Embed Size (px)
Citation preview

Vol. 7, 1651-1660, December 1996 Cell Growth & Differentiation 1651
p53-independent Tumor Growth and in Vitro Cell Survival forF-MuLV-induced Erythroleukemias’
Jeff Howard, Yee Ung, Dena Adachi, andYaacov Ben-David2
Division of Cancer Biology Research, Sunnybrook Health ScienceCentre, Reichmann Research Building, 5-218, 2075 Bayview Avenue,M4N 3M5 Toronto, and Department of Medical Biophysics, Universityof Toronto, Toronto, Ontario, Canada
AbstractRetroviral insertional activation of the FIi-l proto-oncogene is the first genetic event associated with theinduction of erythroleukemias by the Friend munneleukemia virus (F-MuLV). Mutations within p53, whichare only detected in cell lines established fromtransplanted tumors, have been previously shown to beassociated with the immortalization of erythroleukemiccells in culture. In this study, we have demonstratedthat primary erythroleukemic cells grown in liquidculture undergo rapid apoptosis independent of thestabilization of wild-type p53 protein. Furtherconfirmation that the programmed cell death observedfor liquid-cultured F-MuLV-induced primaryerythroleukemic cells is largely p53 independent wasprovided by experimentation with a transgenic mouseline containing multiple copies of the dominantnegative mutant p53����1ee allele. Erythroleukemic cellstaken from tumor-bearing transgenic mice expressinghigh levels of the mutant p53”#{176}� undergoprogrammed cell death in culture in a manner that islargely identical to that observed for tumor cellsderived from nontransgenic littermates. Furthermore,the rate of development of F-MuLV-inducederythroleukemias for both � andnontransgenic littermates are similar. Moreover,cytogenetic analysis indicates that primaryerythroleukemia cells are diploid, whereaschromosomal aberrations were observed in allestablished cell lines. These results are consistent withthe notion that mutations within the p53 tumorsuppressor gene affect genomic stability, subsequentlyleading to changes in gene expression that areassociated with the immortalization of erythroidprogenitor cells.
Received 5/22/96; revised 8/6/96; accepted 9/24/96.The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 1 8 U.S.C. Section 1 734 solely to mdi-cate this fact.1 This work was supported by grants from the National Cancer Institute ofCanada. V. Ben-David is a Research Scientist of the National CancerInstitute of Canada.2 To whom requests for reprints should be addressed, at SunnybrookHealth Science Centre. Phone: (416) 480-6100, Ext. 3359; Fax:(416) 480-5703.
IntroductionFriend virus-induced erythroleukemia has emerged as apowerful animal model system to study the multistage nature
of cancer progression (1 , 2). Similar to human cancers, dis-ease progression in these infected animals involves a multi-stage process that is directly linked to the accumulation ofgenetic changes, including the activation of dominantly act-
ing oncogenes and the inactivation of a tumor suppressor
gene (1). Two separate isolates of Friend leukemia virus,
termed FV-A and FV-P, have been identified. Both FV-A and
FV-P are actual complexes of two distinct viral species,
namely a unique replication-defective SFFV3 strain (SFFV-A
and SFFV-P, respectively) and a common replication-com-
petent F-MuLV. The anemia-inducing (FV-A) and polycythe-mia-inducing (FV-P) strains of the virus produce similar mul-tistage malignancies when injected into susceptible strainsof adult or newbom mice. However, F-MuLV can induce,
independently of SFFV, a number of hematopoietic neo-
plasms, including erythroleukemias, when injected into new-born susceptible mice (3).
Toward understanding the molecular mechanisms associ-
ated with the induction and progression of leukemias, wehave previously identified a common site for retroviral into-
gration, designated Fll-l , that is rearranged in the majority of
F-MuLV-induced erythroleukemias (4). Earlier, Moreau-Gachelin et a!. (5, 6) had identified Sfpi-i as a common sitefor retroviral integration shared by FV-A and FV-P-induced
erythroleukemias. Interestingly, the transcriptional domainsactivated by proviral insertion at these corresponding sitesencode two unique transcription factors, both of which are
members of the ets oncogene family (5, 7, 8). Retroviralinsertional activation of Fll-l and Sfpi-l appears to be mu-tually exclusive with respect to the particular strain of Friend
virus used for infection. Specifically, Fll-i appears to beactivated exclusively in erythroleukemias induced by
F-MuLV, whereas Sfpi-l has only been shown to be acti-
vated in Ri-A/P-induced erythroleukemias.
Upon comparing F-MuLV- and RI-A/P-induced erythro-
leukemias, it has become evident that these primary tumors
display notable biological differences, particularly with re-spect to their ability to grow in culture. Unlike FV-A/P derived
primary tumors, cells derived from F-MuLV-induced primary
erythroleukemias are unable to grow directly in culture, even
in the presence of Epo, an important erythroid survival and
mitogenic factor (9). However, ED cell lines can be estab-
lished following serial in vivo passage of these infected pri-mary tumor cells in syngeneic mice (9). Examination of these
3 The abbreviations used are: SFFV, spleen focus-forming virus; F-MuLV,Friend murine leukemia virus; Fll-l , Friend leukemia mntegration-l ; Epo,erythropoietin; ED, Epo-dependent; El, Epo-independent; PCD, pro-grammed cell death; wt, wild-type; HCV, hematocrit value.

1652 Friend Erythroleukemia, p53 Mutation, and Apoptosis
established ED cell lines revealed that they all containedfunctional inactivating mutations within the p53 tumor sup-
pressor gene, which is strongly associated with the immor-talization of cells in culture. In addition, the large majority ofEl cell lines established following the in vivo passage of EDcell lines constitutively express Epo, resulting in the estab-
lishment of an autocrine loop involving the Epo receptor,
which is also highly expressed in these cells (iO). The selec-tion process operating in vivo enabling these tumor cells toacquire growth factor independence is associated with in-
creased genomic instability, as shown by the frequent rear-rangement of the Epo gene in these El cell lines. This in-creased genetic instability appears to be driven in part by the
lack of a functional p53�.
In the past decade, the study of Friend virus-inducederythroleukemias have shown that p53 mutations appear to
be a prerequisite event for the emergence of clonal tumori-genic erythroleukemia cells. In fact, some of the earliestevidence of the tumor suppressor function of p53 emergedfrom these studies (i 1). Characterization of the function ofthis gene in a number of laboratories have shown that wt p53can trigger G1 arrest and PCD or apoptosis and, in addition,
positively modulate globin gene expression, which is a dis-tinctive marker of erythroid differentiation (1 2). This raises the
intriguing possibility that the cellular death that we observed
for primary F-MuLV erythroleukemias grown in liquid culture
may be attributed to the expression of wt p53 in these clonal
tumor cells. However, this hypothesis is not consistent withthe recent findings of Kelly et a!. (1 3), who have demon-strated that splenic polyclonal proerythroblasts derived fromFV-A postinfected (2 weeks) mice undergo PCD that is in-dependent of the stabilization of wt p53 protein. In this study,we have examined the status of p53 in the context of primaryerythroleukemias induced by F-MuLV in both normal and
ps3Pro-193..transgenic mice. The extensive cell death exhib-
ited by liquid-cultured clonal proerythroblasts derived fromboth normal and p53Pro-l93�transgenic animals is character-istic of apoptosis. The PCD induced in erythroleukemia cells
derived from normal mice was not associated with the sta-bilization of wt p53. The presence of the p53�0�93 transgeneand its subsequent high levels of expression neither accel-erated the onset of the primary disease nor blocked the rapidonset of apoptosis induced in these clonal erythroleukemiacells when placed in culture. The role of p53 with respect toF-MuLV-induced erythroleukemia will be discussed.
Results
Apoptosis in Primary Erythroleukemias Induced by F-MuLV Is p53 Independent. We have previously demon-strated that the clonal primary erythroleukemias induced by
F-MuLV have acquired an activated F/i-i and appear toexpress p53Wt protein (9). Although these primary erythro-leukemias are capable of generating transplantable tumors inmice, they die rapidly when introduced to cell culture. More-
over, other laboratories have shown that exogenous expres-
sion of ts-p53 in a p53-negative FV-P-induced erythroleuke-
mia cell line is associated with the induction of apoptosis (i 2,1 4). These observations raise the possibility that the rapid
cell death observed in F-MuLV-induced primary erythroleu-
kemias may be mediated by the activation of wt p53 duringin vitro cell culture. To test this hypothesis, primary erythro-
leukemic cells derived from a F-MuLV-infected mouse were
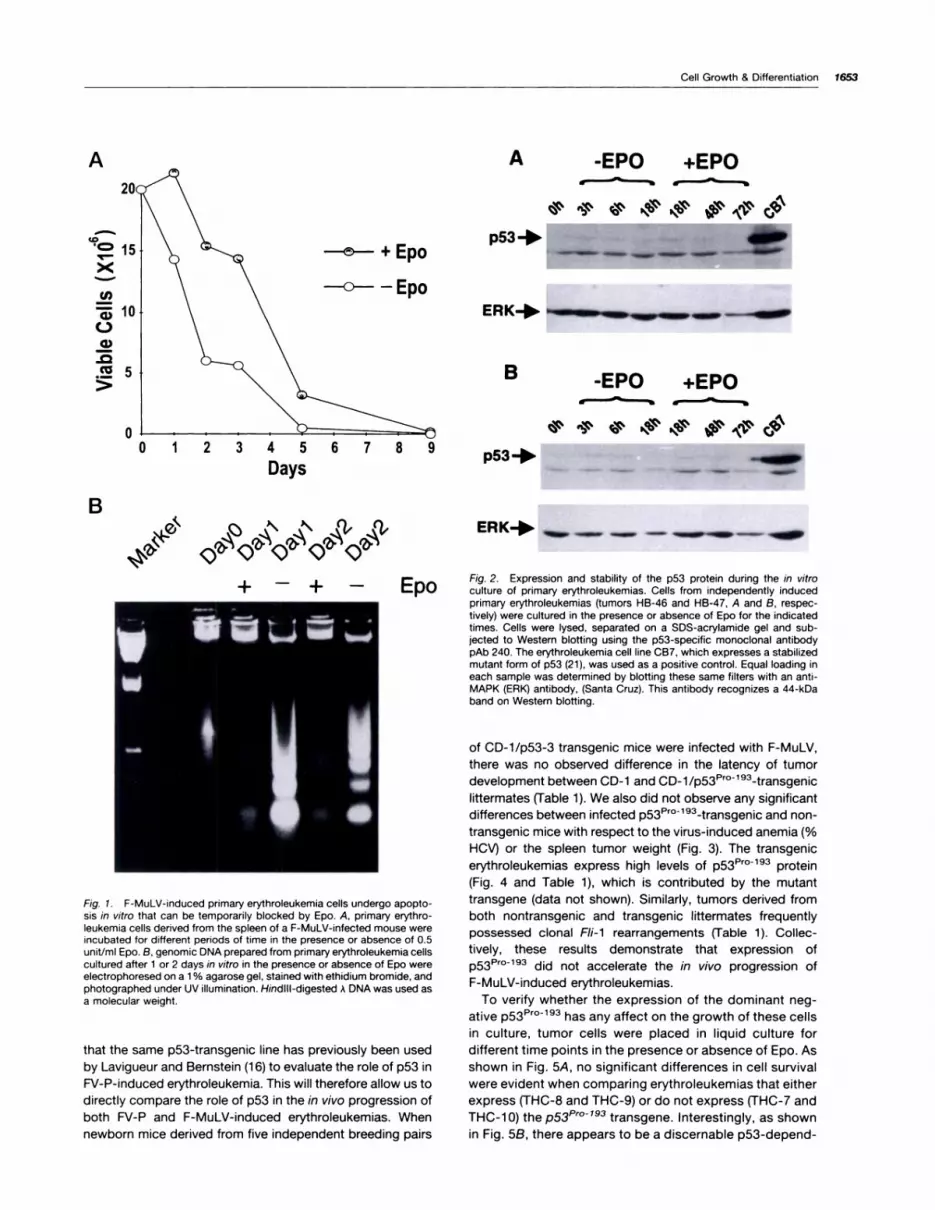
removed and cultured in the absence or presence of 1unit/mI Epo. Rapid loss of cell viability was observed whenthese primary tumor cells were cultured in the absence ofEpo (Fig. 1A). The addition of Epo to the cell culture medium
significantly increased cell survival within the first 3 days of
culture but only delayed the onset of cell death. A similar
pattern of cell survival can be seen when other independentlyderived primary erythroleukemias are grown in culture with or
without the addition of Epo (data not shown). The rapid cell
death observed for these cultured primary erythroleukemiccells is due to apoptosis. This is supported by the ladderpattern of genomic DNA degradation detected within thesecells, which is a hallmark of PCD (Fig. 1B). The addition of
Epo to the cultures markedly increased cell viability andreduced the extent of the DNA fragmentation compared to
cells grown without Epo but could not abrogate the PCD (Fig.
1B).
To examine whether changes in the stability of p53 wereassociated with the Epo-sensitive apoptotic process ob-served in liquid culture, tumor cells derived from two primaryerythroleukemias (HB46 and HB47) were cultured for various
times in the presence or absence of Epo and collected for
Western blot analysis. p53 immunoblot analysis can readilydetect changes in the stability of p53� protein in response toapoptotic stimuli (13). p53�#{176}�93, a stabilized dominant-neg-ative missense mutant of p53 expressed by CB7 cells, has adramatically extended half-life, which translates into itsprominent accumulation and easy detection (Fig. 2). For the
primary tumor cells collected at various time points in thepresence or absence of Epo, there was no detectable sta-bilization of wt p53 protein (Fig. 2). Northern analysis of theseprimary tumor cells demonstrated no change in the RNA
expression of p53 for this same extended time course inculture (data not shown). Although wt p53 is actively tran-scribed in all of these primary tumors (data not shown) theapoptosis observed for these liquid-cultured tumor cells ap-
pears to be independent of p53 RNA regulation. Together,these results suggest that the F-MuLV-induced primaryerythroleukemic cells undergo apoptosis in culture via amechanism that is independent of the active stabilization of
wt p53 protein or changes in the RNA expression of p53.These observations are consistent with the results reportedby Kelly et a!. (1 3), which show that the rapid onset of
apoptosis induced in preleukemic p53’� expressing RI-A
proerythroblasts is not associated with the stabilization of wt
p53 protein.Apoptosis and Tumor Growth in p53�0���transgenic
Mice Infected with F-MuLV. The development of both p53
knockout and ps3muttransgenic mice have provided re-
searchers with an invaluable model system to directly accessthe role of p53 in tumorigenesis. In this study, we have useda CD-l/p53-3 transgenic mouse line expressing a dominantnegative mutant of p53l�r0�93 (Arg-to-Pro-193 mutant) that
have previously been shown to develop a spectrum of spon-taneous tumors similar to that seen in p53-deficient mice(1 5). The rationale for this choice is largely based on the fact
A
-e-- + Epo
-0--- -Epo10
5
0
A -EPO +EPO
p53+ ____-� � - �
ERK+ .-_ -..�-- - -�
B
p53+ _ - - -
0123456789
Days
!
II!
Cell Growth & Differentiation 1653
(0.� 15
Cl)
C)a,
>
BS
Q�� �
+-#{247}
Fig. 1 . F-MuLV-induced primary erythroleukemia cells undergo apopto-sis in vitro that can be temporarily blocked by Epo. A, primary erythro-leukemia cells derived from the spleen of a F-MuLV-infected mouse wereincubated for different periods of time in the presence or absence of 0.5unit/mI Epo. B, genomic DNA prepared from primary erythroleukemia cellscultured after 1 or 2 days in vitro in the presence or absence of Epo wereelectrophoresed on a 1 % agarose gel, stained with ethidium bromide, andphotographed under UV illumination. Hindill-digested A DNA was used asa molecular weight.
that the same p53-transgenic line has previously been used
by Lavigueur and Bernstein (1 6) to evaluate the role of p53 in
RI-P-induced erythroleukemia. This will therefore allow us to
directly compare the role of p53 in the in vivo progression of
both FV-P and F-MuLV-induced erythroleukemias. When
newborn mice derived from five independent breeding pairs
ERK+ � � - � � �
E Fig. 2. Expression and stability of the p53 protein during the in vitropo culture of primary erythroleukemias. Cells from independently inducedprimary erythroleukemias (tumors HB-46 and HB-47, A and B, respec-tively) were cultured in the presence or absence of Epo for the indicatedtimes. Cells were lysed, separated on a SDS-acrylamide gel and sub-jected to Western blotting using the p53-specific monoclonal antibodypAb 240. The erythroleukemia cell line CB7, which expresses a stabilizedmutant form of p53 (21), was used as a positive control. Equal loading ineach sample was determined by blotting these same filters with an anti-MAPK (ERK) antibody, (Santa Cruz). This antibody recognizes a 44-kDaband on Western blotting.
of CD-l/p53-3 transgenic mice were infected with F-MuLV,
there was no observed difference in the latency of tumor
development between CD-i and CD�l/p53�0l93�transgenic
littermates (Table 1). We also did not observe any significantdifferences between infected p53F�r0�93�transgenic and non-
transgenic mice with respect to the virus-induced anemia (%HCV) or the spleen tumor weight (Fig. 3). The transgenic
erythroleukemias express high levels of p53��r0�93 protein
(Fig. 4 and Table 1), which is contributed by the mutanttransgene (data not shown). Similarly, tumors derived from
both nontransgenic and transgenic littermates frequently
possessed clonal F/i-i rearrangements (Table 1). ColIec-
tively, these results demonstrate that expression of
p53Prol93 did not accelerate the in vivo progression of
F-MuLV-induced erythroleukemias.To verify whether the expression of the dominant neg-
ative p53Prol93 has any affect on the growth of these cellsin culture, tumor cells were placed in liquid culture for
different time points in the presence or absence of Epo. As
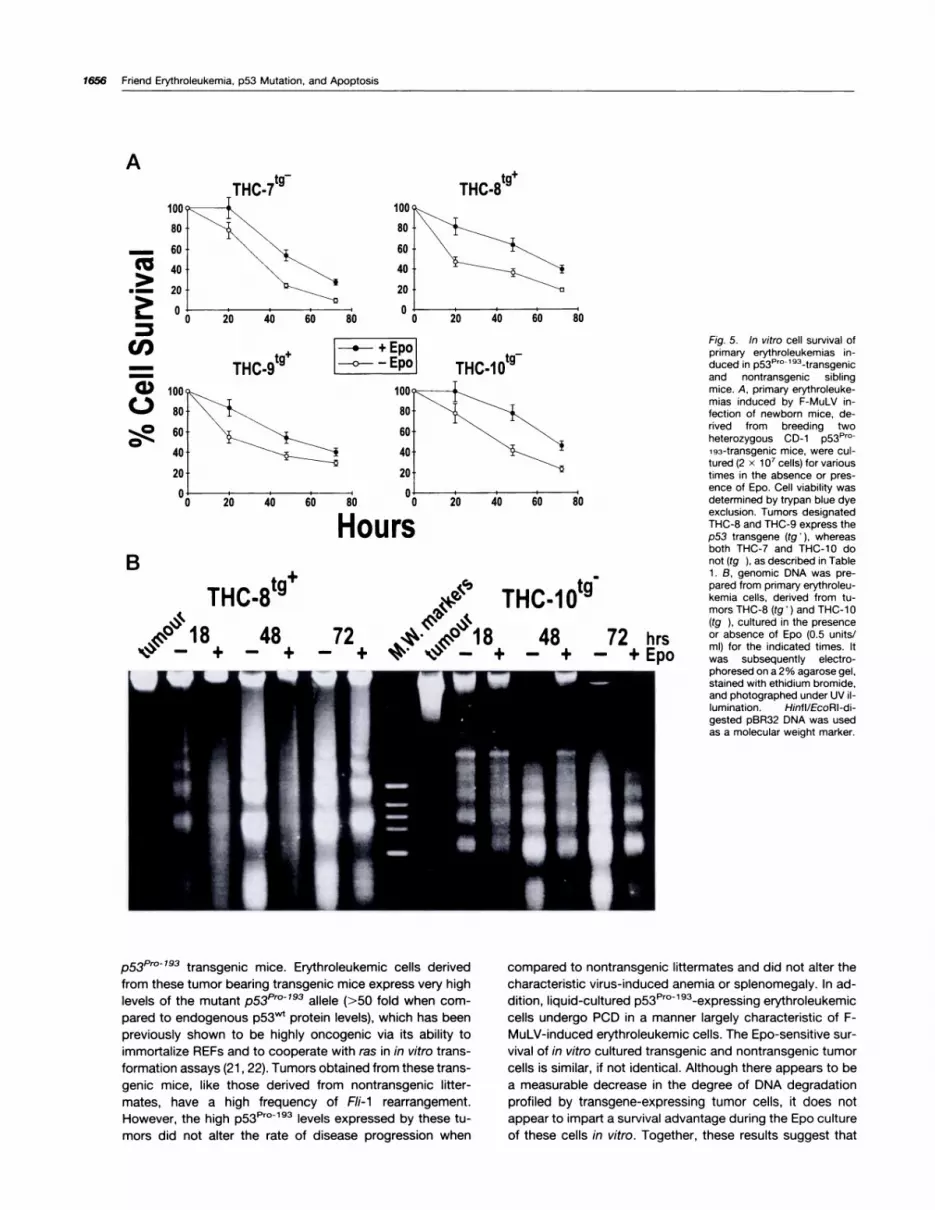
shown in Fig. 5A, no significant differences in cell survival
were evident when comparing erythroleukemias that either
express (THC-8 and THC-9) or do not express (THC-7 and
THC-l0) thep53Pr0l93 transgene. Interestingly, as shownin Fig. SB, there appears to be a discernable p53-depend-

1654 Friend Erythroleukemia, p53 Mutation, and Apoptosis
Table 1 Tumor incidence and in vitro cell survival of primary erythroleukemias induced by F-MuLV in ps3muttransgenmc mice
In experiment 1 , a p53-3 transgenic mouse that expresses a p53’�#{176}1�”mutant transgene was backcrossed with a normal CD-l mouse. The newbornoffspring were then infected with F-MuLV. Tumors THC-1 through ThC-5 represent five erythroleukemias induced in these mice. In experiments 2 and 3,two heterozygous p53�’ml��transgenic mice were bred together, and the resulting newborn offspring infected with F-MuLV. In experiment 4, newbornsfrom two breeding pairs (one pair consisting of two heterozygous p53�’r�l9a�transgenic mice and one pair consisting of one heterozygous p53�#{176}�#{176}3transgenic mouse and one normal CD-l mouse) were infected with F-MuLV. Tumors THC-6 through THC-32 are erythroleukemias derived from these mice.
Ex rim nt T p53�1#{176}’� Tumor latency In vitro FIi-lpe e umor transgene (days)#{176}’ survivaF rearrangement”
1 THC-l - 52 - +
THC-2 - 52 - +
THC-3 - 55 - +
THC-4 - 56 - +
THC-5 + 56 - -
2 THC-6 + 47 - +
THC-7 - 47 - +
THC-8 + 47 - +
THC-9 + 47 - +
THC-lO - 47 - +
THC-110 + 47 - -
THC-l2 + 49 - +
THC-13 - 55 - +
THC-14 + 80 - +
THC-l5 - 80 - +
3 THC-16 + 49 - +
THC-17 - 49 - +
THC-18 + 49 - +
THC-19 + 49 - +
THC-20 - 49 - +
THC-21 + 52 - +
THC-22 - 62 - +
THC-23 + 72 - +
THC-24 - 72 - +
4 THC-25 + 51 - +
ThC-26 + 58 - -
THC-27 + 61 - +
THC-28 - 35 - +
THC-29 - 35 - +
THC-30 + 79 - +
THC-31 - 45 - +
THC-32 - 45 - +
Mean tumor latency ± SE’CD�l/tg�p53�cr�l9a 55.8 ± 2.9CD-l 52.3 ± 2.9
a The transgenic status of the mice was determined by Southern blotting using a p53-specific cDNA probe, which can detect unique and amplified p53
genomic fragments corresponding to transgenic siblings.b Tumor latency was measured as the day the animals were moribund.C In vitro survival of primary tumor cells was determined by culturing these cells (20 x 106) with a-MEM containing 0.5 units/mI rhEpo and 10% FCS.d Fli-l rearrangement was determined by Southern blotting (HindllI and BamHl digests) using a Fll-1 -specific genomic DNA probe as described elsewhere
(4).C Expresses high levels of Fli-1 RNA.
‘P value = 0.39.
ent difference with respect to the extent of DNA degrada-tion associated with this Epo-sensitive PCD. However,none of the erythroleukemias gave rise to established cell
lines in the presence of Epo, despite the high levels ofp53Pml93 transgene expression. The rapid loss of cellviability observed for liquid-cultured p53Pml9a transgenicerythroleukemic cells could not be attributed to a reduc-
tion in the expression of the p53Pr0�9a transgene because
the level of p53Prol93 expressed in these cells did notchange over the extended time course (Fig. 4B). Therefore,the inability of the dominant negative p53Prol93 to altertumor cell survival in vitro, in either the presence or ab-
sence of Epo, suggests that the cell death observed forliquid-cultured F-MuLV-induced erythroleukemias may be
largely independent of the function of wt p53.Karyotypic Analysis of Primary Tumors and Estab-
lished Erythroleukemia Cell Lines. Genomic instability di-
rectly visualized by cytogenetic techniques often accompa-nies the transformation process seen in cancer cells.Changes in chromosomal structure or number have beendocumented previously in established Friend viral complex-
induced erythroleukemia cell lines (1 7-20). We analyzed the
karyotypes of cells derived from several primary erythroleu-
kemias induced in normal or ps3Prol93..transgenic mice and

A B3.5
00
35
30
25
20
%15
2
gm1.5
0
0
000
9 00
Toeo0 00
0�0
8
0 0
0
5
Pmlfl
CD-1/tg.p53
0
A� ?
p53 d� ,� l�
ERK
BTHC-14
EPO _�__ +
c�’ ��q�b45� �#{231}’b#{231}b4�#{149}�4:�
p53 �
Pm193
CD.i/tg-p53 CD-i
Cell Growth & Differentiation 1655
HCV Spleen weight0
0 3
0 2.5
� � I!d�D0
10 10
0 0.500 0
CD-I
Fig. 3. The HCV and spleen tumor weight of p53Pr0�93�transgenic andnontransgenic mice infected with F-MuLV. The tumor descriptions aresummarized in Table 1. The HCVs and spleen tumor weights were meas-ured the day the animals were moribund for experiments 2, 3, and 4 (Table1).
four established F-MuLV-induced erythroleukemia cell lines
(HB9.i -ED, HB39.2-ED, CB3, and CB7). Analysis of 35 cells
collected from a F-MuLV-induced primary erythroleukemia
failed to detect any gross changes in chromosomal struc-
ture; in addition, these cells were diploid (40 chromosomes;
Fig. 6A). Similarly, a normal ploidy was observed in cells
derived from two primary erythroleukemias expressing the
p53Prol93 transgene (tumor THC-6 and THC-8; Fig. 6, F andH), as well as in one tumor derived from a nontransgenic
littermate (THC-7; Fig. 6G). In contrast, all of the cells derived
from F-MuLV-induced erythroleukemia cell lines had either
lost one or more chromosomes (HB9.i-ED, HB39.2-ED, and
CB3) or displayed a hyperdiploid karyotype (CB7). In addi-tion, the appearance of large metacentric chromosomes was
observed in these erythroleukemia cell lines (data not
shown). These results demonstrate that the appearance of
chromosome abnormalities in established F-MuLV-induced
lines is associated with the immortalization of these cells in
culture and the loss of wt p53.
DiscussionIn the past decade, the study of Friend virus-induced eryth-
roleukemias has resulted in the identification of a number of
cellular genes that are frequently targeted for mutation, spe-
cifically F/i-i , Sfpi-l , and p53. Whereas retroviral insertionalactivation of F/i-i has been seen in almost all of the primaryerythroleukemias induced by F-MuLV, p53 mutations have
only been directly identified within cell lines established after
the in vivo transplantation of these primary tumors (9). There-
fore, the role of p53 in the context of in vivo tumorigenesis for
F-MuLV-induced erythroleukemias is not fully understood. In
this study, by using primary erythroleukemia cells derived
Fig. 4. Expression of the p53��0 193 transgene in F-MuLV-induced eryth-roleukemia cells cultured for various times in vitro. A, Western blotting wasperformed with cell lysates derived from primary erythroleukemias THC-6through THC-12 (Table 1) using an anti-p53 (pAb 240) antibody. B, cellsderived from tumor THC-l4 that express the p53�’r0�93 transgene weregrown in culture for the indicated times, lysed, and Western blotted usingan anti-p53 (pAb 240) antibody. These Western blots also included controlcell lysates from two erythroleukemia cell lines, CB3 and CB7, which haveeither lost both alleles of p53 or express the proline 193 mutant of p53,respectively (21 , 45). Equal loading in each sample was determined byblotting these filters with an anti-pan-EAK antibody.
from F-MuLV-infected CD-i and CD-l/p53-3 transgenic
mice and cell lines established from in vivo transplanted
primary tumors, we have attempted to analyze the functional
importance of p53 mutations during the multistage progres-
sion of F-MuLV-induced erythroleukemias.
For primary erythroleukemic cells, low but discernable 1ev-
els of wt p53 protein expression are detectable. The contrastbetween high levels of RNA expression and the rather low
levels of protein expression for p53 is typical of the normalrapid turnover of this protein. Although the continued growth
of these primary tumor cells is permissible in vivo, via trans-plantation into syngeneic mice, they rapidly undergo PCD in
an Epo-sensitive manner when placed in liquid culture. The
mechanism of PCD observed for these F-MuLV-induced
erythroleukemic cells did not involve p53 protein stabiliza-
tion. The same phenomenon has been previously reported
by Kelly et a/. (1 3), who examined PCD in polyclonal FV-A-
induced proerythroblasts, which also express wt p53. Inter-
estingly, these observations are mirrored by experiments
performed with primary erythroleukemic cells obtained from
THC-7� THC-8100
80
60
40
20
0
100
80
60
40
20
0
100
80
60
40
20
I 20 40 60 80
A
>
Cl)
a)C)
B
) 20 40 60 80
-..-- + Epo
THC-9� -#{176}-- - Epo THC-1 0tg
100
80
60
40
20
-�
Hoursu0� 20 40 60 80
I 20 40 60 80
t+THC-8� THC-1 0tg
1656 Friend Erythroleukemia, p53 Mutation, and Apoptosis
Fig. 5. In vitro cell survival ofprimary erythroleukemias in-duced in p53P�o�l93�transgenicand nontransgenic siblingmice. A, primary erythroleuke-mias induced by F-MuLV in-fection of newborn mice, de-rived from breeding twoheterozygous CD-i p53Pro
193-transgenic mice, were cul-tured (2 x 1o� cells) for varioustimes in the absence or pres-ence of Epo. Cell viability wasdetermined by trypan blue dyeexclusion. Tumors designatedTHC-8 and THC-9 express thep53 transgene (tg � ), whereasboth THC-7 and THC-lO donot (tg ), as described in Table1 . B, genomic DNA was pre-pared from primary erythroleu-kemia cells, derived from tu-mors THC-8 (tg� ) and THC-i 0(tg ), cultured in the presenceor absence of Epo (0.5 units/ml) for the indicated times. Itwas subsequently electro-phoresed on a 2% agarose gel,stained with ethidium bromide,and photographed under UV 1-lumination. Hinfl/EcoAI-di-gested pBA32 DNA was usedas a molecular weight marker.
p53Pro 193 transgenic mice. Erythroleukemic cells derived
from these tumor bearing transgenic mice express very high
levels of the mutant p53Pr0193 allele (>50 fold when com-
pared to endogenous p53� protein levels), which has been
previously shown to be highly oncogenic via its ability to
immortalize REFs and to cooperate with ras in in vitro trans-
formation assays (2i , 22). Tumors obtained from these trans-
genic mice, like those derived from nontransgenic litter-
mates, have a high frequency of F/i-i rearrangement.
However, the high p53Prol93 levels expressed by these tu-
mors did not alter the rate of disease progression when
compared to nontransgenic littermates and did not alter the
characteristic virus-induced anemia or splenomegaly. In ad-
dition, liquid-cultured p53Prol93..expressing erythroleukemic
cells undergo PCD in a manner largely characteristic of F-
MuLV-induced erythroleukemic cells. The Epo-sensitive sur-
vival of in vitro cultured transgenic and nontransgenic tumor
cells is similar, if not identical. Although there appears to be
a measurable decrease in the degree of DNA degradation
profiled by transgene-expressing tumor cells, it does not
appear to impart a survival advantage during the Epo culture
of these cells in vitro. Together, these results suggest that

41
Fig. 6. Chromosome numberrange within cells derived fromF-MuLV-induced tumors or es-tablished cell lines. Each histo-gram represents the chromo-some count for cells derivedfrom the following tumors andcell lines: a F-MuLV-induced pri-mary erythroleukemia (A); ED celllines HB9.l-ED and HB 39.2-ED(B and C, respectively); El celllines CB3 and CB7 (D and E, re-spectively); primary tumorsTHC-6 and THC-8, which ex-press the p53 transgene (seeTable 1 : F and H); and primarytumor THC-7, which expresseswt p53 (see Table 1; G).
E
.aa)
E
Cl)
0I-
0
a).�
E
z
40
20
10
40A 40C
30 30
20 20
10 10
0 ‘‘ . . 0 ‘
39 40 39 40
30 30
20
10
0 .� . . U
57 58 59 60 61
G ___ H20 40
15 30
10 20
5 10
�- . 0 . -I--. �
41 39 39
36 37
I38
40
Cell Growth & Differentiation 1657
Number of chromosomes
p53 may have neither a role in the in vivo tumongenesis of
F-MuLV-induced erythroleukemias nor a major role in the
process of PCD observed in liquid culture.
The clear lack of synergy between F-MuLV infection and
the overexpression of mutant p53 may be explained by the
functional redundancy of these two events. This explanation
is analogous to that proposed by Mann et a/. (50), who
observed a similar lack of synergy between p53 and Bcl-2
during in vivo lymphomagenesis. For retroviral mutagenesis,
genetically altered mice, particularly transgenic mice, have
proven to be a very useful system to explore oncogenic
cooperativity during in vivo tumorigenesis (23-26). In thisparticular study there appears to be no discernable synergy
between F-MuLV infection and a mutant p53Prol9a environ-
ment in vivo. However, this property of F-MuLV infection is
not shared by RI-P (1 6). This difference raises an important
point concerning the in vivo mutational environment defined
by these two Friend virus-induced erythroleukemias. It is
within this context that we should consider the importance ofp53 mutations, and likely any other oncogenic event, be-
cause the function of p53 is very much influenced by the cell
type and the transforming events that define the system
being examined (27-30). Specifically, RI-P activates Sfpi-l
and F-MuLV activates Fli-i , two very important but distinct
mutational events that may trigger individual downstream
events. This notion is supported by the observation that F/i-i
and Sfpi-1 appear to recognize distinct DNA-binding se-quences (31) and thereby transactivate discrete primary tar-
get genes. In addition, the Epo mimicry of gp55 in FV-P-
induced erythroleukemias, which is absent in F-MuLV-
induced erythroleukemias, should also be considered. The
mitogenic and survival signaling triggered by the binding
of gp55 to the Epo receptor, coupled with the increased
self-renewal potential likely conferred by the activation of
Sfpi-i (32), may favor the rapid selection of mutations thatimmortalize these erythroid progenitor cells. Alternatively,
these differences may be linked to the fact that theseunique Friend viruses, SFFV-A/P and F-MuLV, may target
distinct cell types (CFU-E and BFU-E, respectively) and
therefore require a different complement of genetic alter-
ations to successfully transform their respective erythroidprogenitor cells. Despite these differences, the ability ofF-MuLV-induced primary erythroleukemic cells to prolifer-ate in vivo suggests that the host spleen microenvironmentlikely supplies the tumor cells with important growth stim-
uli. This may be communicated by the spleen via growth
factors and/or stromal cell interactions (33). The survival of
these erythroleukemia cells in culture may therefore re-
quire additional genetic events to escape all of the nega-
tive growth effects operating in vitro.
The proposed functional redundancy for F-MuLV infection
and mutant p53 during in vivo tumorigenesis suggests thatthe p53 mutations commonly observed in established F-
MuLV-induced erythroleukemia cell lines are a likely conse-quence of the in vitro immortalization process. The karyotypeanalysis of F-MuLV-induced primary tumors and established
erythroleukemia cell lines provides strong support for this
notion. The aneuploidy associated with F-MuLV-induced cell
lines is similar to that which has been previously described
for Friend viral complex-induced cell lines and late-stage
RI-A-induced primary erythroleukemias (1 7-20). However,
the absence of gross karyotypic abnormalities in F-MuLV-
induced primary erythroleukemic cells is striking. This raisesan intriguing question concerning the possible role of p53
mutations during the erythroid immortalization process, spe-
cifically their proposed involvement in generating genetic

1658 Friend Erythroleukemia, p53 Mutation, and Apoptosis
2. Kabat, D. Molecular biology of Friend viral erythroleukemia. Curr. Top.
Microbiol. Immunol., 148: 1-42, 1990.
instability. The transcriptional activity of p53 (34) is capa-
ble of activating or repressing the transcription of genesthat regulate cell cycle progression (35-37), DNA repair
(38, 39), and genomic stability (40, 41). The cell cycle
control believed to be exerted by p53 at the G1-S transi-
tion and the considerable experimental evidence connect-
ing lost control of this specific cell cycle checkpoint and
increased genomic instability and malignant progression
suggests that p53 plays an important role in maintainingthe integrity of the genome (39-43). Therefore, it is pos-
sible that the appearance of p53 mutations in F-MuLV-
induced erythroleukemic cells may be responsible in part
for the emergence of genetic instability. In this respect, a
destabilized genome would conceivably predispose these
erythroid progenitor cells to further genetic changes that
enable them to escape negative growth pressures in cul-
ture. Although primary erythroleukemias derived from
p53Pro-193..transgenic mice retain their normal ploidy, this
may reflect time limitations governed by the morbidity of
the disease. However, major chromosomal abnormalities
may arise subsequent to additional in vivo passaging ofthese F-MuLV-induced primary tumor cells. Therefore, it
remains to be determined whether the in vivo transplan-
tation of p53Prol93 primary erythroleukemia cells results in
an accelerated rate of erythroid immortalization accompa-
nied by aneuploidy. Although the overexpression of v-raf
and c-myc in erythroid progenitor cells derived from the
fetal liver of p53-deficient mice (p53�) has recently been
shown to induce immortalization in the absence of gross
karyotypic changes, this may largely reflect system differ-
ences (44). The specific combination of oncogenicchanges enforced on these p53� erythroid cells is unique
to this system and have not been previously shown to be
associated with the in vivo transformation of Friend virus
target cells. These particular mutational events may there-
fore allow for the immortalization of erythroid cells inde-
pendent of changes in cell ploidy.
In summary, by analyzing the molecular events associated
with F-MuLV-induced erythroleukemias we have shown ev-
idence suggesting that p53 is not involved in the in vivo
progression of this disease. Primary erythroleukemic cells
constitutively expressing Fll-i by way of retroviral insertional
activation undergo PCD in liquid culture independent of the
stabilization of p53 protein. The apoptosis observed during
the Epo culture of these F-MuLV-transformed erythroid cells
appears to be largely mediated by a p53-independent mech-anism, as evidenced by the unperturbed survival kinetics of
F-MuLV-induced p53Prol 93-transgenic erythroleukemic cells.
Conceivably, a subpopulation of transplanted primary eryth-
roleukemic cells possessing p53 mutations emerges display-
ing karyotypic changes and a capacity to be immortalized in
culture.
Materials and MethodsTumors and Cell Lines. The erythroleukemia cell lines CB3, CB7, andHB9.l-ED were previously described elsewhere (9, 45-47). Isolation of
established ED cell lines HB 39.2-ED is described elsewhere (10). Thesecells were maintained in a-MEM supplemented with 10% FCS. The ED
cell lines were maintained in medium containing 0.1 unit/mI Epo (Boeh-
ringer Mannheim).
The primary erythroleukemias were induced by a single i.p. injection
of F-MuLV into newborn BALB/c or CD-i p53-transgenic mice asdescribed previously (9). The generation of the p53-3-transgenic
mouse line containing multiple copies of a mutant p53 allele, bearing anargmnmne-to-prolmne mutation at residue 1 93, has been described pre-
viously (1 5). The majority of these infected animals usually developerythroleukemia between 2 and 3 months postinfection. The F-MuLV-induced murine primary erythroleukemias were cultured for varioustime periods in the presence or absence of 1 unit/mI Epo and used for
DNA, RNA, or protein analysis.Protein Determination. Western blotting was performed by lysing the
cells (l0� cells/mO directly in loading buffer containing 50 m� Tris (pH 6.8),
100 mr�i f3-mercaptoethanol, 2% SDS, 0.1 % bromphenol blue, and 10%glycerol. DNA contained within each of the samples was sheared by
passing the cell lysate twice through 18 and 26 gauge syringes. Afterremovmng the cell debris by centrifugation for 15 mm, 20 jil of protein
extract were loaded on a 1 0% acrylamide gel. The separated protein wasthen blotted onto a nitrocellulose filter and hybridized to 1/1 000 dilution ofpAb24O antibody (Santa Cruz Biotechnology; Ref. 9) using the ECL (en-
hanced chemilummnescence) system (Amersham). Equal loading in eachsample was determined by blotting the filters with 1 /1 000 dilution of an
anti-MAPK (ERK-l) antibody (Santa Cruz). This antibody recognizes a44-kDa band on Western blotting.
Cell Viability Determination. Cells (5 x 10�) were cultured in medium
containing 1 0% FCS in the presence or absence of 1 unit/mI Epo at 37’C.The number of viable cells were determined by staining with trypan blue.
Isolation of DNA. Genomic DNA was prepared as described previ-ously (48). In brief, cells were lysed in TNES buffer consisting oflO m� Tris
(pH 7.5), 10 mM EDTA, 100 m� NaCI, 1 % SDS; digested with proteinase
K; and subjected to organic extraction and ethanol precipitation. DNA (10mg) was loaded onto a 1 % agarose gel and stained with ethidium bro-mide. For Southern blotting, DNA was digested with restriction enzymes
and electrophoresed on agarose gels. The DNA was acid depurinatedbefore denaturation and transferred to nitrocellulose. The filters werehybridized with 2 x 106 cpm of random primed probe per ml of hybrid-ization mixture that contained 1 0% dextran sulfate, 5x SSPE (20x
SSPE = 3 M NaCI, 200 mM NaH2PO4H2O, 20 m� EDTA), 5x Denhardt’s
solution (1 x Denhardt’s solution = 0.02% BSAJO.02% FicoII/0.02% poly-vmnylpyrrolidone), 1-2% SDS, and 100 mg/mI denatured salmon spermDNA at 65’C, overnight. The filters were washed twice at 65’C for 20 mmwith a 0.1 x SSC and 0.1 % SDS solution. Hybridized probe was removed
from the filters by two 20-mm washes with 0.1 % SDS, 10 m� Tris, pH 7.5,and 1 m� EDTA at 95#{176}C.
DNA Probes. The Fli-l probe was an EcoRl genomic fragment sub-
cloned from the FIi-l locus (4). The mouse p53 probe was a 900-bpBgIll-Pstl fragment from mouse p53 cDNA 27.la (49).
Karyotypic Analysis. Erythroleukemia cell lines and cells derived fromF-MuLV-induced primary erythroleukemias were grown in a-MEM sup-plemented with 10% FCS. Epo (1 unit/mI) was added to growth mediumof ED cell lines HB9.l-ED and HB 39.2-ED and primary erythroleukemia
cells. Ten jil of colcemid solution (1 mg/mI) were added to the culturedcells, which were subsequently incubated at 37#{176}Cfor 30 mm. The cells
were then pelleted, resuspended in 1 ml of prewarmed 0.075 M KCL, andthen fixed in methanol-acetic acid (3:1 v/v) for 30 mm at room tempera-
ture. After three changes of fixative, the cells were dropped onto cold dryslides, dried, and stained for 2 mm with a Giemsa solution. Chromosome
numbers were then determined.
AcknowledgmentsWe thank Drs. J. Slingeriand, J. Filmus, Kerbel, and M. Mowat for their
comments on the manuscript and Terrie Walker and Lynda Woodcock forhelp in preparation of the manuscript. We also thank Dr. A. Bernstein forsupplying us with p53-transgenic mice.
References1 . Ben-David, V., and Bernstein, A. Friend virus-induced erythroleukemia
and the multistage nature of cancer. Cell, 66: 831-834, 1991.

Cell Growth & Differentiation 1659
3. Silver, J., and Fredrickson, T. N. A new gene that controls the type of
leukemia induced by Friend murine leukemia virus. J. Exp. Med., 158:493-505, 1983.
4. Ben-David, V., Giddens, E. B., and Bernstein, A. Identification and
mapping of a common proviral integration site FIi-l in erythroleukemia
cells induced by Friend murine leukemia virus. Proc. NatI. Acad. Sci. USA,
87: 1332-1336, 1990.
5. Moreau-Gachelin, F., Tavitian, A., and Tambourin, P. Spi-l is a putative
oncogene in virally induced murine erythroleukemias. Nature (Lond.), 331:
277-280, 1988.
6. Moreau-Gachelin, F., Ray, D., de Both, N. J., van der Feltz, M. J. M.,Tambourin, P., and Tavitian, A. Spi- 1 oncogene activated in Rauscher and
Friend murine virus-induced acute erythroleukemias. Leukemia (Balti-more), 4: 20-23, 1990.
7. Ben-David, V., Giddens, E. G., Letwin, K., and Bernstein, A. Erythro-leukemia induction by Friend murine leukemia virus: insertional activationof a new member of the ets gene family, FIi-l , closely linked to c-ets-l.
Genes Dev. 5: 908-918, 1991.
8. Gobel, M. G., Moreau-Gachelin, F., Ray, D., Tambourin, P., Tavitian, A.,Klemsz, M. J., McKercher, S. C., Van Beveren, C., and Maki, R. A. ThePU.l transcription factor is the product of the putative oncogene Spi-l -
Cell, 61: 1165-1166, 1990.
9. Howard, J. C., Vousefi, S., Cheong, G., Bernstein, A., and Ben-David,V. Temporal order and functional analysis of mutations within the Fli-l andp53 genes during the erythroleukemias induced by F-MuLV. Oncogene, 8:2721-2729, 1993.
1 0. Howard, J. C., Berger, L, Bani, M. R., Hawley, R., and Ben-David, V.Activation of the erythropoietin gene in the majority of F-MuLV-inducederythroleukemias results in growth factor independence and enhancedtumorigenicity. Oncogene, 12: 1405-141 5, 1996.
11. Mowat, M., Cheng, A., Kimura, N., Bernstein, A., and Benchimol, S.Rearrangements of the cellular p53 gene in erythroleukemic cells trans-formed by Friend virus. Nature (Lond.), 314: 633-636, 1985.
12. Johnson, P., Chung, S., and Benchimol, S. Growth suppression of
Friend virus-transformed erythroleukemia cells by p53 protein is accom-panied by hemoglobin production and is sensitive to erythropoietin. Mol.
Cell. Biol., 13: 1456-1463, 1993.
13. Kelly, L L, Green, W. F., Hicks, G. G., Bondurant, M. C., Koury, M. J.,
and Auley, H. E. Apoptosis in erythroid progenitors deprived of erythro-poietin occurs during the G1 and S phase of the cell cycle without growth
arrest or stabilization of wild-type p53. Mol. Cell. Biol., 14: 4183-4192,1994.
14. Ryan, J. J., Danish, R., Gottlieb, C., and Clarke, M. Cell cycle analysis
of p53-induced cell death in murine erythroleukemia cells. Mol. Cell. Biol.,
13: 711-719, 1993.
15. Lavigueur, A., Maltby, V., Mock, D., Rossant, J., Pawson, T., and
Bernstein, A. High incidence of lung, bone, and lymphoid tumors intransgenic mice overexpressing mutant alleles of the p53 oncogene. Mol.
Cell. Biol., 9: 3982-3991, 1989.
1 6. Lavigueur, A., and Bernstein, A. p53 transgenic mice: accelerated
erythroleukemia induction by Friend virus. Oncogene, 6: 2197-2201,1991�
17. Mager, D., MacDonald, M. E., Robson, I. B., Mak, T. W., and Bern-stein, A. Clonal analysis of the late stages of erythroleukemia induced bytwo distinct strains of Friend leukemia virus. Mol. Cell. Biol., 1: 721-730,1981.
18. Miller, D. A., Tantravahi, A., Newman, B., Vaithilingam, G. D., andMiller, 0. J. Karyotype of Friend virus-induced mouse erythroleukemiacells. Cytogenetics (Basel), 1: 103-113, 1979.
19. Ostertag, W., Melderis, H., Steinheider, G., Kluge, N., and Dube, S.Synthesis of mouse haemoglobmn and globin mRNA in leukemic cell
cultures. New Biol., 239: 231-234, 1972.
20. Preisler, H. D., Shiraishi, V., Mori, M., and Sandberg, A. A. Clones ofFriend leukemia cells: differences in karyotypes and responsiveness to
inducers of differentiation. Cell Differ., 5: 207-21 6, 1976.
21 . Munroe, D. G., Peacock, J. W., and Benchimol, S. Inactivation of the
cellular p53 gene is a common feature of Friend erythroleukemia: relationto dominant transforming alleles. Mol. Cell. Biol., 10: 3307-3313, 1990.
22. Rovinski, B., and Benchimol, S. Immortalization of rat embryo fibro-blasts by the cellular p53 oncogene. Oncogene, 2: 445-452, 1988.
23. Haupt, V., Alexander, W. S., Barn, G., Klinken, S. P., and Adams, J. M.Novel zinc finger gene implicated as myc collaborator by retrovirally
accelerated lymphomagenesis in Eu-myc transgenic mice. Cell, 65: 753-763, 1991.
24. Schackleford, G. M., MacArthur, C. A., Kwan, H. C., and Varmus, H.E. Mouse mammary tumor virus infection accelerates mammary carcino-
genesis in Wnt-1 transgenic mice by insertional activation of mnt-1/Fgf-3and hst/Fgf-4. Proc. NatI. Acad. Sci. USA, 90: 740-744, 1993.
25. van Lohuizen, M., Verbeek, S., Krimpenfort, P., Domen, J., Saris, C.,Radaszkiewicz, T., and Bems, A. Predisposition to lymphomagenesis in
pim-l transgenic mice: cooperation with c-myc and N-myc in murine
leukemia virus-induced tumors. Cell, 56: 673-682, 1989.
26. van Lohuizen, M., Verbeek, S., Sceijen, B., Wientjens, E., van derGulden, H., and Bems, A. Identification of cooperating oncogenes inEu-myc transgenic mice by provirus tagging. Cell, 65: 737-752, 1991.
27. Canman, C. E., Gilmer, T. M., Coutts, S. B., and Kastan, M. B. Growth
factor modulation of p53-mediated growth arrest versus apoptosis. GenesDev., 9: 600-611, 1995.
28. Haupt, V., Rowan, S., Shaulian, E., Vousden, K. H., and Oren, M.Induction of apoptosis in HeLa cells by trans-activation-deficient p53.Genes Dev., 9: 2170-2183, 1995.
29. Sabbatini, P., Lin, J., Levine, A. J., and White, E. Essential role for
p53-mediated transcription in E1A-induced apoptosis. Genes Dev., 9:2184-2192, 1995.
30. Soddu, S., Blandino, G., Scardigli, A., Martinelli, R., Rizzo, M. G.,Crescenzi, M., and Sacchi, A. Wild-type p53 induces diverse effects in32D cells expressing different oncogenes. Mol. Cell. Biol., 16: 487-495,
1996.
31. Zhang, V., Jiang, W., Chen, C. J., Lee, C. S., Kahn, S. M., Santella, A.M., and Weinstein, I. B. Amplification and overexpression of cyclin Dl in
human hepatocellular carcinoma. Biochem. Biophys. Res. Comm., 196:1010-1016, 1993.
32. Schuetze, S., Stenberg, P. E., and Kabat, D. The ets-related tran-scription factor PU.l immortalizes erythroblasts. Mol. Cell. Biol., 13:
5670-5678, 1993.
33. Nibbs, R. J. B., Itoh, K., Ostertag, W., and Harrison, P. R. Differenti-ation arrest and stromal cell-independent growth of murine erythroleuke-mia cells are associated with elevated expression of ets-related genes but
not with mutation of p53. Mol. Cell. Biol., 13: 5582-5592, 1993.
34. Raycroft, L, Wu, H., and Lozano, G. Transcriptional activation bywild-type but not transforming mutants of p53 anti-oncogene. Science,
249: 1049-1051, 1990.
35. Gu, V., Turch, C. W., and Morgan, D. 0. Inhibition of CDK2 activity invivo by an associated 20K regulatory subunit. Nature (Lond.), 366: 707-710, 1993.
36. Harper, J. W., Adami, G. R., Wei, N., Keyomarsi, K., and Elledge, S. J.The p21 cdk-interacting protein Cipl is a potent inhibitor of G1 cyclmn-dependent kinases. Cell, 75: 805-816, 1993.
37. Xiong, V., Hannon, G. J., Zhang, H., Casso, D., Kobayashi, A., andBeach, D. P21 is a universal inhibitor of cyclmn kinases. Nature (Lond.),366: 701-704, 1993.
38. Kastan, M. B., Zhan, Q., El-Deiry, W. S., Carrier, F., Jacks, T., Walsh,
W. V., Plunkett, B. S., Vogelstemn, B., and Fomace, A. A mammalian cellcycle checkpoint pathway utilizingp53 and GADD45 is defective in ataxia-telangiectasia. Cell, 71: 587-597, 1992.
39. Kuerbitz, S. J., Plunkett, B. S., Walsh, W. V., and Kastan, M. B.Wild-type p53 is a cell cycle checkpoint determinant following irradiation.
Proc. NatI. Acad. Sci. USA, 89: 7491-7495, 1992.
40. Livingstone, L R., White, A., Sprouse, J., Uvanos, E., Jacks, T., andTlsty, T. D. Altered cell cycle arrest and gene amplification potentialaccompany loss of wild-type p53. Cell, 70: 923-935, 1992.
41 . Vin, V., Tainsky, M. A., Bischoff, F. Z., Strong, L C., and WahI, G. M.Wild-type p53 restores cell cycle control and inhibits gene amplification incells with mutant p53 alleles. Cell, 70: 937-948, 1992.

1� Friend Erythroleukemia, p53 Mutation, and Apoptosis
42. Blount, P. L, Meltzer, S. J., Vin, J., Huang, V., and Krasna, M. J.Clonal ordering of 1 7p and Sq allelic losses in Barrett dysplasia andadenocarcinoma. Proc. NatI. Acad. Sci. USA, 90: 3221-3225, 1993.
43. Donehower, L A., Godley, L A., Aldaz, C. M., Pyle, R., Shi, V., Pinkel,D., Gray, J., Bradley, A., Medina, D., and Varmus, H. E. Deficiency of p53accelerates mammary tumorigenesis in Wnt- 1 transgenic mice and pro-motes chromosomal instability. Genes Dev., 882, 1995.
44. Metz, T., Harris, A. W., and Adams, J. M. Absence of p53 allows direct
immortalization of hematopoietic cells by the myc and rafoncogenes. Cell,82: 29-36, 1995.
45. Ben-David, V., Lavigueur, A., Cheong, G. V., and Bernstein, A. Inser-tional inactivation of the p53 gene during Friend leukemia: a new strategy
for identifying tumor suppressor genes. New Biol., 2: 1015-1023, 1990.
46. Oliff, A., Oliff, I., Schmidt, B., and Famulan, N. Isolation of immortalcell lines from the first stage of murine leukemia virus-induced leukemia.Proc. NatI. Acad. Scm. USA, 81: 5464-5467, 1984.
47. Shibuya, T., and Mak, T. Isolation and induction of erythroleukemic
cell lines with properties of erythroid progenitor burst-forming cell (BFU-E)
and erythroid precursor cell (CFU-E). Proc. NatI. Acad. Sci. USA, 80:3721-3725, 1983.
48. Gross-Bellard, M., Oudet, P., and Chambon, P. Isolation of high
molecularweight DNAfrom mammalian cells. Eur. J. Biochem., 36: 32-38,
1973.
49. Jenkins, J. R., Rudge, K., Redomond, S., and Wade-Evans, A. Clon-
ing and expression analysis of full length mouse cDNA sequences encod-ing the transformation associated protein p53. Nucleic Acids Res., 12:
5609-5626, 1984.
50. Main, M. C., Hsu, B., Meyn, R. E., Donehower, L. A., el-Naggar, A. K.,and McDonnell, T. J. Evidence that p53 and bcl-2 are regulators of acommon cell death pathway important for in vivo lymphomagenesis.
Oncogene, 9: 31 07-31 1 2, 1994.





























