Embed Size (px)
Citation preview

PDF hosted at the Radboud Repository of the Radboud University
Nijmegen
This full text is a publisher's version.
For additional information about this publication click this link.
[http://hdl.handle.net/2066/27457]
Please be advised that this information was generated on 2013-02-07 and may be subject to
change.

Paraoxonase in cardiovascular disease: actions and interactions
Thomas van Himbergen


Paraoxonase in cardiovascular disease: actions and interactions
Een wetenschappelijke proeve op het gebied van de Medische Wetenschappen
Proefschrift
Ter verkrijging van de graad van doctor aan de Radboud Universiteit Nijmegen, op gezag van de Rector Magnificus Prof. dr. C.W.P.M. Blom, volgens besluit van het
College van Decanen in het openbaar te verdedigen op donderdag 29 juni 2006 des namiddags om 1.30 uur precies
door
Thomas Mattheus van Himbergengeboren op 31 mei 1977 te Boston, USA

ISBN: 90-9020733-3
Print: Optima Grafische Communicatie. Rotterdam, The Netherlands
Cover: The author was inspired by the principles of the computer game PAC-MAN
Financial support by the Netherlands Heart Foundation and the Radboud University Nijme-gen for the publication of this thesis is gratefully acknowledged.
The study described in this thesis was supported by a grant of the Netherlands Heart Founda-tion (NHF-2001B038)
Promotor: Prof. dr. A.F.H. Stalenhoef
Co-promotores: Dr. L.J.H. van TitsDr. M. Roest (UMC Utrecht) Dr. H.A.M. Voorbij (UMC Utrecht)
Manuscriptcommissie:Prof. dr. P. Smits Prof. dr. J.F. WetzelsProf. dr. J.J.P. Kastelein (AMC Amsterdam)

Voor mijn ouders, voor Frauke


Contents
1 Introduction and outline of this thesis
2 High-throughput genotyping with infrared Fluorescence Allele Specific Hybridization (iFLASH): a simple, reliable and low-cost alternative
3 Paraoxonase genotype, LDL-oxidation and carotid atherosclerosis in male life-long smokers
4 Indications that paraoxonase-1 contributes to plasma high density lipoprotein levels in familial hypercholesterolemia
5 Paraoxonase-1 and linoleic acid oxidation in familial hypercholesterolemia
6 The effect of statin therapy on plasma HDL-cholesterol levels is modified by paraoxonase-1 in patients with familial hypercholesterolaemia
7 Paraoxonase-1 associates with the Familial Combined Hyperlipidemia phenotype
8 Paraoxonase-1 and the risk for coronary heart disease and myocardial infarction in a general population of Dutch women
9 Discussion
10 Summary
11 Samenvatting
Dankwoord
Curriculum vitae/List of publications/International presentations
9
21
33
49
65
79
93
107
121
133
141
149
155


1Introduction and outline of this thesis
An adapted form of this chapter appeared as editorial in “The Netherlands Journal of Medicine”, Februari 2006, Vol. 64, No. 2


11
Introduction and outline of this thesis
A brief history of paraoxonase
In 1946, Abraham Mazur was the first to report the presence of an enzyme in animal tissue which was able to hydrolyze organophosphate compounds [1]. This led to the initial identifi-cation of the human serum paraoxonase (PON1) enzyme in the early 1950s [2, 3]. PON1 was named after its ability to hydrolyze the organophosphate substrate paraoxon (paraoxonase activity, EC 3.1.8.1), which is the toxic metabolite of the insecticide parathion. Because PON1 could also hydrolyze aromatic esters, like phenylacetate (arylesterase activity, EC 3.1.1.2), the term “A-esterase” was introduced for the enzyme hydrolyzing both compounds [2, 3]. This has led to much discussion during the following years as to whether one enzyme or two were responsible for the paraoxonase and arylesterase activity [4], but finally, conclusive evi-dence was delivered that both paraoxonase activity and arylesterase activity were properties of PON1 [5]. When Mackness and colleagues demonstrated that PON1 could prevent the accumulation of lipoperoxides in low-density lipoprotein (LDL), thus linking PON1 to car-diovascular disease [6], the scientific interest in PON1 has increased immensely. Despite the boom in research, to date the exact physiological function of PON1 remains unclear.
PON1 family
PON1 belongs to the family of serum paraoxonases, consisting of PON1, PON2 and PON3. The genes coding for these enzymes are all located next to each other on the long arm of chromosome 7 (7q21.3-q22.1) [7]. PON1 and PON3 are expressed in the liver and excreted in the blood where they are associated with the high-density lipoprotein (HDL) particle [8, 9]. PON2 is not present in blood, but is expressed widely in a number of tissues, including the liver, lungs, brain and heart [10]. Of the paraoxonase family, PON1 is the most investigated and best understood member. Recently, the crystal structure of a recombinant PON variant was solved, making PON the first HDL-associated protein of which the three-dimensional makeup has been elucidated [11]. PON is a six-bladed β-propeller, each blade consisting of four β-sheets, and contained in the central tunnel of the enzyme are two calcium atoms needed for the stabilization of the structure and the catalytic activity. Three α helices, located at the top of the propeller, are in-volved in the anchoring to the HDL particle. The clarification of the crystal structure led to a better understanding of the catalytical mechanisms underlying PON1’s wide substrate range. Furthermore, the crystal structure provided more information about the binding and orienta-tion of PON1 to the HDL particle, revealing that the PON1 active site was directed towards the surface of the HDL particle.Because the compounds that can be hydrolyzed by PON1 e.g. organophospates (paraoxon and diazoxon), warfare agents (soman and sarin) and aromatic esters (phenyl acetate) are non-

12
CHAPTER 1
physiological substrates [12], these activities are not likely to be the physiological functions of PON1. Recent investigations have suggested that the hydrolytic activity towards lactones (cy-clic esters) is native activity of PON1: structure-activity studies show that lactones are PON1’s preferred substrate for hydrolysis [13]. In addition, all members of the PON family have lac-tonase activity, implying that this activity has been conserved throughout the evolution of the enzyme [14]. In vivo, there is a wide inter-individual variation in PON1 concentration and activity. This variation is for a major part determined by common genetic variants (polymor-phisms) in the PON1 gene. Four polymorphisms in the promoter region of the PON1 gene (-107C>T, -162A>G, -824G>A, -907G>C) have been reported to affect the expression and thus the serum concentration of the enzyme [15-17]. The –107C>T polymorphism has been the most important genetic determinant of PON1 levels [15-17]. The coding region of the PON1 gene contains two polymorphic sites: a leucine (L) to methionine (M) transition at position 55 (55L>M), and a glutamine (Q) to arginine (R) transition at position 192 (192Q>R) [18, 19]. Due to linkage with polymorphisms in the PON1 promoter region, the 55L>M poly-morphism affects the enzyme concentration [16]. In addition, the 55L>M polymorphism is located in the N-terminal side of PON1, which plays a role in the binding of PON-1 with HDL [20], and thus may alter the ability of PON1 to form a complex with HDL [21]. The 192Q>R polymorphism is responsible for a striking substrate specific difference in the hydrolytic ac-tivity of the enzyme [18, 19, 22]. Paraoxon is most efficiently hydrolyzed by the 192R isoform [18, 19], and diazoxon, soman and sarin are more efficiently hydrolyzed by the 192Q isoform [22]. The capacity of blood to hydrolyze paraoxon (paraoxonase activity) is often used as a marker for the PON1 enzyme activity. This enzyme activity reflects the combined effects of the 192Q>R polymorphism and the variation in concentration of the PON1 enzyme. In addi-tion to the paraoxonase activity, the PON1 concentration can be measured directly in serum with an enzyme-linked immunosorbent assay (ELISA) [23]. Otherwise, because PON1 es-terase activity is not polymorphic (i.e. influenced by the 192Q>R polymorphism), the PON1 concentration can be estimated by measuring the arylesterase activity [24].The 192Q>R and –107C>T polymorphisms are responsible for an up to 13-fold inter-individ-ual variation in PON1 enzyme activity and concentration [25]. Life-style factors like smoking and alcohol consumption also influence the PON1 in vivo status. Cigarette smoke inhibits PON1 activity in vitro [26], and in agreement, paraoxonase activity is lower in smokers than in non-smokers [27-29]. Furthermore, moderate consumption of beer, wine or spirits is as-sociated with an increased serum PON1 activity [30, 31].
The role of PON1 in humans
To date, the role of PON1 in vivo is unclear, but in general, PON1 is thought to attenuate the oxidation of LDL. This hypothesis was based on in vitro findings, showing that purified PON1

13
Introduction and outline of this thesis
inhibited the accumulation of lipid peroxides in LDL [6]. In the arterial wall the oxidized LDL particle (oxLDL) is recognized by oxLDL specific receptors on the macrophage and taken up into the cell [32]. Since there is no negative feedback mechanism for this uptake, this process eventually leads to an overload of lipids in the macrophage, which causes the lipid-laden macrophages to aggregate and form a fatty streak characteristic of atherosclerosis [32]. The oxidation of LDL is a key process in the pathophysiology of atherosclerosis and the onset of cardiovascular disease [33], and therefore it is not surprising that PON1 has been the subject of increasing scientific interest since its alleged role in the oxidation of LDL. Apart from inhibition of LDL oxidation, there is evidence from animal and in vitro models that paraoxonase can protect the HDL particle from oxidation and preserve the integrity of HDL [34, 35]. Furthermore, many epidemiological studies found that polymorphisms in the PON1 gene, responsible for the variations in PON1 activity and concentration, also contrib-ute to variation in plasma levels of HDL-C in different populations [36-39]. Because HDL has many athero-protective functions, such as the removal of excess cholesterol from tis-sues (reverse cholesterol transport) and the inhibition of inflammatory processes [40, 41], the preservation of the HDL particle may be a beneficial role of PON1. In blood, PON1 can hydrolyze homocysteine thiolactones, a metabolite of homocysteine [42]. Homocysteine thiolactones can have an adverse effect on protein synthesis and may lead to endothelial dysfunction and vascular damage [43]. The detoxification of the homocysteine thiolactone may therefore be a cardioprotective function of PON1. Other interesting discoveries with respect to PON1 come from the field of pharmacology. The LDL-cholesterol-lowering HMG-CoA reductase inhibitors (statins) were found to affect PON1 activity, concentration and gene expression [44-46]. Reversely, since PON1 signifi-cantly predicted changes of HDL cholesterol during statin treatment [47], PON1 may be an important effect modifier of the success of the statin treatment.
PON1 and cardiovascular disease
As mentioned earlier, the finding that PON1 has properties that inhibit LDL oxidation in vitro implicated that PON1 could have a protective role in the onset of cardiovascular disease. However, the validity of those findings have been questioned, since it could not be excluded that the protection against in vitro oxidation was caused by the detergent used during the preparation or a low molecular mass compound co-purified with PON1 [48]. Still, the results from animal experimental work uniformly show that PON1 is a protective enzyme against atherogenesis: PON1 deficiency in mice results in increased oxidative stress in serum and macrophages[49], and HDL isolated from PON1-deficient mice did not protect LDL from oxidation[50], whereas HDL isolated from human PON1 transgenic mice (having 2- to 4-fold increased PON1 plasma levels) was more protective against LDL oxidation in a dose depen-

14
CHAPTER 1
dent manner [51]. Finally, perhaps the strongest evidence available that PON1 plays a role in atherogenesis is that PON1 deficient mice are more prone to develop atherosclerosis than wild-type mice, when fed a high-fat/high-cholesterol diet [50].In humans, however, the role of PON1 genetic variants, levels and activities and the onset of cardiovascular disease is less clear. Many epidemiological studies report conflicting results (reviewed in [52]), and a recent meta-analysis among 43 investigations studying the 55L>M, 192Q>R and –107C>T polymorphisms in relation to coronary heart disease (CHD) dem-onstrated no effect for the 55L>M and –107C>T polymorphisms, and a slightly increased risk for carriers of the R-allele at position 192 [53]. In general, however, the effects of single genetic variants on the onset of complex diseases (like cardiovascular disease) are often too weak to be detected in studies of relative small sample sizes [54]. It is therefore recommended to measure PON1 activity and concentration in addition to PON1 genotype [25, 55-57]. Until today, there have been only a few studies (the majority being case-control studies) that have measured PON1 activity and concentration (reviewed in [56]). Furthermore, a major limita-tion of measuring PON1 in case-control studies is that blood is drawn after the cardiovascular event has taken place. In this way it is not possible to distinguish whether PON1 activity was the cause of the event or, conversely, a reflection of the event itself. To overcome this problem a prospective study design is needed. Until now, only one prospective investigation on PON1 activity and concentration and CHD outcome has been published. This study showed that low serum PON1 activity toward paraoxon was an independent risk factor for coronary events in men with preexisting CHD [58].
Outline of this thesis
The main research question of this thesis is whether PON1 plays a role in atherogenesis and onset of cardiovascular disease in humans. We investigate PON1 influences on LDL oxida-tion, inflammation, lipid metabolism, atherosclerosis and cardiovascular incidence. In addi-tion, we investigate the interaction of PON1 with statin therapy. Studies were carried out in three different populations at high risk to develop cardiovascular disease: 1.) heavy smokers, 2.) patients with familial hypercholesterolemia (FH) and 3.) a cohort of familial combined hy-perlipidemia (FCH) patients and their unaffected relatives. In addition, a healthy population was followed in time, and monitored for the occurrence of a cardiovascular event (prospec-tive investigation).In Chapter 2, we describe a novel high-throughput genotyping method, which facilitates the typing of PON1 polymorphisms in large population studies. Chapter 3, describes the effects of the 55L>M and 192Q>R genetic variants on LDL oxidation in a population of heavy smokers. LDL oxidation was assessed by the measuring the blood concentration of circulating antibod-ies directed against oxLDL and by monitoring the susceptibility of isolated LDL to oxidation.

15
Introduction and outline of this thesis
In Chapter 4 we investigate the effects of PON1 activity, concentration and genetic variance on plasma HDL-cholesterol level, circulating oxLDL levels, inflammation as reflected by C-reactive protein, and atherosclerosis as assessed by measuring the intima media thickness of the carotid artery. Investigations were carried out in a population of FH patients. Chapter 5 describes the effects of PON1 activity, concentration and genetic variance on lipid oxidation, quantitated by high performance liquid chromatography. Chapter 6 reports the interaction of PON1 with statin therapy in patients with FH. In Chapter 7, the association of PON1 with the FCH phenotype was investigated, in 32 families known for the occurrence of FCH. In Chap-ter 8 we prospectively study the development of CHD in relation to PON1 genotypes and baseline PON1 activities. Finally, in Chapter 9, the main findings of this thesis are discussed in detail and placed in a broader perspective. A brief summary and discussion of this thesis can be found in Chapter 10 (English) and Chapter 11 (Dutch).

16
CHAPTER
References
[1] A. Mazur, An enzyme in animal tissues capable of hydrolyzing the phosphorus-fluorine bond of alkyl fluorophospates, J. Biol. Chem. (1946) 271-289.[2] W.N. Aldridge, Serum esterases. II. An enzyme hy-drolysing diethyl p-nitrophenyl phosphate (E600) and its identity with the A-esterase of mammalian sera, Biochem J 53 (1953) 117-124.[3] W.N. Aldridge, Serum esterases. I. Two types of esterase (A and B) hydrolysing p-nitrophenyl acetate, propionate and butyrate, and a method for their deter-mination, Biochem J 53 (1953) 110-117.[4] B. La Du, Historical Considerations, in: L.G. Costa, Furlong, C.E. (Eds.), Paraoxonase (PON1) in health and disease: basic and clinical aspects, Kluwer aca-demic publishers, 2002, pp. 1-25.[5] R.C. Sorenson, S.L. Primo-Parmo, C.L. Kuo, S. Ad-kins, O. Lockridge, B.N. La Du, Reconsideration of the catalytic center and mechanism of mammalian paraoxonase/arylesterase, Proc Natl Acad Sci U S A 92 (1995) 7187-7191.[6] M.I. Mackness, S. Arrol, P.N. Durrington, Paraoxo-nase prevents accumulation of lipoperoxides in low-density lipoprotein, FEBS Lett 286 (1991) 152-154.[7] S.L. Primo-Parmo, R.C. Sorenson, J. Teiber, B.N. La Du, The human serum paraoxonase/arylesterase gene (PON1) is one member of a multigene family, Genom-ics 33 (1996) 498-507.[8] M.I. Mackness, S.D. Hallam, T. Peard, S. Warner, C.H. Walker, The separation of sheep and human se-rum “A”-esterase activity into the lipoprotein fraction by ultracentrifugation, Comp Biochem Physiol B 82 (1985) 675-677.[9] S.T. Reddy, D.J. Wadleigh, V. Grijalva, C. Ng, S. Hama, A. Gangopadhyay, D.M. Shih, A.J. Lusis, M. Navab, A.M. Fogelman, Human paraoxonase-3 is an HDL-associat-ed enzyme with biological activity similar to paraoxo-nase-1 protein but is not regulated by oxidized lipids,
Arterioscler Thromb Vasc Biol 21 (2001) 542-547.[10] H. Mochizuki, S.W. Scherer, T. Xi, D.C. Nickle, M. Majer, J.J. Huizenga, L.C. Tsui, M. Prochazka, Human PON2 gene at 7q21.3: cloning, multiple mRNA forms, and missense polymorphisms in the coding sequence, Gene 213 (1998) 149-157.[11] M. Harel, A. Aharoni, L. Gaidukov, B. Brumshtein, O. Khersonsky, R. Meged, H. Dvir, R.B. Ravelli, A. Mc-Carthy, L. Toker, I. Silman, J.L. Sussman, D.S. Tawfik, Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic en-zymes, Nat Struct Mol Biol 11 (2004) 412-419.[12] D.I. Draganov, B.N. La Du, Pharmacogenetics of paraoxonases: a brief review, Naunyn Schmiedebergs Arch Pharmacol 369 (2004) 78-88.[13] O. Khersonsky, D.S. Tawfik, Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase, Biochemistry 44 (2005) 6371-6382.[14] D.I. Draganov, J.F. Teiber, A. Speelman, Y. Osawa, R. Sunahara, B.N. La Du, Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities, J Lipid Res 46 (2005) 1239-1247.[15] I. Leviev, R.W. James, Promoter polymorphisms of human paraoxonase PON1 gene and serum paraoxo-nase activities and concentrations, Arterioscler Thromb Vasc Biol 20 (2000) 516-521.[16] V.H. Brophy, R.L. Jampsa, J.B. Clendenning, L.A. McKinstry, G.P. Jarvik, C.E. Furlong, Effects of 5’ regu-latory-region polymorphisms on paraoxonase-gene (PON1) expression, Am J Hum Genet 68 (2001) 1428-1436.[17] S. Deakin, I. Leviev, M.C. Brulhart-Meynet, R.W. James, Paraoxonase-1 promoter haplotypes and serum paraoxonase: a predominant role for polymorphic po-sition - 107, implicating the Sp1 transcription factor, Biochem J 372 (2003) 643-649.[18] S. Adkins, K.N. Gan, M. Mody, B.N. La Du, Molecu-

17
Introduction and outline of this thesis
lar basis for the polymorphic forms of human serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B allozymes, Am J Hum Genet 52 (1993) 598-608.[19] R. Humbert, D.A. Adler, C.M. Disteche, C. Hassett, C.J. Omiecinski, C.E. Furlong, The molecular basis of the human serum paraoxonase activity polymorphism, Nat.Genet. 3 (1993) 73-76.[20] C.E. Furlong, R.J. Richter, C. Chapline, J.W. Crabb, Purification of rabbit and human serum paraoxonase, Biochemistry 30 (1991) 10133-10140.[21] I. Leviev, S. Deakin, R.W. James, Decreased stabil-ity of the M54 isoform of paraoxonase as a contribu-tory factor to variations in human serum paraoxonase concentrations, J.Lipid Res. 42 (2001) 528-535.[22] H.G. Davies, R.J. Richter, M. Keifer, C.A. Broom-field, J. Sowalla, C.E. Furlong, The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin, Nat Genet 14 (1996) 334-336.[23] T. Kujiraoka, T. Oka, M. Ishihara, T. Egashira, T. Fujioka, E. Saito, S. Saito, N.E. Miller, H. Hattori, A sandwich enzyme-linked immunosorbent assay for human serum paraoxonase concentration, J Lipid Res 41 (2000) 1358-1363.[24] H.W. Eckerson, C.M. Wyte, B.N. La Du, The human serum paraoxonase/arylesterase polymorphism, Am J Hum Genet 35 (1983) 1126-1138.[25] R.J. Richter, C.E. Furlong, Determination of paraoxonase (PON1) status requires more than geno-typing, Pharmacogenetics 9 (1999) 745-753.[26] E. Nishio, Y. Watanabe, Cigarette smoke extract inhibits plasma paraoxonase activity by modification of the enzyme’s free thiols, Biochem Biophys Res Com-mun 236 (1997) 289-293.[27] R.W. James, I. Leviev, A. Righetti, Smoking is as-sociated with reduced serum paraoxonase activity and concentration in patients with coronary artery disease, Circulation 101 (2000) 2252-2257.
[28] Senti, M. Tomas, R. Anglada, R. Elosua, J. Marru-gat, M.I. Covas, M. Fito, Interrelationship of smoking, paraoxonase activity, and leisure time physical activity: a population-based study, Eur J Intern Med 14 (2003) 178-184.[29] N. Ferre, J. Camps, J. Fernandez-Ballart, V. Arija, M.M. Murphy, S. Ceruelo, E. Biarnes, E. Vilella, M. Tous, J. Joven, Regulation of serum paraoxonase activity by genetic, nutritional, and lifestyle factors in the general population, Clin Chem 49 (2003) 1491-1497.[30] M.S. van der Gaag, A. van Tol, L.M. Scheek, R.W. James, R. Urgert, G. Schaafsma, H.F. Hendriks, Daily moderate alcohol consumption increases serum paraoxonase activity; a diet-controlled, randomised intervention study in middle-aged men, Atherosclero-sis 147 (1999) 405-410.[31] A. Sierksma, M.S. van der Gaag, A. van Tol, R.W. James, H.F. Hendriks, Kinetics of HDL cholesterol and paraoxonase activity in moderate alcohol consumers, Alcohol Clin Exp Res 26 (2002) 1430-1435.[32] A.J. Lusis, Atherosclerosis, Nature 407 (2000) 233-241.[33] D. Steinberg, S. Parthasarathy, T.E. Carew, J.C. Khoo, J.L. Witztum, Beyond cholesterol. Modifications of low-density lipoprotein that increase its athero-genicity, N Engl J Med 320 (1989) 915-924.[34] M.N. Oda, J.K. Bielicki, T.T. Ho, T. Berger, E.M. Rubin, T.M. Forte, Paraoxonase 1 overexpression in mice and its effect on high-density lipoproteins, Bio-chem Biophys Res Commun 290 (2002) 921-927.[35] M. Aviram, M. Rosenblat, C.L. Bisgaier, R.S. New-ton, S.L. Primo-Parmo, B.N. La Du, Paraoxonase inhib-its high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxo-nase, J Clin Invest 101 (1998) 1581-1590.[36] T.M. van Himbergen, M. Roest, J. de Graaf, E.H. Jansen, H. Hattori, J.J. Kastelein, H.A. Voorbij, A.F. Stalenhoef, L.J. van Tits, Indications that paraoxonase-1 contributes to plasma high density lipoprotein levels

18
CHAPTER
in familial hypercholesterolemia, J Lipid Res 46 (2005) 445-451.[37] J. Ruiz, H. Blanche, R.W. James, M.C. Garin, C. Vaisse, G. Charpentier, N. Cohen, A. Morabia, P. Passa, P. Froguel, Gln-Arg192 polymorphism of paraoxonase and coronary heart disease in type 2 diabetes, Lancet 346 (1995) 869-872.[38] S.R. Srinivasan, S. Li, W. Chen, R. Tang, M.G. Bond, E. Boerwinkle, G.S. Berenson, Q192R polymorphism of the paraoxanase 1 gene and its association with serum lipoprotein variables and carotid artery intima-media thickness in young adults from a biracial community: The Bogalusa Heart Study, Atherosclerosis 177 (2004) 167-174.[39] R.A. Hegele, J.H. Brunt, P.W. Connelly, A polymor-phism of the paraoxonase gene associated with varia-tion in plasma lipoproteins in a genetic isolate, Arte-rioscler Thromb Vasc Biol 15 (1995) 89-95.[40] M. Wang, M.R. Briggs, HDL: the metabolism, function, and therapeutic importance, Chem Rev 104 (2004) 119-137.[41] C. Wadham, N. Albanese, J. Roberts, L. Wang, C.J. Bagley, J.R. Gamble, K.A. Rye, P.J. Barter, M.A. Vadas, P. Xia, High-density lipoproteins neutralize C-reac-tive protein proinflammatory activity, Circulation 109 (2004) 2116-2122.[42] H. Jakubowski, Calcium-dependent human se-rum homocysteine thiolactone hydrolase. A protective mechanism against protein N-homocysteinylation, J Biol Chem 275 (2000) 3957-3962.[43] H. Jakubowski, Anti-N-homocysteinylated protein autoantibodies and cardiovascular disease, Clin Chem Lab Med 43 (2005) 1011-1014.[44] S. Deakin, I. Leviev, S. Guernier, R.W. James, Sim-vastatin modulates expression of the PON1 gene and increases serum paraoxonase: a role for sterol regula-tory element-binding protein-2, Arterioscler Thromb Vasc Biol 23 (2003) 2083-2089.[45] M. Tomas, M. Senti, F. Garcia-Faria, J. Vila, A. Tor-
rents, M. Covas, J. Marrugat, Effect of simvastatin ther-apy on paraoxonase activity and related lipoproteins in familial hypercholesterolemic patients, Arterioscler Thromb Vasc Biol 20 (2000) 2113-2119.[46] C. Gouedard, N. Koum-Besson, R. Barouki, Y. Morel, Opposite regulation of the human paraoxo-nase-1 gene PON-1 by fenofibrate and statins, Mol Pharmacol 63 (2003) 945-956.[47] R. Malin, R. Laaksonen, J. Knuuti, T. Janatuinen, R. Vesalainen, P. Nuutila, T. Lehtimaki, Paraoxonase genotype modifies the effect of pravastatin on high-density lipoprotein cholesterol, Pharmacogenetics 11 (2001) 625-633.[48] J.F. Teiber, D.I. Draganov, B.N. La Du, Purified human serum PON1 does not protect LDL against oxidation in the in vitro assays initiated with copper or AAPH, J Lipid Res 45 (2004) 2260-2268.[49] O. Rozenberg, M. Rosenblat, R. Coleman, D.M. Shih, M. Aviram, Paraoxonase (PON1) deficiency is as-sociated with increased macrophage oxidative stress: studies in PON1-knockout mice, Free Radic Biol Med 34 (2003) 774-784.[50] D.M. Shih, L. Gu, Y.R. Xia, M. Navab, W.F. Li, S. Hama, L.W. Castellani, C.E. Furlong, L.G. Costa, A.M. Fogelman, A.J. Lusis, Mice lacking serum paraoxo-nase are susceptible to organophosphate toxicity and atherosclerosis, Nature 394 (1998) 284-287.[51] A. Tward, Y.R. Xia, X.P. Wang, Y.S. Shi, C. Park, L.W. Castellani, A.J. Lusis, D.M. Shih, Decreased atheroscle-rotic lesion formation in human serum paraoxonase transgenic mice, Circulation 106 (2002) 484-490.[52] L.G. Costa, T.B. Cole, G.P. Jarvik, C.E. Furlong, Functional genomic of the paraoxonase (PON1) poly-morphisms: effects on pesticide sensitivity, cardiovas-cular disease, and drug metabolism, Annu Rev Med 54 (2003) 371-392.[53] J.G. Wheeler, B.D. Keavney, H. Watkins, R. Collins, J. Danesh, Four paraoxonase gene polymorphisms in 11212 cases of coronary heart disease and 12786 con-

19
Introduction and outline of this thesis
trols: meta-analysis of 43 studies, Lancet 363 (2004) 689-695.[54] H.M. Colhoun, P.M. McKeigue, G. Davey Smith, Problems of reporting genetic associations with com-plex outcomes, Lancet 361 (2003) 865-872.[55] B. Mackness, G.K. Davies, W. Turkie, E. Lee, D.H. Roberts, E. Hill, C. Roberts, P.N. Durrington, M.I. Mackness, Paraoxonase status in coronary heart dis-ease: are activity and concentration more important than genotype?, Arterioscler Thromb Vasc Biol 21 (2001) 1451-1457.[56] M. Mackness, B. Mackness, Paraoxonase 1 and atherosclerosis: is the gene or the protein more impor-tant?, Free Radic Biol Med 37 (2004) 1317-1323.[57] G.P. Jarvik, L.S. Rozek, V.H. Brophy, T.S. Hatsukami, R.J. Richter, G.D. Schellenberg, C.E. Furlong, Paraoxo-nase (PON1) phenotype is a better predictor of vascu-lar disease than is PON1(192) or PON1(55) genotype, Arterioscler Thromb Vasc Biol 20 (2000) 2441-2447.[58] B. Mackness, P. Durrington, P. McElduff, J. Yarnell, N. Azam, M. Watt, M. Mackness, Low paraoxonase ac-tivity predicts coronary events in the Caerphilly Pro-spective Study, Circulation 107 (2003) 2775-2779.


2High-throughput genotyping with infrared
Fluorescence Allele Specific Hybridization (iFLASH): a simple, reliable and low-cost alternative
Clinical Biochemistry, 2006 Apr 17, Epub
T.M. van Himbergen H.A. Voorbij
A.D. BarendrechtB.B. van Rijn R. Brambilla
L.J. van TitsM. Roest

Abstract
Our objective was to develop and validate a novel genotyping approach named infrared Fluo-rescence Allele Specific Hybridization (iFLASH), which combines the principle of allele spe-cific oligonucleotide (ASO) hybridization with the advanced possibilities of infrared imag-ing. As an example, we genotyped the 55L>M and the 192Q>R common genetic variants of the paraoxonase-1 gene in 92 DNA samples using the iFLASH technique, and validated the out-comes with the restriction fragment length polymorphism (RFLP) and TAQman genotyping assays. There was a 100 percent agreement in genotype outcome among the three methods. Although we found complete unity in genotype outcome, the iFLASH assay has essential advantages over the RFLP and TAQman genotyping assays. First, the iFLASH technique is capable of handling up to 1536 samples per assay, which makes it a suitable technique for high-through-put genotyping. Secondly, because the costs per assay are lower, high-throughput genotyping with iFLASH is affordable.

23
High-throughput genotyping with iFLASH
Introduction
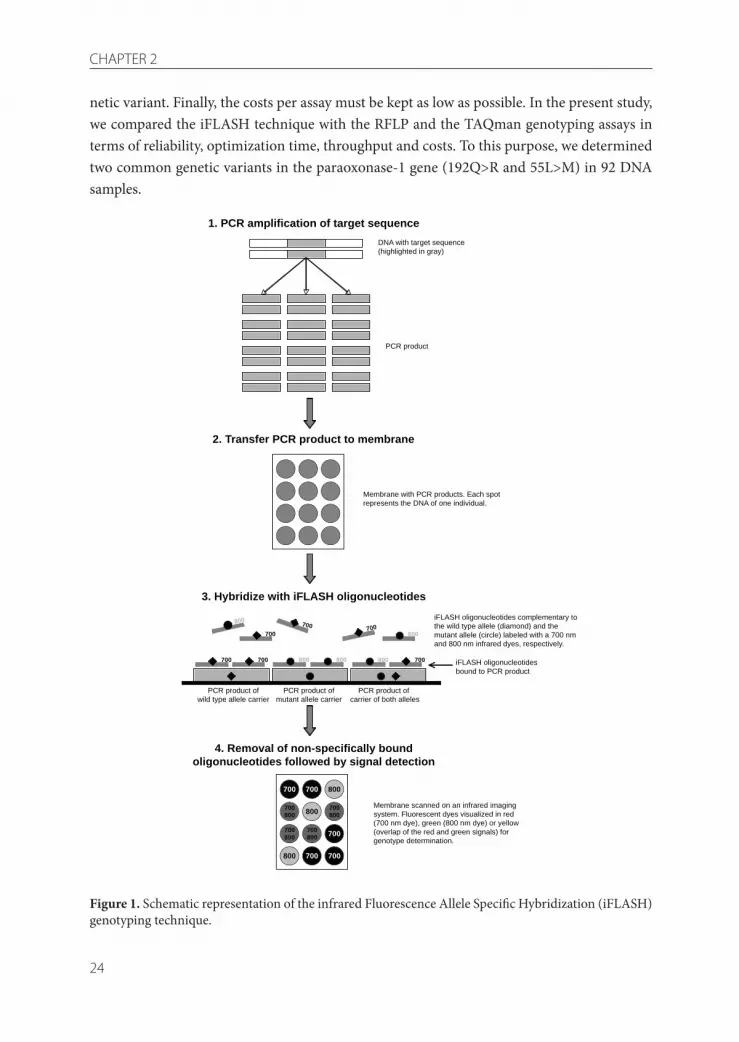
The candidate-gene approach to investigate the involvement of common genetic variants (also known as single nucleotide polymorphisms or SNPs) in the onset of complex diseases, is now commonly used in clinical research and population studies [1]. For the detection of genetic variation in human population studies, different genotyping methods are available. Restriction fragment length polymorphism (RFLP) is the most com-monly used technique, but the laborious sample handling and visual inspection of DNA gels for genotyping makes this technique unsuitable for high-throughput screening. Another widely used method is the TAQman assay, utilizing the 5’-exonuclease activity of the TAQ-polymerase to discriminate alleles that differ by a single base substitution [2]. Although this method is quick and accurate, a major disadvantage of the TAQman assay is that it uses rela-tively expensive dual-labeled allele-specific oligonucleotides (ASOs). This results in substan-tial costs for genotyping large populations. At the moment, the matrix-assisted laser desorp-tion ionization time-of-flight mass spectrometry (MALDI-TOF MS) has gained interest [3]. However, this technique requires highly advanced equipment, not available for the majority of laboratories. In addition to the techniques described above, there is a wide range of com-mercial alternatives for genotype determination, less suitable for high-throughput genotyp-ing in the majority of non-commercial laboratories, because of high costs of equipment and consumables.The infrared Fluorescence Allele Specific Hybridization (iFLASH) technique described in this study provides a solution to the problem of high assay costs by using inexpensive single-la-beled fluorescent oligonucleotides for allelic discrimination. In addition, the iFLASH tech-nique is suitable for handling large amounts of samples. The principle of iFLASH (Figure 1) is based on the classical genotyping technique of ASO hybridization [4], in combination with the new possibilities of high sensitive fluorescence imaging. First, the amplified DNA sequence is immobilized on a nylon membrane (step 1-2). Next, the membrane is hybridized with iFLASH oligonucleotides, i.e. single-strand DNA probes complementary to the wild-type or mutant allele of the SNP of interest which are labeled with infrared dyes for detec-tion (step 3). Finally, the membrane is stringently washed to remove nonspecifically bound iFLASH oligonucleotides, followed by signal detection of the iFLASH oligonucleotides on an infrared imaging system. The determination of the genotypes can be done optically, based on differences in colors, or automatically, based on differences in the intensity of the fluorescent signals (step 4).When considering different techniques for genotyping candidate-genes in large-scale popu-lation studies, four important aspects should be considered. First, the technique should be reliable and give precise outcomes. Second, the optimization of the genotyping assay should be flexible and easy. Third, the technique should be high throughput, i.e. the technique should be able to handle large numbers of samples, as well as automatically detect and type the ge-
24
CHAPTER 2
netic variant. Finally, the costs per assay must be kept as low as possible. In the present study, we compared the iFLASH technique with the RFLP and the TAQman genotyping assays in terms of reliability, optimization time, throughput and costs. To this purpose, we determined two common genetic variants in the paraoxonase-1 gene (192Q>R and 55L>M) in 92 DNA samples.
Figure 1. Schematic representation of the infrared Fluorescence Allele Specific Hybridization (iFLASH) genotyping technique.
700 800700 800 800 700
700 800
700
700
700
700
700700800
800
800
800
700800
700800
700800
1. PCR amplification of target sequence
2. Transfer PCR product to membrane
3. Hybridize with iFLASH oligonucleotides
4. Removal of non-specifically boundoligonucleotides followed by signal detection
DNA with target sequence(highlighted in gray)
PCR product
Membrane with PCR products. Each spotrepresents the DNA of one individual.
PCR product ofwild type allele carrier
PCR product of carrier of both alleles
PCR product ofmutant allele carrier
iFLASH oligonucleotides complementary tothe wild type allele (diamond) and themutant allele (circle) labeled with a 700 nmand 800 nm infrared dyes, respectively.
iFLASH oligonucleotidesbound to PCR product
Membrane scanned on an infrared imagingsystem. Fluorescent dyes visualized in red(700 nm dye), green (800 nm dye) or yellow(overlap of the red and green signals) forgenotype determination.
800700 700

25
High-throughput genotyping with iFLASH
Methods
DNA samplesGenomic DNA was isolated from blood of 92 healthy volunteers using a commercially avail-able kit (Puregene, Gentra Systems, Minneapolis, USA). All participants had given informed consent.
RFLP 55L>M and 192Q>R genotyping RFLP primers, restriction enzymes, PCR- and restriction-conditions are described by Hum-bert et al. [5]. Digested DNA fragments were separated on a 3% agarose gel and visualized with SYBR Green (Molecular Probes, Leiden, The Netherlands). The 55L allele corresponded to the presence of a non-digested 170-bp fragment, the 55M allele to a 44-bp and a 126-bp fragment, the 192Q allele to a non-digested 99-bp fragment, and the 192R allele to a 33-bp and a 66-bp fragment.
TAQman 55L>M and 192Q>R genotyping For the TAQMan assay, PCR primers and FAM and VIC fluorescent dye labeled ASOs were designed by the Assay by-Design service from Applied Biosystems (Applied Biosystems, Nieu-werkerk a/d IJsel, The Netherlands), using the following primers and ASOs to genotype the 55L>M polymorphism: forward 5’-ACAACCTGTACTTTCTGTTCTCTTTTCTG-3’ and re-verse 5’-CAGAGCTAATGAAAGCCAGTCCAT-3’ in combination with the ASOs 5’-[VIC]-AGTATCTCCAAGTCTTC-[NFQ]-3’ for detection of the 55L allele and 5’-[FAM]-CAG-TATCTCCATGTCTTC-[NFQ]-3’ for detection of the 55M allele. Similarly for the 192Q>R polymorphism: forward 5’-CTGAGCACTTTTATGGCACAAATGA-3’ and reverse 5’-AC-CACGCTAAACCCAAATACATCTC-3’ in combination with the ASOs 5’-[VIC]-CCTACT-TACAATCCTG-[NFQ]-3’ for detection of the 192Q allele and 5’-[FAM]-CCCTACTTAC-GATCCTG-[NFQ]-3’ for detection of the 192R allele. The PCR conditions were as follows: initial denaturation at 95 ˚C for 10 minutes, 40 cycles of denaturation for 15 seconds at 92 ˚C and annealing for 1 minute at 60 ˚C, followed by 10 minutes at 72 ˚C. Fluorescence signals were measured on a microplate reader (Fluostar Galaxy, BMG Labtechnologies, Offenburg, Germany).
iFLASH 55L>M and 192Q>R genotyping For the iFLASH-hybridization technique two PCR reactions were performed for the ampli-fication of DNA sequences coding for the 55L>M and the 192Q>R polymorphism. The PCR primers and conditions for the amplification were identical to those used for the RFLP assay. PCRs were performed in 384 wells plates, using approximately 12,5 ng template DNA in an end-volume of 10 μL PCR reaction mix. The amplified fragments were dried by heating and resuspended in 5 μL 0.5 M NaOH. For each polymorphism, an individual array was created

26
CHAPTER 2
by transferring the PCR products from the PCR plate to a Hybond N+ membrane (Amer-sham Pharmacia Biotech, Buckinghamshire, England) by a centrifugation method previously described [6]. In brief, a membrane, pretreated in 10x SSC, was placed over the open wells of the PCR plate and covered by all-purpose filter paper. A clamping device kept the mem-brane and filter paper in place while the PCR products were transferred to the membrane by centrifugation at 1500 rpm in a microplate centrifuge (Mistral 2000, MSE Scientific Instru-ments, Crawley, UK). For each polymorphism, iFLASH-oligonucleotides were designed for detection of the wild-type and the mutant alleles. Discrimination between the wild-type and the mutant iFLASH oligonucleotides was achieved by adding an infrared dye excited at either a wavelength of 700 nm (IRD700) or at a wavelength of 800 nm (IRD800). The iFLASH oli-gonucleotides were commercially obtained from Metabion, Martinsried, Germany. For the genotyping of the 55L>M and 192Q>R polymorphisms we used the following iFLASH-oligo-nucleotides: 5’-IRD800-CTGAAGACATGGAGAT-3’ (55M), 5’-IRD700-CTGAAGACTTG-GAGA-3’ (55L), 5’-IRD800-CTACTTACGATCCTGGG-3’ (192R) and 5’IRD700-CTACT-TACAATCCTGGGA-3’ (192Q). Membranes were pre-hybridized in 30 mL hybridization buffer (6x SSC, 2.5x Denhahardt’s reagent, 0.4% SDS ) at 42 °C for 2 hours. After pre-hy-bridization, 50 pmol of both the wild-type and the mutant iFLASH oligonucleotides were added to the hybridization buffer and the membranes were hybridized for 1 hour at 42 °C. Subsequently, membranes were rinsed in wash buffer (2x SSC, 0.1% SDS) to remove excess iFLASH oligonucleotides, followed by a 30 minute allele-specific temperature wash (at 45 °C for 55L>M and at room temperature for 192Q>R). Fluorescent signals were detected on an Odyssey® Imaging System (LI-COR biosciences, Lincoln, Nebraska, USA). The IRD800 dye was detected by the 800 nm channel and was represented as a green color by the imaging sys-tem, the IRD700 dye was detected by the 700 nm channel and was represented as a red color by the imaging system. A yellow color was visible when both signal intensities were present in equal amounts.
Data analysisThe genotype determination based on the TAQman assay were assigned in a similar fashion as described in detail previously [7]. Briefly, the FAM signal was compared to the VIC sig-nal by calculating the log(FAM/VIC) ratio for each data point. The distribution of the log (FAM/VIC) ratios was displayed in a histogram to arbitrarily determine cut-off values for each genotype group. Genotype assignment for the iFLASH-hybridization assay was similar to the TAQman assay and done by comparing the 800 nm channel signal intensity to the 700 nm channel intensity and calculating the log(800 channel/700 channel) ratios. The iFLASH-hybridization technique was validated by comparing these outcomes to the RFLP and TAQman outcomes. The scatter-plot and the histogram were created with SPSS version 11.5.

27
High-throughput genotyping with iFLASH
Results
Two PCR reactions were performed to amplify the 55L>M and 192Q>R polymorphic regions. PCR products were successfully obtained in 83 and 84 samples for the 55L>M and 192Q>R polymorphism respectively, and these samples were used for further investigations.As depicted in Figure 2, the determination of the 55L>M genotype could be performed opti-cally, based on the differences in color: carriers of the 55L allele displayed a red color during excitation at 700 nm and carriers of the 55M allele displayed a green color during excitation at 800 nm. In the picture of the merged 700 and 800 channels, the 55LL homozygotes were red, 55MM were green and the 55LM heterozygotes were visualized as yellow. The 192Q>R polymorphism showed a similar color pattern (data not shown).
Figure 3 shows the scatter plot of the 700 nm and 800 nm channel signal intensities of the iF-LASH hybridization assay. PCR samples typed as 55LL or 192QQ homozygotes by RFLP, had increased signal intensities at 700 nm, while almost no signal could be detected at 800 nm. In contrast, for PCR samples typed as 55MM or 192RR by RFLP, almost no signal could be detected at 700 nm, while increased signal intensities were obtained at 800 nm. For the 55LM and 192QR heterozygotes, a signal was present at both 700 nm and 800 nm. In order to allow automated (by computer) genotype determination, fixed signal-intensity cut-off values were defined. We assigned cut-off values based on the log ratio of the 800 and 700 channel signal intensities (Figure 4). Log (800 channel / 700 channel) signal intensities below the –0.75 were classified as 55LL and 192QQ homozygotes, between the –0.75 and 0.25 were classified as 55LM and 192QR heterozygotes and above the 0.25 were classified as 55MM and 192RR mutants. To validate the iFLASH assay, we compared the results with the RFLP and TAQman assay. There was a 100 percent similarity of genotype-outcomes measured with the three techniques, and all measured genotype distributions were in Hardy-Weinberg equilibrium.
Figure 2. Typical example of the optical representation of the 55L>M polymorphism as detected in the infrared Fluorescence Allele Specific Hybridization (iFLASH) genotyping assay, per 800 nm channel, 700 nm channel and the combination of the 800 nm and 700nm channel. The genotypes (in duplex) can be read from the combined 800 nm and 700 nm channel, where 55LL, 55LM and 55MM are represented by the red, yellow and green signal respectively. For the interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.

28
CHAPTER 2
Figure 3. Segregation of the 55L>M (A) and 192Q>R (B) genotypes with the 700 nm and the 800 nm channel intensity plotted along the X- and Y-axis, respectively. Genotypes were determined by restriction fragment length polymorphism (RFLP).
log (800 channel / 700 channel)
.75.50
.250.00
-.25-.50
-.75-1.00
-1.25
40
30
20
10
0
log (800 channel / 700 channel)
.75.50
.250.00
-.25-.50
-.75-1.00
-1.25-1.50
-1.75-2.00
50
40
30
20
10
0
55LL 55LM 55MM 192QQ 192QR 192RR
Figure 4. Histograms representing the distribution of the log (800 nm channel intensity / 700 nm channel intensity) within the study population for the 55L>M (A) and 192Q>R (B) genotypes. Subjects with signal intensities below the –0.75 were classified as 55LL and 192QQ homozygotes, between the –0.75 and 0.25 were classified as 55LM and 192QR heterozygotes and above the 0.25 were classified as 55MM and 192RR mutants. The –0.75 and 0.25 cut-off values were used for full automated determination of the genotypes.
700 channel intensity (x1000)
14000
12000
10000
80006000
40002000
0
800
chan
nel i
nten
sity
(x10
00)
7000
6000
5000
4000
3000
2000
1000
0
RFLP55 MM
55 LM
55 LL
700 channel intensity (x1000)
25002000
15001000
5000
800
chan
nel i
nten
sity
(x10
00)
2000
1000
0
RFLP192 RR
192 QR
192 QQ

29
High-throughput genotyping with iFLASH
Discussion
We have developed a novel genotyping approach named iFLASH, which combines the advan-tages of high sensitivity fluorescence imaging with the classic ASO-hybridization technique. Here, we will discuss if the iFLASH assay is an improvement over the RFLP and TAQman techniques with respect to reliability, optimization, throughput and costs.In this population, we obtained complete consensus on genotype outcome for two common polymorphisms in the PON1 gene (55L>M and 192Q>R) among the RFLP, the TAQman and iFLASH technique. This 100 percent unity among the three methods suggests that all these techniques are reliable tools for genotyping. For the optimization, all three methods require PCR primer design to amplify the region of interest followed by 1.) the selection of a restriction enzyme which cuts the polymorphic DNA sequence for the RFLP technique or 2.) the design of ASOs for the TAQman and iF-LASH assay. For the RFLP technique, no further optimizing is required. However, the success of the TAQman and the iFLASH assays, depends on the selection of suitable hybridization oligonucleotides, which give an allele specific signal. The TAQman assay is a single-tube as-say, where the binding of the PCR primers and the hybridization of the TAQman-ASOs to the template DNA take place in the same reaction. This requires that the annealing temperatures of the primers are about the same as the allele specific melting temperatures of the TAQman ASOs. The success of the TAQman assay therefore depends on carefully designed primers and probes. In contrast, for the iFLASH assay, the optimization of the allele specific hybridization temperature can be done separately from the optimization of the PCR conditions. The selec-tion of primers and probes is less critical. This is a benefit of the iFLASH assay over the TAQ-man assay, especially when the polymorphism is located in a DNA region where a design of primers and ASOs with common annealing and melting temperatures is not possible. For the genotype-assay comparison in terms of throughput, RFLP can be excluded as high-throughput method, because the sample handling is labor intensive and determination of genotypes can hardly be automated. TAQman, on the other hand, is a quick method, which has the great advantage that it requires no post-PCR handling. Recently, our group developed a technique based on the TAQman principle using a 384 wells PCR apparatus in combination with a fluorescence plate reader [7]. The throughput of the iFLASH hybridization method is equal or higher than the TAQman method: although we only genotyped 92 samples, the method of centrifugal transfer is capable of creating arrays of 384 or even 1536 samples [6]. A drawback for the throughput of the iFLASH method (when compared to the TAQman as-say) is that it requires post-PCR handling. However, because of the high number of samples (up to 1536 per run) in the initial PCR reaction, it is questionable whether this is a serious limitation. In addition, we show that the iFLASH technique gives an excellent signal intensity discrimination among the 55L>M and 192Q>R genotypes, and that clear cut-points can be defined for automated genotype determination using a computer.

30
CHAPTER 2
Finally, we discuss the cost effectiveness of the genotyping assays. Cost effectiveness depends on the number of samples tested: when genotyping only a few samples, RFLP is the method of choice. Restriction enzymes are relatively cheap and there is no need for expensive equipment other than a PCR machine and a gel electrophoresis set-up. But, when aiming to determine multiple polymorphisms in populations of considerable sample sizes, we recommend using either the TAQman or the iFLASH assay. Both methods require a one time investment for an expensive DNA amplification and detection system: a real-time PCR machine or a PCR machine in combination with a microplate-reader for the TAQman assay, and a PCR machine and an infrared imaging system for the iFLASH assay. The prices for the machinery will be much the same. However, the essential difference in the costs between the TAQman assay and the iFLASH assay is caused by the type of ASO used. The TAQman assay requires dual labeled oligonucleotides (containing a fluorescent reporter dye and a quencher), which are approximately three times more expensive than the single dye labeled oligonucleotides used in the iFLASH assay. Furthermore, in a 384 wells based approach, the TAQman requires ap-proximately 75-fold more labeled oligonucleotides than the iFLASH technique. Therefore, when genotyping numerous polymorphisms in large populations the TAQman assay is con-siderably more expensive than the iFLASH assay. In conclusion, we demonstrate that iFLASH-hybridization is a useful technique to genotype the two common polymorphisms in the PON1 gene. Additionally, due to its flexible opti-mization procedure, it can be used to type virtually any polymorphism desired. Because the iFLASH technique can be used with PCRs performed in 384 (or even 1536) wells format and the genotype detection can easily be automated, the iFLASH technique is suitable for high throughput genotyping. Finally, a major advantage of the iFLASH-hybridization technique is that it is cheaper than most techniques available.
Acknowledgements
The authors thank Fatiha Azouagh for the 192Q>R RFLP genotype determination.

31
High-throughput genotyping with iFLASH
References
[1] H.K. Tabor, N.J. Risch, R.M. Myers, Opinion: Can-didate-gene approaches for studying complex genetic traits: practical considerations, Nat Rev Genet 3 (2002) 391-397.[2] K.J. Livak, Allelic discrimination using fluorogenic probes and the 5’ nuclease assay, Genet Anal 14 (1999) 143-149.[3] L.A. Haff, I.P. Smirnov, Single-nucleotide polymor-phism identification assays using a thermostable DNA polymerase and delayed extraction MALDI-TOF mass spectrometry, Genome Res 7 (1997) 378-388.[4] B.J. Conner, A.A. Reyes, C. Morin, K. Itakura, R.L. Teplitz, R.B. Wallace, Detection of sickle cell beta S-globin allele by hybridization with synthetic oligonu-cleotides, Proc Natl Acad Sci U S A 80 (1983) 278-282.[5] R. Humbert, D.A. Adler, C.M. Disteche, C. Hassett, C.J. Omiecinski, C.E. Furlong, The molecular basis of the human serum paraoxonase activity polymorphism, Nat.Genet. 3 (1993) 73-76.[6] M. Jobs, W.M. Howell, A.J. Brookes, Creating arrays by centrifugation, Biotechniques 32 (2002) 1322-1324, 1326, 1329.[7] B.B. Van Rijn, M. Roest, A. Franx, H.W. Bruinse, H.A. Voorbij, Single step high-throughput determina-tion of Toll-like receptor 4 polymorphisms, J Immunol Methods 289 (2004) 81-87.


3Paraoxonase genotype, LDL-oxidation and carotid
atherosclerosis in male life-long smokers
Free Radic. Res. 2004 Jun;38(6):553-60
T.M. van HimbergenM. Roest
F.G. de WaartJ. de Graaf
H.A. VoorbijL.J. van Tits
A.F. Stalenhoef

Abstract
Paraoxonase (PON-1) is a high-density lipoprotein (HDL) associated enzyme that hydro-lyzes lipid peroxides in vitro, which may therefore protect against the onset of atherosclero-sis. Heavy smokers are more exposed to oxidative stress and hence at high-risk for oxidative modification of LDL.Our hypothesis is that the anti-oxidative properties of paraoxonase inhibit LDL oxidation, especially in populations exposed to high oxidative stress.We have studied the effects of PON-1 genotype and smoking to variation in oxidative status parameters and intima-media thickness (IMT), a surrogate marker of atherosclerosis. The contribution of two common polymorphisms in the PON-1 gene (Q192R and L55M) to LDL oxidizability, autoantibodies directed against oxLDL and IMT were studied in 207 male life-long smokers. Smokers were classified into average, heavy and excessive smokers based on pack years of cigarettes smoked.PON-1 genotype was not associated with autoantibodies to oxLDL, LDL oxidizability or IMT. Smoking was associated with IMT in subgroups with the high levels of LDL, but not in the population at large.The lack of association of PON-1 genotype with oxidative status parameters and IMT suggests that PON-1 is not a major inhibitor of LDL oxidation in a population of life-long smokers.

35
PON1 genotype in life-long smokers
Introduction
Serum paraoxonase (PON-1) is a high-density lipoprotein (HDL) associated enzyme capable of hydrolysing organophosphates [1]. In vitro, PON-1 protects low-density lipoprotein (LDL) from oxidative modification by hydrolysing lipid peroxides. This property argues for a po-tential protective role of PON-1 against atherosclerosis [2]. The hypothesis was supported by observations, that PON-1 deficient mice are more susceptible to develop atherosclerosis than wild-type mice when fed a high-fat/high-cholesterol diet [3].The coding sequence of the PON-1 gene contains two polymorphic sites: a leucine (L) to me-thionine (M) transition at position 55 (L55M) and a glutamine (Q) to arginine (R) transition at position 192 (Q192R). The L55M polymorphism affects the enzyme concentration, partly due to linkage with polymorphims in the PON-1 promoter region [4], and possibly via altered ability of paraoxonase to form a complex with HDL: the L55M polymorphism is located in the N-terminal side of PON-1 which may play a role in the binding of PON-1 with HDL [5]. The Q192R polymorphism determines the catalytic efficiency towards a number of organophos-phate substrates, including paraoxon. The 192R variant hydrolyses paraoxon more efficiently than the 192Q variant, the in vivo substrate of PON-1, however, is not known. Furthermore the Q192R polymorphism is not related to PON-1 levels [6, 7].Results of studies on the contribution of the L55M and Q192R polymorphism to the risk of cardio-vascular disease (CVD) are inconsistent [8-18]. Recent findings from a prospective study have demonstrated the that low paraoxonase activity is a predictor of CVD [19].Previously, our group found a relation between the PON-1 genotype combination LLQQ and increased Intima-media thickness (IMT) in high risk subjects with familial hypercholester-olemia (FH) [20]. IMT is an assessment of the combined thickness of the intima-media layer of the common carotid artery as measured by B-mode ultrasonography. IMT is a powerful predictor for future cardio vascular events and is often used as a surrogate marker for clinical outcomes [21].Smoking is associated with both high oxidative stress and increased risk of CVD [22]. Fur-thermore, cigarette smoke promotes the oxidation of LDL in the presence of peroxidases [23]. Therefore, the anti-oxidative effects the PON-1 polymorphisms are more likely to express in a population consisting of heavy smokers than in the general population.Our hypothesis is that the PON-1 genotype significantly determines the harmful effect of smoking to the oxidative modification of LDL and thus atherosclerosis. For this we deter-mined, the LDL oxidizability, the levels of oxLDL autoantibodies and the IMT among the genetic variants of the L55M and Q192R polymorphisms of the PON-1 gene, in a high risk population of life long smokers. Additionally, the effects of different smoking gradations on LDL oxidizability, levels of oxLDL autoantibodies and the IMT were investigated.

36
CHAPTER 3
Methods
SubjectsThe study population consisted of 218 male chronic smokers who had participated in a clini-cal trial on atherosclerosis progression [24]. Of the 218 participants, 207 participants with complete PON-1 genotype and carotid IMT information were included. Height, weight, blood pressure and the IMT of the common carotid artery (CCA IMT) were measured at baseline (table 1). DNA, plasma and serum samples were stored at -80°C until analysis. All participants gave written informed consent to the use of their blood samples for scientific research. The study was approved by the institutional review board of University Medical Centre Nijmegen and Wageningen University.
Ultrasound Measurement of the Carotid IMTUltrasound scanning of the carotid arteries was performed with a Biosound Phase-two real time scanner (BiosoundEsaote, Indianapolis, IN, USA) equipped with a 10 MHz transducer, as described in detail elsewhere [25]. IMT measurements were done for both anterior and posterior walls of the distal 1.0 cm straight part of both common carotid arteries. Images were analyzed with a semiautomatic software program (Eurequa; TSA company, Meudon, France). The common carotid artery intima-media thickness (CCA IMT) is expressed as the mean of the anterior and posterior walls of the left and right common carotid artery.
PON-1 GenotypingThe L55M and Q192R mutations were determined by PCR RFLP using primers and restric-tion enzymes as described by Humbert et al [6]. Restriction fragments were separated on a 2% agarose gel and visualized with ethidium bromide. The L-55 allele corresponded to non-digested 170-bp fragments, the M-55 allele to 44-bp and 126-bp fragments (figure 1A), the Q-192 allele to non-digested 99-bp fragments, and the R-192 allele to 33-bp and 66-bp frag-ments (figure 1B). Results by two independently working technicians were indistinguishable except for 5 observations, which were reanalyzed until consensus.
Markers of Oxidative Status: LDL OxidizabilitySusceptibility of LDL to in vitro oxidation was determined in 165 suitable samples by moni-toring formation of conjugated dienes at 234 nm on a PE-lambda 12 spectrophotometer (Per-kin Elmer Ltd., Beaconsfield, UK) as described by Esterbauer et al [26], and as modified by Princen et al [27]. The time profile of the absorption pattern shows three distinct phases: the lag time, the propagation phase and the decomposition phase. The lag time is defined as the time interval between the intercept of the linear least-square slope of the absorbance curve with the initial absorbance axis and was taken as a measure of LDL resistance to oxidation. The rate of diene formation is calculated as the slope of the propagation phase and reflects the

37
PON1 genotype in life-long smokers
autocatalytic chain reaction of the lipid peroxidation process. Finally, the net diene concentra-tion is the difference between the absorbance at time zero and the maximum absorbance and reflects the extent of oxidative modification of the isolated LDL.
Markers of Oxidative Status: Antibodies to Oxidized LDLIgG and IgM antibodies to oxidized LDL (oxLDL Ab) were assayed by ELISA as described in detail previously [28]. In short, antibodies were captured by using native and copper-oxidized LDL as antigens and detected with peroxidase-conjugated antibody from goat specific for hu-man IgG or IgM (Sigma-Aldrich). Binding to oxidized LDL over binding to native LDL was taken as a measure of antibodies to oxidized LDL.
Lipids and LipoproteinsCholesterol and triglyceride concentrations in serum were determined by enzymatic methods (Boehringer-Mannheim, Mannheim, Germany) on a Hitachi 747 analyzer (Hitachi,Tokyo, Japan). HDL cholesterol was determined after precipitation of LDL cholesterol, very low-den-sity lipoprotein and chylomicrons using phosphotungstate/Mg2+. LDL cholesterol in serum was calculated using the Friedewald formula. As reported previously, this includes lipoprotein remnant-associated cholesterol [29].
A
B
Figure 1. Representative figures of RFLP of PON-1 L55M (A) and Q192R (B) polymorphisms. The L-55 allele corresponded to non-digested 170-bp fragments and the M-55 allele to 44-bp and 126-bp fragments. The Q-192 allele corresponds to non-digested 99-bp fragments and the R-192 allele to 33-bp and 66-bp fragments. Negative control is indicated by b.

38
CHAPTER 3
Data Analysis and StatisticsSmoking status is expressed in pack years (the number of cigarettes smoked per day mul-tiplied by the years of active smoking divided by twenty) and classified in tertiles (average, heavy and excessive) with cut of points on 23 pack years and 47 pack years.The age adjusted relation between smoking, L55M genotype and Q192R genotype with CCA IMT, LDL oxidizability and oxLDL antibodies was tested with linear regression analysis. Pack years, L55M genotype and Q192R genotype served as independent variable and CCA IMT, lag time, net diene concentration, rate of diene formation, IgG antibodies and IgM antibodies as dependent variable.CCA IMT was adjusted for age by the algorithm: β(mean age population - age smoker) + CCA IMT. Where β is the coefficient derived from the linear regression model with age as independent variable and CCA IMT and dependent variable.The relation between age adjusted CCA IMT and pack years of cigarette smoking, lipoprotein levels, LDL oxidizability and oxLDL antibodies and was tested with the Pearson correlation coefficient.Interaction between smoking status and PON-1 genotype in relation to CCA IMT and the parameters for the oxidative status (lag time, net diene production, rate of diene formation and antibodies to oxLDL) was studied by analysing subgroups.For interaction, tertiles were defined for lipid ratio (total cholesterol/HDL cholesterol) and for plasma LDL cholesterol. Lipid ratio levels lower than 4.7, between 4.7 and 6.0, and higher than 6.0 were assigned to the first, second and third tertile respectively. Plasma LDL choles-terol levels lower than 3.7 mmol/L, between 3.7 and 4.5 mmol/L and higher than 4.5 mmol/L, were assigned to the first, second and third tertile respectively.The significance between subgroups was studied using the Independent-Samples t-test. All analysis were performed with SPSS version10.0.
Results
The population consisted of 207 male smokers with a mean age of 60 (Table 1). The subjects had smoked for a mean of 38 pack years. BMI and systolic blood pressure were increased, while mean total cholesterol, HDL cholesterol and triglyceride levels were within limits for normal.The PON-1 L55M genotypes LL, LM and MM occurred in 77 (37.2 %), 104 (50.2 %) and 26 (12.6 %) subjects, respectively. The PON-1 Q192R genotypes QQ, QR and RR were present in 101 (48.8 %), 92 (44.4 %) and 14 (6.8 %) subjects, respectively. The observed genotype distributions did not significantly differ from the calculated expected distributions, assum-ing a Hardy-Weinberg equilibrium. The Q192R polymorphism was in linkage disequilibrium with the L55M polymorphism: 98% of the carriers of the 192R allele also have an L allele at

39
PON1 genotype in life-long smokers
position 55.Table 2 presents the effects of smoking status and PON-1 genotypes on the oxidation pa-rameters (lag time, net diene production, rate of diene formation and antibodies to oxLDL) and CCA IMT. Smoking status was associated with a statistically significant difference in lag time (p=0.04) and rate of diene formation (p=0.03). No significant difference was observed between the smoking groups and the levels of IgG and IgM antibodies to oxLDL. LDL oxida-tion parameters (lag time, net diene production and rate of diene formation) did not correlate with the levels of IgG and IgM antibodies to oxLDL (data not shown). Age adjusted CCA IMT was not associated with smoking status. Individual PON-1 polymorphisms were not associ-ated with the CCA IMT or with oxidative status. However, a nearly significant trend (p=0.06, Table2) for the IgG antibody titers in the L55M genotype was observed in our data.In Table 3 the correlation between age adjusted CCA IMT and pack years of cigarette smok-ing, lipoprotein levels, LDL oxidizability and oxLDL autoantibodies is presented. There was no correlation between pack years of cigarette smoking and CCA IMT. Plasma HDL choles-terol was associated with decreased CCA IMT values (r=-0.200, p=0.004), while plasma LDL cholesterol was associated with increased CCA IMT values (r=0.208, p=0.003). None of the oxidative status variables were associated with CCA IMT.We observed a trend for the interaction between smoking and LDL levels in relation to age ad-justed CCA IMT (Figure 2). A similar trend was observed for the interaction between smok-ing and ratio HDL cholesterol/total cholesterol (data not shown). The interaction among smoking status, PON-1 genotype, plasma HDL cholesterol and LDL cholesterol levels in rela-tion to lag time, net diene production, rate of diene formation, antibodies to oxLDL and CCA IMT was not significant (data not shown).
Table 1. General characteristics of 207 male smokersCharacteristics Values*Age (y) 60 ± 6Pack years of cigarette smoking 38 ± 21CCA IMT (mm) 0.96 ± 0.15BMI (kg/m2) 26.0 ± 3.3Systolic blood pressure (mm Hg) 142 ± 17Diastolic blood pressure (mm Hg) 84 ± 8Total cholesterol (mmol/L) 6.0 ± 1.0Triglycerides (mmol/L) 1.7 ± 1.0HDL cholesterol (mmol/L) 1.2 ± 0.4LDL cholesterol (mmol/L) 4.1 ± 1.0* Values are means ± standard deviation. CCA IMT, Common carotid artery intima media thickness; BMI, Body mass index; HDL, High-density lipoprotein; LDL, Low-density lipoprotein.

40
CHAPTER 3
Tabl
e 2.
Mea
n ch
arac
teris
tics
for
CC
A I
MT,
oxi
datio
n pa
ram
eter
s an
d ox
LDL
auto
antib
odie
s by
sm
okin
g st
atus
and
PO
N-1
L55
M a
nd Q
192R
ge
noty
peSm
okin
g sta
tus*
Aver
age s
mok
ing
Hea
vy sm
okin
gEx
cess
ive s
mok
ing
p-va
lue*
*C
CA
IMT
(mm
)0.
93±
0.15
690.
96±
0.16
690.
98±
0.16
690.
41La
g tim
e (m
in)
87±
1158
88±
956
92±
951
0.04
Net
die
ne co
ncen
trat
ion
(nm
ol/m
g pr
otei
n)62
7±
6258
622
±55
5660
2±
6351
0.08
Rate
of d
iene
form
atio
n (n
mol
/mg
prot
ein/
min
)15
.2±
2.4
5814
.8±
1.9
5614
.0±
2.3
510.
03ox
LDL
IgG
ant
ibod
ies (
OD
450)
0.37
±0.
1768
0.38
±0.
2067
0.37
±0.
1969
0.84
oxLD
L Ig
M a
ntib
odie
s (O
D45
0)0.
48±
0.29
650.
49±
0.33
640.
54±
0.33
670.
12PO
N-1
L55
M ge
noty
peLL
LMM
MC
CA
IMT
(mm
)0.
96±
0.15
770.
95±
0.16
104
0.98
±0.
1626
0.93
Lag
time
(min
)90
±10
6089
±11
8386
±8
220.
16N
et d
iene
conc
entr
atio
n (n
mol
/mg
prot
ein)
621
±51
6061
3±
6983
626
±53
220.
97Ra
te o
f die
ne fo
rmat
ion
(nm
ol/m
g pr
otei
n/m
in)
14.8
±1.
960
14.6
±2.
683
15.2
±1.
922
0.82
oxLD
L Ig
G a
ntib
odie
s (O
D45
0)0.
34±
0.15
760.
39±
0.21
102
0.41
±0.
1826
0.06
oxLD
L Ig
M a
ntib
odie
s (O
D45
0)0.
52±
0.30
700.
47±
0.32
101
0.59
±0.
3425
0.62
PON
-1 Q
192R
geno
type
QR
RRC
CA
IMT
(mm
)0.
96±
0.15
101
0.97
±0.
1692
0.91
±0.
1314
0.94
Lag
time
(min
)88
±10
8089
±11
7487
±8
110.
76N
et d
iene
conc
entr
atio
n (n
mol
/mg
prot
ein)
616
±66
8061
7±
5874
628
±39
110.
50Ra
te o
f die
ne fo
rmat
ion
(nm
ol/m
g pr
otei
n/m
in)
14.8
±2.
480
14.5
±2.
274
15.7
±1.
411
0.74
oxLD
L Ig
G a
ntib
odie
s (O
D45
0)0.
37±
0.16
100
0.37
±0.
2190
0.38
±0.
1714
0.86
oxLD
L Ig
M a
ntib
odie
s (O
D45
0)0.
52±
0.32
960.
49±
0.32
860.
53±
0.25
140.
65N
ote:
Val
ues r
epre
sent
mea
n ±
stan
dard
dev
iatio
n fo
llow
ed b
y the
num
ber o
f obs
erva
tions
. *Sm
okin
g st
atus
: defi
ned
in d
ata a
naly
sis an
d st
atist
ics s
ectio
n in
the
met
hods
. **p
-val
ues a
re b
ased
on
linea
r reg
ress
ion
anal
ysis
and
are
adju
sted
for a
ge. C
CA
IMT,
Com
mon
car
otid
art
ery
intim
a m
edia
thic
knes
s; ox
LDL,
Oxi
dize
d lo
w-d
ensit
y lip
opro
tein
.

41
PON1 genotype in life-long smokers
Discussion
We have investigated the effects of PON-1 genotypes, smoking and lipid/lipoprotein profile on parameters of oxidative status and variation of CCA IMT, a surrogate marker of athero-sclerosis. Smoking was associated with LDL oxidizability but not with atherosclerosis. There was no relationship between PON-1 genotype and markers of oxidative status or CCA IMT. Furthermore, no relationship was observed between LDL oxidizability and antibodies to ox-LDL and CCA IMT.Smoking is a major pro-oxidative stimulus [22]. Studies on the effects of smoking and LDL oxidation, however, presented controversial results [30-32]. Our findings that smoking was associated with reduced rates of LDL oxidation and prolonged lag times, indicate that pro-found smoking leads to LDL which is more resistant to oxidation. This apparent anti oxida-tive property of smoking may be caused by the antioxidative potential of cigarette smoke, as suggested by a previous report [33], and/or may be related to a continuous oxidative pressure, which results in circulating LDL particles that are less prone to oxidation in vitro. The exact biological meanings of these small differences in LDL oxidizability, remain unclear. However, in this population of life long smokers lag time and the oxidation rate of LDL, do not correlate with CCA IMT and thus may play no role in the development atherosclerosis.PON-1 protects LDL against oxidative modifications, in vitro [2]. The in vivo action of PON-1 in healthy non-smoking subjects is reflected by a decreased ex vivo oxidizability of LDL [34]. Remarkably, however, PON-1 has no influence on the oxidizability of LDL in a population of smokers (present study) or diabetics [35, 36]. The absence of a relation between PON-1 and the oxidizability of LDL suggests that PON-1 does not play a major role in oxidizability of LDL in vivo. Alternatively, the absence of such a relationship may be due to reduced paraox-onase levels and activity in these populations [37, 38], and/or to a masking effect of smoking. In line with this, Sen-Banerjee et al observed an association of PON-1 Q192R genotype with
Table 3. Correlation between age adjusted CCA IMT, smoking, lipoproteins, LDL oxidizability and oxLDL autoantibodies
CCA IMT (mm)*R p-value n
Pack years of cigarette smoking 0.057 0.413 207HDL cholesterol (mmol/L) -0.200 0.004 207LDL cholesterol (mmol/L) 0.208 0.003 202Lag time (min) -0.087 0.266 165Net diene concentration (nmol/mg protein) -0.010 0.903 165Rate of diene formation (nmol/mg protein/min) 0.045 0.569 165oxLDL IgG antibodies (OD450) -0.027 0.706 204oxLDL IgM antibodies (OD450) -0.061 0.394 196 *Values represent the Pearson correlation coefficient (R) followed by the p-value (p) and the number of observations (n). CCA IMT, Common carotid artery intima media thickness; HDL, High-density lipoprotein; LDL, Low-density lipoprotein; oxLDL, Oxidized low-density lipoprotein.

42
CHAPTER 3
an increased risk of myocardial infarction in non-smokers but not in smokers [39]. Unfortu-nately in the present study no serum was collected for PON-1 activity or mass measurements. More research is therefore needed to elucidate the importance of PON-1 on the oxidizability of LDL and its relevance in vivo. Autoantibodies to oxidized LDL have been proposed as marker of pro-oxidative state [40-44]. The absence of a relationship between smoking and oxLDL autoantibody titers in our study suggests that autoantibodies to oxLDL do not reflect smoking induced oxidative stress. Furthermore, it remains to be established whether the contribution of these autoantibodies to oxidized LDL can be used to predict atherosclerosis [45-47].To our knowledge this is the second study examining the effects of PON-1 genotypes on autoantibodies to oxLDL. Recently, Malin et al studied forty-nine healthy men and observed no differences in oxLDL autoantibody levels between the low- and high-active genotypes of PON-1 [48]. We observed a borderline significant trend between IgG autoantibody titers and the L55M polymorphism, the 55MM variant having the highest levels. Since PON-1 55MM homozygotes are more effective than 55LL homozygotes in protecting LDL from oxidation in
averageheavy
excessive
LDL < 3.7
LDL 3.7-4.5
LDL > 4.5
0.85
0.9
0.95
1
1.05
1.1 *
Smoking Status**
LDL cholesterol (mmol/L)
Figure 2. The relationship between the smoking-LDL levels interaction and age-adjusted CCA IMT. *p value of 0.002 for excessive smokers with LDL levels higher than 4.5 mmol/L when compared to the remaining smoking statuses and LDL levels combined. **For classification of smoking status see data analysis and statistics section. CCA IMT, common carotid artery intima-media thickness; LDL, low-density lipoprotein.

43
PON1 genotype in life-long smokers
vitro,[49] our finding is in favour of an inverse correlation of in vivo LDL oxidation with au-toantibodies to oxLDL, as recently shown by Shoji et al [41]. Further studies on PON-1 geno-types and antibodies to oxLDL should include measurement of oxLDL to give more insight in the usefulness of autoantibodies as marker for oxidation in relation to paraoxonase.Large population studies have shown that smoking is a major risk factor for increased CCA IMT [50, 51]. In our population this relationship was present in subgroups with high levels of LDL cholesterol, but not in the population at large, suggesting that the effect of smoking on CCA IMT is strongest in high-risk groups for CVD.The contribution of the L55M and Q192R polymorphism to the risk of CVD has frequently been investigated. If anything, the PON-1 192RR and the 55LL genotype predicted an in-creased risk for CVD [8-12]. In contrast to smoking, there was no clear relation between PON-1 genotype and CCA IMT, indicating that PON-1 genotype is not a strong risk factor for atherosclerosis in smokers. PON-1 phenotype was not investigated in this population since no serum was available for PON-1 activity and concentration measurements.In conclusion, smoking was associated with IMT in subgroups with the high levels of LDL, but not in the population at large. PON-1 genotype does not contribute to the susceptibility of LDL oxidation, in this population of life long smokers. There is no clear relation between PON-1 genotype and autoantibodies to oxLDL and PON-1 genotype has no effect on CCA IMT. These results suggest that PON-1 does not play an important role in atherogenesis in a population of life long smokers.
Acknowledgements
The authors greatly acknowledge Anneke Hijmans and Heidi Hak-Lemmers for assistance in collecting the data.

44
CHAPTER 3
References
[1] H.G. Davies, R.J. Richter, M. Keifer, C.A. Broomfield, J. Sowalla, C.E. Furlong, The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin, Nat Genet 14 (1996) 334-336.[2] M.I. Mackness, S. Arrol, P.N. Durrington, Paraoxo-nase prevents accumulation of lipoperoxides in low-density lipoprotein, FEBS Lett 286 (1991) 152-154.[3] D.M. Shih, L. Gu, Y.R. Xia, M. Navab, W.F. Li, S. Hama, L.W. Castellani, C.E. Furlong, L.G. Costa, A.M. Fogelman, A.J. Lusis, Mice lacking serum paraoxo-nase are susceptible to organophosphate toxicity and atherosclerosis, Nature 394 (1998) 284-287.[4] V.H. Brophy, R.L. Jampsa, J.B. Clendenning, L.A. McKinstry, G.P. Jarvik, C.E. Furlong, Effects of 5’ regu-latory-region polymorphisms on paraoxonase-gene (PON1) expression, Am J Hum Genet 68 (2001) 1428-1436.[5] C.E. Furlong, R.J. Richter, C. Chapline, J.W. Crabb, Purification of rabbit and human serum paraoxonase, Biochemistry 30 (1991) 10133-10140.[6] R. Humbert, D.A. Adler, C.M. Disteche, C. Hassett, C.J. Omiecinski, C.E. Furlong, The molecular basis of the human serum paraoxonase activity polymorphism, Nat.Genet. 3 (1993) 73-76.[7] M.C. Garin, R.W. James, P. Dussoix, H. Blanche, P. Passa, P. Froguel, J. Ruiz, Paraoxonase polymorphism Met-Leu54 is associated with modified serum concen-trations of the enzyme. A possible link between the paraoxonase gene and increased risk of cardiovascular disease in diabetes, J Clin Invest 99 (1997) 62-66.[8] J. Ruiz, H. Blanche, R.W. James, M.C. Garin, C. Vaisse, G. Charpentier, N. Cohen, A. Morabia, P. Passa, P. Froguel, Gln-Arg192 polymorphism of paraoxonase and coronary heart disease in type 2 diabetes, Lancet 346 (1995) 869-872.[9] D.K. Sanghera, C.E. Aston, N. Saha, M.I. Kam-boh, DNA polymorphisms in two paraoxonase genes
(PON1 and PON2) are associated with the risk of coro-nary heart disease, Am J Hum Genet 62 (1998) 36-44.[10] H. Schmidt, R. Schmidt, K. Niederkorn, A. Grad-ert, M. Schumacher, N. Watzinger, H.P. Hartung, G.M. Kostner, Paraoxonase PON1 polymorphism leu-Met54 is associated with carotid atherosclerosis: results of the Austrian Stroke Prevention Study, Stroke 29 (1998) 2043-2048.[11] M. Serrato, A.J. Marian, A variant of human paraoxonase/arylesterase (HUMPONA) gene is a risk factor for coronary artery disease, J Clin Invest 96 (1995) 3005-3008.[12] T. Zama, M. Murata, Y. Matsubara, K. Kawano, N. Aoki, H. Yoshino, G. Watanabe, K. Ishikawa, Y. Ikeda, A 192Arg variant of the human paraoxonase (HUMPONA) gene polymorphism is associated with an increased risk for coronary artery disease in the Japanese, Arterioscler Thromb Vasc Biol 17 (1997) 3565-3569.[13] M. Antikainen, S. Murtomaki, M. Syvanne, R. Pahlman, E. Tahvanainen, M. Jauhiainen, M.H. Frick, C. Ehnholm, The Gln-Arg191 polymorphism of the human paraoxonase gene (HUMPONA) is not associ-ated with the risk of coronary artery disease in Finns, J Clin Invest 98 (1996) 883-885.[14] A. Gardemann, M. Philipp, K. Hess, N. Katz, H. Tillmanns, W. Haberbosch, The paraoxonase Leu-Met54 and Gln-Arg191 gene polymorphisms are not associated with the risk of coronary heart disease, Atherosclerosis 152 (2000) 421-431.[15] S.M. Herrmann, H. Blanc, O. Poirier, D. Arveiler, G. Luc, A. Evans, P. Marques-Vidal, J.M. Bard, F. Cambien, The Gln/Arg polymorphism of human paraoxonase (PON 192) is not related to myocardial infarction in the ECTIM Study, Atherosclerosis 126 (1996) 299-303.[16] Y.L. Ko, Y.S. Ko, S.M. Wang, L.A. Hsu, C.J. Chang, P.H. Chu, N.J. Cheng, W.J. Chen, C.W. Chiang, Y.S. Lee, The Gln-Arg 191 polymorphism of the human paraox-onase gene is not associated with the risk of coronary

45
PON1 genotype in life-long smokers
artery disease among Chinese in Taiwan, Atheroscle-rosis 141 (1998) 259-264.[17] D. Ombres, G. Pannitteri, A. Montali, A. Candeloro, F. Seccareccia, F. Campagna, R. Cantini, P.P. Campa, G. Ricci, M. Arca, The gln-Arg192 polymorphism of hu-man paraoxonase gene is not associated with coronary artery disease in italian patients, Arterioscler Thromb Vasc Biol 18 (1998) 1611-1616.[18] D.K. Sanghera, N. Saha, M.I. Kamboh, The codon 55 polymorphism in the paraoxonase 1 gene is not associated with the risk of coronary heart disease in Asian Indians and Chinese, Atherosclerosis 136 (1998) 217-223.[19] B. Mackness, P. Durrington, P. McElduff, J. Yarnell, N. Azam, M. Watt, M. Mackness, Low paraoxonase ac-tivity predicts coronary events in the Caerphilly Pro-spective Study, Circulation 107 (2003) 2775-2779.[20] F.R. Leus, M.E. Wittekoek, J. Prins, J.J. Kastelein, H.A. Voorbij, Paraoxonase gene polymorphisms are as-sociated with carotid arterial wall thickness in subjects with familial hypercholesterolemia, Atherosclerosis 149 (2000) 371-377.[21] D.H. O’Leary, J.F. Polak, Intima-media thickness: a tool for atherosclerosis imaging and event prediction, Am J Cardiol 90 (2002) 18L-21L.[22] J.D. Morrow, B. Frei, A.W. Longmire, J.M. Gaziano, S.M. Lynch, Y. Shyr, W.E. Strauss, J.A. Oates, L.J. Rob-erts, Increase in circulating products of lipid peroxi-dation (F2- isoprostanes) in smokers. Smoking as a cause of oxidative damage, N.Engl.J.Med. 332 (1995) 1198-1203.[23] N. Santanam, R. Sanchez, S. Hendler, S. Parthasar-athy, Aqueous extracts of cigarette smoke promote the oxidation of low density lipoprotein by peroxidases, FEBS Lett 414 (1997) 549-551.[24] F.G. De Waart, T.J. Smilde, H. Wollersheim, A.F. Stalenhoef, F.J. Kok, Smoking characteristics, antioxi-dant vitamins, and carotid artery wall thickness among life-long smokers, J Clin Epidemiol 53 (2000) 707-714.
[25] T.J. Smilde, H. Wollersheim, H. van Langen, A.F. Stalenhoef, Reproducibility of ultrasonographic measurements of different carotid and femoral artery segments in healthy subjects and in patients with in-creased intima-media thickness, Clin.Sci.(Lond) 93 (1997) 317-324.[26] H. Esterbauer, G. Striegl, H. Puhl, M. Rotheneder, Continuous monitoring of in vitro oxidation of human low density lipoprotein, Free Radic.Res.Commun. 6 (1989) 67-75.[27] H.M. Princen, G. van Poppel, C. Vogelezang, R. Buytenhek, F.J. Kok, Supplementation with vitamin E but not beta-carotene in vivo protects low density lipoprotein from lipid peroxidation in vitro. Effect of cigarette smoking, Arterioscler.Thromb. 12 (1992) 554-562.[28] L.J. van Tits, F. de Waart, H.L. Hak-Lemmers, P. van Heijst, J. de Graaf, P.N. Demacker, A.F. Stalenhoef, Effects of alpha-tocopherol on superoxide production and plasma intercellular adhesion molecule-1 and antibodies to oxidized LDL in chronic smokers, Free Radic.Biol.Med. 30 (2001) 1122-1129.[29] P.N. Demacker, A.G. Hijmans, B.J. Brenninkmeijer, A.P. Jansen, L.A. van ‘t, Five methods for determining low-density lipoprotein cholesterol compared, Clin.Chem. 30 (1984) 1797-1800.[30] D. Harats, M. Ben-Naim, Y. Dabach, G. Hollander, O. Stein, Y. Stein, Cigarette smoking renders LDL sus-ceptible to peroxidative modification and enhanced metabolism by macrophages, Atherosclerosis 79 (1989) 245-252.[31] R. Siekmeier, P. Wulfroth, H. Wieland, W. Gross, W. Marz, Low-density lipoprotein susceptibility to in vitro oxidation in healthy smokers and nonsmokers, Clin.Chem. 42 (1996) 524-530.[32] K. Marangon, B. Herbeth, Y. Artur, H. Esterbauer, G. Siest, Low and very low density lipoprotein compo-sition and resistance to copper-induced oxidation are not notably modified in smokers, Clin.Chim.Acta 265

46
CHAPTER 3
(1997) 1-12.[33] C. Chen, G. Loo, Cigarette smoke extract inhib-its oxidative modification of low density lipoprotein, Atherosclerosis 112 (1995) 177-185.[34] E.Y. Sozmen, B. Sozmen, F.K. Girgin, Y. Delen, E. Azarsiz, D. Erdener, B. Ersoz, Antioxidant enzymes and paraoxonase show a co-activity in preserving low-den-sity lipoprotein from oxidation, Clin Exp Med 1 (2001) 195-199.[35] H. Cao, A. Girard-Globa, A. Serusclat, S. Bernard, P. Bondon, S. Picard, F. Berthezene, P. Moulin, Lack of association between carotid intima-media thickness and paraoxonase gene polymorphism in non-insu-lin dependent diabetes mellitus, Atherosclerosis 138 (1998) 361-366.[36] M.J. Sampson, S. Astley, T. Richardson, G. Willis, I.R. Davies, D.A. Hughes, S. Southon, Increased DNA oxidative susceptibility without increased plasma LDL oxidizability in Type II diabetes: effects of alpha-toco-pherol supplementation, Clin Sci (Lond) 101 (2001) 235-241.[37] R.W. James, I. Leviev, A. Righetti, Smoking is as-sociated with reduced serum paraoxonase activity and concentration in patients with coronary artery disease, Circulation 101 (2000) 2252-2257.[38] C.A. Abbott, M.I. Mackness, S. Kumar, A.J. Boul-ton, P.N. Durrington, Serum paraoxonase activity, concentration, and phenotype distribution in diabe-tes mellitus and its relationship to serum lipids and lipoproteins, Arterioscler Thromb Vasc Biol 15 (1995) 1812-1818.[39] S. Sen-Banerjee, X. Siles, H. Campos, Tobacco smoking modifies association between Gln-Arg192 polymorphism of human paraoxonase gene and risk of myocardial infarction, Arterioscler Thromb Vasc Biol 20 (2000) 2120-2126.[40] J.T. Salonen, S. Yla-Herttuala, R. Yamamoto, S. Butler, H. Korpela, R. Salonen, K. Nyyssonen, W. Palin-ski, J.L. Witztum, Autoantibody against oxidised LDL
and progression of carotid atherosclerosis, Lancet 339 (1992) 883-887.[41] T. Shoji, Y. Nishizawa, M. Fukumoto, K. Shima-mura, J. Kimura, H. Kanda, M. Emoto, T. Kawagishi, H. Morii, Inverse relationship between circulating oxi-dized low density lipoprotein (oxLDL) and anti-oxLDL antibody levels in healthy subjects, Atherosclerosis 148 (2000) 171-177.[42] M. Fukumoto, T. Shoji, M. Emoto, T. Kawagishi, Y. Okuno, Y. Nishizawa, Antibodies against oxidized LDL and carotid artery intima-media thickness in a healthy population, Arterioscler.Thromb.Vasc.Biol. 20 (2000) 703-707.[43] T. Lehtimaki, S. Lehtinen, T. Solakivi, M. Nikkila, O. Jaakkola, H. Jokela, S. Yla-Herttuala, J.S. Luoma, T. Koivula, T. Nikkari, Autoantibodies against oxidized low density lipoprotein in patients with angiographi-cally verified coronary artery disease, Arterioscler.Thromb.Vasc.Biol. 19 (1999) 23-27.[44] J. Hulthe, L. Bokemark, B. Fagerberg, Antibodies to oxidized LDL in relation to intima-media thickness in carotid and femoral arteries in 58-year-old sub-jectively clinically healthy men, Arterioscler.Thromb.Vasc.Biol. 21 (2001) 101-107.[45] A. Boullier, M. Hamon, E. Walters-Laporte, F. Mar-tin-Nizart, R. Mackereel, J.C. Fruchart, M. Bertrand, P. Duriez, Detection of autoantibodies against oxidized low-density lipoproteins and of IgG-bound low den-sity lipoproteins in patients with coronary artery dis-ease, Clin.Chim.Acta 238 (1995) 1-10.[46] J.E. Paiker, F.J. Raal, M. von Arb, Auto-antibodies against oxidized LDL as a marker of coronary artery disease in patients with familial hypercholesterolae-mia, Ann.Clin.Biochem. 37 ( Pt 2) (2000) 174-178.[47] L.P. van de Vijver, R. Steyger, G. van Poppel, J.M. Boer, D.A. Kruijssen, J.C. Seidell, H.M. Princen, Autoan-tibodies against MDA-LDL in subjects with severe and minor atherosclerosis and healthy population controls, Atherosclerosis 122 (1996) 245-253.

47
PON1 genotype in life-long smokers
[48] R. Malin, J. Knuuti, T. Janatuinen, R. Laaksonen, R. Vesalainen, P. Nuutila, H. Jokela, J. Laakso, O. Jaakkola, T. Solakivi, T. Lehtimaki, Paraoxonase gene polymor-phisms and coronary reactivity in young healthy men, J Mol Med 79 (2001) 449-458.[49] B. Mackness, M.I. Mackness, S. Arrol, W. Turkie, P.N. Durrington, Effect of the human serum paraoxo-nase 55 and 192 genetic polymorphisms on the pro-tection by high density lipoprotein against low den-sity lipoprotein oxidative modification, FEBS Lett 423 (1998) 57-60.[50] G. Howard, L.E. Wagenknecht, G.L. Burke, A. Diez-Roux, G.W. Evans, P. McGovern, F.J. Nieto, G.S. Tell, Cig-arette smoking and progression of atherosclerosis: The Atherosclerosis Risk in Communities (ARIC) Study, JAMA 279 (1998) 119-124.[51] G.S. Tell, J.F. Polak, B.J. Ward, S.J. Kittner, P.J. Sav-age, J. Robbins, Relation of smoking with carotid artery wall thickness and stenosis in older adults. The Car-diovascular Health Study. The Cardiovascular Health Study (CHS) Collaborative Research Group, Circula-tion 90 (1994) 2905-2908.


4Indications that paraoxonase-1 contributes to
plasma high density lipoprotein levels in familial hypercholesterolemia
J. Lipid Res. 2005 Mar;46(3):445-51
T.M. van HimbergenM. Roest
J. de GraafE.H. Jansen
H. HattoriJ.J. KasteleinH.A. Voorbij
A.F. StalenhoefL.J. van Tits

Abstract
High-density lipoprotein (HDL) associated paraoxonase type-1 (PON1) can protect low-den-sity lipoprotein (LDL) and HDL against oxidative modification, in vitro, and therefore, may protect against cardiovascular disease. We investigated the effects of PON1 levels, activity and genetic variation on HDL-cholesterol levels, circulating oxidized LDL (oxLDL), subclinical inflammation (High sensitive C-reactive protein, Hs-CRP) and carotid atherosclerosis.PON1 genotypes (L55M, Q192R, -107C/T, -162A/G, -824G/A, and -907G/C) were deter-mined in 302 patients with familial hypercholesterolemia. PON1 activity was monitored by the hydrolysis rate of paraoxon, diazoxon, and phenyl acetate. PON1 levels, oxLDL and Hs-CRP were determined using an immuno assay.The genetic variants of PON1 that were associated with high levels and activity of the enzyme were associated with higher HDL-cholesterol levels (P values for trend: 0.008, 0.020, 0.042, and 0.037 for L55M, Q192R, -107C/T, and -907G/C, respectively). In addition to PON1 geno-type, there was also a positive correlation between PON1 levels and activity and HDL-choles-terol (PON1 levels: r = 0.37, P < 0.001; paraoxonase activity: r = 0.23, P = 0.01; diazoxonase activity: r = 0.29, P < 0.001; arylesterase activity: r = 0.19, P = 0.03).Our observations support the hypothesis that both PON1 levels and activity preserve HDL-cholesterol in plasma.

51
PON1 and HDL-C levels in patients with FH
Introduction
Serum paraoxonase type-1 (PON1) is a high-density lipoprotein (HDL)-associated enzyme capable of hydrolyzing lipid peroxides in low-density lipoprotein (LDL) [1]. Since oxidized LDL (oxLDL) has atherogenic and pro-inflammatory properties [2], PON1 may, therefore, protect against atherosclerosis. This hypothesis is supported by observations in PON1-defi-cient mice; these mice are more prone to develop atherosclerosis than wild-type mice when fed a high-fat/high-cholesterol diet [3]. An additional anti-atherogenic property of PON1 is the inhibition of the oxidative modification of HDL and hence the preservation of HDL func-tion [4]. HDL protects against cardiovascular disease (CVD) by means of reverse cholesterol transport [5]. Even though the physiological role of PON1 in vivo remains to be clarified, the inhibition of both LDL and HDL oxidation may indeed contribute to the protection against CVD. PON1 expression is partly controlled by its molecular variation at the gene locus [6]. Two polymorphic sites have been described in the coding region: a leucine (L)-to-methionine (M) transition at position 55 (L55M) and a glutamine (Q)-to-arginine (R) transition at position 192 (Q192R). The L55M polymorphism affects the enzyme concentration, while the Q192R polymorphism affects the catalytic efficiency, but not the concentration [7, 8]. Four polymor-phisms in the promoter sequence of the PON1 gene (-107C/T, -162A/G, -824G/A, -907G/C) also contribute to the variability in protein expression. During an earlier study, our group found a relation between the PON1 genotype combination LLQQ and an increased intima-media thickness (IMT), a surrogate marker of CVD, in high-risk subjects with familial hy-percholesterolemia (FH) [9]. There is, however, no consensus on the contribution of these genetic variants to the risk of CVD [10]. In addition to genetic influences, PON1 levels and activity could be modified by life-style de-terminants like smoking [11, 12], vitamin C and E consumption [13], and alcohol intake [14]. Therefore, studying PON1 levels and activity, in conjunction with variation at the gene level, gives a more complete view of the role of PON1 in the development of atherosclerosis. The beneficial effects of PON1 on the inhibition of atherosclerosis might be more pronounced in a population that is prone to develop atherosclerosis than in the general population. For this reason, we studied the role of PON1 in patients with FH. These patients are character-ized by substantially elevated serum LDL-cholesterol concentrations and sharply increased CVD risk. Our aim was to investigate the influence of PON1 genotypes and PON1 levels and activity on HDL-cholesterol levels, circulating oxLDL, inflammation markers (High sensitive C-reactive protein, Hs-CRP), and common carotid artery IMT (CCA-IMT) in patients with FH.

52
CHAPTER 4
Methods
SubjectsThe study population consisted of 325 men and women with FH, who participated in a pro-spective, randomized, double-blind, two-center trial as described elsewhere [15]. Of the 325 participants, 23 were excluded because there was no DNA available for PON1 genotyping. After an 8-week placebo run-in (in which all lipid-lowering drugs were discontinued), base-line height, weight, blood pressure, and CCA-IMT were measured. DNA, plasma, and serum samples were stored at -80 °C until analysis. Studies have shown that PON1 levels and activity can only be measured correctly in serum samples [16, 17]. Since only one trial center col-lected additional serum for the determination of PON1 levels and activity, these measure-ments could be performed in only 134 of the 302 participants. All participants gave written informed consent and the ethics committees of both trial centers approved the study.
Lipids and lipoproteinsTotal cholesterol, LDL-cholesterol (LDL-C), HDL-cholesterol (HDL-C), and triglycerides were analyzed as described previously [18].
Ultrasound measurement of the carotid IMTUltrasound scanning of the common carotid arteries (CCAs) was performed using a Bio-sound Phase-two real time scanner (BiosoundEsaote, Indianapolis, IN, USA) equipped with a 10-MHz transducer, as described in detail elsewhere [19]. The IMT was measured in both the anterior and posterior walls of the distal 1.0-cm straight part of both CCAs. Images were ana-lyzed using a semi-automatic software program (Eurequa; TSA company, Meudon, France). A complete set of measurements was available for 288 participants. The CCA-IMT was ex-pressed as the mean of both the anterior and posterior walls of the left and right CCAs.
PON1 genotypeThe L55M and Q192R mutations were determined by means of the polymerase chain reac-tion, followed by restriction fragment length polymorphism (PCR-RFLP) using primers and restriction enzymes as described by Humbert et al [8]. DNA fragments were separated on a 2% agarose gel and visualized with ethidium bromide. The 55L allele corresponded to the presence of a non-digested 170-bp fragment, the 55M allele to a 44-bp and a 126-bp fragment, the 192Q allele to a non-digested 99-bp fragment, and the 192R allele to a 33-bp and a 66-bp fragment. A PCR product was obtained successfully for 299 (L55M) and 298 (Q192R) DNA samples. PCR followed by hybridization with allele-specific oligonucleotides was used to analyze the promoter polymorphisms -107C/T, -162A/G, -824G/A, and -907G/C. Primers for the ampli-fication of a 156-bp fragment coding for the polymorphisms at positions -107 and -162 were

53
PON1 and HDL-C levels in patients with FH
5’GAAAGTGCTGAGCTCCTGCG3’ and 5’CTAGGAGGCTCTGCTGCCTG3’. Primers for the amplification of a 170-bp fragment coding for the polymorphisms at positions -824 and -907 were 5’ACATGGAGCAAATCATTCACAG3’ and 5’ACACATAAAGCAAGAAAGGG-GA3’. The PCR products for polymorphisms -107 and -162 and for polymorphisms -824 and -907 were obtained successfully in 301 and 299 DNA samples, respectively. The fragments were transferred onto Hybond N+ membranes (Amersham Pharmacia Biotech, Buckingham-shire, England) and hybridized with the allele-specific oligonucleotides. The allele-specific oligonucleotides for the -162 and -907 polymorphisms were as follows: 5’GCAAGCCAC-GCCTTCTGT3’ (-162A), 5’GCAAGCCGCGCCTTCTG3’ (-162G), 5’AGAGAAGAGAGA-CATGGTTG3’ (–907G), and 5’AGAGAAGAGACACATGGTTG3’ (–907C). For positions -107 and -824 we used the same allele-specific oligonucleotides as described by Leviev and James [20]. All of the oligonucleotides were hybridized at 42 °C, followed by two washes at room temperature and an allele-specific temperature wash.
Analysis of PON1 levelsPON1 levels were determined at the Department of Advanced Medical Technology and De-velopment, BML, Inc. Saitama, Japan, using an enzyme-linked immunosorbent assay (ELI-SA). This procedure has been described in detail elsewhere [21]. PON1 levels were success-fully measured in 131 serum samples. The intra- and interassay coefficients of variation were 5.5% and 5.1%, respectively.
Analysis of PON1 enzymatic activity Paraoxonase and diazoxonase activities were analyzed spectrophotometrically at the Labora-tory for Toxicology, Pathology and Genetics of the National Institute for Public Health and the Environment (Bilthoven, The Netherlands), using paraoxon and diazoxon as substrates in a Tris buffer (0.1 M, pH 8.5) containing 2 M NaCl and 2 mM CaCl2 [22]. After the addition of the serum sample (diluted tenfold for paraoxonase and twenty-fold for diazoxonase activ-ity), the reaction was monitored in a microtiter plate for 5 minutes at 25 °C. Paraoxonase and diazoxonase activities are expressed as units (U) per liter (L) serum, where one U is 1 µmol substrate hydrolyzed per minute.Arylesterase activity was measured at the Research Laboratory of the Department of Clini-cal Chemistry, UMC Utrecht, Utrecht, The Netherlands, as the rate of hydrolysis of phenyl acetate into phenol. This process can be detected spectrophotometrically [23]. Serum samples were prepared in sample buffer consisting of 20 mM Tris and 0.9 mM CaCl2 (pH 8.0) in a 40-fold dilution. Five µl diluted serum was then added to 200 μl freshly made substrate buf-fer containing 1 mM phenyl acetate, 20 mM Tris, and 0.9 mM CaCl2 (pH 8.0). The reaction was monitored in a microtiter plate at 260 nM on a Fluostar microplate reader (BMG Labtech GmbH, Offenburg, Germany) at 37 oC. The non-enzymatic hydrolysis of phenyl acetate, based on the hydrolysis rate in the absence of serum, was subtracted from the total rate of

54
CHAPTER 4
hydrolysis. The molar extinction coefficient used to calculate the rate of hydrolysis was 1,310 M-1cm-1. A path-length correction was applied for the use of microtiter plates. Arylesterase is expressed as units (U) per milliliter (ml) serum, where one U is 1 µmol phenyl acetate hy-drolyzed per minute.
OxLDL ELISAWe determined concentrations of oxLDL in EDTA-plasma samples that had been supple-mented with saccharose and frozen at –80 °C and not thawed before. Samples of 110 patients fulfilled these criteria. We used a commercially available non-competitive ELISA (Mercodia, Uppsala, Sweden). The intra- and interassay coefficient of variation were 6% and 7% respec-tively.
Hs-CRPHs-CRP was measured by enzyme-immunoassay according to the instructions from the man-ufacturer (Dako, Glastrup, Denmark). The coefficient of variation was 6%.
Statistical analysisThe relations between PON1 genotype, PON1 levels, PON1 activity, HDL-C levels, oxLDL, CRP and CCA-IMT were tested in a linear regression model in which PON1 genotype served as the independent variable and the PON1 levels, PON1 activity, HDL-C levels, oxLDL, Hs-CRP, and CCA-IMT as the dependent variables. The influence of age and gender on this rela-tionship was investigated in a multivariate regression analysis. The Pearson correlation coef-ficient (r) was used to test the relationship between the variables displayed in Tables 4,5 and 7. Hs-CRP had a skewed distribution, the presentation in the tables and statistical analysis were based on log transformed data. All of the analyses were performed using SPSS version 11.5.
Results
The study population consisted of 121 men and 181 women with FH (Table 1). The mean age was 48 years; 98 of the participants were current smokers, 87 were past smokers and 117 were non-smokers. The subjects were slightly overweight, but had normal blood pressure. Levels of triglycerides and HDL-C were normal. The average LDL-C level was higher than 8 mmol/L, which is characteristic for FH. Table 2 shows the relationships between the PON1 genotypes (L55M, Q192R, -107C/T, -162A/G, -824G/A and -907G/C) and PON1 levels and activities. All PON1 genotype distri-butions were in Hardy-Weinberg equilibrium. The 55L variant of the L55M polymorphism contributed to increased PON1 levels (p<0.001) and increased paraoxonase, diazoxonase and arylesterase activities (p<0.001, p=0.018, and p=0.029 respectively). The 192R isoform

55
PON1 and HDL-C levels in patients with FH
of the Q192R polymorphism hydrolyzed paraoxon more efficiently, while the 192Q isoform hydrolyzed diazoxon more efficiently (p<0.001 for both relations). The ratio of paraoxon and diazoxon hydrolyzation segregates with the Q192R polymorphism: there was 99.2% concor-dance between the paraoxon/diazoxon ratio and the Q192R genotypes. The Q192R polymor-phism was not associated with PON1 levels nor with arylesterase activity. The -107C, -162A, -824A, and -907G variants of the -107C/T, -162A/G, -824G/A, and -907G/C polymorphisms in the promoter sequence expressed higher PON1 levels, diazoxonase ac-tivity, and arylesterase activity than the -107T, -162G, -824G, and -907C variants (p<0.001 for all relations except arylesterase activity; the latter has p=0.001 for -107C/T and -162A/G and p=0.002 for -824G/A). The hydrolysis rate of paraoxon was significantly higher for the -107CC and –907GG homozygotes than for the other genetic variants at positions –107 and –907 (p<0.001 and p=0.002, respectively). The ratio of paraoxonase/PON1 levels did not dif-fer among the variants of these polymorphisms (data not shown). Furthermore, the -162A/G and -824G/A polymorphisms were not associated with paraoxonase activity. Finally, there was a strong correlation between PON1 levels and PON1 arylesterase- (r=0.52, p<0.001) and PON1 diazoxonase (r=0.69, p<0.001) activities.The contribution of PON1 genotypes to HDL-C levels is presented in Table 3. The 55L, 192R, -107C, and -907G variants of the L55M, Q192R, -107C/T, and -907G/C polymorphisms pre-dicted increased HDL-C levels (p=0.008, p=0.020, p=0.042, and p=0.037, respectively). Af-ter adjustment for age and gender, the relation between PON1 genotypes and HDL-C levels remained significant. The -162A/G and -824G/A polymorphisms were not associated with HDL-C levels. Like the PON1 genotypes, PON1 levels and paraoxonase, diazoxonase, and arylesterase ac-tivities also contributed to increased HDL-C levels (r=0.37, p<0.001; r=0.23, p=0.008; r=0.29, p=0.001; and r=0.19, p=0.031, respectively; Table 4).
Table 1. Baseline characteristics of FH population (n=302)Characteristics Values*Age (years) 48.5 ± 10.5Sex (M/F) 121 / 181Smoking (current/past/non) 98 / 87 / 117BMI (kg/m2) 25.7 ± 3.5Systolic blood pressure (mmHg) 130.7 ± 15.9Diastolic blood pressure (mmHg) 79 ± 7.9Total cholesterol (mmol/L) 10.14 ± 1.99HDL-C (mmol/L) 1.16 ± 0.31LDL-C (mmol/L) 8.19 ± 1.95Triglycerides (mmol/L) 1.86 ± 1.22
*Values are means ± standard deviation, except for sex, which represents the number of males and females, and smoking, which represents the number of current past and non-smokers. FH, familial hypercholesterolemia; BMI, body mass index; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol.

56
CHAPTER 4
Tabl
e 2.
Rel
atio
nshi
ps b
etw
een
PON
1 ge
noty
pes a
nd P
ON
1 le
vels
and
activ
ityG
enot
ype
PON
1 le
vels
(µg/
ml)
Para
oxon
ase
activ
ity (U
/L)
Dia
zoxo
nase
act
ivity
(U/L
) A
ryle
ster
ase
activ
ity (U
/ml)
mea
n±
sdn
p*m
ean
±sd
np*
mea
n±
sdn
p*m
ean
±sd
np*
L55M
LL87
.3±
22.4
5879
2.6
±35
1.8
5975
61.1
±21
73.2
5976
.4±
31.9
59LM
72.1
±18
.657
<0.0
0153
6.2
±28
2.8
57<0
.001
6840
.7±
1630
.957
0.01
869
.6±
25.2
570.
029
MM
61.9
±16
.815
190.
5±
44.4
1765
63.9
±12
53.9
1758
.9±
32.8
17Q
192R
74.2
±23
.750
246.
5±
79.0
5279
57.9
±18
81.1
5270
.5±
29.4
52Q
R79
.3±
21.1
660.
130
764.
5±
206.
966
<0.0
0169
02.1
±16
43.8
66<0
.001
72.3
±30
.666
0.86
7RR
82.9
±20
.113
1184
.9±
245.
114
5083
.4±
1088
.514
70.7
±28
.614
-107
C/T
CC
92.1
±21
.638
771.
7±
398.
438
8267
.1±
1854
.138
85.9
±31
.338
CT
75.7
±17
.161
<0.0
0159
4.6
±32
0.9
63<0
.001
6912
.2±
1588
.263
<0.0
0165
.6±
25.0
630.
001
TT61
.3±
14.5
3142
1.2
±29
6.0
3260
04.6
±14
04.1
3263
.5±
29.4
32-1
62A
/GA
A10
1.8
±20
.87
791.
0±
483.
47
8657
.3±
1444
.27
103.
5±
29.7
7G
A84
.7±
20.9
46<0
.001
595.
8±
349.
046
0.35
981
51.2
±17
36.4
46<0
.001
74.3
±31
.346
0.00
2G
G70
.2±
18.1
7759
1.4
±35
5.2
8063
27.5
±14
81.2
8066
.1±
26.3
80-8
24G
/AG
G68
.9±
18.4
5755
4.7
±36
7.9
6062
56.0
±13
89.8
6064
.4±
28.5
60G
A81
.1±
20.9
60<0
.001
626.
5±
342.
960
0.09
975
67.8
±19
26.1
60<0
.001
72.0
±28
.460
0.00
1A
A93
.9±
19.6
1372
2.1
±39
0.7
1386
41.2
±12
06.6
1395
.9±
25.4
13-9
07G
/CG
G91
.6±
20.8
3075
0.8
±39
8.0
3080
72.7
±19
91.9
3085
.9±
29.5
30C
G77
.3±
17.9
63<0
.001
605.
3±
321.
965
0.00
273
24.3
±16
95.8
65<0
.001
70.1
±27
.365
<0.0
01C
C64
.9±
19.3
3748
4.1
±35
6.3
3858
81.6
±11
61.3
3860
.3±
28.5
38
Valu
es re
pres
ent t
he m
ean
± st
anda
rd d
evia
tion
(sd)
follo
wed
by
the
num
ber o
f obs
erva
tions
(n).
*p v
alue
s are
bas
ed o
n lin
ear r
egre
ssio
n.

57
PON1 and HDL-C levels in patients with FH
Thus, we have demonstrated an association of PON1 genotypes and intermediate phenotypes with plasma HDL cholesterol. Next we investigated the relation with circulating oxLDL and with Hs-CRP. We observed no significant association of PON1 genotypes with oxLDL or with Hs-CRP (data not shown). As presented in table 5, PON1 levels and activity also did not cor-relate with oxLDL levels. Furthermore, we observed only weak but significant correlations of diazoxonase and arylesterase activities with Hs-CRP (Table 5).Hence, we found remarkable associations of PON1 genotypes, PON1 levels and PON1 ac-tivity with HDL-C levels but not with circulating oxLDL and Hs-CRP. We also investigated the relation of PON1 genotypes and intermediate phenotypes with CCA-IMT. We found no significant relation between PON1 genotypes and CCA-IMT (Table 6) and no correlation of PON1 levels and PON1 activities with CCA-IMT (Table 7). Moreover, HDL-C, oxLDL and Hs-CRP did not correlate with CCA-IMT.
Table 3. HDL-C levels according to PON1 genotypesPON1 genotype HDL-C levels (mmol/L)* p crude† p adjusted‡
mean ± sd nL55MLL 1.20 ± 0.31 137LM 1.15 ± 0.30 133 0.008 0.001MM 1.03 ± 0.25 29Q192RQQ 1.11 ± 0.27 120QR 1.18 ± 0.31 149 0.020 0.033RR 1.24 ± 0.38 29-107C/TCC 1.21 ± 0.31 88CT 1.15 ± 0.30 147 0.042 0.036TT 1.11 ± 0.30 66-162A/GAA 1.11 ± 0.31 16AG 1.20 ± 0.30 101 0.482 0.354GG 1.14 ± 0.31 184-824G/AGG 1.13 ± 0.31 144GA 1.19 ± 0.31 123 0.103 0.087AA 1.19 ± 0.29 32-907G/CGG 1.19 ± 0.30 73GC 1.18 ± 0.30 142 0.037 0.024CC 1.09 ± 0.31 84*Values represent mean ± standard deviation (sd), followed by the number of observations (n). †Values are based on linear regression. ‡Values are based on linear regression and are adjusted for age and gender. HDL-C, high-density lipoprotein cholesterol.

58
CHAPTER 4
Discussion
Previous investigations have demonstrated that high PON1 levels and high PON1 activity contribute to increased levels of HDL-C [17, 24, 25]. The results of the present study rein-force and expand these findings by showing that not only high PON1 levels and high PON1 activity, but also the genotypes associated with high PON1 levels (55L, -107C, and –907C variants), and high PON1 activity (192R variants) were associated with the highest HDL-C levels. These findings support the hypothesis that PON1 is involved in preservation of HDL (4). We speculate that HDL associated lecithin:cholesterol acyltransferase (LCAT) may play an important role in this process. Recently it was shown that in mice overexpressing PON1, LCAT activity is preserved [26]. Activated LCAT prevents catabolism of HDL and therefore may increase HDL-C [27, 28]. Elucidating this mechanism is subject for future studies.In large cohort studies it was established that increased HDL-C levels protect against CVD [29]. Remarkably, several studies, including ours, did not observe a significant relation be-tween HDL-C and CCA-IMT [30-33]. Apparently, HDL-C has only minor effects on CCA-IMT. Similar to HDL-C, the contribution of paraoxonase to IMT is debatable. Some studies have reported a significant association between PON1 and IMT in FH patients [9], in middle-aged women [34], and in a subgroup of non smokers [35], but there are also a number of reports, including the current, that did not find an association between PON1 and IMT [30, 36-38]. These differences in outcome may suggest that there is only a weak relation between
Table 4. HDL-C levels with regard to PON1 levels and activityHDL-C levels (mmol/L)*
r p nPON1 levels (µg/ml) 0.37 <0.001 131Paraoxonase activity (U/L) 0.23 0.008 134Diazoxonase activity (U/L) 0.29 0.001 134Arylesterase activity (U/ml) 0.19 0.031 134*Values represent Pearson correlation coefficient (r) followed by the p value (p) and the number of observations (n). HDL-C, high-density lipoprotein cholesterol.
Table 5. OxLDL levels and Hs-CRP with regard to PON1 levels and activitiesoxLDL (U/L) Hs-CRP (mg/L)
r p n r p nPON1 levels (µg/ml) 0.002 0.98 107 0.16 0.06 129Paraoxonase activity (U/L) 0.054 0.58 110 0.06 0.53 132Diazoxonase activity (U/L) -0.020 0.83 110 0.20 0.02 132Arylesterase activity (U/ml) -0.028 0.77 110 0.20 0.02 132Values represent Pearson correlation coefficient (r), followed by the p value (p) and the number of observations (n). oxLDL, oxidized low-density lipoprotein; Hs-CRP, High sensitive C-reactive protein.

59
PON1 and HDL-C levels in patients with FH
PON1 and IMT that will not be observed in many small studies that lack the power to detect weak effects. Alternatively, PON1 does not play a role in early atherosclerosis (of which IMT is a measurement), but may play a role in a later stage of CVD. As suggested by Cao et al [36], the lack of association of PON1 with IMT does not exclude the possibility of its involvement in the later phases of the atherothrombotic process. We therefore recommend, that future studies on PON1 should be carried out in populations with clinical endpoints.Apart from preserving the function of HDL, PON1 may beneficially influence atherogenesis via inhibition of LDL oxidation [39-41], in that way preventing excessive cholesterol uptake by macrophages and inhibiting foam cell formation [42]. Furthermore, inhibition of LDL oxidation may control inflammation in the arterial wall [43]. Both LDL oxidation and in-flammation are associated with an increased risk for CVD [44, 45]. We studied the relation between PON1 and circulating oxLDL as marker for LDL oxidation and Hs-CRP as marker for inflammation, and could not establish that PON1 is involved in process of atherogenesis
Table 6. CCA-IMT according to PON1 genotypesPON1 genotype CCA-IMT (mm)* p for trend†
mean ± sd nL55MLL 0.86 ± 0.16 132LM 0.87 ± 0.17 126 0.38MM 0.89 ± 0.23 28Q192RQQ 0.88 ± 0.19 110QR 0.85 ± 0.16 146 0.35RR 0.90 ± 0.17 29-107C/TCC 0.85 ± 0.14 85CT 0.86 ± 0.18 139 0.13TT 0.89 ± 0.18 63-162A/GAA 0.85 ± 0.14 15GA 0.87 ± 0.16 96 0.99GG 0.87 ± 0.18 176-824G/AGG 0.88 ± 0.18 137GA 0.86 ± 0.16 118 0.44AA 0.85 ± 0.17 30-907G/CGG 0.85 ± 0.15 70GC 0.86 ± 0.16 134 0.11CC 0.90 ± 0.20 81*Values represent the mean ± standard deviation (sd), followed by the number of observations (n). †p values are based on linear regression analysis and adjusted for age. CCA-IMT, common carotid artery intima-media thickness.

60
CHAPTER 4
via the inhibition of LDL oxidation and later on the induction of inflammation. Some reserv-edness is required, since the role of PON1 on circulating oxLDL and Hs-CRP needs to be confirmed in different populations.Most studies on the role of PON1 and CVD are restricted to PON1 genotypes. However, ad-ditional measurements of PON1 serum levels and activities, give a more complete view of the PON1 status because they reflect the combined genetic and environmental effects of PON1. In the present study, PON1 status was determined using different genotypes (L55M, Q192R, -107C/T, -162A/G, -824G/A, and –907G/C), PON1 levels, and activity towards three different substrates (paraoxon, diazoxon, and phenyl acetate). The PON1 genotypes were determined in all patients, while PON1 serum level and activity measurements were restricted to 134 of the 302 subjects. Fortunately, the power was still sufficient to establish a significant rela-tion between PON1 genotypes and PON1 levels and activity. The associations between the two coding and four promoter region polymorphisms and PON1 levels and activity are in line with previous reports: the high expression alleles 55L, -107C, -162A, -824A, and –907G contributed to the highest PON1 levels [7, 20, 46]. Furthermore, as described in literature, the Q192R polymorphism was responsible for the difference in the hydrolysis rates towards paraoxon and diazoxon and not towards phenyl acetate [47]. Both the activity measurements and the Q192R genotyping were reliable, because there was 99.2% agreement between PON1 phenotyping with the ratio between the hydrolysis rate of paraoxon and diazoxon and Q192R genotyping. However, the physiological substrate of PON1 is unknown and measurements of PON1 activity by monitoring the rates of hydrolysis of phenyl acetate, paraoxon, and di-azoxon may not quantify the degree of anti-atherogenic potential of PON1 and should be regarded as surrogate markers of PON1 protective activity. In this study, PON1 levels and PON1 activities were measured independently at three different laboratories. There was still a strong correlation among the PON1 levels assessed directly with ELISA and the PON1 activ-ity measurements. PON1 activity measurements therefore qualify as suitable surrogate mark-
Table 7. CCA-IMT with regard to PON1 levels, PON1 activities, HDL-C, oxLDL and Hs-CRP
CCA-IMT (mm)*r p n
PON1 levels (µg/ml) -0.04 0.67 130Paraoxonase activity (U/L) -0.03 0.75 129Diazoxonase activity (U/L) -0.06 0.49 129Arylesterase activity (U/ml) -0.10 0.28 129HDL-C (mmol/L) -0.09 0.11 288oxLDL (U/L) -0.03 0.79 110Hs-CRP (mg/L) 0.04 0.62 152*Values represent Pearson correlation coefficient (r), followed by the p value (p) and the number of observations (n). CCA-IMT, common carotid artery intima-media thickness; HDL-C, high-density lipoprotein cholesterol; oxLDL, oxidized low-density lipoprotein; Hs-CRP, High sensitive C-reactive protein.

61
PON1 and HDL-C levels in patients with FH
ers for PON1 levels in serum.We believe that the chance of finding effects of PON1 on CVD increased in a population at high risk for developing atherosclerosis and CVD. For this reason we investigated the effects of PON1 in a population of patients with FH. These patients have elevated lipid levels and present a lipid environment that is different from that of the population at large. Results ob-tained from such a population may not be extrapolated to the general population. In summary, PON1 is involved in the preservation of HDL-C in plasma and may, therefore, protect against CVD. The genetic variants associated with high PON1 levels and activity and the elevated plasma levels and increased activity of PON1 predicted the highest levels of HDL-C. Neither PON1 nor HDL-C was associated with CCA-IMT. Further studies are required to assess the relevance of the HDL-C-preserving activity of PON1 in trials with clini-cal endpoints.
Acknowledgments
The authors greatly acknowledge Arjan Barendrecht, Piet Beekhof, Anneke Hijmans, Heidi Hak-Lemmers, Mayumi Ito, and Takeshi Kujiraoka for their excellent technical assistance.

62
CHAPTER 4
References
[1] Mackness MI, Arrol S, Durrington PN. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 1991;286:152-4.[2] Mertens A, Holvoet P. Oxidized LDL and HDL: an-tagonists in atherothrombosis. FASEB J. 2001;15:2073-2084.[3] Shih DM, Gu L, Xia YR, Navab M, Li WF, Hama S, Castellani LW, Furlong CE, Costa LG, Fogelman AM, Lusis AJ. Mice lacking serum paraoxonase are suscep-tible to organophosphate toxicity and atherosclerosis. Nature. 1998;394:284-287.[4] Aviram M, Rosenblat M, Bisgaier CL, Newton RS, Primo-Parmo SL, La Du BN. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxo-nase. J Clin Invest. 1998;101:1581-1590.[5] Miller GJ, Miller NE. Plasma-high-density-lipo-protein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16-19.[6] Simpson NE. Serum arylesterase levels of ac-tivity in twins and their parents. Am J Hum Genet. 1971;23:375-382.[7] Garin MC, James RW, Dussoix P, Blanche H, Passa P, Froguel P, Ruiz J. Paraoxonase polymorphism Met-Leu54 is associated with modified serum concen-trations of the enzyme. A possible link between the paraoxonase gene and increased risk of cardiovascular disease in diabetes. J Clin Invest. 1997;99:62-6.[8] Humbert R, Adler DA, Disteche CM, Hassett C, Omiecinski CJ, Furlong CE. The molecular basis of the human serum paraoxonase activity polymorphism. Nat Genet. 1993;3:73-76.[9] Leus FR, Wittekoek ME, Prins J, Kastelein JJ, Voor-bij HA. Paraoxonase gene polymorphisms are associ-ated with carotid arterial wall thickness in subjects with familial hypercholesterolemia. Atherosclerosis. 2000;149:371-377.
[10] Wheeler JG, Keavney BD, Watkins H, Col-lins R, Danesh J. Four paraoxonase gene polymor-phisms in 11212 cases of coronary heart disease and 12786 controls: meta-analysis of 43 studies. Lancet. 2004;363:689-695.[11] James RW, Leviev I, Righetti A. Smoking is asso-ciated with reduced serum paraoxonase activity and concentration in patients with coronary artery disease. Circulation. 2000;101:2252-2257.[12] Nishio E, Watanabe Y. Cigarette smoke extract in-hibits plasma paraoxonase activity by modification of the enzyme’s free thiols. Biochem Biophys Res Com-mun. 1997;236:289-93.[13] Jarvik GP, Tsai NT, McKinstry LA, Wani R, Brophy VH, Richter RJ, Schellenberg GD, Heagerty PJ, Hatsu-kami TS, Furlong CE. Vitamin C and E intake is associ-ated with increased paraoxonase activity. Arterioscler Thromb Vasc Biol. 2002;22:1329-33.[14] van der Gaag MS, van Tol A, Scheek LM, James RW, Urgert R, Schaafsma G, Hendriks HF. Daily mod-erate alcohol consumption increases serum paraoxo-nase activity; a diet-controlled, randomised inter-vention study in middle-aged men. Atherosclerosis. 1999;147:405-410.[15] Smilde TJ, Van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet. 2001;357:577-581.[16] Mackness B, Hunt R, Durrington PN, Mackness MI. Increased immunolocalization of paraoxonase, clusterin, and apolipoprotein A-I in the human artery wall with the progression of atherosclerosis. Arterio-scler Thromb Vasc Biol. 1997;17:1233-1238.[17] Blatter Garin MC, Abbott C, Messmer S, Mackness M, Durrington P, Pometta D, James RW. Quantification of human serum paraoxonase by enzyme-linked im-munoassay: population differences in protein concen-

63
PON1 and HDL-C levels in patients with FH
trations. Biochem J. 1994;304 ( Pt 2):549-554.[18] Smilde TJ, Trip MD, Wollersheim H, Van Wissen S, Kastelein JJP, Stalenhoef AFH. Rationale, design and baseline characteristics of a clinical trial comparing the effects of robust vs conventional cholesterol low-ering and intima media thickness in patients with fa-milial hypercholesterolaemia: The atorvastatin versus simvastatin on atherosclerosis progression (ASAP) study. Clinical Drug Investigation. 2000;20:67-79.[19] Smilde TJ, Wollersheim H, van Langen H, Stalen-hoef AF. Reproducibility of ultrasonographic measure-ments of different carotid and femoral artery segments in healthy subjects and in patients with increased in-tima-media thickness. Clin Sci (Lond). 1997;93:317-324.[20] Leviev I, James RW. Promoter polymorphisms of human paraoxonase PON1 gene and serum paraoxo-nase activities and concentrations. Arterioscler Thromb Vasc Biol. 2000;20:516-521.[21] Kujiraoka T, Oka T, Ishihara M, Egashira T, Fujioka T, Saito E, Saito S, Miller NE, Hattori H. A sandwich en-zyme-linked immunosorbent assay for human serum paraoxonase concentration. J Lipid Res. 2000;41:1358-1363.[22] Richter RJ, Furlong CE. Determination of paraox-onase (PON1) status requires more than genotyping. Pharmacogenetics. 1999;9:745-753.[23] Eckerson HW, Wyte CM, La Du BN. The human serum paraoxonase/arylesterase polymorphism. Am J Hum Genet. 1983;35:1126-1138.[24] Saha N, Roy AC, Teo SH, Tay JS, Ratnam SS. Influ-ence of serum paraoxonase polymorphism on serum lipids and apolipoproteins. Clin Genet. 1991;40:277-282.[25] Blatter MC, James RW, Messmer S, Barja F, Pomet-ta D. Identification of a distinct human high-density lipoprotein subspecies defined by a lipoprotein-associ-ated protein, K-45. Identity of K-45 with paraoxonase. Eur J Biochem. 1993;211:871-879.
[26] Oda MN, Bielicki JK, Ho TT, Berger T, Rubin EM, Forte TM. Paraoxonase 1 overexpression in mice and its effect on high-density lipoproteins. Biochem Bio-phys Res Commun. 2002;290:921-927.[27] Brousseau ME, Santamarina-Fojo S, Vaisman BL, Applebaum-Bowden D, Berard AM, Talley GD, Brewer HB, Jr., Hoeg JM. Overexpression of human lecithin:cholesterol acyltransferase in cholesterol-fed rab-bits: LDL metabolism and HDL metabolism are af-fected in a gene dose-dependent manner. J Lipid Res. 1997;38:2537-2547.[28] Wang M, Briggs MR. HDL: the metabolism, function, and therapeutic importance. Chem Rev. 2004;104:119-137.[29] Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR, Jr., Bangdiwala S, Tyroler HA. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8-15.[30] Campo S, Sardo MA, Trimarchi G, Bonaiuto M, Castaldo M, Fontana L, Bonaiuto A, Bitto A, Saitta C, Saitta A. The paraoxonase promoter polymorphism (-107)T>C is not associated with carotid intima-media thickness in Sicilian hypercholesterolemic patients. Clin Biochem. 2004;37:388-394.[31] Schmidt H, Schmidt R, Niederkorn K, Gradert A, Schumacher M, Watzinger N, Hartung HP, Kostner GM. Paraoxonase PON1 polymorphism leu-Met54 is asso-ciated with carotid atherosclerosis: results of the Aus-trian Stroke Prevention Study. Stroke. 1998;29:2043-2048.[32] Koyama H, Maeno T, Fukumoto S, Shoji T, Yamane T, Yokoyama H, Emoto M, Shoji T, Tahara H, Inaba M, Hino M, Shioi A, Miki T, Nishizawa Y. Platelet P-selec-tin expression is associated with atherosclerotic wall thickness in carotid artery in humans. Circulation. 2003;108:524-529.[33] Wohlin M, Sundstrom J, Arnlov J, Andren B, Zethelius B, Lind L. Impaired insulin sensitivity is an

64
CHAPTER 4
independent predictor of common carotid intima-me-dia thickness in a population sample of elderly men. Atherosclerosis. 2003;170:181-185.[34] Fortunato G, Rubba P, Panico S, Trono D, Tinto N, Mazzaccara C, De Michele M, Iannuzzi A, Vitale DF, Salvatore F, Sacchetti L. A paraoxonase gene polymor-phism, PON 1 (55), as an independent risk factor for increased carotid intima-media thickness in middle-aged women. Atherosclerosis. 2003;167:141-148.[35] Malin R, Loimaala A, Nenonen A, Mercuri M, Vuori I, Pasanen M, Oja P, Bond G, Koivula T, Lehti-maki T. Relationship between high-density lipoprotein paraoxonase gene M/L55 polymorphism and carotid atherosclerosis differs in smoking and nonsmoking men. Metabolism. 2001;50:1095-1101.[36] Cao H, Girard-Globa A, Serusclat A, Bernard S, Bondon P, Picard S, Berthezene F, Moulin P. Lack of association between carotid intima-media thick-ness and paraoxonase gene polymorphism in non-insulin dependent diabetes mellitus. Atherosclerosis. 1998;138:361-366.[37] Markus H, Kapozsta Z, Ditrich R, Wolfe C, Ali N, Powell J, Mendell M, Cullinane M. Increased com-mon carotid intima-media thickness in UK African Caribbeans and its relation to chronic inflammation and vascular candidate gene polymorphisms. Stroke. 2001;32:2465-2471.[38] Valabhji J, McColl AJ, Schachter M, Dhanjil S, Richmond W, Elkeles RS. High-density lipoprotein composition and paraoxonase activity in Type I diabe-tes. Clin Sci (Lond). 2001;101:659-670.[39] Mackness MI, Arrol S, Durrington PN. Paraoxo-nase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 1991;286:152-154.[40] Mackness MI, Arrol S, Abbott C, Durrington PN. Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis. 1993;104:129-135.[41] Watson AD, Berliner JA, Hama SY, La Du BN, Faull
KF, Fogelman AM, Navab M. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest. 1995;96:2882-2891.[42] Lusis AJ. Atherosclerosis. Nature. 2000;407:233-241.[43] Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115-126.[44] Holvoet P, Vanhaecke J, Janssens S, Van de WF, Collen D. Oxidized LDL and malondialdehyde-modi-fied LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98:1487-1494.[45] Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density li-poprotein cholesterol levels in the prediction of first cardiovascular events. New England Journal of Medi-cine. 2002;347:1557-1565.[46] Brophy VH, Jampsa RL, Clendenning JB, McKin-stry LA, Jarvik GP, Furlong CE. Effects of 5’ regulatory-region polymorphisms on paraoxonase-gene (PON1) expression. Am J Hum Genet. 2001;68:1428-1436.[47] Davies HG, Richter RJ, Keifer M, Broomfield CA, Sowalla J, Furlong CE. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat Genet. 1996;14:334-6.

5Paraoxonase-1 and linoleic acid oxidation in familial
hypercholesterolemia
Biochem. Biophys. Res. Commun. 2005 Aug 5;333(3):787-93
T.M. van HimbergenL.J. van Tits
M.P. HectorsJ. de Graaf
M. RoestA.F. Stalenhoef

Abstract
Serum paraoxonase-1 (PON1) is a high-density lipoprotein associated enzyme that can in-hibit low-density lipoprotein (LDL) oxidation in vitro. The role of PON1 in vivo, still re-mains to be clarified. We investigated the effect of PON1 genotype (-107C>T and 192Q>R ), concentration, paraoxonase activity and arylesterase activity on the early phase of lipid per-oxidation in plasma samples of 110 patients with heterozygous familial hypercholesterolemia (FH). The degree of lipid oxidation was assessed by quantitation of oxidized-linoleic acid (the most abundant fatty acid present in LDL) using high performance liquid chromatography. We found a significant inverse correlation between paraoxonase activity and the oxidized-linoleic acid concentration (r = -0.22, P = 0.03), independent of baseline linoleic acid levels. These findings support an anti-oxidative role for PON1 in patients with FH, and thus may give in-sight into the functioning of PON1 in vivo.

67
PON1 and linoleic acid oxidation
Introduction
Serum paraoxonase-1 (PON1) is a high-density lipoprotein (HDL)-associated enzyme which can hydrolyze a number of non-physiological substrates [1, 2]. There is a wide inter-indi-vidual variation in PON1 concentration and activity caused by a number of common genetic variants (polymorphisms) in the PON1 gene. A glutamine (Q)-to-arginine (R) transition at position 192 (192Q>R) greatly affects the PON1 hydrolytic activity towards non-physiologi-cal substrates like paraoxon (paraoxonase activity) and diazoxon (diazoxonase activity) [1-3]. Paraoxon is most efficiently hydrolyzed by the 192R isoform [1, 3], and diazoxon is more suc-cessfully hydrolyzed by the 192Q isoform [2]. In addition, the genetic variant –107C>T in the promoter region of the enzyme affects the levels of expression and thus the serum concentra-tion of the enzyme, with the highest PON1 serum concentrations found in individuals who are carrier of the –107CC genetic variant [4]. PON1 concentration can be measured in serum, either directly with an enzyme-linked immunosorbent assay (ELISA) [5], or indirectly by the rate of hydrolysis toward phenylacetate (arylesterase activity) [6]. In vivo, it is suggested that PON1 has a beneficial role in atherogenesis, which is supported by findings in PON1-deficient mice: these animals are more prone to develop atherosclerosis than wild-type mice when fed a high-fat/high-cholesterol diet [7]. Furthermore, low serum PON1 activity was shown to be an independent risk factor for coronary events in a large prospective cohort study [8]. The mechanism underlying these PON1 effects is, however, still unclear.Oxidative modification of LDL is a key event in the process of atherosclerosis [9], and sev-eral studies have suggested that PON1 can prevent the accumulation of oxidatively modified lipids on LDL, in vitro [10, 11]. Another line of evidence that PON1 plays a role in pro-tecting LDL from oxidation are findings of studies in PON1-knockout mice. These studies have demonstrated that PON1 deficiency results in increased oxidative stress in serum and macrophages [12]. Furthermore, HDL isolated from PON1-deficient mice does not protect LDL from oxidation [7], and HDL isolated from human PON1 transgenic mice (having 2- to 4-fold increased PON1 plasma levels) are more protective against LDL oxidation in a dose dependent manner [13]. To date, epidemiological evidence that PON1 protects LDL from oxidation in vivo is limited. In a population of healthy non-smoking men, the 192QQ genotype delayed ex-vivo LDL oxi-dation [14]. In contrast no effect of the 192Q>R polymorphism on ex-vivo LDL oxidation was found in a population of heavy smokers [15] and in a population of type 2 diabetes patients [16]. Studies on the effect of PON1 on the levels of circulating oxidized LDL (oxLDL) are not convincing either: in a population of 155 patients with type 2 diabetes, the PON1 –107C>T genotype (-108C>T, in the original report) contributed to the oxLDL/apoB ratio [17]. On the other hand, no effect for PON1 concentration and/or activity on circulating oxLDL was reported in populations of type 2 diabetes patients [18, 19], or in our population of patients

68
CHAPTER 5
with heterozygous familial hypercholesterolemia (FH) [20]. Interestingly, one study measur-ing oxidation products generated from arachidonic acid (isoprostanes) in a population of patients with type two diabetes showed a protective effect of PON1 against this form of oxida-tive stress [21].Altogether, these findings indicate that more insight in the in vivo action of PON1 can be obtained by direct determination of products of the early phase of lipid peroxidation, rather than measuring markers which 1). indirectly reflect oxidative stress (ex-vivo LDL oxidizabil-ity) or 2). are at a later stage of the LDL oxidation process (circulating oxLDL levels). In the present study we therefore use a high performance liquid chromatography (HPLC) method to quantify the amount of hydroxyl-linoleic acid present in the cholesterol-ester fraction of plasma. Hydroxyl-linoleic acid, (oxidized-linoleic acid or hydroxyl-octadecadienoic acid or HODE) is an oxidation product of linoleic acid, the major substrate for lipid peroxidation in LDL [22], and has been suggested to be an excellent marker for lipid peroxidation [23].We address the hypothesis that PON1 protein levels (ELISA and arylesterase activity), enzyme activity (paraoxonase activity) and genetic variation (–107C>T and 192Q>R) affect the degree of linoleic acid oxidation in blood. Because the effects of PON1 may be more pronounced in a population with an atherogenic lipid profile, the study was carried out in a population of patients with FH, who are characterized by markedly increased LDL cholesterol levels.
Methods
Subjects.We analyzed linoleic acid oxidation in 110 baseline samples of participants from the Atorv-astatin versus Simvastatin on Atherosclerosis Progression (ASAP) study population [24]. The selection was based on the availability of suitable EDTA-plasma samples for HPLC analysis that had 1.) prior to freezing at -80°C been supplemented with saccharose to prevent the structural and functional changes of lipoproteins which may occur due to freezing and thaw-ing and 2.) not been previously thawed before. Study participants were both men and women with heterozygous FH. Smoking status was defined as current-, past- or non-smoker. All pa-tients had plasma LDL cholesterol levels > 5.0 mmol/L and triglycerides levels < 4.5 mmol/L after an 8-week wash-out period of all lipid-lowering medication. Exclusion criteria were coronary heart disease within previous 3 months, active liver disease or hepatic dysfunction, hypertension, untreated or uncontrolled hyperthyroidism, hypothyroidism, diabetes or other endocrinological disease, secondary hyperlipidemia due to any other cause, impaired renal function (plasma creatinine >150 μmol/L) and alcohol or drug abuse, as described extensively previously [25]. Furthermore, patients had no significant biochemical abnormalities and did not use vitamin supplements. The ethical committee of the Radboud University Nijmegen Medical Centre approved the study protocol and all participants gave written informed con-

69
PON1 and linoleic acid oxidation
sent.
Analysis of PON1 polymorphismsThe PON1 polymorphism -107C>T was analyzed with polymerase chain reaction (PCR) fol-lowed by hybridization with allele-specific oligonucleotides as described by Leviev and James [26]. The PON1 common genetic variant 192Q>R was determined by means of PCR, fol-lowed by restriction fragment length polymorphism using primers and restriction enzymes as described by Humbert et al [1]. A PCR product for the determination of the 107C>T and 192Q>R polymorphisms, was obtained successfully for 109 and 108 DNA samples, respec-tively.
Analysis of PON1 protein levels and enzyme activitiesPON1 concentration and activity measurements were performed in serum samples. PON1 levels were measured with an ELISA [5], and were successfully determined in 107 samples. PON1 enzyme activities were determined by spectrophotometric measurement of the hydro-lysis of paraoxon (paraoxonase activity) and phenyl acetate (arylesterase activity), according to the methods of Richter et al. and Eckerson et al, respectively [6, 27].
Plasma lipids and lipoproteins Total cholesterol, triglycerides, LDL-cholesterol and HDL-cholesterol were analyzed using routine clinical chemistry procedures [25].
Preparation of samples for HPLC analysisTen μl of 1 mM desmosterol (Sigma, St. Louis, USA) in isopropanol and 100 μL of 4 mM cho-lesteryl-heptadecanoate (Sigma, St. Louis, USA) in iso-octane were added to 100 μl of plasma sample before extraction with ethanol/chloroform to remove proteins. After a wash with wa-ter, the lipid fraction was collected, dried under nitrogen and dissolved in 500 μL of hexane. The samples were then applied to a Sep-Pak Silica column (Waters, Milford, USA), which was pre-washed subsequently with methanol, 80% acetone, methanol and hexane. Cholesteryl esters were eluted from the column with 40 ml hexane, dried under nitrogen, and dissolved in 800 μl isopropanol. After elution of triglycerides from the Sep-Pak column with 7 ml of 5% ether in hexane, the cholesteryl-hydroxyl linoleic acid containing fraction was eluted from the column with 7 ml 100% ether, dried under nitrogen, and dissolved in 100 μL isopropanol for further HPLC analysis.
HPLC analysis HPLC was performed on a spectraSYSTEM (Thermo Separation Products, San Jose, USA) using a reversed phase pre-column (Chrompack, Varian, Palo Alto, USA) and two 10 cm chromspher 5C18 columns (Chrompack, Varian, Palo Alto, USA). The absorbance from 200

70
CHAPTER 5
nm to 300 nm was monitored with a diode-array detector (UV6000LP, Thermo Separation Products, San Jose, USA). Separation of the cholesteryl-fatty acids was carried out in a flow system using acetonitril/isopropanol (50:50, V:V) as solvent at steady flow rate of 0.6 mL/min-ute. Cholesteryl-heptadecanoate was used as internal recovery standard. The concentration of the lipid components was calculated from calibration curves of known amounts of choles-teryl fatty acids at 210 nm.Analysis of hydroxyl-linoleic acid was carried out using increasing flow rates (from 0.3 mL/minute to 0.4 mL/minute in 24 minutes, followed by 0.5 ml/minute for 1 minute) of a mixture of acetonitril/isopropanol/water (50:50:2.5, V:V:V). Desmosterol was used as internal recov-ery standard and detected at 206 nm. The concentration of the cholesteryl hydroxyl-linoleic acid was calculated from a calibration curve constructed of absorbtion values at 234 nm of known amounts of cholesteryl-hydroxyl linoleic acid standard (Cayman Chemical, Ann Ar-bor, USA).
Statistical analysisThe hydroxyl-linoleic acid measurements showed a skewed distribution. Therefore, corre-lations between all continuous variables were compared with Spearman’s rho and between groups comparisons were based on the Mann-Whitney test. All analyses were performed with SPSS version 11.5.
A B
Figure. A typical HPLC spectrum of the fatty acid cholesterol esters (Panel A) and linoleic acid oxidation products (Panel B). Plasma cholesteryl-linoleic acid can be detected spectrophotometrically at 210 nm (indicated by the arrow in panel A.) and the oxidatively modified hydroxyl-form can be detected at 234 nm (indicated by the arrow in panel B).

71
PON1 and linoleic acid oxidation
Results
The study population consisted of 30 men and 80 women with FH with an average age of 47 years (Table 1). HDL-C levels were normal but the average LDL-C levels were higher than 8 mmol/L, which is characteristic for FH. The genotype distributions of –107C>T and 192Q>R, were in Hardy-Weinberg equilibrium. Mean values for PON1 protein levels, arylesterase ac-tivities and paraoxonase activities are presented in Table 1, and were not associated with age, nor were there differences among male or female patients or among smoking subgroups (data not shown).In the cholesterol-ester fraction of plasma, fatty acids (cholesteryl-fatty acids) were detected spectrophotometrically at 210 nm and the oxidatively modified hydroxyl-form was detected at 234 nm (Figure). As presented in Table 2, linoleic acid is the most abundant fatty acid es-terified to cholesterol with an average plasma concentration of 5.0 mmol/L in the studied FH population. The cholesteryl-hydroxyl linoleic acid concentration of 5 FH patients could not be determined due to a distortion of the HPLC peak by an adjacent compound. There were no significant differences in plasma linoleic acid concentrations and cholesteryl-hydroxyl lin-oleic acid concentrations among male or female patients or among the smoking subgroups (data not shown).The correlations of plasma linoleic acid concentrations and cholesteryl-hydroxyl linoleic acid concentrations and the cholesteryl-hydroxyl linoleic acid to cholesteryl-linoleic acid ratio (percentage of linoleic acid hydroxylized) with plasma levels of LDL-C, HDL-C, PON1 pro-tein and PON1 enzyme activities, are shown in Table 3. The LDL particle is the main carrier of cholesterol esters in blood and, as expected, there is a strong correlation between LDL cho-
Table 1. Baseline characteristics of the patients with familial hypercholesterolemia (n=110)Age (years) 47 ± 10Gender (male/female) 30/80Smoking (current/past/non) 33/33/44Total cholesterol 10.1 ± 1.9Triglycerides 1.8 ± 1.0LDL-C (mmol/L) 8.2 ± 1.9HDL-C (mmol/L) 1.1 ± 0.3PON1 192Q>R (QQ/QR/RR) 42/54/12*PON1 –107C>T (CC/CT/TT) 33/49/27†PON1 levels (µg/ml) 77.8 ± 22.1‡PON1 arylesterase activity (U/mL) 72.0 ± 29.7PON1 paraoxonase activity (U/L) 617.6 (244.9-860.7)Values are presented as mean ± SD, with exception for gender (male/female), PON1 genotype –107C>T (CC/CT/TT) and 192Q>R (QQ/QR/RR). PON1 paraoxonase activity is presented as median value and interquartile range. PON1, paraoxonase-1; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol. Based on *108 observations, †109 observations and ‡107 observations.

72
CHAPTER 5
lesterol and cholesteryl-linoleic acid (r=0.66, P < 0.001). The amount of cholesteryl-hydroxyl linoleic acid (absolute and in proportion to cholesteryl-linoleic acid) did not correlate with LDL-C levels (r=0.10, P = 0.32 and r=-0.07, P=0.49, respectively). Furthermore, HDL-C lev-els, PON1 protein levels and PON1 arylesterase activities did not correlate with the amounts of cholesteryl-linoleic acid and cholesteryl-hydroxyl linoleic acid. There was a small, but sig-nificant inverse correlation between the PON1 hydrolytic activity to paraoxon (paraoxonase activity) and the amount of cholesteryl-hydroxyl linoleic acid (r = -0.22, P = 0.03, Table 3). These findings were independent of cholesteryl-linoleic acid concentrations, as there was no significant relationship between paraoxonase activity and cholesteryl-linoleic acid concentra-tion (r=0.03 and P = 0.80, Table 3), while, PON1 paraoxonase activity also correlated with the percentage of linoleic acid hydroxylized (r=-0.20, P = 0.04, Table 3). After it appeared that serum PON1 paraoxonase activity but not PON1 protein level and PON1 arylesterase activity affected cholesteryl-linoleic acid oxidation, we analyzed the as-sociation with PON1 genotypes. As presented in Table 4, the 192Q>R polymorphism was not associated with variation in plasma cholesteryl-linoleic acid, but RR homozygotes tended to have lower plasma levels of cholesteryl-hydroxyl linoleic acid (P = 0.09) and higher percent-age of linoleic acid hydroxylized in cholesterol ester fractions (P = 0.11), when compared to QQ homozygotes. The –107C>T polymorphism was not significantly related to variation in cholesteryl-linoleic acid or cholesteryl-hydroxyl linoleic acid levels.
Discussion
This is the first study in patients with FH that investigates the effects of PON1 on the amount of oxidized lipid components in plasma measured by HPLC. FH patients with an increased PON1 hydrolytic activity to paraoxon had significantly lower levels of oxidized cholesteryl-linoleic acid levels in their blood. It is well established that paraoxon is most efficient hydro-lyzed by the PON1-192RR homozygotes [1, 2], and, although not reaching statistical signifi-cance, in accordance with the paraoxonase activity measurements, a trend was observed for the association of the PON1-192RR variant with low levels of oxidized cholesteryl-linoleic
Table 2. Plasma concentrations of cholesterol esterified fatty acids in 110 patients with familial hypercholesterolemia (n=110)Cholesteryl-linoleic acid (mmol/L) 5.0 ± 1.0Cholesteryl-arachidonic acid (mmol/L) 0.6 ± 0.2Cholesteryl-oleic acid (mmol/L) 1.5 ± 0.4Cholesteryl-palmitic acid (mmol/L) 1.1 ± 0.2Cholesteryl-hydroxyl linoleic acid (μmol/L) 0.74 (0.44-1.19)*Values are presented as mean ± SD, except for cholesteryl-hydroxyl linoleic acid, which is represented as the median value and interquartile range. *n=105

73
PON1 and linoleic acid oxidation
Tabl
e 3.
Cor
rela
tion
of p
lasm
a lin
olei
c ac
id, h
ydro
xyl-l
inol
eic
acid
and
per
cent
age
of li
nole
ic a
cid
hydr
oxyl
ized
in c
hole
ster
yl
este
r fra
ctio
ns w
ith p
lasm
a le
vels
of H
DL-
C a
nd L
DL-
C a
nd P
ON
1 pr
otei
n le
vels
and
enzy
me
activ
ities
Lino
leic
aci
dH
ydro
xyl-l
inol
eic a
cid
Perc
enta
ge o
f lin
olei
c aci
d hy
drox
yliz
edVa
riabl
er
Pn
rP
nr
Pn
HD
L-C
0.
120.
2111
0-0
.08
0.41
105
-0.0
90.
3810
5LD
L-C
0.
66<0
.001
110
0.10
0.32
105
-0.0
70.
4910
5PO
N1
prot
ein
leve
ls -0
.06
0.56
107
-0.0
40.
6810
2-0
.01
0.91
102
PON
1 ar
yles
tera
se a
ctiv
ity-0
.10
0.30
110
-0.0
70.
4910
5-0
.06
0.55
105
PON
1 pa
raox
onas
e ac
tivity
0.03
0.80
110
-0.2
20.
0310
5-0
.20
0.04
105
Valu
es re
pres
ent t
he Sp
earm
an’s
rho
corr
elat
ion
coeffi
cien
t (r)
, fol
low
ed b
y the
P-v
alue
and
the n
umbe
r of o
bser
vatio
ns. L
inol
eic
acid
and
hyd
roxy
l-lin
olei
c ac
id a
re d
etec
ted
in th
e ch
oles
tero
l est
erifi
ed fo
rm. H
DL-
C, h
igh-
dens
ity li
popr
otei
n ch
oles
tero
l; LD
L-C
, low
-den
sity
lipop
rote
in ch
oles
tero
l; PO
N1,
par
aoxo
nase
-1.
Tabl
e 4.
Pla
sma
chol
este
ryl l
inol
eic
acid
, cho
lest
eryl
hyd
roxy
l-lin
olei
c ac
id a
nd p
erce
ntag
e of
lino
leic
aci
d hy
drox
yliz
ed in
cho
lest
erol
est
er fr
actio
ns
acco
rdin
g to
PO
N1
–107
C>T
and
192
Q>R
gen
otyp
esLi
nole
ic a
cid
(mm
ol/L
)n
PH
ydro
xyl-l
inol
eic
acid
(μm
ol/L
)n
PPe
rcen
tage
of l
inol
eic a
cid
hydr
oxyl
ized
(%)
nP
-107
C>T
C
C4.
81 (3
.95-
5.30
)33
0.66
(0.3
9-1.
14)
290.
015
(0.0
09-0
.026
)29
CT
5.05
(4.2
4-6.
01)
490.
440.
73 (0
.48-
1.25
)49
0.66
0.01
3 (0
.009
-0.0
25)
490.
98TT
4.94
(4.2
3-5.
60)
270.
84 (0
.43-
1.13
)26
0.01
6 (0
.009
-0.0
22)
2619
2Q>R
4.98
(4.3
9-5.
89)
420.
86 (0
.43-
1.28
)42
0.01
5 (0
.010
-0.0
24)
42Q
R 4.
70 (4
.03-
5.54
)54
0.37
0.71
(0.4
2-1.
21)
500.
090.
015
(0.0
08-0
.026
)50
0.11
RR5.
34 (4
.73-
6.06
)12
0.65
(0.3
9-0.
77)
110.
011
(0.0
08-0
.015
)11
Valu
es re
pres
ent t
he m
edia
n ob
serv
atio
n an
d in
terq
uart
ile r
ange
(in
pare
nthe
ses)
, fol
low
ed b
y th
e nu
mbe
r of
obs
erva
tions
and
a P
-val
ue b
ased
on
the
Man
n-W
hitn
ey te
st fo
r the
com
paris
on o
f the
hom
ozyg
ote w
ildty
pe (-
107C
C o
r 192
) with
the h
omoz
ygot
e mut
atio
n (-
107T
T or
192
RR).
Lino
leic
acid
an
d hy
drox
yl-li
nole
ic a
cid
are
dete
cted
in th
e ch
oles
tero
l-est
erifi
ed fo
rm.

74
CHAPTER 5
acid. These data are evidence for anti-oxidative properties of PON1 in vivo.PON1 is well known for its role in protecting LDL from oxidation [10, 11]. However, this evidence was obtained from in vitro experiments. Moreover, recently, there has been some controversy whether this anti-oxidative property is a function of PON1 or perhaps an other enzyme co-purified with PON1 [28-30]. In light of the recent debate, it must be noted that the PON1 specific activity may depend on the presence of additional components or co-factors present on the HDL-particle, and that the PON1 specific activity may decay during the isola-tion process as a result of the loss of these factors. Translation of the in vitro findings of PON1 to in vivo conditions, therefore, requires caution. In the present study, we directly quantified an oxidation product of linoleic acid in choles-terol-ester fractions of plasma samples, and showed that this product was influenced by para-oxonase activity measured in blood. These findings support the assumption that PON1 hy-drolytic activity to paraoxon and not the esterase activity (arylesterase activity) and protein levels, is representative for the PON1 anti-oxidative actions, and are therefore of importance in resolving the physiological role of PON1.Linoleic acid is the most abundant fatty acid present in plasma [31], and oxidation products of linoleic acid have been demonstrated to be highly stable in biological samples, making them suitable markers for studying lipid peroxidation [23]. Products of oxidized linoleic acid are biologically relevant for the process of atherogenesis, since they have been demonstrated to be endogenous activators of the peroxisome proliferator activated receptor γ (PPARγ) [32]. PPARγ is a nuclear receptor which stimulates the expression of ABCA1 [33, 34], a transporter that controls apoA1 mediated cholesterol efflux from macrophages, and thus plays a key role in the reverse cholesterol transport pathway and atherogenesis [35]. In agreement with this, hydroxyl-linoleic acid concentrations correlate with the severity of the atherosclerotic lesion [36], and its levels in LDL are elevated in atherosclerotic patients when compared to healthy controls [37]. Our data obtained from the measurement of cholesteryl-hydroxyl linoleic acid gives a direct representation of the oxidative status of LDL in vivo, which is highly favorable over assessment of LDL oxidizability ex vivo. The latter, as well as measurement of levels of circulating oxLDL, seem to be too crude to detect subtle differences in lipid peroxidation induced by PON1. Though the sample preparation and the analysis of the fatty acid oxidation products using the HPLC method is a laborious and time-consuming procedure that will not be easily applied in large clinical studies, it can be of great value in investigating the physi-ological role of PON1.Finally, as demonstrated in the present study, linoleic acid is present in the milli-molar range while oxidized linoleic acid is present in the micro-molar range. This indicates that less that 0.1 percent of the linoleic acid is oxidized. Whether the up to 30% differences in oxidized linoleic acid levels that we observed between different PON1 genotypes are biological signifi-cant, and, for instance, affect oxLDL particle composition and uptake by macrophages, or ac-tivation of the PPARγ system, is not clear and should be elucidated in future investigations.

75
PON1 and linoleic acid oxidation
In conclusion, we demonstrate that PON1 activity has antioxidative effects on lipid oxidation in vivo and via this mechanism may play a role in atherosclerosis and the onset of cardiovas-cular disease.
Acknowledgments
The authors greatly acknowledge Dr. Hieronymus A.M. Voorbij for the useful discussions, Dr. Hiroaki Hattori for providing the PON1 concentration measurements and Dr. Eugène H.J.M. Jansen for providing the PON1 paraoxonase activity measurements.

76
CHAPTER 5
References
[1] Humbert, R., Adler, D.A., Disteche, C.M., Hassett, C., Omiecinski, C.J., and Furlong, C.E. The molecular basis of the human serum paraoxonase activity poly-morphism. Nat.Genet. 3 (1993) 73-76.[2] Davies, H.G., Richter, R.J., Keifer, M., Broomfield, C.A., Sowalla, J., and Furlong, C.E. The effect of the hu-man serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat Genet. 14 (1996) 334-336.[3] Adkins, S., Gan, K.N., Mody, M., and La Du, B.N. Molecular basis for the polymorphic forms of hu-man serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B al-lozymes. Am J Hum Genet. 52 (1993) 598-608.[4] Brophy, V.H., Jampsa, R.L., Clendenning, J.B., McK-instry, L.A., Jarvik, G.P., and Furlong, C.E. Effects of 5’ regulatory-region polymorphisms on paraoxonase-gene (PON1) expression. Am J Hum Genet. 68 (2001) 1428-1436.[5] Kujiraoka, T., Oka, T., Ishihara, M., Egashira, T., Fu-jioka, T., Saito, E., Saito, S., Miller, N.E., and Hattori, H. A sandwich enzyme-linked immunosorbent assay for human serum paraoxonase concentration. J Lipid Res. 41 (2000) 1358-1363.[6] Eckerson, H.W., Wyte, C.M., and La Du, B.N. The human serum paraoxonase/arylesterase polymor-phism. Am J Hum Genet. 35 (1983) 1126-1138.[7] Shih, D.M., Gu, L., Xia, Y.R., Navab, M., Li, W.F., Hama, S., Castellani, L.W., Furlong, C.E., Costa, L.G., Fogelman, A.M., and Lusis, A.J. Mice lacking serum paraoxonase are susceptible to organophosphate tox-icity and atherosclerosis. Nature. 394 (1998) 284-287.[8] Mackness, B., Durrington, P., McElduff, P., Yarnell, J., Azam, N., Watt, M., and Mackness, M. Low paraoxo-nase activity predicts coronary events in the Caerphilly Prospective Study. Circulation. 107 (2003) 2775-2779.[9] Steinberg, D., Parthasarathy, S., Carew, T.E., Khoo,
J.C., and Witztum, J.L. Beyond cholesterol. Modifica-tions of low-density lipoprotein that increase its ather-ogenicity. N Engl J Med. 320 (1989) 915-924.[10] Mackness, M.I., Arrol, S., and Durrington, P.N. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 286 (1991) 152-154.[11] Watson, A.D., Berliner, J.A., Hama, S.Y., La Du, B.N., Faull, K.F., Fogelman, A.M., and Navab, M. Pro-tective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin In-vest. 96 (1995) 2882-2891.[12] Rozenberg, O., Rosenblat, M., Coleman, R., Shih, D.M., and Aviram, M. Paraoxonase (PON1) deficiency is associated with increased macrophage oxidative stress: studies in PON1-knockout mice. Free Radic Biol Med. 34 (2003) 774-784.[13] Tward, A., Xia, Y.R., Wang, X.P., Shi, Y.S., Park, C., Castellani, L.W., Lusis, A.J., and Shih, D.M. Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation. 106 (2002) 484-490.[14] Bub, A., Barth, S.W., Watzl, B., Briviba, K., and Rechkemmer, G. Paraoxonase 1 Q192R (PON1-192) polymorphism is associated with reduced lipid peroxi-dation in healthy young men on a low-carotenoid diet supplemented with tomato juice. Br J Nutr. 93 (2005) 291-297.[15] Van Himbergen, T., Roest, M., De Waart, F., De Graaf, J., Voorbij, H., Van Tits, L., and Stalenhoef, A. Paraoxonase genotype, LDL-oxidation and carotid atherosclerosis in male life-long smokers. Free Radic Res. 38 (2004) 553-560.[16] Sampson, M.J., Astley, S., Richardson, T., Willis, G., Davies, I.R., Hughes, D.A., and Southon, S. Increased DNA oxidative susceptibility without increased plas-ma LDL oxidizability in Type II diabetes: effects of al-pha-tocopherol supplementation. Clin Sci (Lond). 101

77
PON1 and linoleic acid oxidation
(2001) 235-241.[17] Tsuzura, S., Ikeda, Y., Suehiro, T., Ota, K., Osaki, F., Arii, K., Kumon, Y., and Hashimoto, K. Correlation of plasma oxidized low-density lipoprotein levels to vas-cular complications and human serum paraoxonase in patients with type 2 diabetes. Metabolism. 53 (2004) 297-302.[18] Kopprasch, S., Pietzsch, J., Kuhlisch, E., and Graessler, J. Lack of association between serum paraox-onase 1 activities and increased oxidized low-density lipoprotein levels in impaired glucose tolerance and newly diagnosed diabetes mellitus. J Clin Endocrinol Metab. 88 (2003) 1711-1716.[19] Sampson, M., Braschi, S., Willis, G., and Astley, S. Paraoxonase -1 (PON1) genotype and activity and in vivo oxidised plasma low density lipoprotein [oxLDL] in Type 2 diabetes. Clin Sci (Lond).(2005).[20] van Himbergen, T.M., Roest, M., de Graaf, J., Jansen, E.H., Hattori, H., Kastelein, J.J., Voorbij, H.A., Stalenhoef, A.F., and van Tits, L.J. Indications that paraoxonase-1 contributes to plasma high density lipoprotein levels in familial hypercholesterolemia. J Lipid Res. 46 (2005) 445-451.[21] Malin, R., Laine, S., Rantalaiho, V., Wirta, O., Pas-ternack, A., Jokela, H., Alho, H., Koivula, T., and Lehti-maki, T. Lipid peroxidation is increased in paraoxonase L55 homozygotes compared with M-allele carriers. Free Radic Res. 34 (2001) 477-484.[22] Spiteller, G. Linoleic acid peroxidation--the domi-nant lipid peroxidation process in low density lipopro-tein--and its relationship to chronic diseases. Chemis-try and Physics of Lipids. 95 (1998) 105-162.[23] Spiteller, P., and Spiteller, G. 9-Hydroxy-10,12-oc-tadecadienoic acid (9-HODE) and 13-hydroxy-9,11-octadecadienoic acid (13-HODE): excellent markers for lipid peroxidation. Chemistry and Physics of Lip-ids. 89 (1997) 131-139.[24] Smilde, T.J., van Wissen, S., Wollersheim, H., Trip, M.D., Kastelein, J.J., and Stalenhoef, A.F. Effect of ag-
gressive versus conventional lipid lowering on athero-sclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet. 357 (2001) 577-581.[25] Smilde TJ, T.M., Wollersheim H, Van Wissen S, Kastelein JJP, Stalenhoef AFH. Rationale, design and baseline characteristics of a clinical trial comparing the effects of robust vs conventional cholesterol low-ering and intima media thickness in patients with fa-milial hypercholesterolaemia: The atorvastatin versus simvastatin on atherosclerosis progression (ASAP) study. Clinical Drug Investigation. 20 (2000) 67-79.[26] Leviev, I., and James, R.W. Promoter polymor-phisms of human paraoxonase PON1 gene and serum paraoxonase activities and concentrations. Arterio-scler Thromb Vasc Biol. 20 (2000) 516-521.[27] Richter, R.J., and Furlong, C.E. Determination of paraoxonase (PON1) status requires more than geno-typing. Pharmacogenetics. 9 (1999) 745-753.[28] Connelly, P.W., Draganov, D., and Maguire, G.F. Paraoxonase-1 does not reduce or modify oxidation of phospholipids by peroxynitrite. Free Radic Biol Med. 38 (2005) 164-174.[29] Teiber, J.F., Draganov, D.I., and La Du, B.N. Puri-fied human serum PON1 does not protect LDL against oxidation in the in vitro assays initiated with copper or AAPH. J Lipid Res. 45 (2004) 2260-2268.[30] Marathe, G.K., Zimmerman, G.A., and McIntyre, T.M. Platelet-activating factor acetylhydrolase, and not paraoxonase-1, is the oxidized phospholipid hydrolase of high density lipoprotein particles. J Biol Chem. 278 (2003) 3937-3947.[31] Esterbauer, H., Gebicki, J., Puhl, H., and Jurgens, G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 13 (1992) 341-390.[32] Nagy, L., Tontonoz, P., Alvarez, J.G., Chen, H., and Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma.

78
CHAPTER 5
Cell. 93 (1998) 229-240.[33] Chinetti, G., Lestavel, S., Bocher, V., Remaley, A.T., Neve, B., Torra, I.P., Teissier, E., Minnich, A., Jaye, M., Duverger, N., Brewer, H.B., Fruchart, J.C., Clavey, V., and Staels, B. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 7 (2001) 53-58.[34] Chawla, A., Boisvert, W.A., Lee, C.H., Laffitte, B.A., Barak, Y., Joseph, S.B., Liao, D., Nagy, L., Edwards, P.A., Curtiss, L.K., Evans, R.M., and Tontonoz, P. A PPAR gamma-LXR-ABCA1 pathway in macrophages is in-volved in cholesterol efflux and atherogenesis. Mol Cell. 7 (2001) 161-171.[35] van Dam, M.J., de Groot, E., Clee, S.M., Hovingh, G.K., Roelants, R., Brooks-Wilson, A., Zwinderman, A.H., Smit, A.J., Smelt, A.H., Groen, A.K., Hayden, M.R., and Kastelein, J.J. Association between increased arte-rial-wall thickness and impairment in ABCA1-driven cholesterol efflux: an observational study. Lancet. 359 (2002) 37-42.[36] Harland, W.A., Gilbert, J.D., Steel, G., and Brooks, C.J. Lipids of human atheroma. 5. The occurrence of a new group of polar sterol esters in various stages of human atherosclerosis. Atherosclerosis. 13 (1971) 239-246.[37] Jira, W., Spiteller, G., Carson, W., and Schramm, A. Strong increase in hydroxy fatty acids derived from linoleic acid in human low density lipoproteins of atherosclerotic patients. Chem Phys Lipids. 91 (1998) 1-11.

6The effect of statin therapy on plasma high-
density lipoprotein cholesterol levels is modified by paraoxonase-1 in patients with familial
hypercholesterolaemia
J. Intern. Med. 2005 Nov;258(5):442-9
T.M. van HimbergenL.J. van Tits
H.A. VoorbijJ. de Graaf
A.F. StalenhoefM. Roest

Abstract
Statins reduce low-density lipoprotein cholesterol and can raise high-density lipoprotein cho-lesterol (HDL-C). HDL-bound paraoxonase-1 (PON1) associates with variations in plasma HDL-C, and may, therefore, contribute to changes of HDL-C during statin therapy. We inves-tigate the effects of PON1 to HDL-C changes in familial hypercholesterolemia (FH) patients undergoing statin therapy.134 FH patients were treated with either atorvastatin or simvastatin therapy for two years. PON1 status was determined at baseline with (1) PON1 -107C>T and 192Q>R genotype, (2) PON1 levels and (3) PON1 diazoxonase and arylesterase activity. PON1 levels and activities significantly modified the HDL-C increment (P = 0.002 for PON1 levels and arylesterase ac-tivity and P = 0.001 for diazoxonase activity). The effects were even more evident among sub-group classifications based on PON1 status and baseline HDL-C concentrations: the HDL-C increment was more pronounced in subgroups of -107CT/TT or 192QR/RR genotype com-bined with low baseline HDL-C (+13.9 percent, P < 0.001, respectively +15.4 percent, P < 0.001). In contrast, the -107CC or 192QQ genotype in combination with high baseline HDL-C, did not show a significant increase of HDL-C. PON1 status in conjunction with baseline HDL-C levels predicts HDL-C increment during statin therapy in FH patients.

81
PON1, statins and HDL-C changes
Introduction
HMG-CoA reductase inhibitors (statins) successfully reduce cardiovascular disease (CVD) risk [1]. The primary target of statin therapy is lowering low-density lipoprotein cholesterol (LDL-C), but statins can also raise high-density lipoprotein cholesterol (HDL-C) levels up to 11 percent [2]. HDL-C has anti-atherogenic properties via the removal of cholesterol from cells in the artery wall and initiation of the reverse cholesterol transport (reviewed in [3]), but various other protective properties of HDL-C have been proposed, including anti-oxidative and anti-inflammatory functions (reviewed in [4]). In addition, high HDL-C levels are also associated with a low risk of CVD [5]. The HDL-C raising properties of statin therapy may therefore be an important additional therapeutic benefit.Little is known about the physiological mechanism by which statins raise HDL-C levels. It has been proposed that statins stimulate the synthesis of apoA1 and, in this way, enhance the formation of HDL-C [6-8]. A potential effect modifier of statins therapy is the HDL-associ-ated enzyme Paraoxonase-1 (PON1) [9]. PON1 has antioxidative properties [10], which can protect the HDL particle from oxidation and thus preserve its function [11]. It also can influ-ence variations in HDL-C plasma concentrations [12-17]. This involvement of PON1 in the HDL metabolism pathway may modify the efficacy of statins to elevate HDL-C.The inter-individual variation of PON1 levels is mainly caused by a common genetic variant (polymorphism) at position –107, a cysteine (C) to threonine (T) transition (-107C>T), in the promoter region of the enzyme [18]. The –107C isoform is associated with the highest concentrations of the enzyme [19]. PON1 levels can be measured in serum, either directly with an enzyme-linked immunosorbent assay (ELISA) [20], or indirectly by the rate of hydro-lysis towards phenylacetate (arylesterase activity) [21]. Besides a variation in bioavailability of PON1, there is also a strong inter-individual variation in the catalytic activity towards a number of non-physiological substrates. This variation is determined by a glutamine (Q)- to- arginine (R) transition at position 192 (192Q>R) in the coding region of the PON1 gene. There is a 100 percent correlation between the 192Q>R genotype and the ratio of the hydro-lysis rate of paraoxon and diazoxon. The substrate paraoxon is most efficiently hydrolysed by the 192R isoform [22, 23], whereas diazoxon is more successfully hydrolysed by the 192Q isoform [24].In the present study, we address the hypothesis that PON1 modifies the effect of statins on HDL-C increment. We investigated the contribution of PON1 common genetic variants, PON1 levels and PON1 activity to changes of HDL-C, in a population with familial hyper-cholesterolemia (FH) undergoing two years of statin therapy. Because high serum levels of PON1 are associated with high concentrations of HDL-C[17], we also studied the effect of PON1 genotype, levels and activity on HDL-C changes in subgroups of low and high HDL-C levels at baseline.

82
CHAPTER 6
Methods
Subjects.We selected a subgroup of 134 participants from the Atorvastatin versus Simvastatin on Ath-erosclerosis Progression (ASAP) study population [25]. This selection was based on the avail-ability of serum samples to measure PON1 levels and activity. The ASAP study is a two-centre trial, where additional serum samples were only available from the Nijmegen centre. The design and main results of the ASAP study have been reported previously [25]. In short, the entire study population consisted of 325 men and women with heterozygous FH who par-ticipated in a prospective, randomised, double-blind, trial on the effects of aggressive lipid lowering with atorvastatin (80 mg/day) versus conventional lipid lowering with simvastatin (40 mg/day) on the progression of atherosclerosis. Patients were previously untreated, treated with statins only the previous year, or treated with statins longer than the previous year but with LDL cholesterol remaining above the 4.5 mmol/L. After an 8-week placebo run-in (in which all lipid-lowering drugs were discontinued), baseline height, weight and blood pressure were measured. In addition, smoking status and the use of alcohol and concomitant medica-tion were recorded. Plasma and (in one centre) serum samples were taken and stored at - 80 °C until analysis. During two years of follow up, lipid and lipoprotein levels were determined at 4 weeks, 6 weeks and every 12 weeks thereafter. The ethical committee of the Radboud University Nijmegen Medical Centre approved the study protocol and all participants gave written informed consent.
Analysis of –107C>T and 192Q>RThe polymorphism -107C>T was analysed with polymerase chain reaction (PCR) followed by hybridisation with allele-specific oligonucleotides as described by Leviev and James [19]. The common genetic variant 192Q>R was determined by means of PCR, followed by restriction fragment length polymorphism using primers and restriction enzymes as described by Hum-bert et al [23]. A PCR product was obtained successfully for 133 and 132 DNA samples to determine the genetic make-up for the –107C>T and 192Q>R polymorphisms, respectively.
Analysis of PON1Serum PON1 levels were measured with an ELISA [20], and were successfully determined in 131 samples. PON1 enzyme activities were measured by monitoring the hydrolysis rates of paraoxon (paraoxonase activity), diazoxon (diazoxonase activity) and phenylacetate (aryles-terase activity) on a spectrophotometer, according to the methods previously described [21, 26].
Lipids and lipoproteins Total cholesterol, LDL-C, HDL-C, and triglycerides were analysed using routine clinical

83
PON1, statins and HDL-C changes
chemistry procedures [27].
Statistical analysisThe average HDL-C changes during two years of statin therapy were calculated as the mean HDL-C concentration of eleven consecutive visits during these two years of therapy minus the HDL-C concentration at baseline.The association of baseline lipoproteins, lipids and PON1 levels and activities with the aver-age HDL-C changes during statin therapy was calculated with the Pearson correlation coef-ficient.Multiple linear regression analysis was used to analyse the relation between PON1 genotypes, levels and activities and HDL-C changes. Age (in years), gender (male or female), HDL-C levels at baseline (in mmol/L) and alternately the PON1 -107C>T polymorphism (CC versus CT/TT), the PON1 192Q>R polymorphism (QQ versus QR/RR), PON1 levels (in μg/mL), paraoxonase activity (U/L), diazoxonase activity (in U/L) and arylesterase activity (in U/mL) were entered into the model as explanatory variables. The average HDL-C change during two years of statin therapy (in mmol/L) was used as outcome variable.Baseline PON1 levels, diazoxonase activity, arylesterase activity and HDL-C were classified in two equal groups lower and higher than the median value. Cut-off values were: 76.6 μg/mL for PON1 levels, 6958 U/L for diazoxonase activity, 68.8 U/mL for arylesterase activity and 1.1 mmol/L for HDL-C. P-values and 95% confidence intervals for average HDL-C changes among subgroups were calculated with the One-Sample T-test. Data were checked for normal distributions and outliers. A P value ≤ 0.05 was considered statistical significant. All statistical analyses were performed with SPSS version 11.5.
Results
The study population consisted of 49 men and 85 women with FH, undergoing two years of statin therapy with either atorvastatin (69 patients) or simvastatin (65 patients), Table 1. In both treatment groups there was a significant increase in HDL-C (+7.3 percent, P for change < 0.001 for atorvastatin and +9.7 percent, P for change < 0.001 for simvastatin) during these two years. Because there was no significant difference in this response between both treat-ment groups (2.4 percent difference, P for difference = 0.30), we combined these groups for further analysis. The mean age of the population was 48 years. Levels of triglycerides and HDL-C were normal at baseline. The average LDL-C level was higher than 8 mmol/L, which is characteristic for FH. The genotype distributions of –107C>T and 192Q>R, were in Hardy-Weinberg equilibrium. Baseline values for PON1 levels, diazoxonase- and arylesterase activ-ity are presented in Table 1.The average increase in HDL-C after 2 years of statin therapy in the combined treatment

84
CHAPTER 6
groups was +8.5 percent or +0.08 mmol/L (P for change < 0.001). There was an inverse cor-relation of baseline HDL-C levels, PON1 levels, diazoxonase activity and arylesterase activity with average HDL-C changes during statin therapy (r=-0.201, P=0.02; r=-0.341, P<0.001; r=-0.336, P<0.001 and r=-0.309, P<0.001, respectively), and, apparently, there was no association of paraoxonase activity on HDL-C changes (r=-0.022, P=0.804)(Table 2). Next, we analysed the contribution of PON1 genotypes –107C>T and 192Q>R, PON1 levels, diazoxonase activ-ity and arylesterase activity to HDL-C increment during statin therapy (Table 3), including age, gender and HDL-C levels at baseline in the statistical model as possible effect modifiers. The genetic variants –107C>T and 192Q>R tended to associate with average HDL-C changes (P = 0.07 and P = 0.08 respectively). The associations were stronger for PON1 levels, diazox-onase activity and arylesterase activity. These factors were significantly, and independent of age, gender and HDL levels at baseline, associated with variations in HDL-C increment (P = 0.002 for both PON1 levels and arylesterase activity, and P = 0.001 for diazoxonase activity). Adjustment for smoking status, concomitant medication and baseline levels of LDL-C and triglycerides did not change the association between PON1 genotypes, levels and activities and the HDL-C increment during statin therapy (data not shown). The effect of PON1 genotype, levels and activity on HDL-C increment in subgroups of low baseline HDL-C levels (≤ 1.1 mmol/L) and high baseline HDL-C levels (> 1.1 mmol/L) is presented in Table 4. We observed clear differences in HDL-C increment among subgroups of PON1 –107C>T, 192Q>R, PON1 levels, diazoxonase activity and arylesterase activity in combination with the HDL-C subgroups. In patients with PON1 genotypes characteristic for high PON1 levels (-107CC) and diazoxonase activity (192QQ), there was no significant
Table 1. Baseline characteristics (age, gender, type of statin therapy, lipoproteins, serum lipids, PON1 common genetic variants –107C>T and 192Q>R, PON1 levels and PON1 activities) for 134 patients with familial hypercholesterolemiaAge (years) 48 ± 10Gender (male/female) 49/85Statin therapy (atorvastatin/simvastatin) 69/65Total cholesterol (mmol/L) 10.3 ± 2.0HDL-C (mmol/L) 1.1 ± 0.3LDL-C (mmol/L) 8.4 ± 1.9Triglycerides (mmol/L) 1.8 ± 1.0PON1 –107C>T (CC/CT/TT) (38/63/32)PON1 192Q>R (QQ/QR/RR) (52/66/14)PON1 levels (µg/ml) 77.6 ± 22.1Paraoxonase activity (U/L) 603 ± 359Diazoxonase activity (U/L) 7123 ± 1878Arylesterase activity (U/mL) 71.3 ± 29.5Values are presented as means ± SD, with exception for gender, statin therapy and PON1 common genetic variants –107C>T and 192Q>R. Statin therapy was over a period of two years. PON1 denotes paraoxonase-1; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol.

85
PON1, statins and HDL-C changes
increase of HDL-C in combination with the high baseline HDL-C subgroup (+0.5 percent, P = 0.564 for -107CC and +1.4 percent, P = 0.578 for 192QQ), whereas the HDL-C levels increased significantly in every other subgroup combination. The greatest change was ob-served in patients with PON1 genotypes characteristic for low PON1 levels (-107CT/TT) and diazoxonase activity (192QR/RR) combined with the low baseline HDL-C subgroup (+13.9 percent, P < 0.001 for -107CT/TT, respectively +15.4 percent, P < 0.001 192QR/RR). In line with the findings for the high PON1 levels genetic variant (-107CC), there was no sig-nificant HDL-C increase observed for the high PON1 levels subgroup (above 76.6 μg/mL) or high arylesterase subgroup (above 68.8 U/mL) in combination with the high baseline HDL-C subgroup. Moreover, the strongest increase was observed in subgroups for low PON1 levels or arylesterase activity in combination with the low baseline HDL-C subgroup (for PON1 levels, +14.0 percent, P <0.001 and for arylesterase activity, +14.9 percent, P < 0.001). There was also agreement for the 192Q>R polymorphism with the diazoxonase activity measurements. In concurrence with the 192QQ (high diazoxonase activity) genetic variant, no significant increase was observed for the subgroup combination of high diazoxonase activity (above 6958 U/L) and high baseline HDL-C, while the greatest increase in HDL-C was observed for the low diazoxonase activity and low baseline HDL-C subgroup combination (16.4 percent, P <0.001). Thus, PON1 levels and activity are in agreement with the genetic variants –107C>T and 192Q>R, and show the same effect on HDL-C increment.
Discussion
Our data show that PON1 plays an important role in the HDL-C response to statin therapy in patients with FH. Although baseline HDL-C was associated with HDL-C changes during therapy, surprisingly, PON1 levels, diazoxonase activity and arylesterase activity were stron-ger predictors of this effect. Thus, patients with a low baseline PON1 status (characterized by
Table 2. Pearson correlation coefficients of average HDL-C changes during statin therapy with baseline lipoproteins, lipids and PON1 levels and activities.
Average HDL-C change during therapy
P value
Total cholesterol 0.078 0.370HDL-C -0.201 0.020LDL-C 0.110 0.204Triglycerides -0.019 0.827PON1 levels -0.341 <0.001Paraoxonase activity -0.022 0.804Diazoxonase activity -0.336 <0.001Arylesterase activity -0.309 <0.001HDL-C denotes high-density lipoprotein cholesterol; PON1, paraoxonase-1 LDL-C, low-density lipoprotein cholesterol.
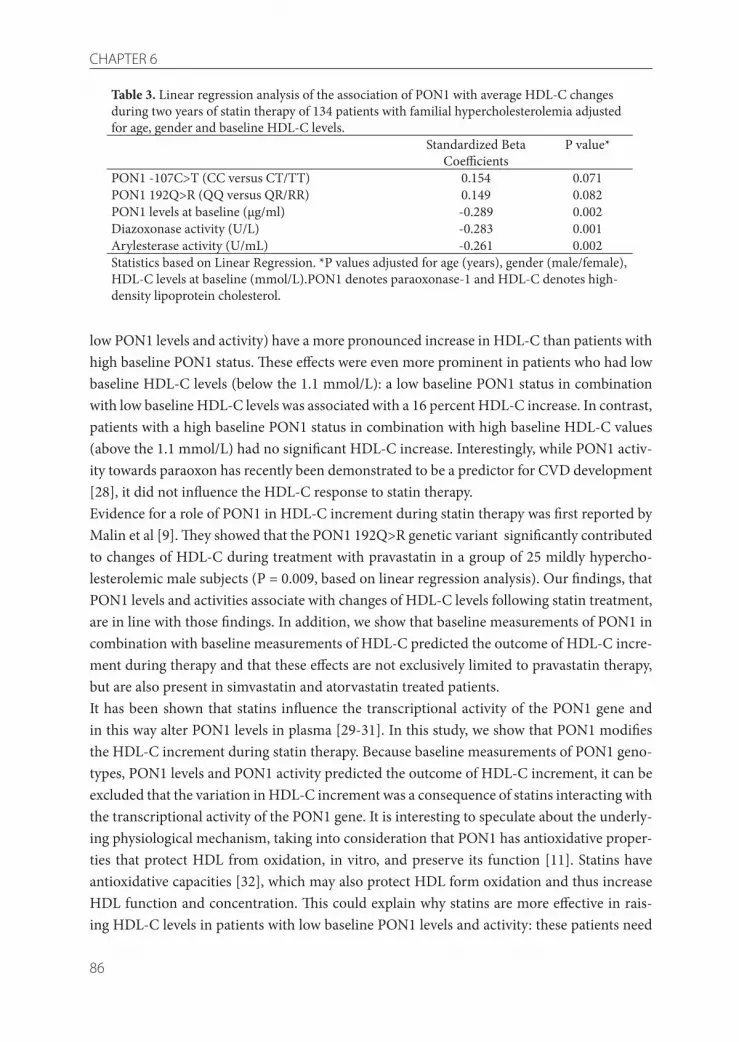
86
CHAPTER 6
low PON1 levels and activity) have a more pronounced increase in HDL-C than patients with high baseline PON1 status. These effects were even more prominent in patients who had low baseline HDL-C levels (below the 1.1 mmol/L): a low baseline PON1 status in combination with low baseline HDL-C levels was associated with a 16 percent HDL-C increase. In contrast, patients with a high baseline PON1 status in combination with high baseline HDL-C values (above the 1.1 mmol/L) had no significant HDL-C increase. Interestingly, while PON1 activ-ity towards paraoxon has recently been demonstrated to be a predictor for CVD development [28], it did not influence the HDL-C response to statin therapy. Evidence for a role of PON1 in HDL-C increment during statin therapy was first reported by Malin et al [9]. They showed that the PON1 192Q>R genetic variant significantly contributed to changes of HDL-C during treatment with pravastatin in a group of 25 mildly hypercho-lesterolemic male subjects (P = 0.009, based on linear regression analysis). Our findings, that PON1 levels and activities associate with changes of HDL-C levels following statin treatment, are in line with those findings. In addition, we show that baseline measurements of PON1 in combination with baseline measurements of HDL-C predicted the outcome of HDL-C incre-ment during therapy and that these effects are not exclusively limited to pravastatin therapy, but are also present in simvastatin and atorvastatin treated patients. It has been shown that statins influence the transcriptional activity of the PON1 gene and in this way alter PON1 levels in plasma [29-31]. In this study, we show that PON1 modifies the HDL-C increment during statin therapy. Because baseline measurements of PON1 geno-types, PON1 levels and PON1 activity predicted the outcome of HDL-C increment, it can be excluded that the variation in HDL-C increment was a consequence of statins interacting with the transcriptional activity of the PON1 gene. It is interesting to speculate about the underly-ing physiological mechanism, taking into consideration that PON1 has antioxidative proper-ties that protect HDL from oxidation, in vitro, and preserve its function [11]. Statins have antioxidative capacities [32], which may also protect HDL form oxidation and thus increase HDL function and concentration. This could explain why statins are more effective in rais-ing HDL-C levels in patients with low baseline PON1 levels and activity: these patients need
Table 3. Linear regression analysis of the association of PON1 with average HDL-C changes during two years of statin therapy of 134 patients with familial hypercholesterolemia adjusted for age, gender and baseline HDL-C levels.
Standardized Beta Coefficients
P value*
PON1 -107C>T (CC versus CT/TT) 0.154 0.071PON1 192Q>R (QQ versus QR/RR) 0.149 0.082PON1 levels at baseline (µg/ml) -0.289 0.002Diazoxonase activity (U/L) -0.283 0.001Arylesterase activity (U/mL) -0.261 0.002Statistics based on Linear Regression. *P values adjusted for age (years), gender (male/female), HDL-C levels at baseline (mmol/L).PON1 denotes paraoxonase-1 and HDL-C denotes high-density lipoprotein cholesterol.

87
PON1, statins and HDL-C changes
Tabl
e 4.
Ave
rage
chan
ge in
HD
L-C
am
ong
subg
roup
s of H
DL-
C le
vels,
PO
N1
com
mon
gen
etic
var
iant
–10
7C>T
and
192
Q>R
, PO
N1
leve
ls, d
iazo
xona
se
activ
ity a
nd a
ryle
ster
ase
activ
ity d
urin
g tw
o ye
ars o
f sta
tin th
erap
y in
134
pat
ient
s with
FH
HD
L-C
leve
ls ≤
1.1
mm
ol/L
HD
L-C
leve
ls >
1.1
mm
ol/L
Base
line
HD
L-C
leve
ls in
mm
ol/L
HD
L-C
ch
ange
in
mm
ol/L
(%)
95%
CI f
or
HD
L-C
chan
ge in
m
mol
/L
P va
lue
for
chan
geN
Base
line
HD
L-C
leve
ls in
mm
ol/L
HD
L-C
ch
ange
in
mm
ol/L
(%)
95%
CI f
or
HD
L-C
chan
ge
in m
mol
/L
P va
lue
for
chan
geN
A. –
107C
>T-1
07 C
T/TT
0.89
0.11
(13.
9)(0
.08
to 0
.14)
<0.0
0152
1.38
0.07
(5.6
)(0
.02
to 0
.13)
0.00
843
-107
CC
0.86
0.09
(10.
6)(0
.08
to 0
.14)
0.00
117
1.43
0.01
(0.5
)(-
0.03
to 0
.05)
0.56
421
B. 1
92Q
>R19
2QR/
RR0.
870.
12 (1
5.4)
(0.0
9 to
0.1
6)<0
.001
381.
430.
07 (4
.9)
(0.0
2 to
0.1
2)0.
010
4219
2QQ
0.90
0.09
(10.
1)(0
.04
to 0
.13)
<0.0
0129
1.34
0.01
(1.4
)(-
0.04
to 0
.07)
0.57
823
C. P
ON
1 le
vels
≤ 76
.6 µ
g/m
l0.
870.
12 (1
4.0)
(0.0
8 to
0.1
6)<0
.001
411.
310.
11 (8
.7)
(0.0
4 to
0.1
9)0.
004
25>
76.6
µg/
ml
0.89
0.09
(12.
3)(0
.05
to 0
.13)
<0.0
0127
1.45
0.01
(0.6
)(-
0.03
to 0
.05)
0.49
838
D. D
iazo
xona
se
activ
ity≤
6958
U/L
0.87
0.13
(16.
4)(0
.10
to 0
.17)
<0.0
0140
1.33
0.10
(7.6
)(0
.03
to 0
.18)
0.01
027
> 69
58 U
/L0.
900.
07 (8
.5)
(0.0
4 to
0.1
1)<0
.001
291.
450.
01 (0
.8)
(-0.
02 to
0.0
5)0.
454
38E.
Ary
lest
eras
e ac
tivity
≤ 68
.8 U
/mL
0.87
0.13
(14.
9)(0
.09
to 0
.16)
<0.0
0141
1.40
0.10
(7.4
)(0
.04
to 0
.17)
0.00
326
> 68
.8 U
/mL
0.90
0.08
(10.
4)(0
.04
to 0
.12)
<0.0
0128
1.40
0.02
(1.1
)(-
0.03
to 0
.06)
0.50
939
P va
lues
for b
etw
een
grou
ps co
mpa
rison
s wer
e ca
lcul
ated
with
the
Inde
pend
ent-
Sam
ples
T te
st, P
val
ues f
or ch
ange
with
the
One
-Sam
ple
T te
st. M
edia
n va
lues
w
ere
used
as c
ut-o
ff po
ints
for t
he cl
assifi
catio
n of
PO
N1,
dia
zoxo
nase
act
ivity
, ary
lest
eras
e ac
tivity
and
HD
L-C
into
subg
roup
s. H
DL-
C d
enot
es h
igh-
dens
ity
lipop
rote
in ch
oles
tero
l; PO
N1,
par
aoxo
nase
-1; F
H, f
amili
al h
yper
chol
este
role
mia
.

88
CHAPTER 6
additional protection against HDL oxidation. In contrast, patients with high baseline PON1 levels and activity already have an optimal protection of HDL and, therefore, statins will have little or no additional effect on HDL-C increment. Several studies have shown that PON1 activity is a stronger predictor of CVD than PON1 genotypes [33, 34], suggesting that PON1 activity is a better tool to investigate the contribu-tion of PON1 to cardiovascular processes than PON1 genotype. However, serum PON1 levels and activity can be modified by life-style determinants like smoking [35, 36], vitamin C and E consumption [37], and alcohol intake [38]. In contrast, genotype is not affected by these confounding variables nor by baseline levels of HDL-C. This makes genotyping a more suit-able marker to study the physiological involvement of PON1 to HDL-C changes. In our study, the relationship between PON1 genotypes and the observed HDL-C increments provides ad-ditional evidence for a causal involvement. Several prospective cohort studies have demonstrated that low HDL-C levels are a powerful predictor of coronary hearth disease [39]. In this study a considerable variation in HDL-C changes was observed, ranging form a 16 percent increase to no increase at all. An increase of 16 percent in HDL-C may have a major clinical significance since a 2 to 3 percent increase in HDL-C is associated with a reduction of 2 to 4 percent in the risk of cardiac events [5]. In addition to the potency of PON1 to interact with HDL-C changes due to statin therapy, PON1 may interact with other drugs aimed at increasing HDL-C levels (e.g. fibrates or niacin). Since these drugs are becoming a new approach to treat cardiovascular disease, further investiga-tions should take PON1 into account as possible effect modifier. Furthermore, if these find-ings also prove to be relevant for patients with more common forms of dyslipidemias, the determination of PON1 status in addition to HDL-C may become a clinically relevant predic-tor of treatment success. In conclusion, PON1 status in conjunction with HDL-C concentration can be used to predict which patients with FH will respond to statin therapy with respect to HDL-C increments. These results may open new doors to unravel the interaction between PON1 and drugs aimed at increasing HDL-C levels.
Acknowledgments
The authors greatly acknowledge, Dr. Hiroaki Hattori for providing the PON1 concentration measurements and Dr. Eugène H.J.M. Jansen for providing the diazoxonase activity measure-ments.

89
PON1, statins and HDL-C changes
References
[1] MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20536 high-risk individu-als: a randomised placebo-controlled trial, The Lancet 360 (2002) 7-22.[2] M.J. Caslake, C.J. Packard, Phenotypes, genotypes and response to statin therapy, Curr Opin Lipidol 15 (2004) 387-392.[3] M. Wang, M.R. Briggs, HDL: the metabolism, func-tion, and therapeutic importance, Chem Rev 104 (2004) 119-137.[4] J.R. Nofer, B. Kehrel, M. Fobker, B. Levkau, G. Ass-mann, A. von Eckardstein, HDL and arteriosclerosis: beyond reverse cholesterol transport, Atherosclerosis 161 (2002) 1-16.[5] D.J. Gordon, J.L. Probstfield, R.J. Garrison, J.D. Nea-ton, W.P. Castelli, J.D. Knoke, D.R. Jacobs, Jr., S. Bangdi-wala, H.A. Tyroler, High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies, Circulation 79 (1989) 8-15.[6] J.R. Schaefer, H. Schweer, K. Ikewaki, H. Stracke, H.J. Seyberth, H. Kaffarnik, B. Maisch, A. Steinmetz, Metabolic basis of high density lipoproteins and apoli-poprotein A-I increase by HMG-CoA reductase inhibi-tion in healthy subjects and a patient with coronary artery disease, Atherosclerosis 144 (1999) 177-184.[7] V. Bonn, R.C. Cheung, B. Chen, C. Taghibiglou, S.C. Van Iderstine, K. Adeli, Simvastatin, an HMG-CoA re-ductase inhibitor, induces the synthesis and secretion of apolipoprotein AI in HepG2 cells and primary ham-ster hepatocytes, Atherosclerosis 163 (2002) 59-68.[8] C. Lahoz, R. Pena, J.M. Mostaza, J. Jimenez, E. Subi-rats, X. Pinto, M. Taboada, A. Lopez-Pastor, Apo A-I promoter polymorphism influences basal HDL-cho-lesterol and its response to pravastatin therapy, Athero-sclerosis 168 (2003) 289-295.[9] R. Malin, R. Laaksonen, J. Knuuti, T. Janatuinen, R. Vesalainen, P. Nuutila, T. Lehtimaki, Paraoxonase
genotype modifies the effect of pravastatin on high-density lipoprotein cholesterol, Pharmacogenetics 11 (2001) 625-633.[10] M.I. Mackness, S. Arrol, P.N. Durrington, Paraoxo-nase prevents accumulation of lipoperoxides in low-density lipoprotein, FEBS Lett 286 (1991) 152-154.[11] M. Aviram, M. Rosenblat, C.L. Bisgaier, R.S. New-ton, S.L. Primo-Parmo, B.N. La Du, Paraoxonase inhib-its high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxo-nase, J Clin Invest 101 (1998) 1581-1590.[12] M.C. Blatter Garin, C. Abbott, S. Messmer, M. Mackness, P. Durrington, D. Pometta, R.W. James, Quantification of human serum paraoxonase by en-zyme-linked immunoassay: population differences in protein concentrations, Biochem J 304 ( Pt 2) (1994) 549-554.[13] N. Saha, A.C. Roy, S.H. Teo, J.S. Tay, S.S. Ratnam, Influence of serum paraoxonase polymorphism on se-rum lipids and apolipoproteins, Clin Genet 40 (1991) 277-282.[14] M.C. Blatter, R.W. James, S. Messmer, F. Barja, D. Pometta, Identification of a distinct human high-den-sity lipoprotein subspecies defined by a lipoprotein-as-sociated protein, K-45. Identity of K-45 with paraoxo-nase, Eur J Biochem 211 (1993) 871-879.[15] C.A. Abbott, M.I. Mackness, S. Kumar, A.J. Boul-ton, P.N. Durrington, Serum paraoxonase activity, concentration, and phenotype distribution in diabe-tes mellitus and its relationship to serum lipids and lipoproteins, Arterioscler Thromb Vasc Biol 15 (1995) 1812-1818.[16] S.R. Srinivasan, S. Li, W. Chen, R. Tang, M.G. Bond, E. Boerwinkle, G.S. Berenson, Q192R polymorphism of the paraoxanase 1 gene and its association with serum lipoprotein variables and carotid artery intima-media thickness in young adults from a biracial community: The Bogalusa Heart Study, Atherosclerosis 177 (2004) 167-174.

90
CHAPTER 6
[17] T.M. van Himbergen, M. Roest, J. de Graaf, E.H. Jansen, H. Hattori, J.J. Kastelein, H.A. Voorbij, A.F. Stalenhoef, L.J. van Tits, Indications that paraoxonase-1 contributes to plasma high density lipoprotein levels in familial hypercholesterolemia, J Lipid Res 46 (2005) 445-451.[18] V.H. Brophy, R.L. Jampsa, J.B. Clendenning, L.A. McKinstry, G.P. Jarvik, C.E. Furlong, Effects of 5’ regu-latory-region polymorphisms on paraoxonase-gene (PON1) expression, Am J Hum Genet 68 (2001) 1428-1436.[19] I. Leviev, R.W. James, Promoter polymorphisms of human paraoxonase PON1 gene and serum paraoxo-nase activities and concentrations, Arterioscler Thromb Vasc Biol 20 (2000) 516-521.[20] T. Kujiraoka, T. Oka, M. Ishihara, T. Egashira, T. Fujioka, E. Saito, S. Saito, N.E. Miller, H. Hattori, A sandwich enzyme-linked immunosorbent assay for human serum paraoxonase concentration, J Lipid Res 41 (2000) 1358-1363.[21] H.W. Eckerson, C.M. Wyte, B.N. La Du, The human serum paraoxonase/arylesterase polymorphism, Am J Hum Genet 35 (1983) 1126-1138.[22] S. Adkins, K.N. Gan, M. Mody, B.N. La Du, Molecu-lar basis for the polymorphic forms of human serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B allozymes, Am J Hum Genet 52 (1993) 598-608.[23] R. Humbert, D.A. Adler, C.M. Disteche, C. Hassett, C.J. Omiecinski, C.E. Furlong, The molecular basis of the human serum paraoxonase activity polymorphism, Nat.Genet. 3 (1993) 73-76.[24] H.G. Davies, R.J. Richter, M. Keifer, C.A. Broom-field, J. Sowalla, C.E. Furlong, The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin, Nat Genet 14 (1996) 334-336.[25] T.J. Smilde, S. van Wissen, H. Wollersheim, M.D. Trip, J.J. Kastelein, A.F. Stalenhoef, Effect of aggressive
versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial, Lancet 357 (2001) 577-581.[26] R.J. Richter, C.E. Furlong, Determination of paraoxonase (PON1) status requires more than geno-typing, Pharmacogenetics 9 (1999) 745-753.[27] Smilde TJ, Wollersheim H, Van Wissen S, Kastelein JJP, Stalenhoef AFH., Rationale, design and baseline characteristics of a clinical trial comparing the effects of robust vs conventional cholesterol lowering and in-tima media thickness in patients with familial hyper-cholesterolaemia: The atorvastatin versus simvastatin on atherosclerosis progression (ASAP) study, Clinical Drug Investigation 20 (2000) 67-79.[28] B. Mackness, P. Durrington, P. McElduff, J. Yarnell, N. Azam, M. Watt, M. Mackness, Low paraoxonase ac-tivity predicts coronary events in the Caerphilly Pro-spective Study, Circulation 107 (2003) 2775-2779.[29] S. Deakin, I. Leviev, S. Guernier, R.W. James, Sim-vastatin modulates expression of the PON1 gene and increases serum paraoxonase: a role for sterol regula-tory element-binding protein-2, Arterioscler Thromb Vasc Biol 23 (2003) 2083-2089.[30] M. Tomas, M. Senti, F. Garcia-Faria, J. Vila, A. Tor-rents, M. Covas, J. Marrugat, Effect of simvastatin ther-apy on paraoxonase activity and related lipoproteins in familial hypercholesterolemic patients, Arterioscler Thromb Vasc Biol 20 (2000) 2113-2119.[31] C. Gouedard, N. Koum-Besson, R. Barouki, Y. Morel, Opposite regulation of the human paraoxo-nase-1 gene PON-1 by fenofibrate and statins, Mol Pharmacol 63 (2003) 945-956.[32] R.S. Rosenson, Statins in atherosclerosis: lipid-lowering agents with antioxidant capabilities, Athero-sclerosis 173 (2004) 1-12.[33] G.P. Jarvik, L.S. Rozek, V.H. Brophy, T.S. Hatsu-kami, R.J. Richter, G.D. Schellenberg, C.E. Furlong, Paraoxonase (PON1) phenotype is a better predictor

91
PON1, statins and HDL-C changes
of vascular disease than is PON1(192) or PON1(55) genotype, Arterioscler Thromb Vasc Biol 20 (2000) 2441-2447.[34] P.N. Durrington, B. Mackness, M.I. Mackness, Paraoxonase and atherosclerosis, Arterioscler Thromb Vasc Biol 21 (2001) 473-480.[35] R.W. James, I. Leviev, A. Righetti, Smoking is as-sociated with reduced serum paraoxonase activity and concentration in patients with coronary artery disease, Circulation 101 (2000) 2252-2257.[36] E. Nishio, Y. Watanabe, Cigarette smoke extract inhibits plasma paraoxonase activity by modification of the enzyme’s free thiols, Biochem Biophys Res Com-mun 236 (1997) 289-293.[37] G.P. Jarvik, N.T. Tsai, L.A. McKinstry, R. Wani, V.H. Brophy, R.J. Richter, G.D. Schellenberg, P.J. Heagerty, T.S. Hatsukami, C.E. Furlong, Vitamin C and E intake is associated with increased paraoxonase activity, Ar-terioscler Thromb Vasc Biol 22 (2002) 1329-1333.[38] M.S. van der Gaag, A. van Tol, L.M. Scheek, R.W. James, R. Urgert, G. Schaafsma, H.F. Hendriks, Daily moderate alcohol consumption increases serum paraoxonase activity; a diet-controlled, randomised intervention study in middle-aged men, Atherosclero-sis 147 (1999) 405-410.[39] P. Barter, J. Kastelein, A. Nunn, R. Hobbs, High density lipoproteins (HDLs) and atherosclerosis; the unanswered questions, Atherosclerosis 168 (2003) 195-211.


7Paraoxonase-1 is associated with Familial Combined
Hyperlipidemia
Submitted
T.M. van HimbergenL.J. van TitsE. ter Avest
M. RoestH.A. Voorbij
J. de GraafA.F. Stalenhoef

Abstract
Familial combined hyperlipidemia (FCH) is a common genetic lipid disorder. The molecular basis of FCH still remains to be elucidated. The HDL-associated enzyme serum paraoxonase-1 (PON1) is associated with variation in serum lipids and lipoproteins. Therefore, we deter-mined whether variation in PON1 contributes to the FCH phenotype. The study population consisted of 32 well-defined families with FCH, including 103 FCH pa-tients and 240 normolipidemic relatives (NLR). In addition to plasma lipids and lipoproteins we determined the hydrolytic activity of PON1 towards phenyl acetate (arylesterase activ-ity) and paraoxon (paraoxonase activity) as well as the common genetic variants –107C>T, 55L>M and 192Q>R in the PON1 gene.Arylesterase activity was significantly higher in the FCH patients when compared to the NLR (P<0.001). In addition, in the total population, the PON1 genetic variants associated with the highest arylesterase activity (-107CC and 55LL) also associated with higher levels of to-tal cholesterol, apolipoprotein B, triglycerides and VLDL-cholesterol and decreased levels of HDL-cholesterol. Furthermore, the combination of the –107CC with the 55LL genotype asso-ciated with a 5-fold increased risk for FCH when compared to the –107TT/55MM genotype combination, independent of age and gender (odds ratio 5.2, 95%CI: 1.5-19.7, P=0.016). In this population of subjects from well-defined families with FCH, PON1 is biochemically and genetically associated with FCH.

95
PON1 is associated with FCH
Introduction
Familial combined hyperlipidemia (FCH) is a common genetic lipid disorder. Affected sub-jects characteristically show elevation of plasma total cholesterol, triglycerides and apolipo-protein (apo) B, and are more prone to develop premature cardiovascular disease (CVD) [1]. The prevalence of FCH in the general population has been reported to be up to 5.7% [2], and FCH has been estimated to cause approximately 10% of premature CVD [3].The pathophysiological mechanism underlying FCH is believed to be a hepatic overproduc-tion of apoB-100 containing lipoprotein particles, i.e. very-low-density lipoproteins (VLDL) and low-density lipoproteins (LDL), resulting in increased plasma cholesterol, triglycerides and apoB levels [4-6]. In addition, lower concentrations of high-density lipoprotein (HDL) cholesterol and increased amounts of small dense LDL (sdLDL) particles can be observed in patients with FCH [7-11]. Despite more than 30 years of research, the genetic origin of FCH remains obscure [12]. The heterogeneous lipid profile, however, indicates that FCH is a multi-factorial disease in which several genes affect the lipid and lipoprotein metabolism. We hypothesized that the HDL-associated enzyme paraoxonase (PON1) contributes to the FCH phenotype. A growing body of evidence shows that PON1 is an important enzyme in the metabolism of serum lipids and lipoproteins and therefore PON1 may also be of importance in the expression of lipid disorders. Many epidemiological studies have shown that PON1 as-sociates with HDL-cholesterol levels [13-18]. In addition, total cholesterol, triglycerides, LDL-cholesterol, VLDL-cholesterol and apoB levels have also been shown to be associated with PON1 [18-22]. Although recent investigations have given much insight into the functioning of PON1, the exact mechanisms underlying these associations of PON1 with the lipids and lipoproteins remain to be elucidated. In addition, the role of PON1 in diseases characterized by lipoprotein disorders has remained largely unexplored, and until today no studies have ad-dressed the effects of PON1 on the FCH phenotype. To study the role of PON1 in humans, serum PON1 activity and concentration can be deter-mined by monitoring the hydrolytic activity towards paraoxon (paraoxonase activity) and phenyl acetate (arylesterase activity), respectively [23, 24]. Although the same active site of the PON1 enzyme is responsible for the hydrolysis of the two substrates, the paraoxonase ac-tivity is greatly affected by a polymorphism, a glutamine (Q)-to-arginine (R) transition at po-sition 192 (192Q>R) in the PON1 gene [25-27]. The arylesterase activity, on the other hand, is not polymorphic and can be used to estimate PON1 serum concentrations [24]. The 55L>M polymorphism in the coding region and the –107C>T polymorphism in the promoter region of the PON1 gene have the strongest impact on PON1 concentrations in blood [28, 29]. We investigated whether the arylesterase and paraoxonase activity of the PON1 enzyme, as well as the PON1 genetic variants 192Q>R, 55L>M and –107C>T are associated with the FCH phenotype. For this we studied the effects of PON1 in a population of 32 well- defined FCH families consisting of 103 patients with FCH and 240 normolipidemic relatives (NLR).

96
CHAPTER 7
In addition, to gain further insight into the role of PON1 in the lipid metabolism, we also in-vestigated the association between PON1 and lipids and lipoproteins in the subgroups of the NLR and the patients with FCH.
Methods
Study population.The study population consists of 343 subjects, from 32 well-defined FCH families. One hun-dred and three subjects were diagnosed with FCH [30]. This diagnosis was based on our recently published nomogram [30]. Briefly, plasma triglycerides and total cholesterol levels, adjusted for age and gender, and absolute apoB levels were included in a nomogram to cal-culate the likelihood of having FCH. A subject was defined to be affected by FCH when the probability was above 60%, provided that the diagnostic phenotype was also present in at least one first degree relative and premature CVD (i.e. before the age of 60) was present in at least one individual in the family. After withdrawal of lipid-lowering medication for four weeks, blood was drawn after an over-night fast. The ethics committee of the Radboud University Nijmegen Medical Centre approved the study protocol and all subjects gave informed consent.
Laboratory measurementsPlasma total cholesterol and triglycerides were determined by enzymatic, commercially avail-able reagents (Boehringer-Mannheim, Germany, catalog No. 237574 and Sera Pak, Miles, Belgium, catalog No. 6639, respectively). LDL-cholesterol was calculated according to the Friedewald formula. VLDL-cholesterol was determined by ultracentrifugation [31], HDL-cholesterol was determined by the polyethylene glycol 6000 method [32], and total plasma apoB concentrations were determined by immunonephelometry [33].Serum PON1 enzyme activity was measured with the hydrolysis rate of paraoxon as previ-ously described [23]. Serum PON1 concentration was estimated with the hydrolysis rate of phenylacetate on a spectrophotometer [24]. The 55L>M and 192Q>R genetic variants were determined by restriction fragment length polymorphism (PCR-RFLP) using primers and restriction enzymes as described by Humbert et al [27]. The –107C>T polymorphism was detected by amplifying the DNA region of interest using PCR primers described by Leviev and James [34], followed by genotype determination according to the previously described iFLASH method [35]. The following infrared-dye (IRD) labeled allele-specific oligonucle-otides were used: 5’-IRD700-GGAGGGGCGGAGCG-3’ for detection of the –107C allele and 5’-IRD800-GGAGGGGTGGGGCG-3’ for detection of the –107T allele. PCR products were successfully obtained in 330, 333 and 324 samples for the 55L>M, 192Q>R and -107C>T,

97
PON1 is associated with FCH
respectively.
Statistical analysesBaseline variables were tested for normal distribution. Comparisons among the FCH patients and the NLR were performed using the Independent Samples t-test for normally distributed continuous variables and the χ2 test for categorical variables. Triglycerides and VLDL-choles-terol showed skewed distribution, baseline comparisons were made with the Mann-Whitney test for non-normally distributed continuous variables and correlations were based on log transformed data. Linear regression analysis was used to evaluate the differences in arylesterase- and paraox-onase activities and plasma lipid and lipoprotein concentrations among the genetic variants of the 192Q>R and –107C>T polymorphisms. Age, gender and –107C>T (CC versus CT/TT), 55L>M (LL versus LM/MM) or 192Q>R (QQ versus QR/RR) were entered into the model as explanatory variables and, alternately, arylesterase activity, paraoxonase activity, total choles-terol, apoB, log transformed triglycerides, HDL-cholesterol and log transformed VLDL-cho-lesterol were entered into the model as dependent variables. Odds ratios for developing FCH among PON1 -107C>T and 55L>M genotype combina-tions were calculated using logistic regression. The homozygote -107TT in combination with 55MM served as reference category for the other genotype combinations. The FCH pheno-type served as outcome variable. Odds ratios were controlled for age and gender. Correlation coefficients for PON1 arylesterase- and paraoxonase activity with plasma lipid and lipoprotein parameters were calculated using the SPSS partial correlation procedure con-trolling for age and gender. In case of a missing value, the correlation pair was excluded.The association between arylesterase and VLDL-cholesterol in the patients with FCH was investigated using a linear regression model with VLDL-cholesterol, age, gender and the -107C>T and 55L>M genotypes as explanatory variable and arylesterase activity as dependent variable. A P-value smaller than 0.05 was considered statistically significant and all calculations were performed with SPSS 11.5.
Results
The characteristics of the 103 patients with FCH and the 240 NLR are described in Table 1. There was no difference in gender distributions among FCH patients and the NLR, but the FCH group was significantly older (P<0.001). Characteristically, plasma total cholesterol, tri-glycerides, apoB, LDL- and VLDL-cholesterol concentrations were significantly higher in the FCH group when compared to the NLR (P<0.001, for all relations). Interestingly, although HDL-cholesterol levels were lower, the PON1 arylesterase activity was higher in individu-

98
CHAPTER 7
als affected with FCH (P<0.001). Paraoxonase activity was also higher in patients with FCH compared to the NLR, but this difference was not significant (Table1).In the total study population, the PON1 arylesterase- and paraoxonase activities were strong-ly influenced by the –107C>T, 55L>M and 192Q>R polymorphisms. The –107CC, 55LL and 192QR/RR genotypes were associated with the highest arylesterase and paraoxonase activity (Table 2). The –107CC variant was also associated with increased total cholesterol (P<0.01), apoB (P<0.05), triglycerides (P<0.05) and VLDL-cholesterol (P<0.01) levels. Additionally, the 55LL variant was associated with lower HDL-cholesterol levels (P<0.01) and higher tri-glycerides (P=0.05) and VLDL-cholesterol (P<0.05) levels. The 192Q>R genotype, which is of strong influence on the paraoxonase activity, was not associated with any of the lipid or lipoprotein traits. When the PON1 genotypes were considered as separate risk factors for FCH, they did not reach statistical significance (data not shown). However, there was significant linkage between the –107C>T and the 55L>M genotypes (χ2 P<0.001), and because the –107CC genotype was associated with higher total cholesterol, apoB, triglycerides and VLDL-cholesterol levels, and the 55LL genotype was associated with higher triglycerides and VLDL-cholesterol levels and lower HDL-cholesterol levels, we investigated whether the combined genotypes contributed to the risk on developing FCH. The low concentration variants 55MM and –107TT were combined and served as reference group. As presented in Figure 1, approximately 20% of the studied population were –107CC/55LL homozygotes, and had a five-fold higher chance to develop FCH when compared to –107TT/55MM homozygotes (OR 5.2, 95%CI 1.4-19.7, P=0.016). Intermediate effects were observed for all other genotype combinations (Figure 1). To understand the basis of elevated PON1 arylesterase- and paraoxonase activities in FCH, we investigated the correlations of the PON1 activities with lipid and lipoprotein parameters in the FCH patients and their unaffected relatives (Table 3). First, in the NLR group there was
Table 1. Baseline characteristics of the normolipidemic relatives (NLR) and patients with familial combined hyperlipidemia (FCH)
NLR(n=240) FCH (n=103)Gender, male/female 117/123 45/58Age, years 44±15 51±15*Total cholesterol, mmol/L 5.2±1.0 6.9±1.2*Triglycerides, mmol/L 1.2 (0.9-1.6) 2.9 (2,0-4.1)*HDL-cholesterol, mmol/L 1.3±0.3 1.1±0.3*LDL-cholesterol, mmol/L 3.5±0.9 4.2±1.2*VLDL-cholesterol, mmol/L 0.4 (0.2-0.6) 1.1 (0.7-1.9)*ApoB, mg/L 960±221 1372±256*Arylesterase activity, U/mL 90.1±19.2 101.3±22.8*Paraoxonase activity, U/L 253.4±182.3 289.4±206.4Values presented as mean ± standard deviation, except for gender, which is presented as the number of male and females, and triglycerides and VLDL-cholesterol, which are presented as median (inter quartile range). *P<0.001 for the comparison with the NLR group.

99
PON1 is associated with FCH
Tabl
e 2.
Ary
lest
eras
e- a
nd p
arao
xona
se a
ctiv
ities
and
pla
sma
lipi
d an
d lip
opro
tein
conc
entr
atio
ns a
mon
g PO
N1
192Q
>R, 5
5L>M
and
–10
7C>T
ge
noty
pes i
n to
tal s
tudy
pop
ulat
ion
-107
C>T
55L>
M19
2Q>R
CC
CT/
TTLL
LM/M
MQ
R/RR
Num
ber o
f sub
ject
s89
235
141
188
187
146
Ary
lest
eras
e ac
tivity
, U/m
L11
0.2±
18.4
†87
.0±1
8.5
100.
2±19
.5†
88.0
±19.
991
.1±1
9.6§
95.8
±21.
5Pa
raox
onas
e ac
tivity
, U/L
339.
3±21
1.2†
237.
3±17
5.7
353.
2±21
2.6†
195.
5±13
7.7
127.
3±44
.2†
439.
1±15
8.7
Tota
l cho
lest
erol
, mm
ol/L
6.0±
1.5‡
5.6±
1.2
5.8±
1.4
5.7±
1.2
5.7±
1.3
5.7±
1.3
Trig
lyce
rides
*, m
mol
/L1.
54 (1
.09-
2.73
)§1.
39 (0
.98-
2.13
)1.
54 (1
.09-
2.45
)§1.
35 (0
.97-
2.10
)1.
42 (1
.0-2
.36)
1.44
(1.0
6-2.
21)
HD
L-ch
oles
tero
l, m
mol
/L1.
24±0
.33
1.27
±0.3
21.
22±0
.28‡
1.30
±0.3
41.
27±0
.34
1.26
±0.2
9LD
L-ch
oles
tero
l, m
mol
/L3.
8±1.
33.
7±0.
93.
7±1.
13.
7±1.
03.
7±1.
03.
8±1.
0V
LDL-
chol
este
rol*
, mm
ol/L
0.62
(0.3
5-1.
12)‡
0.46
(0.2
6-0.
80)
0.53
(0.3
1-0.
96)||
0.46
(0.2
6-0.
80)
0.47
(0.2
8-0.
96)
0.47
(0.2
7-0.
76)
ApoB
, mg/
L11
27±3
31§
1063
±291
1098
±316
1072
±290
1074
±295
1099
±308
Valu
es p
rese
nted
as m
ean
± st
anda
rd d
evia
tion,
exc
ept f
or tr
igly
cerid
es a
nd V
LDL-
chol
este
rol w
hich
are
pre
sent
ed a
s med
ian
(inte
r qua
rtile
rang
e).
*Bet
wee
n gr
oup
com
paris
ons a
re b
ased
on
log
tran
sfor
med
dat
a. P
-val
ues a
re b
ased
on
linea
r reg
ress
ion
and
adju
sted
for a
ge a
nd g
ende
r. †
P<0.
001,
‡P
<0.0
1, §
P<0.
05, |
|P=0
.05
whe
n co
mpa
red
to c
arrie
rs o
f CT/
TT, L
M/M
M o
r QR/
RR.

100
CHAPTER 7
a positive correlation between arylesterase activity and HDL-cholesterol (P<0.001), but also total cholesterol, LDL-cholesterol and apoB correlated positively with arylesterase activity (P<0.001 for all relations). Within the FCH patients, arylesterase activity was not associated with HDL-cholesterol nor with apoB levels, but rather with the total cholesterol, triglycerides and VLDL-cholesterol concentrations (P<0.01, for all relations). The observed correlations between paraoxonase activity and the lipid and lipoprotein parameters were due to the strong association of paraoxonase activity with arylesterase activity which reflects the serum PON1 concentration. The strength of the correlation between paraoxonase activity and lipid param-eters declined after adjusting for arylesterase activity (data not shown). As we have shown, the relationship of PON1 with the lipid profile is different in FCH patients and their NLR. In concordance, there are also differences in the variance in arylesterase ac-
Figure 1. The probability to develop FCH among combinations of the PON1 –107C>T and 55L>M genotypes. The homozygote –107TT in combination with 55MM served as reference category. The number of genotypes are shown in parentheses. Genotype combinations were arranged according to odds ratio (OR). ORs were adjusted for age and gender. *OR, 5.2 (95%CI, 1.4-19.7); P= 0.016. **OR, 3.75 (95%CI, 0.98-14.32) ; P=0.05.
Table 3. Correlation coefficients for PON1 arylesterase and paraoxonase activity with plasma lipid and lipoprotein concentrations in the normolipidemic relatives (NLR) and patients with familial combined hyperlipidemia (FCH)
NLR (n=240) FCH (n=103)AE activity POX activity AE activity POX activity
Total cholesterol 0.337* 0.084 0.297† 0.114ApoB 0.257* 0.152‡ 0.099 0.021HDL-cholesterol 0.227* -0.028 0.022 0.053LDL-cholesterol 0,277* 0,092 -0,057 -0,048Log VLDL-cholesterol 0.047 0.053 0.278† 0.096Log Triglycerides 0.079 0.079 0.275† 0.070Correlations were controlled for age and gender. *P<0.001, †P<0.01, ‡P<0.05. AE, arylesterase and POX, paraoxonase.

101
PON1 is associated with FCH
tivity attributable to the -107C>T and 55L>M polymorphisms. Using a regression model ad-justed for age and gender, we found that in patients with FCH 45% of the variation in aryles-terase activity could be explained by the genetic variants at position -107 and 55, while in the NLR this was only 31% (data not shown). Interestingly, the association between arylesterase activity and VLDL-cholesterol in the FCH patients, was no longer significant after adjustment for the -107C>T and 55L>M polymorphisms, thus suggesting an intermediate effect for the PON1 genotypes (Table 4).
Discussion
This is the first study to investigate the contribution of PON1 to the FCH phenotype. We observed that PON1 arylesterase activity was increased in FCH patients. There was also a ge-netic basis for this observation since the PON1 –107CC and 55LL genotypes, associated with an increased arylesterase activity, were also associated with a 5-fold increased risk on FCH. Finally, the examination of PON1 with the lipid profile in FCH patients and their unaffected relatives, separately, revealed remarkable differences. In the NLR, PON1 specifically associ-ated with higher levels of apoB, HDL- and LDL-cholesterol, while in the FCH patients PON1 specifically associated with the higher levels of VLDL-cholesterol and triglycerides. In general, PON1 is thought to attenuate the oxidation of LDL, based on in vitro findings that purified PON1 inhibits the accumulations of lipid peroxides in LDL [36]. Apart from inhibi-tion of LDL oxidation, there is evidence from animal and in vitro models that paraoxonase can protect the HDL particle from oxidation and preserve the integrity and function of HDL [37, 38]. These findings are supported by epidemiological studies in different populations demonstrating that PON1 contributes to variation in plasma levels of HDL-cholesterol [15-18]. Thus it is hypothesized that the preservation of HDL by PON1 may beneficially influ-ence the lipid metabolism. However, the exact mechanism by which PON1 functions in the lipid metabolism is still not clear. In blood, PON1 is located exclusively on the HDL particle and, as expected, we found a positive correlation between PON1 and HDL-cholesterol in the NLR. In FCH, however, the association of PON1 with lipids and lipoproteins was different. Surprisingly, arylesterase activity was higher in FCH patients, while HDL-cholesterol levels were decreased in these subjects. Apparently, the higher turnover or the reduced production of HDL-cholesterol in FCH patients does not influence PON1, suggesting that there is no in-teraction between PON1 and HDL in these patients. Rather a correlation of arylesterase with VLDL-cholesterol and triglycerides was present in FCH. One could hypothesize that there is a physical displacement of the PON1 particle from the HDL to VLDL. Recently, an in vitro study demonstrated that VLDL not only promoted the secretion of PON1 from cells but also served as an accepting vector for PON1 in the absence of HDL [39]. We investigated whether in FCH the PON1 protein was present on the VLDL particle by fractionating the serum of five

102
CHAPTER 7
FCH patients and measuring PON1 activity in each lipoprotein fraction. We found that the PON1 activity was present only in the HDL fraction, and we could not detect any PON1 activ-ity in the VLDL fractions of these patients (unpublished results). This may implicate that after the initial acceptance by VLDL all the PON1 is quickly transferred to the HDL as seen in vitro [39]. Although HDL-cholesterol levels are lower in the FCH patients, HDL is still abundantly present to serve as a carrier for PON1. VLDL may enhance the secretion of PON1 into blood, but does not carry PON1 in vivo.Context-dependent transcriptional regulation of PON1 may underly the differential associa-tions of PON1 with lipids and lipoproteins observed in FCH patients and the NLR. Transcrip-tion factors, which play an important role in the regulation of the cholesterol homeostasis [40], were also found to regulate the expression of PON1 in vitro [41, 42]. In the NLR the PON1 genotypes only explain 31% of the variation in arylesterase activity while in FCH 45% of the variation could be explained by these genotypes. These data indicate that after PON1 is expressed, in the NLR it is more prone to influences by environmental factors than in the FCH patients. Furthermore, our data show that the association between VLDL and arylester-ase activity diminishes after adjustment for the effects of PON1 genotypes. This indicates that PON1 genotype is an intermediate step in the association between VLDL and arylesterase activity and that serum PON1 and VLDL levels may be the result of a regulation by the same mechanism. Our data indicate that PON1 plays a role in the disturbed lipoprotein metabolism in FCH patients. In support, as mentioned earlier, we find differences in the association of PON1 with lipids and lipoproteins among FCH and NLR: arylesterase activity correlated with tri-glycerides and VLDL-cholesterol in the FCH subgroup, these correlations were not found in the NLR subgroup. Instead, in the NLR subgroup, arylesterase activity correlated with apoB, LDL- and HDL-cholesterol levels. The consistency of these findings with recent data pre-
Table 4. Multiple regression models for variables associated with arylesterase activity in patients with FCH
Standardized beta coefficients
P value
Model 1*Age, years -0.312 0.003Gender, M/F 0.242 0.023Log VLDL, mmol/L 0.325 0.005
Model 2†Age, years -0.202 0.019Gender, M/F 0.126 0.152Log VLDL, mmol/L 0.087 0.367-107C>T (CC/CT/TT) -0.553 <0.00155L>M (LL/LM/MM) -0.141 0.121Arylesterase activity (U/mL) served as outcome variable. *Variance explained 10%, †Variance explained 45%.

103
PON1 is associated with FCH
sented by Rozek et al is remarkable [22]. They found that in a control population consisting of healthy men without severe coronary artery disease, arylesterase activity correlated with HDL-cholesterol and ApoA1, whereas in men with severe carotid artery disease, a signifi-cant correlation was observed for arylesterase activity with total cholesterol, triglycerides and VLDL-cholesterol but not with HDL-cholesterol [22]. Apparently, in the normal situation PON1 associates HDL-cholesterol, and in case of FCH or carotid artery, disease PON1 associ-ates with non-HDL particles. In contrast to Rozek et al, we also found that PON1 genotypes associate with variation in lipids and lipoprotein. In general, the effect in the human body of a single genetic variant is the result of complex interactions with the environment, and therefore in order to detect such effects, well-powered study populations are needed. When a genetic association is not found, this may very well be the result of a lack of power. Rozek et al speculated that the differences in patients and controls might be the result of context-de-pendent transcription regulation of PON1 and lipoprotein components, but did not find the effects of PON1 genotype to support this hypothesis. In the present study we do find these effects and therefore our data confirm that the observed differences in association with lipo-proteins may be the result of a common regulation in transcription. In summary, we show evidence that PON1 is biochemically and genetically associated with FCH. In addition, we find that PON1 associates differentially with lipids and lipoproteins in FCH and NLR. These data suggest that PON1 is involved in the lipoprotein metabolism. The mechanisms of such an involvement require further investigation in well-controlled experi-mental studies.
Acknowledgements
The authors thank Anneke Hijmans and Arjan Barendrecht for their technical assistance.

104
CHAPTER 7
Reference
[1] J.L. Goldstein, H.G. Schrott, W.R. Hazzard, E.L. Bierman, A.G. Motulsky, Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia, J Clin Invest 52 (1973) 1544-1568.[2] P.N. Hopkins, G. Heiss, R.C. Ellison, M.A. Province, J.S. Pankow, J.H. Eckfeldt, S.C. Hunt, Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: a case-control compari-son from the National Heart, Lung, and Blood Institute Family Heart Study, Circulation 108 (2003) 519-523.[3] J.D. Brunzell, H.G. Schrott, A.G. Motulsky, E.L. Bier-man, Myocardial infarction in the familial forms of hypertriglyceridemia, Metabolism 25 (1976) 313-320.[4] A. Chait, J.J. Albers, J.D. Brunzell, Very low density lipoprotein overproduction in genetic forms of hyper-triglyceridaemia, Eur J Clin Invest 10 (1980) 17-22.[5] A.H. Kissebah, S. Alfarsi, D.J. Evans, Low density lipoprotein metabolism in familial combined hyperli-pidemia. Mechanism of the multiple lipoprotein phe-notypic expression, Arteriosclerosis 4 (1984) 614-624.[6] S. Venkatesan, P. Cullen, P. Pacy, D. Halliday, J. Scott, Stable isotopes show a direct relation between VLDL apoB overproduction and serum triglyceride levels and indicate a metabolically and biochemically coher-ent basis for familial combined hyperlipidemia, Arte-rioscler Thromb 13 (1993) 1110-1118.[7] M. Castro Cabezas, T.W. de Bruin, H.W. de Valk, C.C. Shoulders, H. Jansen, D. Willem Erkelens, Impaired fatty acid metabolism in familial combined hyperlipi-demia. A mechanism associating hepatic apolipopro-tein B overproduction and insulin resistance, J Clin Invest 92 (1993) 160-168.[8] A.F. Ayyobi, S.H. McGladdery, M.J. McNeely, M.A. Austin, A.G. Motulsky, J.D. Brunzell, Small, dense LDL and elevated apolipoprotein B are the common char-
acteristics for the three major lipid phenotypes of fa-milial combined hyperlipidemia, Arterioscler Thromb Vasc Biol 23 (2003) 1289-1294.[9] S.J. Bredie, L.A. Kiemeney, A.F. de Haan, P.N. Demacker, A.F. Stalenhoef, Inherited susceptibility determines the distribution of dense low-density lipo-protein subfraction profiles in familial combined hy-perlipidemia, Am J Hum Genet 58 (1996) 812-822.[10] J.E. Hokanson, R.M. Krauss, J.J. Albers, M.A. Aus-tin, J.D. Brunzell, LDL physical and chemical proper-ties in familial combined hyperlipidemia, Arterioscler Thromb Vasc Biol 15 (1995) 452-459.[11] J. Vakkilainen, M. Jauhiainen, K. Ylitalo, I.O. Nuo-tio, J.S. Viikari, C. Ehnholm, M.R. Taskinen, LDL par-ticle size in familial combined hyperlipidemia: effects of serum lipids, lipoprotein-modifying enzymes, and lipid transfer proteins, J Lipid Res 43 (2002) 598-603.[12] C.C. Shoulders, E.L. Jones, R.P. Naoumova, Genet-ics of familial combined hyperlipidemia and risk of coronary heart disease, Hum Mol Genet 13 Spec No 1 (2004) R149-160.[13] M.C. Blatter Garin, C. Abbott, S. Messmer, M. Mackness, P. Durrington, D. Pometta, R.W. James, Quantification of human serum paraoxonase by en-zyme-linked immunoassay: population differences in protein concentrations, Biochem J 304 ( Pt 2) (1994) 549-554.[14] M.C. Blatter, R.W. James, S. Messmer, F. Barja, D. Pometta, Identification of a distinct human high-den-sity lipoprotein subspecies defined by a lipoprotein-as-sociated protein, K-45. Identity of K-45 with paraoxo-nase, Eur J Biochem 211 (1993) 871-879.[15] T.M. van Himbergen, M. Roest, J. de Graaf, E.H. Jansen, H. Hattori, J.J. Kastelein, H.A. Voorbij, A.F. Stalenhoef, L.J. van Tits, Indications that paraoxonase-1 contributes to plasma high density lipoprotein levels in familial hypercholesterolemia, J Lipid Res 46 (2005) 445-451.[16] J. Ruiz, H. Blanche, R.W. James, M.C. Garin, C.

105
PON1 is associated with FCH
Vaisse, G. Charpentier, N. Cohen, A. Morabia, P. Passa, P. Froguel, Gln-Arg192 polymorphism of paraoxonase and coronary heart disease in type 2 diabetes, Lancet 346 (1995) 869-872.[17] S.R. Srinivasan, S. Li, W. Chen, R. Tang, M.G. Bond, E. Boerwinkle, G.S. Berenson, Q192R polymorphism of the paraoxanase 1 gene and its association with serum lipoprotein variables and carotid artery intima-media thickness in young adults from a biracial community: The Bogalusa Heart Study, Atherosclerosis 177 (2004) 167-174.[18] R.A. Hegele, J.H. Brunt, P.W. Connelly, A polymor-phism of the paraoxonase gene associated with varia-tion in plasma lipoproteins in a genetic isolate, Arte-rioscler Thromb Vasc Biol 15 (1995) 89-95.[19] R.A. Hegele, P.W. Connelly, A.J. Hanley, F. Sun, S.B. Harris, B. Zinman, Common genomic variants associ-ated with variation in plasma lipoproteins in young aboriginal Canadians, Arterioscler Thromb Vasc Biol 17 (1997) 1060-1066.[20] A.P. Boright, P.W. Connelly, J.H. Brunt, S.W. Scher-er, L.C. Tsui, R.A. Hegele, Genetic variation in paraoxo-nase-1 and paraoxonase-2 is associated with variation in plasma lipoproteins in Alberta Hutterites, Athero-sclerosis 139 (1998) 131-136.[21] N. Saha, A.C. Roy, S.H. Teo, J.S. Tay, S.S. Ratnam, Influence of serum paraoxonase polymorphism on se-rum lipids and apolipoproteins, Clin Genet 40 (1991) 277-282.[22] L.S. Rozek, T.S. Hatsukami, R.J. Richter, J. Ran-chalis, K. Nakayama, L.A. McKinstry, D.A. Gortner, E. Boyko, G.D. Schellenberg, C.E. Furlong, G.P. Jarvik, The correlation of paraoxonase (PON1) activity with lipid and lipoprotein levels differs with vascular disease sta-tus, J Lipid Res 46 (2005) 1888-1895.[23] K.N. Gan, A. Smolen, H.W. Eckerson, B.N. La Du, Purification of human serum paraoxonase/aryleste-rase. Evidence for one esterase catalyzing both activi-ties, Drug Metab Dispos 19 (1991) 100-106.
[24] H.W. Eckerson, C.M. Wyte, B.N. La Du, The human serum paraoxonase/arylesterase polymorphism, Am J Hum Genet 35 (1983) 1126-1138.[25] S. Adkins, K.N. Gan, M. Mody, B.N. La Du, Molecu-lar basis for the polymorphic forms of human serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B allozymes, Am J Hum Genet 52 (1993) 598-608.[26] H.G. Davies, R.J. Richter, M. Keifer, C.A. Broom-field, J. Sowalla, C.E. Furlong, The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin, Nat Genet 14 (1996) 334-336.[27] R. Humbert, D.A. Adler, C.M. Disteche, C. Hassett, C.J. Omiecinski, C.E. Furlong, The molecular basis of the human serum paraoxonase activity polymorphism, Nat.Genet. 3 (1993) 73-76.[28] M.C. Garin, R.W. James, P. Dussoix, H. Blanche, P. Passa, P. Froguel, J. Ruiz, Paraoxonase polymorphism Met-Leu54 is associated with modified serum concen-trations of the enzyme. A possible link between the paraoxonase gene and increased risk of cardiovascular disease in diabetes, J Clin Invest 99 (1997) 62-66.[29] V.H. Brophy, R.L. Jampsa, J.B. Clendenning, L.A. McKinstry, G.P. Jarvik, C.E. Furlong, Effects of 5’ regu-latory-region polymorphisms on paraoxonase-gene (PON1) expression, Am J Hum Genet 68 (2001) 1428-1436.[30] M.J. Veerkamp, J. de Graaf, J.C. Hendriks, P.N. Demacker, A.F. Stalenhoef, Nomogram to diagnose fa-milial combined hyperlipidemia on the basis of results of a 5-year follow-up study, Circulation 109 (2004) 2980-2985.[31] P.N. Demacker, H.E. Vos-Janssen, A.P. Jansen, A. van ‘t Laar, Evaluation of the dual-precipitation meth-od by comparison with the ultracentrifugation method for measurement of lipoproteins in serum, Clin Chem 23 (1977) 1238-1244.[32] P.N. Demacker, A.G. Hijmans, H.E. Vos-Janssen, A.

106
CHAPTER 7
van’t Laar, A.P. Jansen, A study of the use of polyeth-ylene glycol in estimating cholesterol in high-density lipoprotein, Clin Chem 26 (1980) 1775-1779.[33] P.N. Demacker, M.J. Veerkamp, S.J. Bredie, S.M. Marcovina, J. de Graaf, A.F. Stalenhoef, Comparison of the measurement of lipids and lipoproteins versus assay of apolipoprotein B for estimation of coronary heart disease risk: a study in familial combined hyper-lipidemia, Atherosclerosis 153 (2000) 483-490.[34] I. Leviev, R.W. James, Promoter polymorphisms of human paraoxonase PON1 gene and serum paraoxo-nase activities and concentrations, Arterioscler Thromb Vasc Biol 20 (2000) 516-521.[35] T.M. Van Himbergen, H.A. Voorbij, A.D. Baren-drecht, B.B. Van Rijn, R. Brambilla, L.J. Van Tits, M. Roest, High-throughput genotyping with infrared Fluorescence Allele Specific Hybridization (iFLASH): a simple, reliable and low-cost alternative, Clinical Bio-chemistry In press (2006).[36] M.I. Mackness, S. Arrol, P.N. Durrington, Paraoxo-nase prevents accumulation of lipoperoxides in low-density lipoprotein, FEBS Lett 286 (1991) 152-154.[37] M.N. Oda, J.K. Bielicki, T.T. Ho, T. Berger, E.M. Rubin, T.M. Forte, Paraoxonase 1 overexpression in mice and its effect on high-density lipoproteins, Bio-chem Biophys Res Commun 290 (2002) 921-927.[38] M. Aviram, M. Rosenblat, C.L. Bisgaier, R.S. New-ton, S.L. Primo-Parmo, B.N. La Du, Paraoxonase inhib-its high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxo-nase, J Clin Invest 101 (1998) 1581-1590.[39] S. Deakin, X. Moren, R.W. James, Very low density lipoproteins provide a vector for secretion of paraoxo-nase-1 from cells, Atherosclerosis 179 (2005) 17-25.[40] M.S. Brown, J.L. Goldstein, The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor, Cell 89 (1997) 331-340.[41] S. Deakin, I. Leviev, M.C. Brulhart-Meynet, R.W.
James, Paraoxonase-1 promoter haplotypes and serum paraoxonase: a predominant role for polymorphic po-sition - 107, implicating the Sp1 transcription factor, Biochem J 372 (2003) 643-649.[42] S. Deakin, I. Leviev, S. Guernier, R.W. James, Sim-vastatin modulates expression of the PON1 gene and increases serum paraoxonase: a role for sterol regula-tory element-binding protein-2, Arterioscler Thromb Vasc Biol 23 (2003) 2083-2089.

8Paraoxonase-1 and the risk for coronary heart disease
and myocardial infarction in a general population of Dutch women
Submitted
T.M. van HimbergenY.T. van der Schouw
H.A. VoorbijL.J. van Tits
A.F. StalenhoefP.H. Peeters
M. Roest

Abstract
There is strong evidence from both animal- and in vitro-models that PON1 is involved in the onset of cardiovascular disease. In humans, however, there is no consensus on this issue. We investigated the effect of PON1 genotype, arylesterase- and paraoxonase activity on the incidence of coronary heart disease (CHD) and acute myocardial infarction (AMI) in a large prospective cohort of 17,357 middle-aged women.We applied a case-cohort design using the CHD (n=211) and AMI cases (n=71) and a ran-dom sample from the baseline cohort (n=1527). A weighted Cox proportional hazards model was used to estimate age- and multivariate-adjusted hazard ratios (HR) for the PON1 genetic variants and tertiles of the PON1 activities.Neither the PON1 genetic variants, nor the PON1 activities affected the incidence of CHD in general, but, an increased paraoxonase activity was associated with a higher risk of AMI: the second and third tertile HR were 1.31 and 2.07 respectively (P-trend=0.029, multivariate model). In the subgroup of never-smokers, paraoxonase activity was also associated with an increased risk for AMI: the second and third tertile HR were 4.13 and 4.72 respectively (P-trend=0.009, multivariate model). In addition, when compared to the lowest paraoxonase tertile in never-smokers, the highest paraoxonase tertile in current-smokers showed a 19,2 fold higher risk for AMI (95%CI: 5.3-69.5, P<0.0001, multivariate model).This study shows that in middle-aged women, paraoxonase activity was associated with an increased risk for AMI and that this risk was amplified by the effects of smoking.

109
PON1 and the risk for CHD and AMI
Introduction
Serum paraoxonase-1 (PON1) is a high-density lipoprotein (HDL)-associated enzyme which can hydrolyze lactones [1], and several non-physiological substrates, including arylesters and organophosphates [2, 3]. Animal experimental work indicates that PON1 may be involved in atherogenesis: PON1-deficient mice are more prone to develop atherosclerosis than wild-type mice, when fed a high-fat/high-cholesterol diet [4]. Several lines of evidence suggest, that PON1 involvement in atherogenesis is due to its ability to attenuate the oxidative modification of lipoprotein particles (reviewed in [5]), but conclusive evidence remains to be delivered. In vivo, there is a wide inter-individual variation in PON1 concentration and activity. This variation is for a major part determined by common genetic variants (also called poly-morphisms) in the PON1 gene. A glutamine (Q)-to-arginine (R) transition at position 192 (192Q>R) affects the PON1 hydrolytic activity towards paraoxon (paraoxonase activity) and diazoxon (diazoxonase activity), where paraoxon is most efficiently hydrolyzed by the 192R isoform [2, 6], and diazoxon is more efficiently hydrolyzed by the 192Q isoform [3]. In addi-tion, the genetic variant –107C>T in the promoter region of the enzyme affects the expression and thus the serum concentration of the enzyme. The highest PON1 serum concentrations have been found in individuals who are carrier of the –107CC genetic variant [7]. PON1 con-centration can be measured directly in serum with an enzyme-linked immunosorbent assay (ELISA) [8], or estimated by the rate of hydrolysis toward phenylacetate (arylesterase activity) [9]. The 192Q>R and –107C>T polymorphisms are the bases for an up to 13-fold inter-indi-vidual variation in PON1 enzyme activity and concentration [10]. Epidemiological studies on the role of PON1 in cardiovascular disease have reported conflict-ing results (reviewed in [11]). A recent meta-analysis among 43 investigations studying the 192Q>R and –107C>T polymorphisms in relation to coronary heart disease (CHD), demon-strated no effect for the –107C>T polymorphism and a slightly increased risk for carriers of the R-allele at position 192 [12]. The main recommendation of this meta-analysis was, that the relation of PON1 genotype to CHD should be studied in large and prospective popula-tions. Furthermore, it has been recommended to measure PON1 activity and concentration in addition to genotype [10, 13-15], because PON1 activity and concentrations in serum are also influenced by lifestyle factors like smoking and alcohol consumption [16-18]. Up to now, only one prospective investigation on PON1 activity and concentration and CHD outcome has been published in which low serum PON1 activity towards paraoxon was shown to be an independent risk factor for coronary events [19].We studied the effects of PON1 genotypes, as well as paraoxonase- and arylesterase activity on the risk for developing CHD in a large prospective population-based cohort of middle-aged women. In this cohort, we separately investigated the relation between PON1 and the occurrence of an acute myocardial infarction (AMI) as the most acute and serious clinical manifestation of CHD.

110
CHAPTER 8
Methods
PopulationThe study population consisted of participants of the Prospect-EPIC cohort, which is one of the two Dutch contributions to the European Prospective Investigation into Cancer and nutrition (EPIC). A detailed description of the design, sampling strategies and examination techniques of the cohort has been published previously [20]. Participants were recruited be-tween 1993 and 1997 among women living in Utrecht and vicinity who attended the regional population-based breast cancer-screening program. The potential full cohort consisted of 17,357 women aged 49-70, who were followed up for subsequent development of a clinical event. At enrolment all women underwent a physical examination and filled out a general questionnaire relating to lifestyle and medical factors and a food frequency questionnaire. In addition, women donated a 30-ml non-fasting blood sample, which was fractionated into serum, citrated plasma, buffy coat and erythrocyte aliquots of 0.5 ml each. The samples were stored under liquid nitrogen for future research. Data on morbidity were obtained from the Dutch Centre for Health Care Information, which holds a standardized computerized register of hospital discharge diagnoses. Admission files are filed continuously from all general and university hospitals in the Netherlands since 1990. Whenever a patient was discharged from a hospital, data on sex, date of birth, dates of admis-sion and discharge, one mandatory principal diagnosis, and up to nine optional additional diagnoses were recorded. All diagnoses were coded according to the International Classifica-tion of Diseases, ninth Revision (ICD-9). Using the ICD-9 codes, we categorized cardiovas-cular disease (ICD-9 390-459) as CHD (ICD-9 410-414) including AMI (ICD-9 410), the latter was also analyzed as a separate disease outcome. The diagnosis of CHD or AMI was the primary reason for hospitalisation. Whenever multiple events occurred during follow-up, the first diagnosis was taken as endpoint. Follow-up was complete until January 1, 2000. The database was linked to the cohort on the basis of birth date, gender, postal code, and general practitioner with a validated probabilistic method [21]. Information on vital status was gained through linkage with the national municipal adminis-tration database. Causes of death were obtained from the women’s general practitioners.All women signed an informed consent form prior to study inclusion. The study was ap-proved by the Institutional Review Board of the University Medical Center Utrecht.
Design To reduce costs and preserve valuable biological material we applied the case-cohort design introduced by Prentice [22].We selected all 303 first fatal and non-fatal CHD events that arose during follow-up until January 1st 2000. From the 17,357 women in the total cohort we randomly selected a sample of 10% (n=1,736). Women who did not consent to linkage with vital status registries or who

111
PON1 and the risk for CHD and AMI
were not traceable (cases n=3/subcohort n=38), were not included. Women who reported a diagnosis of cardiovascular disease (ICD-9; 390-459) at baseline (cases n=73/subcohort n=107), who had missing questionnaires or blood or DNA samples (cases n=24/subcohort n=71), or a daily energy intake below 500 Kcal/day (cases n=1/subcohort n=2) were excluded from the analyses. For some women multiple reasons applied for exclusion resulting in a total number of 1,527 women in the sub-cohort, that remained in the analyses and 211 CHD cases of which 71 were AMI cases. For all case subjects follow up ended at the date of diagnosis or at the date of death due to cardiovascular disease. Moving out of the Netherlands (n=2) and death due to causes other than cardiovascular disease (n=16) were considered censor-ing events. All others (n=1,422) were censored on January 1st 2000. Overall eleven first fatal CHDs occurred during follow-up.
General questionnaire and anthropometric measurementsThe general questionnaire contained questions on demographic characteristics, lifestyle hab-its, obstetric and gynecological history and past and current morbidity. Women were classi-fied according to their smoking habits as current, past or never smokers. Alcohol intake was expressed as grams of daily alcohol consumption. Systolic and diastolic blood pressures were measured in duplicate at the left arm with the subjects in sitting position after 10 minutes of rest with an automated and calibrated oscil-lomat (Bosch & Son, Jungingen, Germany). Subsequently, the mean systolic and diastolic blood pressures were calculated. Body height was measured to the nearest 0.5 cm with a wall-mounted stadiometer (Lameris, Utrecht, The Netherlands). Body weight was measured in light indoor clothing without shoes to the nearest 0.5 kg with a floor scale (Seca, Atlanta, GA, USA). Body mass index was calculated as weight divided by height squared (kg/m2).
Laboratory measurementsBiochemical measurements were performed for all sub-cohort members and CHD cases us-ing standard laboratory procedures. There was an overlap between the CHD cases (n=23) and the sub-cohort, therefore the total number of analyzed samples was 1,715. Sera of cases were randomly distributed among those of the sub-cohort, and all biochemical analyses were car-ried out without knowledge of disease status. Total cholesterol and glucose were determined using an automated enzymatic procedure on a Vitros 250 (Johnson & Johnson, Rochester, New York, USA). Serum iron, low density lipoprotein (LDL)- and HDL cholesterol were mea-sured using a colorimetric assay on a Hitachi 904 (Roche Diagnostics, Indianapolis, USA). Genomic DNA was extracted from buffy coats with the use of the QIAamp Blood Kit (Qiagen Inc., Valencia, California, USA). Serum PON1 enzyme activities were measured by monitoring the hydrolysis rates of para-oxon and phenylacetate on a spectrophotometer, according to the methods previously de-scribed [9, 23]. The -107C>T polymorphism was analyzed with PCR followed by hybridiza-

112
CHAPTER 8
tion with the allele-specific oligonucleotides as described by Leviev and James [24], with the modification that for the detection of the –107C allele the following allele-specific oligonucle-otides with artificial mismatch was used: 5’GGGAGGGGCGGAGCGG3’. Genotyping of the 192Q>R polymorphism was performed using a multilocus genotyping assay for candidate markers of cardiovascular disease risk [25]. Briefly, each DNA sample is amplified using two multiplex PCRs, and the alleles are genotyped simultaneously using an array of immobilized, sequence-specific oligonucleotide probes. This array of probes is blotted on plastic strips and, after staining, genotypes can be scored based on blue (positive) and white (negative) bands. Each blue band, representing a specific genotype, was scored by specific software (counting the pixel intensity of each band) and checked manually.
Statistical analysesBaseline variables were tested for a normal distribution and comparisons among the random cohort and disease outcome were performed using the two-sample T-test for normal distrib-uted continuous variables, the Mann-Whitney test for non-normal distributed continuous variables and the χ2 test for categorical variables. The χ2 test was used both to test if genotype frequencies deviated from Hardy-Weinbergequi-librium expectations and to evaluate the significance of the linkage disequilibrium between the two polymorphisms. The hazard ratios (HR) for paraoxonase- and arylesterase activity with CHD and AMI was evaluated in Cox-proportional-hazard models with an estimation procedure adapted for case-cohort designs according to unweighted method by Prentice [22], using the SAS macro written by Barlow and Ichikawa [26]. Results were summarized as HR with 95% confidence intervals (95%CI). Paraoxonase- and arylesterase activity were divided in to tertiles, where the lowest activity tertile was used as the reference group. Cox-proportional-hazard models were used to asses the effects of paraoxonase- and arylesterase activity on CHD and AMI outcome, adjusting for age and HDL-cholesterol, smoking status and alcohol consumption. In the test for trend analysis (P-trend), the –107C>T and 192Q>R polymorphisms and the tertiles of paraoxonase activity were added to the model as a continuous variable. Smoking status strongly predicted AMI incidence, for this reason, we also investigated the effects of paraoxonase activity on AMI incidence in subgroups of current-smokers, past-smokers and never-smokers, controlling for age, HDL-cholesterol and alcohol consumption. All statistical analyses were performed using SAS version 9.1.

113
PON1 and the risk for CHD and AMI
Results
Table 1 shows the baseline characteristics of the random cohort, the CHD and the AMI cases. When compared with the random cohort, the CHD and the AMI cases were older, had higher blood pressures and LDL-cholesterol levels and had lower HDL-cholesterol levels. The body mass index was only significantly higher in the CHD cases. There was a graded increase for the percentage of smokers with the severity of CHD (23% in the random cohort, 34% in the CHD cases and 45% in the AMI cases), and the consumption of alcohol was lower in both the CHD and AMI cased. Finally, paraoxonase activity was increased in the AMI cases and arylesterase activity was lower in the CHD cases, when compared to the random cohort.In the random cohort, the distribution of the 192Q>R and –107C>T polymorphisms did not deviate from the Hardy-Weinbergequilibrium. Furthermore, there was significant linkage disequilibrium between these polymorphisms (χ2 P-value < 0.001). HDL cholesterol levels and the PON1 activities among PON1 genotypes in the random cohort are shown in Table 2. No differences in HDL cholesterol levels were observed among the 192Q>R and –107C>T polymorphisms. There was an evident relationship between the PON1 genotypes and the PON1 activity measurements. The –107C>T polymorphism had the strongest influence on the arylesterase activity, with the highest arylesterase activity found in individuals with the –107CC genotype. The 192Q>R polymorphism had the strongest influence on the paraoxon hydrolysis, with the highest paraoxon hydrolytic activity found in carriers of the 192RR ge-netic variant. There was also an association of the 192Q>R polymorphism with the arylester-
Table 1. Baseline characteristicsVariable Random
Cohort n=1527
Coronary heart disease
n=211
Acute myocardial infarction
n=71
Pvalue* Pvalue†
Age, years 57 ± 6 61 ± 6 62 ± 6 <0.001 <0.001Body mass index, kg/m2 25.8 ± 3.9 26.8 ± 3.9 26.4 ± 4.0 0.001 0.245Systolic blood pressure, mm Hg 133 ± 20 143 ± 22 146 ± 25 <0.001 <0.001Diastolic blood pressure, mm Hg 79 ± 11 82 ± 11 84 ± 13 0.001 <0.001LDL cholesterol, mmol/L 3.9 ± 0.9 4.4 ± 1.0 4.4 ± 1.0 <0.001 <0.001HDL cholesterol, mmol/L 1.6 ± 0.4 1.4 ± 0.3 1.4 ± 0.4 <0.001 0.001Smoking, current/past/never 351/529/652 72/55/84 32/16/23 <0.001 <0.001Alcohol consumption, g/day 3.9 (0.4-14.1) 1.4 (0.03-9.5) 1.7 (0.03-8.2) <0.001 0.004Paraoxonase activity, U/L 114 (62-240) 115 (64-211) 152 (73-235) 0.466 0.241Arylesterase activity, U/mL 87 ± 25 82 ± 23 83 ± 24 0.005 0.144Age, body mass index, systolic blood pressure, diastolic blood pressure, LDL cholesterol, HDL cholesterol and PON1 arylesterase activity are reported as means ± standard deviation. Alcohol consumption and paraoxonase activity are presented as median values and the interquartile range. *For the comparison of the coronary heart disease cases with the random cohort. †For the comparison of the acute myocardial infarction cases with the random cohort.

114
CHAPTER 8
Tabl
e 2.
Dist
ribut
ion
HD
L ch
oles
tero
l and
PO
N1
para
oxon
ase
and
aryl
este
rase
act
iviti
es a
mon
g PO
N1
–107
C>T
and
19
2Q>R
gen
otyp
es in
the
rand
om co
hort
(n=1
527) -107
C>T
192Q
>RC
CC
TTT
QR
RRN
umbe
r of s
ubje
cts
412
752
341
785
589
140
HD
L ch
oles
tero
l, m
mol
/L1.
6 ±
0.4
1.6
± 0.
41.
6 ±
0.4
1.6
± 0.
41.
6 ±
0.4
1.6
± 0.
4Pa
raox
onas
e ac
tivity
, U/L
206
(85-
291)
102
(63-
237)
71 (4
0-15
6)*
65 (4
6-84
)22
4 (1
69-2
67)
382
(316
-460
)*A
ryle
ster
ase
activ
ity, U
/mL
104
± 22
86 ±
21
68 ±
21*
84 ±
24
89 ±
25
94 ±
25*
HD
L ch
oles
tero
l and
ary
lest
eras
e ar
e re
port
ed a
s mea
ns ±
stan
dard
dev
iatio
n. P
arao
xona
se a
ctiv
ity is
pre
sent
ed a
s the
m
edia
n va
lue
and
the
inte
rqua
rtile
rang
e. *P
-tre
nd<
0.00
1.
Tabl
e 4.
Cor
onar
y he
art d
iseas
e an
d ac
ute
myo
card
ial i
nfar
ctio
n ha
zard
ratio
s (w
ith 9
5% co
nfide
nce
inte
rval
s) b
y te
rtile
s of p
arao
xona
se- a
nsd
aryl
este
rase
act
ivity
.C
oron
ary
hear
t dise
ase
haza
rd ra
tios (
95%
CI)
Acu
te m
yoca
rdia
l inf
arct
ion
haza
rd ra
tios (
95%
CI)
Mod
el 1
Mod
el 2
Mod
el 3
Mod
el 1
Mod
el 2
Mod
el 3
Para
oxon
ase
activ
ity<7
3 U
/L1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)73
-204
U/L
1.08
(0.7
6-1.
54)
1.10
(0.7
7-1.
56)
1.07
(0.7
5-1.
55)
1.34
(0.7
1-2.
54)
1.34
(0.7
1-2.
53)
1.31
(0.6
9-2.
50)
>204
U/L
1.04
(0.7
2-1.
50)
1.14
(0.7
8-1.
65)
1.18
(0.8
0-1.
72)
1.74
(0.9
4-3.
24)*
1.88
(1.0
1-3.
50)†
,‡,
2.07
(1.1
1-3.
84)§
,||A
ryle
ster
ase
activ
ity
<76
U/m
L1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)1.
00 (R
efer
ence
)76
-97
U/m
L0.
98 (0
.70-
1.38
)1.
08 (0
.76-
1.52
)1.
07 (0
.75-
1.51
)0.
87 (0
.49-
1.54
)0.
92 (0
.51-
1.64
)0.
91 (0
.50-
1.64
)>9
7 U
/mL
0.74
(0.5
0-1.
08)¶
0.90
(0.6
0-1.
34)
1.01
(0.6
7-1.
51)
0.85
(0.4
6-1.
56)
0.92
(0.4
9-1.
74)
1.17
(0.6
2-2.
19)
Mod
el 1
was
adj
uste
d fo
r age
, mod
el 2
was
adj
uste
d fo
r age
and
HD
L-ch
oles
tero
l and
mod
el 3
was
adj
uste
d fo
r age
, HD
L-ch
oles
tero
l, sm
okin
g st
atus
an
d al
coho
l con
sum
ptio
n. *P
-tre
nd=0
.062
, †P=
0.04
6 w
hen
com
pare
d to
the
refe
renc
e gr
oup,
‡ P-
tren
d=0.
045,
§P=
0.02
2 w
hen
com
pare
d to
the
refe
renc
e gr
oup,
||P-
tren
d=0.
029,
¶P-
tren
d=0.
107.

115
PON1 and the risk for CHD and AMI
ase activity (P-trend<0.001), and the –107C>T polymorphism with the paraoxonase activity (P-trend<0.001).Table 3 shows the relationship of the PON1 genetic variants –107C>T and 192Q>R with the HR for CHD and AMI. Overall, the effects of both genotypes on CHD and AMI incidence were too weak to draw conclusions. The hazard ratios for tertiles of paraoxonase- and arylesterase activity to develop CHD or AMI, are presented in Table 4. Although not reaching statistical significance, the increased arylesterase activity tended to associate with a lower incidence of CHD in the age adjusted model (second and third tertile HR: 0.98 and 0.74, P-trend=0.107). This effect , however, de-clined in the multivariate adjusted models. In general, there was no apparent effect of PON1 paraoxonase- and arylesterase activity on the incidence of CHD. When investigating the role of PON1 on the incidence of AMI, paraoxonase activity tended to associate with a higher in-cidence of AMI in the age adjusted model, hazard ratios for the second and third tertile were 1.34 and 1.74 respectively (P-trend=0.062). The strength of the association of paraoxonase activity with AMI became stronger after additional adjustments for HDL-cholesterol, smok-ing status and alcohol consumption, with the highest tertile significantly associated with a two-fold increased incidence of AMI (HR: 2.07, 95% CI:1.11-3.84, P=0.022). The combined effects of smoking and paraoxonase activity on AMI incidence adjusted for age, HDL-cholesterol and alcohol are presented in Figure 1. In people who had never smoked, increased paraoxonase activity was a risk factor for a higher incidence of AMI: the second and third tertile of paraoxonase activity HR were 4.1 (95%CI: 0.9-18.5, P=0.063) and 4.7 (95%CI: 1.2-18.5, P=0.026) respectively (P-trend=0.009). In addition, the high-paraoxonase/cur-rent-smokers subgroup had a 19.2 fold increased risk for AMI incidence (95%CI: 5.3-69.5, P<0.0001) when compared to the low-paraoxonase/never-smokers subgroup.
Table 3. Coronary heart disease and acute myocardial infarction hazard ratios (with 95% confidence intervals) by PON1 –107C>T and 192Q>R genotype
Coronary heart disease hazard ratios (95% CI)
Acute myocardial infarction hazard ratios (95% CI)
–107C>T CC 1.00 (Reference) 1.00 (Reference)CT 0.93 (0.66-1.31) 0.97 (0.56-1.69)TT 0.79 (0.51-1.20) 0.67 (0.32-1.39)192Q>RQQ 1.00 (Reference) 1.00 (Reference)QR 1.06 (0.78-1.44) 1.39 (0.83-2.32)RR 0.78 (0.43-1.42) 1.37 (0.59-3.21)Hazard models were adjusted for age.

116
CHAPTER 8
Discussion
In this cohort of women, we found no evidence for a relationship between PON1 genetic vari-ants, activity and CHD in general, or between the PON1 genetic variants and AMI. Increased paraoxonase activity, however, was an AMI risk factor. In addition, when the highest para-oxonase activity tertile in current-smokers was compared to the lowest paraoxonase activity tertile in never-smokers, the combined effects of paraoxonase activity with smoking status predicted a more than 19 fold increased risk for AMI incidence. Many population studies have shown that smoking is a major risk-factor for cardiovascular disease, and, in agreement, we found that when compared to the random cohort, the per-centage of smokers was higher among people who developed an AMI. We also found that paraoxonase activity was a risk factor for AMI. This effect appeared to be influenced by smok-ing, because the risk for AMI incidence was strikingly higher for high-paraoxonase/current-smokers when compared to low-paraoxonase/never-smokers. Although these findings must viewed with some reservedness since they were based on a small number of cases per sub-group, they do underline the importance of stratifying for smoking status when investigating the effect of PON1 on cardiovascular disease. Studies on a direct relationship between PON1 paraoxonase activity and the incidence of cardiovascular disease are scarce [19]. In contrast, there are many studies on the effects of the 192Q>R polymorphism and cardiovascular disease (reviewed in [11]). In this respect, it is important to realize that paraoxon is most efficiently hydrolyzed by the PON1-192RR genetic variant [2, 3], and, as shown in a recent meta-analysis among 43 studies involving a total of 11212 CHD cases and 12786 controls, subjects with the 192R-allele have a significant 1.12 fold increased risk for CHD [12]. Although it is questionable whether this slight increase in relative risk found for the 192R-allele has a relevant impact on CHD, it does support our findings, that elevated paraoxonase activities at baseline were associated with an increased risk to develop AMI. We found that paraoxonase activity was a risk factors for AMI, but there was no strong evi-dence that PON1 polymorphisms were associated with the onset of CHD or AMI. Many previous reports on PON1 and CHD conclude that PON1 phenotype (i.e. activity, concen-tration measurements and/or the combination of both) is a stronger predictor of CHD than PON1 genotype [10, 13-15]. Because a large number of cases is required to study the effects of genetic variation on disease outcome [27], the absence of a gene-disease relationship in this study (as in many other studies) may be attributed to a lack of power to detect significant effects. These findings once more implicate, that studies on the role of PON1 and the onset of CHD are only meaningful when also measurements of PON1 concentrations and activities have been included. Relative to the number of PON1 genetic studies on the onset of cardiovascular disease, there have been a few case-control studies that have included a quantification of PON1 activity and

117
PON1 and the risk for CHD and AMI
concentration (reviewed in [14]). Before drawing firm conclusions from these kind of stud-ies, it must be stressed that a major limitation of the case-control study design is that blood is drawn after the cardiovascular event has taken place. One can not distinguish whether PON1 activity contributed to the manifestation of the event or, conversely, the event itself influenced the PON1 activity. To overcome this problem a prospective study design is needed. The pres-ent study is the second prospective investigation on the effects of PON1 activity and con-centration to cardiovascular endpoints. Mackness et al. previously reported, that low serum PON1 activity toward paraoxon is an independent risk factor for coronary events in men with preexisting CHD, while no relationship is observed in men without preexisting CHD [19]. We have excluded all women with preexisting CHD at baseline, and, similarly to the results of Mackness et al., we found no relationship between PON1 activity and the incidence of CHD in the general population. Remarkably, we found that an elevated paraoxonase activity was a risk factor for AMI. This seems to be in conflict with the findings of Mackness et al. in the men with preexisting CHD. However, PON1 levels and/or activity diminish after an inflammatory acute phase response, as seen in animals [28, 29], or in humans after a myocardial infarction [30]. Consequently, a decrease of PON1 levels and/or activity due to a previous CHD event, may explain the association between low PON1 activity and recurrent CHD in the population studied by Mackness et al [19].
Figure 1. Combined effects of paraoxonase activity (in tertiles) and smoking status (current-, past- and never smoking) on the incidence of AMI. The hazard ratios (HR) and 95% confidence intervals (95% CI), are displayed in the bars and represent the comparison with the low paraoxonase/non smoking reference group. HR are controlled for age, HDL cholesterol and alcohol consumption. *HR: 4.7 (1.2-18.5), P=0.026. **HR:1.6 (0.4-7.0), P=0.532.
CurrentPast
Never
Low (<73)
Intermediate (73-204)
High (>204)
0
2
4
6
8
10
12
14
16
18
20
Smoking status
Paraoxonaseactivity (U/L)
Acu
te m
yoca
rdia
l inf
arct
ion
haza
rd r
atio
s (9
5% C
I)
Reference
19.2(5.3-69.5)P<0.0001
11.1(3.1-39.5)P=0.0002
7.7(2.3-26.0)P=0.0009
4.1(0.9-18.5)P=0.0634.2
(1.0-16.9)P=0.046
3.9(1.1-14.3)P=0.038
*
**

118
CHAPTER 8
In this study, paraoxonase activity was a risk factor for AMI only, and not for CHD in gen-eral. CHD is an umbrella-term covering different stages of (chronic stable and acute) heart problems. One of the first steps in atherogenesis is the formation of a lipid-rich plaque, which forms the basis for all forms of CHD [31]. At a later stage in the atherosclerotic process, the physiological disruption of the atherosclerotic plaque and, as a result, the formation of a thrombotic occlusion, causes the most acute and serious clinical manifestations of CHD, i.e. AMI [32]. Our findings that paraoxonase was a risk factor for AMI, suggest that PON1 may be of importance in the later stages of atherosclerosis. This does not only warrant further investigation on the role of PON1 in plaque instability and thrombosis, but also suggests that important effects of PON1 will be missed when all forms of CHD are grouped as a common CHD outcome. In a previous investigation in patients with familial hypercholesterolemia we found an asso-ciation between PON1 genotypes and HDL cholesterol levels, and hypothesized that PON1 enhances the function of the HDL particle [33]. In this general population of women, we observed a positive correlation for both arylesterase- and paraoxonase activity with HDL-cholesterol levels, however, the strength of the association of paraoxonase activity with the incidence of AMI was only marginally affected by adjustments for HDL-cholesterol levels, thus suggesting that the mechanism by which paraoxonase activity forms a risk factor for AMI incidence, was not primarily via an interaction with the function of the HDL-cholesterol particle. It must be noted that the exact physiological function of PON1 is still unclear, but the supposed anti-oxidative properties of PON1 [34], and a secondary inflammatory response [35], may be important parameters for the occurrence of AMI. Finally, we will address the strengths and weaknesses of our investigation. We were aware that the small number of CHD cases was a limitation to our study. At the time of investigation, there were only 211 cases of CHD (of which 71 cases of AMI) in the total cohort of 17,357 women, and it can be expected that more events will occur during a longer period of follow-up. Nevertheless, our prospective study design was still highly favorable over findings from case-control studies, because we could be sure that the effects of PON1 preceded the onset of CHD, and so, we could assess a causal role of PON1 in this process. In conclusion, several animal models and in vitro studies have suggested a protective function of PON1 against the onset of cardiovascular disease. On the other hand, we demonstrated that rather than being protective, paraoxonase activity was a risk factor for the incidence of AMI, and that this risk seems to be amplified by smoking. Future (prospective) investigations are needed to confirm this hypothesis.
Acknowledgments
The authors thank J.H. van Kats-Renaud for technical assistance.

119
PON1 and the risk for CHD and AMI
References
[1] D.I. Draganov, J.F. Teiber, A. Speelman, Y. Osawa, R. Sunahara, B.N. La Du, Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities, J Lipid Res 46 (2005) 1239-1247.[2] R. Humbert, D.A. Adler, C.M. Disteche, C. Hassett, C.J. Omiecinski, C.E. Furlong, The molecular basis of the human serum paraoxonase activity polymorphism, Nat.Genet. 3 (1993) 73-76.[3] H.G. Davies, R.J. Richter, M. Keifer, C.A. Broomfield, J. Sowalla, C.E. Furlong, The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin, Nat Genet 14 (1996) 334-336.[4] D.M. Shih, L. Gu, Y.R. Xia, M. Navab, W.F. Li, S. Hama, L.W. Castellani, C.E. Furlong, L.G. Costa, A.M. Fogelman, A.J. Lusis, Mice lacking serum paraoxo-nase are susceptible to organophosphate toxicity and atherosclerosis, Nature 394 (1998) 284-287.[5] C.J. Ng, D.M. Shih, S.Y. Hama, N. Villa, M. Navab, S.T. Reddy, The paraoxonase gene family and athero-sclerosis, Free Radic Biol Med 38 (2005) 153-163.[6] S. Adkins, K.N. Gan, M. Mody, B.N. La Du, Molecu-lar basis for the polymorphic forms of human serum paraoxonase/arylesterase: glutamine or arginine at position 191, for the respective A or B allozymes, Am J Hum Genet 52 (1993) 598-608.[7] V.H. Brophy, R.L. Jampsa, J.B. Clendenning, L.A. McKinstry, G.P. Jarvik, C.E. Furlong, Effects of 5’ regu-latory-region polymorphisms on paraoxonase-gene (PON1) expression, Am J Hum Genet 68 (2001) 1428-1436.[8] T. Kujiraoka, T. Oka, M. Ishihara, T. Egashira, T. Fujioka, E. Saito, S. Saito, N.E. Miller, H. Hattori, A sandwich enzyme-linked immunosorbent assay for human serum paraoxonase concentration, J Lipid Res 41 (2000) 1358-1363.[9] H.W. Eckerson, C.M. Wyte, B.N. La Du, The human
serum paraoxonase/arylesterase polymorphism, Am J Hum Genet 35 (1983) 1126-1138.[10] R.J. Richter, C.E. Furlong, Determination of paraoxonase (PON1) status requires more than geno-typing, Pharmacogenetics 9 (1999) 745-753.[11] L.G. Costa, T.B. Cole, G.P. Jarvik, C.E. Furlong, Functional genomic of the paraoxonase (PON1) poly-morphisms: effects on pesticide sensitivity, cardiovas-cular disease, and drug metabolism, Annu Rev Med 54 (2003) 371-392.[12] J.G. Wheeler, B.D. Keavney, H. Watkins, R. Collins, J. Danesh, Four paraoxonase gene polymorphisms in 11212 cases of coronary heart disease and 12786 con-trols: meta-analysis of 43 studies, Lancet 363 (2004) 689-695.[13] B. Mackness, G.K. Davies, W. Turkie, E. Lee, D.H. Roberts, E. Hill, C. Roberts, P.N. Durrington, M.I. Mackness, Paraoxonase status in coronary heart dis-ease: are activity and concentration more important than genotype?, Arterioscler Thromb Vasc Biol 21 (2001) 1451-1457.[14] M. Mackness, B. Mackness, Paraoxonase 1 and atherosclerosis: is the gene or the protein more impor-tant?, Free Radic Biol Med 37 (2004) 1317-1323.[15] G.P. Jarvik, L.S. Rozek, V.H. Brophy, T.S. Hatsu-kami, R.J. Richter, G.D. Schellenberg, C.E. Furlong, Paraoxonase (PON1) phenotype is a better predictor of vascular disease than is PON1(192) or PON1(55) genotype, Arterioscler Thromb Vasc Biol 20 (2000) 2441-2447.[16] E. Nishio, Y. Watanabe, Cigarette smoke extract inhibits plasma paraoxonase activity by modification of the enzyme’s free thiols, Biochem Biophys Res Com-mun 236 (1997) 289-293.[17] R.W. James, I. Leviev, A. Righetti, Smoking is as-sociated with reduced serum paraoxonase activity and concentration in patients with coronary artery disease, Circulation 101 (2000) 2252-2257.[18] M.S. van der Gaag, A. van Tol, L.M. Scheek, R.W.

120
CHAPTER 8
James, R. Urgert, G. Schaafsma, H.F. Hendriks, Daily moderate alcohol consumption increases serum paraoxonase activity; a diet-controlled, randomised intervention study in middle-aged men, Atherosclero-sis 147 (1999) 405-410.[19] B. Mackness, P. Durrington, P. McElduff, J. Yarnell, N. Azam, M. Watt, M. Mackness, Low paraoxonase ac-tivity predicts coronary events in the Caerphilly Pro-spective Study, Circulation 107 (2003) 2775-2779.[20] L.K. Boker, P.A. van Noord, Y.T. van der Schouw, N.V. Koot, H.B. Bueno de Mesquita, E. Riboli, D.E. Grobbee, P.H. Peeters, Prospect-EPIC Utrecht: study design and characteristics of the cohort population. European Prospective Investigation into Cancer and Nutrition, Eur J Epidemiol 17 (2001) 1047-1053.[21] R.M. Herings, A. Bakker, B.H. Stricker, G. Nap, Pharmaco-morbidity linkage: a feasibility study com-paring morbidity in two pharmacy based exposure cohorts, J Epidemiol Community Health 46 (1992) 136-140.[22] R.L. Prentice, A case-cohort design for epidemio-logic cohort studies and disease prevention trials., Bi-ometrika 73 (1986) 1-11.[23] K.N. Gan, A. Smolen, H.W. Eckerson, B.N. La Du, Purification of human serum paraoxonase/aryleste-rase. Evidence for one esterase catalyzing both activi-ties, Drug Metab Dispos 19 (1991) 100-106.[24] I. Leviev, R.W. James, Promoter polymorphisms of human paraoxonase PON1 gene and serum paraoxo-nase activities and concentrations, Arterioscler Thromb Vasc Biol 20 (2000) 516-521.[25] S. Cheng, M.A. Grow, C. Pallaud, W. Klitz, H.A. Er-lich, S. Visvikis, J.J. Chen, C.R. Pullinger, M.J. Malloy, G. Siest, J.P. Kane, A multilocus genotyping assay for can-didate markers of cardiovascular disease risk, Genome Res 9 (1999) 936-949.[26] W.E. Barlow, L. Ichikawa, D. Rosner, S. Izumi, Anal-ysis of case-cohort designs, J Clin Epidemiol 52 (1999) 1165-1172.
[27] H.M. Colhoun, P.M. McKeigue, G. Davey Smith, Problems of reporting genetic associations with com-plex outcomes, Lancet 361 (2003) 865-872.[28] V.G. Cabana, C.A. Reardon, N. Feng, S. Neath, J. Lukens, G.S. Getz, Serum paraoxonase: effect of the apolipoprotein composition of HDL and the acute phase response, J Lipid Res 44 (2003) 780-792.[29] K.R. Feingold, R.A. Memon, A.H. Moser, C. Grun-feld, Paraoxonase activity in the serum and hepatic mRNA levels decrease during the acute phase response, Atherosclerosis 139 (1998) 307-315.[30] A. Ayub, M.I. Mackness, S. Arrol, B. Mackness, J. Patel, P.N. Durrington, Serum paraoxonase after myo-cardial infarction, Arterioscler.Thromb.Vasc.Biol. 19 (1999) 330-335.[31] D. Steinberg, Atherogenesis in perspective: hy-percholesterolemia and inflammation as partners in crime, Nat Med 8 (2002) 1211-1217.[32] P.K. Shah, Mechanisms of plaque vulnerability and rupture, J Am Coll Cardiol 41 (2003) 15S-22S.[33] T.M. van Himbergen, M. Roest, J. de Graaf, E.H. Jansen, H. Hattori, J.J. Kastelein, H.A. Voorbij, A.F. Stalenhoef, L.J. van Tits, Indications that paraoxonase-1 contributes to plasma high density lipoprotein levels in familial hypercholesterolemia, J Lipid Res 46 (2005) 445-451.[34] T.M. van Himbergen, L.J. van Tits, M.P. Hectors, J. de Graaf, M. Roest, A.F. Stalenhoef, Paraoxonase-1 and linoleic acid oxidation in familial hypercholeste-rolemia, Biochem Biophys Res Commun 333 (2005) 787-793.[35] A. Mertens, P. Holvoet, Oxidized LDL and HDL: antagonists in atherothrombosis, Faseb J 15 (2001) 2073-2084.

9General discussion


123
General discussion
In vivo et in vitro!
Although paraoxonase (PON1) has been the subject of different fields of research, there is little understanding about the role of PON1 in the human body. Indeed, several functional properties of PON1 are clarified in controlled in vitro experiments. But still, it remains to be established whether findings from those controlled experiments, are applicable to the com-plex biological system of the human body. In addition, experiments with transgenic animals give insight into the in vivo functions of PON1, but caution is needed when extrapolating these findings to humans. Therefore, to determine the true in vivo effects of PON1, further support from population studies in defined cohorts of human subjects is needed. If an effect is found repeatedly in independent investigations carried out by different investigators, it can be concluded that the observed effect is indeed a true one. In this thesis we investigate the role of PON1 on the development of cardiovascular disease (CVD) in humans. We have studied the effects of PON1 genetic variation as well as the ef-fects of PON1 concentration and activities in different study populations posing the follow-ing research questions: does PON1 inhibit the formation of oxidized LDL in vivo? If so, does this then lead to a reduced inflammatory response? What is the influence of PON1 on the metabolism of lipoproteins? And finally, does PON1 play a role in the onset of cardiovascular disease?
PON1, LDL oxidation, and inflammation
It has been shown in vitro, that the inhibition of the oxidation of low-density lipoprotein (LDL) is a potential biological function of PON1 [1]. This is unambiguously supported by animal experimental work: high-density lipoprotein (HDL) isolated from PON1-deficient mice does not protect LDL from oxidation [2], while HDL isolated from PON1 transgenic mice (having increased plasma levels of human PON1) is protective against LDL oxidation [3]. To test the hypothesis whether the oxidation of LDL is influenced by PON1 in vivo, data from human population studies are essential. In Chapter 3, we investigate the effects of PON1 genotype on LDL oxidation in heavy smokers. We demonstrated that the PON1 genetic vari-ants 55L>M and 192Q>R were not associated with LDL oxidizability or levels of circulating antibodies directed against oxLDL. This suggests that PON1 does not influence the oxidation of LDL in vivo, but it could also mean that the effect of PON1 is too small to result in altered in vitro oxidizability of LDL. Chapters 4 and 5 describe a more direct approach to study the effects of PON1 on concentrations of LDL-oxidation products in patients with familial hy-percholesterolemia (FH). For a better understanding of our study design, it is necessary to explain that LDL oxidation is initiated by the seeding of oxidized lipids in the LDL particle [4]. As a result of a continuous oxidative stimulus, the protein moiety of the LDL particle, the

124
CHAPTER 9
apolipoprotein (apo) B, becomes modified [4]. Oxidatively modified apo B can be detected and measured by monoclonal antibodies, resulting in a quantitative measurement of circulat-ing oxidized LDL (oxLDL) in blood [5]. In patients with FH, however, there was no associa-tion of PON1 with the concentration of circulating oxLDL particles (Chapter 4). On the other hand, in the same population, the paraoxonase activity of the PON1 enzyme (determined by the 192R genetic variant) correlated negatively with oxidized linoleic acid (Chapter 5), the most abundant fatty acid present in LDL. The oxidation products of linoleic acid are stable markers of lipid oxidation, and reflect the early stages of LDL oxidation [6]. Furthermore, oxidized linoleic acid has a significant biological role as an activator of the peroxisome pro-liferator-activated receptor (PPAR) gamma [7]. This receptor is involved in the regulation of oxLDL uptake and cholesterol efflux from macrophages [8,9]. The effect of PON1 on linoleic acid oxidation products could provide an important link with lipid metabolism via the reverse cholesterol transport and the local inflammatory response of oxLDL uptake by macrophages. Thus, it can be hypothesized that PON1 prevents the early stages of the LDL oxidation (lin-oleic acid oxidation), but this antioxidative potential did not result in protecting circulating LDL from complete oxidative modification, as measured by oxLDL. An overall limitation of our approach is that we are restricted to LDL oxidation measurements in the blood. This does not necessarily translate to local antioxidative effects of PON1 in the vessel wall. The proportion of oxLDL particle re-entering the circulation may be an underes-timated representation of the oxidative processes occurring in the vessel wall, and therefore, the local antioxidative effects of PON1 may, in fact, be stronger than what can be concluded from our measurement of oxidation products in the circulation.Chapter 4 describes the association of PON1 with inflammation. OxLDL promotes inflam-matory processes which may lead to increased levels of inflammatory markers, like C-reac-tive protein (CRP). Although there was no relation between PON1 and LDL oxidizability or oxLDL levels in the FH patient population, PON1 levels did (weakly) correlate with CRP levels in these patients. Possibly, PON1 exhibits local effects which are not of influence on cir-culating LDL or oxLDL, but do result in increased inflammatory markers. To our knowledge, no association between PON1 levels and CRP has been reported before, and therefore, these findings will be explored in future studies.
PON1 and the interaction with lipid metabolism
In addition to genotype, it is generally assumed that HDL cholesterol levels are the major determinant for PON1 bioavailability in blood. This assumption is based on knowledge that in blood, the PON1 protein is attached to HDL particles, and accordingly, several studies have reported positive correlations between PON1 and HDL-cholesterol levels [10, 11]. On the other hand, as the following calculation will indicate, the contribution of HDL to PON1

125
General discussion
bioavailability is weaker than we initially expected. In the ASAP study population [12], the average concentration of PON1 was 78 μg/mL [13], and the average apoA1 concentration 114 mg/dL [14]. The molecular weights of PON1 and apoA1 are approximately 42 kDa and 28 kDa, respectively. Thus, the molar PON1 concentration is approximately 22-fold lower than the molar apoA1 concentration. It is known that stable HDL particles contain, on average, 3 apoA1 proteins [15], and functional PON1 is a dimer [16], this means that per 14 functional HDL particles there is one functional PON1 dimer. Because the majority of HDL particles do not contain PON1, a fluctuation in the number of HDL particles leads to variations in over-capacity with little effect on the PON1 concentration.There is a growing support for the theory that PON1 can influence the function of HDL. From the 3D crystal structure of PON1 it has been extrapolated that the active site is oriented toward the surface of the HDL particle [17]. This suggests that PON1’s actions could involve the HDL particle itself rather than another particle. Accordingly, PON1 was able to protect the HDL particle against oxidation in vitro, and preserve an essential function of the HDL-particle, namely, the uptake of cholesterol from macrophages [18]. For this reason we looked at the role of PON1 in the metabolism of HDL. Our data show that not only PON1 levels and activities, but also PON1 genotype contributes to HDL cholesterol levels in patients with FH, suggesting a causal relationship (Chapter 4). In support, a number of different investigations have reported similar effects of PON1 genotype on HDL-cholesterol levels [13,19-24]. And therefore, it is becoming increasingly clear that PON1 determines HDL-cholesterol levels rather than merely reflecting them. When the patients with FH, as discussed in this thesis, were treated with statins, PON1 showed a clear effect on the metabolism of HDL (Chapter 6). Apart from the primary de-crease in LDL-cholesterol levels, statin treatment leads to an increase of HDL-cholesterol lev-els in FH patients. The PON1 and HDL concentration at baseline (i.e. before the start of the statin therapy) predicted the HDL response, ranging from a 16 % increase to no increase at all. Surprisingly, the paraoxonase activity (determined by measurement of the 192R genetic variant) had no effect on the HDL changes in the statin treated patients. This suggests that the bioavailability of PON1 rather than the paraoxonase activity of PON1, has an important function in the HDL metabolism. In patients with familial combined hyperlipidemia (FCH), PON1 arylesterase activity (a sur-rogate measurement of PON1 concentration) was associated with the atherogenic lipopro-teins (LDL, VLDL and TG) and not with HDL (chapter). These data may indicate that PON1 contributes to the dyslipidemia found in FCH patients. Similar observations were made very recently in patients with severe carotid artery disease, by an other group of investigators: PON1 correlated with HDL in the healthy controls and with LDL and VLDL in the cases [25]. Therefore, it seems that in certain diseased states, the association of PON1 with lipoproteins has shifted from the anti-inflammatory HDL to atherogenic lipoprotein particles, like LDL and VLDL. We investigated whether there was a physical transfer of the PON1 particle from

126
CHAPTER 9
the HDL particles to non-HDL particles. This was done by measuring PON1 activity in sepa-rate lipoprotein fractions isolated from the serum of 5 FCH patients. We could only detect PON1 activity in the HDL fraction, indicating that there is no transfer of PON1 particles to other lipoproteins. Further studies are needed to understand the mechanism underlying the altered association of PON1 with other lipids and lipoproteins and its clinical consequences.
PON1 and cardiovascular disease
The intima-media thickness (IMT) of the carotid artery is a marker of carotid atherosclerosis and has been used as a pre-clinical cardiovascular endpoint in a number of PON1 investiga-tions [20,26-39]. Although a number of studies reported a significant association of PON1 genetic variants with IMT [20,27,29,30,33,34,37], a large number of negative studies have also been published [26,28,31,32,35, 36,38,39]. Moreover, as result of a negative publication bias, the number of studies reporting no PON1 effect on IMT is very likely an underestima-tion of the true number of negative studies. In this thesis, the effects of PON1 on IMT were investigated in populations of smokers (Chapter 3) and patients with FH (chapter). In both populations we did not find an association of the PON1 genetic variants with measures of ca-rotid IMT. In addition, in the patients with FH, no correlation between PON1 concentration or the activity measurements and IMT was observed. These negative findings are not surpris-ing because in these patients, even HDL cholesterol levels which are powerful predictors of cardiovascular disease had only a marginal effect on carotid IMT (chapter). We therefore believe that, if anything, the effects of PON1 are too weak to significantly influence IMT in the studied populations. The lack of an association between PON1 and carotid IMT does not exclude a relationship of PON1 with cardiovascular disease. The IMT measurement reflects the accumulation of lipids in the artery wall, which is an early step in atherogenesis. It is possible that PON1 plays a role in the later stages of CVD, characterized by acute processes such as the rupture of an atherosclerotic plaque and as a result, blockage of the vessel by a thrombotic plug. Our prospective investigation indeed suggests that PON1 predominantly plays a causal role in the onset of the acute forms of CVD. We observe that an increased paraoxonase activity of PON1 (determined by the 192R genetic variant) was a risk factor for developing an acute myocardial infarction. These results are in line with a recently published meta-analysis summarizing the outcomes of 43 investigations on the 192Q>R polymorphism and CVD [40]. In this meta-analysis, the 192R variant demonstrated to be a (weak) risk factor for CVD [40]. Still, the out-come of our prospective investigation looks controversial: rather than being anti-atherogenic (which could be expected based on in vitro studies [1], and animal models [2]) an increased paraoxonase activity of the PON1 enzyme in blood, was found to be a risk factor for devel-opment of an acute form of CVD. A dual functionality of PON1, i.e. one enzyme possessing

127
General discussion
multiple activities toward different substrates, may explain this apparent contradiction (Fig-ure). There is a growing body of evidence that PON1 is a lactonase [16, 41], which most likely has an important physiological function and, therefore, is conserved throughout evolution. Lactonase activity is not polymorphic, i.e. not influenced by genetic variants in the coding region of the enzyme. The inter-individual variation in PON1’s lactonase activity is therefore a result of variations in the PON1 concentration. PON1’s activity towards paraoxon (paraox-onase activity), is influenced by the 192Q>R polymorphism. Although paraoxon is not the in vivo substrate for PON1, this activity measure reflects a polymorphic action of PON1 in vivo, which leads to an increased risk for acute forms of CVD (chapter). We suggest that the overall effect of PON1 in vivo depends on the sum of beneficial (lactonase) and harmful (paraox-onase) activities (Figure). In people who are 192QQ homozygotes (resulting in PON1 with a low paraoxonase activity), the beneficial (lactonase) activity prevails over the harmful effects of the paraoxonase activity. However, in carriers of one or two 192R alleles, the paraoxonase activity is more prominently present and overshadows the beneficial activity. This increased paraoxonase activity may cause the PON1 particle to have negative effects in the human body, with the severity of distress depending on the number of 192R alleles.
PON1 and smoking
Our prospective investigation showed that increased PON1 paraoxonase activity and smok-ing had synergistic effects on the risk for acute CVD (Chapter). This means that the com-bined effect (a 19 fold increased risk) was higher than the sum of the individual effects of smoking (more than 7 fold increased risk) and increased paraoxonase activity (more than 4 fold increased risk). Our prospective investigation is in agreement with several observational studies reporting different PON1 effects in smokers and non-smokers [30,42-44]. Because the underlying mechanism for these observations is not well understood, the interaction between smoking and PON1 deserves the attention of future research.
Final conclusions and future perspectives
Based on the findings presented in this thesis, we suggest that PON1 plays multiple roles in the human body (Figure). Possibly, PON1’s lactonase activity is a beneficial activity which is an important factor for the metabolism of lipids and lipoproteins. In contrast, PON1’s ability to hydrolyze paraoxon, is associated with an in vivo action that has detrimental effects and results in an increased risk for acute forms of CVD.

128
CHAPTER 9
Future studies should focus on:The physiological function of PON1’s lactonase activityThe underlying mechanism by which PON1 is involved in the metabolism of
serum lipids and lipoproteinsThe interaction of PON1 with statin therapy, and its clinical importanceThe role of PON1 in FCH patientsThe mechanism by which PON1 is cardioprotective as well as a risk factor for acute
coronary events The interaction of PON1 with smoking, and its consequences for the human body
••
•••
•
Figure. Hypothetical model of PON1’s dual functionality. Circles indicate the total pool of PON1 paraoxonase (moon-shaped) and lactonase activities. The size of the arrows and the shades of grey indicate the rate of the paraoxonase and lactonase activity, respectively.
Harmful (paraoxonase) activity
Beneficial (lactonase) activity
PON1 activitiesof a 192QQsubject
PON1 activitiesof a 192QRsubject
PON1 activitiesof a 192RRsubject

129
General discussion
References
[1] M.I. Mackness, S. Arrol, P.N. Durrington, Paraoxo-nase prevents accumulation of lipoperoxides in low-density lipoprotein, FEBS Lett 286 (1991) 152-154.[2] D.M. Shih, L. Gu, Y.R. Xia, M. Navab, W.F. Li, S. Hama, L.W. Castellani, C.E. Furlong, L.G. Costa, A.M. Fogelman, A.J. Lusis, Mice lacking serum paraoxo-nase are susceptible to organophosphate toxicity and atherosclerosis, Nature 394 (1998) 284-287.[3] A. Tward, Y.R. Xia, X.P. Wang, Y.S. Shi, C. Park, L.W. Castellani, A.J. Lusis, D.M. Shih, Decreased atheroscle-rotic lesion formation in human serum paraoxonase transgenic mice, Circulation 106 (2002) 484-490.[4] S. Parthasarathy, N. Santanam, S. Ramachandran, O. Meilhac, Oxidants and antioxidants in atherogen-esis. An appraisal, J Lipid Res 40 (1999) 2143-2157.[5] P. Holvoet, J. Donck, M. Landeloos, E. Brouwers, K. Luijtens, J. Arnout, E. Lesaffre, Y. Vanrenterghem, D. Collen, Correlation between oxidized low density li-poproteins and von Willebrand factor in chronic renal failure, Thromb Haemost 76 (1996) 663-669.[6] P. Spiteller, G. Spiteller, 9-Hydroxy-10,12-octadeca-dienoic acid (9-HODE) and 13-hydroxy-9,11-octa-decadienoic acid (13-HODE): excellent markers for lipid peroxidation, Chemistry and Physics of Lipids 89 (1997) 131-139.[7] L. Nagy, P. Tontonoz, J.G. Alvarez, H. Chen, R.M. Evans, Oxidized LDL regulates macrophage gene ex-pression through ligand activation of PPARgamma, Cell 93 (1998) 229-240.[8] P. Tontonoz, L. Nagy, J.G. Alvarez, V.A. Thomazy, R.M. Evans, PPARgamma promotes monocyte/macro-phage differentiation and uptake of oxidized LDL, Cell 93 (1998) 241-252.[9] A. Chawla, W.A. Boisvert, C.H. Lee, B.A. Laffitte, Y. Barak, S.B. Joseph, D. Liao, L. Nagy, P.A. Edwards, L.K. Curtiss, R.M. Evans, P. Tontonoz, A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in
cholesterol efflux and atherogenesis, Mol Cell 7 (2001) 161-171.[10] M.C. Blatter Garin, C. Abbott, S. Messmer, M. Mackness, P. Durrington, D. Pometta, R.W. James, Quantification of human serum paraoxonase by en-zyme-linked immunoassay: population differences in protein concentrations, Biochem J 304 ( Pt 2) (1994) 549-554.[11] M.C. Blatter, R.W. James, S. Messmer, F. Barja, D. Pometta, Identification of a distinct human high-den-sity lipoprotein subspecies defined by a lipoprotein-as-sociated protein, K-45. Identity of K-45 with paraoxo-nase, Eur J Biochem 211 (1993) 871-879.[12] T.J. Smilde, S. van Wissen, H. Wollersheim, M.D. Trip, J.J. Kastelein, A.F. Stalenhoef, Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial, Lancet 357 (2001) 577-581.[13] T.M. van Himbergen, M. Roest, J. de Graaf, E.H. Jansen, H. Hattori, J.J. Kastelein, H.A. Voorbij, A.F. Stalenhoef, L.J. van Tits, Indications that paraoxonase-1 contributes to plasma high density lipoprotein levels in familial hypercholesterolemia, J Lipid Res 46 (2005) 445-451.[14] G.J. de Grooth, T.J. Smilde, S. Van Wissen, A.H. Klerkx, A.H. Zwinderman, J.C. Fruchart, J.J. Kastelein, A.F. Stalenhoef, J.A. Kuivenhoven, The relationship be-tween cholesteryl ester transfer protein levels and risk factor profile in patients with familial hypercholestero-lemia, Atherosclerosis 173 (2004) 261-267.[15] K.A. Rye, M.A. Clay, P.J. Barter, Remodelling of high density lipoproteins by plasma factors, Athero-sclerosis 145 (1999) 227-238.[16] D.I. Draganov, J.F. Teiber, A. Speelman, Y. Osawa, R. Sunahara, B.N. La Du, Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities, J Lipid Res 46 (2005) 1239-1247.

130
CHAPTER 9
[17] M. Harel, A. Aharoni, L. Gaidukov, B. Brumshtein, O. Khersonsky, R. Meged, H. Dvir, R.B. Ravelli, A. Mc-Carthy, L. Toker, I. Silman, J.L. Sussman, D.S. Tawfik, Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic en-zymes, Nat Struct Mol Biol 11 (2004) 412-419.[18] M. Aviram, M. Rosenblat, C.L. Bisgaier, R.S. New-ton, S.L. Primo-Parmo, B.N. La Du, Paraoxonase inhib-its high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxo-nase, J Clin Invest 101 (1998) 1581-1590.[19] J. Ruiz, H. Blanche, R.W. James, M.C. Garin, C. Vaisse, G. Charpentier, N. Cohen, A. Morabia, P. Passa, P. Froguel, Gln-Arg192 polymorphism of paraoxonase and coronary heart disease in type 2 diabetes, Lancet 346 (1995) 869-872.[20] S.R. Srinivasan, S. Li, W. Chen, R. Tang, M.G. Bond, E. Boerwinkle, G.S. Berenson, Q192R polymorphism of the paraoxanase 1 gene and its association with serum lipoprotein variables and carotid artery intima-media thickness in young adults from a biracial community: The Bogalusa Heart Study, Atherosclerosis 177 (2004) 167-174.[21] R.A. Hegele, J.H. Brunt, P.W. Connelly, A polymor-phism of the paraoxonase gene associated with varia-tion in plasma lipoproteins in a genetic isolate, Arte-rioscler Thromb Vasc Biol 15 (1995) 89-95.[22] S.H. Hong, J. Song, W.K. Min, J.Q. Kim, Genetic variations of the paraoxonase gene in patients with coronary artery disease, Clin Biochem 34 (2001) 475-481.[23] S. Campo, M.A. Sardo, G. Trimarchi, M. Bonaiuto, L. Fontana, M. Castaldo, A. Bonaiuto, C. Saitta, A. Bitto, B. Manduca, S. Riggio, A. Saitta, Association between serum paraoxonase (PON1) gene promoter T(-107)C polymorphism, PON1 activity and HDL levels in healthy Sicilian octogenarians, Exp Gerontol 39 (2004) 1089-1094.[24] M.C. Blatter Garin, X. Moren, R.W. James, Paraox-
onase-1 and serum concentrations of HDL-cholesterol and apoA-I, J Lipid Res 47 (2006) 515-520.[25] L.S. Rozek, T.S. Hatsukami, R.J. Richter, J. Ran-chalis, K. Nakayama, L.A. McKinstry, D.A. Gortner, E. Boyko, G.D. Schellenberg, C.E. Furlong, G.P. Jarvik, The correlation of paraoxonase (PON1) activity with lipid and lipoprotein levels differs with vascular disease sta-tus, J Lipid Res 46 (2005) 1888-1895.[26] H. Cao, A. Girard-Globa, A. Serusclat, S. Bernard, P. Bondon, S. Picard, F. Berthezene, P. Moulin, Lack of association between carotid intima-media thickness and paraoxonase gene polymorphism in non-insu-lin dependent diabetes mellitus, Atherosclerosis 138 (1998) 361-366.[27] T. Sakai, B. Matsuura, M. Onji, Serum paraoxonase activity and genotype distribution in Japanese patients with diabetes mellitus, Intern Med 37 (1998) 581-584.[28] M. Dessi, A. Gnasso, C. Motti, A. Pujia, C. Irace, S. Casciani, F. Staffa, G. Federici, C. Cortese, Influence of the human paraoxonase polymorphism (PON1 192) on the carotid-wall thickening in a healthy population, Coron Artery Dis 10 (1999) 595-599.[29] F.R. Leus, M.E. Wittekoek, J. Prins, J.J. Kastelein, H.A. Voorbij, Paraoxonase gene polymorphisms are as-sociated with carotid arterial wall thickness in subjects with familial hypercholesterolemia, Atherosclerosis 149 (2000) 371-377.[30] R. Malin, A. Loimaala, A. Nenonen, M. Mercuri, I. Vuori, M. Pasanen, P. Oja, G. Bond, T. Koivula, T. Lehti-maki, Relationship between high-density lipoprotein paraoxonase gene M/L55 polymorphism and carotid atherosclerosis differs in smoking and nonsmoking men, Metabolism 50 (2001) 1095-1101.[31] H. Markus, Z. Kapozsta, R. Ditrich, C. Wolfe, N. Ali, J. Powell, M. Mendell, M. Cullinane, Increased common carotid intima-media thickness in UK African Carib-beans and its relation to chronic inflammation and vascular candidate gene polymorphisms, Stroke 32 (2001) 2465-2471.

131
General discussion
[32] J. Valabhji, A.J. McColl, M. Schachter, S. Dhanjil, W. Richmond, R.S. Elkeles, High-density lipoprotein com-position and paraoxonase activity in Type I diabetes, Clin Sci (Lond) 101 (2001) 659-670.[33] G. Fortunato, P. Rubba, S. Panico, D. Trono, N. Tinto, C. Mazzaccara, M. De Michele, A. Iannuzzi, D.F. Vitale, F. Salvatore, L. Sacchetti, A paraoxonase gene polymor-phism, PON 1 (55), as an independent risk factor for increased carotid intima-media thickness in middle-aged women, Atherosclerosis 167 (2003) 141-148.[34] Y. Hu, H. Tian, R. Liu, Gln-Arg192 polymorphism of paraoxonase 1 is associated with carotid intima-me-dia thickness in patients of type 2 diabetes mellitus of Chinese, Diabetes Res Clin Pract 61 (2003) 21-27.[35] S. Campo, M.A. Sardo, G. Trimarchi, M. Bonaiuto, M. Castaldo, L. Fontana, A. Bonaiuto, A. Bitto, C. Saitta, A. Saitta, The paraoxonase promoter polymorphism (-107)T>C is not associated with carotid intima-media thickness in Sicilian hypercholesterolemic patients, Clin Biochem 37 (2004) 388-394.[36] J. Karvonen, H. Kauma, M. Paivansalo, Y.A. Ke-saniemi, Paraoxonase-1 gene Leu-Met55 and Gln-Arg192 polymorphisms are not associated with carot-id artery atherosclerosis in a population-based cohort, Eur J Cardiovasc Prev Rehabil 11 (2004) 511-512.[37] M. Roest, A.C. Jansen, A. Barendrecht, F.R. Leus, J.J. Kastelein, H.A. Voorbij, Variation at the paraoxonase gene locus contributes to carotid arterial wall thick-ness in subjects with familial hypercholesterolemia, Clin Biochem 38 (2005) 123-127.[38] D.N. Kiortsis, S. Tsouli, E.S. Lourida, V. Xydis, M.I. Argyropoulou, M. Elisaf, A.D. Tselepis, Lack of associa-tion between carotid intima-media thickness and PAF-acetylhydrolase mass and activity in patients with pri-mary hyperlipidemia, Angiology 56 (2005) 451-458.[39] K.P. Burdon, C.D. Langefeld, S.R. Beck, L.E. Wagenknecht, J.J. Carr, B.I. Freedman, D. Herrington, D.W. Bowden, Association of genes of lipid metabolism with measures of subclinical cardiovascular disease in
the Diabetes Heart Study, J Med Genet 42 (2005) 720-724.[40] J.G. Wheeler, B.D. Keavney, H. Watkins, R. Collins, J. Danesh, Four paraoxonase gene polymorphisms in 11212 cases of coronary heart disease and 12786 con-trols: meta-analysis of 43 studies, Lancet 363 (2004) 689-695.[41] O. Khersonsky, D.S. Tawfik, Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase, Biochemistry 44 (2005) 6371-6382.[42] R. Rontu, P.J. Karhunen, E. Ilveskoski, J. Mikkels-son, O. Kajander, M. Perola, A. Penttila, A.M. Koivisto, T. Lehtimaki, Smoking-dependent association between paraoxonase 1 M/L55 genotype and coronary athero-sclerosis in males: an autopsy study, Atherosclerosis 171 (2003) 31-37.[43] K.S. Robertson, E. Hawe, G.J. Miller, P.J. Talmud, S.E. Humphries, Human paraoxonase gene cluster pol-ymorphisms as predictors of coronary heart disease risk in the prospective Northwick Park Heart Study II, Biochim Biophys Acta 1639 (2003) 203-212.[44] S. Sen-Banerjee, X. Siles, H. Campos, Tobacco smoking modifies association between Gln-Arg192 polymorphism of human paraoxonase gene and risk of myocardial infarction, Arterioscler Thromb Vasc Biol 20 (2000) 2120-2126.


10Summary


135
Summary
Paraoxonase type 1 (PON1) is an enzyme that belongs to the family of paraoxonases. Other members of the paraoxonase family are PON2 and PON3. In the human body, PON1 is lo-cated exclusively on the high-density lipoprotein (HDL) particle. As the name “paraoxonase” indicates, PON1 can hydrolyze the highly toxic organophosphate insecticide “paraoxon”. Since paraoxon is a chemical compound, it is not plausible that the hydrolysis of paraoxon is the primary biological function of PON1. Despite many years of re-search the exact physiological function of PON1 remains unclear. Many research groups have previously suggested that the prevention of oxidation of low-density lipoprotein (LDL) is an important beneficial function of PON1 in the human body. Because the oxidation of LDL is one of the first steps in the development of atherosclerosis, the inhibition of LDL oxidation by PON1 may be an important step in the prevention of cardiovascular disease (CVD). A second important function of PON1 may be an interaction with the HDL particle. There are indica-tions that PON1 can also prevent the oxidation of the HDL particle. As a result, PON1 may preserve HDL and enhance its in the reverse cholesterol transport, i.e. the cholesterol efflux from macrophages and via this pathway play a role in the inhibition of atherogenesis. In this thesis, we have addressed the following questions regarding PON1. 1.) To what extent does PON1 inhibit the oxidation of LDL? 2.) Does PON1 influence the functioning of the HDL particle? 3.) To what extent does PON1 contribute to, or protect against, the onset of CVD? To answer these questions we have conducted a number of epidemiological studies in different groups of people. In these populations, we investigated the effects of PON1 concen-tration, activity, and genetic variations (polymorphisms) in the PON1 DNA on LDL oxida-tion products, cholesterol levels in the blood and the onset of CVD. For large-scale epidemiological studies, reliable (and affordable) measurement techniques are important. The first study of this thesis describes the so-called iFLASH technique (Chapter 2), which we have recently developed. The iFLASH technique is capable of typing polymor-phisms in large-scale populations at a fraction of the cost of many commercial techniques cur-rently available. For this purpose, the iFLASH technique uses small pieces of DNA (probes), which can recognize a specific polymorphism. These probes are labelled with a fluorescent dye, which enables the detection. Because the iFLASH technique uses single-labelled fluores-cent probes, it is much cheaper than many other techniques, which use dual-labelled probes. We used iFLASH to genotype the PON1 polymorphisms in a number of the following stud-ies.We expect that the protective effects of PON1 on development of CVD are more pronounced in study populations that are prone to develop CVD, like cigarette smokers, who are exposed to a high degree of oxidative stress. For this reason we have investigated the effects of PON1 polymorphisms on LDL oxidation (measured by in vitro LDL oxidizability) in a population of male life-long smokers (Chapter 3). In addition, we have investigated whether PON1 was of influence on the development of CVD by measuring the accumulation of lipids in the ar-

136
CHAPTER 10
tery wall. This process thickens the artery wall and marks the beginning of atherosclerosis. An ultra-sound measurement of the intima-media thickness (IMT) of the wall of the carotid artery (CA), is commonly used as a marker of preclinical atherosclerosis. In the population of life-long smokers we found no indications that PON1 could inhibit the oxidation of LDL, or that PON1 was of influence on the CA IMT. Thus, in this population we found no support for a protective effect of PON1 on LDL oxidation or on development of CVD. In addition to the exposure to higher oxidative stress, the protective effects of PON1 on CVD may also be more pronounced in subjects with hereditary elevated cholesterol levels in the blood, as seen in patients with familial hypercholesterolemia (FH). For this reason we inves-tigated the effects of PON1 on LDL oxidation (measured by circulating oxidized LDL parti-cles), lipid metabolism and CA IMT in a population of patients with FH (Chapter 4). Despite the fact that these patients have an increased IMT and increased risk to develop CVD, we found no evidence that PON1 inhibited LDL oxidation or reduced CA IMT in these patients. This was in agreement with our findings in the life-long smokers. On the other hand, we did observe an association between PON1 and serum HDL-cholesterol levels of these patients. There was a positive correlation between PON1 levels, activity and HDL-cholesterol. The strongest correlation was observed for PON1 levels (r=0.37, P<0.001). In support, the genetic variants of PON1 that were associated with high levels and activities of the enzyme were also associated with higher levels of HDL-cholesterol. These findings have been confirmed by dif-ferent research groups and suggest that the interaction with HDL is an important function of PON1.
In Chapter 5 we used High Performance Liquid Chromatography (HPLC) to further study the effects of PON1 on lipid oxidation in patients with FH. The advantage of the HPLC tech-nique is that it is considerably more sensitive when compared to the previously used methods to measure LDL oxidation. We used the HPLC technique to specifically measure the degree of linoleic acid oxidation. Because oxidized linoleic acid is very stable, it is an excellent marker for studying lipid oxidation in the human body. In this study the PON1 paraoxonase activity was specifically protective against linoleic acid oxidation as indicated by a negative correla-tion between paraoxonase activity and oxidized linoleic acid (r=-0.22, P=0.03). However, the effects of PON1 were marginal, and therefore, the biological relevance of these findings is still unclear.
There is a growing body of evidence that PON1 plays a role in the metabolism of certain li-poproteins. For this reason PON1 may also be of importance in situations where changes in lipoprotein metabolism occur. An example of such a situation is observed in patients using lipid-lowering medication (statins). An often observed effect of statin therapy is an increase in serum HDL-cholesterol levels. This phenomenon is not completely understood. In Chapter 6

137
Summary
we have investigated whether PON1 was associated with HDL-cholesterol changes in patients with FH who were on statins therapy. We observed that in addition to basal HDL-cholesterol levels, the PON1 concentration at baseline was also an important predictor of the HDL-cho-lesterol changes during statin therapy. In patients with high baseline levels of PON1 and high baseline levels of HDL-cholesterol, as a result of statin therapy, only marginal non-significant changes in HDL-cholesterol were observed (on average 1%). In patients with low baseline levels of PON1 in addition to high baseline levels of HDL-cholesterol, a significant HDL-cho-lesterol increment of about 9% was observed. The strongest changes in HDL-cholesterol were observed in patients with both low baseline levels of PON1 and HDL-cholesterol (on average 15%). In comparison, changes between the 9-11% were observed in patients high baseline PON1 levels in addition to low baseline HDL-cholesterol levels. These findings not only con-firm a role for PON1 in HDL metabolism, but also suggest that PON1 could be a predictor of the beneficial effect of statin therapy with respect to HDL-cholesterol.
Its involvement in HDL metabolism may also suggest that PON1 plays a role in diseases caused by lipid impairment. The familial combined hyperlipidemia (FCH) syndrome for example, is characterized by an atherogenic lipid profile resulting in an elevated CVD risk. In Chapter 7 we have investigated whether PON1 contributes to the onset of FCH by investigating the dif-ferences in PON1 concentration, activity and genetic make-up among FCH patients and their healthy family members. We found that in comparison with the healthy family members, PON1 arylesterase activity (a measure of PON1 concentrations) was significantly higher in FCH patients (about 11%, P<0.001), despite the fact that HDL-cholesterol levels were lower in these patients (about 15%, P<0.001). We also found that carriers of a certain combination of PON1 genes, which associate with higher levels of the enzyme, had a 5 times higher risk of being affected with FCH, indicated by an odds ratio of 5.2 (95%CI, 1.4-19.7; P=0.016). These PON1 genes were also associated with higher serum cholesterol levels and lower HDL-choles-terol levels. This latter observation seems in conflict with our previous observation that PON1 in particular is associated with increased HDL-cholesterol levels in FH. Thus, in this study we can confirm that PON1 may affect the metabolism of serum lipids and lipoproteins, however this involvement appears to be more complex than initially thought, since it is not clear why PON1 concentration is higher in the FCH patients and why especially in these patients a higher PON1 concentration is associated with elevated cholesterol levels. In many epidemiological studies, associations are found among multiple variables. However, for a better understanding of the development of a disease, one must understand how these variables influence each other, and therefore, a prospective study design is necessary. The most important aspect of a prospective study is that PON1 levels and activity are measured in blood that has been collected before the cardiovascular event has taken place. In Chapter 8 we conducted a prospective investigation to establish whether PON1 is a risk factor for the

138
CHAPTER 10
onset of CVD. For this purpose, we measured PON1 levels and paraoxonase activity in a large population of healthy women and followed these women in time to monitor the occurrence of cardiovascular events. In general, we observed that women who had the highest paraoxonase activity in their blood, were 2-times more likely to get an acute coronary event which was in-dicated by a hazard ratio of 2.07 (95%CI, 1.1-3.8; P= 0.022). Remarkably, when investigating the effects of PON1 in subgroups of smokers and non-smokers, we observed that in women who smoked in addition to having the highest paraoxonase activity had a 19-fold higher risk for an acute coronary event (hazard ratio: 19.2, 95%CI 5.3-69.5; P<0.001), whereas women who only had a high paraoxonase activity or who only smoked had about a 5- respectively 8-fold increased risk (hazard ratios: 4.7, 95%CI, 1.2-18.5; P=0.026 for paraoxonase activity and 7.7, 95%CI 2.3-26.0; P=0.001 for smoking). It appears that in women who smoke a high paraoxonase activity is more harmful. This is an interesting observation, which merits further investigation.
In conclusion, in contrast to what has been frequently suggested by other investigators, our studies do not confirm that PON1 plays an important role in the oxidation of LDL in the hu-man body. We do find that PON1 may have other effects, which relate to CVD. We found that higher PON1 levels contribute to higher serum HDL-cholesterol levels. This finding demon-strates a beneficial role for PON1 in the metabolism of lipoproteins and is in contrast with our finding that an increased paraoxonase activity of the PON1 particle is a risk factor for an acute coronary event. We therefore hypothesize that PON1 may have differential roles in the human body, depending on the genetic make-up of PON1 and on external factors like HDL-cholesterol levels and smoking. As we mentioned in the introduction of this summary, PON1 is an enzyme, which possesses paraoxonase activity. Recent studies have demonstrated that in addition to paraoxonase activity, PON1 also possesses lactonase activity. Unlike paraoxon, lactones are present in the human body and could play an important role in lipid metabolism. The physiological function of PON1’s lactonase activity, therefore, merits further research. In addition, confirming that increased paraoxonase activity is a risk factor for acute coronary events and understanding the interaction of PON1 with smoking will be important research themes for the future.
This thesis has shown that:In patients with FH, PON1 is involved in lipid metabolism by increasing HDL-
cholesterol levels PON1 influences the effect of statin therapy in terms of HDL-cholesterol changesPON1 is involved in the manifestation of the atherogenic lipid profile associated
with FCHIn a general population of women, an increased PON1 paraoxonase activity is a
risk factor for an acute coronary event
•
••
•

139
Summary
The effects of PON1 on the risk for CVD differ in smokers and non-smokers
Future studies should focus on:The physiological function of PON1’s lactonase activityThe underlying mechanism by which PON1 is involved in the metabolism of
serum lipids and lipoproteinsThe interaction of PON1 with statin therapy, and its clinical importanceThe role of PON1 in FCH patientsThe mechanism by which PON1 is cardioprotective as well as a risk factor for acute
coronary events The interaction of PON1 with smoking, and its consequences for the human body
•
••
•••
•


11Samenvatting
(ook voor niet ingewijden)


143
Samenvatting
Paraoxonase type 1 (PON1) is een enzym dat behoort tot de familie van de paraoxonases. Naast PON1 bestaan er ook PON2 en PON3. In het menselijke lichaam bevindt PON1 zich exclusief op het hoge-dichtheids lipoproteine (HDL) deeltje, beter bekend als het “goede cho-lesterol”. Zoals de naam “paraoxonase” aangeeft, katalyseert PON1 de afbraak van het landbouwgif paraoxon. Maar omdat paraoxon geen natuurlijke verbinding is, is het niet aannemelijk dat deze afbraakreactie de primaire functie van PON1 is. Door meerdere onderzoeksgroepen is in de afgelopen jaren al gesuggereerd dat remming van de oxidatie van het lage-dichtheids lipoproteine (LDL) deeltje, ook wel bekend als het “slechte cholesterol”, een belangrijke func-tie is van PON1 in het menselijk lichaam. Hierdoor is PON1 misschien wel een belangrijke beschermende factor tegen hart- en vaatziekten, omdat de oxidatie van LDL één van de eerste stappen is in het ontstaan van atherosclerose (aderverkalking). Daarnaast zijn er aanwijzingen dat PON1 de oxidatie van het HDL-deeltje kan remmen, met als gevolg dat het HDL-deeltje beter kan functioneren. Omdat het HDL-deeltje mede verantwoordelijk is voor de lipiden-huishouding in het lichaam, zou dit een tweede belangrijke functie van PON1 kunnen zijn.In dit proefschrift hebben we de volgende vragen rondom PON1 onderzocht. 1.) In welke mate remt PON1 de oxidatie van LDL? 2.) Heeft PON1 invloed op het HDL-deeltje en de lip-idenhuishouding? 3.) In welke mate speelt PON1 een rol bij het ontstaan van hart- en vaartz-iekten? Om een antwoord te krijgen op deze vragen hebben wij epidemiologisch onderzoek verricht in verschillende studiepopulaties. Dit houdt in dat we bij specifieke groepen mensen hebben gekeken naar de effecten van PON1 concentratie, activiteit en genetische variaties in het PON1-DNA (zogenaamde polymorfismen), op LDL oxidatie producten, cholesterolspie-gels in het bloed en op het ontstaan van hart- en vaatziekten.
Voor grootschalig epidemiologisch onderzoek zijn betrouwbare (en betaalbare) meetmethodes essentieel. De eerste studie in dit proefschrift beschrijft de zogenaamde iFLASH techniek, (Hoofdstuk 2). Deze nieuwe techniek is door ons ontwikkeld en is in staat om op grote schaal polymorfismen te typeren. Hiervoor maakt de iFLASH techniek gebruik van stukjes DNA die een specifiek polymorfisme kunnen herkennen (zogenaamde probes). Een fluoriserend label dat aan de probes vastzit maakt de detectie mogelijk. Doordat de iFLASH techniek gebruik maakt van enkelvoudig gelabelde fluoriserende probes is deze techniek aanzienlijk goedkoper dan veel van de vergelijkbare technieken die gebruik maken van tweevoudig gelabelde probes. De iFLASH techniek is daarom uitermate aantrekkelijk voor gebruik in niet-commerciële laboratoria. We hebben de iFLASH techniek toegepast om PON1 polymorfismen te bepalen in enkele van de hieronder beschreven studies.
Over het algemeen verwachten we dat het beschermende effect van PON1 op hart- en vaatz-iekten sterker tot uiting komt in populaties die al een verhoogd risico hebben op hart- en vaatziekten, zoals rokers, van wie bekend is dat ze extra worden blootgesteld aan oxidatieve

144
CHAPTER 11
stress. Daarom hebben we in een populatie mannen die bijna hun hele leven hebben gerookt, de effecten van PON1 polymorfismen op LDL-oxidatie onderzocht (Hoofdstuk 3). Daarnaast hebben we gekeken of PON1 van invloed was op het ontstaan van hart- en vaatziekten door de ophoping van lipiden in de vaatwand te meten. Stapeling van vetten maakt de vaatwand dikker en is het begin van atherosclerose. De vaatwanddikte van de halsslagader wordt daar-om vaak gebruikt als maat voor preklinische atherosclerose. In deze rokerspopulatie vonden we geen aanwijzing dat PON1 van grote invloed was op de oxidatie van LDL. Ook vonden we dat PON1 niet van invloed was op de vaatwanddikte van de halsslagader. In deze rokers vonden we dus geen duidelijke effecten van PON1 op hart- en vaatziekten of daaraan gerela-teerde parameters.
Behalve bij verhoogde blootstelling aan oxidatieve stress zullen de effecten van PON1 op hart- en vaatziekten ook sterker tot uiting komen in mensen met erfelijk verhoogde cholesterol spie-gels in het bloed, zoals in patiënten met familiaire hypercholesterolemie (FH). Vandaar dat wij in een populatie FH patiënten de effecten van PON1 op LDL-oxidatie, lipidenhuishouding en de vaatwanddikte van de halsslagader hebben bestudeerd (Hoofdstuk 4). Ondanks het hoge risicoprofiel vonden we in deze patiënten, net als in de rokerspopulatie, geen aanwijzingen dat PON1 de LDL-oxidatie beïnvloedt of dat PON1 effect heeft op de vaatwanddikte van de halss-lagader. Er was wel een gunstig effect van PON1 op de hoogte van HDL-cholesterolspiegels in het bloed: naast een positieve correlatie tussen PON1 activiteit en HDL-cholesterol vonden we ook een associatie tussen de PON1 polymorfismen en de HDL-cholesterolspiegels. Deze bevindingen zijn bevestigd door een aantal andere groepen en suggereren dat in het lichaam er daadwerkelijk een functionele interactie tussen PON1 en HDL is.
In Hoofdstuk 5 gebruiken we de zogenaamde High Performance Liquid Chromatography (HPLC) techniek om de effecten van PON1 op lipiden-oxidatie in het bloed van de FH patiënt-en verder in kaart te brengen. Het voordeel van de HPLC techniek is dat deze, in vergelijking met de voorgaande technieken om LDL-oxidatie te meten, een stuk gevoeliger is. Wij hebben de HPLC-techniek toegepast om specifiek linolzuur-oxidatie te meten. De linolzuur-oxidatie is namelijk een uitstekende marker voor lipiden-oxidatie in het lichaam, omdat het oxidatie-product van linolzuur uitermate stabiel is. In deze studie vonden we dat specifiek de PON1 paraoxonase activiteit een beschermend effect had op linolzuur-oxidatie. Maar, omdat het een relatief zwak effect betreft, is de biologische relevantie ervan nog onduidelijk.
Er zijn steeds meer aanwijzingen dat PON1 betrokken is in de lipidenhuishouding. Om deze reden kan PON1 ook betrokken zijn bij situaties waar een sterke verandering in de lipiden-huishouding plaatsvindt. Een voorbeeld van een sterke verandering in het lipiden-profiel zien we in mensen die cholesterolverlagende medicatie (statines) gebruiken. Een vaak gezien (en gewenst) neveneffect van statine-gebruik is een toename van de HDL-cholesterol spiegel in

145
Samenvatting
het bloed. In Hoofdstuk 6 hebben we in FH patiënten onderzocht of PON1 van invloed is op veranderingen in HDL-cholesterol als gevolg van statine gebruik. In deze studie vonden we dat naast basale HDL-cholesterol spiegels ook de basale PON1 spiegel een belangrijke voorspeller is voor de mate van toename van het HDL-cholesterol tijdens de behandeling met statines. Bij patiënten met hoge PON1 en hoge HDL spiegels werd bij statine behan-deling nauwelijks een toename van het HDL-cholesterol geconstateerd (gemiddeld 1%). Bij de patiënten met lage PON1 spiegels in combinatie met hoge HDL spiegels werd als gevolg van de statine behandeling een toename van het HDL-cholesterol van gemiddeld 9% gezien. De sterkste HDL-cholesterol toenames werden gevonden in patienten met zowel basaal laag HDL als PON1 (gemiddeld 15%), terwijl in patienten met de combinatie hoog PON1 en laag HDL, toenames tussen de 9-11% werden gezien. Deze bevindingen bevestigen niet alleen een betrokkenheid van PON1 in de lipidenhuishouding, maar suggereren ook dat PON1, wat betreft HDL-cholesterol, een voorspeller zou kunnen zijn voor de mate van succes van de statine behandeling.
Door de betrokkenheid in de lipidenhuishouding zou PON1 ook een rol kunnen spelen bij het ontstaan van stoornissen in de lipidenhuishouding. Het familiaire gecombineerde hyper-lipidemie (FCH) syndroom wordt gekenmerkt door een erfelijk verstoord lipidenprofiel, en als gevolg daarvan een verhoogd risico op hart- en vaatziekten. In Hoofdstuk 7 hebben we onderzocht of PON1 een bijdrage levert aan het ontstaan van FCH. Om dit te onderzoeken, hebben wij gekeken of er verschillen waren in PON1 concentratie, activiteit en genetische op-maak tussen FCH patiënten en gezonde familieleden van deze patiënten. Wij vonden, dat in vergelijking met de gezonde familieleden, de PON1 concentraties significant hoger waren in de patiënten met FCH. Dit ondanks het feit dat de HDL-cholesterol concentraties lager waren in deze patiënten. Ook vonden we, dat de dragers van een bepaalde combinatie PON1 genen een 5 keer hoger risico hadden om aangedaan te zijn door het FCH syndroom. Deze PON1-genen waren ook geassocieerd met hogere cholesterolspiegels in het lichaam. Dit laatste lijkt in tegenspraak met wat we eerder vonden, namelijk dat PON1 specifiek het HDL-cholesterol verhoogt. In deze studie wordt de rol van PON1 in het lipidenhuishouding verder bevestigd, maar de betrokkenheid blijkt complexer dan aanvankelijk werd gedacht. Het is namelijk nog niet duidelijk waarom de PON1 concentratie hoger is in de FCH patiënten en waarom juist in deze patiënten een hogere PON1 concentratie samenhangt met hogere cholesterol spiegels. In epidemiologisch onderzoek worden vaak verbanden gevonden tussen diverse waarnemi-ngen. Voor een beter inzicht in het ontstaan van een ziekte is het belangrijk om te weten hoe die verbanden zijn ontstaan. Dit kan worden vergeleken met de “kip-of-het-ei”-vraag. Als er een verband gevonden wordt tussen afwijkende PON1 spiegels en hart- en vaatziekten, zijn de afwijkende PON1 spiegels dan de oorzaak of het gevolg van hart- en vaatziekten? Om deze vraag te beantwoorden is een prospectief onderzoek nodig. Het belangrijkste aspect van een

146
CHAPTER 11
prospectief onderzoek is, dat PON1 spiegels en activiteiten worden bepaald in bloed dat is verzameld voordat hart- en vaatziekten zijn opgetreden. Hoofdstuk 8 beschrijft een prospec-tief onderzoek waarin bestudeerd wordt of PON1 daadwerkelijk een risicofactor is voor het ontstaan van hart- en vaatziekten. Dit hebben we onderzocht door bij een grote groep van ge-zonde vrouwen PON1 activiteiten te bepalen en vervolgens deze vrouwen in de tijd te volgen en te registreren bij wie er zich cardiovasculaire gebeurtenissen voordoen. Uit deze studie bli-jkt dat vrouwen met een hoge PON1 paraoxonase-activiteit ook een hoger risico lopen op het krijgen van een hartaanval. Opmerkelijk was dat vrouwen, die een hoge paraoxonase-activit-eit hadden en daarnaast ook nog eens rookten, een 19 keer hoger risico liepen op het krijgen van een hartaanval, terwijl vrouwen met alleen maar een hoge paraoxonase-activiteit of die alleen maar rookten, een 4 keer respectievelijk 8 keer verhoogd risico hadden op het krijgen van een hartaanval. Blijkbaar is een verhoogde paraoxonase-activiteit in rokers gevaarlijker dan in niet-rokers. Dit is een interessante observatie die verder onderzocht dient te worden.
Samenvattend, we kunnen met onze studies niet bevestigen dat PON1 een sleutelrol speelt bij de oxidatie van LDL in het lichaam, zoals reeds werd gesuggereerd door veel andere onder-zoekers. Wel lijkt PON1 andere functies in het lichaam te vervullen die samenhangen met hart- en vaatziekten. Zo zagen we in FH patiënten, dat hogere PON1 concentraties correleren met hogere HDL-cholesterolspiegels. PON1 lijkt wat HDL betreft dus een gunstige rol te spelen in de lipidenhuishouding. Het is daarom paradoxaal dat een verhoogde paraoxonase-activiteit van het PON1 deeltje een risicofactor is voor het krijgen van hart- en vaatziekten. PON1 lijkt in het menselijke lichaam dus meerdere gezichten te hebben. De mogelijke verk-laring ligt in de wijze waarin PON1 zich uiteindelijk manifesteert in het lichaam. Dit hangt niet alleen af van de PON1 polymorfismen, maar ook van externe factoren zoals bijvoorbeeld HDL-cholesterolspiegels en roken. Zoals in de inleiding werd besproken is PON1 een enzym met paraoxonase-activiteit. Uit recente studies blijkt dat PON1 behalve paraoxonase-activ-iteit ook lactonase-activiteit heeft. Lactonen komen in tegenstelling tot paraoxon wel in ons lichaam voor en kunnen een belangrijke rol in de lipidenhuishouding spelen. Toekomstig onderzoek dient zich daarom vooral te richten op de fysiologische functie van PON1’s lac-tonase-activiteit en het ophelderen van de onderliggende mechanismen van de betrokken-heid van PON1 in de lipidenhuishouding. Daarnaast zullen het bevestigen dat een verhoogde paraoxonase-activiteit een risicofactor is voor het krijgen van een hartaanval en het ophel-deren van de interactie van PON1 met roken ook belangrijke onderzoeksthema’s voor de toekomst zijn.
In dit proefschrift komt naar voren:Dat door HDL cholesterol spiegels te verhogen PON1 betrokken is bij de
lipidenhuishouding in FH patiëntenDat PON1 het succes van statine therapie beïnvloedt wat betreft HDL cholesterol
•
•

147
Samenvatting
Dat PON1 ook betrokken is bij het ontstaan een verstoord lipiden-profiel (FCH) Dat in gezonde vrouwen een verhoogde paraoxonase-activiteit van PON1 een
verhoogd risico geeft op het krijgen van een hartaanval Dat de effecten van PON1 op het krijgen van een hartaanval verschillen in rokers
en niet-rokers
Toekomstige studies dienen zicht te richten op:De fysiologische functie van PON1’s lactonase-activiteitHet onderliggende mechanisme waardoor PON1 een rol speelt in de
lipidenhuishoudingHet mechanisme en de klinische betekenis van PON1 in relatie met het effect van
statines De rol van PON1 bij de ontwikkeling van het FCH syndroomHet mechanisme waardoor PON1 zowel een risicoverhogende als verlagende factor
kan zijn voor het krijgen van hart- en vaatziektenDe interactie tussen PON1 en roken, en wat de gevolgen zijn voor het lichaam
••
•
••
•
••
•


Dankwoord


151
Dankwoord
“That was that!” Time flies when you’re having fun, not? Promoveren is een mooie tijd en er zijn veel mensen die daar een belangrijke bijdrage aan hebben geleverd. Mede door hun inzet en inspiratie is dit proefschrift geworden wat het nu is. Daarom wil ik graag de laatste woor-den die ik in het kader van dit paraoxonase onderzoek type, gebruiken om hen te bedanken.
Als eerste mijn promotor, Anton. Dank je voor de mogelijkheid die jij mij hebt gegeven voor het doen van dit onderzoek. Ik denk dat dit onderzoek een mooi voorbeeld is geweest van een succesvolle samenwerking tussen twee labs in verschillende steden. Je enthousiasme en no-nonsense manier van handelen hebben zeker bijgedragen aan dit succes.
Mijn Nijmeegse co-promotor, Berry. Het is goed samenwerken met jou. Ook jou no-nonsense mentaliteit, je duidelijke inzichten en eerlijke commentaar hebben mij altijd verder geholpen. En ondanks het feit dat er hemelsbreed 80 kilometer tussen onze werkplekken zat (of 2 uur en 10 minuten in termen van de NS, sorry dat ik vaak te laat was voor een afspraak) was het toch net alsof je in de “office down the hall” zat. Dank je voor alles, Berry!
Mijn Utrechtse co-pomoter en kamergenoot, Mark. Wij hebben 4 jaar zeer intensief sa-mengewerkt. En ook al hebben wij vaak zeer uiteenlopend visies gehad op de interpretatie van onderzoeksdata, lekker eten (vega bbq?) en de charme van Amerika, ik heb veel van je geleerd in die afgelopen 4 jaar. Verder maakte je grenzeloze enthousiasme om nieuwe dingen ontdekken en je gevoel voor humor het ASL research lab een mooie plek om te werken. Dank je Mark!
Mijn andere Utrechtse co-promoter, Ron. Toch wel ‘The Don’ van het Utrecht lab. Het is geen makkelijke taak om in dezelfde ruimte zowel een research als een diagnostiek lab te managen, maar jij deed dat met grote charme, vandaar dat ook elk ASL-estafetteteam (indirect) jou naam droeg (“Flits Voorbij”, “Snel Passé” en “Ronnies Angels”). Als ik ergens mee zat kon ik altijd bij je binnenstappen, en je crash-course terrasassertiviteit zal mij altijd bij blijven!
Jacqueline, jij weet als geen ander de zwakke plekken in de data te vinden, ik denk dat veel van mijn manuscripten sterk verbeterd zijn door jouw commentaar. Dank daarvoor! Daarnaast is het altijd leuk om jou op een congres (of al in het vliegtuig) tegen te komen!
Pierre, uit het oog maar niet uit het hart. Je nuchtere manier om tegen onderzoek aan te kijken is voor mij een inspiratie geweest. Dank.
Arjan, mijn lab-maat van het eerste uur. Jij bent een absolute allrounder, zonder Arjan geen lab! Het was (en is nog steeds) goeie humor om over alles te klagen: de vissen, het lab, het UMC, en alles op de weg tot Primus. Dank je voor je hulp en je kennis, en helemaal bedankt

152
Dankwoord
dat je mijn paranimf wil zijn.
Bas, halverwege de rit versterkte jij het ASL team. Het was altijd goed om met jou (onderzoeks)ervaringen uit te wisselen, succes met je promotie! (gebruik je trouwens al stiekem iFLASH?)
De dames en heer van het ASL diagnostiek lab. Het was altijd gezellig om even bij te praten tijdens het printen van al die SAS-output bij jullie op het lab. En echt waar, ik heb een lichte vorm van bloedprik-angst en daarom wordt ik rood. (maar dit heb ik al heel vaak uitgelegd!)
Mijn oud collega’s Agnes, Barbara en Sven, de “Julius mensen” en alle oud-studenten. Jullie gaven het lab extra kleur! Roberta en Fatiha, bedankt voor jullie bijdrage aan de iFLASH studie. (why? learn Dutch ;-)
Het Nijmegen lab. Het was niet vaak dat ik bij jullie over de vloer kwam maar het was voor mij altijd interessant om een kijkje in de andere keuken te nemen. In het speciaal wil ik Anneke, Heidi en Magda bedanken voor jullie inzet en de goede samenwerking.
Mijn collega AIO’s in Nijmegen. Gerly, het is altijd goed om de AIO-weetjes uit te wisselen en Ewoud, dank voor het beschikbaar stellen van de FCH studie. En succes met jullie promotie!
Alle co-auteurs van mijn artikelen. Dank jullie wel voor jullie bijdrage en de altijd goede samenwerking.
Richard, mijn andere kamergenoot. Ook al begrijpen we werkelijk niets van elkaars onder-zoek, we hadden altijd genoeg stof om over te praten (dus niet roddelen, Annet!). Succes met je verdere onderzoek!
Jeroen, over personen gesproken die onmisbaar zijn voor het slagen van een promotie. Je computer-expertise, je connecties met de verstrekkers van mailboxruimte, je spam en natu-urlijk je rosé, ik zal het niet snel vergeten!
De Haematologie hoogleraren, Flip en Jan Willem, dank jullie dat ik als semi-haemato-aio mocht meedraaien in de werkbesprekingen en de biomembranen.
Mijn collega’s van de Haematologie en de Rode Cellen Groep. Jullie zijn te veel om allemaal persoonlijk op te noemen, maar jullie hebben allemaal op een eigen wijze voor mij wat betek-end bij het tot stand komen van dit proefschrift.

153
Dankwoord
Toch even in het speciaal Bahram, my main man on the oxidation. Cool dat ik je promotie van dicht bij heb mogen meemaken.
Mijn collega’s bij de RvB, mijn (semi-)vrienden van het medisch biologisch heren gezelschap villa virorum en de nog altijd actieve roeiploeg famous adcie. Dank voor jullie oprechte inter-esse in mijn onderzoek. En Anwar, ik heb altijd tijd voor koffie, dat weet je!
De familie natuurlijk! Ooms, tantes, neefjes, nichtjes, de schoonfamilie en de Oma’s. Allen bedankt voor jullie medeleven! En zet in de agenda: 29 juni promotie in Nijmegen, ‘s avonds feest in Utrecht. Maarten, nog bedankt voor je InDesign expertise.
Tot slot, my precious loved ones!
Mijn broertje Thijs. Emiel! Ik ben trots op je hoe je het allemaal in Barca flikt, heel Canada is nu al aan de Visa heb ik gehoord! Naast het zijn van een (bijna) native Catalano, ben je (and you’ve got the the damn papers to prove it, huh!) ook een echte native English speaker. Dank je dat je mijn manuscripten hebt nagelopen, en cool dat je mijn paranimf wil zijn. Hug!
Mijn ouders. Mieps en Hans. Puk and Dad! Ik heb veel steun gehad aan jullie relaxte kijk op dingen en jullie relativerende vermogen, dankzij jullie is een promotieonderzoek, maar een promotieonderzoek. Jullie (artistieke en bouwkundige) creativiteit zullen mij altijd inspireren in “the pursue of creativity”. Thanx, Hug!
Tot slot mijn vriendin, Frauke. Vanaf de eerste dag van mijn carrière als medisch bioloog was jij er bij, en nog steeds! Lieve Frauke, met jouw steun heb ik het allemaal gered. I luv you! The future has much in store, I can feel it!
Thomas


Curriculum vitae
List of publications
International presentations


157
CV, publications and presentations
Curriculum vitae
Thomas van Himbergen werd geboren op 31 mei 1977 te Boston, USA. Na het behalen van het VWO diploma aan Het Nieuwe Lyceum te Bilthoven begon hij in 1995 met de studie Me-dische Biologie aan de Universiteit Utrecht. Onderzoekservaring werd opgedaan in het UMC Utrecht tijdens een hoofdvakstage bij het Eijkman-Winkler instituut voor medische microbi-ologie, infectie en inflammatie (onder begeleiding van dr. Sylvain Brisse) en een bijvakstage bij de vakgroep Endocrinologie (onder begeleiding van drs. Joost van der Ven). Naast onder-zoekservaring vergaarde hij kennis op het gebied van beleid en management door extra cur-riculaire vakken te volgen aan het Centrum voor Beleid en Management van de Universiteit Utrecht. Het doctoraalexamen Medische Biologie werd behaald in 2001. Van november 2001 tot december 2005 heeft hij gewerkt aan het in dit proefschrift beschreven promotie-onder-zoek onder supervisie van Prof. dr. Anton Stalenhoef, Dr. Berry van Tits, Dr. Ron Voorbij en Dr. Mark Roest. Het promotie onderzoek werd uitgevoerd bij de Research Afdeling ASL Klinische Chemie van het UMC Utrecht in samenwerking met de afdeling Algemeen Interne Geneeskunde van het UMC St Radboud te Nijmegen. Momenteel is hij werkzaam als staf-medewerker onderzoek bij de Raad van Bestuur van het UMC Utrecht.
List of publications
Van Himbergen TM, Van der Schouw YT, Voorbij HA, Van Tits LJ, Stalenhoef AF, Peeters PH, Mark Roest M. Paraoxonase-1 and the risk for coronary heart disease and myocardial infarction in a general population of Dutch women. Submitted
Van Himbergen TM, Van Tits LJ, Ter Avest E, Roest M, Voorbij HA, De Graaf J, Stalenhoef AF. Paraoxonase-1 associates with the Familial Combined Hyperlipidemia phenotype. Submitted
Van Himbergen TM, Voorbij HA, Barendrecht AD, Van Rijn BB, Brambilla R, Van Tits LJ, Roest M. High-throughput genotyping with infrared Fluorescence Allele Specific Hybridization (iFLASH): a simple, reliable and low-cost alternative. Clinical Biochemistry, 2006 Apr 17, Epub
Van Himbergen TM, van Tits LJ, Roest M, Stalenhoef AF. The story of PON1: how an organophosphate-hydro-lysing enzyme is becoming a player in cardiovascular medicine. Neth J Med. 2006 Feb;64(2):34-8
Van Himbergen TM, van Tits LJ, Voorbij HA, de Graaf J, Stalenhoef AF, Roest M. The effect of statin therapy on plasma high-density lipoprotein cholesterol levels is modified by paraoxonase-1 in patients with familial hyperc-holesterolaemia. J Intern Med. 2005 Nov;258(5):442-9

158
CV, publications and presentations
Van Tits LJ, Van Himbergen TM, Lemmers HL, de Graaf J, Stalenhoef AF. Proportion of oxidized LDL relative to plasma apolipoprotein B does not change during statin therapy in patients with heterozygous familial hyperc-holesterolemia. Atherosclerosis. 2006 Apr;185(2):307-12. Epub 2005 Jul 11
Van Himbergen TM, van Tits LJ, Hectors MP, de Graaf J, Roest M, Stalenhoef AF. Paraoxonase-1 and linoleic acid oxidation in familial hypercholesterolemia. Biochem Biophys Res Commun. 2005 Aug 5;333(3):787-93
Van Himbergen TM, Roest M, de Graaf J, Jansen EH, Hattori H, Kastelein JJ, Voorbij HA, Stalenhoef AF, van Tits LJ. Indications that paraoxonase-1 contributes to plasma high density lipoprotein levels in familial hypercholes-terolemia. J Lipid Res. 2005 Mar;46(3):445-51. Epub 2004 Dec 1
Brisse S, Van Himbergen TM, Kusters K, Verhoef J. Development of a rapid identification method for Kleb-siella pneumoniae phylogenetic groups and analysis of 420 clinical isolates. Clin Microbiol Infect. 2004 Oct;10(10):942-5
Van Himbergen TM, Roest M, De Waart FG, De Graaf J, Voorbij HA, Van Tits LJ, Stalenhoef AF. Paraoxo-nase genotype, LDL-oxidation and carotid atherosclerosis in male life-long smokers. Free Radic Res. 2004 Jun;38(6):553-60
International presentations
Paraoxonase-1 and the Risk for coronary heart disease in a general population of dutch women. Presentation, American Heart Association, Scientific Sessions 2005
The effect of statin therapy on plasma high-density lipoprotein cholesterol levels is modified by paraoxonase-1 in patients with familial hypercholesterolaemia. Poster presentation, European Atherosclerotic Society Meeting. 2005
Paraoxonase genotype and phenotype contribute to plasma high-density lipoprotein levels in familial hypercho-lesterolemia. Presentation, First International Conference on “Paraoxonases - Basic and Clinical Directions of Current Research”, Ann Arbor, USA 2004
Paraoxonase-1 contributes to plasma high density lipoprotein levels in familial hypercholesterolemia. Poster presentation, American Heart Association, Scientific Sessions 2003































