Embed Size (px)
Citation preview

Bri!ish Journal of Haematology, 1973, 24, 703.
Haematological Data in 3 12 Cases of P-Thalassaemia Trait in Thailand
P. POOTRAKUL, P. WASI AND S. NA-NAKORN
Faculty of Medical Technology and Department of Medicine, F a c d t y of Medicine Siriraj Hospital , Mahidol University, Bangkok
(Received 3 0 October 1972; acceptedfor publication 14 November 1972)
SUMMARY. The haematological data are presented in 312 individuals of Thai or Chinese extraction with the high A2 p-thalassaemia trait. The mean haemoglobin concentrations were 12.1 g x among males and 10.8 g”/;: among fcmalcs; 54% of thc subjccts wcre not anacmic and all wcrc asyniptomatic. Low MCV, MCH and MCHC values were found in 67%, 81 % aiid 6 ”/, rcspcctively; erythrocytosis was detected in 34 %. Variable hypochromia and microcytosis were usually present, but leptocytosis and red cell basophilic stippling were often absent. Decreased red ccll osmotic fragility was always detected. Fifteen cases were found to have normal red cell morphology and 17 normal red cell osmotic fragility. The reticulocyte counts, although ranged up to IZ%, were normal in the majority. The serum iron levels and unsaturated iron binding capacity were normal except in 11 % of the cases whcre the scrum iron lcvels were lower than 50 p g % . Haemoglobin A, levels varied from 3.4 to 7.8% with a normal distribution and a mean of 5.2%. Haerno- globin F lcvels varied from o to 7.8% with 5 8 % elevated values. Analysis of variance revealed intrafamilial segregation of both haemoglobin A, and hacmo- globin F levels. The literature is reviewed and findings are compared.
Findings in the heterozygous state of p-thalassaemia vary considerably. Generally carriers of thc P-thalassaemia gene are asymptomatic, but cases with anaemia and splenomegaly have becn reported, especially in Italians (Bianco et al, 1952 ; Chini & Valeri, 1949; Silvestrorii & Bianco, 1949, 1959) aiid Grccks (Fcssas, 1959) but not in American Negroes (Weatherall, 1964). Even the degree of red cell basophilic stippling in p-thalassaemia trait is stated to be racially different. Although p-thalassaemia has been known to be prevalent in Thailand, detailed haematological findings have never before been reported. This paper presents findings in 3 12 cases of p-thalassaemia trait, probably the largest series examiiied in detail and by modern techniques. The large body of data allows examination for correlation be- tween various parameters.
MATERIALS AND METHODS
The subjects were Thai and Chinese. Of the 3 12 individuals, 277 were members of faniilics
Corrcspondcnce: U r P. Wasi, Division of Haematology, Department of Medicine, Siriraj Hospital, Bangkok 7, Thailand.
703
704 P. Pootrakul, P. Wusi and S . Nu-Nakorn
with classical P-thalassaemia/haemoglobin (Hb) E disease (Hbs E+ F) who were found to have elevatcd Hb A,. The remaining 35 cases were examined for other reasons and found to have clevatcd Hb A,.
Standard hacmatological tcchniques were employed. The haemoglobin concentration was measured by the cyanmethaemoglobin method. The red cell counts were originally carricd out in counting chambers but later by a Coulter electronic cell counter. The volume of packed red cells (PCV) was obtained by the inicrohaeinatocrit method. The red cell osmotic fragility was detcrinincd by the standard buffered saline method (Dacie, 1960) and on a Danon’s fragiligraph (Danon, 1963). Haemoglobin A, was quantitated by DEAE-Sephadex chromatography and by cellulose acetate electrophoresis in Tris-borate EDTA buffer as previously described (Wasi et al, 1968). Haemoglobin F was quantitated by the I min alkali denaturation technique (Singer et al, 1951). The serum iron and unsaturated iron binding capacity (UIBC) were determined according to Schade et a1 (1954).
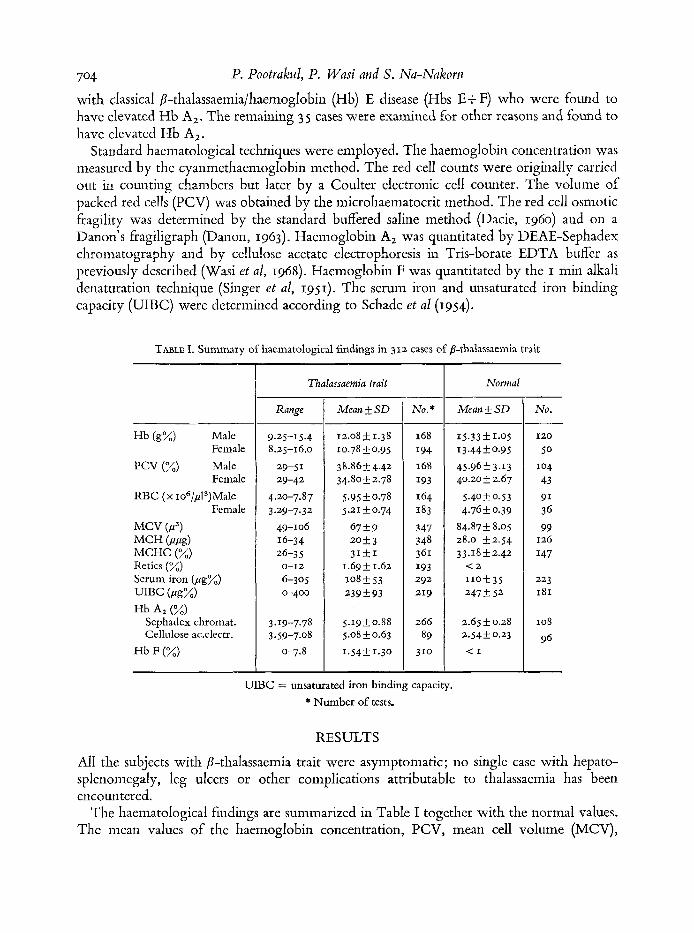
TABLE I. Summary of haematological findings in 312 cases of B-thalassaemia trait
Hb (g%) Male
PCV (%) Male
RBC (x io6/pI3)Male
Female
Female
Female
MCV(P3) M C H ( P P d MCHC (%I
UIBC (Peg%) Hb A2 (%I
Hb F (%I
Retics (%) Scrum iron (@ex)
Sephadex chromat. Cellulose acdcctr.
Thalassaemia trait
Range
9.25-15.4 8.25-16.0
29-51 29-42
4.20-7.8 7 3.2Y-7.32
49-106 16-34 26-3 5 0-12
6-305 0-400
3.19-7.78 3.59-7.08
0-7.8
Mean SD
12.08 5 1.38 10.78 f 0.95 38.86-1: 4.42 34.80 f 2.78
5.95 k 0.78
67+9
5.21 50.74
2 0 k 3 3IkI
1.695 1.62
239kY3 108 5 53
5.19*0.88 5.08 f 0.63 1.54f 1.30
Nu. *
Nurnral
Mean k SD
15.33 f 1.05 13.44 k 0 .Y 5 45.9653.13 40.20 f 2.67
5.4OkO.53 4.762 0.39
84.87f 8.05 28.0 k2.54 33 .I 8 f 2.42 < 2
I Iok3S 247 f 52
2.65 f 0.28 2.54f 0.23 < I
Nu.
I20
SO
104 43
91 36 99
126 I47
223 I81
108
96
- UIBC = unsaturated iron binding capacity.
* Number of tests.
RESULTS
All the subjects with /?-thalassaemia trait werc asymptomatic; no single case with hepato- splcnomegaly, leg ulcers or other complications attributable to thalassaemia has been encountered.
The liaematological findings are summarized in Table I together with the normal values. The mean values of the haemoglobin concentration, PCV, mean cell volume (MCV),
fl-TIzalassaciiiia Trait in Thailaiid
Saline concentrotion (96)
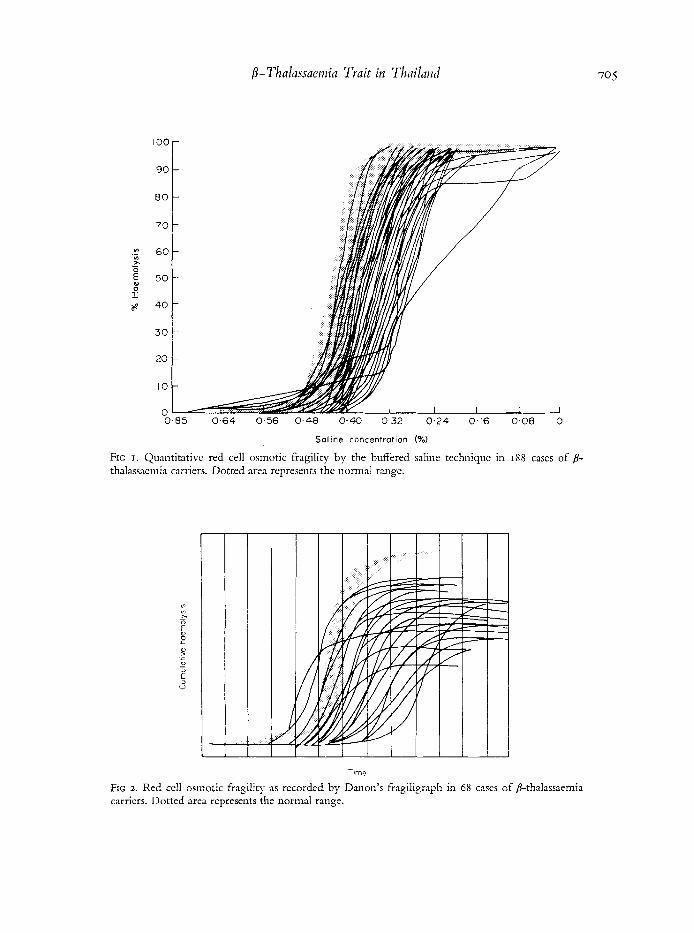
FIG I . Quantitative red cell osmotic fragility by the buffered saline techniquc in 188 cases of /3- thalassaemia carriers. Dotted area represents the normal range.
FIG 2 . Red cell osmotic fragility as recorded by Danon's fragiligraph in 68 cases of 8-thalassaemia carriers. Dotted area represents the normal range.
706 P. Pootrakul, P. Wasi and S. Na-Nakorn mean cell haemoglobin (MCH) and mean cell haemoglobin concentration (MCHC) were significantly lower than the normal means. The mean red cell count was significantly higher than the normal mean. However, among the 3 12 cases of p-thalassaemia trait, 54 % were not anaemic and among the anaemic group 83 % had haemoglobin concentrations above 10 g/Ioo ml. Low indices were found in 67% for the MCV, 81 % for the MCH and 6% for the MCHC; 34% had erythrocytosis and 74% had reticulocyte counts of 2% or lower. The mean values of serum iron levels and UIBC were not different from normal. 11 % of the P-thalassaemia carriers had serum iron levels lower than 50 ,ug % and 6 % had values higher than 200 pg %.
The red cells showed variable degree of hypochromia, microcytosis, anisocytosis, poikilo- cytosis and leptocytosis. Slight spherocytosis and basophilic stippling were detected in the minority of cases. No nucleated red cells were observed. The red cell morphology was
E n - 0 4 -
r" 3 -
0 7 E 2 -
a .
* . - . . . . . *. .- . . . .. . . .. ' . . . . . ... . . .t. . . ........ . . . . * . . 2 . : . ..: ,,
.. ... . .*.:? ;... %.. :*-:* .e
. . . . . . . - . . . . . . . . &*,. :;.... .... . . . . . . . . . . . . +-:. ................. . ::. . I .
I I I ' . I I I .. 1 . . . . I -
normal in 15 cases. Osmotic fragility of the red cells as determined by the usual quantitative technique (Fig I) and by Danon's fragiligraph (Fig 2) was decreased. Seventeen cases were found to have normal red cell osmotic fragility. The normal red cell morphology and the normal red cell osomotic fragility in these few cases of p-thalassaemia trait did not segregate in the families.
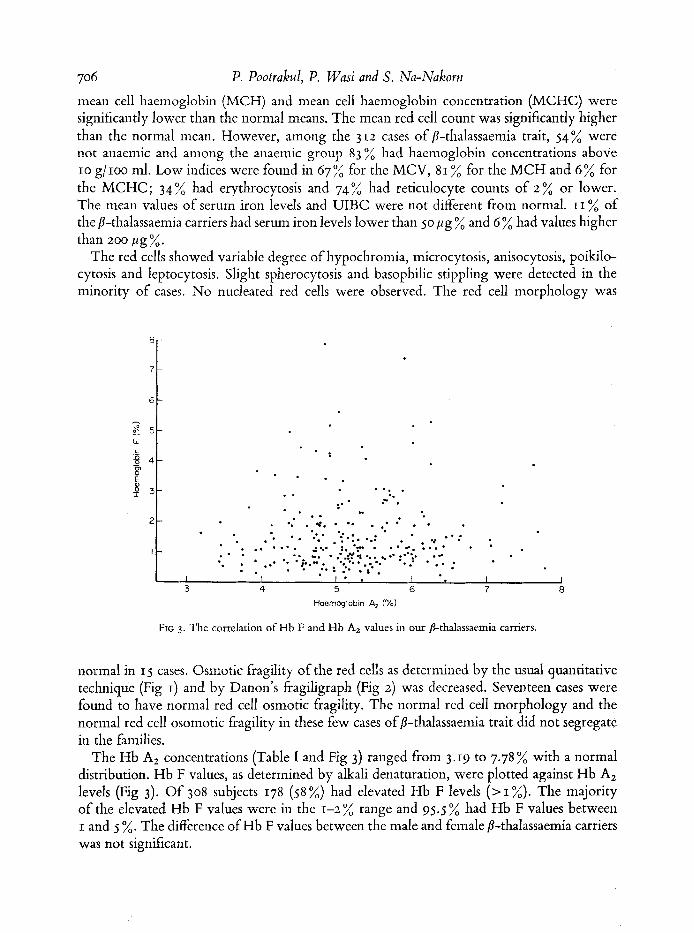
The Hb A2 concentrations (Table I and Fig 3) ranged from 3.19 to 7.78 % with a normal distribution. Hb F values, as determined by alkali denaturation, were plotted against Hb A2 levels (Fig 3). Of 308 subjects 178 (58 %) had elevated Hb F levels ( > I %). The majority of the elevated Hb F values were in the 1-2 % range and 95.5 % had Hb F values between I and 5 %. The difference of Hb F values between the male and female 8-thalassaemia carriers was not significant.

p- Thalassncnzia Trait iri T / la i lad 707 A summary of computations for analysis of variance between interfamilial and intrafainilial
variation of Hb A, and of Hb F is presciited in Table 11. Tlie correlation coefficients were: 0.0178 between Hb A, and Hb F levels, 0.0209 between Hb A, levels and haematocrit,
- 0.1183 between Hb F levels and haematocrit (P<o.o2), and 0.0936 between Hb A, levels and MCH.
TABLE 11. Summary of computations for analysis of variancc of data on Hbs A, and F iu 25 families
Between family means Within families
Hb A, i
I I
I I p<O.OO1
HL F
Ai~oi int cg- Drgreer Estirnated varintiori cffreedom variance
40.2839 24 1.6784 41.2112 60 0.6868
I I
DISCUSSION
Both high Hb A, p-thalassaemia and high F (normal A2) P-thalassaemia (‘6P’-thalassaemia) have been detected in Thailand (Wasi et al , 1969). The most common subtype is the classical P-thalassaemia which is associated with elevated Hb A, levels in heterozygotes and in which there is a complete inhibition ofp-chain synthesis. In the high A, p-thalassaemia group there is a mild allele which partially inhibits p-chain synthesis. This is demonstrated by the presence of Hb A when this type of P-thalassacmia gene is prcseiit in double dose or when it interacts with one of the P-chain structural haemoglobin variants. Of the subjects in this series 277 most likely had the classical p-thalassaemia trait since they were members of families of patients with classical P-thalassaemia/Hb E disease (Hbs E + F). All known cases of high A,-thalassaemia trait who were relatives of patients with mild p-thalassaemia/Hb E disease (Hbs A+ E+ F) were excluded. Probably all or most of the 35 cases without family data also had classical P-thalassaemia trait, since the mild P-thalassaemia gene is much less frequent They were thus included in this series.
Our P-thalassaemia carriers are uniformly symptomless, and hcpatosplenomegaly attri- butable to this condition and complications such as gallstones and leg ulcers have not been found. This is unlike its counterpart in the Mediterranean area whose manifestation is irregular, some patients being asymptomatic while others have hepatosplenomegaly and severe anaemia (Chin; & Valeri, 1949; Silvcstroni & Bianco, 1949, 1959; Bianco et al, 1952; Zuelzer et al, 1956; Fessas, 1959; Weatherall, 1965; Aksoy & Erdem, 1969). In Greece, half of the p-thalassaemia carriers had palpable spleens (Fessas, 1959). In Italy, 23-25 % of the indi- viduals with microcythacmia (thalassaemia minima) were pale and asthenic (Silvestroni & Bianco, 1949). Complications such as bone changes, gall stones (Weatherall, 1965) and leg ulcers (Samitz et a\, 1964) have been reported in Mediterraneanp-thalassaemia trait. In contrast,
C

708 P. Pootrakd, P. Wasi and S. Nu-Nakorn
54 cases of p-thalassaemia trait in the Negro wcrc uniformly asymptomatic (Weatherall,
Of our 3 12 cases with P-thalassaemia trait, 54% were not anaemic and in 83 2 of the anaemic group the haemoglobin levels were above 10 g/Ioo ml. Iron deficiency, much pre- valent here, was probably responsible for a significant reduction of the haemoglobin con- centration among these cases. In support of this coiiclusion was the observation that most of the cases of P-thalassacmia trait with less than 10 g/Ioo ml haemoglobin concentration werc fcmalc; in 11 % of 292 cases with serum iron determinations the serum iron level was lower than 50 p g x , aiid in a few of these who were given iron orally the haemoglobin levels had riscn.
The incidence of a-thalassaemia trait in Thailand is 20-30% (Na-Nakorn & Wasi, 1970; Pootrakul et ul, 1970). The 8-thalassaemia trait in association with a-thalassaemia trait is not apparently different from P-thalassaemia alone (Wasi et al, 1969). However, whether the additional presence of a-thalassaeniia trait affects the haemoglobin level in fl-thalassaemia trait is not known.
Of the red ccll indices, the MCHC was reduced in only 6 x of cases and is thus not useful for the diagnosis of this condition. The incidence of reduced MCV and MCH values was less than that of red cell morphological and osmotic fragility alterations. This was probably due to the great variation of our red cell counts. This may also be responsible for erythrocytosis being found in only 34% of our cases with p-thalassaemia trait. Others have reported ery- throcytosis in from 50 ”/, of cases (Hammond et ul, 1964) to almost all cases of p-thalassaemia trait (Silvestroni & Bianco, 1949).
Absence of leptocytes aiid red cell basophilic stippling was frequent in our cases. The lattcr appears to be more common in the Mediterranean than in the Negro p-thalassaemia trait (Weatherall, 1965).
The red cell osmotic fragility, determined by either the conventional technique or by the fragiligraph, was decreased in practically all cases, with a varying degree of alteration (Figs I and 2). In a few cases the fragility curves wcre skewed, crossing the normal curves near their bases, suggesting the preseiice of spherocytcs.
Although iron deficiency and thalassacmia share many red cell morphological alterations, it is not difficult to distinguish the two. If morphological and osmotic fragility changes are associated with haemoglobin concentrations of 10 g/Ioo ml or above, they are most likely due to thalassaemia trait. At this haelnoglobin level iron deficiency is associated with normal red cell morphology (Beutler, 1959) and normal red cell osmotic fragility (personal observa- tion).
In this series the red cell morphology was normal in 15 cases and the red cell osmotic fragility was normal in 17 cases ofp-thalassaemia trait. Our data showed that these observa- tions did not run true in families, thus suggesting different expressivity rather than their being due to a different gene; the latter explanation for this finding has been suggested by some authors (Beaven & White, 1962; Ceppelini, 1959).
For Hb A2 determination, both cellulose-acetate electrophoresis in a tris-borate-EDTA buffer and DEAE-Scphadcx chromatography have been satisfactory. The upper limit of normal for Hb A, levels in this laboratory is 3.2 %. In this series of 3 IZ cases one borderline value, 3.19 % by DEAE-Sephadex chromatography, was included because of targctting of
J964).
Auth
ors
Kun
kel e
t nl(
19g)
Si
lves
troni
et a
l(195
7)
Muz
zolii
i (19
57)
Ger
ald
& D
iam
ond
(195
8)
Jose
phso
n et
al(
r958
) Fe
ssas
(19 j
9)
Bea
vcn
et al
(196
1)
Mal
amos
el n
l(19
62)
Han
imon
d et
a2 (
1964
) W
eath
eral
l (19
64)
Aks
oy &
Erd
eni (
1969
) Pr
esen
t rep
ort
Rac
e
ItaIia
ii 8(
G
reek
Ita
lian
Italia
n M
ainl
y Ita
lian
Gre
ek
-
Mai
nly
Med
iterr
anea
n G
reek
G
reek
Neg
ro
Tur
k T
hai a
nd C
hine
se
-
Hb
A,
Rang
e
2.4
t-9.5
3.
0-5.
8
3.3-
6.8
-
3.9-
10.5
3.
3-12
.3
-
3.6-
6.6
3.7-
7.2
3.0-
8.5
3.4-
6.9
4.0-
7.1
3.2-
7.8
Hb
F
Rattg
e (%
)
-
-
0.45
-4.4
8 1.
7-4.
0 1.
0-84
.0
%
Incr
ease
d va
lues
*
-
-
2s 53
> 2%
in
20 c
ases
V
alue
s 3-
7%
in s
ix c
ases
0- >
6.0
-
0.y-
8.3
N 0 r
m a 1
- 6
0-10
0-4.5
0-7.
8
* Var
iabl
e va
lues
wer
e us
ed a
s no
rmal
lev
els b
y di
ffer
ent l
abor
ator
ies.
? T
wo
norm
al v
alue
s mos
t lik
ely
belo
nged
to
diff
eren
t typ
es o
f b-th
alas
saem
ia.
Tw
o ca
m w
ith
hom
ozyg
ous
thal
assa
emia
from
ori
gina
l tab
le w
ere
excl
uded
.

710 P. Pootrakul, P. Wasi arid S. Na-Nakoyrz
the red cells, a slightly elevated Hb F, and an Hb A, value 3.59% which was obtained on the same sample by cellulose-acetate electrophoresis. Otherwise Hb A, lcvcls ranged from 3.4 %, to 7.8 % with an average of 5.16 % and their distribution was almost normal. By the criteria used for case selection in this study, /S-thalassaemia carriers with a norinal Hb A, lcvel would havc bccn excluded. However, this condition belongs to a /S-thalassaemia group othcr than that with which wc arc dealing in this report.
Iron deficiency decreases Hb A2 levcls cvcn in P-thalassaeniia trait (Wasi ct nl, 1968). However, the levels of Hb A, in this series were probably not appreciably affected by iron deficiency, since iron deficiency anaemia of a sufficient magnitude to produce this effect was not ciicountcred in these cases. Furthermore, the correlation coefficient between the hacmoglobin and haenioglobin A2 values was not significant.
P. vivax malaria increases Hb A2 levels and the effect persists for a long time after an attack of malaria (Arends, 1967). Our study in P. fa2cQmwn malaria revealed no increase in Hb A, lcvels (Wasi et al, 1971). The chance that elevation of Hb A? in some of our cases was due to malaria and not to thalassacmia must be very small. Most of these were relatives of patients with /S-thalassaemia and they had other findings of thalassaemia, such as changes in red cell morphology and osmotic fragility and elevated Hb F levels. In this area, malaria due to P. vivax is less common than that following P..falciparum infection.
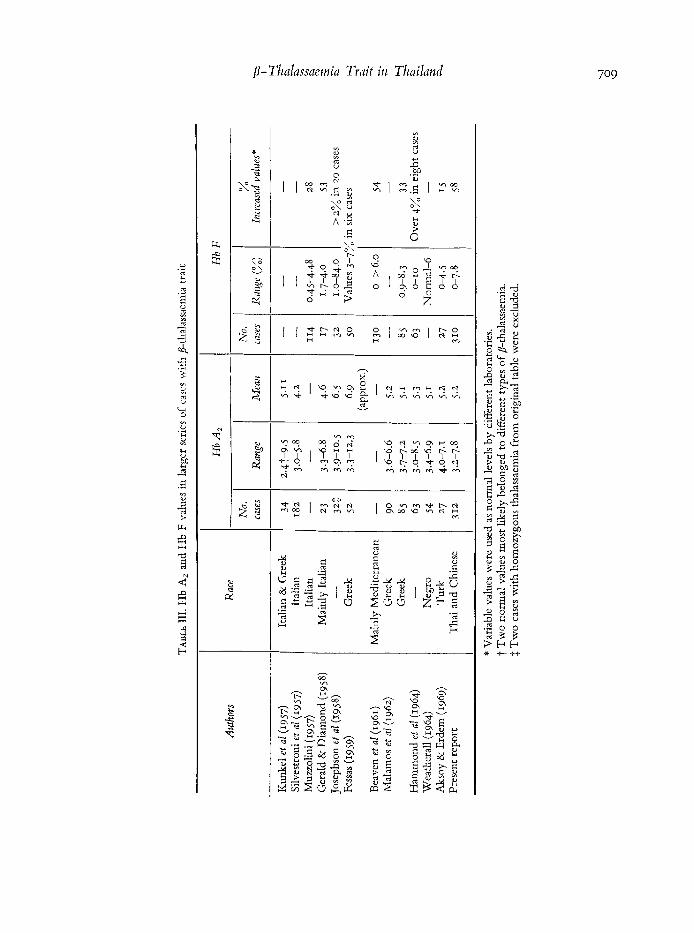
€hemoglobin A2 values from reported series are summarized in Table 111. Some of these were recalculated from graphic presentation, and from the series of Josephson et al (1958); two cases with homozygous Cooley’s disease were excluded. In older series, the valucs were more variable but various subtypes of /S-thalassaemia were not then known and may well have bccn included. In the series ofJosephsoil et al (1958) and of Fessas (1959), higher values of Hb A, up to 10% or over were more frequent than in other series. We have never encountered such high values in any of our cases with p-thalassaemia trait. It is remarkable that in the six more recent series (Table HI), in spite of different ethnic backgrounds of the subjects, the mean Hb A, values were almost identical, 5.1-5.3 %. Probably correct values of Hb A, in classical 8-thalassaemia trait stretch from 3.4 to 8 % with a normal distribution.
Intrafainilial segregation of Hb A, in 8-thalassaemia trait was earlier suggested by Carcassi (Weatherall, 1965) and by Gerald & Diamond (~958). This has been strongly supported by analysis of variance in Weatherall’s data (Weatherall, 1964) and in this paper (Table 11).
Haemoglobin F levels as determined by the alkali-denaturation mcthod varied from o to 7.8 averaging 1.54%. 58 % of the values were considered elevated (> I %) and 95.5 % of these the values were between I and 5 %. The reciprocal correlation between Hb F levels and PCV (7 = - 0.1~83 ; P<0.02) was just significant. Hb F values in p-thalassaemia trait reported in the literature are summarized in Table 111. The Hb A, levels were not known in the series of Muzzolini (1957) and of Bcavcii et a2 (r961), but presumably their subjects had p-thalas- saemia trait. Different laboratories used variable values as normal levels of Hb F, hencc the difference in the proportion of valucs considered as elevated. However, it appears that in p-thalassaemia trait from many parts of the world, Hb F values range from o to 8 % while half of them are below I % and most of the other half are between I and 4%. It is likely that whcn a niorc accurate determination of Hb F at low levels, especially below I % becomes available, the proportion of cases of p-tlialassaemia trait with elevated Hb F will bc incrcascd.
The correlation cocfficieiit between the Hb A, and haematocrit levels was -0.1183

/3-Thalassaeinia Truit in Thailand 71 I
(P<o.oa). Those ‘between Hb A, and Hb F levels, Hb A, and PCV values and Hb A, levels and MCH, were 0.0178,0.0209 and 0.0936, respectively-. Weatherall (1964) has shown a vcry significant negative correlation between Hb A, levels and MCH. The absence of this in our data could be due to the great variation of our red ccll counts.
Recently cases of p-thalassaemia trait with normal haematological findings including rcd cell morphology, osmotic fragility, Hb A, and F levels have been reported (Heller et al , 1966; Schwartz, 1969). In the absence of a globin synthesis study such cases ofB-thalassaemia trait cannot bc recognized. In view of the above, alleged cases of p-thalassaeinia trait with very high Hb F (Aksoy, 1959; Aksoy et al, 1961; Josephson et of, 1958) and even some ofthc symptomatic D-thalassaernia carriers may actually carry a silent p-thalassaemia gene in addi- tion to the classical 8-thalassaemia gene. That symptomatic cases of p-thalassaemia trait may carry a modifier gene was envisaged earlier (March et al , 195.2). With the discovery of the silent P-thalassaeniia gene, cases with atypical findings must be carefully investigated by inodcrn tcchniquc s before a diagnosis of /?-thalassaemia trait is made.
ACKNOWLEDGMENT
This iiives,tigation was supported by United States Public Hcalth rcsearch grant AM 09805 from the Institute of Arthritis and Metabolic Diseases.
REFERENCES
AKSOY, M. (1959) Thalassaemia rninor with large amount of fctal haemoglobin. Acta Haeniatologica, 22, 188.
AKSOY, M., EGRIBOZLU, A. & ALPUSTUN, H. (1961) The thalassaeniia syndromes. I. Thalassaernia minor with large amounts of foetal haemoglobin. Study of a family. Acta Haematologicn, 25, 136.
AKSOY, M. 8: ERDEM, S. (1969) Some problems of hemoglobin pattcms in different thalasscmic syndromes showing the heterogeneity of beta- thalassemia genes. Annals of the New York Academy of Scierzces, 165, I 3.
ARENDS, T. (1967) High concentration of haemo- globin A, in malaria patients. Nature, 215, 1517.
BEAVEN, C.H., ELLIS, M.J. & WHITE, J.C. (1961) Studies on human foetal hacmoglobin. 111. The hereditary liacnioglobinopatliies and thalassaemias. British Joiirrtal of Haernafology, 7, 169.
BEAVEN, G.H. Sr WHITE, J.C. (1962) Variability of thalassaemic red cell characters. Nature, 193, 448.
BEUTLER, E. (1959) The red cell indices in the diagnosis of iron-deficiency anemia. Annals of Internal hfedicirze, 50, 3 I 3 .
BIANCO, I., MONTALENTI, G., SILVEFTKONI, E. Sr SINISCALCO, M. (1952) Further data on genetics of microcythaemia or ithalassaemia minor and Coolcy’s disease or thalassaemia major. AmLalx qf Eigcizics,
CEPPELLINI, R. (1959) 1,’ernoglobina nornialc lcnta A, : 16, 299.
Sttoi rapporti con una nuova frazioiie emoglobinica lenta, B,, e sua iniportanza per il riconoscinieiito di varianti talassemiche che compaiono nelle famiglic di portatori di Thalassemia media e di emoglobinopatia H. Actu Gerzeficae Medicae ef Gemel- lologiae, 8, Suppl. 2, 47.
CHINI, V. & VALERI, C.M. (1949) Mediterranean heinopathic syndromes. Blood, 4, 989.
DACIE, J.V. (1960) The ITaetnolytic Anaemias, Corigeizital afzd Acquired. Part r. The Congerzital Anaemias, 2nd edn, p 36. Grune 8: Stratton, Ncw York.
DANON, D. (1963) A rapid micro inethod for recording rcd cell osmotic fragility by continuous decrease o f salt concentration. joi./rnal oJ Clinical Patho/o,qy, 16, 377-
FESSAS, PH. (1959) Thalassaemia and the alterations ofthe haenioglobiri pa ttcrn. Ahizortnnl Haetm,q/ubins (Ed. by J. 11. P. Jonxis and J. F. Delafresiiaye), p 134. Blackwell Scientific Publications, Oxfnrd.
GER4LD, P.S. 81 DIAMOND, L.K. (1958) The diagno5is of thalas<eiiiia trait by starch block clcctrophoresis ofthe hemoglobin. Blood, 13, 61.
HAMMOND, D., STURGEON, P., BERGREN, W. 8r CAVILES, A., JR (1964) Defmition of Cooley’s trait or thalasseiiiia minor : classical, clinical and routuic laboratory hernatology. Aiinals oj ‘ the Neio York 2 4 ~ ~ d r t t ~ y of Sciences, 119, 372.
HBLLER, P., YAKUI~IS, V.J., ROSENLWFIG, A.I., ABILII- GAARD, C.F. 8( RUCKNAGEL, D.L. (1966) Mild

P. Pootrakul, P. Wasi and S. Nu-Nakorn homozygous beta-thalassemia : further evidence for the heterogeneity of beta-thalassemia genes. Annals of Internal Medicine, 64, 52.
JOSEPHSON, A.M., MASRI, M.S., SINGER, L., DWORKIN, D. & SINGER, K. (1958) Starch block electrophoretic studies of human hemoglobin solutions. 11. Results in cord blood, thalassemia and other hematologic disorders : comparison with Tiselius electrophoresis.
KUNKEL, H.G., CEPPELLINI, R., MULLER-EBERHARD, U. & WOLF, J. (19.57) Observations on the minor basic hemoglobin component in the blood of normal individuals and patients with thalassemia. Journal of Clinical Investigation, 36, 1615.
MALAMOS, B., FESSAS, PH. & STAMATOYANNOPOULOS, G. (1962) Types of thalassaemia-trait carriers as revealed by a study of their incidence in Greece. BritishJournal of Haematology, 8, 5 .
ARCH, H.W., SCHLYEN, S.M. & SCHWARTZ, S.E. (1952) Mediterranean hemopathic syndromes (Cooley’s anemia) in adults: Study of a family with unusual complications. American Journal of Medicine, 13, 46.
MUZZOLINI, M. (1957) Alkali-resistant hemoglobin in normal subjects and in healthy microcythemic subjects. Gazetta Internazionafe di Medicina e Chirurgia, 61, 1199.
NA-NAJIORN, S. & WASI, P. (1970) Alpha-thalassemia in Northern Thailand. American Journal of Human Genetics, 22, 645.
NAKORN, S. (1970) Incidence of alpha thalassemia in Bangkok. Journal of the Medical Association of Thailand, 53, 250.
SMTZ, M.H., WALDORP, D.S. & SHRAGER, J. (1964) Leg ulccrs in Mediterranean anemia. Archives Pf
Dermatology, 90, 567. SCHADE, A.L., OYAMA, J., REINHART, R.W. & MILLER,
J.R. (1954) Bound iron and unsaturated iron- binding capacity of serum; rapid and reliablc quantitative determination. Proceedings of the Society for Experimental Biology and Medicine, 87, 443.
Blood, I37 543.
POOTRAKUL, s., WASI, P., PORNPATKUL, M, & NA-
SCHWARTZ, E. (1969) The silent carrier of beta thalas- semia. New England Journal of Medicine, 281, 1327.
SILVESTRONI, E. & BIANCO, I. (1949) Microcytemia, constitutional microcytic anemia, and Cooley’s anemia (Mediterranean anemia or thalassemia). American Journal of Human Genetics, I, 83.
SILVESTRONI, E. & BIANCO, I. (1959) The distribution of microcythaemias (or thalassaernias) in Italy. Abnormnl Haemoglobins (Ed. by J. H. P. Jonxis and J. F. Delafresnaye), p 242. Blackwell Scientific Publications, Oxford.
SILVESTRONI, E., BIANCO, I., MUZZOLINI, M., MODIANO, G. & VALLISNW, E. (1957) Studio biochimico, elettroforetico e spettrofotometrico dell’ emoglobina di malati di anemia microcitica constituzionale e di morbo di Cooley. Progress0 Medico (Napoli), 13, 70s.
Studies on abnormal hemoglobins. I. Their demon- stration in sickle cell anemia and other hematologic disorders by means of alkali denaturation. Blood,
WASI, P., DISTHASONGCHAN, P. & NA-NAKORN, S. (1968) The effect of iron deficiency on the levels of hemoglobins A, and E. Journal of Laboratory and Clinical Medicine, 71, 85.
WASI, P., KRUATRACHUE, M., PIANKIJAGUM, A. S. PRAVATMEUNC, P. (1971) Hemoglobins Az and E levels in malaria. JournaZ of the Medical Association of Thailand, 54, 559.
WASI, P., NA-NAKORN, S., POOTRAKUL, S., SOOKANEK, M., DISTHASONGCHAN, P., PORNPATKUL, M. & PANICH, V. (1969) Alpha- and beta-thalassemia in Thailand. Annals ofthe New York Academy of Sciences, 165, 60.
W~THERALL, D.J. (1964) Biochemical phenotypes of thalassemia in the American Negro population. Annals of the New Yo& Academy of Sciences, 119,450.
WEATHERALL, D. J. (1965) The Thalassaemia Syndromes. Blackwell Scientific Publications, Oxford.
ZUELZER, W.W., NEEL, J.V. & ROBINSON, A.R. (1956) Abnormal hemoglobins. Progress in Haematology, I, 91.
SJNGER, K., CHERNOFP, A.I. & SINGER, L. (1951)
6, 413-










![[Incidence of haematological neoplasms in Castilla y Leon.]](https://img.pdfslide.net/doc/110x75/634c4a087e26824f4400ef99/incidence-of-haematological-neoplasms-in-castilla-y-leon.jpg)


















