Embed Size (px)
Citation preview

Clinical Neurophysiology 124 (2013) 2079–2090
Contents lists available at SciVerse ScienceDirect
Clinical Neurophysiology
journal homepage: www.elsevier .com/locate /c l inph
Review
Mechanisms of axonal dysfunction in diabetic and uraemic neuropathies
1388-2457/$36.00 � 2013 International Federation of Clinical Neurophysiology. Published by Elsevier Ireland Ltd. All rights reserved.http://dx.doi.org/10.1016/j.clinph.2013.04.012
⇑ Corresponding author Address: Translational Neuroscience Facility, School ofMedical Sciences, University of New South Wales, Sydney, NSW 2052, Australia.Tel.: +61 2 93852756; fax: +61 2 93851485.
E-mail address: [email protected] (A.V. Krishnan).
Ria Arnold, Natalie C.G. Kwai, Arun V. Krishnan ⇑Translational Neuroscience Facility, School of Medical Sciences, University of New South Wales, Sydney, Australia
a r t i c l e i n f o h i g h l i g h t s
Article history:Accepted 13 April 2013Available online 15 May 2013
Keywords:Diabetic neuropathyDiabetesUraemic neuropathyChronic kidney diseaseDialysisNerve excitability
� Nerve excitability techniques have provided important insights into the mechanisms underlying axo-nal dysfunction in diabetic and uraemic neuropathy.� Excitability studies in diabetes have suggested that axonal ion channel dysfunction may contribute to
the development of neuropathic symptoms.� Membrane depolarization due to hyperkalemia may underlie the development of uraemic
neuropathy.
a b s t r a c t
The global burden imposed by metabolic diseases and associated complications continue to escalate.Neurological complications, most commonly peripheral neuropathy, represent a significant cause of mor-bidity and disability in patients with diabetes and chronic kidney disease. Furthermore, health care costsare substantially increased by the presence of complications making investigation into treatment a mat-ter of high priority. Over the last decade nerve excitability techniques have entered the clinical realm andenabled in vivo assessment of biophysical properties and function of peripheral nerves in health and dis-ease. Studies of excitability in diabetic neuropathy have demonstrated alteration in biophysical proper-ties, including changes in Na+ conductances and Na+/K+ pump function, which may contribute to thedevelopment of neuropathic symptoms. Interventional studies have demonstrated that these changesare responsive to pharmacological agents. Excitability studies in patients with chronic kidney diseasehave demonstrated prominent changes that may contribute to the development of uraemic neuropathy.In particular, these studies have demonstrated strong correlation between hyperkalaemia and the devel-opment of nerve dysfunction. These studies have provided a basis for future work assessing the benefitsof potassium restriction as a therapeutic strategy in this condition.� 2013 International Federation of Clinical Neurophysiology. Published by Elsevier Ireland Ltd. All rights
reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2080
1.1. Nerve excitability assessment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20812. Diabetic neuropathy. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2081
2.1. Biophysical abnormalities in animal models of diabetic neuropathy. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20822.2. Altered Na+ conductances in diabetic neuropathy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20822.3. Altered Na+/K+ pump function . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20832.4. Relationship to neuropathic symptoms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20842.5. Nerve excitability abnormalities prior to diabetic neuropathy onset . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20843. Uraemic neuropathy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2085
3.1. Pathophysiology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2086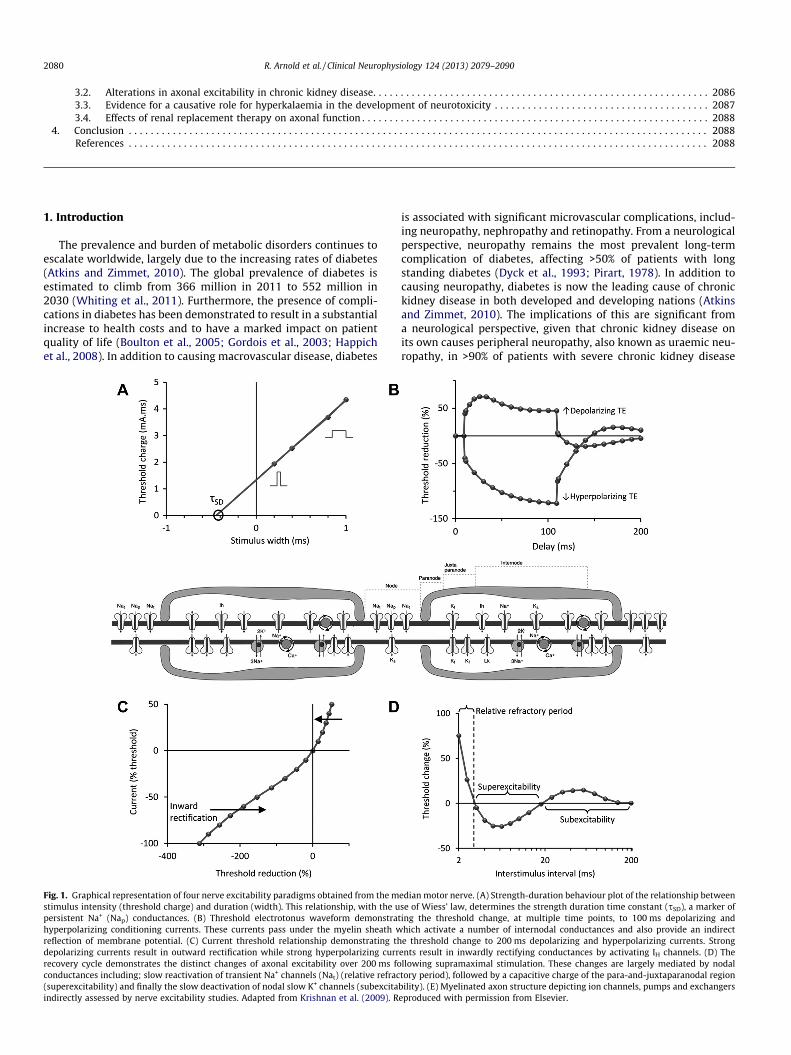
Fig. 1.stimulupersistehyperpreflectidepolarrecoverconduc(supereindirec
2080 R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090
3.2. Alterations in axonal excitability in chronic kidney disease. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20863.3. Evidence for a causative role for hyperkalaemia in the development of neurotoxicity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20873.4. Effects of renal replacement therapy on axonal function . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2088
4. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2088References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2088
1. Introduction
The prevalence and burden of metabolic disorders continues toescalate worldwide, largely due to the increasing rates of diabetes(Atkins and Zimmet, 2010). The global prevalence of diabetes isestimated to climb from 366 million in 2011 to 552 million in2030 (Whiting et al., 2011). Furthermore, the presence of compli-cations in diabetes has been demonstrated to result in a substantialincrease to health costs and to have a marked impact on patientquality of life (Boulton et al., 2005; Gordois et al., 2003; Happichet al., 2008). In addition to causing macrovascular disease, diabetes
Graphical representation of four nerve excitability paradigms obtained from the ms intensity (threshold charge) and duration (width). This relationship, with the unt Na+ (Nap) conductances. (B) Threshold electrotonus waveform demonstra
olarizing conditioning currents. These currents pass under the myelin sheath won of membrane potential. (C) Current threshold relationship demonstrating thizing currents result in outward rectification while strong hyperpolarizing curry cycle demonstrates the distinct changes of axonal excitability over 200 ms fotances including; slow reactivation of transient Na+ channels (Nat) (relative refracxcitability) and finally the slow deactivation of nodal slow K+ channels (subexcita
tly assessed by nerve excitability studies. Adapted from Krishnan et al. (2009). R
is associated with significant microvascular complications, includ-ing neuropathy, nephropathy and retinopathy. From a neurologicalperspective, neuropathy remains the most prevalent long-termcomplication of diabetes, affecting >50% of patients with longstanding diabetes (Dyck et al., 1993; Pirart, 1978). In addition tocausing neuropathy, diabetes is now the leading cause of chronickidney disease in both developed and developing nations (Atkinsand Zimmet, 2010). The implications of this are significant froma neurological perspective, given that chronic kidney disease onits own causes peripheral neuropathy, also known as uraemic neu-ropathy, in >90% of patients with severe chronic kidney disease
edian motor nerve. (A) Strength-duration behaviour plot of the relationship betweense of Wiess’ law, determines the strength duration time constant (sSD), a marker of
ting the threshold change, at multiple time points, to 100 ms depolarizing andhich activate a number of internodal conductances and also provide an indirecte threshold change to 200 ms depolarizing and hyperpolarizing currents. Strong
ents result in inwardly rectifying conductances by activating IH channels. (D) Thellowing supramaximal stimulation. These changes are largely mediated by nodaltory period), followed by a capacitive charge of the para-and-juxtaparanodal regionbility). (E) Myelinated axon structure depicting ion channels, pumps and exchangerseproduced with permission from Elsevier.

R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090 2081
(Laaksonen et al., 2002; Tilki et al., 2009; Van den Neucker et al.,1998).
Metabolic polyneuropathies due to diabetes and chronic kidneydisease share similar clinical features, typically manifesting as aprogressive, symmetrical, length dependent neuropathy that com-mences in lower limb nerves. In its initial stages, patients presentwith sensory symptoms with motor involvement occurring inmore severe cases (Krishnan et al., 2009b). Consequently, earlysymptoms include pain, paraesthesia and numbness in the distallower limbs, and clinical examination reveals loss of distal sensoryfunction and a reduction in ankle deep tendon reflexes. With dis-ease progression distal motor involvement may develop resultingin muscle atrophy and weakness.
Neurophysiological techniques have provided important in-sights into the mechanisms that underlie nerve dysfunction inmetabolic neuropathies. Early neurophysiological studies utilisednerve conduction methods to assess important nerve parameters,including action potential amplitude and latency. While thesemethods continue to provide important diagnostic informationregarding neuropathy, they provide only limited informationregarding pathophysiological mechanisms that may underlie thedevelopment of axonal dysfunction in these disorders.
Over the last decade, nerve excitability methods have beendeveloped as clinical techniques for the investigation of the patho-physiological processes involved in the development of peripheralneuropathy. These methods enable the indirect assessment of axo-nal membrane potential and the activity of ion channels, energydependent pumps and ion exchange processes involved in impulseconduction in peripheral nerve axons (Bostock et al., 1998; Krish-nan et al., 2009a). In recent years these techniques have enabledthe assessment of biophysical properties and function of peripheralnerves in health and disease (Krishnan et al., 2009a). While studiesof excitability have been undertaken in inflammatory, toxic, meta-bolic and degenerative disorders of peripheral nerve (Cheah et al.,2012; Krishnan et al., 2006b; Park et al., 2009), it may be arguedthat the greatest clinical mileage has been gained in the investiga-tion of metabolic neuropathies, particularly those relating to diabe-tes and chronic kidney disease. This review will focus on recentstudies of nerve excitability that have been undertaken in diabeticand uraemic neuropathies. The Review will address the potentialmechanisms of axonal dysfunction in these disorders and the clin-ical relevance of these findings in terms of the development of fu-ture diagnostic methods and novel treatments.
1.1. Nerve excitability assessment
The concept of nerve excitability assessment as a method ofinvestigating axonal function was pioneered by the work of Berg-mans, who demonstrated that changes in the ‘threshold’ or currentrequired to elicit a target response reflected physiological altera-tions of the axon (Bergmans, 1970). Bergmans discovered that bymeasuring changes in the threshold of axons induced by impulseactivity or artificial polarisation, considerable information aboutaxonal physiology could be inferred (Bergmans, 1970; Bostocket al., 1998). The recent development of automated excitabilitysoftware has enabled the rapid acquisition of multiple excitabilityparameters in a short period of time (Kiernan et al., 2000). Thesedevelopments have resulted in significant clinical translation, withstudies of excitability having been conducted in a range of neuro-logical disorders (Krishnan et al., 2009a).
Automated excitability protocols consist of four discrete testingparadigms; strength-duration time constant, threshold electroto-nus, the current-threshold relationship and the recovery cycle.
Strength-duration behaviour is calculated from the relationshipbetween the stimulus intensity and its duration (Fig. 1A). Thestrength duration properties of the nerve reflect the properties of
the nodal membrane (Mogyoros et al., 1996). Specifically,strength-duration time constant provides an assessment of nodalpersistent Na+ currents (Bostock and Rothwell, 1997). Thoughthese currents only contribute to �2.5% of the total Na+ current,these conductances have an important role in repetitive and spon-taneous activity, making this measurement a clinically relevantexcitability parameter (Kiernan et al., 2000). However, thestrength-duration time constant may also be influenced by passivemembrane properties and membrane potential; as such measure-ments of latent addition may help separate persistent Na+ currentfrom the passive nodal time constant (Nodera and Kaji, 2006). La-tent addition uses subthreshold polarising currents of short dura-tion to enable an in depth assessment of axonal strengthduration properties (Bostock and Rothwell, 1997).
Threshold electrotonus (TE) is assessed by measuring the changein threshold at multiple time points during and after prolonged(100 ms) subthreshold, depolarizing (TEd) and hyperpolarizing(TEh) conditioning currents (Fig. 1B). Although these currents arenot sufficient to produce an action potential, they enable a slowspread of current under the myelin sheath into the internode, con-sequently altering the membrane potential and activating a num-ber of internodal accommodative conductances (Baker et al.,1987; Bostock et al., 1998; Burke et al., 2001).
Current/threshold relationship (I/V) assesses the effects of longerduration polarising currents by measuring the change in thresholdfollowing polarising currents of 200 ms duration, ranging from+50% (depolarising) to �100% (hyperpolarising) of the uncondi-tioned threshold intensity (Fig 1C). This parameter is particularlyuseful for examining the rectifying properties of the axon (Kiernanet al., 2000). Strong depolarising currents result in outward rectifi-cation achieved by activation of fast and slow K+ channels (Kiernanand Bostock, 2000). Strong hyperpolarising currents result in acti-vation of inwardly rectifying (IH) conductances (Kiernan et al.,2000; Pape, 1996).
Recovery cycle (RC) measures the changes in threshold that oc-cur over 200 ms following a supramaximal stimulus (Fig. 1D).These parameters provide insights into the behaviour of nodaland juxtaparanodal Na+ and K+ channels. With short condition-ing-test intervals, a supramaximal stimulus causes a brief period(0.5–1 ms) of absolute refractoriness, during which the axon is com-pletely inexcitable (Hodgkin and Huxley, 1952). This period is fol-lowed by �3 ms of relative refractoriness while Na+ channelsrecover from inactivation. (Hodgkin and Huxley, 1952). During thistime the axon is less excitable and an action potential can only begenerated with greater than usual stimuli. Following the recoveryof Na+ channel inactivation, a passive capacitive charge stored inthe internodal membrane produces a depolarising afterpotential,effectively increasing membrane excitability (Barrett and Barrett,1982; Kiernan et al., 1996). This is referred to as superexcitabilityand peaks between 5–7 ms following impulse conduction. Finally,there is a second period of reduced excitability due to activation ofslow K+ channels at the node of Ranvier during depolarisation (Ba-ker et al., 1987; Schwarz et al., 1995). These channels are slow todeactivate and produce hyperpolarisation as the final stage of therecovery cycle known as subexcitability.
Current excitability software enables the acquisition of theseparameters from a single nerve in a ten minute period. This tech-nological advance has enabled detailed assessment of axonal func-tion in a clinical setting, thus providing a valuable tool for clinicaltranslation.
2. Diabetic neuropathy
The most common neurological complication of diabetes (DM)is the development of a distal symmetric polyneuropathy affecting

2082 R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090
�50% of patients at long-term follow-up (Dyck et al., 1993; Pirart,1978). Diabetic neuropathy has a profound negative impact on pa-tient morbidity and quality of life (Kwai et al., 2013), with the pres-ence of diabetic neuropathy accounting for 60–70% of diabetic footulceration and 50–75% of non-traumatic foot amputations (Gonz-alez and Oley, 2000). Patients typically present with symptomssuch as hyperalgesia, allodynia, paraesthesias, burning and numb-ness in distal regions of the lower limbs, with symptoms later pro-gressing proximally (Vileikyte et al., 2003). While a wide range oftherapeutic approaches have been proposed for the treatment ofdiabetic neuropathy, including antioxidants, aldose reductaseinhibitors and nerve growth factors (Habib and Brannagan,2010), clinical results have been disappointing. One of the majorhurdles that these clinical trials have faced is the absence of anobjective test that permits the detection of mild neuropathy (Viniket al., 2005). Standard techniques, such as nerve conduction studiesand quantitative sensory testing, are based on the detection of ax-onal loss, which renders them unsuitable for early diagnosis and,consequently, for prevention of disability (Vinik et al., 2005). As aresult, diabetic neuropathy frequently remains undiagnosed untilthere is irreversible nerve injury, which may lead to foot infection,ulceration and even amputation (Manes and Papazolglou, 2002;Veves et al., 1993).
Pathologically, diabetic neuropathy is characterised by multifo-cal loss of myelinated and unmyelinated fibres, which is mostmarked in the distal regions of the lower limbs (Dyck et al.,1986; Johnson et al., 1986). Biopsy results demonstrate a varietyof abnormalities including axonal degeneration and regeneration,demyelination and remyelination, Schwann cell abnormalitiesand hypertrophy of the basal lamina (Behse et al., 1977; Dycket al., 1986; Johnson et al., 1986; Said, 2007). Studies of teased fibrepreparations have suggested that axonal degeneration may be theprimary abnormality in diabetic neuropathy, with demyelinationoccurring as a secondary or separate process (Behse et al., 1977;Dyck et al., 1986). Nerve conduction studies of diabetic neuropathydemonstrate reduction in sensory and motor amplitudes, that aremost marked in the lower limbs, consistent with a length-depen-dent axonal process (Said, 2007). Changes in nerve conductionvelocity often accompany amplitude loss as a result of demyelina-tion and dying back of large myelinated nerve fibres. Experimentalstudies also suggest nerve conduction slowing may be attributed tometabolic derangement rather than structural causes (Greeneet al., 1984).
2.1. Biophysical abnormalities in animal models of diabeticneuropathy
Animal models of diabetic neuropathy have demonstratedprominent changes in axonal function that may contribute to thedevelopment of both neuropathic symptoms and axonal loss (Cra-ner et al., 2002; Hong et al., 2004; Hong and Wiley, 2006). Thedevelopment of diabetic neuropathy is currently viewed as occur-ring in two separate phases, namely an initial reversible stage thatis responsive to intervention and a second stage of irreversiblenerve injury that is characterised by nerve conduction slowingand reduction in action potential amplitude (Brismar et al., 1987;Sugimoto et al., 2000).
During the reversible stage, there are prominent changes inaxonal biophysical properties that may be amenable to pharmaco-logical intervention (Kamiya et al., 2004). Of note, reduced functionof the energy-dependent axonal Na+/K+ pump has long been recog-nised as a potential contributor to the development of diabeticneuropathy (Graf et al., 1981; Greene and Lattimer, 1986; Greeneet al., 1984). Altered Na+/K+ pump activity may be due to C-peptidedeficiency (Sima et al., 2004; Wahren et al., 2000) or metabolicalterations resulting from hyperglycaemia. These include the
formation of advanced glycation end products (AGE) which initiateoxidative stress and upregulation of the polyol pathway, causingan increase in intra-axonal sorbitol levels and subsequent myoino-sitol depletion (Greene et al., 1988). These changes in turn result ina deleterious reduction in protein kinase C activation (Zhu andEichberg, 1990b) which is required for Na+/K+ pump function. Re-duced Na+/K+ pump activity has also been postulated to causestructural changes in nodal and paranodal regions of the axon,(Sima and Brismar, 1985). At the nodal region, there exists a highdensity of voltage gated Na+ channels which have been the focusof a number of studies in diabetic neuropathy. More recently, al-tered expression of voltage gated Na+ channels and associatedmRNA have been found in animal models of diabetic neuropathyand these results have suggested that Na+ channel dysregulationmay play a role in the development of neuropathic pain in diabeticneuropathy (Craner et al., 2002; Hong et al., 2004). Specifically,modification of Na+ channels by methylglyoxal, a reactive dicar-bonyl formed in the presence of hyperglycaemia (Thornalley,2005), and by phosphorylation have been linked to increased firingof nociceptive neurones and heightened pain states in diabeticmice models (Bierhaus et al., 2012; Hong et al., 2004).
2.2. Altered Na+ conductances in diabetic neuropathy
There have been numerous recent studies of nerve excitability,undertaken in both type 1 and type 2 diabetic patients, and thesehave provided further support for the role of biophysical changesas important contributors to the development of diabetic neurop-athy. Excitability studies in diabetic neuropathy have demon-strated significant changes in parameters of thresholdelectrotonus and the recovery cycle (Fig 2) (Kitano et al., 2004;Krishnan and Kiernan, 2005; Kwai et al., 2013; Misawa et al.,2006a; Sung et al., 2012). The first comprehensive study of axonalexcitability in diabetic neuropathy was undertaken by Kitano andcolleagues (2004) who assessed median motor nerve excitabilityin a cohort of 21 diabetic patients, comprised of patients with bothtype 1 and type 2 diabetes. Enrolled patients had poorly controlleddiabetes with a mean glycosylated haemoglobin of 10.9% (nor-mal < 6.5% (Gillett, 2009)). Baseline excitability studies demon-strated reductions in refractoriness, duration of the relativerefractory period (RRP) and strength-duration time constant, find-ings that were interpreted as evidence of reduced trans-axonal Na+
gradients. Patients were then re-assessed following a four weekperiod of intensive insulin treatment. Post-treatment excitabilitystudies demonstrated prominent improvements in Na+ channeldependent parameters, with an increase in refractoriness andstrength-duration time constant as well as improvement in stan-dard nerve conduction parameters including median nerve con-duction velocity and F wave latency. The authors concluded thathyperglycaemia was associated with a reduced trans-axonal Na+
gradient, which was subsequently restored with strict glycaemiccontrol. These conclusions were supported by further studies thatdemonstrated a significant relationship between hyperglycaemiaand the presence of excitability abnormalities (Misawa et al.,2005b). In a cohort of 79 patients, those with poor glycaemic con-trol (HbA1c% > 9) had significantly shorter strength-duration timeconstant values compared to patients with good glycaemic control(HbA1c% < 7%) (Misawa et al., 2005b). A similar reduction in Na+
conductances was suggested by a second study of 58 diabetic pa-tients, in which poor glycaemic control was associated with short-er relative refractory period (Misawa et al., 2004). The authorsinterpreted these findings as evidence that poor glycaemic controlmay reduce inward Na+ conductances.
The potential benefits of pharmacological intervention inrestoring Na+ channel conductances in diabetic neuropathy wasfurther emphasised in a study of 30 diabetic patients enrolled in
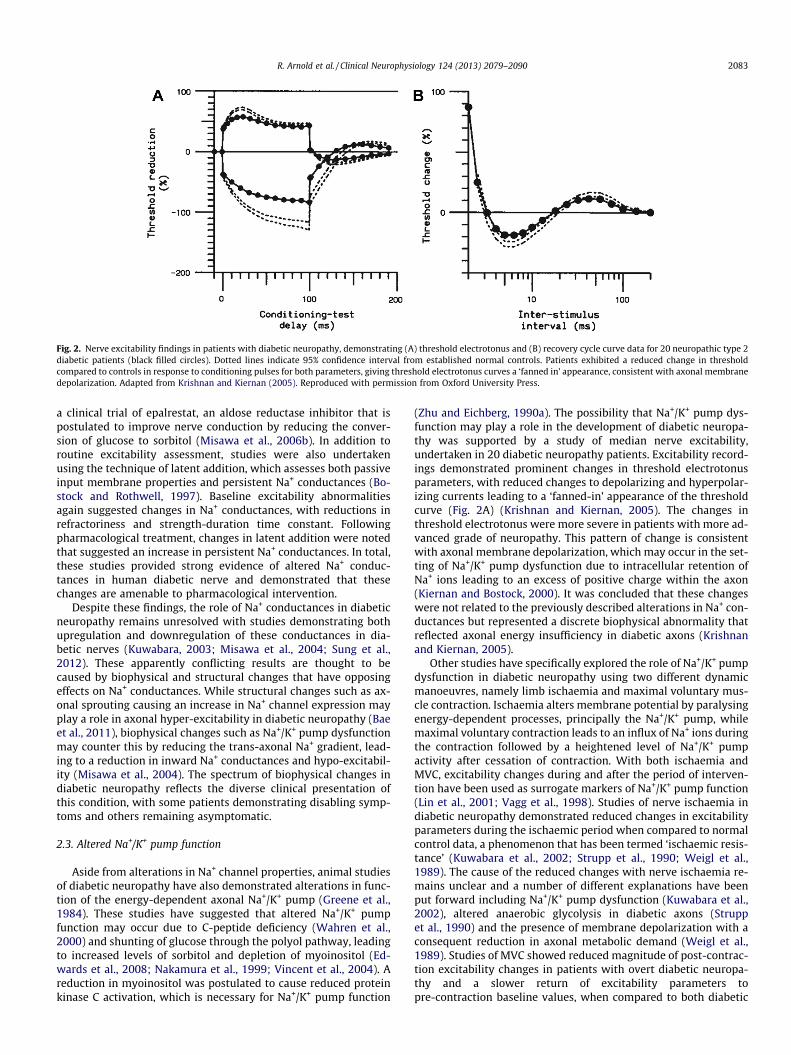
Fig. 2. Nerve excitability findings in patients with diabetic neuropathy, demonstrating (A) threshold electrotonus and (B) recovery cycle curve data for 20 neuropathic type 2diabetic patients (black filled circles). Dotted lines indicate 95% confidence interval from established normal controls. Patients exhibited a reduced change in thresholdcompared to controls in response to conditioning pulses for both parameters, giving threshold electrotonus curves a ‘fanned in’ appearance, consistent with axonal membranedepolarization. Adapted from Krishnan and Kiernan (2005). Reproduced with permission from Oxford University Press.
R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090 2083
a clinical trial of epalrestat, an aldose reductase inhibitor that ispostulated to improve nerve conduction by reducing the conver-sion of glucose to sorbitol (Misawa et al., 2006b). In addition toroutine excitability assessment, studies were also undertakenusing the technique of latent addition, which assesses both passiveinput membrane properties and persistent Na+ conductances (Bo-stock and Rothwell, 1997). Baseline excitability abnormalitiesagain suggested changes in Na+ conductances, with reductions inrefractoriness and strength-duration time constant. Followingpharmacological treatment, changes in latent addition were notedthat suggested an increase in persistent Na+ conductances. In total,these studies provided strong evidence of altered Na+ conduc-tances in human diabetic nerve and demonstrated that thesechanges are amenable to pharmacological intervention.
Despite these findings, the role of Na+ conductances in diabeticneuropathy remains unresolved with studies demonstrating bothupregulation and downregulation of these conductances in dia-betic nerves (Kuwabara, 2003; Misawa et al., 2004; Sung et al.,2012). These apparently conflicting results are thought to becaused by biophysical and structural changes that have opposingeffects on Na+ conductances. While structural changes such as ax-onal sprouting causing an increase in Na+ channel expression mayplay a role in axonal hyper-excitability in diabetic neuropathy (Baeet al., 2011), biophysical changes such as Na+/K+ pump dysfunctionmay counter this by reducing the trans-axonal Na+ gradient, lead-ing to a reduction in inward Na+ conductances and hypo-excitabil-ity (Misawa et al., 2004). The spectrum of biophysical changes indiabetic neuropathy reflects the diverse clinical presentation ofthis condition, with some patients demonstrating disabling symp-toms and others remaining asymptomatic.
2.3. Altered Na+/K+ pump function
Aside from alterations in Na+ channel properties, animal studiesof diabetic neuropathy have also demonstrated alterations in func-tion of the energy-dependent axonal Na+/K+ pump (Greene et al.,1984). These studies have suggested that altered Na+/K+ pumpfunction may occur due to C-peptide deficiency (Wahren et al.,2000) and shunting of glucose through the polyol pathway, leadingto increased levels of sorbitol and depletion of myoinositol (Ed-wards et al., 2008; Nakamura et al., 1999; Vincent et al., 2004). Areduction in myoinositol was postulated to cause reduced proteinkinase C activation, which is necessary for Na+/K+ pump function
(Zhu and Eichberg, 1990a). The possibility that Na+/K+ pump dys-function may play a role in the development of diabetic neuropa-thy was supported by a study of median nerve excitability,undertaken in 20 diabetic neuropathy patients. Excitability record-ings demonstrated prominent changes in threshold electrotonusparameters, with reduced changes to depolarizing and hyperpolar-izing currents leading to a ‘fanned-in’ appearance of the thresholdcurve (Fig. 2A) (Krishnan and Kiernan, 2005). The changes inthreshold electrotonus were more severe in patients with more ad-vanced grade of neuropathy. This pattern of change is consistentwith axonal membrane depolarization, which may occur in the set-ting of Na+/K+ pump dysfunction due to intracellular retention ofNa+ ions leading to an excess of positive charge within the axon(Kiernan and Bostock, 2000). It was concluded that these changeswere not related to the previously described alterations in Na+ con-ductances but represented a discrete biophysical abnormality thatreflected axonal energy insufficiency in diabetic axons (Krishnanand Kiernan, 2005).
Other studies have specifically explored the role of Na+/K+ pumpdysfunction in diabetic neuropathy using two different dynamicmanoeuvres, namely limb ischaemia and maximal voluntary mus-cle contraction. Ischaemia alters membrane potential by paralysingenergy-dependent processes, principally the Na+/K+ pump, whilemaximal voluntary contraction leads to an influx of Na+ ions duringthe contraction followed by a heightened level of Na+/K+ pumpactivity after cessation of contraction. With both ischaemia andMVC, excitability changes during and after the period of interven-tion have been used as surrogate markers of Na+/K+ pump function(Lin et al., 2001; Vagg et al., 1998). Studies of nerve ischaemia indiabetic neuropathy demonstrated reduced changes in excitabilityparameters during the ischaemic period when compared to normalcontrol data, a phenomenon that has been termed ‘ischaemic resis-tance’ (Kuwabara et al., 2002; Strupp et al., 1990; Weigl et al.,1989). The cause of the reduced changes with nerve ischaemia re-mains unclear and a number of different explanations have beenput forward including Na+/K+ pump dysfunction (Kuwabara et al.,2002), altered anaerobic glycolysis in diabetic axons (Struppet al., 1990) and the presence of membrane depolarization with aconsequent reduction in axonal metabolic demand (Weigl et al.,1989). Studies of MVC showed reduced magnitude of post-contrac-tion excitability changes in patients with overt diabetic neuropa-thy and a slower return of excitability parameters topre-contraction baseline values, when compared to both diabetic
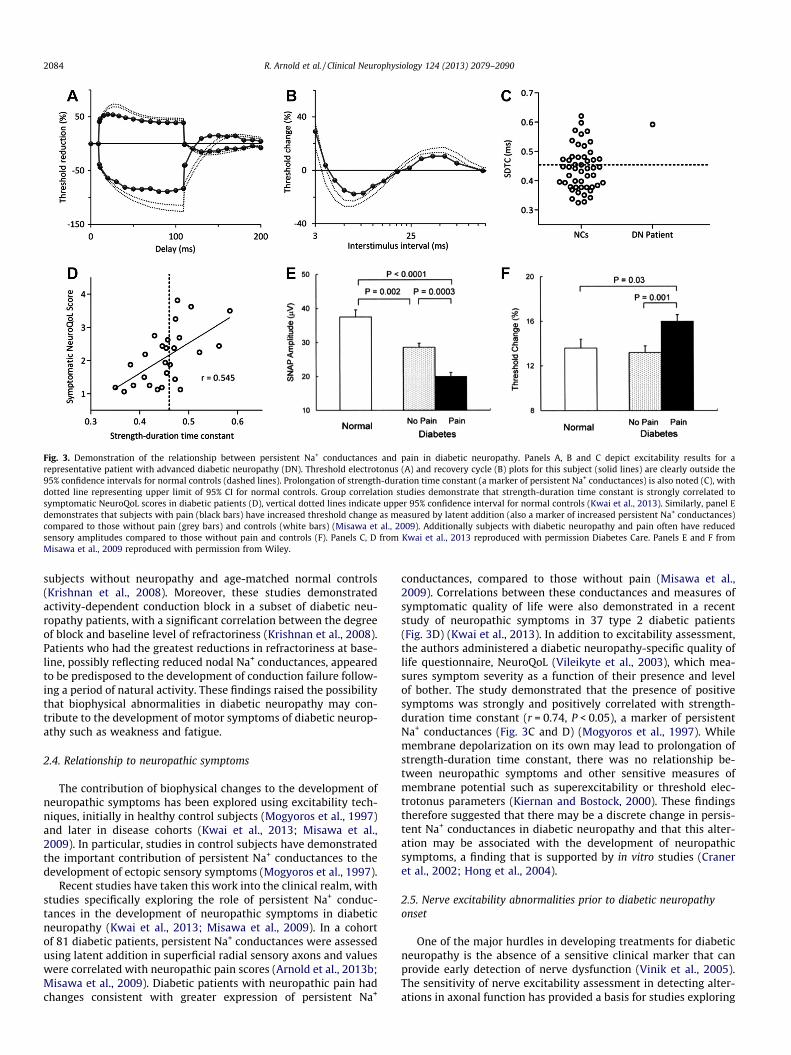
Fig. 3. Demonstration of the relationship between persistent Na+ conductances and pain in diabetic neuropathy. Panels A, B and C depict excitability results for arepresentative patient with advanced diabetic neuropathy (DN). Threshold electrotonus (A) and recovery cycle (B) plots for this subject (solid lines) are clearly outside the95% confidence intervals for normal controls (dashed lines). Prolongation of strength-duration time constant (a marker of persistent Na+ conductances) is also noted (C), withdotted line representing upper limit of 95% CI for normal controls. Group correlation studies demonstrate that strength-duration time constant is strongly correlated tosymptomatic NeuroQoL scores in diabetic patients (D), vertical dotted lines indicate upper 95% confidence interval for normal controls (Kwai et al., 2013). Similarly, panel Edemonstrates that subjects with pain (black bars) have increased threshold change as measured by latent addition (also a marker of increased persistent Na+ conductances)compared to those without pain (grey bars) and controls (white bars) (Misawa et al., 2009). Additionally subjects with diabetic neuropathy and pain often have reducedsensory amplitudes compared to those without pain and controls (F). Panels C, D from Kwai et al., 2013 reproduced with permission Diabetes Care. Panels E and F fromMisawa et al., 2009 reproduced with permission from Wiley.
2084 R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090
subjects without neuropathy and age-matched normal controls(Krishnan et al., 2008). Moreover, these studies demonstratedactivity-dependent conduction block in a subset of diabetic neu-ropathy patients, with a significant correlation between the degreeof block and baseline level of refractoriness (Krishnan et al., 2008).Patients who had the greatest reductions in refractoriness at base-line, possibly reflecting reduced nodal Na+ conductances, appearedto be predisposed to the development of conduction failure follow-ing a period of natural activity. These findings raised the possibilitythat biophysical abnormalities in diabetic neuropathy may con-tribute to the development of motor symptoms of diabetic neurop-athy such as weakness and fatigue.
2.4. Relationship to neuropathic symptoms
The contribution of biophysical changes to the development ofneuropathic symptoms has been explored using excitability tech-niques, initially in healthy control subjects (Mogyoros et al., 1997)and later in disease cohorts (Kwai et al., 2013; Misawa et al.,2009). In particular, studies in control subjects have demonstratedthe important contribution of persistent Na+ conductances to thedevelopment of ectopic sensory symptoms (Mogyoros et al., 1997).
Recent studies have taken this work into the clinical realm, withstudies specifically exploring the role of persistent Na+ conduc-tances in the development of neuropathic symptoms in diabeticneuropathy (Kwai et al., 2013; Misawa et al., 2009). In a cohortof 81 diabetic patients, persistent Na+ conductances were assessedusing latent addition in superficial radial sensory axons and valueswere correlated with neuropathic pain scores (Arnold et al., 2013b;Misawa et al., 2009). Diabetic patients with neuropathic pain hadchanges consistent with greater expression of persistent Na+
conductances, compared to those without pain (Misawa et al.,2009). Correlations between these conductances and measures ofsymptomatic quality of life were also demonstrated in a recentstudy of neuropathic symptoms in 37 type 2 diabetic patients(Fig. 3D) (Kwai et al., 2013). In addition to excitability assessment,the authors administered a diabetic neuropathy-specific quality oflife questionnaire, NeuroQoL (Vileikyte et al., 2003), which mea-sures symptom severity as a function of their presence and levelof bother. The study demonstrated that the presence of positivesymptoms was strongly and positively correlated with strength-duration time constant (r = 0.74, P < 0.05), a marker of persistentNa+ conductances (Fig. 3C and D) (Mogyoros et al., 1997). Whilemembrane depolarization on its own may lead to prolongation ofstrength-duration time constant, there was no relationship be-tween neuropathic symptoms and other sensitive measures ofmembrane potential such as superexcitability or threshold elec-trotonus parameters (Kiernan and Bostock, 2000). These findingstherefore suggested that there may be a discrete change in persis-tent Na+ conductances in diabetic neuropathy and that this alter-ation may be associated with the development of neuropathicsymptoms, a finding that is supported by in vitro studies (Craneret al., 2002; Hong et al., 2004).
2.5. Nerve excitability abnormalities prior to diabetic neuropathyonset
One of the major hurdles in developing treatments for diabeticneuropathy is the absence of a sensitive clinical marker that canprovide early detection of nerve dysfunction (Vinik et al., 2005).The sensitivity of nerve excitability assessment in detecting alter-ations in axonal function has provided a basis for studies exploring
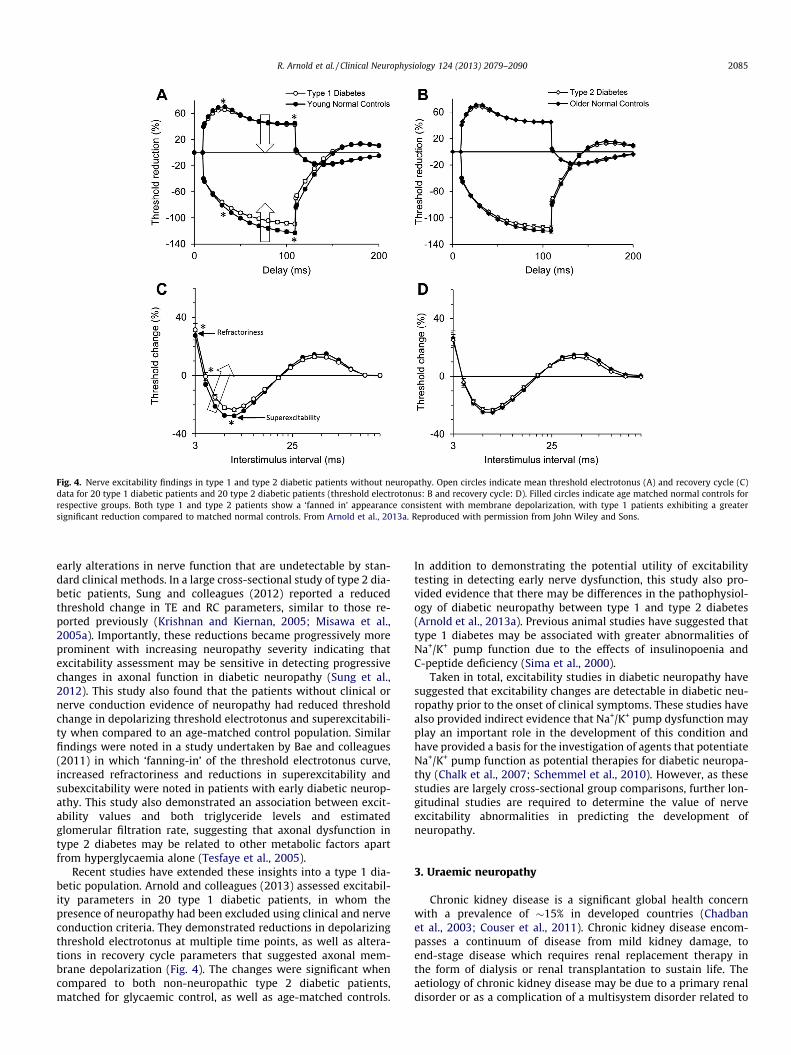
Fig. 4. Nerve excitability findings in type 1 and type 2 diabetic patients without neuropathy. Open circles indicate mean threshold electrotonus (A) and recovery cycle (C)data for 20 type 1 diabetic patients and 20 type 2 diabetic patients (threshold electrotonus: B and recovery cycle: D). Filled circles indicate age matched normal controls forrespective groups. Both type 1 and type 2 patients show a ‘fanned in’ appearance consistent with membrane depolarization, with type 1 patients exhibiting a greatersignificant reduction compared to matched normal controls. From Arnold et al., 2013a. Reproduced with permission from John Wiley and Sons.
R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090 2085
early alterations in nerve function that are undetectable by stan-dard clinical methods. In a large cross-sectional study of type 2 dia-betic patients, Sung and colleagues (2012) reported a reducedthreshold change in TE and RC parameters, similar to those re-ported previously (Krishnan and Kiernan, 2005; Misawa et al.,2005a). Importantly, these reductions became progressively moreprominent with increasing neuropathy severity indicating thatexcitability assessment may be sensitive in detecting progressivechanges in axonal function in diabetic neuropathy (Sung et al.,2012). This study also found that the patients without clinical ornerve conduction evidence of neuropathy had reduced thresholdchange in depolarizing threshold electrotonus and superexcitabili-ty when compared to an age-matched control population. Similarfindings were noted in a study undertaken by Bae and colleagues(2011) in which ‘fanning-in’ of the threshold electrotonus curve,increased refractoriness and reductions in superexcitability andsubexcitability were noted in patients with early diabetic neurop-athy. This study also demonstrated an association between excit-ability values and both triglyceride levels and estimatedglomerular filtration rate, suggesting that axonal dysfunction intype 2 diabetes may be related to other metabolic factors apartfrom hyperglycaemia alone (Tesfaye et al., 2005).
Recent studies have extended these insights into a type 1 dia-betic population. Arnold and colleagues (2013) assessed excitabil-ity parameters in 20 type 1 diabetic patients, in whom thepresence of neuropathy had been excluded using clinical and nerveconduction criteria. They demonstrated reductions in depolarizingthreshold electrotonus at multiple time points, as well as altera-tions in recovery cycle parameters that suggested axonal mem-brane depolarization (Fig. 4). The changes were significant whencompared to both non-neuropathic type 2 diabetic patients,matched for glycaemic control, as well as age-matched controls.
In addition to demonstrating the potential utility of excitabilitytesting in detecting early nerve dysfunction, this study also pro-vided evidence that there may be differences in the pathophysiol-ogy of diabetic neuropathy between type 1 and type 2 diabetes(Arnold et al., 2013a). Previous animal studies have suggested thattype 1 diabetes may be associated with greater abnormalities ofNa+/K+ pump function due to the effects of insulinopoenia andC-peptide deficiency (Sima et al., 2000).
Taken in total, excitability studies in diabetic neuropathy havesuggested that excitability changes are detectable in diabetic neu-ropathy prior to the onset of clinical symptoms. These studies havealso provided indirect evidence that Na+/K+ pump dysfunction mayplay an important role in the development of this condition andhave provided a basis for the investigation of agents that potentiateNa+/K+ pump function as potential therapies for diabetic neuropa-thy (Chalk et al., 2007; Schemmel et al., 2010). However, as thesestudies are largely cross-sectional group comparisons, further lon-gitudinal studies are required to determine the value of nerveexcitability abnormalities in predicting the development ofneuropathy.
3. Uraemic neuropathy
Chronic kidney disease is a significant global health concernwith a prevalence of �15% in developed countries (Chadbanet al., 2003; Couser et al., 2011). Chronic kidney disease encom-passes a continuum of disease from mild kidney damage, toend-stage disease which requires renal replacement therapy inthe form of dialysis or renal transplantation to sustain life. Theaetiology of chronic kidney disease may be due to a primary renaldisorder or as a complication of a multisystem disorder related to

2086 R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090
comorbidities such as diabetes which is now the leading cause ofchronic kidney disease worldwide (Atkins and Zimmet, 2010).Neurological complications are a major cause of morbidity inchronic kidney disease and affect all levels of the nervous systemcausing encephalopathy, cognitive impairment, restless leg syn-drome and a range of neuromuscular disorders, including mono-neuropathy, polyneuropathy and myopathy (Brouns and DeDeyn, 2004; Krishnan and Kiernan, 2009). Of the many neurologi-cal complications that can occur in chronic kidney disease, periph-eral neuropathy (uraemic neuropathy) remains the most commonlong-term disorder, particularly in end-stage disease. Improve-ments in dialysis care have failed to significantly reduce neuropa-thy prevalence in chronic kidney disease, with several recentstudies demonstrating rates of 70–100% in dialysis populations(Krishnan et al., 2005; Laaksonen et al., 2002; Tilki et al., 2009;Van den Neucker et al., 1998).
Early histological studies provided evidence that uraemic neu-ropathy was primarily an axonal neuropathy, with secondarydemyelination occurring in some cases (Dyck et al., 1971). Thesefindings were supported by electrophysiological studies utilisingnerve conduction techniques, that demonstrated prominent reduc-tions in sural amplitudes, prolongation of soleus H reflexes and tib-ial nerve F waves and alterations in peroneal motor nerveconduction velocity (Ackil et al., 1981; Angus-Leppan and Burke,1992; Bazzi et al., 1991; Laaksonen et al., 2002; Mansouri et al.,2001; Mitz et al., 1980; Panayiotopoulos and Lagos, 1980; Vanden Neucker et al., 1998). A recent study indicated that a changein amplitude of the sural sensory potential was the most sensitivenerve conduction abnormality in chronic kidney disease patients,with abnormalities noted in 50% of cases (Krishnan et al., 2005).The systemic effects of uraemia were emphasised by the general-ised nature of nerve conduction slowing which was demonstratedin upper and lower extremities, and in both sensory and motornerves (Nielsen, 1973). Additionally, nerve conduction slowingwas demonstrated in patients with both clinical and subclinicalneuropathy and often correlated with the severity of renal impair-ment (Nielsen, 1971; Preswick and Jeremy, 1964). As such, it wassuggested that though structural changes occur in a length depen-dent manner, there was a universal neurotoxic effect caused by anunknown uraemic toxin (Nielsen, 1973).
3.1. Pathophysiology
The earliest insights into the possible mechanisms underlyingneuropathy development came from observational studies thatdemonstrated a reduction in neuropathy prevalence with increas-ing dialysis frequency and efficiency (Asbury, 1971; D’Amouret al., 1984). Furthermore, dialysis regimens with longer durationand thinner dialysis membranes were shown to reduce the inci-dence of severe neuropathy (Ginn et al., 1971). This observationgave rise to the hypothesis that uraemic neuropathy be causedby a dialyzable neurotoxin or toxins (Hegstrom et al., 1962). Fur-ther studies demonstrated that peritoneal dialysis was associatedwith a reduced prevalence of neuropathy compared to haemodi-alysis, despite higher serum concentrations of small moleculessuch as creatinine and urea (Babb et al., 1973, 1981). Thisimprovement was attributed to better clearance of molecules inthe middle molecular weight range of 500–2000 Daltons andled to a greater focus on the ‘middle molecule hypothesis’ (Babbet al., 1981). However, studies exploring the potential toxicity ofthese compounds failed to provide any compelling evidence ofneurotoxicity for any of the putative toxins in this molecularweight range (Bostock et al., 2004). Kjellstrand and colleaguesfurther demonstrated middle molecule levels had no significanteffects on neuropathic complications (Kjellstrand et al., 1975). In-stead, they demonstrated that patients who experienced the
greatest fluctuations in electrolyte levels and osmolality weremost likely to develop neuropathy (Kjellstrand et al., 1975). Assuch, they proposed that avoiding harsh peaks and volleys inblood chemistry would help preserve neural homeostasis and im-prove patient outcomes, a hypothesis that remains the major fo-cus of current research efforts into the pathophysiology of thiscondition.
The complex accumulation and removal of a multitude ofuraemic toxins in dialysis patients has complicated the interpre-tation of in vivo studies. As such, the initial proposal that uraemicneurotoxicity may have been due to an alteration in membraneexcitability (Nielsen, 1973) was based on in vitro studies of mus-cle and red blood cells in dialysis patients (Bittar, 1967; Weltet al., 1964). It was hypothesised that one or more of the toxinsknown to accumulate in uremia could inhibit the activity of theNa+/K+ pump (Nielsen, 1973). Blockade of the Na+/K+ pump wouldresult in excess intracellular positive charge creating a depolar-ised state. The consequent disruption to normal ionic gradientsmay result in the reversal of the Na+/Ca2+ exchanger and activa-tion of apoptotic processes leading to cell death (Lehning et al.,1996).
3.2. Alterations in axonal excitability in chronic kidney disease
Recent studies exploring the mechanisms of uraemic neuropa-thy have largely focussed on the use of nerve excitability tech-niques. In contrast to standard nerve conduction measures whichhave shown little change across a dialysis session (Mansouriet al., 2001), studies of excitability have demonstrated significantshifts in excitability that occur during and after a single dialysissession (Fig. 5A and B) (Kiernan et al., 2002; Krishnan et al.,2005, 2006b). Initial studies of excitability in dialysis patients wereconducted in median motor axons (Kiernan et al., 2002). While theclinical features of uraemic neuropathy are lower limb predomi-nant, the systemic distribution of uraemic toxins led the authorsof that study to postulate that alterations in excitability would oc-cur in a generalised manner. These studies demonstrated signifi-cant alterations in excitability prior to commencement of adialysis session, with the changes suggestive of axonal membranedepolarization. (Kiernan and Bostock, 2000; Kiernan et al., 2002).Furthermore pre-dialysis abnormalities had improved at the mid-point of dialysis and were virtually abolished at the post-dialysistest (Kiernan et al., 2002; Krishnan et al., 2005, 2006b). Pre-dialysisnerve excitability changes included reduced superexcitability, pro-longed relative refractory periods and increased accommodation todepolarising and hyperpolarising currents (Fig. 5A and B). To inves-tigate the basis for pre-dialysis excitability abnormalities routineblood chemistry was collected at corresponding time-points tonerve excitability tests. This analysis revealed significant correla-tions between measures of membrane depolarisation and pre-dial-ysis serum K+. As such, the conclusion of this study was that theaxons of dialysis patients are chronically depolarised and that thisdepolarisation could be accounted for by hyperkalaemia (Kiernanet al., 2002).
In order to investigate a region of greater clinical involvement,further studies were conducted in lower limb motor nerves (Krish-nan et al., 2005). Nerve excitability recordings obtained from tibi-alis anterior (TA) and extensor digitorum brevis (EDB)demonstrated that lower limb motor axons manifested the samepattern of change in pre- and-post dialysis studies as median mo-tor axons (Krishnan et al., 2005). Similarly, pre-dialysis nerve excit-ability abnormalities were shown to be significantly correlatedwith pre-dialysis serum K+. Of clinical relevance, this studyrevealed significant correlations between nerve excitability param-eters and increasing total neuropathy symptom scores, suggestingthat altered axonal membrane potential may be directly related to
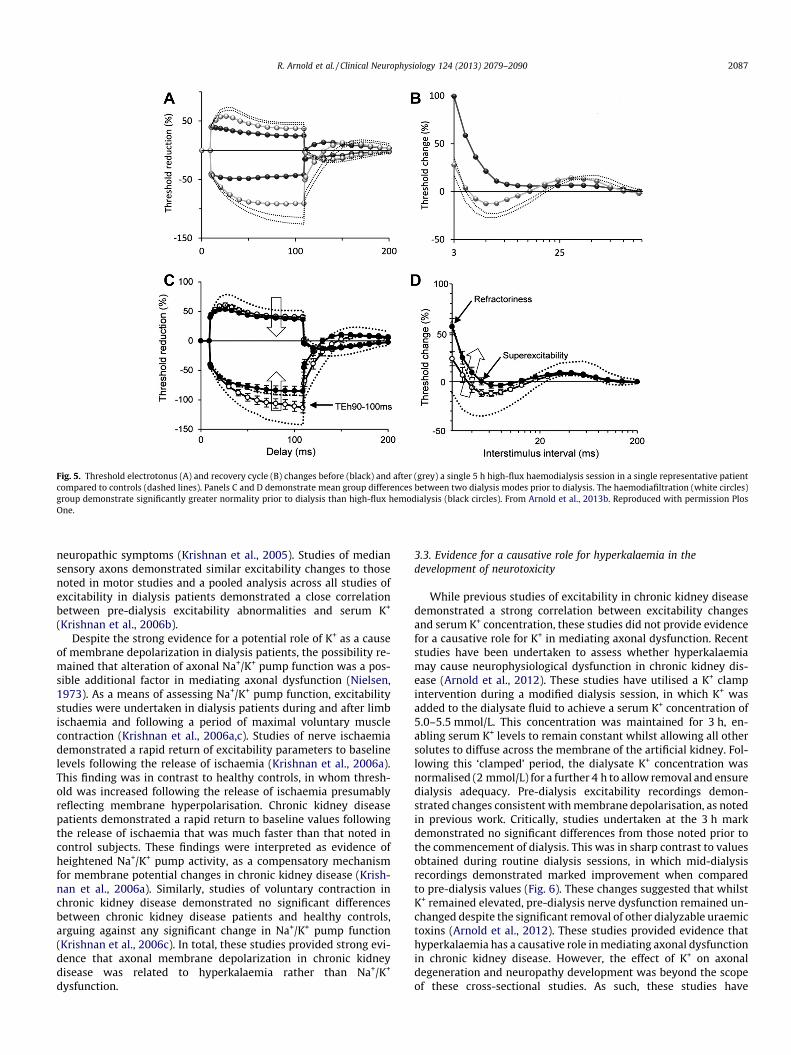
Fig. 5. Threshold electrotonus (A) and recovery cycle (B) changes before (black) and after (grey) a single 5 h high-flux haemodialysis session in a single representative patientcompared to controls (dashed lines). Panels C and D demonstrate mean group differences between two dialysis modes prior to dialysis. The haemodiafiltration (white circles)group demonstrate significantly greater normality prior to dialysis than high-flux hemodialysis (black circles). From Arnold et al., 2013b. Reproduced with permission PlosOne.
R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090 2087
neuropathic symptoms (Krishnan et al., 2005). Studies of mediansensory axons demonstrated similar excitability changes to thosenoted in motor studies and a pooled analysis across all studies ofexcitability in dialysis patients demonstrated a close correlationbetween pre-dialysis excitability abnormalities and serum K+
(Krishnan et al., 2006b).Despite the strong evidence for a potential role of K+ as a cause
of membrane depolarization in dialysis patients, the possibility re-mained that alteration of axonal Na+/K+ pump function was a pos-sible additional factor in mediating axonal dysfunction (Nielsen,1973). As a means of assessing Na+/K+ pump function, excitabilitystudies were undertaken in dialysis patients during and after limbischaemia and following a period of maximal voluntary musclecontraction (Krishnan et al., 2006a,c). Studies of nerve ischaemiademonstrated a rapid return of excitability parameters to baselinelevels following the release of ischaemia (Krishnan et al., 2006a).This finding was in contrast to healthy controls, in whom thresh-old was increased following the release of ischaemia presumablyreflecting membrane hyperpolarisation. Chronic kidney diseasepatients demonstrated a rapid return to baseline values followingthe release of ischaemia that was much faster than that noted incontrol subjects. These findings were interpreted as evidence ofheightened Na+/K+ pump activity, as a compensatory mechanismfor membrane potential changes in chronic kidney disease (Krish-nan et al., 2006a). Similarly, studies of voluntary contraction inchronic kidney disease demonstrated no significant differencesbetween chronic kidney disease patients and healthy controls,arguing against any significant change in Na+/K+ pump function(Krishnan et al., 2006c). In total, these studies provided strong evi-dence that axonal membrane depolarization in chronic kidneydisease was related to hyperkalaemia rather than Na+/K+
dysfunction.
3.3. Evidence for a causative role for hyperkalaemia in thedevelopment of neurotoxicity
While previous studies of excitability in chronic kidney diseasedemonstrated a strong correlation between excitability changesand serum K+ concentration, these studies did not provide evidencefor a causative role for K+ in mediating axonal dysfunction. Recentstudies have been undertaken to assess whether hyperkalaemiamay cause neurophysiological dysfunction in chronic kidney dis-ease (Arnold et al., 2012). These studies have utilised a K+ clampintervention during a modified dialysis session, in which K+ wasadded to the dialysate fluid to achieve a serum K+ concentration of5.0–5.5 mmol/L. This concentration was maintained for 3 h, en-abling serum K+ levels to remain constant whilst allowing all othersolutes to diffuse across the membrane of the artificial kidney. Fol-lowing this ‘clamped’ period, the dialysate K+ concentration wasnormalised (2 mmol/L) for a further 4 h to allow removal and ensuredialysis adequacy. Pre-dialysis excitability recordings demon-strated changes consistent with membrane depolarisation, as notedin previous work. Critically, studies undertaken at the 3 h markdemonstrated no significant differences from those noted prior tothe commencement of dialysis. This was in sharp contrast to valuesobtained during routine dialysis sessions, in which mid-dialysisrecordings demonstrated marked improvement when comparedto pre-dialysis values (Fig. 6). These changes suggested that whilstK+ remained elevated, pre-dialysis nerve dysfunction remained un-changed despite the significant removal of other dialyzable uraemictoxins (Arnold et al., 2012). These studies provided evidence thathyperkalaemia has a causative role in mediating axonal dysfunctionin chronic kidney disease. However, the effect of K+ on axonaldegeneration and neuropathy development was beyond the scopeof these cross-sectional studies. As such, these studies have
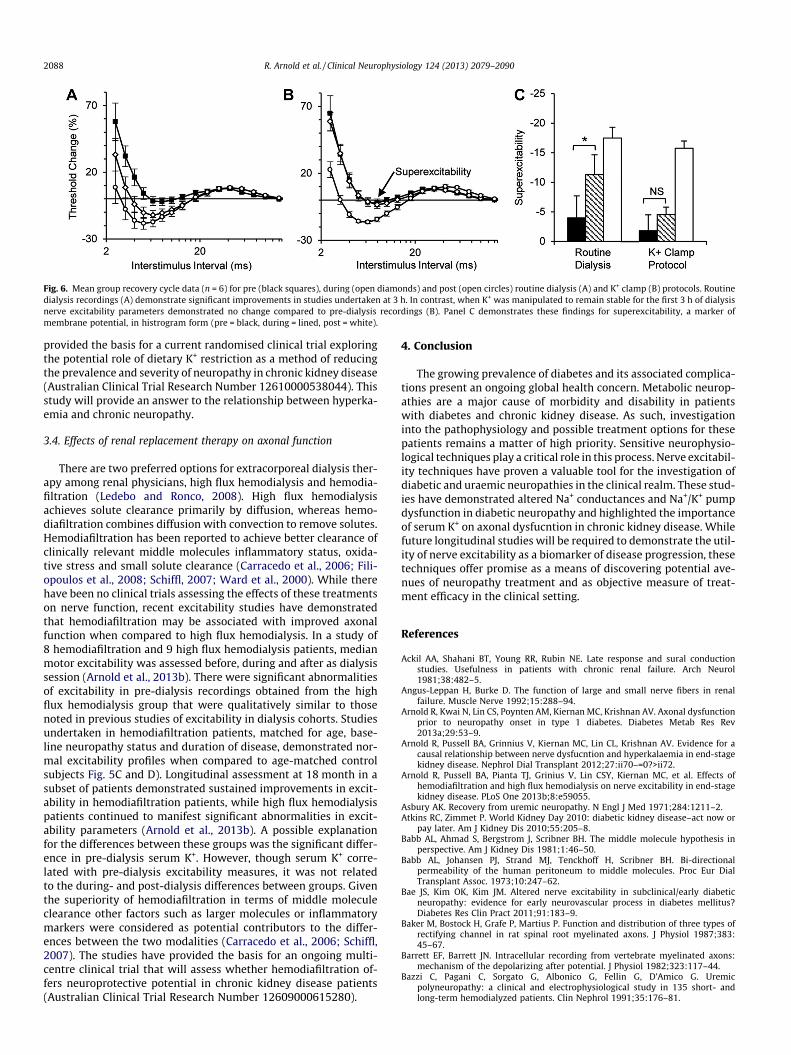
Fig. 6. Mean group recovery cycle data (n = 6) for pre (black squares), during (open diamonds) and post (open circles) routine dialysis (A) and K+ clamp (B) protocols. Routinedialysis recordings (A) demonstrate significant improvements in studies undertaken at 3 h. In contrast, when K+ was manipulated to remain stable for the first 3 h of dialysisnerve excitability parameters demonstrated no change compared to pre-dialysis recordings (B). Panel C demonstrates these findings for superexcitability, a marker ofmembrane potential, in histrogram form (pre = black, during = lined, post = white).
2088 R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090
provided the basis for a current randomised clinical trial exploringthe potential role of dietary K+ restriction as a method of reducingthe prevalence and severity of neuropathy in chronic kidney disease(Australian Clinical Trial Research Number 12610000538044). Thisstudy will provide an answer to the relationship between hyperka-emia and chronic neuropathy.
3.4. Effects of renal replacement therapy on axonal function
There are two preferred options for extracorporeal dialysis ther-apy among renal physicians, high flux hemodialysis and hemodia-filtration (Ledebo and Ronco, 2008). High flux hemodialysisachieves solute clearance primarily by diffusion, whereas hemo-diafiltration combines diffusion with convection to remove solutes.Hemodiafiltration has been reported to achieve better clearance ofclinically relevant middle molecules inflammatory status, oxida-tive stress and small solute clearance (Carracedo et al., 2006; Fili-opoulos et al., 2008; Schiffl, 2007; Ward et al., 2000). While therehave been no clinical trials assessing the effects of these treatmentson nerve function, recent excitability studies have demonstratedthat hemodiafiltration may be associated with improved axonalfunction when compared to high flux hemodialysis. In a study of8 hemodiafiltration and 9 high flux hemodialysis patients, medianmotor excitability was assessed before, during and after as dialysissession (Arnold et al., 2013b). There were significant abnormalitiesof excitability in pre-dialysis recordings obtained from the highflux hemodialysis group that were qualitatively similar to thosenoted in previous studies of excitability in dialysis cohorts. Studiesundertaken in hemodiafiltration patients, matched for age, base-line neuropathy status and duration of disease, demonstrated nor-mal excitability profiles when compared to age-matched controlsubjects Fig. 5C and D). Longitudinal assessment at 18 month in asubset of patients demonstrated sustained improvements in excit-ability in hemodiafiltration patients, while high flux hemodialysispatients continued to manifest significant abnormalities in excit-ability parameters (Arnold et al., 2013b). A possible explanationfor the differences between these groups was the significant differ-ence in pre-dialysis serum K+. However, though serum K+ corre-lated with pre-dialysis excitability measures, it was not relatedto the during- and post-dialysis differences between groups. Giventhe superiority of hemodiafiltration in terms of middle moleculeclearance other factors such as larger molecules or inflammatorymarkers were considered as potential contributors to the differ-ences between the two modalities (Carracedo et al., 2006; Schiffl,2007). The studies have provided the basis for an ongoing multi-centre clinical trial that will assess whether hemodiafiltration of-fers neuroprotective potential in chronic kidney disease patients(Australian Clinical Trial Research Number 12609000615280).
4. Conclusion
The growing prevalence of diabetes and its associated complica-tions present an ongoing global health concern. Metabolic neurop-athies are a major cause of morbidity and disability in patientswith diabetes and chronic kidney disease. As such, investigationinto the pathophysiology and possible treatment options for thesepatients remains a matter of high priority. Sensitive neurophysio-logical techniques play a critical role in this process. Nerve excitabil-ity techniques have proven a valuable tool for the investigation ofdiabetic and uraemic neuropathies in the clinical realm. These stud-ies have demonstrated altered Na+ conductances and Na+/K+ pumpdysfunction in diabetic neuropathy and highlighted the importanceof serum K+ on axonal dysfucntion in chronic kidney disease. Whilefuture longitudinal studies will be required to demonstrate the util-ity of nerve excitability as a biomarker of disease progression, thesetechniques offer promise as a means of discovering potential ave-nues of neuropathy treatment and as objective measure of treat-ment efficacy in the clinical setting.
References
Ackil AA, Shahani BT, Young RR, Rubin NE. Late response and sural conductionstudies. Usefulness in patients with chronic renal failure. Arch Neurol1981;38:482–5.
Angus-Leppan H, Burke D. The function of large and small nerve fibers in renalfailure. Muscle Nerve 1992;15:288–94.
Arnold R, Kwai N, Lin CS, Poynten AM, Kiernan MC, Krishnan AV. Axonal dysfunctionprior to neuropathy onset in type 1 diabetes. Diabetes Metab Res Rev2013a;29:53–9.
Arnold R, Pussell BA, Grinnius V, Kiernan MC, Lin CL, Krishnan AV. Evidence for acausal relationship between nerve dysfucntion and hyperkalaemia in end-stagekidney disease. Nephrol Dial Transplant 2012;27:ii70–=0?>ii72.
Arnold R, Pussell BA, Pianta TJ, Grinius V, Lin CSY, Kiernan MC, et al. Effects ofhemodiafiltration and high flux hemodialysis on nerve excitability in end-stagekidney disease. PLoS One 2013b;8:e59055.
Asbury AK. Recovery from uremic neuropathy. N Engl J Med 1971;284:1211–2.Atkins RC, Zimmet P. World Kidney Day 2010: diabetic kidney disease–act now or
pay later. Am J Kidney Dis 2010;55:205–8.Babb AL, Ahmad S, Bergstrom J, Scribner BH. The middle molecule hypothesis in
perspective. Am J Kidney Dis 1981;1:46–50.Babb AL, Johansen PJ, Strand MJ, Tenckhoff H, Scribner BH. Bi-directional
permeability of the human peritoneum to middle molecules. Proc Eur DialTransplant Assoc. 1973;10:247–62.
Bae JS, Kim OK, Kim JM. Altered nerve excitability in subclinical/early diabeticneuropathy: evidence for early neurovascular process in diabetes mellitus?Diabetes Res Clin Pract 2011;91:183–9.
Baker M, Bostock H, Grafe P, Martius P. Function and distribution of three types ofrectifying channel in rat spinal root myelinated axons. J Physiol 1987;383:45–67.
Barrett EF, Barrett JN. Intracellular recording from vertebrate myelinated axons:mechanism of the depolarizing after potential. J Physiol 1982;323:117–44.
Bazzi C, Pagani C, Sorgato G, Albonico G, Fellin G, D’Amico G. Uremicpolyneuropathy: a clinical and electrophysiological study in 135 short- andlong-term hemodialyzed patients. Clin Nephrol 1991;35:176–81.

R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090 2089
Behse F, Buchthal F, Carlsen F. Nerve biopsy and conduction studies in diabeticneuropathy. J Neurol Neurosurg Psychiatry 1977;40:1072–82.
Bergmans J. The physiology of single human nerve fibres. Vander,Belgium: University of Louvain; 1970.
Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, et al. Methylglyoxalmodification of Nav1.8 facilitates nociceptive neuron firing and causeshyperalgesia in diabetic neuropathy. Nat Med 2012;18:926–33.
Bittar EE. Maia muscle fibre as a model for the study of uraemic toxicity. Nature1967;214:310–2.
Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of humanperipheral nerve. Muscle Nerve 1998;21:137–58.
Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of humanperipheral nerve. J Physiol 1997;498:277–94.
Bostock H, Walters RJ, Andersen KV, Murray NM, Taube D, Kiernan MC. Haspotassium been prematurely discarded as a contributing factor to thedevelopment of uraemic neuropathy? Nephrol Dial Transplant 2004;19:1054–7.
Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, Apelqvist J. The global burden ofdiabetic foot disease. Lancet 2005;366:1719–24.
Brismar T, Sima AA, Greene DA. Reversible and irreversible nodal dysfunction indiabetic neuropathy. Ann Neurol 1987;21:504–7.
Brouns R, De Deyn PP. Neurological complications in renal failure: a review. ClinNeurol Neurosurg 2004;107:1–16.
Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin Neurophysiol2001;112:1575–85.
Carracedo J, Merino A, Nogueras S, Carretero D, Berdud I, Ramirez R, et al. On-linehemodiafiltration reduces the proinflammatory CD14+CD16+ monocyte-derived dendritic cells: a prospective, crossover study. J Am Soc Nephrol2006;17:2315–21.
Chadban SJ, Briganti EM, Kerr PG, Dunstan DW, Welborn TA, Zimmet PZ, et al.Prevalence of kidney damage in Australian adults: the AusDiab kidney study. JAm Soc Nephrol 2003;14:S131–8.
Chalk C, Benstead TJ, Moore F. Aldose reductase inhibitors for the treatment ofdiabetic polyneuropathy. Cochrane Database Syst Rev. 2007: CD004572.
Cheah BC, Lin CSY, Park SB, Vucic S, Krishnan AV, Kiernan MC. Progressive axonaldysfunction and clinical impairment in amyotrophic lateral sclerosis. ClinNeurophysiol 2012;123:2460–7.
Couser WG, Remuzzi G, Mendis S, Tonelli M. The contribution of chronic kidneydisease to the global burden of major noncommunicable diseases. Kidney Int2011;80:1258–70.
Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodiumchannel expression in experimental painful diabetic neuropathy. Ann Neurol2002;52:786–92.
D’Amour ML, Dufresne LR, Morin C, Slaughter D. Sensory nerve conduction inchronic uremic patients during the first six months of hemodialysis. Can JNeurol Sci 1984;11:269–71.
Dyck PJ, Johnson WJ, Lambert EH, O’Brien PC. Segmental demyelination secondaryto axonal degeneration in uremic neuropathy. Mayo Clin Proc 1971;46:400–31.
Dyck PJ, Karnes JL, O’Brien P, Okazaki H, Lais A, Engelstad J. The spatial distributionof fiber loss in diabetic polyneuropathy suggests ischemia. Ann Neurol 1986;19:440–9.
Dyck PJ, Kratz KM, Karnes JL, Litchy WJ, Klein R, Pach JM, et al. The prevalence bystaged severity of various types of diabetic neuropathy, retinopathy, andnephropathy in a population-based cohort: the rochester diabetic neuropathystudy. Neurology 1993;43:817–24.
Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: mechanismsto management. Pharmacol Ther 2008;120:1–34.
Filiopoulos V, Hadjiyannakos D, Metaxaki P, Sideris V, Takouli L, Anogiati A, et al.Inflammation and oxidative stress in patients on hemodiafiltration. Am JNephrol 2008;28:949–57.
Gillett MJ. International expert committee report on the role of the A1c assay in thediagnosis of diabetes. Diabetes Care 2009;32(7):1327–34 [Clin Biochem Rev.2009; 30: 197–200].
Ginn HE, Bugel HJ, James L, Hopkins P. Clinical experience with small surface areadialyzers (SSAD). Proc Clin Dial Transplant Forum. 1971;1:53–60.
Gonzalez ER, Oley MA. The management of lower-extremity diabetic ulcers.Manage Care Interface 2000;13:80–7.
Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs ofdiabetic peripheral neuropathy in the US. Diabetes Care 2003;26:1790–5.
Graf RJ, Halter JB, Pfeifer MA, Halar E, Brozovich F, Porte Jr D. Glycemic control andnerve conduction abnormalities in non-insulin-dependent diabetic subjects.Ann Intern Med 1981;94:307–11.
Greene DA, Lattimer SA. Altered sorbitol and myo-inositol metabolism as the basisfor defective protein kinase C and (Na, K)-ATPase regulation in diabeticneuropathy. Ann N Y Acad Sci 1986;488:334–40.
Greene DA, Lattimer SA, Sima AA. Are disturbances of sorbitol, phosphoinositide,and Na+–K+-ATPase regulation involved in pathogenesis of diabeticneuropathy? Diabetes 1988;37:688–93.
Greene DA, Yagihashi S, Lattimer SA, Sima AA. Nerve Na+–K+-ATPase, conduction,and myo-inositol in the insulin-deficient BB rat. Am J Physiol 1984;247:E534–9.
Habib AA, Brannagan 3rd TH. Therapeutic strategies for diabetic neuropathy. CurrNeurol Neurosci 2010;10:92–100.
Happich M, John J, Stamenitis S, Clouth J, Polnau D. The quality of life and economicburden of neuropathy in diabetic patients in Germany in 2002–results from theDiabetic Microvascular Complications (DIMICO) study. Diabetes Res Clin Pract2008;81:223–30.
Hegstrom RM, Murray JS, Pendras JP, Burnell JM, Scribner BH. Two year’s experiencewith periodic hemodialysis in the treatment of chronic uremia. Trans Am SocArtif Intern Organs 1962;8:266–80.
Hodgkin AL, Huxley AF. A quantitative description of membrane current and itsapplication to conduction and excitation in nerve. J Physiol 1952;117:500–44.
Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW. Early painful diabeticneuropathy is associated with differential changes in tetrodotoxin-sensitiveand -resistant sodium channels in dorsal root ganglion neurons in the rat. J BiolChem 2004;279:29341–50.
Hong S, Wiley JW. Altered expression and function of sodium channels in large DRGneurons and myelinated A-fibers in early diabetic neuropathy in the rat.Biochem Biophys Res Commun 2006;339:652–60.
Johnson PC, Doll SC, Cromey DW. Pathogenesis of diabetic neuropathy. Ann Neurol1986;19:450–7.
Kamiya H, Zhang W, Sima AA. C-peptide prevents nociceptive sensory neuropathyin type 1 diabetes. Ann Neurol 2004;56:827–35.
Kiernan MC, Bostock H. Effects of membrane polarization and ischaemia on theexcitability properties of human motor axons. Brain 2000;123:2542–51.
Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonalexcitability: a new approach in clinical testing. Muscle Nerve 2000;23:399–409.
Kiernan MC, Mogyoros I, Burke D. Differences in the recovery of excitability insensory and motor axons of human median nerve. Brain 1996;119:1099–105.
Kiernan MC, Walters RJL, Andersen KV, Taube D, Murray NMF, Bostock H. Nerveexcitability changes in chronic renal failure indicate membrane depolarizationdue to hyperkalaemia. Brain 2002;125:1366–78.
Kitano Y, Kuwabara S, Misawa S, Ogawara K, Kanai K, Kikkawa Y, et al. The acuteeffects of glycemic control on axonal excitability in human diabetics. AnnNeurol 2004;56:462–7.
Kjellstrand CM, Evans RL, Petersen RJ, Shideman JR, von Hartitzsch B, Buselmeier TJ.The unphysiology of dialysis: a major cause of dialysis side effects? Kidney IntSuppl 1975;7:30–4.
Krishnan AV, Kiernan MC. Altered nerve excitability properties in establisheddiabetic neuropathy. Brain 2005;128:1178–87.
Krishnan AV, Kiernan MC. Neurological complications of chronic kidney disease. NatRev Neurol 2009;5:542–51.
Krishnan AV, Lin CS, Park SB, Kiernan MC. Axonal ion channels from bench tobedside: a translational neuroscience perspective. Prog Neurobiol 2009a;89:288–313.
Krishnan AV, Lin CSY, Kiernan MC. Activity-dependent excitability changes suggestNa(+)/K(+) pump dysfunction in diabetic neuropathy. Brain 2008;131:1209–16.
Krishnan AV, Phoon RK, Pussell BA, Charlesworth JA, Bostock H, Kiernan MC. Alteredmotor nerve excitability in end-stage kidney disease. Brain 2005;128:2164–74.
Krishnan AV, Phoon RK, Pussell BA, Charlesworth JA, Bostock H, Kiernan MC.Ischaemia induces paradoxical changes in axonal excitability in end-stagekidney disease. Brain 2006a;129:1585–92.
Krishnan AV, Phoon RK, Pussell BA, Charlesworth JA, Kiernan MC. Sensory nerveexcitability and neuropathy in end stage kidney disease. J Neurol NeurosurgPsychiatry 2006b;77:548–51.
Krishnan AV, Phoon RKS, Pussell BA, Charlesworth JA, Bostock H, Kiernan MC.Neuropathy, axonal Na+/K+ pump function and activity-dependent excitabilitychanges in end-stage kidney disease. Clin Neurophysiol 2006c;117:992–9.
Krishnan AV, Pussell BA, Kiernan MC. Neuromuscular disease in the dialysis patient:an update for the nephrologist. Semin Dial 2009b;22:267–78.
Kuwabara S. Shortened refractory periods in human diabetic neuropathy. ClinNeurophysiol 2003;114:169–70.
Kuwabara S, Ogawara K, Harrori T, Suzuki Y, Hashimoto N. The acute effects ofglycemic control on axonal excitability in human diabetic nerves. Intern Med2002;41:360–5.
Kwai NC, Arnold R, Wickremaarachchi C, Lin CS, Poynten AM, Kiernan MC, KrishnanAV. Effects of axonal ion-channel dysfunction on quality of life in type 2diabetes. Diabetes Care 2013;36:1272–7.
Laaksonen S, Metsarinne K, Voipio-Pulkki LM, Falck B. Neurophysiologic parametersand symptoms in chronic renal failure. Muscle Nerve 2002;25:884–90.
Ledebo I, Ronco C. The best dialysis therapy? Results from an international surveyamong nephrology professionals. NDT Plus 2008;1:403–8.
Lehning EJ, Doshi R, Isaksson N, Stys PK, LoPachin Jr RM. Mechanisms of injury-induced calcium entry into peripheral nerve myelinated axons: role of reversesodium–calcium exchange. J Neurochem 1996;66:493–500.
Lin CS, Mogyoros I, Kuwabara S, Cappelen-Smith C, Burke D. Differences inresponses of cutaneous afferents in the human median and sural nerves toischemia. Muscle Nerve 2001;24:1503–9.
Manes C, Papazolglou N, Sossidou E, Soulis K, Milarakis D, Satsoglou A, et al.Prevalence of diabetic neuropathy and foot ulceration: identification ofpotential risk factors – a population-based study. Wounds 2002;14:11–5.
Mansouri B, Adybeig B, Rayegani M, Yasami S, Behshad V. Uremic neuropathy andthe analysis of electrophysiological changes. Electromyogr Clin Neurophysiol2001;41:107–15.
Misawa S, Kuwabara S, Kanai K, Tamura N, Hiraga A, Nakata M, et al. Axonalpotassium conductance and glycemic control in human diabetic nerves. ClinNeurophysiol 2005;116:1181–7.
Misawa S, Kuwabara S, Kanai K, Tamura N, Nakata M, Ogawara K, et al. Nodalpersistent Na+ currents in human diabetic nerves estimated by the technique oflatent addition. Clin Neurophysiol 2006a;117:815–20.
Misawa S, Kuwabara S, Kanai K, Tamura N, Nakata M, Sawai S, et al. Aldosereductase inhibition alters nodal Na+ currents and nerve conduction in humandiabetics. Neurology 2006b;66:1545–9.

2090 R. Arnold et al. / Clinical Neurophysiology 124 (2013) 2079–2090
Misawa S, Kuwabara S, Ogawara K, Kitano Y, Hattori T. Strength-duration propertiesand glycemic control in human diabetic motor nerves. Clin Neurophysiol2005;116:254–8.
Misawa S, Kuwabara S, Ogawara K, Kitano Y, Yagui K, Hattori T. Hyperglycemiaalters refractory periods in human diabetic neuropathy. Clin Neurophysiol2004;115:2525–9.
Misawa S, Sakurai K, Shibuya K, Isose S, Kanai K, Ogino J, et al. Neuropathic pain isassociated with increased nodal persistent Na(+) currents in human diabeticneuropathy. J Peripher Nerv Syst 2009;14:279–84.
Mitz M, Prakash AS, Melvin J, Piering W. Motor nerve conduction indicators inuremic neuropathy. Arch Phys Med Rehabil 1980;61:45–8.
Mogyoros I, Kiernan MC, Burke D. Strength-duration properties of humanperipheral nerve. Brain 1996;119:439–47.
Mogyoros I, Kiernan MC, Burke D, Bostock H. Excitability changes in human sensoryand motor axons during hyperventilation and ischaemia. Brain 1997;120:317–25.
Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S, Nakashima E, et al. A proteinkinase C-beta-selective inhibitor ameliorates neural dysfunction instreptozotocin-induced diabetic rats. Diabetes 1999;48:2090–5.
Nielsen VK. The peripheral nerve function in chronic renal failure. II.Intercorrelation of clinical symptoms and signs and clinical grading ofneuropathy. Acta Med Scand 1971;190:113–7.
Nielsen VK. The peripheral nerve function in chronic renal failure. V. Sensory andmotor conduction velocity. Acta Med Scand 1973;194:445–54.
Nodera H, Kaji R. Nerve excitability testing and its clinical application toneuromuscular diseases. Clin Neurophysiol 2006;117:1902–16.
Panayiotopoulos CP, Lagos G. Tibial nerve H-reflex and F-wave studies in patientswith uremic neuropathy. Muscle Nerve 1980;3:423–6.
Pape HC. Queer current and pacemaker: the hyperpolarization-activated cationcurrent in neurons. Annu Rev Physiol 1996;58:299–327.
Park SB, Goldstein D, Lin CS, Krishnan AV, Friedlander ML, Kiernan MC. Acuteabnormalities of sensory nerve function associated with oxaliplatin-inducedneurotoxicity. J Clin Oncol 2009;27:1243–9.
Pirart J. Diabetes mellitus and its degenerative complications: a prospective study of4,400 patient observed between 1947 and 1973. Diabetes Care 1978;1:168.
Preswick G, Jeremy D. Subclinical polyneuropathy in renal insufficiency. Lancet1964;2:731–2.
Said G. Diabetic neuropathy – a review. Nat Clin Pract Neurol 2007;3:331–40.Schemmel KE, Padiyara RS, D’Souza JJ. Aldose reductase inhibitors in the treatment
of diabetic peripheral neuropathy: a review. J Diabetes Complications2010;24:354–60.
Schiffl H. Prospective randomized cross-over long-term comparison of onlinehaemodiafiltration and ultrapure high-flux haemodialysis. Eur J Med Res2007;12:26–33.
Schwarz JR, Reid G, Bostock H. Action potentials and membrane currents in thehuman node of Ranvier. Pflugers Arch 1995;430:283–92.
Sima AA, Zhang W, Grunberger G. Type 1 diabetic neuropathy and C-peptide. ExpDiabesity Res 2004;5:657–77.
Sima AA, Zhang W, Xu G, Sugimoto K, Guberski D, Yorek MA. A comparison ofdiabetic polyneuropathy in type II diabetic BBZDR/Wor rats and in type Idiabetic BB/Wor rats. Diabetologia 2000;43:786–93.
Sima AAF, Brismar T. Reversible diabetic nerve dysfunction: structural correlates toelectrophysiological abnormalities. Ann Neurol 1985;18:21–9.
Strupp M, Bostock H, Weigl P, Piwernetz K, Renner R, Grafe P. Is resistance toischaemia of motor axons in diabetic subjects due to membrane depolarization?J Neurol Sci 1990;99:271–80.
Sugimoto K, Murakawa Y, Sima AAF. Diabetic neuropathy – a continuing enigma.Diabetes Metab Res Rev 2000;16:408–33.
Sung JY, Park SB, Liu YT, Kwai N, Arnold R, Krishnan AV, et al. Progressive axonaldysfunction precedes development of neuropathy in type 2 diabetes. Diabetes2012;61:1592–8.
Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-Tirgoviste C, et al.Vascular risk factors and diabetic neuropathy. N Engl J Med 2005;352:341–50.
Thornalley PJ. Dicarbonyl intermediates in the maillard reaction. Ann N Y Acad Sci2005;1043:111–7.
Tilki HE, Akpolat T, Coskun M, Stalberg E. Clinical and electrophysiologic findings indialysis patients. J Electromyogr Kinesiol 2009;19:500–8.
Vagg R, Mogyoros I, Kiernan MC, Burke D. Activity-dependent hyperpolarization ofhuman motor axons produced by natural activity. J Physiol 1998;507:919–25.
Van den Neucker K, Vanderstraeten G, Vanholder R. Peripheral motor and sensorynerve conduction studies in haemodialysis patients. A study of 54 patients.Electromyogr Clin Neurophysiol 1998;38:467–74.
Veves A, Manes C, Murray HJ, Young MJ, Boulton AJ. Painful neuropathy and footulceration in diabetic patients. Diabetes Care 1993;16:1187–9.
Vileikyte L, Peyrot M, Bundy C, Rubin RR, Leventhal H, Mora P, et al. Thedevelopment and validation of a neuropathy- and foot ulcer-specific qualityof life instrument. Diabetes Care 2003;26:2549–55.
Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis ofdiabetic neuropathy. Endocr Rev 2004;25:612–28.
Vinik AI, Bril V, Litchy WJ, Price KL, Bastyr 3rd EJ. Sural sensory action potentialidentifies diabetic peripheral neuropathy responders to therapy. Muscle Nerve2005;32:619–25.
Wahren J, Ekberg K, Johansson J, Henriksson M, Pramanik A, Johansson BL, et al. Roleof C-peptide in human physiology. Am J Physiol – Endoc M 2000;278:E759–68.
Ward RA, Schmidt B, Hullin J, Hillebrand GF, Samtleben W. A comparison of on-linehemodiafiltration and high-flux hemodialysis: a prospective clinical study. J AmSoc Nephrol 2000;11:2344–50.
Weigl P, Bostock H, Franz P, Martius P, Muller W, Grafe P. Threshold trackingprovides a rapid indication of ischaemic resistance in motor axons of diabeticsubjects. Electroencephalogr Clin Neurophysiol 1989;73:369–71.
Welt LG, Sachs JR, McManus TJ. An ion transport defect in erythrocytes fromuraemic subjects. Trans Ass Am Phys 1964;77:169–81.
Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes atlas: global estimates of theprevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract 2011;94:311–21.
Zhu X, Eichberg J. 1,2-diacylglycerol content and its arachidonyl-containingmolecular species are reduced in sciatic nerve from streptozotocin-induceddiabetic rats. J Neurochem 1990a;55:1087–90.
Zhu X, Eichberg J. A myo-inositol pool utilized for phosphatidylinositol synthesis isdepleted in sciatic nerve from rats with streptozotocin-induced diabetes. ProcNatl Acad Sci USA 1990b;87:9818–22.









![[Diabetic nephropathy and cardiac disease]](https://img.pdfslide.net/doc/110x75/634dfd2d92e70b3087022c72/diabetic-nephropathy-and-cardiac-disease.jpg)
![[Purines and neuroregeneration in neuropathies] Пурини и неврорегенерация при невропатии](https://img.pdfslide.net/doc/110x75/63618beea514f501cd0cd230/purines-and-neuroregeneration-in-neuropathies-purini-i-nevroregeneratsiya.jpg)


















