Embed Size (px)
Citation preview

Q4
Q1
Journal of the American Society of Hypertension -(-) (2012) 1–10
123456789
1011121314151617181920212223242526272829303132333435363738394041424344454647484950515253
54555657585960616263646566
Research Article
Impaired vasomotor function induced by the combination of hypertensionand hypercholesterolemia
Hizir Kurtel, MDa, Stephen F. Rodrigues, PhDb, Cigdem E. Yilmaz, MD, PhDc,Alper Yildirim, PhDb, and D. Neil Granger, PhDb,*
aDepartment of Physiology, Marmara University School of Medicine, Istanbul, Turkey;bDepartment of Molecular and Cellular Physiology, LSU Health Sciences Center, Shreveport, Louisiana; and
cDepartment of Cell Biology and Anatomy, Sophie Davis School of Biomedical Education, City University of New York, New York
Manuscript received September 10, 2012 and accepted November 14, 2012
6768
69 Abstract 707172737475767778798081828384Although it is well known that endothelial function is compromised in the presence of either hypertension (HTN) or hyper-cholesterolemia (HCh), less is known about whether and how the combination of these risk factors (HTNþHCh) results inimpaired endothelium-dependent dilation (EDD). The aims of this study were to evaluate the influence of HTNþHCh onvasomotor function and to identify the mechanisms that underlie the altered vascular reactivity elicited by HTNþHCh.Endothelium-dependent and -independent vasomotor responses of aortic vessels were studied in mice with diet-inducedHCh and/or HTN induced by chronic administration of either angiotensin II (AngII) or deoxycorticosterone acetate-salt.HTNþHCh elicited an impairment of EDD that appeared between each risk factor alone. Incubation with catalase resultedin more severe EDD impairment. Each risk factor enhanced vascular H2O2 production, but a larger response was noted withHTNþHCh. An attenuated EDD was not observed in AngII type 1a receptor deficient (AT1r�/�) mice, but AT1r�/� bonemarrow chimeras exhibited more profound impairment compared with wild-type. HTNþHCh does not exert an additiveeffect of vasomotor dysfunction compared with either risk factor alone, and both H2O2 and blood cell–associated AT1rcontribute to the impaired EDD responses in mice with HTNþHCh. J Am Soc Hypertens 2012;-(-):1–10. � 2012American Society of Hypertension. All rights reserved.Keywords: Angiotensin II type-1 receptors; endothelium-dependent vasodilation; hydrogen peroxide; risk factors.
8586
87 88899091929394959697Introduction
Hypertension (HTN) and hypercholesterolemia (HCh) aretwo major factors that increase the risk of cardiovasculardisease. Long-term exposure to either of these risk factorsleads to changes in the structure and function of large andmicroscopic blood vessels.1 Although the vascular responsesvary somewhat between the two risk factors, a characteristicfeature of both responses is endothelial dysfunction.Increased leukocyte adhesion, accelerated thrombus
Supported by a grant from the National Heart Lung and BloodInstitute (HL26441).
*Corresponding author: D. Neil Granger, PhD, Department ofMolecular & Cellular Physiology, LSU Health Science Center –Shreveport, 1501 Kings Hwy, Shreveport, LA 71130-3932.
E-mail: [email protected]
1933-1711/$ - see front matter � 2012 American Society of Hypertenshttp://dx.doi.org/10.1016/j.jash.2012.11.005
FLA 5.1.0 DTD � JASH348_proof � 1
9899
100101102103104105
formation, diminished endothelial barrier function, andimpaired vasomotor responses are common manifestationsof the risk factor–induced endothelial cell dysfunction.1–3
Although the mechanisms underlying these endothelium-dependent vascular alterations remain poorly understood,evidence points to reduced nitric oxide (NO) bioavailabilitysecondary to enhanced generation of superoxide as a criticalcomponent of the endothelial cell dysfunction that accom-panies HTN and HCh.4 The capacity of macroscopic andmicroscopic arteries to dilate in response to acetylcholine,bradykinin, and other endothelium-dependent vasodilatorsis significantly impaired in humans and experimentalanimals with either HTN or HCh.5 This impairment hasbeen linked to different factors, including oxidative stress,circulating immune cells, and angiotensin II (AngII) type 1receptors (AT1r).6 Among the various sources of reactiveoxygen species (ROS) induced by the risk factors, NADPHoxidase has received the most attention. The engagement
ion. All rights reserved.
3 December 2012 � 2:50 pm � ce

2 H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
106107108109110111112113114115116117118119120121122123124125126127128129130131132133134135136137138139140141142143144145146147148149150151152153154155156157158159160
161162163164165166167168169170171172173174175176177178179180181182183184185186187188189190191192193194195196197198199200201202203204205206207208209210211212213214
of angiotensin II with AT1r appears to result in NADPHoxidase–dependent ROS production that has been linked toboth the vasomotor dysfunction and inflammatory compo-nent (leukocyte adhesion, endothelial barrier failure) of thevascular response to HTN and HCh.7,8 Similarly, theinvolvement of T lymphocytes in HCh and HTN inducedvasomotor dysfunction has also been linked tolymphocyte-associated AT1r and ROS production.1,9
Although much has been learned about the responses ofthe vasculature to individual risk factors, less is knownabout whether, and how, combinations of risk factors(eg, HTNþHCh) for cardiovascular disease result inendothelium-dependent vascular dysfunction. Epidemiolog-ical evidence on the incidence of ischemic diseases of theheart and brain in humans suggest that combinations of riskfactors exert a synergistic effect relative to each risk factoralone.10–12 Because HTN and HCh are known to individuallyinduce similar phenotypic changes (eg, endothelial celldysfunction, oxidative stress) in the vasculature and appearto use some common signaling pathways (eg, AT1rsignaling), it may be expected that the combination of theserisk factors would yield neither additive nor synergisticvascular responses. However, the limited available evidencein the literature is inconsistent in this regard, with somestudies demonstrating synergism between HTN and HChon vascular reactivity,12,13 whereas other reports14,15 describean absence of synergism. For example, in pigs with renovas-cular HTN and diet-induced HCh, the two risk factors exerta synergistic deleterious effect on endothelium-dependentdilation and oxidative stress in coronary arteries, comparedwith either risk factor alone.12 A comparison of the vaso-motor responses to both a vasodilator (acetylcholine) anda vasoconstrictor (norepinephrine) between hypercholesterol-emia apoE�/� mice, without or without angiotensin II–dependent renovascular hypertension, did not reveal additiveeffects on vessel function.14 It remains unclear whether theseinconsistent responses to risk factor combinations result fromthe vascular bed examined and/or models used to induceHTN or HCh. Furthermore, it is uncertain whether the mech-anisms that underlie the responses to multiple risk factorsdiffer from the mechanisms that mediate the vasculardysfunction elicited by individual risk factors.
The overall objective of this study was to test the hypoth-esis that the combination of HTNþHCh alters vascularreactivity to a greater degree than either risk factor alonevia a mechanism that involves the activation of AT1r andincreased ROS production. Three specific issues were ad-dressed: (1) whether HTNþHCh alters vasomotor functionin a manner that is different from each risk factor alone, (2)the role of ROS in the risk factor-induced alterations invascular reactivity, and (3) whether AT1a receptorsexpressed on circulating blood cells vs the vessel wallcontributes to the vascular responses elicited byHTNþHCh.
FLA 5.1.0 DTD � JASH348_proof � 1
Methods
Animals
All experimental procedures were performed on malewild-type C57Bl/6 (WT) and AT1a-R�/� mice (JacksonLaboratory, Bar Harbor, Maine). The animal experimentswere performed according to the criteria outlined by theNational Institutes of Health and approved by the LSUHealth Sciences Center Institutional Animal Care and UseCommittee.
Hypercholesterolemia
Mice (6–8 weeks old) were placed either on a normal(ND) or high-cholesterol (HC) diet with (Teklad 90221,containing 1.25% cholesterol, 0.5 % sodium choline,15.8% fat, Harlan Teklad) or without cholate (Teklad94059, containing 1.25% cholesterol, 15.8% fat, Harlan Te-klad) for 2 (AngII groups) or 3 (DOCA-salt groups) weeks,respectively.
Angiotensin II–Induced Hypertension
In some mice, an osmotic minipump (Alzet) was im-planted subcutaneously between the scapulas. The pumpcontained either saline (sham) or angiotensin II (SigmaChemical Co.) dissolved in saline. The AngII infusionrate was 1.0 ug/kg/min for 2 weeks. Separate groups ofmice were treated with the AT1r antagonist losartan (25mg/kg/per day) in drinking water for 7 days, beginning atday 8 on HCD or Ang II.
Hypertension Induced by DOCA-Salt
In C57BL6 mice, deoxycorticosterone acetate-salt(DOCA-salt) hypertension was induced by subcutaneousimplantation of a 50-mg slow release DOCA pellet. A rightflank incision was made and the right kidney removed.Drinking water was replaced by 1.0% saline/0.2% potas-sium chloride. Control animals underwent nephrectomy,sham implantation of a pellet, and were given water adlibitum.
Blood Pressure Measurement
On day 14 of Alzet pump (AngII groups) implantation orday-21 of pellet implantation (DOCA-salt groups), bloodpressures were measured by tail-cuff plethysmography(model SC-1000, Hatteras Instruments, Inc., North Caro-lina) in nonanesthetized animals. The average of eightsuccessive measurements was taken as the mean systolicblood pressure in each animal.
Wire Myography
The reactivity of mouse aortic segments to different dila-tors and constrictors was quantified using wire myography.
3 December 2012 � 2:50 pm � ce
215

3H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
216217218219220221222223224225226227228229230231232233234235236237238239240241242243244245246247248249250251252253254255256257258259260261262263264265266267268269270
271272273274275276277278279280281282283284285286287288289290291292293294295296297298299300301302303304305306307308309310311312313314315316317318319320321322323324325
Briefly, mice were anesthetized with xylazine (7.5 mg/kgbody weight intraperitoneally) and ketamine chloride(150 mg/kg body weight intraperitoneally) and the thoracicaorta was quickly removed by central sternotomy. Theaortic segment was gently washed through the lumenwith ice-cold saline and placed in ice-cold physiologicalsalt solution (PSS). Two-millimeter-long segments of aortawere mounted on an eight-channel wire myograph (RandotiGlass, Monrovia, California). Vessel rings were maintainedin 15-mL organ baths with oxygenated PSS (95% O2 and5% CO2) at 37.1
�C. The resting tension was increased step-wise to 1.4 g, followed by a 40-minute equilibration period.An eight-channel octal bridge and data-acquisition softwarewere used to record all force measurements. After equili-bration, aortic rings were rinsed with 120 mM potassiumchloride for vascular smooth muscle activation and to deter-mine the maximal contractile response. The aortic ringswere then precontracted with 10�6 M phenylephrine (PE)to obtain submaximal contraction (60–80% of potassiumchloride–induced maximum response), and after obtaininga stable plateau phase of contraction, the integrity of theendothelium was assessed with acetylcholine (Ach; 3 �10�9 – 10�6). Endothelium-dependent dilation was ex-pressed as the percent dilation from the precontractionresponse to 10�6 M PE. Endothelium independent vasore-laxation was evoked by sodium nitroprusside (SNP; 10�9
– 10�4 M) or papaverine (10�9 – 10�4 M), whereasendothelium-independent contraction was induced withPE (10�9 – 10�4 M). In some experiments, vascular reac-tivity to ACh was assessed in the presence (5 minutes’ pre-incubation) of catalase (1200 U/mL; Sigma C-100, SigmaAldrich) to determine the contribution of H2O2 to the vaso-relaxation response.
Production of Bone Marrow Chimeras
Chimeras were produced by transplanting bone marrowderived from AT1r�/� mice into WT recipients, as previ-ously described.8 This procedure normally yields >90%penetrance of the transferred marrow at 6 weeks after trans-plant. This bone marrow transfer protocol allowed for thecreation of mice in which the genetic deficiency of AT1areceptors is confined to the circulating blood cells.8
Measurement of H2O2
Extracellular H2O2 was determined with a fluorometrichorseradish peroxidase-linked assay kit (Amplex red assay,Molecular Probes, Eugene, Oregon). Aortic segments wereincubated for 60 minutes at 37�C in PSS containing 50 mMAmplex red and 0.1 U/mL horseradish peroxidase protectedfrom light. The tissue was removed from the buffer andbuffer fluorescence was detected at 590 nm, using an exci-tation of 530 nm. Background fluorescence, determinedusing a reaction without tissue, was subtracted from eachvalue. H2O2 release, calculated using H2O2 standards was
FLA 5.1.0 DTD � JASH348_proof � 1
expressed as mM per millimeter of vessel. The Amplexred assay is highly specific and sensitive, with detectionlimit of approximately 5 pmol of H2O2.
16 It detects H2O2
released by cells and tissues and can be applied to cell-free systems. Resorufin, the end product that, when excitedat 530 nm, strongly emits light at 590 nm17 is a very stableproduct that allows detection of H2O2 both in oxidative andreductive conditions. The specificity of the assay is evi-denced by the fact that catalase abolishes the assay signal.16
Serum Cholesterol Levels
Serum samples were frozen for subsequent measurementof cholesterol levels using a spectrophotometric assay(Sigma Chemicals Co.).
Statistical Analysis
Each experimental group included seven to nine mice.All values are reported as means � SE. Comparisonsbetween groups of animals or treatments were made byone-way analysis of variance. When significance was indi-cated, a Student-Newman-Keuls post hoc analysis was usedwith statistical significance set at P < .05.
Results
All mice placed on a cholesterol-enriched diet exhibitedan increased serum cholesterol concentration (Table 1).Similarly, all mice with an implanted AngII-loaded pumpor a DOCA pellet exhibited a significantly elevated systolicblood pressure compared with WT mice. In both hyperten-sion models, the elevated serum cholesterol did not alter theblood pressure response. The AngII–induced hypertensionresponse was significantly attenuated in HCh mice treatedwith losartan (Los) and in AT1r�/� mice, while AT1r�/�
chimeras showed no attenuation of blood pressurecompared with either WT or WT-HCh mice.
Figure 1 demonstrates that acetylcholine-induced vasodi-lation was significantly blunted in mice placed on a choles-terol-enriched diet. The HCh-induced impairment ofacetylcholine mediated vascular relaxation was largely pre-vented by treatment with the AT1r antagonist losartan.
Figure 2 summarizes the dose-dependent vasodilationresponses to acetylcholine (A, B), sodium nitroprusside(C), and papaverine (D). The data shown in Figure 2A illus-trate that aortic segments from mice with AngII–inducedhypertension exhibit a blunted relaxation response to acetyl-choline and that HTNþHCh exhibited an attenuatedresponse that lies between that produced by either risk factoralone. Losartan completely prevented the blunted relaxationresponse in AngII hypertensive mice. Figure 2B illustratesthat the blunted acetylcholine-induced relaxation notedwith HTNþHCh is more profoundly impaired in the pres-ence of catalase. Figure 2C demonstrates that, althoughHCh is not associated with an altered relaxation response
3 December 2012 � 2:50 pm � ce
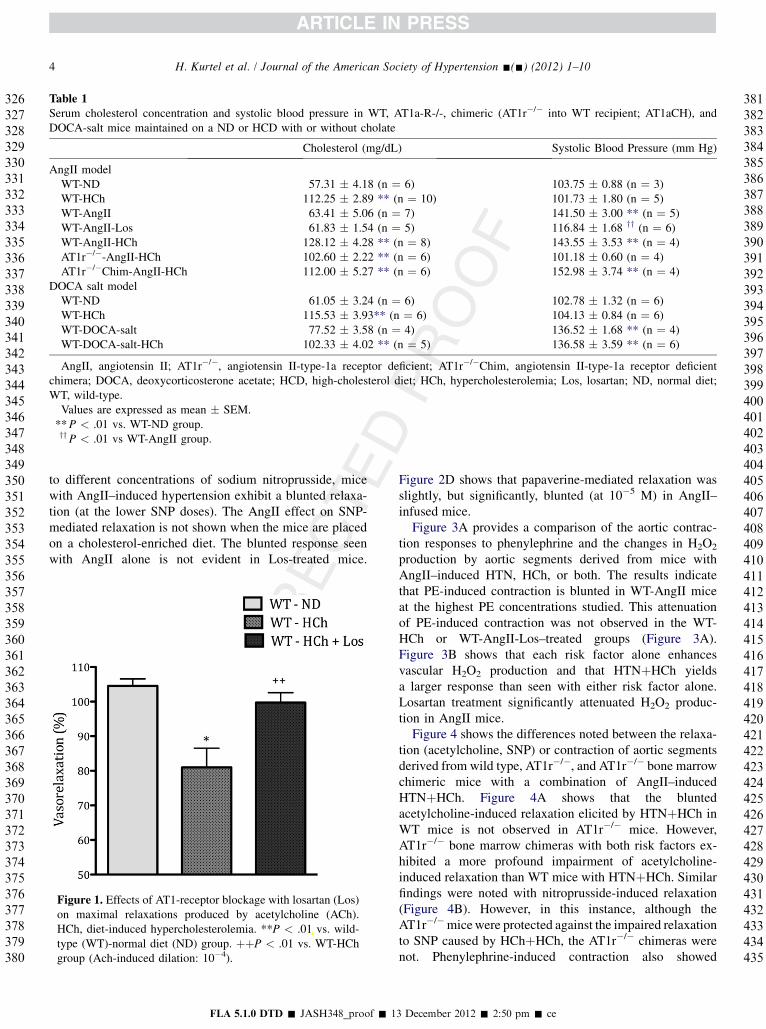
Table 1Serum cholesterol concentration and systolic blood pressure in WT, AT1a-R-/-, chimeric (AT1r�/� into WT recipient; AT1aCH), andDOCA-salt mice maintained on a ND or HCD with or without cholate
Cholesterol (mg/dL) Systolic Blood Pressure (mm Hg)
AngII modelWT-ND 57.31 � 4.18 (n ¼ 6) 103.75 � 0.88 (n ¼ 3)WT-HCh 112.25 � 2.89 ** (n ¼ 10) 101.73 � 1.80 (n ¼ 5)WT-AngII 63.41 � 5.06 (n ¼ 7) 141.50 � 3.00 ** (n ¼ 5)WT-AngII-Los 61.83 � 1.54 (n ¼ 5) 116.84 � 1.68 yy (n ¼ 6)WT-AngII-HCh 128.12 � 4.28 ** (n ¼ 8) 143.55 � 3.53 ** (n ¼ 4)AT1r�/�-AngII-HCh 102.60 � 2.22 ** (n ¼ 6) 101.18 � 0.60 (n ¼ 4)AT1r�/�Chim-AngII-HCh 112.00 � 5.27 ** (n ¼ 6) 152.98 � 3.74 ** (n ¼ 4)
DOCA salt modelWT-ND 61.05 � 3.24 (n ¼ 6) 102.78 � 1.32 (n ¼ 6)WT-HCh 115.53 � 3.93** (n ¼ 6) 104.13 � 0.84 (n ¼ 6)WT-DOCA-salt 77.52 � 3.58 (n ¼ 4) 136.52 � 1.68 ** (n ¼ 4)WT-DOCA-salt-HCh 102.33 � 4.02 ** (n ¼ 5) 136.58 � 3.59 ** (n ¼ 6)
AngII, angiotensin II; AT1r�/�, angiotensin II-type-1a receptor deficient; AT1r�/�Chim, angiotensin II-type-1a receptor deficientchimera; DOCA, deoxycorticosterone acetate; HCD, high-cholesterol diet; HCh, hypercholesterolemia; Los, losartan; ND, normal diet;WT, wild-type.Values are expressed as mean � SEM.**P < .01 vs. WT-ND group.yyP < .01 vs WT-AngII group.
4 H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
326327328329330331332333334335336337338339340341342343344345346347348349350351352353354355356357358359360361362363364365366367368369370371372373374375376377378379380
381382383384385386387388389390391392393394395396397398399400401402403404405406407408409410
to different concentrations of sodium nitroprusside, micewith AngII–induced hypertension exhibit a blunted relaxa-tion (at the lower SNP doses). The AngII effect on SNP-mediated relaxation is not shown when the mice are placedon a cholesterol-enriched diet. The blunted response seenwith AngII alone is not evident in Los-treated mice.
Figure 1. Effects of AT1-receptor blockage with losartan (Los)on maximal relaxations produced by acetylcholine (ACh).HCh, diet-induced hypercholesterolemia. **P < .01 vs. wild-type (WT)-normal diet (ND) group. þþP < .01 vs. WT-HChgroup (Ach-induced dilation: 10�4).
FLA 5.1.0 DTD � JASH348_proof � 1
411412413414415416417418419420421422423424425426427428429430431432433434435
Figure 2D shows that papaverine-mediated relaxation wasslightly, but significantly, blunted (at 10�5 M) in AngII–infused mice.
Figure 3A provides a comparison of the aortic contrac-tion responses to phenylephrine and the changes in H2O2
production by aortic segments derived from mice withAngII–induced HTN, HCh, or both. The results indicatethat PE-induced contraction is blunted in WT-AngII miceat the highest PE concentrations studied. This attenuationof PE-induced contraction was not observed in the WT-HCh or WT-AngII-Los–treated groups (Figure 3A).Figure 3B shows that each risk factor alone enhancesvascular H2O2 production and that HTNþHCh yieldsa larger response than seen with either risk factor alone.Losartan treatment significantly attenuated H2O2 produc-tion in AngII mice.
Figure 4 shows the differences noted between the relaxa-tion (acetylcholine, SNP) or contraction of aortic segmentsderived from wild type, AT1r�/�, and AT1r�/� bone marrowchimeric mice with a combination of AngII–inducedHTNþHCh. Figure 4A shows that the bluntedacetylcholine-induced relaxation elicited by HTNþHCh inWT mice is not observed in AT1r�/� mice. However,AT1r�/� bone marrow chimeras with both risk factors ex-hibited a more profound impairment of acetylcholine-induced relaxation than WT mice with HTNþHCh. Similarfindings were noted with nitroprusside-induced relaxation(Figure 4B). However, in this instance, although theAT1r�/�micewere protected against the impaired relaxationto SNP caused by HChþHCh, the AT1r�/� chimeras werenot. Phenylephrine-induced contraction also showed
3 December 2012 � 2:50 pm � ce
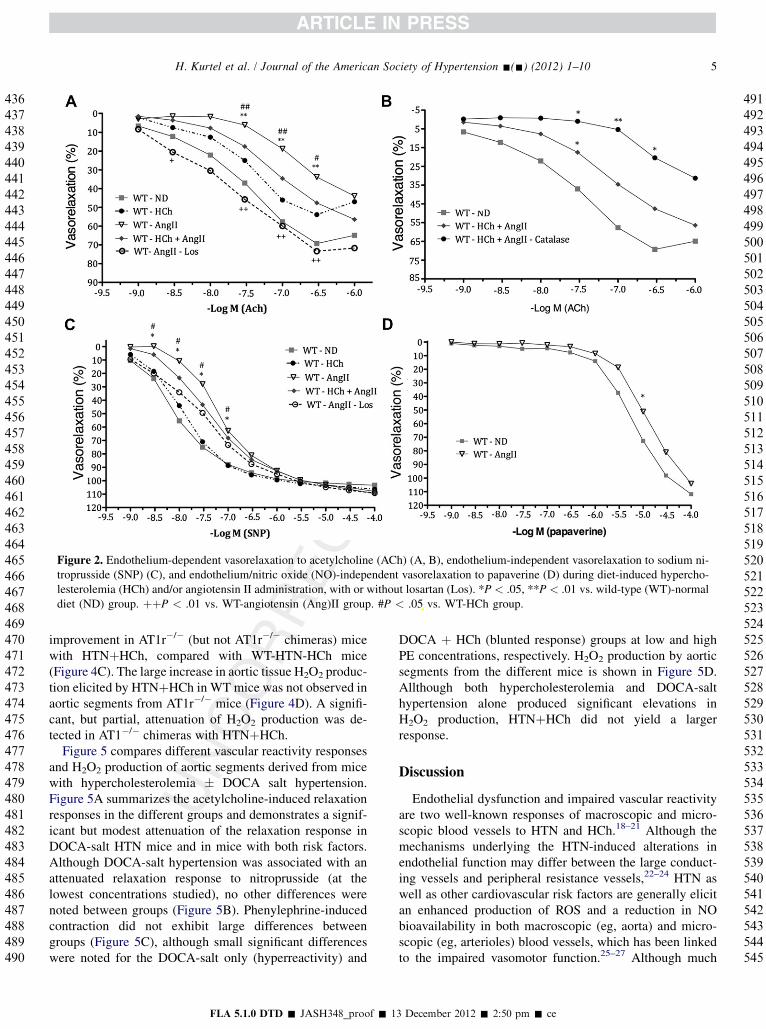
Figure 2. Endothelium-dependent vasorelaxation to acetylcholine (ACh) (A, B), endothelium-independent vasorelaxation to sodium ni-troprusside (SNP) (C), and endothelium/nitric oxide (NO)-independent vasorelaxation to papaverine (D) during diet-induced hypercho-lesterolemia (HCh) and/or angiotensin II administration, with or without losartan (Los). *P < .05, **P < .01 vs. wild-type (WT)-normaldiet (ND) group. þþP < .01 vs. WT-angiotensin (Ang)II group. #P < .05 vs. WT-HCh group.
5H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
436437438439440441442443444445446447448449450451452453454455456457458459460461462463464465466467468469470471472473474475476477478479480481482483484485486487488489490
491492493494495496497498499500501502503504505506507508509510511512513514515516517518519520521522523524525526527528529530531532533534535536537538539540541542543544545
improvement in AT1r�/� (but not AT1r�/� chimeras) micewith HTNþHCh, compared with WT-HTN-HCh mice(Figure 4C). The large increase in aortic tissue H2O2 produc-tion elicited by HTNþHCh in WT mice was not observed inaortic segments from AT1r�/� mice (Figure 4D). A signifi-cant, but partial, attenuation of H2O2 production was de-tected in AT1�/� chimeras with HTNþHCh.
Figure 5 compares different vascular reactivity responsesand H2O2 production of aortic segments derived from micewith hypercholesterolemia � DOCA salt hypertension.Figure 5A summarizes the acetylcholine-induced relaxationresponses in the different groups and demonstrates a signif-icant but modest attenuation of the relaxation response inDOCA-salt HTN mice and in mice with both risk factors.Although DOCA-salt hypertension was associated with anattenuated relaxation response to nitroprusside (at thelowest concentrations studied), no other differences werenoted between groups (Figure 5B). Phenylephrine-inducedcontraction did not exhibit large differences betweengroups (Figure 5C), although small significant differenceswere noted for the DOCA-salt only (hyperreactivity) and
FLA 5.1.0 DTD � JASH348_proof � 1
DOCA þ HCh (blunted response) groups at low and highPE concentrations, respectively. H2O2 production by aorticsegments from the different mice is shown in Figure 5D.Allthough both hypercholesterolemia and DOCA-salthypertension alone produced significant elevations inH2O2 production, HTNþHCh did not yield a largerresponse.
Discussion
Endothelial dysfunction and impaired vascular reactivityare two well-known responses of macroscopic and micro-scopic blood vessels to HTN and HCh.18–21 Although themechanisms underlying the HTN-induced alterations inendothelial function may differ between the large conduct-ing vessels and peripheral resistance vessels,22–24 HTN aswell as other cardiovascular risk factors are generally elicitan enhanced production of ROS and a reduction in NObioavailability in both macroscopic (eg, aorta) and micro-scopic (eg, arterioles) blood vessels, which has been linkedto the impaired vasomotor function.25–27 Although much
3 December 2012 � 2:50 pm � ce
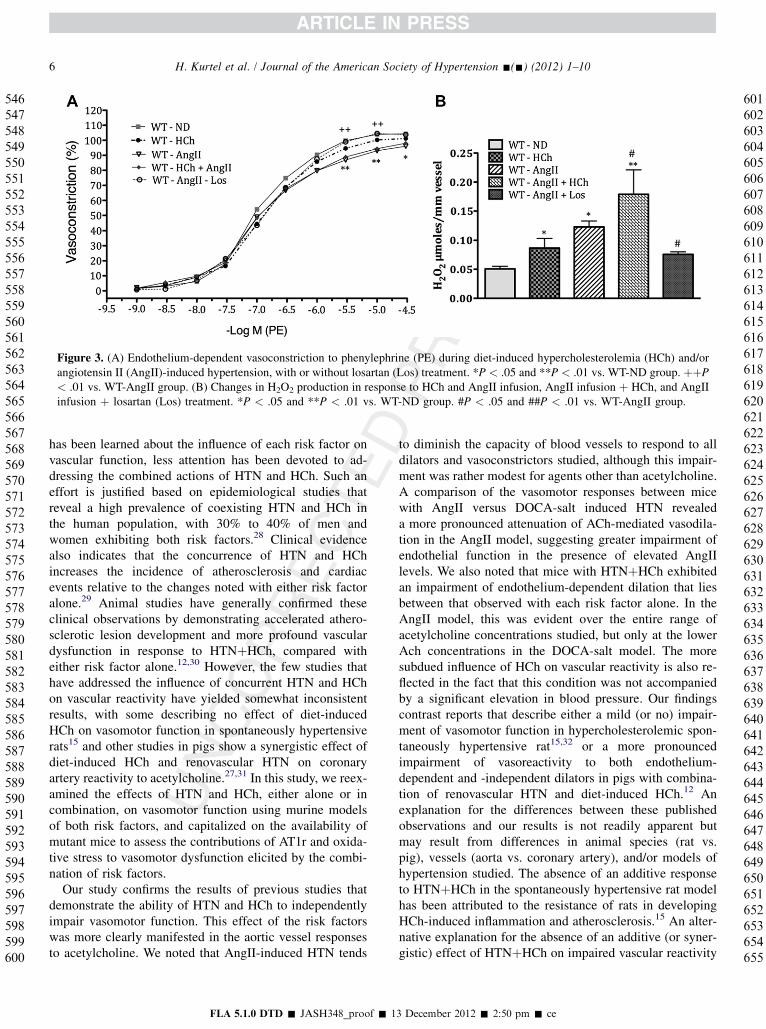
Figure 3. (A) Endothelium-dependent vasoconstriction to phenylephrine (PE) during diet-induced hypercholesterolemia (HCh) and/orangiotensin II (AngII)-induced hypertension, with or without losartan (Los) treatment. *P < .05 and **P < .01 vs. WT-ND group. þþP< .01 vs. WT-AngII group. (B) Changes in H2O2 production in response to HCh and AngII infusion, AngII infusion þ HCh, and AngIIinfusion þ losartan (Los) treatment. *P < .05 and **P < .01 vs. WT-ND group. #P < .05 and ##P < .01 vs. WT-AngII group.
6 H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
546547548549550551552553554555556557558559560561562563564565566567568569570571572573574575576577578579580581582583584585586587588589590591592593594595596597598599600
601602603604605606607608609610611612613614615616617618619620621622623624625626627628629630631632633634635636637638639640641642643644645646647648649650651652653654655
has been learned about the influence of each risk factor onvascular function, less attention has been devoted to ad-dressing the combined actions of HTN and HCh. Such aneffort is justified based on epidemiological studies thatreveal a high prevalence of coexisting HTN and HCh inthe human population, with 30% to 40% of men andwomen exhibiting both risk factors.28 Clinical evidencealso indicates that the concurrence of HTN and HChincreases the incidence of atherosclerosis and cardiacevents relative to the changes noted with either risk factoralone.29 Animal studies have generally confirmed theseclinical observations by demonstrating accelerated athero-sclerotic lesion development and more profound vasculardysfunction in response to HTNþHCh, compared witheither risk factor alone.12,30 However, the few studies thathave addressed the influence of concurrent HTN and HChon vascular reactivity have yielded somewhat inconsistentresults, with some describing no effect of diet-inducedHCh on vasomotor function in spontaneously hypertensiverats15 and other studies in pigs show a synergistic effect ofdiet-induced HCh and renovascular HTN on coronaryartery reactivity to acetylcholine.27,31 In this study, we reex-amined the effects of HTN and HCh, either alone or incombination, on vasomotor function using murine modelsof both risk factors, and capitalized on the availability ofmutant mice to assess the contributions of AT1r and oxida-tive stress to vasomotor dysfunction elicited by the combi-nation of risk factors.
Our study confirms the results of previous studies thatdemonstrate the ability of HTN and HCh to independentlyimpair vasomotor function. This effect of the risk factorswas more clearly manifested in the aortic vessel responsesto acetylcholine. We noted that AngII-induced HTN tends
FLA 5.1.0 DTD � JASH348_proof � 1
to diminish the capacity of blood vessels to respond to alldilators and vasoconstrictors studied, although this impair-ment was rather modest for agents other than acetylcholine.A comparison of the vasomotor responses between micewith AngII versus DOCA-salt induced HTN revealeda more pronounced attenuation of ACh-mediated vasodila-tion in the AngII model, suggesting greater impairment ofendothelial function in the presence of elevated AngIIlevels. We also noted that mice with HTNþHCh exhibitedan impairment of endothelium-dependent dilation that liesbetween that observed with each risk factor alone. In theAngII model, this was evident over the entire range ofacetylcholine concentrations studied, but only at the lowerAch concentrations in the DOCA-salt model. The moresubdued influence of HCh on vascular reactivity is also re-flected in the fact that this condition was not accompaniedby a significant elevation in blood pressure. Our findingscontrast reports that describe either a mild (or no) impair-ment of vasomotor function in hypercholesterolemic spon-taneously hypertensive rat15,32 or a more pronouncedimpairment of vasoreactivity to both endothelium-dependent and -independent dilators in pigs with combina-tion of renovascular HTN and diet-induced HCh.12 Anexplanation for the differences between these publishedobservations and our results is not readily apparent butmay result from differences in animal species (rat vs.pig), vessels (aorta vs. coronary artery), and/or models ofhypertension studied. The absence of an additive responseto HTNþHCh in the spontaneously hypertensive rat modelhas been attributed to the resistance of rats in developingHCh-induced inflammation and atherosclerosis.15 An alter-native explanation for the absence of an additive (or syner-gistic) effect of HTNþHCh on impaired vascular reactivity
3 December 2012 � 2:50 pm � ce
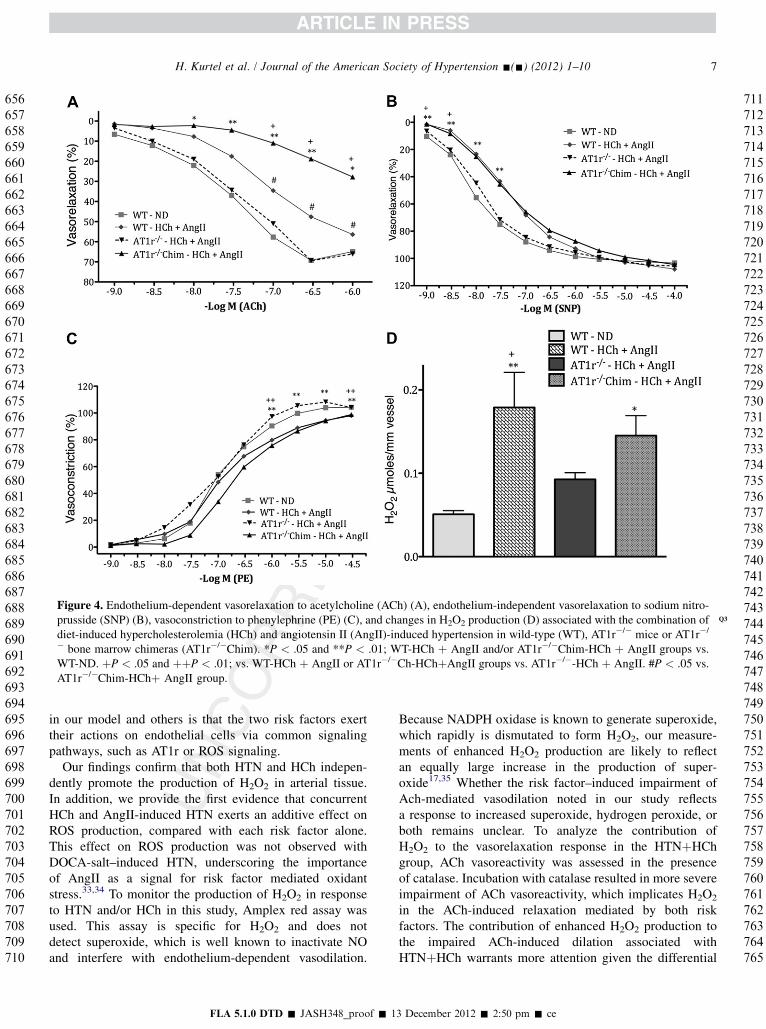
Figure 4. Endothelium-dependent vasorelaxation to acetylcholine (ACh) (A), endothelium-independent vasorelaxation to sodium nitro-prusside (SNP) (B), vasoconstriction to phenylephrine (PE) (C) Q3, and changes in H2O2 production (D) associated with the combination ofdiet-induced hypercholesterolemia (HCh) and angiotensin II (AngII)-induced hypertension in wild-type (WT), AT1r�/� mice or AT1r�/
� bone marrow chimeras (AT1r�/�Chim). *P < .05 and **P < .01; WT-HCh þ AngII and/or AT1r�/�Chim-HCh þ AngII groups vs.WT-ND. þP < .05 and þþP < .01; vs. WT-HCh þ AngII or AT1r�/�Ch-HChþAngII groups vs. AT1r�/�-HCh þ AngII. #P < .05 vs.AT1r�/�Chim-HChþ AngII group.
7H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
656657658659660661662663664665666667668669670671672673674675676677678679680681682683684685686687688689690691692693694695696697698699700701702703704705706707708709710
711712713714715716717718719720721722723724725726727728729730731732733734735736737738739740741742743744745746747748749750751752753754755756757758759760761762763764765
in our model and others is that the two risk factors exerttheir actions on endothelial cells via common signalingpathways, such as AT1r or ROS signaling.
Our findings confirm that both HTN and HCh indepen-dently promote the production of H2O2 in arterial tissue.In addition, we provide the first evidence that concurrentHCh and AngII-induced HTN exerts an additive effect onROS production, compared with each risk factor alone.This effect on ROS production was not observed withDOCA-salt–induced HTN, underscoring the importanceof AngII as a signal for risk factor mediated oxidantstress.33,34 To monitor the production of H2O2 in responseto HTN and/or HCh in this study, Amplex red assay wasused. This assay is specific for H2O2 and does notdetect superoxide, which is well known to inactivate NOand interfere with endothelium-dependent vasodilation.
FLA 5.1.0 DTD � JASH348_proof � 1
Because NADPH oxidase is known to generate superoxide,which rapidly is dismutated to form H2O2, our measure-ments of enhanced H2O2 production are likely to reflectan equally large increase in the production of super-oxide17,35 Whether the risk factor–induced impairment ofAch-mediated vasodilation noted in our study reflectsa response to increased superoxide, hydrogen peroxide, orboth remains unclear. To analyze the contribution ofH2O2 to the vasorelaxation response in the HTNþHChgroup, ACh vasoreactivity was assessed in the presenceof catalase. Incubation with catalase resulted in more severeimpairment of ACh vasoreactivity, which implicates H2O2
in the ACh-induced relaxation mediated by both riskfactors. The contribution of enhanced H2O2 production tothe impaired ACh-induced dilation associated withHTNþHCh warrants more attention given the differential
3 December 2012 � 2:50 pm � ce
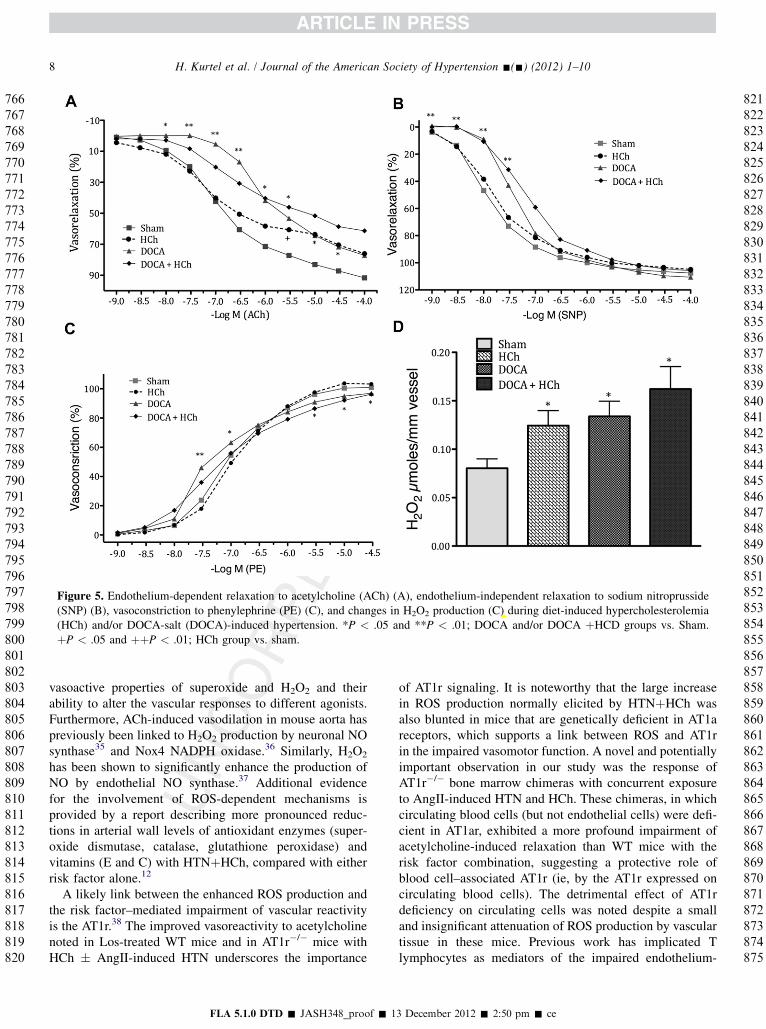
Figure 5. Endothelium-dependent relaxation to acetylcholine (ACh) (A), endothelium-independent relaxation to sodium nitroprusside(SNP) (B), vasoconstriction to phenylephrine (PE) (C), and changes in H2O2 production (C) during diet-induced hypercholesterolemia(HCh) and/or DOCA-salt (DOCA)-induced hypertension. *P < .05 and **P < .01; DOCA and/or DOCA þHCD groups vs. Sham.þP < .05 and þþP < .01; HCh group vs. sham.
8 H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
766767768769770771772773774775776777778779780781782783784785786787788789790791792793794795796797798799800801802803804805806807808809810811812813814815816817818819820
821822823824825826827828829830831832833834835836837838839840841842843844845846847848849850851852853854855856857858859860861862863864865866867868869870871872873874875
vasoactive properties of superoxide and H2O2 and theirability to alter the vascular responses to different agonists.Furthermore, ACh-induced vasodilation in mouse aorta haspreviously been linked to H2O2 production by neuronal NOsynthase35 and Nox4 NADPH oxidase.36 Similarly, H2O2
has been shown to significantly enhance the production ofNO by endothelial NO synthase.37 Additional evidencefor the involvement of ROS-dependent mechanisms isprovided by a report describing more pronounced reduc-tions in arterial wall levels of antioxidant enzymes (super-oxide dismutase, catalase, glutathione peroxidase) andvitamins (E and C) with HTNþHCh, compared with eitherrisk factor alone.12
A likely link between the enhanced ROS production andthe risk factor–mediated impairment of vascular reactivityis the AT1r.38 The improved vasoreactivity to acetylcholinenoted in Los-treated WT mice and in AT1r�/� mice withHCh � AngII-induced HTN underscores the importance
FLA 5.1.0 DTD � JASH348_proof � 1
of AT1r signaling. It is noteworthy that the large increasein ROS production normally elicited by HTNþHCh wasalso blunted in mice that are genetically deficient in AT1areceptors, which supports a link between ROS and AT1rin the impaired vasomotor function. A novel and potentiallyimportant observation in our study was the response ofAT1r�/� bone marrow chimeras with concurrent exposureto AngII-induced HTN and HCh. These chimeras, in whichcirculating blood cells (but not endothelial cells) were defi-cient in AT1ar, exhibited a more profound impairment ofacetylcholine-induced relaxation than WT mice with therisk factor combination, suggesting a protective role ofblood cell–associated AT1r (ie, by the AT1r expressed oncirculating blood cells). The detrimental effect of AT1rdeficiency on circulating cells was noted despite a smalland insignificant attenuation of ROS production by vasculartissue in these mice. Previous work has implicated Tlymphocytes as mediators of the impaired endothelium-
3 December 2012 � 2:50 pm � ce

9H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
876877878879880881882883884885886887888889890891892893894895896897898899900901902903904905906907908909910911912913914915916917918919920921922923924925926927928929930
931932933934935936937938939940941942943944945946947948
dependent dilation induced by either AngII-induced HTN39
or HCh40 in mice. In both instances, evidence was providedto implicate pro-inflammatory cytokines (tumor necrosisfactor-a, interferon-g) as potential chemical mediators ofthe T cell–dependent responses. Our findings would suggestthat, in the presence of HTNþHCh, an AT1r-dependentprotective agent is liberated by circulating blood cells,perhaps T cells. A candidate molecule is interleukin-10,which has been shown to preserve endothelium-dependentvasodilation in diabetic mice via a superoxide-dependentmechanism.41 Interleukin-10 appears to counteract impairedendothelium-dependent relaxation and upregulation ofNADPH oxidase induced by AngII in murine aortic rings.42
Regardless of the AT1r-dependent chemical mediator liber-ated from blood cells in the presence of HTNþHCh, ourdata lend support to the view that circulating cells playa role in mediating the vasomotor dysfunction associatedwith risk factors for cardiovascular disease.
949950951952953954955956957958959960961962963964965966967968969970971972973974975976977978979980981982983984985
References
1. Granger DN, Rodrigues SF, Yildirim A,Senchenkova EY. Microvascular responses to cardiovas-cular risk factors. Microcirculation 2010;17:192–205.
2. Brunner H, Cockcroft JR, Deanfield J, Donald A,Ferrannini E, Halcox J, et al. Endothelial function anddysfunction. Part II: association with cardiovascularrisk factors and diseases. A statement by the workinggroup on endothelins and endothelial factors of theEuropean Society of Hypertension. J Hypertens 2005;23:233–46.
3. Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunc-tion: cardiovascular risk factors, therapy, and outcome.Vasc Health Risk Manage 2005;1:183–98.
4. Forstermann U, Munzel T. Endothelial nitric oxide syn-thase in vascular disease: from marvel to menace.Circulation 2006;113:1708–14.
5. Granger DN, Senchenkova E. Inflammation and themicrocirculation. Integrated Systems Physiology—From Cell to Function. San Rafael (CA): Morgan &Claypool Life Sciences; 2010.
6. Stokes KY, Granger DN. The microcirculation: a motorfor the systemic inflammatory response and large vesseldisease induced by hypercholesterolaemia? J Physiol2005;562:647–53.
7. Cannon RO 3rd. Role of nitric oxide in cardiovasculardisease: focus on the endothelium. Clin Chem 1998;44:1809–19.
8. Vital SA, Terao S, Nagai M, Granger DN. Mechanismsunderlying the cerebral microvascular responses toangiotensin II-induced hypertension. Microcirculation2010;17:641–69.
9. Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA,Gratze P, et al. Regulation of t-cell function by
FLA 5.1.0 DTD � JASH348_proof � 1
endogenously produced angiotensin ii. Am J PhysiolRegul Integr Comp Physiol 2009;296:R208–16.
10. Lopez ADM, CD. Ezzati, M. Jamison, DT. Murray,CJL. Global burden of disease and risk factors. Wash-ington (DC): World Bank; 2006.
11. Rodrigues SF, Granger DN. Role of blood cells inischaemia-reperfusion induced endothelial barrierfailure. Cardiovasc Res 87:291–9
12. Rodriguez-Porcel M, Lerman LO, Herrmann J,Sawamura T, Napoli C, Lerman A. Hypercholesterol-emia and hypertension have synergistic deleteriouseffects on coronary endothelial function. ArteriosclerThromb Vasc Biol 2003;23:885–91.
13. Javeshghani D, Schiffrin EL, Sairam MR, Touyz RM.Potentiation of vascular oxidative stress and nitricoxide-mediated endothelial dysfunction by high-fatdiet in a mouse model of estrogen deficiency and hy-perandrogenemia. J Am Soc Hypertens 2009;3:295–305.
14. Arruda RM, Peotta VA, Meyrelles SS, Vasquez EC.Evaluation of vascular function in apolipoprotein eknockout mice with angiotensin-dependent renovas-cular hypertension. Hypertension 2005;46:932–6.
15. Lorkowska B, Bartus M, Franczyk M, Kostogrys RB,Jawien J, Pisulewski PM, et al. Hypercholesterolemiadoes not alter endothelial function in spontaneouslyhypertensive rats. J Pharmacol Exp Ther 2006;317:1019–126.
16. Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP.A stable nonfluorescent derivative of resorufin for thefluorometric determination of trace hydrogen peroxide:applications in detecting the activity of phagocyteNADPH oxidase and other oxidases. Anal Biochem1997;253:162–18.
17. Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH,Harrison DG, Griendling KK. Distinct roles of nox1and nox4 in basal and angiotensin ii-stimulated super-oxide and hydrogen peroxide production. Free RadicalBiol Med 2008;45:1340–151.
18. Ghiadoni L, Taddei S, Virdis A. Hypertension andendothelial dysfunction: therapeutic approach. CurrVasc Pharmacol 2012;10:42–60.
19. Petnehazy T, Stokes KY, Russell JM, Granger DN.Angiotensin II type-1 receptor antagonism attenuatesthe inflammatory and thrombogenic responses tohypercholesterolemia in venules. Hypertension 2005;45:209–15.
20. Stapleton PA, Goodwill AG, James ME, Brock RW,Frisbee JC. Hypercholesterolemia and microvasculardysfunction: interventional strategies. J Inflamm(Lond) 2010;18:54.
21. Virdis A, Ghiadoni L, Versari D, Giannarelli C,Salvetti A, Taddei S. Endothelial function assessmentin complicated hypertension. Curr Pharm Des 2008;14:1761–70.
3 December 2012 � 2:50 pm � ce

2
10 H. Kurtel et al. / Journal of the American Society of Hypertension -(-) (2012) 1–10
986
987
988
989
990
991
992
993
994
995
996
997
998
999
1000
1001
1002
1003
1004
1005
1006
1007
1008
1009
1010
1011
1012
1013
1014
1015
1016
1017
1018
1019
1020
1021
1022
1023
1024
1025
1026
1027
1028
1029
1030
1031
1032
1033
1034
1035
1036
1037
1038
1039
1040
1041
1042
1043
1044
1045
1046
1047
1048
1049
1050
1051
1052
1053
1054
1055
1056
1057
1058
1059
1060
1061
1062
1063
1064
1065
1066
1067
1068
1069
1070
1071
1072
1073
1074
1075
1076
1077
1078
1079
1080
1081
1082
1083
1084
1085
1086
22. Luscher TF, Vanhoutte PM. Endothelium-dependentcontractions to acetylcholine in the aorta of the spontane-ously hypertensive rat. Hypertension 1986;8:344–38.
23. Koga T, Takata Y, Kobayashi K, Takishita S,Yamashita Y, Fujishima M. Age and hypertensionpromote endothelium-dependent contractions to acetyl-choline in the aorta of the rat. Hypertension 1989;14:542–58.
24. Konishi M, Su C. Role of endothelium in dilatorresponses of spontaneously hypertensive rat arteries.Hypertension 1983;5:881–6.
25. Bonetti PO, Lerman LO, Lerman A. Endothelialdysfunction: a marker of atherosclerotic risk. Arte-rioscle Thromb Vasc Biol 2003;23:168–75.
26. Luscher TF, Tanner FC, Dohi Y. Age, hypertension andhypercholesterolaemia alter endothelium-dependentvascular regulation. Pharmacol Toxicol 1992;70:S32–S9.
27. Versari D, Gossl M, Mannheim D, Daghini E, Galili O,Napoli C, et al. Hypertension and hypercholesterolemiadifferentially affect the function and structure of pigcarotid artery. Hypertension 2007;50:1063–108.
28. De Bacquer D, De Backer G. The prevalence ofconcomitant hypertension and hypercholesterolaemiain the general population. Int J Cardiol 2006;110:217–23.
29. Smith GD, Shipley MJ, Marmot MG, Rose G. Plasmacholesterol concentration and mortality. The Whitehallstudy. JAMA 1992;267:70–6.
30. Chobanian AV, Lichtenstein AH, Nilakhe V,Haudenschild CC, Drago R, Nickerson C. Influenceof hypertension on aortic atherosclerosis in the wata-nabe rabbit. Hypertension 1989;14:203–29.
31. Shimokawa H, Vanhoutte PM. Impaired endothelium-dependent relaxation to aggregating platelets andrelated vasoactive substances in porcine coronaryarteries in hypercholesterolemia and atherosclerosis.Circ Res 1989;64:900–94.
32. Cappelli-Bigazzi M, Rubattu S, Battaglia C, Russo R,Enea I, Ambrosio G, et al. Effects of high-cholesteroland atherogenic diets on vascular relaxation in sponta-neously hypertensive rats. Am J Physiol 1997;273:H647–54.
FLA 5.1.0 DTD � JASH348_proof � 1
33. Berry C, Brosnan MJ, Fennell J, Hamilton CA,Dominiczak AF. Oxidative stress and vascular damagein hypertension. Curr Opin Nephrol Hypertens 2001;10:247–55.
34. Ushio-Fukai M, Alexander RW. Reactive oxygenspecies as mediators of angiogenesis signaling: roleof NAD(P)H oxidase. Mol Cell Biochem 2004;264:85–97.
35. Capettini LS, Cortes SF, Gomes MA, Silva GA,Pesquero JL, Lopes MJ, et al. Neuronal nitric oxidesynthase-derived hydrogen peroxide is a majorendothelium-dependent relaxing factor. Am J PhysiolHeart Circ Physiol 2008;295:H2503–11.
36. Ray R, Murdoch CE, Wang M, Santos CX, Zhang M,Alom-Ruiz S, et al. Endothelial nox4 NADPH oxidaseenhances vasodilatation and reduces blood pressurein vivo. Arterioscl Thromb Vasc Biol 31:1368–76 Q
37. Cai H, Li Z, Davis ME, Kanner W, Harrison DG,Dudley SC Jr. Akt-dependent phosphorylation of serine1179 and mitogen-activated protein kinase kinase/ex-tracellular signal-regulated kinase 1/2 cooperativelymediate activation of the endothelial nitric-oxide syn-thase by hydrogen peroxide. Molec Pharmacol 2003;63:325–31.
38. Nickenig G. Central role of the at(1)-receptor in athero-sclerosis. J Hum Hypertens 2002;16(Suppl 3):S26–33.
39. Guzik TJ, Hoch NE, Brown KA, McCann LA,Rahman A, Dikalov S, et al. Role of the t cell in thegenesis of angiotensin ii induced hypertension andvascular dysfunction. J Exp Med 2007;204:2449–260.
40. Stokes KY, Gurwara S, Granger DN. T-cell derivedinterferon-gamma contributes to arteriolar dysfunctionduring acute hypercholesterolemia. ArteriosclerThromb Vasc Biol 2007;27:1998–2004.
41. Gunnett CA, Heistad DD, Faraci FM. Interleukin-10protects nitric oxide-dependent relaxation during dia-betes: role of superoxide. Diabetes 2002;51:1931–197.
42. Zemse SM, Hilgers RH, Webb RC. Interleukin-10counteracts impaired endothelium-dependent relaxa-tion induced by ANG II in murine aortic rings.Am J Physiol Heart Circ Physiol 2007;292:H3103–H318.
3 December 2012 � 2:50 pm � ce
1087

Our reference: JASH 348 P-authorquery-v9
AUTHOR QUERY FORM
Journal: JASH
Article Number: 348
Please e-mail or fax your responses and any corrections to:
E-mail: [email protected]
Fax: +91 44 4596 4899
Dear Author,
Please check your proof carefully and mark all corrections at the appropriate place in the proof (e.g., by using on-screen
annotation in the PDF file) or compile them in a separate list. Note: if you opt to annotate the file with software other than
Adobe Reader then please also highlight the appropriate place in the PDF file. To ensure fast publication of your paper please
return your corrections within 48 hours.
For correction or revision of any artwork, please consult http://www.elsevier.com/artworkinstructions.
Any queries or remarks that have arisen during the processing of your manuscript are listed below and highlighted by flags in
the proof.
Location
in articleQuery / Remark: Click on the Q link to find the query’s location in text
Please insert your reply or correction at the corresponding line in the proof
Q1 If there are any drug dosages in your article, please verify them and indicate that you have done so by
initialing this query
Q2 In reference 36, please provide year of publication.
Q3 In the Figure 4 and 5 legends, “(D)” is used twice; please verify that change of first “(D)” to “(C)” is correct.
Q4 Please confirm that given names and surnames have been identified correctly.
Please check this box if you have no
corrections to make to the PDF file ,
Thank you for your assistance.










![[Familial hypercholesterolemia] Фамилна хиперхолестеролемия](https://img.pdfslide.net/doc/110x75/635c923c87785f4426063817/familial-hypercholesterolemia-familna-khiperkholesterolemiya.jpg)


















