Embed Size (px)
Citation preview

The FASEB Journal • Research Communication
Genetically determined angiotensin converting enzymelevel and myocardial tolerance to ischemia
Erij Messadi,* Marie-Pascale Vincent,* Violaine Griol-Charhbili,* Chantal Mandet,*,†
Juliana Colucci,* John H. Krege,‡ Patrick Bruneval,*,†,§ Nadine Bouby,*Oliver Smithies,� Francois Alhenc-Gelas,*,†,§ and Christine Richer*,§,¶,1
*INSERM U872, Centre de Recherche des Cordeliers, Paris, France; †Universite Paris Descartes,Paris, France; ‡Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, Indiana, USA;§Assistance Publique, Hopitaux de Paris, Paris, France; �Department of Pathology, University of NorthCarolina, Chapel Hill, North Carolina, USA; and ¶Universite Paris-Sud, Le Kremlin-Bicetre, France
ABSTRACT Angiotensin I-converting enzyme (ACE; kini-nase II) levels in humans are genetically determined.ACE levels have been linked to risk of myocardialinfarction, but the association has been inconsistent,and the causality underlying it remains undocu-mented. We tested the hypothesis that genetic varia-tion in ACE levels influences myocardial tolerance toischemia. We studied ischemia-reperfusion injury inmice bearing 1 (ACE1c), 2 (ACE2c, wild type), or 3(ACE3c) functional copies of the ACE gene anddisplaying an ACE level range similar to humans.Infarct size in ACE1c was 29% lower than in ACE2c(P<0.05). Pretreatment with a kinin B2 receptorantagonist suppressed this reduction. In ACE3c, in-farct size was the same as in ACE2c. But ischemicpreconditioning, which reduced infarct size in ACE2c(�63%, P<0.001) and ACE1c (�52%, P<0.05), wasnot efficient in ACE3c (�2%, NS, P<0.01 vs. ACE2c).In ACE3c, ischemic preconditioning did not decreasemyocardial inflammation or cardiomyocyte apopto-sis. Pretreatment with a renin inhibitor had no car-dioprotective effect in ACE2c, but in ACE3c partiallyrestored (38%) the cardioprotection of ischemicpreconditioning. Thus, a modest genetic increase inACE impairs myocardial tolerance to ischemia. ACElevel plays a critical role in cardiac ischemia, throughboth kinin and angiotensin mediated mechanisms.—Messadi, E., Vincent, M.-P., Griol-Charhbili, V., Man-det, C., Colucci, J., Krege, J. H., Bruneval, P., Bouby,N., Smithies, O., Alhenc-Gelas, F., Richer, C. Genet-ically determined angiotensin converting enzymelevel and myocardial tolerance to ischemia. FASEB J.24, 4691– 4700 (2010). www.fasebj.org
Key Words: renin inhibition � kinins � genetically modifiedmice
Angiotensin converting enzyme (ACE; kininase II)plays an important role in cardiovascular homeostasisby catalyzing the conversion of inactive angiotensin Iinto active angiotensin II and the inactivation of brady-kinin (1). ACE is an ectoenzyme of vascular endothelial
cells, also released into the circulation on cleavage of itsmembrane anchor. ACE levels vary largely among sub-jects. These levels are under strong genetic influence.Studies in nuclear families have documented genetictransmission of plasma ACE levels (2). Polymorphismof the ACE gene, especially an insertion/deletion (I/D)polymorphism located in intron 16, has been associatedwith plasma levels of ACE (3). ACE concentrations intissues are also genetically determined, and associatedwith the ACE gene I/D polymorphism, including in theheart (4, 5).
Variation in plasma and tissue ACE concentrationwas generally considered to have no or little conse-quence for cardiovascular homeostasis, contrary tovariation in renin secretion, because of high availabilityof endothelial ACE for angiotensin I conversion. How-ever, this concept has been challenged following obser-vation of heterogeneity in vascular ACE content amongperipheral organs. In the kidney and the heart, endo-thelial ACE content is low, and moderate variation inACE levels might influence the rate of angiotensin IIformation (6, 7). In addition, ACE is the main enzymeinactivating kinins in the circulation. Theoretical andexperimental evidence suggests that the ACE levelinfluences kinin concentration (8).
In pathological situations, angiotensin II formationand/or kinin depletion may play a deleterious role,resulting in heart, artery, or kidney damage duringhemodynamic, ischemic, or metabolic injury (6, 9).The effect of genetic variation in ACE levels on prog-nosis of cardiac and renal diseases has been studied inpopulations and cohorts of patients. Thus, genetic ACElevels are causally linked to severity of renal involve-ment in type 1 diabetes. Association between ACE levelsand diabetic nephropathy has been replicated in majorclinical studies, and causality between genetically highACE levels and diabetic renal damage has been docu-mented in genetically modified mice (10, 11). The ACE
1 Correspondence: INSERM U872, 15, rue de l’Ecole deMedecine, 75270 Paris, France. E-mail: [email protected]
doi: 10.1096/fj.10-165902
46910892-6638/10/0024-4691 © FASEB

gene polymorphism and ACE levels have also beenassociated with risk of myocardial infarction (12, 13).Individuals harboring the deletion allele of this geneand having high ACE levels have been found to have anincreased prevalence of myocardial infarction (11–15).However, association of the ACE gene with myocardialinfarction has not been consistent across all studies (16,17). In addition, the mechanism underlying the associ-ation remains unclear and causality has not beenaddressed.
As an attempt to clarify the effect of genetic variabil-ity in ACE levels in ischemic heart disease, we usedgenetically engineered mice carrying either an inacti-vation or a duplication of the ACE gene that wesubmitted to myocardial ischemia reperfusion (IR)injury and ischemic preconditioning (IPC). These micedisplay an ACE level range similar to humans (10, 18).IR injury has relevance to the human disease, andseveral studies have documented the role of at least oneACE substrate, kinins, in myocardial tolerance to isch-emia (19–22). We especially studied the effect of amodest genetic increase in ACE levels above physiolog-ical value on outcome of experimental cardiac isch-emia, and assessed the mechanism underlying thiseffect.
MATERIALS AND METHODS
Generation and characterization of mice with 1–3 copies ofthe ACE gene
Genetically engineered mice carrying either an inactivationor a duplication of the ACE gene on chromosome 11 wereused for the study (18). Adult male mice carrying either 1(ACE1c), 2 (ACE2c, identical to wild type), or 3 (ACE3c)functional ACE gene copies were studied. All the experimen-tal procedures were performed in accordance with the Euro-pean regulations for the care and use of laboratory animals (L358-86/609/EEC) and were approved by the Universite ParisAnimal Care and Use Committee.
Blood pressure response to angiotensin I and angiotensin II
Blood pressure changes triggered by angiotensin II (300ng/kg) or increasing doses of angiotensin I (0.3–30 �g/kg)(1 �l/g body weight bolus injected at 5-min intervals via thejugular vein) were measured in pentobarbital-anesthetizedACE1c (n�5–9), ACE2c (n�7–15), or ACE3c (n�7–10). Atsacrifice, the animals were used for quantification of plasmaACE activity and ACE mRNA in the lungs and hearts.
Quantification of plasma ACE activity and ACE mRNAabundance in the lung and heart
Plasma ACE activity was measured spectrophotometricallywith hippuryl-histidine-leucine as substrate (23). Relativechanges in gene expression of ACE in heart and lung werequantified using real-time PCR (24).
Total RNA was isolated from the heart and lung (Trizol;Invitrogen/Life Technologies, Cergy Pontoise, France). cDNAwas synthesized from 2 �g total RNA using 1 �g randomhexamers and 40 U Superscript II MMLV-reverse transcriptase(Invitrogen/Life Technologies) in the presence of 200 U RNase-
OUT in a 20 �l final volume. Real-time PCR was performed onthe ABI PRISM 7000 Sequence Detection System by using SYBRgreen PCR Master Mix and assay-on-demand gene expressionprobes (Applied Biosystems; Applera France, Courtaboeuf,France). Reverse transcribed RNA (15 ng) was submitted to PCRin a 20-�l final volume. Each sample was tested in triplicate.DNA contamination was excluded by performing PCR amplifi-cation without the RT step for each RNA sample, and blankwithout sample but with all reagents. Data were normalized toGAPDH mRNA (24). Changes in the target gene relative to themean expression in the wild-type control group were calculatedby the 2���Ct comparative method for each sample (25).
In vivo mouse model of myocardial IR injury and IPC:effect of ACE gene titration
Surgical preparation
Mice were anesthetized with sodium pentobarbital (60mg/kg, i.p.). The animals were intubated and ventilatedwith 100% oxygen (200 �l/breath at a rate of 170 breaths/min), using a Harvard rodent ventilator (Model 845,Harvard Apparatus, Les Ulis, France). Drugs were admin-istered via a catheter inserted into the jugular vein. Bodytemperature was monitored with a rectal probe connectedto a digital thermometer, and maintained at 37°C using aheating pad. The electrocardiogram (ECG) was recordedthroughout the experiments on a Gould TA240 recorder(ECG biotech; Gould Instruments, Cleveland, OH, USA). Aleft thoracotomy was performed to expose the heart, andthe pericardium was removed. The left anterior descend-ing coronary artery was occluded with an 8.0 prolenesuture, 2 mm from the tip of the left atrium for 30 min.Successful coronary occlusion was verified by the develop-ment of a pale color in the distal myocardium and by STsegment elevation and QRS widening on the ECG. After 30min of sustained ischemia, coronary blood flow was re-stored by loosening the suture. Successful reperfusion wasconfirmed by visualization of hyperaemic response and resto-ration of normal ECG. The lungs were then reinflated byincreasing positive end expiratory pressure, and the chest wasclosed. Reperfusion was maintained for a 3-h period (19). IPCwas produced by a sequence of 3 successive cycles, eachconsisting of 3 min of coronary occlusion followed by 5 min ofreperfusion, according to Yang et al. (26). Immediately afterIPC, all the animals were subjected to IR injury (30 min ofcoronary occlusion followed by reperfusion). ACE1c, ACE2c(wild type), or ACE3c mice were subjected to the samemyocardial IR, alone (ACE1c, n�8; ACE2c, n�12; ACE3C,n�8) or after IPC (ACE1c, n�8; ACE2c, n�14; ACE3C n�9).
Measurement of infarct size (IS)
After reperfusion, the chest was reopened, the coronaryartery was reoccluded, and 0.5 ml of a 5% Evans blue solutionwas injected as a bolus into the jugular vein in order todelineate the area at risk (AR), which remained unstained bythe Evans blue. The heart was excised, and the left ventricle(LV) was isolated, weighed, and sliced into 4 transverse piecesfrom base to apex, the first cutter blade being positioned atthe site of the coronary occlusion. The slices were weighed,and color digital images of both sides of each slice wereobtained with a Power Shot S50 zoom digital camera(Canon, Tokyo, Japan) connected to a microscope (LeicaMZ 75; Leica Microsystems, Rueil-Malmaison, France),using the Adobe Photoshop software (Adobe Systems, SanJose, CA, USA). The slices were then incubated at 37°Cwith buffered 1% 2,3,5-triphenyltetrazolium chloride
4692 Vol. 24 December 2010 MESSADI ET AL.The FASEB Journal � www.fasebj.org

(TTC) solution for 20 min. Viable myocardium, whichcontained dehydrogenases, reacted with TTC and wasstained brick red, whereas any necrotic tissue remainedunstained due to the lack of active enzymes. The tissuesections were then fixed in a buffered 10% formalinsolution for 24 h before being photographed again todelineate the IS (19). The cross-sectional area, the lumenarea, the AR (unstained by Evans blue), and IS (unstainedby TTC) of the LV were outlined on each color image andquantified by a masked observer using the Scion Imagesoftware (Scion Image for Windows; http://www.scioncorp.com). The absolute weights of AR and IS were then calculatedfor each slice. The sum of the absolute weight values of AR andIS of the 3 ischemic slices of each heart was calculated andexpressed as a percentage of the total weight of the slice. Theratio of IS to AR was calculated from these absolute weightevaluations and expressed as a percentage of AR.
Plasma troponin determination
As an additional readout for myocardial infarct severity, wemeasured cardiac TroponinI (cTnI) levels in the plasma ofmice with or without IPC after a 24-h reperfusion period sothat IR lesions were full blown at this time (n�6–13/group).Blood samples were obtained from carotid artery at the endof reperfusion. Plasma cTnI levels were determined with amouse cardiac quantitative cTnI assay (Life Diagnostics, WestChester, PA, USA) (27). Then, the hearts were excised andfixed for AR and IS determination, and assessment of myo-cardial apoptosis and inflammation.
Histological analysis of the heart: assessment of myocardialapoptosis and inflammation
The hearts were divided into several slices, apex and upper-medium slices, for paraffin embedding. Paraffin sections werestained with hematoxylin-eosin (H&E) for histological deter-mination of necrosis. Myocardial apoptosis was assessed bythe terminal deoxynucleotidyltransferase (TdT)-mediateddUTP nick-end labeling (TUNEL) technique (ApoTag kit;Chemicon-Millipore, Molsheim, France) as described previ-ously (28).The percentage of positive apoptotic nuclei incardiomyocytes was counted along the whole border zonesurrounding the myocardial infarction with a light micro-scope at �40 in the 2 sections from the heart of each mouse.The mean percentage of apoptotic cells per group wascalculated. For inflammation, leukocyte infiltration was de-tected by immunohistochemistry using an anti-mouse CD45antibody (BD Biosciences-Pharmingen, San Diego, CA, USA)reacting against most of leukocytes, polymorphonuclearsbeing prominent at 24 h reperfusion time. The CD45 labelingwas evaluated semiquantitatively in the infarcted area of theLV using a score from 0 (no leukocyte infiltration) to 5(highest leukocyte infiltration in density and extension) witha light microscope at �10 in the 2 sections from the heart ofeach mouse. The mean inflammation score per group wascalculated.
Role of kinins in the cardioprotective effect of low ACElevel: effect of B2 receptor blockade by icatibant
To investigate the role of kinins through the B2 receptoractivation in the cardioprotective effect observed in ACE1cmice after IR, additional ACE1c (n�12) and ACE2c (n�17)mice were pretreated with the B2 receptor antagonist icati-bant (HOE140; Aventis Pharma Deutschland GmbH, Frank-furt, Germany; 500 �g/kg i.v.) (ACE1c, n�4; ACE2c, n�5) or
with saline (ACE1c, n�8; ACE2c, n�12) 5 min before theonset of ischemia.
Role of angiotensin II in myocardial IR injury and in IPC:effect of renin inhibition by aliskiren
To investigate the role of angiotensin II in the loss ofcardioprotection observed in ACE3c mice, additional exper-imental groups of ACE2c and ACE3c mice were subjected toeither IR alone (ACE2c, n�19; ACE3c n�14), or IR after IPC(ACE2c, n�18; ACE3c, n�12). The mice were pretreatedwith aliskiren (Novartis Pharma AG, Basel, Switzerland), adirect renin inhibitor (1 mg/kg, i.v. bolus 5 min beforestarting ischemia followed by an infusion 10 �g/kg/min)(ACE2c, n�23; ACE3c, n�13). Other mice received saline(ACE2c, n�14; ACE3c, n�13).
Preliminary experiments were designed to determine theproper dose of aliskiren. In a first set of experiments, theblood pressure-lowering effect of increasing doses of aliskirenfrom 0.5 to 10 mg/kg was assessed in pentobarbital anesthe-tized wild-type mice (n�8/group). Blood pressure was re-corded in these mice up to 45 min after aliskiren or salineadministration, and the maximal blood pressure variationswere determined. Blood samples were collected into heparin-ized tubes, and plasma renin activity (PRA) was determinedby radioimmunoassay of angiotensin I generated after incu-bating the plasma for 1 h at pH 7.4 (29). In a second set ofexperiments, systemic and coronary and renal vascular effectsof aliskiren administered at the previously determined dose(1 mg/kg, i.v. bolus 5 min before starting ischemia followedby an infusion 10 �g/kg/min), were quantified in anesthe-tized wild-type mice (n�6–7/group) using the fluorescentmicrosphere technique (30).
Statistical analysis
Results are expressed as means � se. Comparisons betweengenotypes and between treatments in each genotype wereperformed by 2-way ANOVA followed by post hoc analysis usingthe JMP software system (JMP, SAS Institute Inc., Cary, NC,USA). The effects of icatibant or aliskiren were assessed byunpaired Student’s t test. Angiotensin I-pressor response[variation in mean blood pressure (MBP) expressed as abso-lute value] curves in genotype groups were compared byANOVA for repeated measurements with the Greenhouse-Geisser adjustment (31). AUCs for pressor responses vs. logdose angiotensin I were compared by ANOVA. Values of P �0.05 were considered to be statistically significant.
RESULTS
Characterization of mice with 1–3 copies of the ACEgene
Plasma ACE activity was directly related to the ACE genecopy number (genotypic effect P�0.001, Fig. 1A) andACE mRNA in the lung and in the LV were alsodependent on ACE gene copy number (Fig. 1B, C;genotypic effect P�0.05). MBP and heart rate (HR)were not significantly affected by ACE gene copy num-ber. ACE1c: MBP � 59.0 � 7.8 mm Hg, HR � 365 � 9bpm; ACE2c: MBP � 59.2 � 2.9 mm Hg, HR � 374 �14 bpm; and ACE3c: MBP � 65.0 � 3.3 mm Hg, HR �381 � 13 bpm. The increases in blood pressure trig-gered by increasing doses of angiotensin I were signif-
4693ACE GENE TITRATION AND MYOCARDIAL ISCHEMIA

icantly enhanced in ACE3c mice (P�0.05) and unaf-fected in ACE1c mice compared to ACE2c mice.Maximal pressor responses to angiotensin II did notsignificantly differ among the three genotype groups(Fig. 2).
Effect of ACE gene titration on IR injury and IPC
There was no difference in body weight, LV weight, ARto LV ratio, or HR before occlusion of the coronaryartery or after reperfusion between ACE1c, ACE2c, orACE3c mice, regardless of whether they had beensubjected to IPC (Table 1).
Effect on IS
In basal conditions (IR without IPC), IS/AR in ACE1c(27 � 3%) was significantly lower than IS/AR in ACE2c(38 � 3%, �29%, P�0.05) but IS/AR in ACE3c (41 �4%) did not differ from that in ACE2c (Fig. 3).
After IPC (IPC�IR), a marked cardioprotective ef-fect of IPC was observed in the IPC-ACE2c and IPC-ACE1c groups. IS/AR fell to 14 � 1% (�63% vs. ACE2c
without IPC, P�0.001) and 13 � 1% (�52% vs. ACE1cwithout IPC, P�0.05, NS vs. IPC-ACE2c), respectively.In ACE3c mice, IPC no longer exerted a significantcardioprotective effect (40�3% after IPC vs. 41�4%without IPC, �2%, NS; Fig. 3A). The loss of thecardioprotection afforded by IPC in IPC-ACE3c micewas also documented by histology (Fig. 3B).
Effect on plasma troponin I levels
The loss of the cardioprotection afforded by IPC inIPC-ACE3c mice was further documented by measuringcTnI plasma levels. The cTnI plasma levels were signif-icantly reduced by IPC in IPC-ACE2c mice (P�0.01),but not in IPC-ACE3c mice (Fig. 4).
Apoptosis and inflammation
In ACE2c mice, IPC induced a reduction of 49%(P�0.05) in cardiomyocyte apoptosis compared to IRwithout IPC. No significant effect of IPC was observedin IPC-ACE3c (Fig. 5A, B).The number of CD45-posi-tive cells in the infarcted area of the LV was reduced byIPC in IPC-ACE2c mice (�72% compared to IR-ACE2c,P�0.05), whereas no difference was found in IPC-ACE3c mice as compared to IR-ACE3c mice withoutIPC (Fig. 5C, D).
Mechanism of cardioprotection in ACE1c mice; roleof kinins: effect of B2 receptor blockade by icatibant
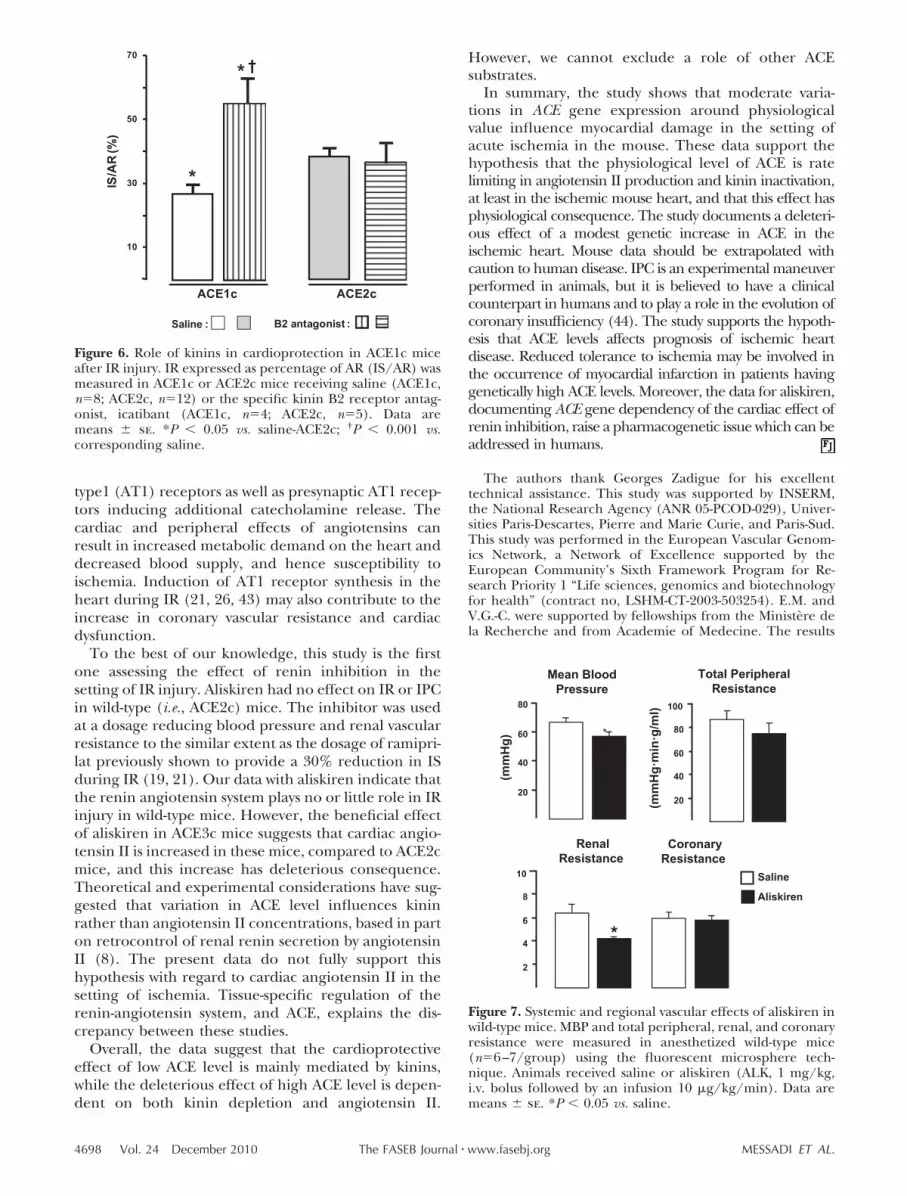
Pretreatment with icatibant, a selective kinin B2 recep-tor antagonist, did not affect IS/AR in ACE2c mice, butcompletely suppressed the reduction in IS/AR reduc-tion in pretreated ACE1c mice (Fig. 6).
Mechanism of loss of cardioprotection by IPC inACE3c mice; role of angiotensins: effect of renininhibition by alikiren
Hemodynamic effects of aliskiren
Preliminary experiments indicated that in wild-type anes-thetized mice, aliskiren, a direct renin inhibitor, at the
Figure 1. Plasma ACE activity (A, effect of genotype P�0.001) and ACE mRNA in lung (B, effect of genotype P�0.05) and LV(C, effect of genotype P�0.05) in ACE1c (n�9), ACE2c (n�10), or ACE3c (n�10) mice. Results are normalized to GAPDHmRNA level. Data are means � se. *P � 0.05 vs. ACE2c.
Figure 2. ACE gene titration and pressor responses to angio-tensins. A) Maximal pressor responses (�MBP, absolute variation ofMBP) to angiotensin I (Ang I) or angiotensin II (Ang II) in ACE1c,ACE2c, or ACE3c mice under basal conditions. Area under curve(AUC) of pressor responses (absolute increases in MBP) vs. logdose Ang I; data are means � se. *P � 0.05 vs. ACE2c.
4694 Vol. 24 December 2010 MESSADI ET AL.The FASEB Journal � www.fasebj.org

dose used, induced a mild hypotensive response that wasmaximal between 10 and 20 min after administration andaveraged 15% 5 min after administration. Blood pressureand systemic, renal, and coronary vascular resistance werereduced by 15% (P�0.05), 18% (NS), 35% (P�0.001),and 11% (NS), respectively (Fig. 7). PRA was decreased by60% (from 1304�104 to 516�46 pg/ml/h, P�0.001).
Effect of aliskiren on myocardial IR injury
After IR, pretreatment with aliskiren did not signifi-cantly affect IS/AR either in ACE2c or in ACE3c mice.However, after IPC, the loss of cardioprotection ob-served in IPC-ACE3c mice was partially restored by 38%
in aliskiren-pretreated ACE3c mice (P�0.01, Fig. 8).Interestingly, no effect of aliskiren was observed inIPC-ACE2c mice (Fig. 8).
DISCUSSION
We report here findings relating genetically deter-mined ACE level to myocardial tolerance to ischemia.To do this, we used an original in vivo experimentalmodel of IR injury in mice having 1, 2, or 3 functionalcopies of the ACE gene. Through backcrossing of theACE gene mutations to an inbred mouse strain(C57BL/6), these mice differed only at the ACE locus,
IS/A
R (%
)
10
30
50
*
†
†
ACE3cACE2cACE1c
IR : IPC + IR:
A B
ACE2c +IPC+IR
ACE3c +IPC+IR
ACE2c + IR
ACE3c + IR
**
Figure 3. Effect of ACE gene titration on IR after IR injury without or with IPC. A) IR determined by TTC staining expressedas percentage of AR (IS/AR) was measured in ACE1c, ACE2c, or ACE3c mice. Data are means � se. For n, see Table 1. *P �0.05, **P � 0.001 vs. corresponding IR; †P � 0.05 vs. corresponding ACE2c. B) Representative H&E staining of cross sectionsof the hearts from ACE2c or ACE3c mice after ischemia and 24-h reperfusion period. Left panel: original view, �2.5; black linesurrounds infarcted area. Right panel: high-magnification view, �40; black line shows border between living area (above line)and infarcted area (below line). ACE2c mice after IPC�IR had a smaller infarct compared to ACE2c mice with IR without IPC.ACE3c mice had a similar infarct after IPC�IR compared to ACE3c mice after IR without IPC.
TABLE 1. Basal characteristics of groups of mice with 1–3 copies of the ACE gene subsequently subjected to IR without or with IPC
Genotype Group n BW (g) LVW/BW AR (%LV)
HR (bpm)
Before occlusion At reperfusion
ACE1c IR 8 31.3 � 1.1 3.4 � 0.2 26.8 � 2.0 420 � 18 446 � 22IPC�IR 8 32.9 � 1.0 3.6 � 0.1 26.2 � 2.2 398 � 16 413 � 18
ACE2c IR 12 30.9 � 0.7 3.6 � 0.1 28.8 � 1.6 340 � 13 440 � 15IPC�IR 14 32.4 � 0.6 3.8 � 0.1 29.4 � 1.2 411 � 14 425 � 17
ACE3c IR 8 29.5 � 0.5 3.5 � 0.1 27.1 � 2.2 383 � 19 405 � 15IPC�IR 9 29.7 � 0.6 3.8 � 0.1 27.9 � 1.8 386 � 18 437 � 17
Data are means � se. BW, body weight; LVW/BW, left ventricle weight to body weight ratio; AR, area at risk; HR, heart rate; bpm, beatsper minute.
4695ACE GENE TITRATION AND MYOCARDIAL ISCHEMIA

providing an opportunity to study changes in the ACEgene as a single experimental variable. Tissue patternof ACE gene expression, and ACE gene regulation aremaintained in the genetically modified mice allowingextrapolation to physiological situations. The mice dis-played moderate differences in ACE mRNA level andan ACE activity range in plasma and tissue of the sameorder of magnitude as that found in humans (Fig. 1)(10, 18, 32, 33). This is the first study assessing the roleof ACE level and its physiological variation in cardiacischemia. This role, and the effect of a genetic increasein ACE activity, could not be inferred from data ob-tained with ACE inhibitors. We found that a geneticdecrease in ACE activity reduced IS after IR, and that agenetic increase in ACE activity level did not worsen IR,but suppressed the cardioprotection afforded by IPC.
There were no preexisting cardiac hemodynamic oranatomical differences among the mice, which couldhave influenced the results of IR and IPC. Specifically,in agreement with previous observations (10, 18),blood pressure values measured under basal conditionswere not affected by ACE gene copy number. We alsofound that HR, LV mass and morphology, and AR afterIR or IPC�IR were not affected by ACE gene copynumber. A similar observation has been made forcardiomyocyte size and morphology (33).
A genetically induced 35% decrease in ACE activityin ACE1c mice was sufficient to reduce IS by 29%,supporting the hypothesis that ACE level is an impor-tant determinant of myocardial tolerance to ischemia.In the ACE1c mice, reduced angiotensin II formationand increased kinin availability could both participatein this effect. Kinins are induced in the ischemic heart(22) along with the synthesis of kinin B1 and B2receptor (19, 34, 35). The kinin system may protectagainst IR injury by eliciting coronary vasodilatation,inhibiting platelet aggregation, promoting fibrinolysis,
and decreasing the production of free radicals, whileother actions such as leukocyte recruitment and activa-tion may not result in cardioprotection (19, 36, 37).The reduction of IS in ACE1c mice was completelysuppressed by pretreatment with icatibant, a specifickinin B2 receptor antagonist. Potentiation of endoge-nous kinins through B2 receptor activation may thus bethe most important pathway by which genetically deter-mined decrease in ACE activity in the ACE1c micemediates cardiac protection against ischemia. Interest-ingly, in icatibant-treated ACE1c mice submitted to IR,IS was higher than in ACE2c mice. This effect may bedue to enhanced effect of vasoconstrictor agents inabsence of kinin action (38). Our current findings inthe ACE1c mice are consistent with observations madein wild-type mice treated with ACE inhibitors andsubmitted to IR (19, 39). However, data obtained aftercomplete pharmacological ACE inhibition cannot bepredictive of consequences of low amplitude physiolog-ical variation in ACE activity. The present study showsthat a modest decrease in ACE activity is sufficient toinfluence the outcome of cardiac IR injury.
ACE3c mice that have genetically increased ACElevels did not show increased IS after IR compared towild-type mice but did show that the beneficial conse-quence of IPC was lost. In ACE2c mice, it is possiblethat IS may already be maximal. Additionally, in theabsence of IPC, kinin concentration and/or B2 recep-tor density may be insufficient to significantly affect IR.This is supported by previous studies documenting lackof effect of B2 receptor blockade in wild-type mice, anobservation confirmed in the present study, or lack ofeffect of genetic deficiency in tissue kallikrein andkinins, in IR without IPC (19, 21). The reduction in ISafforded by IPC in ACE2c mice was not observed inACE3c mice. In ACE3c mice, IPC did not reduceplasma troponin levels, myocardial apoptosis, or in-
ACE2c
Plas
ma
cTnI
(ng/
ml)
5
15
25A B
��
*
ACE3c
C
Plas
ma
cTnI
(ng/
ml)
5
15
25
Plas
ma
cTnI
(ng/
ml)
IS/AR (%)ACE2c
IR:
IPC+IR:
r2=0.729
IR:IPC+IR:
Figure 4. Measurement of cTnI plasma levels in ACE2c or ACE3c mice after ischemia and 24-h reperfusion period without IPC(IR) or with IPC (IPC�IR). A) IPC-ACE2c mice had significantly lower cTnI plasma levels compared to ACE2c mice without IPC.B) Correlation between IR (IS/AR) and cTnI plasma levels in ACE2c mice with or without IPC (r2� 0.729, P�0.01). C) IPC nolonger affected cTnI plasma levels in IPC-ACE3c mice as compared to ACE3c mice without IPC. Data are means � se. *P � 0.01vs. corresponding IR.
4696 Vol. 24 December 2010 MESSADI ET AL.The FASEB Journal � www.fasebj.org

flammation. This effect of a genetically increased ACElevel on IPC could result from an increase in theconversion of angiotensin I to angiotensin II or from adecrease in the concentration of kinins. Some evidencesuggests that kinin depletion may at least partiallyexplain these observations. Kinin production and B2receptor synthesis are strongly stimulated by IPC (19,22). Blockade of the B2 receptor reduces the cardio-protective effect of IPC (20). In addition, tissue kal-likrein-deficient mice do not produce kinins, and whiletheir response to IR is similar to that of wild-type mice,IPC has a reduced effect to suppress IS in these mice(19, 21). Thus, the effect on IPC of increased ACE geneexpression in ACE3c mice is similar to the effect ofkallikrein and kinin deficiency in tissue kallikrein geneinactivated mice (19). However, pretreatment of IPC-
ACE3c mice with aliskiren, a direct renin inhibitor,partially restored the beneficial effect of IPC, suggest-ing that angiotensin II is also involved in the loss of IPCin ACE3c mice. A stimulatory effect of an increasedACE level on angiotensin II production is furtherdocumented in the present study by monitoring bloodpressure during angiotensin I injection in ACE3c mice,and has also been observed in other experimentalsettings (40, 41). Thus, our present data suggest thatkinin depletion and elevated angiotensin II formationare both involved in decreasing the protective effect ofIPC in ACE3c mice. Coronary artery occlusion bothincreases sympathetic tone and activates the reninangiotensin system, resulting in increased angiotensinII formation (42). Angiotensin II causes coronary vaso-constriction by activation of postsynaptic angiotensin
10
8
6
4
2
0
5
4
3
2
1
0
**
†
ACE2c +IR ACE2c +IPC+IR ACE2c +IR ACE2c +IPC+IR
ACE3c +IR ACE3c +IPC+IR ACE3c +IR ACE3c +IPC+IR
ACE2c ACE3c ACE2c ACE3c
apop
totic
cel
ls (%
)
apop
totic
cel
ls (%
)
TUNEL CD45C
DB
A
IR: IPC + IR:
Figure 5. Assessment of myocardial apoptosis and inflammation in ACE2c or ACE3c mice. Mice were studied after ischemia anda 24-h reperfusion period without IPC (IR) or with IPC (IPC�IR). A) Quantification of TUNEL-positive cardiomyocytes (%) inthe border zone surrounding the myocardial infarction. Data are means � se. *P � 0.05 vs. corresponding IR. B) Representativesections of hearts from ACE2c or ACE3c IR and IPC�IR mice showing the variable density of apoptic nuclei (arrows) inmyocytes in the border zone. Black line delineates the infarcted area. TUNEL, original view �40. C) Semiquantification ofCD45-positive cells (score) in the infarcted area. Data are means � se. *P � 0.05 vs. corresponding IR; †P � 0.05 vs. IPC-ACE2c.D) Representative sections of hearts from ACE2c or ACE3c IR and IPC�IR mice showing variable density of the leukocytes:inflammation is mainly limited to infarcted area. Immunohistochemistry with anti-CD45 antibody labeling all the types ofleukocytes. Original view �10.
4697ACE GENE TITRATION AND MYOCARDIAL ISCHEMIA
type1 (AT1) receptors as well as presynaptic AT1 recep-tors inducing additional catecholamine release. Thecardiac and peripheral effects of angiotensins canresult in increased metabolic demand on the heart anddecreased blood supply, and hence susceptibility toischemia. Induction of AT1 receptor synthesis in theheart during IR (21, 26, 43) may also contribute to theincrease in coronary vascular resistance and cardiacdysfunction.
To the best of our knowledge, this study is the firstone assessing the effect of renin inhibition in thesetting of IR injury. Aliskiren had no effect on IR or IPCin wild-type (i.e., ACE2c) mice. The inhibitor was usedat a dosage reducing blood pressure and renal vascularresistance to the similar extent as the dosage of ramipri-lat previously shown to provide a 30% reduction in ISduring IR (19, 21). Our data with aliskiren indicate thatthe renin angiotensin system plays no or little role in IRinjury in wild-type mice. However, the beneficial effectof aliskiren in ACE3c mice suggests that cardiac angio-tensin II is increased in these mice, compared to ACE2cmice, and this increase has deleterious consequence.Theoretical and experimental considerations have sug-gested that variation in ACE level influences kininrather than angiotensin II concentrations, based in parton retrocontrol of renal renin secretion by angiotensinII (8). The present data do not fully support thishypothesis with regard to cardiac angiotensin II in thesetting of ischemia. Tissue-specific regulation of therenin-angiotensin system, and ACE, explains the dis-crepancy between these studies.
Overall, the data suggest that the cardioprotectiveeffect of low ACE level is mainly mediated by kinins,while the deleterious effect of high ACE level is depen-dent on both kinin depletion and angiotensin II.
However, we cannot exclude a role of other ACEsubstrates.
In summary, the study shows that moderate varia-tions in ACE gene expression around physiologicalvalue influence myocardial damage in the setting ofacute ischemia in the mouse. These data support thehypothesis that the physiological level of ACE is ratelimiting in angiotensin II production and kinin inactivation,at least in the ischemic mouse heart, and that this effect hasphysiological consequence. The study documents a deleteri-ous effect of a modest genetic increase in ACE in theischemic heart. Mouse data should be extrapolated withcaution to human disease. IPC is an experimental maneuverperformed in animals, but it is believed to have a clinicalcounterpart in humans and to play a role in the evolution ofcoronary insufficiency (44). The study supports the hypoth-esis that ACE levels affects prognosis of ischemic heartdisease. Reduced tolerance to ischemia may be involved inthe occurrence of myocardial infarction in patients havinggenetically high ACE levels. Moreover, the data for aliskiren,documenting ACE gene dependency of the cardiac effect ofrenin inhibition, raise a pharmacogenetic issue which can beaddressed in humans.
The authors thank Georges Zadigue for his excellenttechnical assistance. This study was supported by INSERM,the National Research Agency (ANR 05-PCOD-029), Univer-sities Paris-Descartes, Pierre and Marie Curie, and Paris-Sud.This study was performed in the European Vascular Genom-ics Network, a Network of Excellence supported by theEuropean Community’s Sixth Framework Program for Re-search Priority 1 “Life sciences, genomics and biotechnologyfor health” (contract no, LSHM-CT-2003-503254). E.M. andV.G.-C. were supported by fellowships from the Ministere dela Recherche and from Academie of Medecine. The results
Figure 6. Role of kinins in cardioprotection in ACE1c miceafter IR injury. IR expressed as percentage of AR (IS/AR) wasmeasured in ACE1c or ACE2c mice receiving saline (ACE1c,n�8; ACE2c, n�12) or the specific kinin B2 receptor antag-onist, icatibant (ACE1c, n�4; ACE2c, n�5). Data aremeans � se. *P � 0.05 vs. saline-ACE2c; †P � 0.001 vs.corresponding saline.
Figure 7. Systemic and regional vascular effects of aliskiren inwild-type mice. MBP and total peripheral, renal, and coronaryresistance were measured in anesthetized wild-type mice(n�6–7/group) using the fluorescent microsphere tech-nique. Animals received saline or aliskiren (ALK, 1 mg/kg,i.v. bolus followed by an infusion 10 �g/kg/min). Data aremeans � se. *P � 0.05 vs. saline.
4698 Vol. 24 December 2010 MESSADI ET AL.The FASEB Journal � www.fasebj.org

were presented in part at the High Blood Pressure ResearchConference 2008 (Atlanta, GA, USA).
REFERENCES
1. Erdos, E. G. (1990) Angiotensin I converting enzyme and thechanges in our concepts through the years. Lewis, K. Dahlmemorial lecture. Hypertension 16, 363–370
2. Cambien, F., Alhenc-Gelas, F., Herbeth, B., Andre, J. L., Rako-tovao, R., Gonzales, M. F., Allegrini, J., and Bloch, C. (1988)Familial resemblance of plasma angiotensin-converting enzymelevel: the Nancy study. Am. J. Hum. Genet. 43, 774–780
3. Rigat, B., Hubert, C., Alhenc-Gelas, F., Cambien, F., Corvol, P., andSoubrier, F. (1990) An insertion/deletion polymorphism in theangiotensin I-converting enzyme gene accounting for half thevariance of serum enzyme levels. J. Clin. Invest. 86, 1343–1346
4. Danser, A. H., Schalekamp, M. A., Bax, W. A., van den Brink,A. M., Saxena, P. R., Riegger, G. A., and Schunkert, H. (1995)Angiotensin-converting enzyme in the human heart. Effect ofthe deletion/insertion polymorphism. Circulation 92, 1387–1388
5. Costerousse, O., Allegrini, J., Lopez, M., and Alhenc-Gelas, F.(1993) Angiotensin I-converting enzyme in human circulatingmononuclear cells: genetic polymorphism of expression inT-lymphocytes. Biochem. J. 290(Pt. 1), 33–40
6. Alhenc-Gelas, F., and Corvol, P. (2000) Molecular and physio-logical aspects of angiotensinI-converting enzyme. In Handbookof Physiology, Sec. 7, Vol. 3 (Fray, J. C. S, and Goodman, H., eds)pp. 81–103, American Physiological Society and Oxford Univer-sity Press, New York
7. Falkenhahn, M., Franke, F., Bohle, R. M., Zhu, Y. C., Stauss,H. M., Bachmann, S., Danilov, S., and Unger, T. (1995) Cellulardistribution of angiotensin-converting enzyme after myocardialinfarction. Hypertension 25, 219–226
8. Takahashi, N., Hagaman, J. R., Kim, H. S., and Smithies, O.(2003) Minireview: computer simulations of blood pressureregulation by the renin-angiotensin system. Endocrinology 144,2184–2190
9. Dzau, V. J., Bernstein, K., Celermajer, D., Cohen, J., Dahlof, B.,Deanfield, J., Diez, J., Drexler, H., Ferrari, R., van Gilst, W.,Hansson, L., Hornig, B., Husain, A., Johnston, C., Lazar, H.,Lonn, E., Luscher, T., Mancini, J., Mimran, A., Pepine, C.,Rabelink, T., Remme, W., Ruilope, L., Ruzicka, M., Schunkert,H., Swedberg, K., Unger, T., Vaughan, D., and Weber, M.
(2001) The relevance of tissue angiotensin-converting enzyme:manifestations in mechanistic and endpoint data. Am J Cardiol.88, 1L–20L
10. Huang, W., Gallois, Y., Bouby, N., Bruneval, P., Heudes, D.,Belair, M. F., Krege, J. H., Meneton, P., Marre, M., Smithies, O.,and Alhenc-Gelas, F. (2001) Genetically increased angiotensinI-converting enzyme level and renal complications in the dia-betic mouse. Proc. Natl. Acad. Sci. U. S. A. 98, 13330–13334
11. Sayed-Tabatabaei, F. A., Oostra, B. A., Isaacs, A., van Duijn,C. M., and Witteman, J. C. (2006) ACE polymorphisms. Circ. Res.98, 1123–1133
12. Cambien, F., Poirier, O., Lecerf, L., Evans, A., Cambou, J. P.,Arveiler, D., Luc, G., Bard, J. M., Bara, L., Ricard, S., Tiret, L.,Amouyel, P., Alhenc-Gelas, F., and Soubrier, F. (1992) Deletionpolymorphism in the gene for angiotensin-converting enzyme isa potent risk factor for myocardial infarction. Nature 359,641–644
13. Cambien, F., Costerousse, O., Tiret, L., Poirier, O., Lecerf, L.,Gonzales, M. F., Evans, A., Arveiler, D., Cambou, J. P., Luc, G.,Rakotovao, R., Ducimetiere, P., Soubrier, F., and Alhenc-Gelas,F. (1994) Plasma level and gene polymorphism of angiotensin-converting enzyme in relation to myocardial infarction. Circula-tion 90, 669–676
14. Mattu, R. K., Needham, E. W., Galton, D. J., Frangos, E.,Clark, A. J., and Caulfield, M. (1995) A DNA variant at theangiotensin-converting enzyme gene locus associates withcoronary artery disease in the Caerphilly Heart Study. Circu-lation 91, 270 –274
15. Samani, N. J., Thompson, J. R., O’Toole, L., Channer, K., andWoods, K. L. (1996) A meta-analysis of the association of thedeletion allele of the angiotensin-converting enzyme gene withmyocardial infarction. Circulation 94, 708–712
16. Lindpaintner, K., Pfeffer, M. A., Kreutz, R., Stampfer, M. J.,Grodstein, F., LaMotte, F., Buring, J., and Hennekens, C. H.(1995) A prospective evaluation of an angiotensin-converting-enzyme gene polymorphism and the risk of ischemic heartdisease. N. Engl. J. Med. 332, 706–711
17. Zintzaras, E., Raman, G., Kitsios, G., and Lau, J. (2008) Angio-tensin-converting enzyme insertion/deletion gene polymorphicvariant as a marker of coronary artery disease: a meta-analysis.Arch. Intern. Med. 168, 1077–1089
18. Krege, J. H., Kim, H. S., Moyer, J. S., Jennette, J. C., Peng, L.,Hiller, S. K., and Smithies, O. (1997) Angiotensin-convertingenzyme gene mutations, blood pressures, and cardiovascularhomeostasis. Hypertension 29, 150–157
Figure 8. Role of renin and angiotensins in loss of IPC in ACE3c mice. IR expressed as percentage of AR (IS/AR) was measuredin ACE2c or ACE3c mice receiving saline or aliskiren (ALK). Data are means � se. *P � 0.01, **P � 0.001 vs. correspondingIR � saline; †P � 0.001 vs. ACE2c after IPC � saline; $P � 0.01 vs. ACE2c after IPC � ALK.
4699ACE GENE TITRATION AND MYOCARDIAL ISCHEMIA

19. Griol-Charhbili, V., Messadi-Laribi, E., Bascands, J. L., Heudes,D., Meneton, P., Giudicelli, J. F., Alhenc-Gelas, F., and Richer,C. (2005) Role of tissue kallikrein in the cardioprotective effectsof ischemic and pharmacological preconditioning in myocar-dial ischemia. FASEB J. 19, 1172–1174
20. Goto, M., Liu, Y., Yang, X. M., Ardell, J. L., Cohen, M. V., andDowney, J. M. (1995) Role of bradykinin in protection ofischemic preconditioning in rabbit hearts. Circ. Res. 77, 611–621
21. Messadi-Laribi, E., Griol-Charhbili, V., Pizard, A., Vincent, M. P.,Heudes, D., Meneton, P., Alhenc-Gelas, F., and Richer, C.(2007) Tissue kallikrein is involved in the cardioprotective effectof AT1-receptor blockade in acute myocardial ischemia. J. Phar-macol. Exp. Ther. 323, 210–216
22. Pan, H. L., Chen, S. R., Scicli, G. M., and Carretero, O. A. (2000)Cardiac interstitial bradykinin release during ischemia is en-hanced by ischemic preconditioning. Am. J. Physiol. Heart Circ.Physiol. 279, H116–H121
23. Costerousse, O., Allegrini, J., Huang, H., Bounhik, J., andAlhenc-Gelas, F. (1994) Regulation of ACE gene expression andplasma levels during rat postnatal development. Am. J. Physiol.267, E745–753
24. Bodin, S., Chollet, C., Goncalves-Mendes, N., Gardes, J., Pean,F., Heudes, D., Bruneval, P., Marre, M., Alhenc-Gelas, F., andBouby, N. (2009) Kallikrein protects against microalbuminuriain experimental type I diabetes. Kidney Int. 76, 395–403
25. Livak, K. J., and Schmittgen, T. D. (2001) Analysis of relativegene expression data using real-time quantitative PCR and the2(-Delta Delta C(T)) method. Methods 25, 402–408
26. Yang, X. P., Liu, Y. H., Scicli, G. M., Webb, C. R., and Carretero,O. A. (1997) Role of kinins in the cardioprotective effect ofpreconditioning: study of myocardial ischemia/reperfusion in-jury in B2 kinin receptor knockout mice and kininogen-defi-cient rats. Hypertension 30, 735–740
27. Eckle, T., Grenz, A., Kohler, D., Redel, A., Falk, M., Rolauffs, B.,Osswald, H., Kehl, F., and Eltzschig, H. K. (2006) Systematicevaluation of a novel model for cardiac ischemic precondition-ing in mice. Am. J. Physiol. Heart Circ. Physiol. 291, H2533–H2540
28. Aouam, K., Tissier, R., Bruneval, P., Mandet, C., Berdeaux, A.,and Ghaleh, B. (2005) Preconditioning of salvaged myocardiumin conscious rabbits with postinfarction dysfunction. Am. J.Physiol. Heart Circ. Physiol. 288, H2763–H2769
29. Menard, J., and Catt, K. J. (1972) Measurement of renin activity,concentration and substrate in rat plasma by radioimmunoassayof angiotensin I. Endocrinology 90, 422–430
30. Pons, S., Griol-Charhbili, V., Heymes, C., Fornes, P., Heudes, D.,Hagege, A., Loyer, X., Meneton, P., Giudicelli, J. F., Samuel,J. L., Alhenc-Gelas, F., and Richer, C. (2008) Tissue kallikreindeficiency aggravates cardiac remodelling and decreases sur-vival after myocardial infarction in mice. Eur. J. Heart Fail. 10,343–351
31. Ludbrook, J. (1994) Repeated measurements and multiplecomparisons in cardiovascular research. Cardiovasc. Res. 28,303–311
32. Tian, B., Meng, Q. C., Chen, Y. F., Krege, J. H., Smithies, O., andOparil, S. (1997) Blood pressures and cardiovascular homeosta-
sis in mice having reduced or absent angiotensin-convertingenzyme gene function. Hypertension 30, 128–133
33. Evangelista, F. S., and Krieger, J. E. (2006) Small gene effect andexercise training-induced cardiac hypertrophy in mice: an Acegene dosage study. Physiol. Genomics 27, 231–236
34. Tschope, C., Heringer-Walther, S., Koch, M., Spillmann, F.,Wendorf, M., Leitner, E., Schultheiss, H. P., and Walther, T.(2000) Upregulation of bradykinin B1-receptor expression aftermyocardial infarction. Br. J. Pharmacol. 129, 1537–1538
35. Tschope, C., Heringer-Walther, S., Koch, M., Spillmann, F.,Wendorf, M., Hauke, D., Bader, M., Schultheiss, H. P., andWalther, T. (2000) Myocardial bradykinin B2-receptor expres-sion at different time points after induction of myocardialinfarction. J. Hypertens. 18, 223–228
36. Brown, N. J., Gainer, J. V., Murphey, L. J., and Vaughan, D. E.(2000) Bradykinin stimulates tissue plasminogen activator re-lease from human forearm vasculature through B(2) receptor-dependent, NO synthase-independent, and cyclooxygenase-in-dependent pathway. Circulation 102, 2190–2196
37. Oldenburg, O., Qin, Q., Krieg, T., Yang, X. M., Philipp, S., Critz,S. D., Cohen, M. V., and Downey, J. M. (2004) Bradykinininduces mitochondrial ROS generation via NO, cGMP, PKG,and mitoKATP channel opening and leads to cardioprotection.Am. J. Physiol. Heart Circ. Physiol. 286, H468–H476
38. Bergaya, S., Meneton, P., Bloch-Faure, M., Mathieu, E., Alhenc-Gelas, F., Levy, B. I., and Boulanger, C. M. (2001) Decreasedflow-dependent dilation in carotid arteries of tissue kallikrein-knockout mice. Circ. Res. 88, 593–599
39. Yang, X. P., Liu, Y. H., Shesely, E. G., Bulagannawar, M., Liu, F.,and Carretero, O. A. (1999) Endothelial nitric oxide geneknockout mice: cardiac phenotypes and the effect of angioten-sin-converting enzyme inhibitor on myocardial ischemia/reper-fusion injury. Hypertension 34, 24–30
40. Xiao, H. D., Fuchs, S., Bernstein, E. A., Li, P., Campbell, D. J.,and Bernstein, K. E. (2008) Mice expressing ACE only in theheart show that increased cardiac angiotensin II is not associ-ated with cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol.294, H659–H667
41. Muller, D. N., Bohlender, J., Hilgers, K. F., Dragun, D., Coster-ousse, O., Menard, J., and Luft, F. C. (1997) Vascular angioten-sin-converting enzyme expression regulates local angiotensin II.Hypertension 29, 98–104
42. Sato, M., Engelman, R. M., Otani, H., Maulik, N., Rousou, J. A.,Flack, J. E., 3rd, Deaton, D. W., and Das, D. K. (2000) Myocar-dial protection by preconditioning of heart with losartan, anangiotensin II type 1-receptor blocker: implication of bradyki-nin-dependent and bradykinin-independent mechanisms. Circu-lation 102, III346-III351
43. Sun, Y., and Weber, K. T. (1994) Angiotensin II receptorbinding following myocardial infarction in the rat. Cardiovasc.Res. 28, 1623–1628
44. Yellon, D. M., Alkhulaifi, A. M., and Pugsley, W. B. (1993)Preconditioning the human myocardium. Lancet 342, 276–277
Received for publication June 15, 2010.Accepted for publication July 15, 2010.
4700 Vol. 24 December 2010 MESSADI ET AL.The FASEB Journal � www.fasebj.org





























