Embed Size (px)
Citation preview

European Journal of Pharmacology 651 (2011) 41–50
Contents lists available at ScienceDirect
European Journal of Pharmacology
j ourna l homepage: www.e lsev ie r.com/ locate /e jphar
Molecular and Cellular Pharmacology
Metformin reduces cisplatin-mediated apoptotic death of cancer cells throughAMPK-independent activation of Akt
Kristina Janjetovic a,b,1, Ljubica Vucicevic a,b,1, Maja Misirkic a,b,1, Urosh Vilimanovich c, Gordana Tovilovic b,Nevena Zogovic b, Zoran Nikolic d, Svetlana Jovanovic e, Vladimir Bumbasirevic c,Vladimir Trajkovic a,⁎, Ljubica Harhaji-Trajkovic b,⁎a Institute of Microbiology and Immunology, School of Medicine, University of Belgrade, Dr. Subotica 1, 11000 Belgrade, Serbiab Institute of Biological Research, Bulevar Despota Stefana 142, 11000 Belgrade, Serbiac Institute of Histology and Embryology, School of Medicine, University of Belgrade, Belgrade, Serbiad Faculty of Physics, University of Belgrade, Belgrade, Serbiae Vinca Institute of Nuclear Sciences, University of Belgrade, Belgrade, Serbia
⁎ Corresponding authors.E-mail addresses: [email protected] (V. Trajkovic)
(L. Harhaji-Trajkovic).1 These authors contributed equally to this work.
0014-2999/$ – see front matter © 2010 Published by Edoi:10.1016/j.ejphar.2010.11.005
a b s t r a c t
a r t i c l e i n f oArticle history:Received 4 May 2010Received in revised form 30 September 2010Accepted 7 November 2010Available online 27 November 2010
Keywords:MetforminCisplatinCancerApoptosisAMPKAkt
Metformin is an antidiabetic drug with anticancer properties, which mainly acts through induction of AMP-activated protein kinase (AMPK). In the present study we investigated the influence of metformin on thein vitro anticancer activity of the well-known chemotherapeutic agent cisplatin. Cell viability was determinedby MTT and LDH release assay, oxidative stress and apoptosis (caspase activation, DNA fragmentation, andphosphatidylserine exposure) were assessed by flow cytometry, while activation of AMPK and Akt wasanalyzed by immunoblotting. Althoughmetformin reduced the number of tumour cells when applied alone, itsurprisingly antagonized the cytotoxicity of cisplatin towards U251 human glioma, C6 rat glioma, SHSY5Yhuman neuroblastoma, L929 mouse fibrosarcoma and HL-60 human leukemia cell lines. Only in B16 mousemelanoma cells metformin augmented the cytotoxicity of cisplatin. In U251 glioma cells metforminsuppressed cisplatin-induced apoptotic cell death through inhibition of oxidative stress and caspaseactivation. The observed cytoprotection was apparently AMPK-independent, as metformin did not furtherincrease cisplatin-induced AMPK activation in U251 cells and other pharmacological AMPK activators failed toblock cisplatin-mediated apoptosis. On the other hand, metformin induced Akt activation in cisplatin-treatedcells and Akt inhibitor 10-DEBC hydrochloride or phosphoinositide 3-kinase/Akt inhibitor LY294002abolished metformin-mediated antioxidant and antiapoptotic effects. In conclusion, the antidiabetic drugmetformin reduces cisplatin in vitro anticancer activity through AMPK-independent upregulation of Aktsurvival pathway. These data warrant caution when considering metformin for treatment of diabetic cancerpatients receiving cisplatin or as a potential adjuvant in cisplatin-based chemotherapeutic regimens.
lsevier B.V.
© 2010 Published by Elsevier B.V.
1. Introduction
Metformin (N,N-dimethylbiguanide) is an oral anti-diabetic drugregularly used for the treatment of type 2 diabetes, particularly inoverweight and obese people (Correia et al., 2008a,b). The principalmode of its action is the stimulation of the intracellular enzyme AMP-activated protein kinase (AMPK), leading to inhibition of gluconeogen-esis in liver and increase in glucose uptake byperipheral tissues (Correiaet al., 2008a,b; Zhou et al., 2001). In addition to its antidiabeticproperties, metformin has also been shown to exert beneficial effect inthe treatment of polycystic ovary syndrome (Moll et al., 2007) and is
currently being considered for anticancer therapy (Gonzalez-AnguloandMeric-Bernstam, 2010). Namely, it has been shown that metformininhibits in vitro growth of various types of cancer cells, including breastcancer, prostate cancer and glioma cells (Zakikhani et al., 2006; BenSahra et al., 2008; Isakovic et al., 2007), as well as tumourigenesis andtumour progression in vivo (Ben Sahra et al., 2008; Buzzai et al., 2007;Huang et al., 2008; Anisimov et al., 2005). Moreover, population studieshave suggested that metformin decreases the incidence of cancer andcancer-relatedmortality in diabetic patients (Evans et al., 2005; Bowkeret al., 2006),while a recent report revealed thatmetformin improves theresponse to chemotherapy in diabetic patients with breast cancer(Jiralerspong et al., 2009).
Cisplatin [cis-diamminedichloroplatinum (II)] is one of the mostpotent chemotherapeutics used for treatment of testis, ovary, head andneck tumours (Kelland, 2007), as well as in adjuvant or neoadjuvanttherapy of various cancers, including glioma (Díaz et al., 2005; Wolf

42 K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50
et al., 2005). The major mechanism of cisplatin anticancer activity isinteraction with purine bases in DNA, leading to the formation of DNA–protein and DNA–DNA interstrand and intrastrand crosslinks that causetumour cell proliferation block and apoptosis (Jordan and Carmo-Fonseca, 2000). In order to circumvent cisplatin resistance and toxicity,the drug can be combined with other chemotherapeutics, such asgemcitabine, paclitaxel or Gleevec, which have all been found tosensitize different tumour cell types to cisplatin (Smith et al., 2006;Locke et al., 2003; Zhang et al., 2003). The ability of metformin toimprove the response to cisplatin-free chemotherapy (Díaz et al., 2005)and to potentate cisplatin-mediated killing of epithelial ovarian cancercells in vitro (Gotlieb et al., 2008) makes it a plausible candidate forcombination with cisplatin-based therapy. However, the cytotoxicefficiency of cisplatin/metformin combination has not been assessedin other types of cancer cells.
In the present study, we investigated the influence of metforminon cisplatin-mediated killing of various cancer cell lines. Surprisingly,we have found that metformin significantly reduced the cisplatintoxicity in most of the tested tumour cell lines. A more detailedanalysis performed on U251 glioma cells revealed that the cytopro-tective action of metformin was mediated through AMPK-indepen-dent activation of pro-survival signaling molecule Akt.
2. Materials and methods
2.1. Cells and cell culture
The human glioma cell line U251 and rat glioma cell line C6 werekindly donated by Dr. Pedro Tranque (Universidad de Castilla-LaMancha, Albacete, Spain). The mouse fibrosarcoma L929, humanneuroblastoma SHSY5Y and human leukemia HL-60 cell lineswere obtained from the European Collection of Animal Cell Cultures(Salisbury, UK), while B16 mouse melanoma cell line was akind gift from Dr. Sinisa Radulovic (Institute for Oncology andRadiology of Serbia, Belgrade, Serbia). Cells were maintained at 37 °C ina humidified atmosphere with 5% CO2, in a HEPES-bufferedRPMI 1640 cell culture medium supplemented with 5% fetal calfserum, L-glutamine, pyruvate and antibiotics (all from Sigma-Aldrich,St. Louis, MO). The cells were incubated in 96-well flat bottom plates(1×104 cells/well) for the cell viability assays, 24-well plates(1×105cells/well) for the flow cytometry analysis, or 90 mm Petridishes (2×106 cells) for theWestern blotting. Cells were rested for 24 hand then treated with cisplatin (Sigma-Aldrich) and/or metforminhydrochloride (99.9%; Hemofarm, Vrsac, Serbia), in the absence orpresence of an Akt inhibitor 10-DEBC hydrochloride (10-[4′-(N,N-diethylamino)butyl]-2-chlorophenoxazine hydrochloride), phosphoi-nositide 3-kinase (PI3K) inhibitor LY294002 (2-(4-morpholino)-8-phenyl-4H-1-benzopyran-4-one) (both from Tocris Bioscience, Ellis-ville, MO), antioxidant agent N-acetylcysteine, autophagy inhibitorbafilomycin A1, extracellular signal-regulated kinase (ERK) antagonistPD98059 (2′-amino-3′-methoxyflavone), AMP-activated protein kinaseinhibitor compound C (6-[4-(2-Piperidin-1-ylethoxy)phenyl]-3-pyri-din-4-ylpyrazolo[1,5-a]pyrimidine), AMP-activated protein kinase ag-onist aminoimidazole carboxamide ribonucleotide (AICAR) or aninhibitor of mitochondrial F1F0-ATPase oligomycin A (all from Sigma-Aldrich). Metformin and all other pharmacological modulators wereadded to cells 30 min before cisplatin.
2.2. Cell viability assays
Mitochondrial dehydrogenase-dependent reduction of 3-4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide (MTT) to for-mazan reflects the mitochondrial activity of cultured cells, while therelease of cytosolic lactate dehydrogenase (LDH) indicates the loss ofmembrane integrity that occurs in dead cells. After 24 h of incubationin culture medium containing different agents, the assays were
performed exactly as previously described (Raicevic et al., 2005).Mitochondrial-dependent production of formazan was assessed byan automated microplate reader at 570 nm, while the pyruvate-mediated conversion of 2,4-dinitrophenylhydrazine into visiblehydrazone precipitate in the LDH assay was measured at 492 nm.The results of the MTT are presented as percentage of controlvalues obtained in untreated cell cultures. The cytotoxicity in LDHrelease test was calculated using the formula: (E−C)/(T−C)×100,where E is the experimental absorbance of cell cultures, C is thecontrol absorbance of cell culture medium and T is the absorbancecorresponding to themaximal (100%) LDH release of TritonX-100-lysedcells.
2.3. Flow cytometry analysis of apoptosis and reactive oxygen speciesproduction
DNA fragmentation as amarker of apoptotic cell death was assessedbyflow cytometry analysis of DNA content in ethanol-fixed cells stainedwith the DNA-binding dye propidium iodide (PI) exactly as previouslydescribed (Harhaji et al., 2007). Alternatively, apoptotic cell death wasanalyzed by double staining with fluorescein-isothiocyanate (FITC)-conjugated annexin V and PI, in which annexin binds to the early andlate apoptotic cells withmembrane-exposed phosphatidylserine, whilePI labels only the late apoptotic/necrotic cells with membrane damage.Staining was performed according to the manufacturer's instructions(BD Pharmingen, San Diego, CA). Activation of caspases was measuredby flow cytometry after labeling the cells with a cell-permeable, FITC-conjugated pan-caspase inhibitor (ApoStat; R&D Systems, Minneapolis,MN) according to themanufacturer's instructions. Intracellular produc-tion of reactive oxygen species was determined by measuring theintensity of green fluorescence emitted by the redox-sensitive dyedihydrorhodamine 123 (Invitrogen, Paisley, UK), which was added tocell cultures (2 μM) at the beginning of treatment. The green (FL1) orred (FL2) fluorescence of cells stained with PI, annexin/PI, Apostat ordihydrorhodamine was analyzed with a FACSCalibur flow cytometer(BD, Heidelberg, Germany), and the numbers of early apoptotic(annexin+/PI−) and late apoptotic/necrotic (annexin+/PI+) cells, aswell as the percentage of cells displaying DNA fragmentation, caspaseactivation and increased production of reactive oxygen species weredetermined with the CellQuest Pro software (BD). A peak fluorescencegate was used to exclude cell aggregates from the DNA fragmentationanalysis.
2.4. Western blot analysis
The cells were lysed in a lysis buffer (30 mM Tris–HCl pH 8.0,150 mM NaCl, 1% NP-40, 1 mM phenylmethylsulfonylfluoride andprotease inhibitor cocktail; all from Sigma-Aldrich) on ice for 30 min,centrifuged at 14,000 g for 15 min at 4 °C, and the cell lysates werecollected. Equal amounts of protein from each sample were separatedby SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad,Marnes-la-Coquette, France). Following incubation with antibodiesrecognizing caspase-3 cleavage fragment, phospho-Akt, Akt, phos-pho-AMPK, AMPK,microtubule-associated protein light chain 3 (LC3),phospho-ERK1/2, ERK1/2 (Cell Signaling Technology, Beverly, MA) orβ-actin (Abcam, Cambridge, MA) as primary antibodies and perox-idase-conjugated goat anti-rabbit IgG (Jackson IP Laboratories, WestGrove, PA) as a secondary antibody, specific bands corresponding toinvestigated proteins were visualized using enhanced chemilumines-cence reagents for Western blot analysis (Amersham PharmaciaBiotech, Piscataway, NJ). The signal intensities were determined bydensitometry using NIH image software. The results are presentedrelative to the total levels of the specific protein (phospho-Akt,phospho-AMPK, phospho-ERK1/2), relative to actin (caspase-3 cleav-age fragment), or as LC3-II/LC3-I ratio, which increases duringautophagy due to the accumulation of LC3-II in autophagosomes
43K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50
(Eskelinen, 2005). The control values obtained in untreated cells werearbitrarily set to 1.
2.5. Mathematical analysis of metformin/cisplatin antagonism
To confirm the antagonistic interaction of metformin and cisplatinin inducing tumour cell death, U251 cells were treated with differentconcentrations of each agent alone and in combination and thecombination index was calculated using the approach by Chou andTalalay (1984). The values of combination indexN1 indicate antago-nistic interaction, while the valuesb1 or not significantly differentfrom 1 specify synergistic or additive interaction, respectively.
2.6. Statistical analysis
The statistical significance of the differences was analyzed by t-test(two independent samples) or one-way analysis of variance (ANOVA)followed by Newman–Keuls test for multiple comparisons. The valueof Pb0.05 was considered significant.
3. Results
3.1. Metformin inhibits in vitro anticancer activity of cisplatin
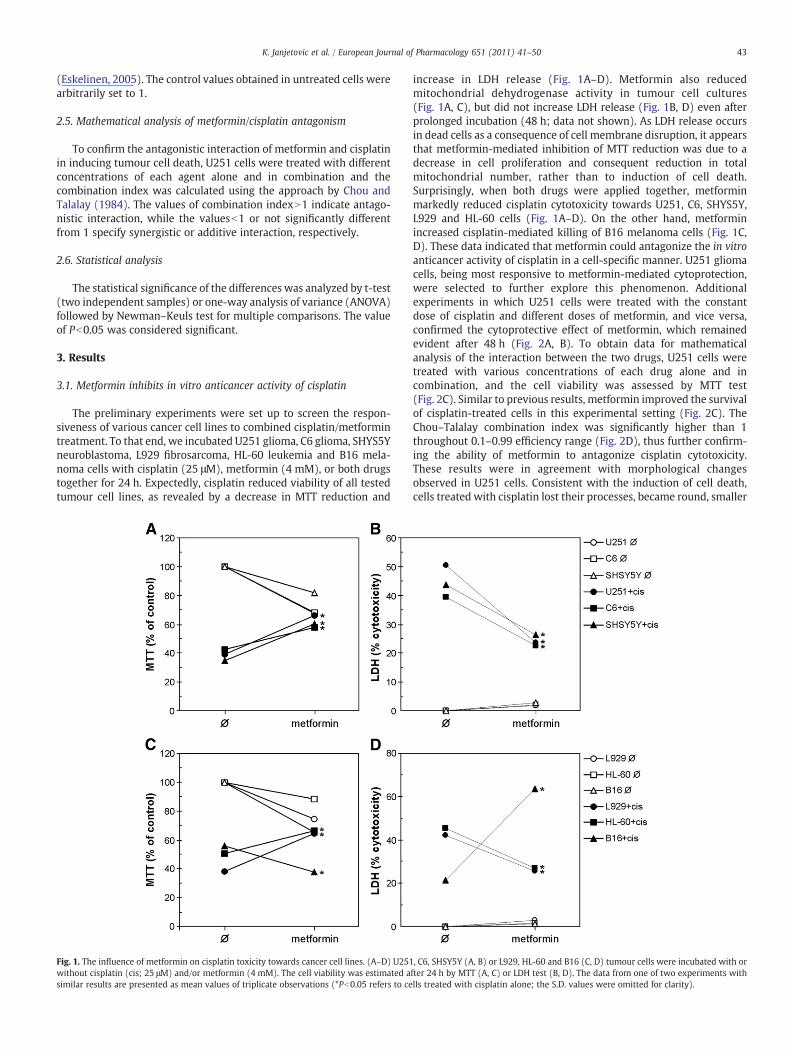
The preliminary experiments were set up to screen the respon-siveness of various cancer cell lines to combined cisplatin/metformintreatment. To that end, we incubated U251 glioma, C6 glioma, SHYS5Yneuroblastoma, L929 fibrosarcoma, HL-60 leukemia and B16 mela-noma cells with cisplatin (25 μM), metformin (4 mM), or both drugstogether for 24 h. Expectedly, cisplatin reduced viability of all testedtumour cell lines, as revealed by a decrease in MTT reduction and
Fig. 1. The influence of metformin on cisplatin toxicity towards cancer cell lines. (A–D) U251without cisplatin (cis; 25 μM) and/or metformin (4 mM). The cell viability was estimated asimilar results are presented as mean values of triplicate observations (*Pb0.05 refers to ce
increase in LDH release (Fig. 1A–D). Metformin also reducedmitochondrial dehydrogenase activity in tumour cell cultures(Fig. 1A, C), but did not increase LDH release (Fig. 1B, D) even afterprolonged incubation (48 h; data not shown). As LDH release occursin dead cells as a consequence of cell membrane disruption, it appearsthat metformin-mediated inhibition of MTT reduction was due to adecrease in cell proliferation and consequent reduction in totalmitochondrial number, rather than to induction of cell death.Surprisingly, when both drugs were applied together, metforminmarkedly reduced cisplatin cytotoxicity towards U251, C6, SHYS5Y,L929 and HL-60 cells (Fig. 1A–D). On the other hand, metforminincreased cisplatin-mediated killing of B16 melanoma cells (Fig. 1C,D). These data indicated that metformin could antagonize the in vitroanticancer activity of cisplatin in a cell-specific manner. U251 gliomacells, being most responsive to metformin-mediated cytoprotection,were selected to further explore this phenomenon. Additionalexperiments in which U251 cells were treated with the constantdose of cisplatin and different doses of metformin, and vice versa,confirmed the cytoprotective effect of metformin, which remainedevident after 48 h (Fig. 2A, B). To obtain data for mathematicalanalysis of the interaction between the two drugs, U251 cells weretreated with various concentrations of each drug alone and incombination, and the cell viability was assessed by MTT test(Fig. 2C). Similar to previous results, metformin improved the survivalof cisplatin-treated cells in this experimental setting (Fig. 2C). TheChou–Talalay combination index was significantly higher than 1throughout 0.1–0.99 efficiency range (Fig. 2D), thus further confirm-ing the ability of metformin to antagonize cisplatin cytotoxicity.These results were in agreement with morphological changesobserved in U251 cells. Consistent with the induction of cell death,cells treated with cisplatin lost their processes, became round, smaller
, C6, SHSY5Y (A, B) or L929, HL-60 and B16 (C, D) tumour cells were incubated with orfter 24 h by MTT (A, C) or LDH test (B, D). The data from one of two experiments withlls treated with cisplatin alone; the S.D. values were omitted for clarity).
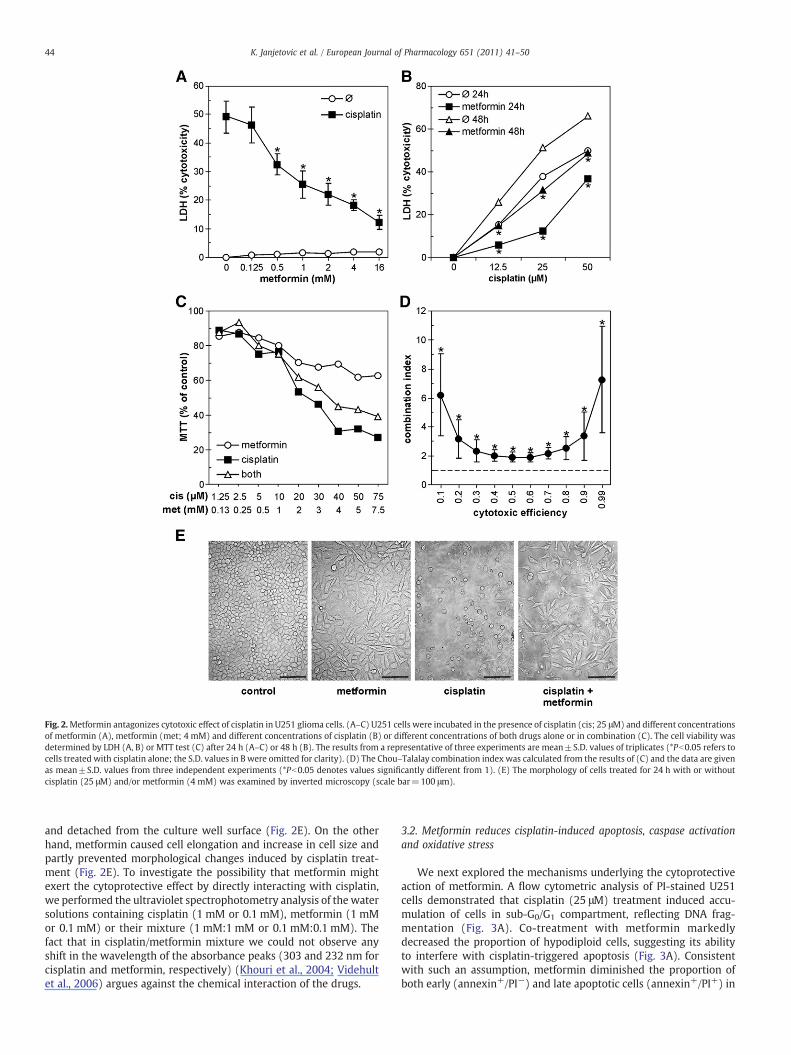
Fig. 2.Metformin antagonizes cytotoxic effect of cisplatin in U251 glioma cells. (A–C) U251 cells were incubated in the presence of cisplatin (cis; 25 μM) and different concentrationsof metformin (A), metformin (met; 4 mM) and different concentrations of cisplatin (B) or different concentrations of both drugs alone or in combination (C). The cell viability wasdetermined by LDH (A, B) or MTT test (C) after 24 h (A–C) or 48 h (B). The results from a representative of three experiments are mean±S.D. values of triplicates (*Pb0.05 refers tocells treated with cisplatin alone; the S.D. values in B were omitted for clarity). (D) The Chou–Talalay combination index was calculated from the results of (C) and the data are givenas mean±S.D. values from three independent experiments (*Pb0.05 denotes values significantly different from 1). (E) The morphology of cells treated for 24 h with or withoutcisplatin (25 μM) and/or metformin (4 mM) was examined by inverted microscopy (scale bar=100 μm).
44 K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50
and detached from the culture well surface (Fig. 2E). On the otherhand, metformin caused cell elongation and increase in cell size andpartly prevented morphological changes induced by cisplatin treat-ment (Fig. 2E). To investigate the possibility that metformin mightexert the cytoprotective effect by directly interacting with cisplatin,we performed the ultraviolet spectrophotometry analysis of the watersolutions containing cisplatin (1 mM or 0.1 mM), metformin (1 mMor 0.1 mM) or their mixture (1 mM:1 mM or 0.1 mM:0.1 mM). Thefact that in cisplatin/metformin mixture we could not observe anyshift in the wavelength of the absorbance peaks (303 and 232 nm forcisplatin and metformin, respectively) (Khouri et al., 2004; Videhultet al., 2006) argues against the chemical interaction of the drugs.
3.2. Metformin reduces cisplatin-induced apoptosis, caspase activationand oxidative stress
We next explored the mechanisms underlying the cytoprotectiveaction of metformin. A flow cytometric analysis of PI-stained U251cells demonstrated that cisplatin (25 μM) treatment induced accu-mulation of cells in sub-G0/G1 compartment, reflecting DNA frag-mentation (Fig. 3A). Co-treatment with metformin markedlydecreased the proportion of hypodiploid cells, suggesting its abilityto interfere with cisplatin-triggered apoptosis (Fig. 3A). Consistentwith such an assumption, metformin diminished the proportion ofboth early (annexin+/PI−) and late apoptotic cells (annexin+/PI+) in
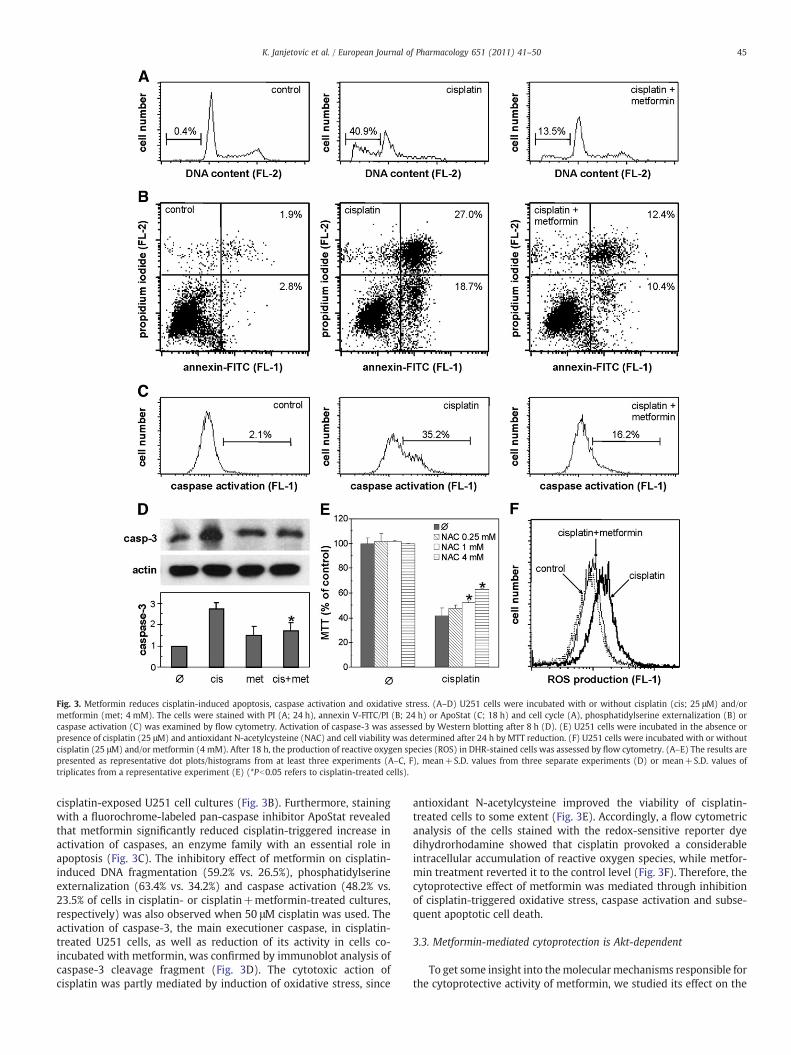
Fig. 3. Metformin reduces cisplatin-induced apoptosis, caspase activation and oxidative stress. (A–D) U251 cells were incubated with or without cisplatin (cis; 25 μM) and/ormetformin (met; 4 mM). The cells were stained with PI (A; 24 h), annexin V-FITC/PI (B; 24 h) or ApoStat (C; 18 h) and cell cycle (A), phosphatidylserine externalization (B) orcaspase activation (C) was examined by flow cytometry. Activation of caspase-3 was assessed by Western blotting after 8 h (D). (E) U251 cells were incubated in the absence orpresence of cisplatin (25 μM) and antioxidant N-acetylcysteine (NAC) and cell viability was determined after 24 h by MTT reduction. (F) U251 cells were incubated with or withoutcisplatin (25 μM) and/or metformin (4 mM). After 18 h, the production of reactive oxygen species (ROS) in DHR-stained cells was assessed by flow cytometry. (A–E) The results arepresented as representative dot plots/histograms from at least three experiments (A–C, F), mean+S.D. values from three separate experiments (D) or mean+S.D. values oftriplicates from a representative experiment (E) (*Pb0.05 refers to cisplatin-treated cells).
45K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50
cisplatin-exposed U251 cell cultures (Fig. 3B). Furthermore, stainingwith a fluorochrome-labeled pan-caspase inhibitor ApoStat revealedthat metformin significantly reduced cisplatin-triggered increase inactivation of caspases, an enzyme family with an essential role inapoptosis (Fig. 3C). The inhibitory effect of metformin on cisplatin-induced DNA fragmentation (59.2% vs. 26.5%), phosphatidylserineexternalization (63.4% vs. 34.2%) and caspase activation (48.2% vs.23.5% of cells in cisplatin- or cisplatin+metformin-treated cultures,respectively) was also observed when 50 μM cisplatin was used. Theactivation of caspase-3, the main executioner caspase, in cisplatin-treated U251 cells, as well as reduction of its activity in cells co-incubated with metformin, was confirmed by immunoblot analysis ofcaspase-3 cleavage fragment (Fig. 3D). The cytotoxic action ofcisplatin was partly mediated by induction of oxidative stress, since
antioxidant N-acetylcysteine improved the viability of cisplatin-treated cells to some extent (Fig. 3E). Accordingly, a flow cytometricanalysis of the cells stained with the redox-sensitive reporter dyedihydrorhodamine showed that cisplatin provoked a considerableintracellular accumulation of reactive oxygen species, while metfor-min treatment reverted it to the control level (Fig. 3F). Therefore, thecytoprotective effect of metformin was mediated through inhibitionof cisplatin-triggered oxidative stress, caspase activation and subse-quent apoptotic cell death.
3.3. Metformin-mediated cytoprotection is Akt-dependent
To get some insight into themolecularmechanisms responsible forthe cytoprotective activity of metformin, we studied its effect on the
46 K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50
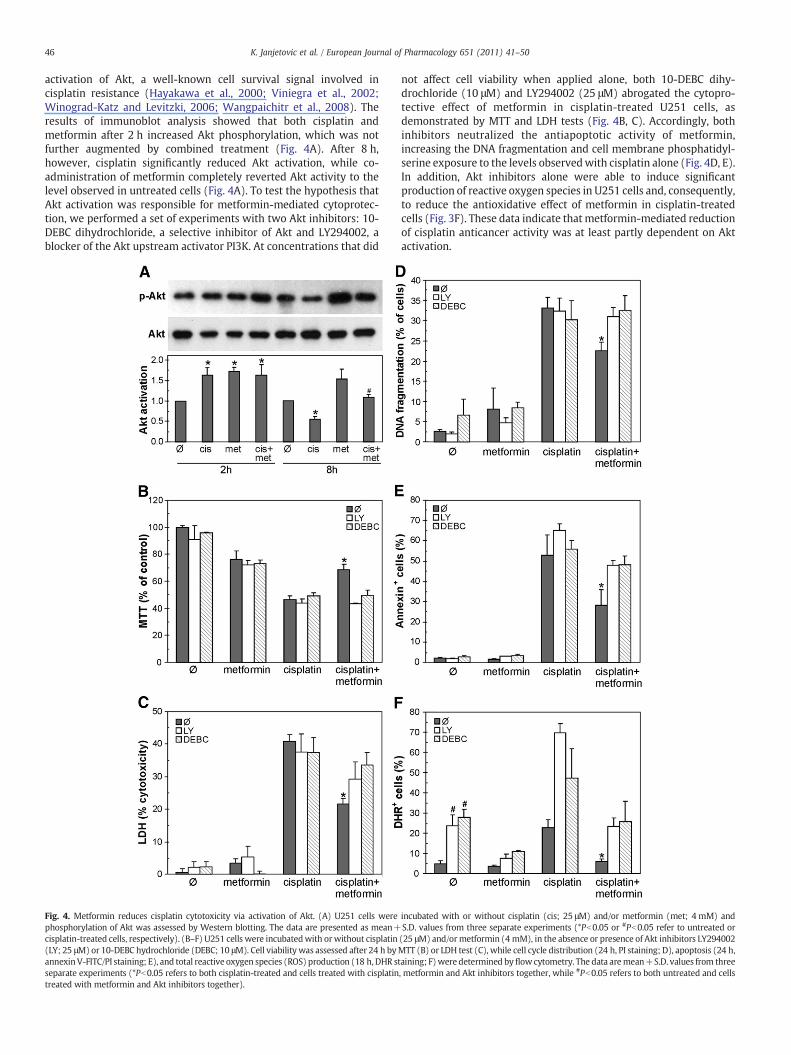
activation of Akt, a well-known cell survival signal involved incisplatin resistance (Hayakawa et al., 2000; Viniegra et al., 2002;Winograd-Katz and Levitzki, 2006; Wangpaichitr et al., 2008). Theresults of immunoblot analysis showed that both cisplatin andmetformin after 2 h increased Akt phosphorylation, which was notfurther augmented by combined treatment (Fig. 4A). After 8 h,however, cisplatin significantly reduced Akt activation, while co-administration of metformin completely reverted Akt activity to thelevel observed in untreated cells (Fig. 4A). To test the hypothesis thatAkt activation was responsible for metformin-mediated cytoprotec-tion, we performed a set of experiments with two Akt inhibitors: 10-DEBC dihydrochloride, a selective inhibitor of Akt and LY294002, ablocker of the Akt upstream activator PI3K. At concentrations that did
Fig. 4. Metformin reduces cisplatin cytotoxicity via activation of Akt. (A) U251 cells werephosphorylation of Akt was assessed by Western blotting. The data are presented as mean+cisplatin-treated cells, respectively). (B–F) U251 cells were incubatedwith orwithout cisplatin(LY; 25 μM) or 10-DEBC hydrochloride (DEBC; 10 μM). Cell viability was assessed after 24 h byannexinV-FITC/PI staining; E), and total reactive oxygen species (ROS) production (18 h, DHR stseparate experiments (*Pb0.05 refers to both cisplatin-treated and cells treated with cisplatintreated with metformin and Akt inhibitors together).
not affect cell viability when applied alone, both 10-DEBC dihy-drochloride (10 μM) and LY294002 (25 μM) abrogated the cytopro-tective effect of metformin in cisplatin-treated U251 cells, asdemonstrated by MTT and LDH tests (Fig. 4B, C). Accordingly, bothinhibitors neutralized the antiapoptotic activity of metformin,increasing the DNA fragmentation and cell membrane phosphatidyl-serine exposure to the levels observedwith cisplatin alone (Fig. 4D, E).In addition, Akt inhibitors alone were able to induce significantproduction of reactive oxygen species in U251 cells and, consequently,to reduce the antioxidative effect of metformin in cisplatin-treatedcells (Fig. 3F). These data indicate that metformin-mediated reductionof cisplatin anticancer activity was at least partly dependent on Aktactivation.
incubated with or without cisplatin (cis; 25 μM) and/or metformin (met; 4 mM) andS.D. values from three separate experiments (*Pb0.05 or #Pb0.05 refer to untreated or(25 μM) and/ormetformin (4 mM), in the absence or presence of Akt inhibitors LY294002MTT (B) or LDH test (C), while cell cycle distribution (24 h, PI staining; D), apoptosis (24 h,aining; F)were determined byflowcytometry. The data aremean+S.D. values from three, metformin and Akt inhibitors together, while #Pb0.05 refers to both untreated and cells

47K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50
3.4. Modulation of autophagy, ERK and AMPK activation is not involvedin metformin-mediated cytoprotection
It has previously been demonstrated that cisplatin toxicity can bereduced by inhibition of ERK, or augmented by inhibition of autophagyand AMPK (Mijatovic et al., 2005; Schweyer et al., 2004a,b; Kim et al.,2008; Harhaji-Trajkovic et al., 2009). Indeed, an ERK activation blockerPD 98059 reduced, while bafilomycin A1 and compound C, theinhibitors of autophagy and AMPK, respectively, significantly increasedthe cisplatin toxicity towards U251 glioma cells (Fig. 5A). We furtherinvestigated if the modulation of autophagy, ERK or AMPK activity wasresponsible for the cytoprotective effect of metformin.Metformin failedto reduce the phosphorylation of ERK induced after 8 h of cisplatintreatment (Fig. 5B), thus arguing against the involvement of ERKinhibition in metformin-mediated cell protection. Although metforminalone was apparently able to induce autophagy-associated conversionof LC3-I to LC3-II, its inability to further increase cisplatin-triggered LC3conversion (Fig. 5C) indicated that autophagy could not account for thecytoprotective activity of metformin. Finally, we performed a set ofexperiments to delineate the role AMPK, a key intracellular mediator ofmetformin action, in the reduction of cisplatin anticancer activity bymetformin. In contrast to metformin, two other AMPK activators—AICAR and oligomycin A—completely failed to rescue U251 cells fromcisplatin-mediated toxicity (Fig. 5D). Although AICAR readily activatedAMPK in U251 cells (Fig. 5E), it did not increase Akt phosphorylation(Fig. 5F), thus arguing against the involvement of AMPK in Aktactivation and improved survival of metformin-treated cells. Thisassumption was further supported by the finding that metformin,although able to induce AMPK by itself, could not further increasecisplatin-stimulated AMPK activation (Fig. 5E). It therefore appears thatreduction of cisplatin anticancer activity by metformin was notmediated through modulation of autophagy, ERK or AMPK activation.
4. Discussion
The present report for the first time demonstrates the ability ofmetformin to antagonize anticancer effect of cisplatin in a variety ofcancer cell lines. The observed ability of metformin to reduceapoptotic death of cisplatin-exposed U251 cells was apparentlymediated via up-regulation of Akt, inhibition of oxidative stress andblockade of caspase activation.
In light of the known anticancer action of metformin (Ben Sahraet al., 2008; Buzzai et al., 2007; Huang et al., 2008; Anisimov et al., 2005;Evans et al., 2005; Bowker et al., 2006), its ability to improve survival ofcisplatin-treated tumour cells might seem surprising. However, oneshould have in mind that the cytoprotective role of metformin hasalready been demonstrated in normal cells and tissues. Namely, it hasbeen shown thatmetformin can prevent high glucose-induced death ofbovine endothelial cell (Detaille et al., 2005), reduce the lipotoxicity inhuman pancreatic islets (Lupi et al., 2002) and protect brain, heart andliver tissues in rodent models of CNS oxidative damage (Correia et al.,2008a,b), cardiac ischemia (Bhamra et al., 2008) and hepatotoxicity(Poon et al., 2003). The protective effect of metformin in the presentstudy was not limited to U251 human glioma cells, since it was alsoobserved in C6 rat glioma, SHSY5Y human neuroblastoma, HL-60human leukemia and L929 mouse fibrosarcoma cell lines. On the otherhand, metformin markedly increased cisplatin toxicity in B16 mousemelanoma cells. Having in mind previously demonstrated ability ofmetformin to potentiate cisplatin-induced killing of epithelial ovariancancer cells (Gotlieb et al., 2008), it therefore appears that the effect ofmetformin on cisplatin-mediated death of tumour cells might dependon the tumour cell type.
Our data suggest that the cytoprotective action ofmetformin inU251cells was mediated through activation of serine/threonine kinase Akt, aPI3K downstream target that plays a critical role in controlling cellsurvival and apoptosis. Akt inhibits apoptosis directly through phos-
phorylation and inactivation of several pro-apoptotic targets, includingBad, forkhead transcription factors, c-Raf and caspase-9 (Datta et al.,1997; Brunet et al., 1999; Zimmermann and Moelling, 1999; Cardoneet al., 1998), or indirectly via activation of NF-κB and subsequenttranscription of pro-survival genes (Romashkova and Makarov, 1999).Consequently, many components of PI3K/Akt pathway are amplified intumours resistant to chemotherapy (Yuan andCantley, 2008), and it hasbeen demonstrated that inhibition of Akt sensitizes cancer cells tocisplatin (Hayakawa et al., 2000; Viniegra et al., 2002; Winograd-Katzand Levitzki, 2006). In accordance with these findings, we heredemonstrated that the antiapoptotic activity of metformin in U251cells was associated with prevention of cisplatin-triggered Akt down-regulation. The hypothesis that the cytoprotective activity ofmetforminwasmediated viaAkt survival pathway is also supportedby the ability ofAkt inhibitors to abrogate the antiapoptotic effect of metformin.
The present study confirms the previous findings that reactiveoxygen species are involved in cisplatin-mediated tumour cell death(Simons et al., 2007; Schweyer et al., 2004a,b). Although we did notdirectly explore the possible link between Akt activity and cisplatin-triggered oxidative stress, the ability of Akt inhibitors to inducegeneration of reactive oxygen species suggests that downregulation ofAkt contributed to oxidative stress and apoptosis induced by cisplatin.Such an assumption is consistent with the finding that metformin-mediated Akt activation was accompanied by a decrease in reactiveoxygen species production in cisplatin-treated cells. Accordingly, Akthas previously been reported to prevent oxidative stress-inducedmitochondrial permeability transition pore opening and protectcardiomyocytes, mesenchimal stem cells and prostate cancer cellsagainst oxidative stress (Bhamra et al., 2008; Wang et al., 2009;Muders et al., 2009). Nevertheless, having in mind the complexity ofAkt pro-survival functions, it is conceivable to assume that, in additionto antioxidant activity, some other Akt-dependent mechanism(s)could also contribute to the reduction of cisplatin anticancer activityby metformin.
Since cisplatin-induced AMPK activation has been proposed as asurvival signal (Kimet al., 2008; Harhaji-Trajkovic et al., 2009) andmostof themetformin actions aremediated by AMPK (Correia et al., 2008a,b;Zhou et al., 2001), it seemed plausible to assume that AMPK might beinvolved in Akt-dependent metformin protection from cisplatincytotoxicity. Moreover, although AMPK and Akt signaling pathwaysare usually regarded as independent signaling routes, AMPK wasapparently involved in vascular endothelial growth factor-induced Aktphosphorylation in breast cancer cells (Levine et al., 2007). However,several lines of evidence presented here, including the inability ofmetformin to increase cisplatin-induced AMPK activation and failure ofAMPK activators to mimic metformin-mediated Akt activation andcytoprotection, clearly indicate that Akt-mediated antiapoptotic actionof metformin was independent of AMPK. The inability of AMPKknockdown with small interfering RNA to affect Akt activation inU251 cells further supports such an assumption (Vucicevic et al.,unpublished data). This is not surprising having in mind previouslyreported AMPK-independent effects of metformin, including suppres-sion of glucose-6-phosphatase expression in rat hepatoma cells (Otaet al., 2009), inhibition of HER2 oncoprotein overexpression in breastcancer cells (Vazquez-Martin et al., 2009) and fatty acid oxidation andglucose utilization in the heart (Saeedi et al., 2008). We have recentlyreported that AMPK-dependent autophagy, a catabolic process respon-sible for the removal of long-lived proteins and damaged organellesthrough the lysosomal machinery, protects cancer cells from cisplatin-induced apoptotic death (Harhaji-Trajkovic et al., 2009). In agreementwith the proposed AMPK-independent antiapoptotic action of metfor-min, our finding that metformin was unable to augment autophagy incisplatin-treated cells indicates that autophagy was not involved in thereduction of cisplatin cytotoxicity. Finally, activation of variousmembers of the mitogen-activated protein kinase (MAP) signalingpathway, including ERK, is one of themajor intracellular events induced
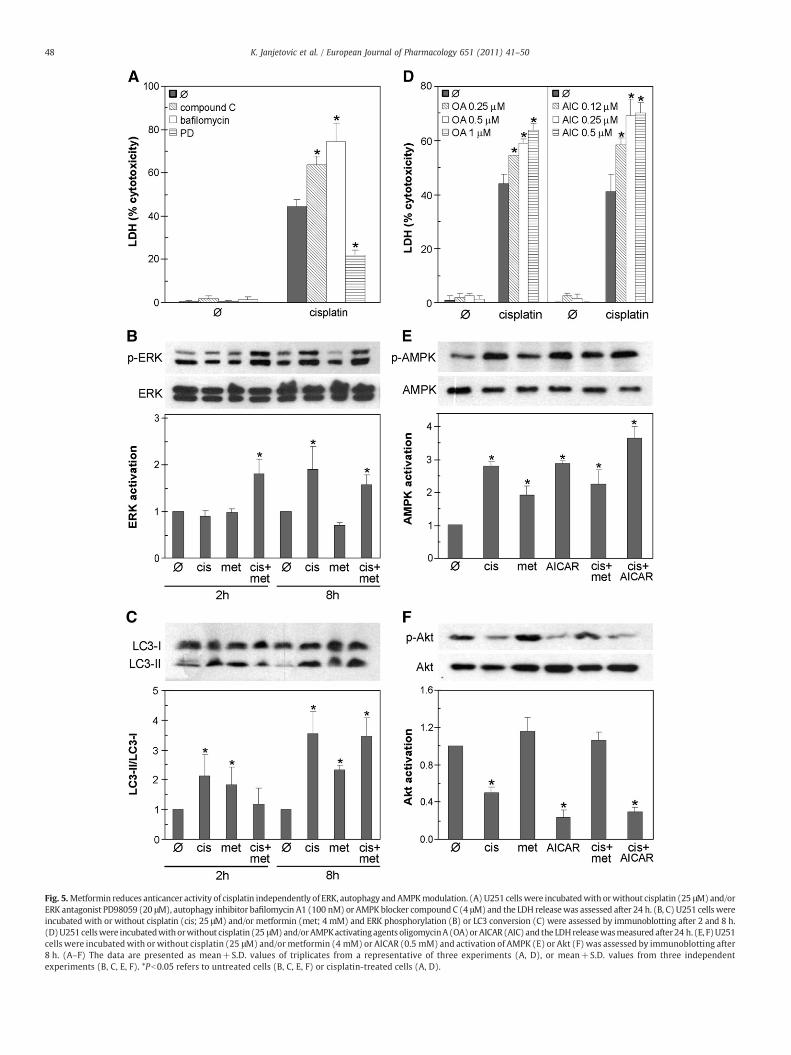
Fig. 5.Metformin reduces anticancer activity of cisplatin independently of ERK, autophagy andAMPKmodulation. (A) U251 cellswere incubatedwith orwithout cisplatin (25 μM)and/orERK antagonist PD98059 (20 μM), autophagy inhibitor bafilomycin A1 (100 nM) or AMPK blocker compound C (4 μM) and the LDH releasewas assessed after 24 h. (B, C) U251 cells wereincubated with or without cisplatin (cis; 25 μM) and/or metformin (met; 4 mM) and ERK phosphorylation (B) or LC3 conversion (C) were assessed by immunoblotting after 2 and 8 h.(D)U251cellswere incubatedwithorwithout cisplatin (25 μM)and/orAMPKactivatingagents oligomycinA (OA)orAICAR (AIC) and the LDHreleasewasmeasured after24 h. (E, F)U251cells were incubatedwith or without cisplatin (25 μM) and/or metformin (4 mM) or AICAR (0.5 mM) and activation of AMPK (E) or Akt (F) was assessed by immunoblotting after8 h. (A–F) The data are presented as mean+S.D. values of triplicates from a representative of three experiments (A, D), or mean+S.D. values from three independentexperiments (B, C, E, F). *Pb0.05 refers to untreated cells (B, C, E, F) or cisplatin-treated cells (A, D).
48 K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50

49K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50
in tumour cells by cisplatin treatment and is responsible for its toxicity(Mijatovic et al., 2005; Schweyer et al., 2004a,b). Nevertheless,since metformin failed to inhibit cisplatin-induced ERK activation,we concluded that the observed cytoprotective activity was ERKindependent.
In conclusion, our data demonstrate that the antidiabetic drugmetformin can rescue cancer cells from cisplatin-induced oxidativestress and apoptosis through Akt-dependent mechanisms not involvingmodulation of AMPK, autophagy or ERK. Although the fact that theseresults were obtained in cultured tumour cell lines calls for caution intheir interpretation, it nevertheless seems thatmetforminmightnot be aplausible candidate to enhance cisplatin sensitivity, at least in sometypes of cancer. Moreover, this study brings into question the benefit ofparallel usage of both drugs in patients who simultaneously suffer fromcancer and diabetes, a not-so-rare occurrence considering that morethan 7% of adults in developed countries have type 2 diabetes andtherefore increased risk from cancer (Wolff et al., 2010). On the otherhand, it would be interesting for further studies to test if the side effectsof cisplatin treatment, suchasnephrotoxicity andneurotoxicity (Barabaset al., 2008), could be mitigated by metformin co-administration.
Acknowledgements
The study was supported by the Ministry of Science and Technolog-ical Development of the Republic of Serbia (grant nos. 145073 and145058). The authors thank Prof. Dragan Micic and Dr. MirjanaSumarac-Dumanovic (Institute of Endocrinology, Diabetes andDiseasesof Metabolism, School of Medicine, Belgrade, Serbia) for providingAICAR, Dr. Dragomir Marisavljevic (Hemofarm, Vrsac, Serbia) forproviding metformin and Dr. Zoran Markovic for the help in UV–visanalysis.
References
Anisimov, V.N., Berstein, L.M., Egormin, P.A., Piskunova, T.S., Popovich, I.G., Zabezhinski,M.A., Kovalenko, I.G., Poroshina, T.E., Semenchenko, A.V., Provinciali, M., Re, F.,Franceschi, C., 2005. Effect of metformin on life span and on the development ofspontaneous mammary tumours in HER-2/neu transgenic mice. Exp. Gerontol. 40,685–693.
Barabas, K., Milner, R., Lurie, D., Adin, C., 2008. Cisplatin: a review of toxicities andtherapeutic applications. Vet. Comp. Oncol. 6, 1–18.
Ben Sahra, I., Laurent, K., Loubat, A., Giorgetti-Peraldi, S., Colosetti, P., Auberger, P., Tanti,J.F., Le Marchand-Brustel, Y., Bost, F., 2008. The antidiabetic drug metformin exertsan antitumoural effect in vitro and in vivo through a decrease of cyclin D1 level.Oncogene 27, 3576–3586.
Bhamra, G.S., Hausenloy, D.J., Davidson, S.M., Carr, R.D., Paiva, M., Wynne, A.M., Mocanu,M.M., Yellon, D.M., 2008. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. BasicRes. Cardiol. 103, 274–284.
Bowker, S.L., Majumdar, S.R., Veugelers, P., Johnson, J.A., 2006. Increased cancer-relatedmortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diab.Care 29, 254–258.
Brunet, A., Bonni, A., Zigmond, M.J., Lin, M.Z., Juo, P., Hu, L.S., Anderson, M.J., Arden, K.C.,Blenis, J., Greenberg, M.E., 1999. Akt promotes cell survival by phosphorylating andinhibiting a Forkhead transcription factor. Cell 96, 857–868.
Buzzai, M., Jones, R.G., Amaravadi, R.K., Lum, J.J., DeBerardinis, R.J., Zhao, F., Viollet, B.,Thompson, C.B., 2007. Systemic treatment with the antidiabetic drug metforminselectively impairs p53-deficient tumour cell growth. Cancer Res. 67, 6745–6752.
Cardone, M.H., Roy, N., Stennicke, H.R., Salvesen, G.S., Franke, T.F., Stanbridge, E., Frisch,S., Reed, J.C., 1998. Regulation of cell death protease caspase-9 by phosphorylation.Science 282, 1318–1321.
Chou, T.C., Talalay, P., 1984. Quantitative analysis of dose–effect relationships: thecombined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22,27–55.
Correia, S., Carvalho, C., Santos, M.S., Proença, T., Nunes, E., Duarte, A.I., Monteiro, P.,Seiça, R., Oliveira, C.R., Moreira, P.I., 2008a. Metformin protects the brain against theoxidative imbalance promoted by type 2 diabetes. Med. Chem. 4, 358–364.
Correia, S., Carvalho, C., Santos, M.S., Seiça, R., Oliveira, C.R., Moreira, P.I., 2008b.Mechanisms of action of metformin in type 2 diabetes and associated complica-tions: an overview. Mini Rev. Med. Chem. 8, 1343–1354.
Datta, S.R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y., Greenberg, M.E., 1997. Aktphosphorylation of BAD couples survival signals to the cell-intrinsic deathmachinery.Cell 91, 231–241.
Detaille, D., Guigas, B., Chauvin, C., Batandier, C., Fontaine, E.,Wiernsperger, N., Leverve, X.,2005. Metformin prevents high-glucose-induced endothelial cell death through amitochondrial permeability transition-dependent process. Diabetes 54, 2179–2187.
Díaz, R., Jordá, M.V., Reynés, G., Aparicio, J., Segura, A., Amador, R., Calderero, V., Beltrán,A., 2005. Neoadjuvant cisplatin and etoposide, with or without tamoxifen, prior toradiotherapy in high-grade gliomas: a single-center experience. Anticancer Drugs16, 323–329.
Eskelinen, E.L., 2005.Maturation of autophagic vacuoles inmammalian cells. Autophagy 1,1–10.
Evans, J.M., Donnelly, L.A., Emslie-Smith, A.M., Alessi, D.R., Morris, A.D., 2005.Metformin and reduced risk of cancer in diabetic patients. BMJ 330, 1304–1305.
Gonzalez-Angulo, A.M., Meric-Bernstam, F., 2010. Metformin: a therapeutic opportu-nity in breast cancer. Clin. Cancer Res. 16, 1695–1700.
Gotlieb, W.H., Saumet, J., Beauchamp, M.C., Gu, J., Lau, S., Pollak, M.N., Bruchim, I., 2008.In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol.Oncol. 110, 246–250.
Harhaji, L., Isakovic, A., Raicevic, N., Markovic, Z., Todorovic-Markovic, B., Nikolic, N.,Vranjes-Djuric, S., Markovic, I., Trajkovic, V., 2007. Multiple mechanisms underly-ing the anticancer action of nanocrystalline fullerene. Eur. J. Pharmacol. 568, 89–98.
Harhaji-Trajkovic, L., Vilimanovich, U., Kravic-Stevovic, T., Bumbasirevic, V., Trajkovic,V., 2009. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumourcells. J. Cell. Mol. Med. 13, 3644–3654.
Hayakawa, J., Ohmichi, M., Kurachi, H., Kanda, Y., Hisamoto, K., Nishio, Y., Adachi, K.,Tasaka, K., Kanzaki, T., Murata, Y., 2000. Inhibition of BAD phosphorylation either atserine 112 via extracellular signal-regulated protein kinase cascade or at serine 136via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res. 60,5988–5994.
Huang, X., Wullschleger, S., Shpiro, N., McGuire, V.A., Sakamoto, K., Woods, Y.L., McBurnie,W., Fleming, S., Alessi, D.R., 2008. Important role of the LKB1-AMPK pathway insuppressing tumourigenesis in PTEN-deficient mice. Biochem. J. 412, 211–221.
Isakovic, A., Harhaji, L., Stevanovic, D., Markovic, Z., Sumarac-Dumanovic, M., Starcevic,V., Micic, D., Trajkovic, V., 2007. Dual antiglioma action of metformin: cell cyclearrest and mitochondria-dependent apoptosis. Cell. Mol. Life Sci. 2007 (64),1290–1302.
Jiralerspong, S., Palla, S.L., Giordano, S.H., Meric-Bernstam, F., Liedtke, C., Barnett, C.M.,Hsu, L., Hung, M.C., Hortobagyi, G.N., Gonzalez-Angulo, A.M., 2009. Metformin andpathologic complete responses to neoadjuvant chemotherapy in diabetic patientswith breast cancer. J. Clin. Oncol. 27, 3297–3302.
Jordan, P., Carmo-Fonseca, M., 2000. Molecular mechanisms involved in cisplatincytotoxicity. Cell. Mol. Life Sci. 57, 1229–1235.
Kelland, L., 2007. The resurgence of platinum-based cancer chemotherapy. Nat. Rev.Cancer 7, 573–584.
Khouri, H., Collin, F., Bonnefont-Rousselot, D., Legrand, A., Jore, D., Gardès-Albert, M.,2004. Radical-induced oxidation of metformin. Eur. J. Biochem. 271, 4745–4752.
Kim, H.S., Hwang, J.T., Yun, H., Chi, S.G., Lee, S.J., Kang, I., Yoon, K.S., Choe, W.J., Kim, S.S.,Ha, J., 2008. Inhibition of AMP-activated protein kinase sensitizes cancer cells tocisplatin-induced apoptosis via hyper-induction of p53. J. Biol. Chem. 283,3731–3742.
Levine, Y.C., Li, G.K., Michel, T., 2007. Agonist-modulated regulation of AMP-activatedprotein kinase (AMPK) in endothelial cells. Evidence for an AMPK -N Rac1 -N Akt -Nendothelial nitric-oxide synthase pathway. J. Biol. Chem. 282, 20351–20364.
Locke, V., Davey, R., Davey, M., 2003. Paclitaxel sensitization of multidrug-resistant cellsto chemotherapy is independent of the cell cycle. Cytometry 43, 170–174.
Lupi, R., Del Guerra, S., Fierabracci, V., Marselli, L., Novelli, M., Patanè, G., Boggi, U.,Mosca, F., Piro, S., Del Prato, S., Marchetti, P., 2002. Lipotoxicity in human pancreaticislets and the protective effect of metformin. Diabetes 51 (Suppl 1), 134–137.
Mijatovic, S., Maksimovic-Ivanic, D., Radovic, J., Miljkovic, D., Kaludjerovic, G.N., Sabo, T.J.,Trajkovic, V., 2005. Aloe emodin decreases the ERK-dependent anticancer activity ofcisplatin. Cell. Mol. Life Sci. 62, 1275–1282.
Moll, E., van der Veen, F., van Wely, M., 2007. The role of metformin in polycystic ovarysyndrome: a systematic review. Hum. Reprod. Update 13, 527–537.
Muders, M.H., Zhang, H., Wang, E., Tindall, D.J., Datta, K., 2009. Vascular endothelialgrowth factor-C protects prostate cancer cells from oxidative stress by theactivation of mammalian target of rapamycin complex-2 and AKT-1. Cancer Res.69, 6042–6048.
Ota, S., Horigome, K., Ishii, T., Nakai, M., Hayashi, K., Kawamura, T., Kishino, A., Taiji, M.,Kimura, T., 2009. Metformin suppresses glucose-6-phosphatase expression by acomplex I inhibition and AMPK activation-independent mechanism. Biochem.Biophys. Res. Commun. 388, 311–316.
Poon, M.K., Chiu, P.Y., Mak, D.H., Ko, K.M., 2003. Metformin protects against carbontetrachloride hepatotoxicity in mice. J. Pharmacol. Sci. 93, 501–504.
Raicevic, N., Mladenovic, A., Perovic, M., Harhaji, L., Miljkovic, D., Trajkovic, V., 2005.Iron protects astrocytes from 6-hydroxydopamine toxicity. Neuropharmacology48, 720–731.
Romashkova, J.A., Makarov, S.S., 1999. NF-κB is a target of AKT in anti-apoptotic PDGFsignalling. Nature 401, 86–90.
Saeedi, R., Parsons, H.L., Wambolt, R.B., Paulson, K., Sharma, V., Dyck, J.R., Brownsey, R.W.,Allard, M.F., 2008. Metabolic actions of metformin in the heart can occur by AMPK-independent mechanisms. Am. J. Physiol. Heart Circ. Physiol. 294, 2497–2506.
Schweyer, S., Soruri, A., Heintze, A., Radzun, H.J., Fayyazi, A., 2004a. The role of reactiveoxygen species in cisplatin-induced apoptosis in human malignant testicular germcell lines. Int. J. Oncol. 25, 1671–1676.
Schweyer, S., Soruri, A., Meschter, O., Heintze, A., Zschunke, F., Miosge, N., Thelen, P.,Schlott, T., Radzun, H.J., Fayyazi, A., 2004b. Cisplatin-induced apoptosis in humanmalignant testicular germ cell lines depends on MEK/ERK activation. Br. J. Cancer91, 589–598.

50 K. Janjetovic et al. / European Journal of Pharmacology 651 (2011) 41–50
Simons, A.L., Ahmad, I.M., Mattson, D.M., Dornfeld, K.J., Spitz, D.R., 2007. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidativestress in human head and neck cancer cells. Cancer Res. 67, 3364–3370.
Smith, J.A., Gaikwad, A., Ramondetta, L.M., Wolf, J.K., Brown, J., 2006. Determination ofthe mechanism of gemcitabine modulation of cisplatin drug resistance in panel ofhuman endometrial cancer cell lines. Gynecol. Oncol. 103, 518–522.
Vazquez-Martin, A., Oliveras-Ferraros, C., Menendez, J.A., 2009. The antidiabetic drugmetformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition ofthe mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle 8, 88–96.
Videhult, P., Laurell, G., Wallin, I., Ehrsson, H., 2006. Kinetics of cisplatin and itsmonohydrated complex with sulfur-containing compounds designed for localotoprotective administration. Exp. Biol. Med. (Maywood) 231, 1638–1645.
Viniegra, J.G., Losa, J.H., Sánchez-Arévalo, V.J., Parada Cobo, C., Soria, V.M., Ramón yCajal, S., Sánchez-Prieto, R., 2002. Modulation of PI3K/Akt pathway by E1amediatessensitivity to cisplatin. Oncogene 21, 7131–7136.
Wang, X., Zhao, T., Huang, W., Wang, T., Qian, J., Xu, M., Kranias, E.G., Wang, Y., Fan, G.C.,2009. Hsp20-engineered mesenchymal stem cells are resistant to oxidative stressvia enhanced activation of Akt and increased secretion of growth factors. Stem Cells27, 3021–3031.
Wangpaichitr, M., Wu, C., You, M., Kuo, M.T., Feun, L., Lampidis, T., Savaraj, N., 2008.Inhibition of mTOR restores cisplatin sensitivity through down-regulation ofgrowth and anti-apoptotic proteins. Eur. J. Pharmacol. 591, 124–127.
Winograd-Katz, S.E., Levitzki, A., 2006. Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene 25, 7381–7390.
Wolf, I., Sadetzki, S., Catane, R., Karasik, A., Kaufman, B., 2005. Diabetes mellitus andbreast cancer. Lancet Oncol. 6, 103–111.
Wolff, J.E., Driever, P.H., Erdlenbruch, B., Kortmann, R.D., Rutkowski, S., Pietsch, T.,Parker, C., Metz, M.W., Gnekow, A., Kramm, C.M., 2010. Intensive chemotherapyimproves survival in pediatric high-grade glioma after gross total resection: resultsof the HIT-GBM-C protocol. Cancer 116, 705–712.
Yuan, T.L., Cantley, L.C., 2008. PI3K pathway alterations in cancer: variations on a theme.Oncogene 27, 5497–5510.
Zakikhani, M., Dowling, R., Fantus, I.G., Sonenberg, N., Pollak, M., 2006. Metformin is anAMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 66,10269–10273.
Zhang, P., Gao, W.Y., Turner, S., Ducatman, B.S., 2003. Gleevec (STI-571) inhibits lungcancer cell growth (A549) and potentiates the cisplatin effect in vitro. Mol. Cancer2, 1.
Zhou, G., Myers, R., Li, Y., Chen, Y., Shen, X., Fenyk-Melody, J., Wu,M., Ventre, J., Doebber,T., Fujii, N., Musi, N., Hirshman, M.F., Goodyear, L.J., Moller, D.E., 2001. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108,1167–1174.
Zimmermann, S., Moelling, K., 1999. Phosphorylation and regulation of Raf by Akt(protein kinase B). Science 286, 1741–1744.





























