Embed Size (px)
Citation preview

MUTATION IN BRIEF
HUMAN MUTATION Mutation in Brief #716 (2004) Online
© 2004 WILEY-LISS, INC. DOI: 10.1002/humu.9245
Received 30 July 2003; accepted revised manuscript 1 March 2004.
Novel and Recurrent Mutations in the NF1 Gene in Italian Patients with Neurofibromatosis Type 1 Alessandro De Luca1,4*, Annalisa Schirinzi1,4, Anna Buccino1, Irene Bottillo1, Lorenzo Sinibaldi1,4, Isabella Torrente1, Angela Ciavarella1, Tania Dottorini2, Roberto Porciello3, Sandra Giustini3, Stefano Calvieri3, and Bruno Dallapiccola1,4
1IRCCS-CSS, San Giovanni Rotondo and CSS-Mendel Institute, Rome, Italy; 2Department of Sperimental Medicine and Biochemical Sciences, University of Perugia, Perugia, Italy; 3Department of Dermatology-Venereology and Plastic and Reconstructive Surgery, University of Rome “La Sapienza”, Rome, Italy; 4Department of Experimental Medicine and Pathology, University of Rome “La Sapienza”, Rome, Italy
*Correspondence to: Alessandro De Luca, CSS-Mendel Institute, Viale Regina Margherita 261, 00198 Rome, Italy; Tel.: +39-06-44160543 ; Fax: +39-06-44160548. E-mail: [email protected] Grant sponsor: Italian Ministry of Health; Grant numbers: RC0304MD38, RF02MD13; Grant sponsor: Italian Ministry of Instruction, University and Research; Grant number: RBNE01JJ45 Communicated by Richard G.H. Cotton
Neurofibromatosis type 1 (NF1) is one of the most common autosomal dominant disorders in humans, affecting 1 in 3500 individuals. NF1 is a fully penetrant exhibiting a mutation rate some 10-fold higher compared to most other disease genes. As a consequence, a high number of cases (up to 50%) are sporadic. Mutation detection is complex due to the large size of NF1 gene, the presence of pseudogenes and the great variety of lesions. In the present study we attempted to delineate the NF1 mutational spectrum in the Italian population reporting four-year experience with the direct analysis of the whole NF1 coding region in 110 unrelated subjects affected by NF1. For each patient, the whole coding sequence and all splice sites were studied for mutations, either by the protein truncation test (PTT), or, most often, by denaturing high performance liquid chromatography (DHPLC). Mutations were identified in 75 (68%) patients. Twenty-two mutations were found to be novel. The detection rate for the different methods was 7/18 (39%) for PTT, and 68/103 (66%) for DHPLC. The mutations were evenly distributed along the NF1 coding sequence. Thirty-two of the 75 unrelated NF1 patients in which germline mutations were identified (32/75, 43%) harbour 23 different recurrent mutations. Fifteen sequence variants likely to represent non-pathogenic polymorphisms were observed at the NF1 locus. Genotype-phenotype analysis was unable to detect any obvious correlation. © 2004 Wiley-Liss, Inc.
KEY WORDS: neurofibromatosis type 1; NF1; DHPLC; PTT; genotype-phenotype; mutation detection
INTRODUCTION
Neurofibromatosis type 1 (NF1; MIM# 162200) is one of the most common autosomal dominant disorders in man, affecting 1 in 3500 people. The condition appears to be fully penetrant but has a highly variable expression, even within families (Huson SM and Hughes R, 1994). Diagnosis is based on the clinical criteria recommended by

2 De Luca et al.
an NIH Consensus Conference (Stumpf et al., 1988), which include multiple cafè-au-lait spots, cutaneous or subcutaneous neurofibromas, plexiform neurofibromas, axillary or inguinal freckling, optic gliomas, and iris Lisch nodules. Complications occur in some patients and include learning difficulties or mental retardation, focal neurological deficits, dysplastic skeletal lesions, hypertension, and, rarely, malignancy (Huson SM and Hughes R, 1994). The disease is fully penetrant and exhibits a mutation rate some 10-fold higher than that reported for most other disease genes (Huson et al., 1989). As a consequence, a high number of sporadic cases (up to 50%) is observed (Upadhyaya and Cooper, 1998). The disease is caused by mutations in the NF1 gene, one of the largest human genes, composed of 60 exons and spanning more than 300 kb of genomic DNA. Due to the large number of coding exons and the considerable mutational heterogeneity, the determination of the NF1 mutational spectrum has been complex. However, despite no true “hotspots” have been found in NF1 (Fahsold et al., 2000), recent data suggest that recurrence of several mutations are not so uncommon in NF1 patients (Ars et al., 2003).
In the present study we attempted to delineate the NF1 mutational spectrum in Italian patients by reporting our four years experience with the direct analysis of the whole NF1 coding region in 110 unrelated NF1 individuals. Mutations were identified in 75 (68%) patients, 32 (43%) of them bearing recurrent mutations. For each patient, the whole coding sequence and all splice sites were studied for mutations, either using protein truncation test (PTT), or, most often, denaturing high performance liquid chromatography (DHPLC). We have also tried to discern any possible correlation between genotype and phenotype.
PATIENTS AND METHODS
Patients One hundred-ten unrelated subjects (53 men; 57 women) were analysed in this study, including also 58 NF1
patients previously reported (Origone et al., 2002; De Luca et al., 2003). Clinical data confirming NF1 diagnostic criteria were available in 73 (66%) patients, while in the remaining cases either no clinical data were provided or patients fulfilled only one diagnostic criterion. All patients were of Italian origin except patient no. 51, who was of Indian descent. DNA from peripheral blood was extracted from each patient, applying one of several standard procedures. DNA samples from 150 healthy individuals (300 chromosomes) of Caucasian origin were screened to determine whether the missense/splicing mutations identified in this study were present in the normal population.
PTT RNA was extracted from fresh lymphocyte according to standard techniques (Chomczynski et al., 1987). Two
to five micrograms of RNA were reverse transcribed and the entire NF1 cDNA was then amplified with five pairs of primers that generate five overlapping fragments, as described elsewhere (Heim et al., 1995). After in vitro transcription/translation incorporating 35S methionine, the samples were electrophoresed on a 12.5% SDS-polyacrylamide gel. The gel was then dried and subjected to autoradiography.
DHPLC Fifty-six of the 60 exons of NF1 gene were amplified as described elsewhere (De Luca et al., 2003), whereas
four primers (4c, 5, 22 and 23-1) were redesigned according to Han et al. (2001). Denaturing high-performance liquid chromatography was carried out on a 3500HT WAVE DNA fragment analysis system (Transgenomic, Crewe, UK) equipped with a DNASep column (Transgenomic, Crewe, UK). PCR products were examined for heteroduplexes by subjecting 6 to 8 µl of each PCR product to a denaturation step (5 min at 95°C), followed by a cooling period at room temperature of 60 min. The PCR products were then separated (flow rate of 1.5 ml/min) through a 5% linear acetonitrile gradient. Commercially available WAVE OptimizedTM Buffers (A, B, D) (Transgenomic, Crewe, UK) and Syringe Solution (Transgenomic, Crewe, UK) were used to provide highly reproducible retention times with WAVE System instrumentation. Amplicons length, annealing temperature for PCR amplification, resolution temperature for DHPLC analysis have been previously reported (De Luca et al., 2003). Exons 4c, 5, 22 and 23-1 have been analysed according to Han et al. (2001).
Sequencing Direct DNA sequencing of the single exons were carried out using a dRhodamine Terminator Cycle Sequencing
Kit (PE Applied Biosystem, Foster City, CA, USA) and an ABI-PRISM 3100 Genetic Analyzer (PE Applied

Novel and Recurrent Mutations in Italian NF1 Patients 3
Biosystem, Foster City, CA, USA), according to the manufacturer’s instructions. Forward and reverse sequences were analysed and compared with the mRNA reference sequence (GenBank accession no. M82814.1) and with the chromosome 17 genomic contig reference sequence (GenBank accession no. NT010799.1). The first base (position +1) of the initiator methionine is taken as the start of the cDNA.
Comparative Molecular Modelling Comparative molecular modelling of the mutant K1423N protein was performed with the SWISS-MODEL and
the Swiss-Pdb Viewer (Guex et al., 1997), according to the PDB-Brookhaven crystal structure coordinates of neurofibromin (NF1) (Scheffzek et al., 1999). Secondary structure prediction were done using PSIpred (McGuffin et al., 2000) database.
RESULTS
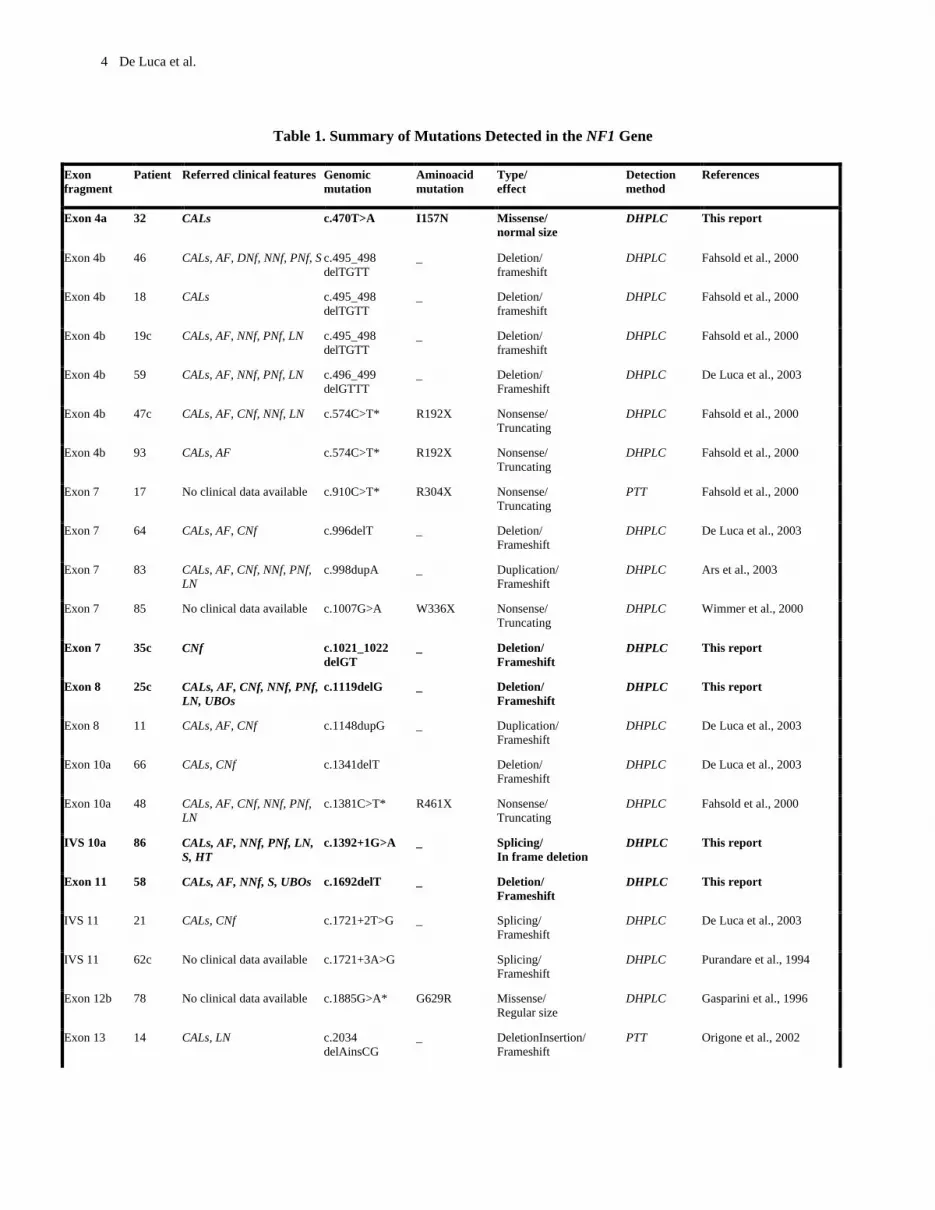
One hundred-ten unrelated NF1 patients were screened for mutations in the 60 exons and splice sites of the NF1 gene. A total of ninety sequence changes were identified (Table 1 and Table 2), 75 of which were considered pathogenic (Table 1). Seven of these were found by PTT, whereas 68 by DHPLC (Table 1). Eleven patients were analysed by both PTT and DHPLC. The detection rate for the different methods was 7/18 (39%) for PTT, and 68/103 (66%) for DHPLC. All mutations were characterized by repeat amplification and sequencing. Twenty-two mutations were found to be novel (Figure 1; Table 1).
Description of mutations Among the 75 mutations identified, we observed 25 (33%) nonsense mutations and 36 (48%) frameshift
mutations, including 20 deletions, 5 insertions, 6 out-of-frame exon skipping events, 4 complex rearrangements and 1 tandem duplication. Thus, as many as 61 (81%) mutations caused, directly or indirectly, a premature termination codon (PTC). Three mutations (4%) caused in-frame exon skipping. In addition, 10 (13%) missense mutations and 1 (1%) small in-frame deletion was also observed. Thirty-two patients (32/75, 43%) harbour 23 recurrent mutations (table 2) and the remaining 43 patients bear unique mutations (43/75, 57%).
The mutations were evenly distributed along the NF1 coding sequence (Figure 2). However, 51 (68%) of the 75 mutations clustered more commonly to 12 exons/flanking introns (4b, 7, 10a, 11, 15, 16, 23-1, 29, 31, 36, 37 and 45), which included only 23% of the coding region. Seventeen (23%) of the 75 mutations, were due to a C>T or G>A transition at CpG dinucleotides (Table 1). Of the eleven missense and/or small in-frame deletions, seven [c.1885G>A (G629R), c.2330G>C (W777S), c.2339C>A (T780K), c.2350T>C (W784R), c.2540T>C (L847P), c.2543G>A (G848E) and c.2903T>G (M968R)] were located within the putative cysteine/serine-rich domain (exons 11-17), one [c.4269G>T (K1423N)] in the GAP-related domain (exons 21-27a), and two [c.5794_5796delCTG (1932delL) and [c.5938G>A (G1980R)] in exon 31. The mutations c.470T>A (I156N), c.2330G>C (W777S), c.2543G>A (G848E) and c.5794_5796delCTG (1932delL) segregated with the disorder in familial cases, whereas the mutations c.1885G>A (G629R), c.2339C>A (T780K) and c.2350T>C (W784R) were de novo in a sporadic case. No all family members were available to confirm the de novo origin of c.2540T>C (L847P), c.2903T>G (M968R) and c.4269G>T (K1423N) mutations. Except for one patient (n=103), who carried two missense mutations, c.5938G>A (G1980R) and c.528T>A (D176E), all other eleven missense and/or small in-frame deletion mutations turned out to be the only sequence change present in the entire NF1 gene. Because the unaffected father of patient 103 was also a carrier of c.528T>A (D176E), we were able to determine that this amino acid exchange was not pathogenetic. All missense and/or small in-frame deletions were absent on 300 control chromosomes. Mutations c.470T>A (I156N), c.2339C>A (T780K), c.2350T>C (W784R), c.2540T>C (L847P), c.2543G>A (G848E), c.2903T>G (M968R), c.4269G>T (K1423N), c.5794_5796delCTG (1932delL) and c.5938G>A (G1980R) affected amino acids conserved in rattus (D45201), mouse (L10370), fugu (AF064564), and Drosophila (L26501), whereas mutation c.1885G>A (G629R) was conserved in rattus (D45201) and mouse (L10370), but not in fugu (AF064564) and Drosophila (L26501). Whether mutations c.1392+1G>A, c.6084-1G>A, c.2326-2A>G, c.6084-1G>A, c.6641+1G>A, c.6756+2T>A, c.7907+5G>A and c.7258+1G>T altered typical splice sites, c.7907+5G>A mutations did not pertain to the group of typical splicing mutations. However, c.7907+5G>A change was not detected in patient’s parents (originated de novo), and it was not found in 150 healthy subjects. Minigene assay (Baralle et al., 2003) is on going to test for the influence of c.7907+5G>A on the splicing process (data not shown).
4 De Luca et al.
Table 1. Summary of Mutations Detected in the NF1 Gene
Exon fragment
Patient Referred clinical features Genomic mutation
Aminoacid mutation
Type/ effect
Detection method
References
Exon 4a 32 CALs c.470T>A I157N Missense/ normal size
DHPLC This report
Exon 4b
46
CALs, AF, DNf, NNf, PNf, S c.495_498 delTGTT
_ Deletion/ frameshift
DHPLC
Fahsold et al., 2000
Exon 4b 18 CALs c.495_498 delTGTT
_ Deletion/ frameshift
DHPLC Fahsold et al., 2000
Exon 4b
19c
CALs, AF, NNf, PNf, LN c.495_498 delTGTT
_ Deletion/ frameshift
DHPLC
Fahsold et al., 2000
Exon 4b
59
CALs, AF, NNf, PNf, LN c.496_499 delGTTT
_ Deletion/ Frameshift
DHPLC
De Luca et al., 2003
Exon 4b 47c CALs, AF, CNf, NNf, LN c.574C>T* R192X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 4b 93 CALs, AF c.574C>T* R192X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 7 17 No clinical data available c.910C>T* R304X Nonsense/ Truncating
PTT Fahsold et al., 2000
Exon 7 64 CALs, AF, CNf c.996delT _ Deletion/ Frameshift
DHPLC De Luca et al., 2003
Exon 7 83 CALs, AF, CNf, NNf, PNf, LN
c.998dupA _ Duplication/ Frameshift
DHPLC Ars et al., 2003
Exon 7 85 No clinical data available c.1007G>A W336X Nonsense/ Truncating
DHPLC Wimmer et al., 2000
Exon 7 35c CNf c.1021_1022 delGT
_ Deletion/ Frameshift
DHPLC This report
Exon 8 25c CALs, AF, CNf, NNf, PNf, LN, UBOs
c.1119delG _ Deletion/ Frameshift
DHPLC This report
Exon 8 11 CALs, AF, CNf c.1148dupG _ Duplication/ Frameshift
DHPLC De Luca et al., 2003
Exon 10a 66 CALs, CNf c.1341delT Deletion/ Frameshift
DHPLC De Luca et al., 2003
Exon 10a 48 CALs, AF, CNf, NNf, PNf, LN
c.1381C>T* R461X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
IVS 10a 86 CALs, AF, NNf, PNf, LN, S, HT
c.1392+1G>A _ Splicing/ In frame deletion
DHPLC This report
Exon 11 58 CALs, AF, NNf, S, UBOs c.1692delT _ Deletion/ Frameshift
DHPLC This report
IVS 11 21 CALs, CNf c.1721+2T>G _ Splicing/ Frameshift
DHPLC De Luca et al., 2003
IVS 11 62c No clinical data available c.1721+3A>G Splicing/ Frameshift
DHPLC Purandare et al., 1994
Exon 12b 78 No clinical data available c.1885G>A* G629R Missense/ Regular size
DHPLC Gasparini et al., 1996
Exon 13 14 CALs, LN c.2034 delAinsCG
_ DeletionInsertion/ Frameshift
PTT Origone et al., 2002
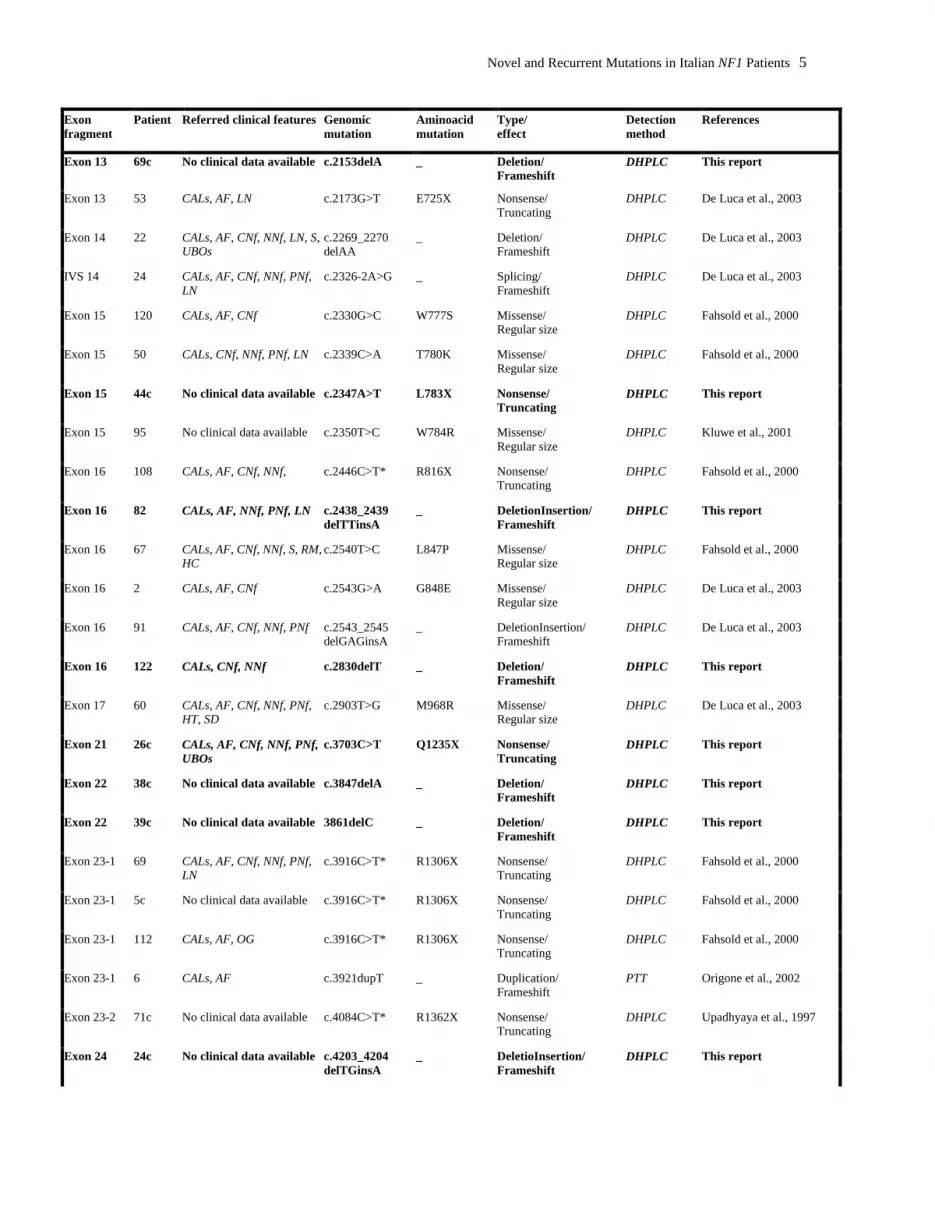
Novel and Recurrent Mutations in Italian NF1 Patients 5
Exon fragment
Patient Referred clinical features Genomic mutation
Aminoacid mutation
Type/ effect
Detection method
References
Exon 13 69c No clinical data available c.2153delA _ Deletion/ Frameshift
DHPLC This report
Exon 13 53 CALs, AF, LN c.2173G>T E725X Nonsense/ Truncating
DHPLC De Luca et al., 2003
Exon 14 22 CALs, AF, CNf, NNf, LN, S, UBOs
c.2269_2270 delAA
_ Deletion/ Frameshift
DHPLC De Luca et al., 2003
IVS 14 24 CALs, AF, CNf, NNf, PNf, LN
c.2326-2A>G _ Splicing/ Frameshift
DHPLC De Luca et al., 2003
Exon 15 120 CALs, AF, CNf c.2330G>C W777S Missense/ Regular size
DHPLC Fahsold et al., 2000
Exon 15 50 CALs, CNf, NNf, PNf, LN c.2339C>A T780K Missense/ Regular size
DHPLC Fahsold et al., 2000
Exon 15 44c No clinical data available c.2347A>T L783X Nonsense/ Truncating
DHPLC This report
Exon 15 95 No clinical data available c.2350T>C W784R Missense/ Regular size
DHPLC Kluwe et al., 2001
Exon 16 108 CALs, AF, CNf, NNf, c.2446C>T* R816X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 16 82 CALs, AF, NNf, PNf, LN c.2438_2439 delTTinsA
_ DeletionInsertion/ Frameshift
DHPLC This report
Exon 16 67 CALs, AF, CNf, NNf, S, RM, HC
c.2540T>C L847P Missense/ Regular size
DHPLC Fahsold et al., 2000
Exon 16 2 CALs, AF, CNf c.2543G>A G848E Missense/ Regular size
DHPLC De Luca et al., 2003
Exon 16 91 CALs, AF, CNf, NNf, PNf c.2543_2545 delGAGinsA
_ DeletionInsertion/ Frameshift
DHPLC De Luca et al., 2003
Exon 16 122 CALs, CNf, NNf c.2830delT _ Deletion/ Frameshift
DHPLC This report
Exon 17 60 CALs, AF, CNf, NNf, PNf, HT, SD
c.2903T>G M968R Missense/ Regular size
DHPLC De Luca et al., 2003
Exon 21 26c CALs, AF, CNf, NNf, PNf, UBOs
c.3703C>T Q1235X Nonsense/ Truncating
DHPLC This report
Exon 22 38c No clinical data available c.3847delA _ Deletion/ Frameshift
DHPLC This report
Exon 22 39c No clinical data available 3861delC _ Deletion/ Frameshift
DHPLC This report
Exon 23-1 69 CALs, AF, CNf, NNf, PNf, LN
c.3916C>T* R1306X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 23-1 5c No clinical data available c.3916C>T* R1306X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 23-1 112 CALs, AF, OG c.3916C>T* R1306X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 23-1 6 CALs, AF c.3921dupT _ Duplication/ Frameshift
PTT Origone et al., 2002
Exon 23-2 71c No clinical data available c.4084C>T* R1362X Nonsense/ Truncating
DHPLC Upadhyaya et al., 1997
Exon 24 24c No clinical data available c.4203_4204 delTGinsA
_ DeletioInsertion/ Frameshift
DHPLC This report

6 De Luca et al.
Exon fragment
Patient Referred clinical features Genomic mutation
Aminoacid mutation
Type/ effect
Detection method
References
Exon 24 39 CALs, AF, CNf, NNf, PNf, LN, S, HT, PA
c.4269G>T K1423N Missense/ Normal size
DHPLC De Luca et al., 2003
Exon 26 73 CALs c.4481_4482 delAG
_ Deletion/ Frameshift
DHPLC De Luca et al., 2003
Exon 27a 65 CALs, AF, NNf c.4537C>T* R1513X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 29 25 CALs, CNf c.5224C>T Q1742X Nonsense/ Truncating
DHPLC De Luca et al., 2003
Exon 29 107 No clinical data available c.5264C>G S1755X Nonsense/ Truncating
DHPLC Messiaen et al., 2000
Exon 29 70c No clinical data available c.5339T>G L1780X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 30 119 CALs, AF, CNf, NNf, LN, S
c.5556_5578 dup
TandemDuplication/ Frameshift
DHPLC This report
Exon 30 116 No clinical data available c.5656delT _ Deletion/ Frameshift
DHPLC This report
Exon 31 9 CALs, AF, CNf c.5794_5796 delCTG
1932delL Deletion/ In frame deletion
DHPLC De Luca et al., 2003
Exon 31 103 CALs, AF, LN, S c.5938G>A G1980R Missense/ Regular size
DHPLC De Luca et al., 2003
Exon 31 104 CALs, CNf, NNf, PNf c.5839C>T* R1947X Nonsense/ Truncating
DHPLC Klose et al., 1999
Exon 31 37 No clinical data available c.5839C>T* R1947X Nonsense/ Truncating
DHPLC Klose et al., 1999
Exon 31 96 CALs, AF, PNf c.5893A>T K1965X Nonsense/ Truncating
DHPLC De Luca et al., 2003
Exon 32 13c No clinical data available c.6084+1G>A _ Splicing/ in frame deletion
DHPLC This report
Exon 35 128 No clinical data available c.6641+1G>A _ Splicing/ Frameshift
DHPLC This report
Exon 36 7 CALs c.6709C>T* R2237X Nonsense/ Truncating
PTT Fahsold et al., 2000
Exon 36 33 CALs, AF, CNf, NNf, PNf, LN, SD
c.6709C>T* R2237X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 36 36 CALs, AF, NNf, S c.6709C>T* R2237X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 36 28 CALs, AF, CNf, NNf, PNf c.6756+2T>A Splicing/ Frameshift
DHPLC De Luca et al., 2003
Exon 37 5 CALs c.6772delT _ Deletion/ Frameshift
PTT Origone et al., 2002
Exon 37 4 No clinical data available c.6789_6792 delTTAC
_ Deletion/ Frameshift
PTT Robinson et al., 1995
Exon 37 73c No clinical data available c.6791dupA _ Duplication/ Frameshift
DHPLC This report
Exon 37 121 CALs c.6791dupA _ Duplication/ Frameshift
DHPLC This report
Exon 37 1 CALs, AF, CNf c.6792C>A Y2264X Nonsense/ Truncating
PTT Robinson et al., 1995

Novel and Recurrent Mutations in Italian NF1 Patients 7
Exon fragment
Patient Referred clinical features Genomic mutation
Aminoacid mutation
Type/ effect
Detection method
References
IVS 40 20 CALs, CNf c.7258+1G>T _ Splicing/ In frame deletion
DHPLC De Luca et al., 2003
Exon 44 99 CALs, NNf, PNf c.7682_7683 delAG
_ Deletion/ Frameshift
DHPLC This report
Exon 45 109 CALs, AF, CNf, NNf, PNf, LN, S, UBOs
c.7846C>T* R2616X Nonsense/ Truncating
DHPLC Fahsold et al., 2000
Exon 45 51 CALs, AF, NNf, PNf c.7900delC _ Deletion/ Frameshift
DHPLC This report
Exon 45 12c CALs c.7907+5G>A _ Splicing/ Frameshift
DHPLC This report
Legend: DHPLC: Denaturing High Performance Liquid Chromatography; PTT: Protein Truncation Test; CALs: Cafè-au-Lait Spots, AF: Axillary Freckling; DNf: Dermal Neurofibromas; NNf: Nodular Neurofibromas; LN: Lish Nodules; PNf: Plexiform Neurofibromas; S: Scoliosis, HT: Hypertension, PA: Pseudoarthrosis; SD: Sphenoid Displasia; MR: Mental Retardation; UBOs: Unidentified Bright Objects; HC: Hydrocephalus. Nucleotide numbering is based on cDNA sequence (GenBank accession no. M82814.1) and genomic reference sequence (GenBank accession no. NT010799.1). The first base (position +1) of the initiator methionine is taken as the start of the cDNA. *Single base pair substitutions due to C>T transition at CpG dinucleotides. Heterozygosity is based on the number of patients. ^ heterozygosity is based on the number of controls. Nucleotide numbering is based on cDNA sequence (GenBank accession no. M82814.1) and on genomic reference sequence (GenBank accession no. NT010799.1). The first base (position +1) of the initiator methionine is taken as the start of the cDNA.
Recurrent mutations Thirty-two of the 75 NF1 patients in which germline mutations were identified (32/75, 43%) harboured 23 different
recurrent mutations (Table 1). These mutations have been found in different unrelated patients of our set and/or have been previously published by other groups (Table 1). Seventeen of the 32 (53%) recurrent mutation were due to a C>T or G>A transition at CpG dinucleotides, known to be prone to mutation if methylated. Among the recurrent mutations, the three most common were c.495_498delTGTT in exon 4b (three patients), c.3916C>T (R1306X) in exon 23-1 (three patients), and c.6709C>T (R2237X) in exon 36 (three patients). These mutations represent the 28% of recurrent mutations and the 12% of the overall identified mutations. c.3916C>T (R1306X) and c.6709C>T (R2237X) are C>T substitutions at CpG dinucleotides, which may explain their recurrence.
Modelling analysis of K1423N Comparative molecular modelling was performed to determine the putative effects of K1423N on the
neurofibromin function. The analysis revealed that the introduced asparagine (N1423) determines the loss of the hydrogen bond between K1423 and E1437, and instead, the new introduced aminoacid is bended toward the aminoacid K1419 establishing an additive hydrogen bond with the latter (Figure 3).
Polymorphisms Fifteen sequence variants likely to represent non-pathogenic polymorphisms were observed at the NF1 locus (Table
2). They included one missense c.528T>A (D176E), 3 silent (c.846G>A; c.4866G>C and c.5172G>A), and 11 intronic changes. Two of these have not been reported previously (Table 2). With the exception of c.(730-6)A>C and c.1163-24)delT, all polymorphisms were found in either controls and/or in patients in association with a lesion deemed to be of pathological significance. The variant c.(730-6)A>C was tested for its influence on the splicing process by mRNA sequencing, but no exon skipping was observed (data not shown). Consequently, c.(730-6)A>C variant was included in the polymorphisms’ list.

8 De Luca et al.
Clinical signs versus type of mutation Fifty-one NF1 patients were scored for the presence or absence of cafè-au-lait spots, axillary freckling, dermal and nodular neurofibromas, iris Lisch nodules, plexiform neurofibromas, scoliosis, hypertension, pseudoarthrosis, sphenoid dysplasia, and mental retardation (Table 1; Table 3). Mutation detection rate, frequency of truncating mutations and frequency of mutation’s type (nonsense, frameshift, splicing and missense mutations) were analysed for each specific clinical feature (Table 3). No obvious correlation could be detected.
Table 2. Summary of Polymorphisms Detected in the NF1 gene
Location Nucleotide/ (aminoacid) change
Heterozygosity Type of change Reference
IVS 3 c.288+41G>A 8/40 (21%)* Intronic De Luca et al., 2003
Exon 4b c.528T>A/ (D176E) 1/103 (1%)* Missense Fashold et al., 2000
IVS 5 c.730-6A>C 1/40 (2%)* Intronic De Luca et al., 2003
Exon 6 c.846G>A 1/40 (2%)* Silent De Luca et al., 2003
IVS 7 c.1163-24delT 1/40 (2%)* Intronic De Luca et al., 2003
IVS 7 c.1063-28G>C 3/40 (8%)* Intronic De Luca et al., 2003
IVS 20 c.3496+33C>A 13/40 (33%)* Intronic De Luca et al., 2003
IVS 25 c.4368-46G>C 3/40 (8%)* Intronic Fashold et al., 2000
Exon 28 c.4866G>C 2/72 (3%)^ Silent This report
Exon 28 c.5172G>A 2/72 (3%)^ Silent Fashold et al., 2000
IVS 29 c.5546+18T>A 16/40 (41%)* Intronic De Luca et al., 2003
IVS 31 c.5943+114A>T 1/72 (2%)^ Intronic This report
IVS 34 c.6579+45T>A 3/72 (5%)* Intronic Fahsold et al., 2000
IVS 39 c.7126+37G>C 2/40 (5%)* Intronic Rodenhiser and Hovland, 1995
IVS 41 c.7395-29G>A 4/40 (10%)* Intronic Fahsold et al., 2000

Novel and Recurrent Mutations in Italian NF1 Patients 9
Figure 1. DHPLC chromatograms for novel NF1 mutations (wild type vs. mutation). Most often, the presence of a mutation in the sample was detected as a change in the number of peaks (i. e. mutation c.3847delA in exon 22, or mutation c.7682_7683delAG in exon 44). Some mutations appeared as a slight broadening of the single peak (mutation c.5656delT in exon 30), or as to a shoulder on the peak (mutation c.4203_4204delTGinsA in exon 24), or as a subtle change in the retention time [mutation c.3703C>T (Q1235X) in exon 21].

10 De Luca et al.
Figure 2. Distribution of NF1 mutations in 75 neurofibromatosis type 1 patients. Exons are represented consecutively on the X axis and the number of mutations identified on the Y axis.
Figure 3. Surface representation of neurofibromin protein molecular model. Utilizing the Brookhaven crystal structure coordinates of neurofibromin (Scheffzek et al., 1999) as a structural reference and employing comparative molecular modeling with the SWISS-MODEL and the Swiss-Pdb Viewer (Guex et al., 1997), a proposed model structure of the mutant NF1 (K1423N) was generated (right) and compared to the native protein (left). Aminoacids K1423 (α-helix), E1437 (loop) and 1419 (α-helix) are shown. Hydrogen bonds are shown as broken yellow lines.

Novel and Recurrent Mutations in Italian NF1 Patients 11
*Frequency of truncating mutations and frequency of different types of mutation (nonsense, frameshift, splicing and missense mutations) are reported for each specific clinical feature.
DISCUSSION
Here we report the NF1 mutational analysis of 110 unrelated subjects suspected of having NF1 in whom we have detected 75 mutations (68%). Patients were analysed by means of PTT, and or DHPLC, for mutations in the whole coding sequence and the splice sites of the NF1 gene. The mutation detection rate between PTT and DHPLC methods differed markedly (39% vs 66). The 66% detection rate observed in the present DHPLC study is lower, but in line with our previous reports (De Luca et al., 2003). One should take into account that in our previous study only patients fulfilling NF1 consensus diagnostic criteria were included, whereas in present, clinical data confirming NF1 diagnostic criteria were available in 73/110 (66%) of the subjects studied. No differences in clinical presentation were observed between patients with or without mutations. Unfortunately, we cannot exclude the possibility that patients who did not have NF1 were included in the screening, because some of the patients were diagnosed by others. However, the DHPLC protocol used here might have failed to detect some mutations, since no technique is 100% sensitive. Large deletions, comprising the whole NF1 gene or a major part of it, multi-exon deletions, large duplications or inversions, which together represent more than 15-25% of the total spectrum of NF1 mutations, would have escaped the DHPLC scanning (Cowley et al., 1998; Riva et al., 2000). Incomplete assessment of intronic and regulatory sequences may account for other unidentified mutations. However, we found DHPLC-based heteroduplex analysis more efficient compared to PTT, in terms of mutation detection. Probably, the inability of PTT to identify missense mutations, the occurrence of nonsense-mediated mRNA decay leading to allelic exclusion (Osborn and Upadhyaya, 1999), and the relatively low level of expression of the NF1 transcript in lymphocytes are limiting the sensitivity of PTT screening in NF1 patients.
Table 3. Mutation detection rate*
Detection rate
Truncating
Nonsense
Frameshift
Splicing
Missense
Cafè-au-lait spots (CALs) 30/50 24/30 11/30 11/30 3/30 5/30
Axillary freckling (AF) 26/44 21/26 10/26 9/26 3/26 4/26
Dermal neurofibromas (DNf) 20/34 16/20 8/20 6/20 2/20 4/20
Nodular neurofibromas (NNf) 28/44 23/28 10/28 11/28 3/28 4/28
Lish nodules (LN) 15/26 12/15 6/15 5/15 2/15 2/15
Plexiform neurofibromas (PNf) 20/33 16/20 6/20 8/20 3/20 3/20
Scoliosis (S) 9/16 5/9 2/9 3/9 1/9 3/9
Hypertension (HT) 4/7 1/4 1/4 0 1/4 2/4
Pseudoarthrosis (PA) 1/2 0 0 0 0 1/1
Sphenoid displasia(SD) 2/2 1/2 1/2 0 0 1/2
Mental retardation (MR) 1/2 0 0 0 0 1/1

12 De Luca et al.
The mutations were evenly distributed along the NF1 coding sequence. However, exons 4b, 7, 10a, 11, 15, 16, 23-1, 29, 31, 36, 37 and 45 appeared to be more mutation rich, as would be expected if mutations were distributed at random, suggesting that these exons, together with exons 10b, 10c, 13, 23-2, 27a and 39, reported by others to contain recurrent mutations (Messiaen et al., 2000), should be implemented with priority in NF1 mutation analysis. Twenty-three mutations were recurrent in our study and each account for ~2% of the germline mutations. The high number of recurrent mutations was unexpected. Interestingly, seventeen of the 32 (53%) recurrent mutation were due to a C>T or G>A transition at CpG dinucleotides. These data are in accordance to the earlier comprehensive study of Fahsold et al., (2000) in which 17% of alterations occurred within a CpG dinucleotide. We did not find a large number of splicing errors, as indicated by Ars et al., (2000). This may be due to our smaller patient cohort.
K1423 appears to be critical for the maintenance of GAP activity (Upadhyaya et al., 1997). This residue is conserved in the GAP domains of human and murine neurofibromin and was initially thought to be involved in protein interaction and catalysis. Possible mutant mechanism explained with molecular modelling is that the K1423N does not determine a change in charge in the RAS interface-binding region, which could be unfavourable for the mostly acidic effector region of Ras entering the surface groove. Instead it appears to disrupt a favourable internal interaction with E1437 and a change in the status of aminoacid K1419 that are both important for successful interaction with Ras. Possibly, the mutation is destabilizing the protein.
Because no exon skipping was associated to c.730-6A>C intronic change (data not shown), we decided to include this variant in the polymorphisms (Table 2), and not in the mutation’s (Table 1) list. However, unequal expression of both NF1 alleles is a common observation in NF1, and c.730-6A>C change was not detected in multiple controls. Thus, minigene assay (Baralle et al., 2003) is on going to test for the influence of both c.(730-6)A>C nucleotide change on the splicing process.
As in previous studies (Castle et al., 2003), we were not able to find any clear relationship between a NF1 mutation and distinct clinical features. The phenotypic differences in NF1 patients are more likely to be caused by mechanisms such as a second hit, modifying genes and stochastic events (Wiest et al., 2003). However, in the present study, the correlation analysis was quite limited because we were able to collect adequate clinical data only for the 46% of the patients.
The present study adds to the growing number of reports showing the usefulness of DHPLC for mutation screening in disorders where disease-causing mutations are heterogeneous (Colosimo et al., 2002; Flex et al., 2002; De Luca et al., 2003). Nevertheless, DHPLC analysis of the entire NF1 gene in the present group of Italian patients disclosed a large number (32/75, 43%) of recurrent mutations suggesting the possibility of developing rapid tests focused on searching for these recurrent mutations as a first step in the routine diagnostic procedure. However, new approaches should be developed in order to identify mutations in the remaining patients. In this respect, sequencing by hybridization (SbH), a technique which involves hybridization of the DNA of unknown sequence with an enormous set of short oligonucleotides seems particoularly promising (Drmanac et al., 2002). In addition, real time and multiplex PCR technique (Zhou et al., 2003), a method designed for the detection of multi-exon deletions, should also be considered in order to assess the role of this type of mutations as cause of NF1.
ACKNOWLEDGMENTS
We thank the NF1 patients and their parents, for participation to the study. We thank the clinicians who referred their patients to us. The financial support of the Italian Ministry of Health (RC0304MD38 and RF02MD13) and of the Italian Ministry of Instruction, University and Research (RBNE01JJ45) is gratefully acknowledged.
REFERENCES
Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, Estivill X. 2000. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet 9:237-247.
Ars E, Kruyer H, Morell M, Pros E, Serra E, Ravella A, Estivill X, Lazaro C. 2003. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet 40:e82.
Baralle M, Baralle D, De Conti L, Mattocks C, Whittaker J, Knezevich A, Ffrench-Constant C, Baralle FE. 2003. Identification of a mutation that perturbs NF1 agene splicing using genomic DNA samples and a minigene assay. J Med Genet 40:220-222.

Novel and Recurrent Mutations in Italian NF1 Patients 13
Castle B, Baser ME, Huson SM, Cooper DN, Upadhyaya M. 2003. Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J Med Genet 40:e109.
Chomczynski P, Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analyt Biochem 162:156-159.
Colosimo A, Guida V, De Luca A, Cappabianca MP, Bianco I, Palka G, Dallapiccola B. 2002. Reliability of DHPLC in mutational screening of beta-globin (HBB) alleles. Hum Mutat 19:287-295.
Cowley GS, Murthy AE, Parry DM, Schneider G, Korf B, Upadhyaya M, Harper P, MacCollin M, Bernards A, Gusella JF. 1998. Genetic variation in the 3' untranslated region of the neurofibromatosis 1 gene: application to unequal allelic expression. Somat Cell Mol Genet 24:107-119.
De Luca A, Buccino A, Gianni D, Mangino M, Giustini S, Richetta A, Divona L, Calvieri S, Mingarelli R, Dallapiccola B. 2003. NF1 gene analysis based on DHPLC. Hum Mutat 21:171-172.
Drmanac R, Drmanac S, Chui G, Diaz R, Hou A, Jin H, Jin P, Kwon S, Lacy S, Moeur B, Shafto J, Swanson D, Ukrainczyk T, Xu C, Little D. 2002. Sequencing by hybridization (SBH): advantages, achievements, and opportunities. Adv Biochem Eng Biotechnol 77:75-101.
Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, Buske A, Tinschert S, Nurnberg P. 2000. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet 66:790-818.
Flex E, De Luca A, D'Apice MR, Buccino A, Dallapiccola B, Novelli G. 2002. Rapid scanning of myotubularin (MTM1) gene by denaturing high-performance liquid chromatography (DHPLC). Neuromuscul Disord 12:501-505.
Gasparini P, D'Agruma L, Pio de Cillis G, Balestrazzi P, Mingarelli R, Zelante L. 1996. Scanning the first part of the neurofibromatosis type 1 gene by RNA-SSCP: identification of three novel mutations and of two new polymorphisms. Hum Genet 97:492-495
Guex, N., and M. C. Peitsch.. 1997. SWISS-MODEL and the Swiss-Pdb viewer: an environment for comparative protein modelling. Electrophoresis 18:2714–2723.
Han SS, Cooper DN, Upadhyaya MN. 2001. Evaluation of denaturing high performance liquid chromatography (DHPLC) for the mutational analysis of the neurofibromatosis type 1 ( NF1) gene. Hum Genet 109:487-497.
Heim RA, Kam-Morgan LN, Binnie CG, Corns DD, Cayouette MC, Farber RA, Aylsworth AS, Silverman LM, Luce MC. 1995. Distribution of 13 truncating mutations in the neurofibromatosis 1 gene. Hum Mol Genet 4: 975-981.
Huson SM, Compston DA, Clark P, Harper PS. 1989. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet 26:704-711.
Huson SM and Hughes R. 1994. The neurofibromatoses: a clinical and pathogenetic overview. London: Chapman and Hall.
Klose A, Peters H, Hoffmeyer S, Buske A, Luder A, Hess D, Lehmann R, Nurnberg P, Tinschert S. 1999. Two independent mutations in a family with neurofibromatosis type 1 (NF1). Am J Med Genet 83:6-12.
Kluwe L, Friedrich RE, Korf B, Fahsold R, Mautner VF. 2002. NF1 mutations in neurofibromatosis 1 patients with plexiform neurofibromas. Hum Mutat 19:309.
McGuffin LJ, Bryson K, Jones, D.T. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16:404-405.
Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, Paepe AD. 2000. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat 15:541-555.
Origone P., De Luca A., Bellini C., Buccino A., Mingarelli R., Costabel S., La Rosa C., Garrè C., Coviello D.A., Ajmar F., Dallapiccola B., Bonioli E. 2002. Ten novel mutations in the human neurofibromatosis type 1 (NF1) gene in Italian patients. Hum Mutat 20:74-75.
Osborn MJ, Upadhyaya M. 1999. Evaluation of the protein truncation test and mutation detection in the NF1 gene: mutational analysis of 15 known and 40 unknown mutations. Hum Genet 105:327-332.

14 De Luca et al.
Purandare SM, Lanyon WG, Connor JM. 1994. Characterisation of inherited and sporadic mutations in neurofibromatosis type-1. Hum Mol Genet 3:1109-1115.
Riva P, Corrado L, Natacci F, Castorina P, Wu BL, Schneider GH, Clementi M, Tenconi R, Korf BR, Larizza L. 2000. NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am J Hum Genet 66:100-109.
Robinson PN, Boddrich A, Peters H, Tinschert S, Buske A, Kaufmann D, Nurnberg P. 1995. Two recurrent nonsense mutations and a 4 bp deletion in a quasi-symmetric element in exon 37 of the NF1 gene. Hum Genet 96:95-98.
Rodenhiser D, Hovland K. 1995. A novel RsaI polymorphism within intron 39 of the neurofibromatosis type 1 (NF1) gene. Hum Genet 95:241-242.
Scheffzek, K., Ahmadian, M. R., Wiesmueller, L., Kabsch, W., Stege, P., Schmitz, F., Wittinghofer, A. 1999. Structural Analysis of the Gap-Related Domain from Neurofibromin and its Implications. Embo J 17:4313-4327.
Stumpf DA, Alksne JF, Annegers JF, Brown SS, Conneally PM, Housman D, Leppert MF, Miller JP, Moss ML, Pileggi AJ, Rapin I, Strohman RC, Swanson LW, Zimmerman A. 1988. Neurofibromatosis: conference statement. Arch Neurol 45:575-578.
Upadhyaya M, Osborn MJ, Maynard J, Kim MR, Tamanoi F, Cooper DN. 1997. Mutational and functional analysis of the neurofibromatosis type 1 (NF1) gene. Hum Genet 99:88-92.
Upadhyaya M and Cooper DN. 1998. The mutational spectrum in neurofibromatosis 1 and its underlying mechanisms. In: Upadhyaya M, Cooper DN (editors) Neurofibromatosis type 1: from genotype to phenotype. Oxford: BIOS Scientific Publishers. p 65–88.
Wiest V, Eisenbarth I, Schmegner C, Krone W, Assum G. 2003. Somatic NF1 mutation spectra in a family with neurofibromatosis type 1: toward a theory of genetic modifiers. Hum Mutat 22:423-427.
Wimmer K, Muhlbauer M, Eckart M, Callens T, Rehder H, Birkner T, Leroy JG, Fonatsch C, Messiaen L. 2002. A patient severely affected by spinal neurofibromas carries a recurrent splice site mutation in the NF1 gene. Eur J Hum Genet 10:334-338.
Zhou XP, Waite KA, Pilarski R, Hampel H, Fernandez MJ, Bos C, Dasouki M, Feldman GL, Greenberg LA, Ivanovich J, Matloff E, Patterson A, Pierpont ME, Russo D, Nassif NT, Eng C. 2003. Germline PTEN promoter mutations and deletions in Cowden-Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase Akt pathway. Am J Hum Genet 73:404-411.





























