Animal Models for Idiosyncractic DILIRobert A. Roth, Ph.D., DABT, Professor, Pharmacology and Toxicology, Director, Graduate Program in Environmental and Integrative Toxicological Sciences, Michigan State UniversityAmodestinflammatorystressinanimalmodelscanrendertheliversensitiveto injury from numerous drugs that cause idiosyncratic toxicity in humans. Thisenhancedsensitivityisassociatedwithexpressionofproinflammatorycytokines,othermediatorsofinflammationandwithenhancedsensitivityofhepatocytestotheirdamaginginfluence.Inparticular,prolongedgeneration of tumor necrosis factor-alpha (TNF) appears to be important for thedevelopmentofinjury.Understandingmechanismsofdrug-inflammation
JuNE 8 - 9, 2011

WorldPharmaCongress.com World Pharma Congress | 7
3rd Annual
New Assays and Tools for Predicting HepatotoxicityJuNE 8 - 9, 2011
interactions could lead to models to predict preclinically the potential of some drug candidates to cause idiosyncratic liver injury in humans.
10:00 Networking Coffee Break in the Exhibit Hall
MECHANISMS UNDErLyINg HEPATOTOxICITy
10:45 Prediction of Immune-Mediated Drug-Induced Liver Injury in Pre-Clinical Drug DevelopmentTsuyoshi Yokoi, Ph.D., Professor, Drug Metabolism and Toxicology, Kanazawa UniversityDrug-induced liver injury (DILI) is a major problem in drug development and clinical drug therapy. The pathogenesis of DILI usually involves the participation of the parent drug or metabolites that either directly affect the cell biochemistry or elicit an immune response. However, in most cases the mechanisms are still unknown, thus it is difficult to predict and prevent these reactions. Recently, we demonstrated that halothane- and alpha-naphythylisothocyanete (ANIT)-induced liver injury is mediated by interleukin-17 in mice, and carbamazepine-induced liver injury also mediatedbyIL-17.Dicloxacillin-,methimazole-,andflutamide-inducedliver injury is mediated by IL-4. Continued advances in our understanding of immune-mediated DILI will lead to earlier prediction of hepatotoxic potential of drug under development.
11:15 Hepatoprotective Effect of Peroxisome Proliferators is Associated with Induction of Vanin-1 gene Expression José E. Manautou, Ph.D., Associate Professor of Toxicology, Pharmaceutical Sciences, University of Connecticut Clofibrate (CFB) is a peroxisome proliferator, hypolipidemic drug that affords protection against acetaminophen (APAP) hepatotoxicity. The mechanism of this protection is still unknown but thought to be dependent on peroxisome proliferator-activated receptor alpha (PPARα) function. A gene array analysis revealed that the expression of Vanin 1 (Vnn1) is greatly increased in animals exhibiting CFB-mediated resistance to APAP toxicity. In this presentation in vivo and in vitro approaches to examine the role of Vnn1 in APAP hepatotoxicity and hepatoprotection by CFB, and for the development of new therapeutics approaches to minimize acute liver failure produced by APAP overdose will be discussed.
11:45 Luncheon Presentations (Sponsorship Opportunities Available) or Lunch on your Own
EArLy PrE-CLINICAL PrEDICTIONS OF LIVEr INJUry
(Joint session for Hepatotoxicity and ADME/DMPK tracks)
1:15 pm Chairperson’s remarksEric Blomme, D.V.M., Ph.D., D.A.C.V.P., Sr Project Leader, Abbott Labs
1:25 Current Toolbox for the Prediction of Hepatotoxicity Eric Blomme, D.V.M., Ph.D., D.A.C.V.P., Senior Project Leader, Abbott Laboratories Hepatotoxicity represents an important cause of failure in drug discovery and development, and improved approaches to predict and characterize this toxicity could significantly impact pharmaceutical R&D. This presentation will provide an overview of the current status of several technologies used to improve hepatotoxicity prediction during drug discovery. Examples will
be used to illustrate the strengths, limitations and optimal application of these technologies during lead optimization and candidate selection.
1:55 In vitro Strategies: High Content Mechanistic Screening, Mitochondrial Toxicity, and Transporter Assessment Yvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D In recent years we have developed several assays that can be deployed early in the drug discovery process. These include 1. An oxygen consumption assayformitochondrialtoxicity,2.SeveralassaysforERstressand3.AHigh Content Mechanistic Screening approach for multiple assay endpoints in the presence and absence of cytokine addition. Here, I will describe theses screens using examples of hepatotoxic drugs. I will discuss advantages and limitations of each in vitro screen with respect to predicting hepatotoxicity.
2:25 Ice Cream refreshment Break in the Exhibit Hall
3:05 Approaching Hepatotoxicity in Drug Discovery as a Lead Optimization Problem Dylan P. Hartley, Ph.D., Senior Scientist, Investigative Toxicology, Genentech, Inc. Giventhedemandsondrugdiscoveryteamstoproducemoleculesdevoid of hepatotoxicity, toxicologists are now integral members of these teams with new responsibilities geared toward lead optimization. Toxicologists are now expected to assess risk for hepatotoxicity in a compound in predictive manner, or ascribe a mechanism to early hepatoxicity findings, link the findings to an offending moiety within the structure, and define the chemical lead optimization path. Structure-based strategiestopredictand/orattenuatehepatotoxicitywillbepresentedwith respect to various hepatotoxic signals.
3:35 The Utility of Emerging Biomarkers of Liver Injury in Pre-Clinical and Clinical Drug DevelopmentShelli Schomaker, Principal Scientist, Drug Safety R&D, Pfizer, Inc. While alanine aminotransferase (ALT) activity remains the gold standard biomarker of liver injury, the correlation between increased ALT levels and morphological liver findings is rather imperfect. These discrepancies could be due to adaptive responses, altered liver membrane permeability, extrahepatic injury, or treatment-related effects on ALT enzyme activity. This presentation will focus on an evaluation of malate dehydrogenase, purine nucleoside phosphorylase and glutamate dehydrogenase for the detection of liver injury when ALT activity is limited and will demonstrate the utility of this alternative biomarker approach for improving confidence.
4:05 From Mild Pre-Clinical Transaminase Elevations to Idiosyncratic Liver Injury in One Easy Lesson Paul Vancutsem, D.V.M., Ph.D., Director, Pre-Clinical Safety; Senior Member, Novartis Internal Liver Experts Team, Novartis Pharmaceuticals Even if we do not fully understand idiosyncratic liver injury (IDILI), it appears to necessitate events involving an often subtle local liver insult, an immune system imbalance and a specific genetic make-up (HLA alleles). Integration of these traditionally unconnected areas of toxicology will lead to an improved prediction of IDILI. This presentation proposes a template for integration using a pragmatic approach.
4:35 End of Conference
Present a poster and save $50! Cambridge Healthtech Institute encourages attendees to gain further exposure by presenting their work in the poster sessions. To secure a poster board and inclusion in the conference materials, your abstract must be submitted, approved and your registration paid in fullbyApril22,2011.
Reasons you should present your research poster at this conference:
• Yourposterwillbeexposedtoourinternationaldelegation• Receive$50offyourregistration• Yourposterabstractwillbepublishedinourconferencematerials• Yourresearchwillbeseenbyleadersfromtoppharmaceutical,
biotech, academic and government institutes

8 | World Pharma Congress WorldPharmaCongress.com
JuNE 7 - 8, 20112nd Annual
Detecting Nephrotoxicity using Early Markers and Imaging ToolsTUESDAY, JUNE 7
7:45 am registration and Morning Coffee
UNDErSTANDINg MECHANISMS OF NEPHrOTOxICITy
8:45 Chairperson’s Opening remarksStephen Furlong, Ph.D., Safety Science Lead, U.S., Patient Safety, AstraZeneca
8:55 Cisplatin Nephrotoxicity and renal Protective Strategies Zheng Dong, Ph.D., Professor, Department of Cellular Biology and Anatomy, Medical College of Georgia Cisplatin, a widely used chemotherapy drug, has major side effects in normal tissues, notably nephrotoxicity in kidneys. Research during last few years has delineated several signaling pathways leading to renal tubular damage and kidney injury in cisplatin nephrotoxicity, including a rapid DNA damage response. Importantly, cisplatin may activate different signaling pathways in normal tissues and tumors. Targeting these pathways may uncover clinically effective strategies for kideny protection without diminishing the chemotherapy efficacy of cisplatin in tumors.
9:25 Primary Cell Cultures from Human and rat Proximal Tubule as Models to Study Mechanisms of Acute kidney Injury Lawrence H. Lash, Ph.D., Professor, Associate Chair, Pharmacology, School of Medicine, Wayne State UniversityOur laboratory has developed several in vitro models from rat and human kidney, using both non-specific toxic chemicals and specific nephrotoxicants, including halogenated solvents, analgesics, and antibiotics. One focus has been the metabolism and toxicity of the halogenated solvent trichloroethylene (TRI), which is a significant environmental contaminant that has the kidney as one target organ. Molecular approaches and proteomics have been integrated into our in vitro toxicology models to explore the role of specific proteins andexaminetheinfluenceofdiseaseprocesses,suchasdiabeticnephropathy and reduced nephron mass, on renal toxicological responses.
9:55 Networking Coffee Break
MONITOrINg AND ASSESSINg kIDNEy INJUry10:25 Current Use of renal Biomarkers in Early Drug Development
Diann Weddle, Ph.D., D.V.M., Senior Pathologist, Pre-Clinical Safety, Abbott Laboratories An overview of renal toxicology will be presented including a discussion of use and limitations of current biomarkers. Key concepts will be further emphasized through case examples. Within the last year, the Predictive SafetyTestingConsortium’sNephrotoxicityWorkingGroupsubmittedaqualification package for multiple renal biomarkers to the FDA and EMEA and received clearance for limited use in nonclinical and clinical drug development. Conclusions and recommendations from the submission will be summarized.
10:55 Pre-Clinical Biomarkers of Nephrotoxicity: Applications in Drug Discovery and Development Eric Blomme, D.V.M., Ph.D., D.A.C.V.P., Senior Project Leader, Abbott LaboratoriesThis presentation will build upon the preceding presentation discussing the current use of renal biomarkers in early drug development. Specifically, several case examples will be used to illustrate how biomarkers of renal toxicity can be applied in drug discovery and development to increase probability of success and make better decisions
on compounds at earlier stages. Data on performance characteristics for several of these biomarkers in rats will be presented.
11:25 Integrative Assessment of Drug-Induced kidney Function Changes and Acute Injury Using an Automated Blood Sampling and Telemetry (ABST) System Yafei Chen, M.D., M.S., Scientist Safety Pharmacology, Global Safety Assessment, AstraZeneca Pharmaceuticals~20%ofhospitaladmissionsarecausedbynephrotoxicdrugsdueto acute kidney injury. Current preclinical methods are insufficient todetectandpredictdrug-inducedchangesinkidneyfunctionand/or kidney injury (DIKI). We will describe an integrated pharma-cology platform representing a convergence of automated blood sampling with telemetry (ABST) for simultaneous assessment of cardiovascular, renal hemodynamic and excretory functions, and nephron site-specific DIKI biomarkers in surgically prepared conscious rats. This integrated preclinical approach provides multiple translational markers for risk management in early clinical development.
11:55 Luncheon Presentations (Sponsorship Opportunities Available) or Lunch on your Own
kIDNEy MArkErS: FrOM BENCH TO BEDSIDE 1:30 pm Chairperson’s remarksW. Brian Reeves, M.D., F.A.C.P., Chief, Division of Nephrology, Professor and Vice Chair, Medicine, Penn State College of Medicine
» 1:35 FEATURED pRESENTATION: Establishing the Context for Introducing New Safety Biomarkers into Clinical Trials
Stephen Furlong, Ph.D., Safety Science Lead, U.S., Patient Safety, AstraZeneca
There has been rapid progress in developing new safety biomarkers for monitoring organ toxicity. Some of these new markers have already been qualified for use in
supporting pre-clinical studies. Efforts to qualify these new safety biomarkers for use in human clinical trials are also underway. This presentation will explore recent experiences with organ-specific markers and strategies and challenges for progressing these markers for support of clinical trials.
2:05 Clinical Evaluation and Qualification of kidney Safety Biomarkers: A Collaboration between Two ConsortiaMaria Vassileva, Ph.D., Scientific Program Manager, The Biomarkers Consortium, Foundation for the NIHThe Biomarkers Consortium (BC), a public private partnership managed by the Foundation for the NIH, is preparing to launch an important project expected to generate the data needed to advance the regulatory acceptance of new biomarkers appropriate for monitoring kidney safety in the clinic, and reaching alignment on how these biomarkers could improve clinical diagnoses of drug-induced acute kidney injury during drug development and patient therapy with aminoglycosides in patients with cystic fibrosis and cisplatin in patients with head and neck cancer. This project is a collaboration between the BC and the Predictive Safety Testing Consortium of the Critical Path Institute.
2:35 Urinary Cytokines as Biomarkers of Nephrotoxicity W. Brian Reeves, M.D., F.A.C.P., Chief, Division of Nephrology, Professor and Vice Chair, Medicine, Penn State College of Medicine Inflammationisacriticalcomponentofdrug-inducedacutekidneyinjury. In response to injury, renal epithelial cells elaborate a variety of chemokines and cytokines which alter epithelial cell function and also lead totherecruitmentand/oractivationofinflammatorycellsinthekidney.Levels of cytokines increase in the urine early after kidney injury and may

WorldPharmaCongress.com World Pharma Congress | 9
JuNE 7 - 8, 20112nd Annual
Detecting Nephrotoxicity using Early Markers and Imaging Toolsprovideawindowtotheinflammatoryprocessesongoingwithinthekidney.
3:05 Sponsored Presentations (Opportunities Available)
3:35 grand Opening refreshment Break in the Exhibit Hall 4:35 rapid Point-of-Care gFr: Technique, Diagnostic and Therapeutic Advantages Bruce A. Molitoris, M.D., Director, Division of Nephrology and Professor of Medicine, Indiana UniversityGlomerularfiltrationrate(GFR)isthemostimportantmeasureofkidneyfunction.AlthoughmanywaysexisttomeasureGFR,thereisnoacceptable rapid point-of-care technique. Therefore, we have developed afluorescentbasedratiometrictechniqueindogsandpigsusinganLED-based detection device and an intravenous optical catheter by using anon-filterablelargeredfluorescentdextranandasmallkidneyfreelyfilterableFITCdextran.InlessthanonehouraccurateGFRsareobtainedin many conditions including acute kidney injury (AKI). We believe this has clinically important diagnostic and therapeutic advantages in AKI and chronic kidney disease.
5:05 PANEL DISCUSSION: renal Injury Markers and How Effectively Can They Be Used?Moderator: W. Brian Reeves, M.D., F.A.C.P., Chief, Nephrology; Professor and Vice Chair, Medicine, Penn State College of Medicine
5:35 - 6:30 Happy Hour in the Exhibit Hall
WEDNESDAY, JUNE 8
7:30 am Continental Breakfast Breakout Discussions Breakout Discussion Topics:l Biomarkers for Organ Toxicty and Their Effective Use in Pre-Clinical and
Clinical Development l Monitoring Mitochondria and Their Impact on Organ Toxicities
MITOCHONDrIAL INVOLVEMENT IN CArDIAC, rENAL AND LIVEr TOxICITy
(Joint session for Cardiotoxicity and Nephrotoxicity tracks)
8:30 am Chairperson’s remarksYvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D
8:40 Introduction to Mitochondrial Function and Drug-Induced DysfunctionJames Dykens, Ph.D., CEO, EyeCyte Therapeutics Mitochondria typically produce more than 90% of the ATP in aerobically poised cells, plus the majority of potentially injurious, free radicals. Inhibition of the electron transfer system, or the uncoupling of the membrane potential it generates from phosphorylation, reduce the bioenergetic capacity of the cell. Many drugs have direct and deleterious mitochondrial effects that contribute to the etiology of idiosyncratic drug-induced organ toxicity. These off-target effects are a function of compound chemistry, but also of genetic diversity in plasma membrane transporters that facilitate bio-accumulate of drugs into the mitochondria. Emerging animal models will be discussed with an emphasis on hepatotoxicity.
9:10 Tales of Broken Mitochondria: Drug-Induced Cardiac MitochondrionopathyPaulo Oliveira, Ph.D., Group Leader, Mitochondrial Toxicology and Disease, Center for Neuroscience and Cell Biology, University of Coimbra, Portugal and Visiting Research Associate, University of Minnesota Medical School Drug-induced cardiac mitochondrial dysfunction can progressively result in organ degeneration. Classical examples of drug-induced cardiac mitochondrionopathy include nucleoside reverse transcriptase inhibitors, local anesthetics and anthracyclines. Doxorubicin (adriamycin, DOX) is a clear case study of a well known pharmaceutical that can lead to progressive degeneration of cardiac mitochondrial function. DOX is a potent anthracycline anti-neoplastic agent, whose clinical use is limited by a dose-dependent and cumulative cardiotoxicity, with a clear mitochondrial component. In this case, protection of cardiac mitochondrial function during DOX treatment appears to be critical for preventing the maintenance of the myocyte bioenergetics.
9:40 Sponsored Presentations (Opportunities Available)
10:10 Networking Coffee Break in the Exhibit Hall
10:50 Mitochondrial Homeostasis in Acute kidney InjuryRick Schnellmann, Ph.D., Professor, Chair, Pharmaceutical and Biomedical Sciences, South Carolina College of Pharmacy Mitochondrial damage is a major contributor to the initiation of tubular cell injury and the progression of acute kidney injury (AKI) produced by drugs, toxicants, and ischemia. To understand the role of mitochondria in organ damage and repair, we think that mitochondria need to be examined holistically by measuring mitochondrial homeostasis. This includes changesinmitochondrialloss,fission/fusion,mitophagy,andbiogenesisover time. Using this approach, temporal differences in mitochondrial loss, dynamics and biogenesis were observed with mitochondrial lossoccurringearlyandchangesinmitochondrialfission/fushionandbiogenesis occurring later after AKI.
11:20 Mechanistic Insights into Mitochondrial-Based Organ Toxicity Yvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D Previous speakers have elucidated in detail on the contribution of mitochondrial impairment to different organ toxicities. It is apparent that in order to ovoid late stage attrition due to mitochondrial toxicity, early predictive screens need to be deployed early in the drug discovery process. Here I will show organelle and cell-based HTS applicable screens to detect such liabilities. I will describe the screens using examples from differentdrugclassessuchasantidiabetics/antilipidemics,antivirals,antibiotics, and NSAIDs.
11:50 PANEL: Strategies for Assessing Mitochondrial Involvement in Drug-Induced Organ ToxicitiesModerator: Yvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D
12:20 pm End of Conference
Sponsoring Organizations
Corporate Support Sponsor
Corporate Sponsor
M e s o S c a l e D i s c o v e ry
®

10 | World Pharma Congress WorldPharmaCongress.com
Inaugural
Early ADME and DMPK Predictions for Better Lead OptimizationJuNE 8 - 9, 2011
WEDNESDAY, JUNE 8
12:30 pm registration
PrEDICTIVE ADME AND DMPk ASSESSMENTS
1:55 Chairperson’s Opening remarksAlan G.E. Wilson, Ph.D., Vice President, Drug Metabolism, Pharmacokinetics, Toxicology and Pathology, Lexicon Pharmaceuticals
2:00 Transforming Drug Discovery and Development: The Strategic role of ADME, Pk and Toxicology Alan G.E. Wilson, Ph.D., Vice President, Drug Metabolism, Pharmacokinetics, Toxicology and Pathology, Lexicon PharmaceuticalsOver the past decade, or so, we have seen the implementation of early screening for ADME and PK become a standard approach in the drug discovery armamentarium. In addition, there is growing interest intheearlierapplicationofpredictivePKandPK/PDmodeling.Morerecently, we have seen and increasing focus on improving the screening approaches for toxicity. This presentation will discuss our strategy for the earlyapplicationofADME/PKandtoxicologyscreeningandtheapplicationof this data to the rapid transition from Discovery into Development.
2:30 Pharmacokinetic Drivers of Toxicity for Small MoleculesDolores Diaz, Ph.D., DABT, Investigative Toxicology, Genentech, Inc.Plasma drug exposures are routinely used in the interpretation of animal toxicity data, but target tissue exposures can be much higher than plasma exposures, which could have implications in the understanding of in vivo toxicities and in the determination of in vivo-in vitro correlations. Examples will be presented in which in vitro-in vivo correlations and differences in species sensitivity can be better understood by considering target organ exposures. Systematic analysis of tissue drug levels for a group of structurally diverse small molecules and consideration of their pharmacokinetic parameters indicates that tissue distribution can be roughly predicted from the volume of distribution (Vss) and the clearance (CLp) of a particular molecule. These findings suggest that consideration of these pharmacokinetic parameters could help provide relevant and translatable information.
3:00 Sponsored Presentation (Opportunity Available)
3:30 Networking refreshment Break in the Exhibit Hall
4:30 Strategies for Transporter Studies in Discovery and Development Praveen Balimane, Ph.D., Senior Research Investigator, Metabolism and Pharmacokinetics Group, Bristol-Myers Squibb Co. InadditiontoCYP’s,membranetransportershavebeendemonstratedtobe major determinants of pharmacokinetics, efficacy and safety profiles of drugs. Appropriate use of in vitro/in vivo transporter tools for assessing the risk of transporter-mediated clinical effects has the potential to significantly reduce the attrition rates and help progress the right compounds through development. The talk will highlight current industry strategies for transporter studies-whichtransporters,whichmodels,timingofthestudies,result/interpretations, translation of data etc. A few case studies underscoring the impact of these transporter strategies will also be discussed.
5:00 Predicting in vivo Drug Interactions from in vitro Data Jan Wahlstrom, Ph.D., Principal Scientist, Pharmacokinetics and Drug Metabolism, Amgen, Inc. Early assessment of the potential for metabolism-based drug-drug interactions (DDI) is critical for designing safer therapeutics and reducing clinical attrition rates. This presentation will focus on identifying the key featuresthatinfluenceexperimentaldesignforin vitro DDI experiments, comparing DDI assay design in the discovery and development
environments, interpreting DDI results according to regulatory guidance, and predicting the magnitude of in vivo DDIs based upon in vitro data.
5:30 End of Day
ThURSDAY, JUNE 9
7:20 am Continental Breakfast Breakout Discussions Breakout Discussion Topics:l UtilizationofGenotoxicDataforEffectiveRiskAssessmentsl EmergingTrendsinADME/DMPKTesting
TACkLINg gENOTOxICITy ISSUES
8:20 Chairperson’s remarksMartha Moore, Ph.D., Director, Division of Genetic and Molecular Toxicology, U.S. FDA, National Center for Toxicological Research
8:30 genetic Toxicity Concepts and Strategies for Small Molecule Lead Optimization Dolores Diaz, Ph.D., DABT, Investigative Toxicology, Genentech, Inc. Genotoxicityisacriticalcomponentofthesafetyassessmentstrategyfor small molecule drug development. As a field, genotoxicity is a complex discipline with unique challenges. This presentation will provide an overview of relevant genotoxicity concepts and mechanism and a discussion of genotoxicity assays (including limitations), with emphasis in practical applications in pharmaceutical drug development. Screening strategies for genotoxicity lead optimization and their rationale will be discussed, as well as investigative approaches around positive findings.
9:00 Early genotoxicity Assessment: From HTS to regulatory Testing Stephan Kirchner, Ph.D., Predictive Toxicology & Emerging Technologies, Lab Head, Genotoxicology, Phototoxicity, F. Hoffmann-La Roche Ltd. Indicator assays to predict genotoxic liabilities have high throughput and require very small amounts of compound. However, they are limited by lower predictivity for the outcome of regulatory OECD guideline assays compared to the miniaturized versions. In order to screen more products at the low quantities, while keeping excellent predictivity, an integrated strategy relying on in silico assessment, high throughput approaches and automation of regulatory-like assays will be discussed and contrasted within the context of supporting the early development of drug candidates.
9:30 going Beyond the Standard genetic Toxicology Battery Pamela L. Heard, Ph.D., Principal Scientist, Drug Safety R&D, Genetic Toxicology CoE, Pfizer Global Research & Development The standard in vitro genotoxicity test battery was designed to be a sensitive screen to assess the genotoxic hazard of a chemical or drug. However, the tests often show limited specificity. Positive results in these assays demonstrate the intrinsic genotoxic activity of a chemical or drug but do not provide insight into its genotoxic mechanism of action. Over the past several years our laboratory has focused on establishing a series of follow-up assays designed to demonstrate a possible mechanism of action for a drug candidate that has tested positive in the standard tests. This presentation will discuss the follow-up options.
10:00 Networking Coffee Break in the Exhibit Hall

WorldPharmaCongress.com World Pharma Congress | 11
Inaugural
Early ADME and DMPK Predictions for Better Lead OptimizationJuNE 8 - 9, 2011
»10:45 FEATURED pRESENTATIONWeight of the Evidence Integration of genetic Toxicology Data for risk Assessment
Martha Moore, Ph.D., Director, Division of Genetic and Molecular Toxicology, U.S. FDA, National Center for Toxicological Research
The currently used genetic toxicology battery includes both in vitro and in vivo assays which have different characteristics and different abilities to detect chemically induced genetic damage. Often there are data from several of these assays that can be used as a part of a risk assessment. It is important to understand the characteristics of the various assays and use that information in conducting a weight of the evidence assessment. An analysis of the available genetic toxicity data is now, more than ever, a central key component to consider in the derivation of a mechanism of action for characterizing observed adverse health outcomes such as cancer and for making decisions on the “safety” of various chemicals proposed for registration.
11:15 PANEL DISCUSSION: New Strategies for genotoxic risk AssessmentsModerator: Martha Moore, Ph.D., Director, Division of Genetic and Molecular Toxicology, U.S. FDA, National Center for Toxicological Research
11:45 Luncheon Presentations (Sponsorship Opportunities Available) or Lunch on your Own
EArLy PrE-CLINICAL PrEDICTIONS OF LIVEr INJUry
1:15 pm Chairperson’s remarksEric Blomme, D.V.M., Ph.D., D.A.C.V.P., Sr Project Leader, Abbott Labs
1:25 Current Toolbox for the Prediction of Hepatotoxicity Eric Blomme, D.V.M., Ph.D., D.A.C.V.P., Sr Project Leader, Abbott Labs This presentation will provide an overview of the current status of several technologies used to improve hepatotoxicity prediction during drug discovery. Examples will be used to illustrate the strengths, limitations and optimal application of these technologies during lead optimization and candidate selection.
1:55 In vitro Strategies: High Content Mechanistic Screening, Mitochondrial Toxicity, and Transporter Assessment Yvonne Will, Ph.D., Associate Research Fellow, Compound Safety Prediction, Pfizer Global R&D
2:25 Ice Cream refreshment Break in the Exhibit Hall 3:05 Approaching Hepatotoxicity in Drug Discovery as a Lead Optimization Problem Dylan P. Hartley, Ph.D., Senior Scientist, Investigative Toxicology, Genentech, Inc. Giventhedemandsondrugdiscoveryteamstoproducemoleculesdevoid of hepatotoxicity, toxicologists are now integral members of these teams with new responsibilities geared toward lead optimization. Toxicologists are now expected to assess risk for hepatotoxicity in a compound in predictive manner, or ascribe a mechanism to early hepatoxicity findings, link the findings to an offending moiety within the structure, and define the chemical lead optimization path. Structure-based strategiestopredictand/orattenuatehepatotoxicitywillbepresentedwith respect to various hepatotoxic signals.
3:35 The Utility of Emerging Biomarkers of Liver Injury in Pre-Clinical and Clinical Drug DevelopmentShelli Schomaker, Principal Scientist, Drug Safety R&D, Pfizer, Inc.While alanine aminotransferase (ALT) activity remains the gold standard biomarker of liver injury, the correlation between increased ALT levels and morphological liver findings is rather imperfect. These discrepancies could be due to adaptive responses, altered liver membrane permeability, extrahepatic injury, or treatment-related effects on ALT enzyme activity. This presentation will focus on an evaluation of malate dehydrogenase, purine nucleoside phosphorylase and glutamate dehydrogenase for the detection of liver injury when ALT activity is limited and will demonstrate the utility of this alternative biomarker approach for improving confidence.
4:05 From Mild Pre-Clinical Transaminase Elevations to Idiosyncratic Liver Injury in One Easy Lesson Paul Vancutsem, D.V.M., Ph.D., Director, Pre-Clinical Safety and Senior Member of Novartis Internal Liver Experts Team, Novartis Pharmaceuticals Even if we do not fully understand idiosyncratic liver injury (IDILI), it appears to necessitate events involving an often subtle local liver insult, an immune system imbalance and a specific genetic make-up (HLA alleles). Integration of these traditionally unconnected areas of toxicology will lead to an improved prediction of IDILI. This presentation proposes a template for integration using a pragmatic approach.
4:35 End of Conference
Conference Hotel:
Sheraton Philadelphia City Center17th and Race StreetsPhiladelphia, PA 19103www.Sheraton.comPhone:215-448-2000
Discounted Room Rate: $159 s/dDiscounted Cut-off Date: May 9, 2011
Please visit our website (worldpharmacongress.com) or call the hotel directly to reserve your sleeping accommodations. Identify yourself as a Cambridge Healthtech Institute conference attendee to receive the discounted room rate. Reservations made after the cut-off date or after the group room block has been filled (whichever comes first) will be accepted on a space- and rate-availability basis. Rooms are limited, so please book early.
HOTEL & TRAVEL INFORMATION
Flight Discounts:To receive a 5% or greater discount on all American Airlineflightspleaseuseoneofthefollowingmethods:
•Call1-800-433-1790useConferencecode1361AV
•Gotowww.aa.com enter Conference code 1361AV in promotion discount box
•ContactWendyLevine,GreatInternationalTravel1-800-336-5248ext.137
Car rental Discounts:Special discount rentals have been established with Hertz for this conference.•CallHertzdirectlyat800-654-3131andreference
Discount Number 04kL0002

REGISTER 3 - 4th IS FREE: Individuals must register for the same conference or conference combination and submit completed registration form together for discount to apply.
GROUP DISCOUNTS AVAILABLE! Special rates are available for multiple attendees from the same organization. For more information on group discounts contact David Cunningham at +1-781-972-5472, [email protected]
I cannot attend but would like to purchase the World Pharma Congress conference CD for $750 (plus shipping) Massachusetts delivery will include sales tax.
CONFERENCE DISCOUNTS Poster Discount ($50 Off) Poster SubmissionPoster abstracts are due by April 22, 2011. Once your registration has been fully processed, we will send an email containing a unique link allowing you to submit your poster abstract. If you do not receive your link within 5 business days, please contact [email protected]. CHI reserves the right to publish your poster title and abstract in various marketing materials
ADDITIONAL REGISTRATION DETAILS
Each registration includes all conference sessions, posters and exhibits, food functions, and access to the conference proceedings link.
Handicapped Equal Access: In accordance with the ADA, Cambridge Healthtech Institute is pleased to arrange special accommodations for attendees with special needs. All requests for such assistance must be submitted in writing to CHI at least 30 days prior to the start of the meeting.
To view our Substitutions/Cancellations Policy, go to http://www.healthtech.com/regdetails
Video and or audio recording of any kind is prohibited onsite at all CHI events.
How to Register WorldPharmaCongress.comP: 781.972.5400 or Toll-free in the U.S. 888.999.6288Please use keycode WPC F1 when registering
Cambridge Healthtech Institute 250 First Avenue Suite 300Needham, MA 02494
Register by March 4 and Save up to $350!
Pricing and Registration Information
Program Selection When registering please indicate the program(s) you will attend:
SHORT COURSE PRICINGChoose 1 Short CourseCommercial ________________________ $695Academic, Gov’t, Hospital Affiliated _______ $395Choose 2 Short CoursesCommercial ________________________ $995 Academic, Gov’t, Hospital Affiliated ______ $695
Monday, June 6th (Morning)Animal Models of Pain: Progress and Challenges Use of Stem Cells for Safety ScreeningAdvanced Topics in Drug Metabolism
Monday, June 6th (Afternoon)Translating Safety Biomarkers from the Lab to the ClinicAddressing Safety Concerns for Biological DrugsPractical Application of Plate-Based Label-Free Biosensors: Getting the Basics & Details You Need for Decision Making
BASIC PACKAGE Single Conference Pricing (Includes access to 1 conference, excludes short courses)
STANDARD PACKAGE Multiple Conference Pricing (Includes access to 2 conferences, excludes short courses)
CONFERENCE PRICING
DISCOVERy SUMMITJune 7 - 8PainAlzheimer’s DiseaseJune 8 - 9Parkinson’s DiseaseIn Vivo Molecular Imaging
SAFETy SUMMITJune 7 - 8CardiotoxicityNephrotoxicityJune 8 - 9HepatotoxicityADME/DMPK Predictions
SCREENING SUMMITJune 7 - 8Tools and Technologies for HTS
June 8 - 9Cell-Based Screening
Wednesday, June 8th (Evening)Pre-Clinical In Vivo Molecular Imaging: Back to BasicsMechanistic Insights into Hepatotoxicity
CommercialAcademic, Government,
Hospital-AffiliatedEarly Registration Discount until March 4 $2290 $945Advance Registration Discount until April 22 $2450 $1025Registrations After April 22, and on-site $2595 $1095
CommercialAcademic, Government,
Hospital-AffiliatedEarly Registration Discount until March 4 $1395 $695Advance Registration Discount until April 22 $1545 $775Registrations After April 22, and on-site $1745 $875
Please refer to the registration Code below:
Recieve free a FREE eNewsletter by signing up at chimediagroup.com
The latest industry news, commentary and highlights from BiolIT World
Innovative management in clinical trials
A series of diverse reports designed to keep life science professionals informed of the salient trends in pharmaceutical technology, business, clinical development, and therapeutic disease markets.For a detailed list of reports, visit InsightPharmaReports.com, or contact Rose LaRaia, [email protected], +1-781-972-5444.
Barnett is a recognized leader in clinical education, training, and reference guides for life science professionals involved in the drug development process. For more information, visit barnettinternational.com.
Drug Safety SummitJune 7 - 9, 2011 Sheraton Philadelphia City Center Philadelphia, PA
Part of WPCWorld Pharma Congress
Promising Assays and Technologies forBetter Pre-Clinical Predictions
































