Embed Size (px)
Citation preview

1
1
This is a Post-print of a manuscript, which appeared in:
Thin Solid Films
Please cite this publication as follows:
M. Müller, B. Torger, E. Bittrich, E. Kaul, L. Ionov, P. Uhlmann, M. Stamm,
In-situ ATR-FTIR for characterization of thin biorelated polymer films,
Thin Solid Films 2014, 556, 1-8.
You can download the published version at:
http://dx.doi.org/10.1016/j.tsf.2013.12.025

2
2
In-situ ATR-FTIR for characterization of thin biorelated polymer films
M. Müllera b
, B. Torger a, b
, E. Bittricha, E. Kaul
a,b, L. Ionov
a ,b, P. Uhlmann
a, M. Stamm
a, b
aLeibniz Institute of Polymer Research Dresden (IPF Dresden), Hohe Straße 6, 01069
Dresden, Germany
bTechnical University of Dresden (TUD), Department of Chemistry and Food Chemistry,
01062 Dresden, Germany
E-mail: [email protected]
Abstract
We present and review in-situ-attenuated total reflection Fourier transform infrared (ATR-
FTIR) spectroscopic data from thin biorelated polymer films useful for the modification and
functionalization of polymer and inorganic materials and discuss their applications related to
life sciences. A special ATR mirror attachment operated by the single-beam-sample-reference
(SBSR) concept and housing a homebuilt thermostatable flow cell was used, which allows for
appropriate background compensation and signal to noise ratio.
ATR-FTIR data on the reactive deposition of dopamine on inorganic model surfaces are
shown. Information on the structure and deposition pathway for such bioinspired melanin-like
films is provided. ATR-FTIR data on thermosensitive polymer brushes of poly(N-
isopropylacrylamide) (PNIPAAM) is then presented. The thermotropic hydration and
hydrogen bonding behaviour of PNIPAAM brush films is described. Finally, ATR-FTIR data
on biorelated polyelectrolyte multilayers (PEM) are given together with details on PEM
growth and detection. Applications of these latter films for biopassivation/activation and local
drug delivery are addressed.
Keywords
ATR-FTIR, SFM, polymer film, polydopamine, poly(N-isopropylacrylamide) brush,
polyelectrolyte multilayer, protein adsorption, drug release
Introduction
Surface modification using thin biorelated polymer films is an important issue for the
research, development and production of biomedical materials and devices including bone
implants, contact lenses, blood contacting stents, and catheters. Polymer coatings are aimed at

3
3
biopassivation such as the prevention of unspecific protein adsorption or cell adhesion as was
shown by polyethylene glycol modified surfaces [1]
and bioactivation like the exposure of
targeting molecules at the surface [2]
or the inclusion of elutable drugs.[3]
The characterization
of both deposition and function of these coatings at the model, in-vitro, and in-vivo levels are
important tasks for their future application in biomedicine.
Here, we present and review in-situ ATR-FTIR data on three examples of thin biorelated
polymer coatings on model substrates such as silicon and germanium. These are
polydopamine, poly(N-isopropylacrylamide), and polyelectrolyte multilayers of
poly(etyhleneimine) and sodium alginate, which are all usable for the modification and
functionalization of biomedical polymer as well as inorganic materials in applications related
to life sciences. A dedicated ATR mirror attachment (OPTISPEC, Zürich) housing a
homebuilt transparent and thermostatable flow cell is operated by the single-beam-sample-
reference (SBSR) concept,[4]
which allows for appropriate background compensation and
signal/noise ratio.
Experimental details
1.1. Polydopamine films
Polydopamine (PDA) films were prepared by injecting solutions of 2 mg dopamine (Sigma)
in 1 ml 0.001M Tris(hydroxymethyl)-aminomethan (TRIS) buffer (pH = 8.5) into the sample
(S) compartment of the in-situ-ATR-FTIR cell (see below) housing a Ge internal reflection
element (IRE) and allowed to react as described in reference.[5]
1.2. PNIPAAM brush films
Poly(N-isopropylacrylamide) (PNIPAAM) brush films were prepared by two approaches.
According to the grafting-to-approach described in reference [6] Si internal reflection
elements (IRE, see below) were spincoated by a 0.01% (w/w) poly(glycidylmethacrylate)
(PGMA) solution in tetrahydrofuran (THF) and dried for 20 min in a vacuum oven at 100°C.
Onto this PGMA precursor, a 1% (w/w) solution of carboxylic acid terminated PNIPAAM
(50 kg/mol) was spin coated and surface reacted by drying at 150°C in a vacuum oven.
According to the grafting-from-approach on silicon wafers described in reference [7] the
surface of the Si IREs was treated by alkaline hydrogen peroxide solution

4
4
(NH4OH/H2O2/H2O-1:1:1), thoroughly rinsed and dried. Thereafter, the IRE surface was
reacted with a 2% (v/v) ethanol solution of 3-aminopropyltriethoxysilane (APTES) (2h, RT).
The modification to the initiator was achieved using 0.5% (v/v) -bromo-isobutyrylbromide
and 1 % (v/v) triethylamine in dichloromethane (2h, RT) according to Matyjaszewski.[8]
PNIPAAM brushes were grafted via a modified ATRP using an ethanol solution of NIPAAM
(1g/1ml) and CuBr2/PMDTA as a catalyst and ascorbic acid as a reducing agent (2 h, RT).
1.3. Polyelectrolyte multilayer films
Polyelectrolyte multilayer (PEM) films were prepared by consecutively injecting
poly(ethyleneimine) (PEI) solution, pure water and sodium alginate (ALG) or poly(acrylic
acid) (PAC) solution, into the in-situ-ATR cell (see below) as described for other PEL
systems in reference [9]. We used the polyelectrolytes (PEL) branched-PEI (750kg/mol,
BASF, Germany), linear-ALG (460kg/mol, Kelco, USA), and linear-PAC (50kg/mol,
Polysciences, USA) based on 0.005M PEL solutions.
1.4. Proteins, drugs
Human serum albumin (HSA, 66kg/mol, Sigma, Germany) was the standard protein and
streptomycin (STRP, Carl Roth, Germany) the standard drug in these experiments. HSA was
applied as 1 mg/ml solutions in phosphate buffered saline (PBS) and STRP was applied at the
concentration of 0.001M solutions in the unbuffered original 0.005M PEL (PEI, ALG, PAC)
solution.
1.5. in-situ ATR-FTIR spectroscopy
In Fig. 1. the in-situ ATR-FTIR concept, and in Fig. 2 the transparent thermostatable flow cell
(M.M. IPF Dresden) with separate sample and reference compartments around Si, Ge or ZnSe
internal reflection element are shown.

5
5
Fig. 1. Scheme of the in-situ ATR-FTIR setup
including polarized light.
Fig. 2. Transparent in-situ ATR-FTIR cell
housing the Ge (Si, ZnSe) IRE used in the
deposition experiments. B: Back cell
component, F: Front cell component, S:
Upper sample (liquid) compartment, R:
Lower reference (liquid) compartment
(from [5], Supporting Information).
The flow cell is situated in an ATR mirror attachment (OPTISPEC, Zürich), which is
integrated into an FTIR spectrometer (Bruker Optics, Ettlingen, Germany). The flow cell is
shuttled between two different heights with respect to the fixed IR beam according to the
single-beam-sample-reference (SBSR) concept,[4]
which enables the quasi-simultaneous
measurement of sample (S) and reference (R) intensity spectra. Processing these S- and R-
intensity spectra (A = log IS/IR) provides in-situ ATR-FTIR spectra featuring flat baselines by
appropriate background compensation with a convenient signal/noise ratio. These in-situ
ATR-FTIR spectra can be quantitatively evaluated.
1.5.1. Surface concentration
From band integrals A of in-situ ATR-FTIR spectra, the surface concentration = c d (c:
concentration, d: geometric thickness) of IR-active functional groups in polymer films can be
calculated based on the formalisms of Harrick[10]
and Fringeli[4]
using eqns. 1-3. For this
calculation, the number of reflections N, absorption coefficient , the effective thickness dE
which is a function of refractive index ratio n21 = n2/n1, depth of penetration dP, electric field
amplitude E with its relative electric field components Ex, Ey, Ez, the incident angle , and d
must be known.

6
6
A = N c dE (1)
dE = n21dPE2/(2 cos ) [1-exp(-2d/dP)] (2)
= c d (MIN = 0.1 nM/cm2) (3)
Strictly, the equations hold only for band integrals measured by parallel (A║) or vertical (A)
polarized IR light. However, for randomly-oriented polymer samples, there is an
approximately constant factor F between, for example, A and band integrals measured by
unpolarized light (A). Therefore, for the calculation of eq. 3), it can be empirically
approximated that A = F A and E = Ey (Ex, Ey, Ez values are known from Harrick [10]
).
Scanning force microscopy (SFM)
SFM images were measured from polymer films using Nanostation II of Bruker Nano GmbH
(Karlsruhe, Germany). Silicon probe tips from Nanosensors (Darmstadt, Germany) with apex
radii of around 10 nm were used. Images were measured in topography, error and phase mode
and scanning parameters were optimized by minimizing the error mode signal. Images and
surface profiles were generated from SFM raw data by the SISCANPro software (Bruker
Nano GmbH, Karlsruhe, Germany). Thicknesses of polymer films were determined by
imaging the surface profile around careful scalpel cuts into the polymer film not damaging the
substrate. The distance between the bottom of the cut and the undamaged film surface was
taken as film thickness.
Results and discussion
In the following sections, results on the characterization (1.) and application (2.) of thin
biorelated polymer coatings based on in-situ ATR-FTIR spectroscopy, are presented and
reviewed.
1. In-situ ATR-FTIR characterization of biorelated polymer films
1.1. Reactive dopamine deposition
Mussels adhere to surfaces via secreted byssus threads, which are composed of proteins
containing a large amount of amino acid L-dihydroxyphenylalanin (L-DOPA). L-DOPA is
able to form reactive crosslinking biopolymerisates. Recently it was found by Messersmith,[11]
that dopamine (DA, Fig. 1B), which is the structural analogue of L-DOPA, also forms thin

7
7
crosslinked polymerisate films with thicknesses d 50 nm simply by adsorption from alkaline
solutions onto various material surfaces. Such bioinspired polydopamine (PDA) films are
appealing for biomedical, cosmetic (UV protection) and even electronic applications.[11]
The
chemistry is claimed to be known on the monomer level, where after a sequence of pH-
dependent oxidation steps, DA is transformed into the reactive monomer indole-5,6-quinone
(IQ)[12]
as shown in Fig. 3a. However, neither the subsequent surface-mediated
polymerisation mechanism, nor the exact structure of PDA is fully resolved. Actually, the
structure of PDA is claimed to be similar to the skin pigment melanin.[12]
We studied DA deposition by in-situ ATR-FTIR spectroscopy as a function of time under a
variety of parameters: pH, concentration, ionic strength, and substrate type.[5]
In Fig. 3b ATR-
FTIR spectral data concerning the pH dependence of DA deposition on Ge model substrates
are shown. To our knowledge this is the first in-situ-ATR-FTIR spectral data. At pH = 8.5,
ATR-FTIR spectra with increasing signal intensities at 1490, 1435 and 1250 cm-1
(polyphenoles) were obtained, while for pH = 6.2, no significant peaks were recorded.
However, ATR-FTIR spectra for the former case differ very much from that of synthetic
melanin (data not shown).
Fig. 3a. Proposed
reaction mechanism at
the monomer level.
Fig. 3b. In-situ ATR-FTIR spectra for the time-dependent deposition
of PDA layers on Ge model substrates for pH = 8.5 and pH = 6.2
(spectra correspond to times from bottom to top: 5, 10, 15, 20, 30,
60, 120, 180, 1200 min) (from [5]
with kind permission of ACS)
NH2
OH
OH
HN
O
O

8
8
In Fig. 3c, the integrated areas under the peaks are plotted versus time. The kinetic DA
deposition data (A(t)) can be represented by a simple exponentially damped function of the
type A(t) = A0 (1 - exp(-k1 t)), as is also shown in Fig 3c.
Fig. 3c. Plot of the IR band integrals at 1490, 1435 und 1250 cm-1
versus time and fits based
on the Langmuir adsorption model. (from [5]
with kind permission of ACS)
A kinetic rate constant of k1 0.013 min-1
was determined, which qualifies as a rather slow
process compared to protein adsorption (k1 0.08 min-1
, unpublished data). Furthermore, the
concentration (c) dependence of the DA deposition can be fit by a simple Langmuir
adsorption model [5] to A(c) = A0/(a + c) (see Fig. 4a). Scanning force microscopy (SFM)
images of the PDA film suggest granular individual particles[5]
as shown in Fig. 4b. Thus,
preformed PDA particles in solution appear to be the film-forming species. Such preformed
PDA particles are obvious by visual inspection of the reactive DA solution, which becomes
blackish with time and by dynamic light scattering studies revealing particle sizes around 50
nm. Since PDA film thicknesses in this range were reported [11]
and also measured by us using
SFM[5]
, we suggest monolayers of preformed reactive PDA particles as the film architecture.
PDA coatings will be considered for use in drug delivery applications.

9
9
Fig. 4a. Adsorption isotherms for DA
deposition based on plots of ATR-FTIR
peaks at 1490, 1435 and 1250 cm-1
versus the
DA concentration. (from [5]
with kind
permission of ACS)
Fig. 4b. SFM image (phase, 2 x 2 m2) of
PDA film deposited onto Ge IRE substrates.
(from [5]
with kind permission of ACS)
1.2. Swelling of ultrathin PNIPAAM brush films
In the second example, results for thermosensitive polymer brushes based on poly(N-
isopropylacrylamide) (PNIPAAM) chemically attached to silicon substrates are presented.
Chemical attachment was achieved by both the grafting-to[6]
and the grafting-from[7]
approach
(see Experimental Details). In Fig. 5a, ATR-FTIR spectra of a 0.05M PNIPAAM solution and
of a PNIPAAM brush film (grafting-to) on silicon in the dry state are shown. Given a
thickness of around 10 nm for the brush film based upon ellipsometry data, a well-resolved
ATR-FTIR spectrum with appropriate baseline and signal/noise ratio was obtained. However,
the ATR-FTIR spectrum of a 0.05M PNIPAAM solution did not reveal such high spectral
quality. This illustrates the general sensitivity of the ATR method for macromolecules located
directly on optical denser media, where the interacting electric field amplitude of the
evanescent wave is maximum. In addition, the concentration of PNIPAAM molecules in a
0.05 M solution is less than the concentration of PNIPAAm molecules in the brush.
Otherwise, the ATR-FTIR spectrum in the solution should be more intense than that of the
brush due to the higher refractive index of water (n3 = 1.33) compared to air (n3 = 1).
Nevertheless, from the ATR-FTIR spectrum of the 0.05 M PNIPAAM solution, the
0.0
0.5
1.0
1.5
0 0.005 0.01 0.015 0.02
Ban
d i
nte
gra
l / [c
m-1
]
Concentration / [Mol/L]
1250/TRIS1490/TRIS1435/TRISFIT

10
10
absorption coefficient of the Amide II band (Amide I interferes with the (OH) of bound
water) can be determined using eq. 1. Based on this value of = 7.13 x 107 cm/mol for the
Amide II band, a surface concentration of = 0.90 g/cm2 was determined, which is close to
the value of = 1.08 g/cm2 obtained by ellipsometry.
[6] In Fig. 5b, in-situ ATR-FTIR
spectra from ultrathin PNIPAAM brush films in contact with a PBS buffer are shown at
temperatures T = 22°C - 44°C.[13]
Fig. 5a. In-situ-ATR-FTIR spectra from
0.05M PNIPAAM solution (bottom) and on a
PNIPAAM (grafted-to) brush film on silicon
in the dry state (top).
Fig. 5b. In-situ-ATR-FTIR spectra from
amide bands centered at around 1631 and
1555 cm-1
of PNIPAAM (132kg/mol,
grafted-to) brush film in contact with PBS
recorded at temperatures T = 22 (bottom),
26, 28, 30, 32, 34, 35, 38 to 44°C (top).
(from [13]
with kind permission of ACS).
Fig. 5b shows that the Amide I band shifts up in wavenumber with temperature and the
Amide II band shifts down. Hence, PNIPAAM brushes show similar thermotropic behaviour
as PNIPAAM solutions. The wavenumber positions of the peak maxima are plotted versus
temperature in Fig. 6a and the respective integrated peak areas of the Amide I, Amide II and
additionally the a(CH3) band of the isopropyl group (spectral data not shown) are shown
versus temperature in Fig. 6b. Amide I and II band positions of PNIPAAM are diagnostic of
the type and degree of hydrogen bonding, which is described for PNIPAAM solutions in
references [14]
and [15]
. For temperatures T < LCST (lower critical solution temperature), where

11
11
PNIPAAM is claimed to be in the coiled (soluble) state, the Amide I band position is lower
and the Amide II band position higher than for T > LCST, where PNIPAAM is claimed to be
in a globular (less soluble) state. Lower Amide I band positions are due to hydrogen bonding
between the amide C=O and water (C=O …
H …
O-H), weakening the C=O double bond
character and decreasing its force constant. Higher Amide II band positions are due to
hydrogen bonding between amide N-H and water (N-H …
O-H2) restricting the (NH)
bending mode and increasing its force constant. Obviously, Amide I band positions shift
continuously with increasing T over the entire T range (Fig. 6a, top), while Amide I and II
intensities were rather invariant from T = 22 - 32°C, but step-like increases were found for T
> 32°C (Fig. 6a, bottom). These step-like intensity changes of Amide I and Amide II bands
are due to a sudden increase of the PNIPAAM segment concentration in the brush for T >
LCST, in contrast to the rather continuous hydrogen bonding changes. Therefore, we suggest
that a step-like coil-to-globule transition of PNIPAAM brushes takes place when the
continuous hydrogen bonding changes have reached a certain critical point at T = LCST.
Fig. 6a. Wavenumber positions of the Amide I
and II bands from the ATR-FTIR spectra in Fig.
5b plotted as a function of T. (from [13]
with
kind permission of ACS).
Fig. 6b. Integrated peak intensities of
Amide I, Amide II and a(CH3) bands
from the ATR-FTIR spectra given in Fig.
5b as a function of T. (from [13]
with kind
permission of ACS).
Alternatively, Amide I and Amide II band shifts can be analyzed by lineshape analysis (LSA).
Fig. 7 compares LSA results for spectra obtained from PNIPAAM solutions by Maeda [15]
(Fig. 7a) and our own LSA results on ATR-FTIR spectra of PNIPAAM brush films (Fig. 7b).

12
12
Fig. 7a. Results of line shape analysis on
FTIR spectra of PNIPAAM solution in D2O
at T < LCST (“Tp”) and T > LCST (“Tp”).
Solid lines correspond to original spectral
data and fitted components, dashed ones to
simulated ones. (from [15]
with kind
permission of Wiley)
Fig. 7b. Results of line shape analysis on
ATR-FTIR spectra of PNIPAAM brush
(grafted-to) films at T < LCST (22°C, top)
and T > LCST (40°C, bottom) in contact
with H2O. Original spectra are indicated by
solid lines, fitted components and simulated
spectra by dashed lines.
The peak centered at 1625/1627 cm-1
is assigned to external (segment/water) hydrogen
bonding, while the peak centered at 1650/1652 cm-1
is due to internal segment/segment
hydrogen bonding. For the spectrum of PNIPAAM in D2O solution at T < LCST, only one
Amide I peak at around 1625 cm-1
is required to fit the data, while for T > LCST two peaks
centered at 1650, 1625 cm-1
(D2O) are required. In contrast, in ATR-FTIR spectra from
PNIPAAM brush films in H2O, two peaks at 1652 and 1627 cm-1
are required for T both
below and above LCST. This shows clearly, that for T < LCST in the solution state,
PNIPAAM is fully dissolved and in a coiled conformation, while for T > LCST PNIPAAM is
less soluble and in a globular state as expected. However, since for the brush-state films two
components are required for both T < LCST and T > LCST, we conclude that in the brush
state the attached PNIPAAM chains are never in a fully dissolved or coiled state, but always
contain globular portions, a larger concentration at T > LCST and a lower at T < LCST.

13
13
1.3.Layer-by-layer deposition of polyelectrolyte complex films
In this section, we review in-situ ATR-FTIR data on polyelectrolyte multilayers (PEM),
which are deposited by the layer-by-layer (LbL) technique reported by Decher.[16]
ATR-FTIR
spectroscopy of PEM films provides information concerning deposition mechanism,[17]
composition,[18]
and orientation.[19]
Here, ATR-FTIR data on LbL deposited
poly(ethyleneimine)/alginate (PEI/ALG) multilayers on silicon substrates [20]
are reviewed.
Fig. 8a. shows in-situ ATR-FTIR spectra over the range 1800-1300 cm-1
for PEI/ALG
multilayers for odd layer numbers: 1, 3, 5 … 17.[20]
Fig. 8a. in-situ ATR-FTIR spectra of PEM-1
to PEM-17 of PEI/ALG multilayers
deposited from 0.005 M PEL solutions on
silicon in contact with water (rinsing). (from [20]
with kind permission of Wiley)
Fig. 8b. aCOO-) integrated peak intensities
(ALG) from in-situ ATR-FTIR spectra of
Fig. 8a and corresponding thicknesses (SFM,
dry state) of PEM of PEI/ALG as a function
of adsorption step z. (from [20]
with kind
permission of Wiley)
Thus, spectra were obtained from the first adsorbed PEI layer (“PEM-1”); the next adsorbed
PEI/ALG/PEI layer (PEM-3) in the multilayer stack, up to PEM-17 were acquired and
compared. The a(COO-) band at 1593 cm
-1 and s(COO
-) band at 1410 cm
-1 of ALG (see Fig.
8a) were used as diagnostic markers for the PEM deposition process. In Fig. 8b, the
respective integrals of the a(COO-) band are plotted versus the adsorption step z = 1, 3, 5 …
17 (cube symbols). ATR-FTIR spectroscopy is characterized by an exponential decay of
sensitivity for processes taken place at larger distances from the ATR crystal surface. Thus,
for film growth processes, there is a certain film thickness at which the evanescent wave is no
0
50
100
150
200
0
2
4
6
8
10
0 2 4 6 8 10 12 14 16 18
Th
ick
ne
ss
/ [
nm
]
(C
OO
- )-I
nte
gra
l / [c
m-1
]
Adsorption step z

14
14
longer sensitive to further deposition. For large thicknesses, the integrated peak intensity of,
for example, a(COO-) versus the adsorption step reaches a plateau following a damped
exponential curve (1 – exp(-d/dP)), where d and dP are the geometric thickness and the depth
of penetration. However, for small thicknesses d < 200 nm, ATR-FTIR spectroscopy senses
film growth in a linear fashion such that the measured band integral A is approximately
proportional to d. In Fig. 8b, an exponential increase is observed for the a(COO-) band. That
is, the polysaccharide band integral was found to scale exponentially with the adsorption step
according to a function of type A = A0 exp(a z), in which a is a growth parameter and z is the
adsorption step number. The fit is also shown in Fig. 8b (black line).
The geometric film thickness of PEI/ALG-17 was measured by SFM (see Experimental
Details). In Fig. 8b these thicknesses of PEM-PEI/ALG (dry state) are also plotted as round
red symbols versus z. An exponential increase of d with increasing z and, for PEM-17,
d = 180 nm was obtained. Thus, our ATR-FTIR data varied approximately linearly with the
SFM data and therefore the exponential growth found by ATR-FTIR is confirmed.
Exponential growth of PEM has been observed for various PEM types by Picart[21]
and
Garza[22]
and they explained this behaviour based on high PEL diffusivity and a 3-zone-
model, especially when weakly-charged PEL or higher salinity was used. According to a
recent compilation of this model,[20]
zone I is the initial zone of the first few layers situated
directly above the substrate and zone III is the diffusion zone, in which after build up of
zone I, PEM grows exponentially. In zone III, the supplied PELs can “diffuse in and out” to
compensate charges at the surface by complexation. All PEL can participate in complexation
(“Reservoir Effect”) and thickness depends on the thickness increment. Zone II between I and
III is the so called “restructuration zone” in which PEL can no longer diffuse. It is formed
when new sorbed PELs on top of zone III cause a release of PELs at the bottom of zone III
into zone II (at the interface II/III).[20]
2. Applications of selected polymer films
Application examples of the functional coatings analyzed in Section 1 are considered for
binding/repulsion of proteins (2.1) and the controlled release of drugs (2.2). In addition, the
potential of in-situ-ATR-FTIR spectroscopy as a screening tool to optimize protein interaction
or drug formulation is discussed.

15
15
2.1. Protein interaction of polymer coatings
All polymer coatings described in Section 1 have potential for either bioinert or bioactive
functionalization of biomedical materials or surfaces. Using ATR-FTIR spectroscopy results
concerning protein composition and bound amount,[9]
location of the adsorbed proteins[23]
and
secondary structure[24]
are possible based on the integrated areas of the diagnostic Amide I
and Amide II bands. Example results are presented for the adsorption of the model protein
human serum albumin (HSA, isoelectrical point IEP = 4.7).
In Fig. 9a, HSA adsorption on PDA films is shown in comparison to adsorption on the bare
Ge substrate. The maximum Amide II band integral recorded for HSA adsorption on Ge was
taken as 100%. Significantly, HSA adsorption on PDA decreased by approximately 50%.
Fig. 9a. Relative adsorbed
amounts of HSA versus time
at bare Ge and at PDA film.
Fig. 9b. Relative adsorbed
amounts of HSA versus time
at bare Si and at PNIPAAM
(grafted-from) brush films for
T = 25°C and T = 40°C.
Fig. 9c. Relative adsorbed
amounts of HSA versus time
at PEM-4 and PEM-5 of
PEI/PAC and at PEM-8 and
PEM-9 of PEI/ALG.
HSA adsorption on PNIPAAM brush films was compared to that on Si substrates at both
temperatures T of 25 °C and 40 °C. As shown in Fig. 9b, PNIPAAM brush films exhibit
remarkably reduced HSA adsorption compared to bare Si substrate at both temperatures. In
these series on HSA adsorption, PNIPAAM brush films prepared by the grafting-from
concept (see Experimental Details) were used, which showed similar thermotropic behaviour
compared to those prepared by the grafting-to concept. While a slight effect of temperature
was observed for bare Si (higher HSA adsorption at T = 40°C), no such effect was seen on
PNIPAAM films. This effect was interpreted in terms of the strongly bound water on
PNIPAAM films, which prevents further attractive interactions between proteins and
PNIPAAM. This hydration shell is so strong, that it is still intact even at T = 40 °C, although

16
16
it is known, that at this temperature PNIPAAM brush films lose considerable amounts of
water.[13]
The significant influence of the outermost PEL layer on the adsorbed HSA concentration is
illustrated in Fig. 9c, where the bound HSA amount at PEM terminated by polycations like
PEI (i.e., odd-numbered PEM layers) is compared to PEM terminated by polyanions such as
ALG or poly(acrylic acid) (PAC). Significantly, at PEM-8 of PEI/ALG and PEM-4 of
PEI/PAC, less HSA is adsorbed compared to PEM-9 of PEI/ALG and PEM-5 of PEI/PAC.
This can be explained by the anionic nature of HSA (isoelectric point IEP = 4.8) being bound
under either electrostatic attraction (odd PEM) or repulsion (even PEM). PAC seems to be
even more effective than ALG. Presumably, polysaccharidic ALG undergoes additional
interaction forces like H-bonding or van der Waals towards proteins.
3.2.2. Drug interactions on PEM films
Recently, PEM systems based on PEI and the polyanions ALG or PAC loaded with the
antiobiotic streptomycin (STRP) were reported.[25]
These model studies are relevant for the
functionalization of bone-substituting material by interfacial drug-eluting polyelectrolyte-
based material. PEM build-up, and STRP loading and release, was monitored using in-situ-
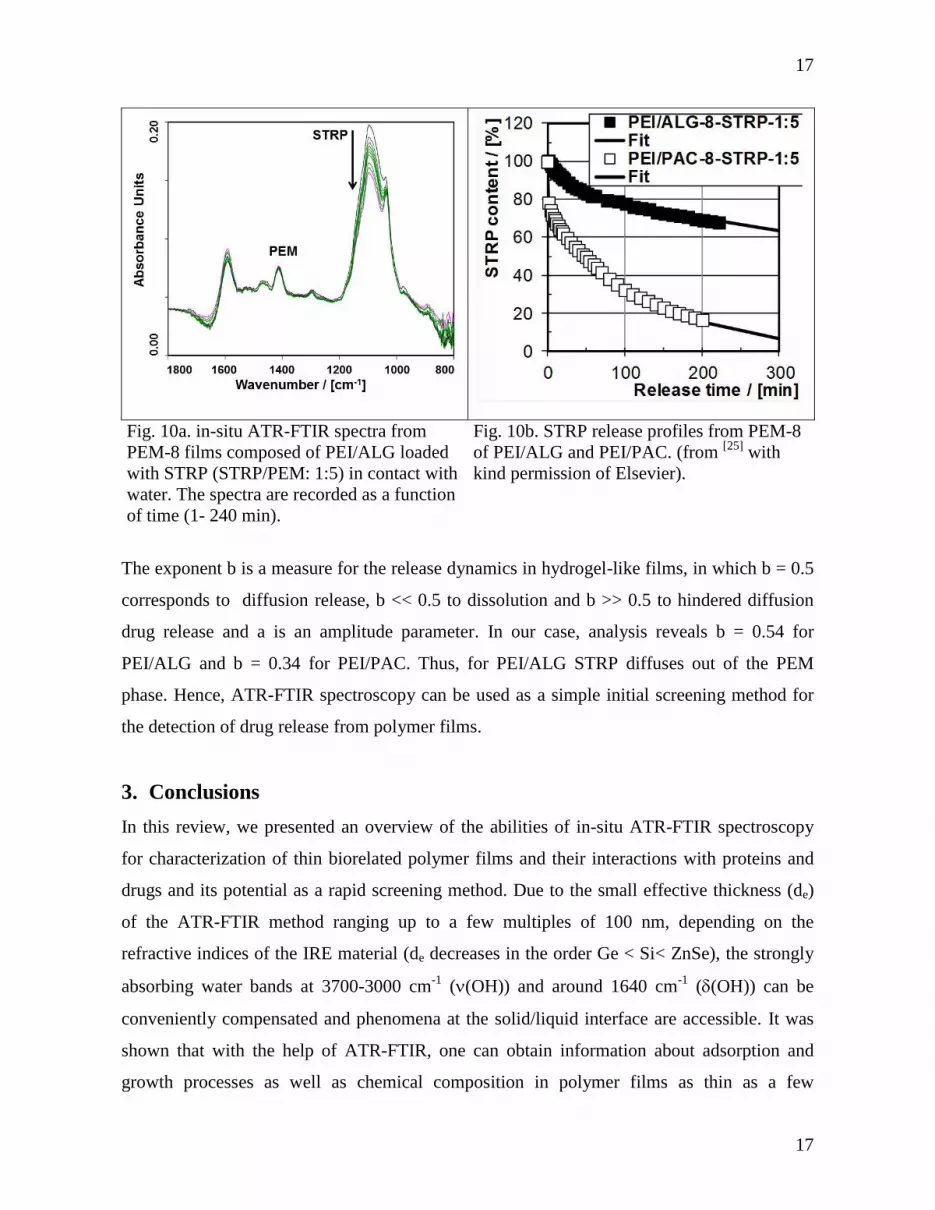
ATR-FTIR spectroscopy. Typical ATR-FTIR spectra of a PEM-PEI/ALG-8 loaded with
STRP (STRP/PEM: 1:5) in contact with water are shown in Fig. 10a. The (C-O) band of
STRP at approximately 1100 cm-1
can be used as a diagnostic marker for the STRP content
and the s(COO-) band at 1400 cm
-1 for the PEM film. The ratio between the former and the
latter was used to determine the percentage content of STRP (relative to the initial content).[25]
Fig. 10b shows release curves for PEI/ALG in comparison to PEI/PAC. Interestingly, a
smaller initial burst (IB, percentage of STRP released after 1 min) IB = 2% for PEI/ALG
compared to IB = 22% for PEI/PAC was obtained and also the residual amount (RA) of STRP
after 200 min was RA = 70 % for PEI/ALG and RA = 17% for PEI/PAC.
Since STRP has a tetrasaccharidic structure, we assume that it interacts stronger to the
polysaccharide-based ALG compared to synthetic PAC. Moreover, in Fig. 10b fits of the
ATR-FTIR release data to a simple Ritger/Peppas model[26]
according to A(t) = 100 - a tb were
performed, where A(t) is the integrated peak intensity of the (C-O) band.
17
17
Fig. 10a. in-situ ATR-FTIR spectra from
PEM-8 films composed of PEI/ALG loaded
with STRP (STRP/PEM: 1:5) in contact with
water. The spectra are recorded as a function
of time (1- 240 min).
Fig. 10b. STRP release profiles from PEM-8
of PEI/ALG and PEI/PAC. (from [25]
with
kind permission of Elsevier).
The exponent b is a measure for the release dynamics in hydrogel-like films, in which b = 0.5
corresponds to diffusion release, b << 0.5 to dissolution and b >> 0.5 to hindered diffusion
drug release and a is an amplitude parameter. In our case, analysis reveals b = 0.54 for
PEI/ALG and b = 0.34 for PEI/PAC. Thus, for PEI/ALG STRP diffuses out of the PEM
phase. Hence, ATR-FTIR spectroscopy can be used as a simple initial screening method for
the detection of drug release from polymer films.
3. Conclusions
In this review, we presented an overview of the abilities of in-situ ATR-FTIR spectroscopy
for characterization of thin biorelated polymer films and their interactions with proteins and
drugs and its potential as a rapid screening method. Due to the small effective thickness (de)
of the ATR-FTIR method ranging up to a few multiples of 100 nm, depending on the
refractive indices of the IRE material (de decreases in the order Ge < Si< ZnSe), the strongly
absorbing water bands at 3700-3000 cm-1
((OH)) and around 1640 cm-1
((OH)) can be
conveniently compensated and phenomena at the solid/liquid interface are accessible. It was
shown that with the help of ATR-FTIR, one can obtain information about adsorption and
growth processes as well as chemical composition in polymer films as thin as a few

18
18
nanometers. As FTIR spectroscopy in general is sensitive to internal or external hydrogen
bonding, especially to the carbonyl oxygen or the amide nitrogen, the intramolecular
interaction between related polymer segments or the intermolecular one to water in thin films
can be nicely studied by surface sensitive ATR-FTIR. These analytical options were
demonstrated in the examples of (i) reactive deposition of dopamine at inorganic model
surfaces, (ii) switching and adsorption of proteins on thermosensitive polymer brushes of
poly(N-isopropylacrylamide) (PNIPAAM) as well as (iii) adsorption of drugs to biorelated
polyelectrolyte multilayer (PEM). Sensitivity to polypeptide and protein conformation based
on diagnostic positions of the Amide I and Amide II band[27, 28]
is a further classical analytical
option for the FTIR method, which however is a topic of its own and was not treated herein.
Finally, limitations of the ATR-FTIR method should also be mentioned. These are mainly
related to the nonlinear relationship (damped exponential) between measured absorbance and
thickness d of thicker polymer films (d >> 300 nm). While for thin films (d < 300 nm), this
relationship can be approximated by a linear function (A(d) = k d) and processes at the
outermost zone of thin films are still observable by ATR-FTIR, for increasingly thick films,
the ATR-FTIR method becomes increasingly insensitive to processes occurring at the
outermost zone. Furthermore, by ATR-FTIR, from the measured absorbance A there is not a
direct access to the absolute value of the geometrical thickness d as it is possible by
ellipsometry, which however, is rather insensitive to the chemical composition. Thus, there is
a possibility based on the combination of ATR-FTIR and transmission-FTIR to access even
absolute values of the thickness of a polymer or organic film within a certain error range,
which will be the subject of a forthcoming paper.
Acknowlegements
Part of this work is related to the Transregio SFB TRR 79 (M7) of German Research
Foundation (DFG) "Materials for tissue regeneration in the systemically diseased bone"
(University Giessen, University Heidelberg, Technical University Dresden, and cooperating
research institutes) as well as DFG Grant IO 68/1-1.

19
19
References
1. M.Q. Zhang, T. Desai, M. Ferrari, Biomaterials 19 (10) (1998) 953-960.
2. Z.W. Mao, L. Ma, J. Zhou et al., Bioconjugate Chemistry 16 (5) (2005) 1316-1322.
3. A.N. Zelikin, ACS Nano 4 (5) 2010 2494-2509.
4. U.P. Fringeli, in Internal Reflection Spectroscopy: Theory and Applications,
F.M.Mirabella ed., M.Dekker, N.Y. (1992)
5. M. Müller, B. Keßler, Langmuir 27 (2011) 12499.
6. S urkert, E ittrich, M untzsch, M M ller, Eichhorn, P hlmann, M Stamm,
Langmuir 26 (2010) 1786−1795
7. S. Minko, A. Sidorenko, M. Stamm, G. Gafijchuk, V. Senkovsky, S. Voronov,
Macromolecules 32(14) (1999) 4532-4538.
8. J. Wang und K. Matyjaszewski, J. Amer. Chem. Soc. 117 (1995) 5614–5615.
9. M. Müller, B. Keßler, N. Houbenov, K. Bohata, Z. Pientka, E. Brynda,
Biomacromolecules 7 (4) (2006) 1285-1294.
10. N.J. Harrick, in Internal Reflection Spectroscopy : Review and Supplement, F.M.
Mirabella, N.J. Harrick eds, Harrick Scientific Corporation, Ossining, NY (1985)
11. H. Lee, S.M. Dellatore, W.M. Miller, P.B. Messersmith, Science 318 (2007) 426-430.
12. S.H. Ku, J.S. Lee, C.B. Park, Langmuir 26 (2010) 15104–15108.
13. E. Bittrich, S. Burkert, M. Müller, K.J. Eichhorn, M. Stamm, P. Uhlmann, Langmuir 28
(7) (2012) 3439–3448.
14. Y. Maeda, T. Higuchi, I. Ikeda, Langmuir 16 (2000) 7503.
15. Y. Maeda, Macromol. Symp. 303 (2011) 63-70.
16. G. Decher, Science 277 (1997) 1232.

20
20
17. M. Müller, S. Paulik, Macromol. Symp. 265 (2008) 77-88.
18. M. Müller, M. Briššová, T. Rieser, A.C. Powers ; K. Lunkwitz, Materials Science &
Engineering C 8-9 (1999) 167-173.
19. M. Müller, W. Ouyang, B. Keßler, Spectrochimica Acta A 77 (2010) 709.
20. M. Müller, B. Torger, B. Keßler, Advanced Biomaterials 12 (12) (2010) 676.
21. C. Picart, P. Lavalle, P. Hubert, F.J.G. Cuisinier,G . Decher, P. Schaaf and J.C. Voegel,
Langmuir 17 (2001) 7414-7424.
22. J.M. Garza, P. Schaaf, S. Muller, V. Ball, J.F. Stoltz, J.C. Voegel and P. Lavalle,
Langmuir 20 (2004) 7298-7302.
23. M. Müller, B. Keßler, W. Ouyang, Z. Phys. Chem. 221 (2007) 127-138.
24. H.H. Bauer, M. Müller, J. Goette, H.P. Merkle and U.P. Fringeli, Biochemistry 33 (1994)
12276-12282.
25. B. Torger, M. Müller, Spectrochim. Acta A 104 (2013) 546-553.
26. P.L. Ritger, N.A. Peppas, J. Controlled. Release 5 (1987) 23–36.
27. T. Miyazawa, E.R. Blout, J. Am. Chem. Soc. 83 (1960) 712.
28. S. Krimm, J. Mol. Biol. 4 (1962) 528-540.





























