Embed Size (px)
Citation preview

Human MutationRESEARCH ARTICLE
Predicting Functional Significance of Cancer-Associatedp16INK4a Mutations in CDKN2A
Heather A. McKenzie, Carina Fung, Therese M. Becker, Mal Irvine, Graham J. Mann, Richard F. Kefford,and Helen Rizos�
Westmead Institute for Cancer Research and Melanoma Institute of Australia, University of Sydney at Westmead Millennium Institute,
Westmead Hospital, Westmead NSW 2145, Australia
Communicated by David E. GoldgarReceived 14 July 2009; accepted revised manuscript 4 March 2010.
Published online 25 March 2010 in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/humu.21245
ABSTRACT: Inherited mutations affecting the INK4a/ARFlocus (CDKN2A) are associated with melanoma suscept-ibility in 40% of multiple case melanoma families. Over60 different germline INK4a/ARF mutations have beendetected in more than 190 families worldwide. Themajority of these alterations are missense mutationsaffecting p16INK4a, and only 25% of these have beenfunctionally assessed. There is therefore a need for anaccurate and rapid assay to determine the functionalsignificance of p16INK4a mutations. We reviewed theperformance of several in vivo functional assays thatmeasure critical aspects of p16INK4a function, includingsubcellular location, CDK binding and cell cycle inhibi-tion. In this report the function of 28 p16INK4a variants,many associated with melanoma susceptibility werecompared. We show that assessment of CDK4 bindingand subcellular localization can accurately and rapidlydetermine the functional significance of melanoma-associated p16INK4a mutations. p16INK4a-CDK6 bindingaffinity was unhelpful, as no disease-associated mutationshowed reduced CDK6 affinity while maintaining theability to bind CDK4. Likewise, in silico analyses did notcontribute substantially, with only 12 of 25 melanoma-associated missense variants consistently predicted asdeleterious. The ability to determine variant functionalactivity accurately would identify disease-associatedmutations and facilitate effective genetic counselling ofindividuals at high risk of melanoma.Hum Mutat 31:692–701, 2010. & 2010 Wiley-Liss, Inc.
KEY WORDS: CDKN2A; p16INK4a; CDK4; CDK6; mel-anoma; cancer
Introduction
The INK4a/ARF locus on chromosome band 9p21 (HUGO-approved symbol CDKN2A; MIM] 600160) encodes the p16INK4a
and p14ARF tumor suppressor proteins in alternate readingframes. Somatic alterations affecting this genomic sequence occur
frequently in human tumors; the locus is homozygously deleted inup to 67% of breast cancers and altered in 37% of pancreaticadenocarcinomas (reviewed in [Foulkes et al., 1997]). Moreover,inherited INK4a/ARF mutations are associated with mela-noma susceptibility in 40% of multiple case melanoma families[Goldstein et al., 2006b]. p16INK4a promotes cell cycle arrest bybinding to and inhibiting the kinase activities of the cyclinD-dependent kinases, CDK4 and CDK6. This activity maintainsthe retinoblastoma protein, pRb in its hypophosphorylated,antiproliferative state [Serrano et al., 1993]. The progressiveaccumulation of p16INK4a is also associated with the onset ofreplicative senescence [Alcorta et al., 1996; Brenner et al., 1998]and p16INK4a expression induces growth arrest that resemblescellular senescence in human cells [Haferkamp et al., 2008;McConnell et al., 1999; Zhu et al., 1998].
More than 60 different germline INK4a/ARF mutations have sofar been detected in over 190 families worldwide [Goldstein et al.,2006a]. The majority of these alterations are missense mutationsaffecting p16INK4a, rather than p14ARF, which is specificallyaltered in only 2% of high-risk melanoma families. No p16INK4a
mutational ‘‘hot spots’’ have been observed [Goldstein et al.,2006a] and only 25% of p16INK4a missense mutation have beenfunctionally assessed (https://biodesktop.uvm.edu/perl/p16) usingassays that determine p16INK4a subcellular distribution, CDK4/6binding affinity, cyclin D-CDK kinase inhibitory activity, cell cycleinhibitory action, and/or senescence-inducing function. Theseassays have not been universally applied, and no single study hasperformed a comprehensive comparison of relevant functionaltests. There have also been diverse and serious discrepancies in thefunctional assessment of the same p16INK4a variants by differentmethods. For instance, the common p.G101W mutation, whichhas been identified in 14 melanoma-prone kindreds, has beenreported to have 70% wild-type CDK4 binding in a yeast two-hybrid screen, significant CDK4 binding in vivo using immuno-precipitations, variable CDK4 binding in vitro at 301C, and noCDK4 binding in vitro at 421C [Parry and Peters, 1996; Ranadeet al., 1995; Walker et al., 1999; Yang et al., 1995]. Assays thatrequire the production of recombinant p16INK4a protein inbacteria are vulnerable to artefactual protein aggregation [Zhangand Peng, 1996], and may be exposed to nonphysiologicalposttranslational modifications. In addition, in vitro assays cannotaccurately reflect the in vivo environment. For instance, cyclinD-dependent kinase activity is associated with high molecular weightcomplexes in vivo rather than the simple cyclin D-CDK binarycomplexes used for in vitro kinase assays [Mahony et al., 1998].
Considering that over 65% of p16INK4a mutations have beendetected only once and the risk of developing melanoma in
OFFICIAL JOURNAL
www.hgvs.org
& 2010 WILEY-LISS, INC.
Additional Supporting Information may be found in the online version of this article.�Correspondence to: Helen Rizos, Westmead Institute for Cancer Research,
University of Sydney at Westmead Millennium Institute, Westmead Hospital,
Westmead NSW 2145, Australia. E-mail: [email protected]

carriers may vary widely depending on the type of p16INK4a
mutation [Bishop et al., 2002; Walker et al., 1995], there is acritical need for accurate and rapid assays to determine thefunctional significance of p16INK4a mutations. In silico predictionsare useful, but are limited to simple missense mutations and aloneare not conclusive evidence of pathogenicity [Kannengiesser et al.,2009]. The ability to determine variant functional activityaccurately might allow population polymorphisms to be distin-guished from disease-associated mutations, and would facilitatemore informed estimates of melanoma risk in mutation carriers.
In this study we sought to develop a set of functional assays thatrapidly and accurately estimate the biochemical severity ofp16INK4a mutations. We chose to focus on in vivo assays thatmeasure key features of p16INK4a function, including subcellularlocation, CDK binding and cell cycle inhibition. Our analysesincluded 28 p16INK4a variants, including a small duplication andinsertion [Koh et al., 1995; Parry and Peters, 1996; Ranade et al.,1995; Reymond and Brent, 1995]. We propose that the combinedassessment of CDK4 binding affinity and subcellular distributionprovides an effective predictive tool for p16INK4a function.
Materials and Methods
p16INK4a Mutations
Mutation nomenclature follows the numbering recommendedby the journal (www.hgvs.org/mutnomen) with 11 nucleotide asthe A of the ATG initiation codon, GenBank accession numberNM_000077.2.
In Silico Analyses
Amino acid alignments of p16INK4a sequences were constructedusing the M-Coffee tool suite (www.igs.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/index.cgi) [Wallace et al., 2006] followed by minorchanges in alignment to remove insertion sequences with nohomology to human p16INK4a [Chan et al., 2007]. Sequences forp15INK4b, p18INK4c, and p19INK4d were omitted from sequencealignments as suggested by Chan et al. [2007]. Accession numbersand alignments are shown in Supp. Figure S1.
To assess the potential impact of missense mutations, weapplied a combination of methods, to improve predictive accuracy[Chan et al., 2007]. BLOSUM62 values were classified conservative(Z0) or non conservative (o0) [Henikoff and Henikoff, 1992].The BLOSUM62 matrix was also used to measure the evolutionaryvariation at each codon. The average score for every possiblesequence pair at each codon was calculated, and variants wereconsidered tolerated if they occurred at an amino acid withBLOSUM62 pairwise scores lower than 3.5 [Chan et al., 2007;Greenblatt et al., 2003]. Sorting Intolerant from Tolerant (SIFT)SIFT version 4.0 (http://sift.jcvi.org/) was used to analyze the setof aligned p16INK4a sequences. PolyPhen was performed with theProtein Quaternary (PQS) database using default settings (http://genetics.bwh.harvard.edu/pph). Results for each variant wereclassified as ‘‘Benign,’’ ‘‘Possibly Damaging,’’ or ‘‘ProbablyDamaging,’’ and the latter two were considered deleterious in thisstudy. The Grantham chemical score was used to classify theamino acid substitutions as either tolerated (scoreo60) ordeleterious (scoreZ60) [Chan et al., 2007; Grantham, 1974]. Inaddition the Grantham difference (GD) relative to the evolu-tionary difference (Grantham variation, GV) was also determinedusing Align-GVGD (http://agvgd.iarc.fr). The output from Align-GVGD is an ordered series of grades ranging from C65 (most
likely deleterious) to C0 (most likely neutral) [Tavtigian et al.,2008a,b]. Align-GVGD grades of ZC25 were considered likelydeleterious [Tavtigian et al., 2008a,b].
Cell Culture and Transfections
The WMM1175 human melanoma cell line, originally isolatedfrom a subcutaneous metastatic tumor of an individual with a familyhistory of melanoma, contains a homozygous deletion encompassingthe INK4a/ARF region on chromosome band 9p21 [Rizos et al.,1999]. An inducible mammalian expression system (Lac-switchsystem; Stratagene, La Jolla, CA) was used to obtain WMM1175cell clones stably expressing inducible forms of wild-type p16INK4a,p.R24P, or p.G101W under IPTG-inducible expression [Becker et al.,2001]. p16INK4a inducible cells were maintained in Dulbecco’smodified Eagle’s medium supplemented with 10% fetal bovine serum(DMEM/10% FBS) with 250mg/ml hygromycin and 500mg/mlgeneticin (Gibco BRL, Carlsbad, CA). Stable cells were seeded 24 hprior to induction in the absence of antibiotics and were induced with4 mM IPTG. NM39 melanoma cells, Saos-2, and U20S osteosarcomacells were also cultured in DMEM/10% FBS.
For ectopic p16INK4a transfections, cells (1� 105) were trans-fected with 1–4 mg DNA using Lipofectamine 2000 (InvitrogenLife Technologies, Carlsbad, CA).
Immunofluorescence
Cultured cells (1�105) seeded on coverslips in six-well plateswere fixed in PBS/3.7% formaldehyde and permeabilized with PBS/0.2% Triton X-100, 40 h posttransfection. Cells were immunos-tained for 50 min with rabbit a-FLAG (Sigma, St. Louis, MO),rabbit a-p16INK4a (N20, Santa Cruz, Santa Cruz, CA), or mousea-Ki67 (MIB-1; DAKO, Hamburg, Germany), followed by a50-min exposure to FITC-, Alexa Fluor 488, or Alexa Fluor 594-conjugated secondary IgG (Sigma and Molecular Probes, Carlsbad,CA). Subcellular distribution was determined from a total of atleast 400 cells from two independent experiments.
Mammalian-Two-Hybrid Assay
p16INK4a was cloned in frame with the GAL4 nuclear localizationsignal in the pM vector and CDK4 and CDK6 were each tagged withthe SV40 nuclear localization sequence in the pVP16 vector(Clontech, Mountain View, CA). Each assay was performed at leasttwice, in duplicate using Saos-2 cells seeded in six-well plates(1.5� 105 cells/well). Each well was transfected with a total of 0.5mgDNA consisting of the pM, pVP16, the pG5luc reporter vector(Promega, Madison, WI) and CMV-galactosidase vector in a5:5:1:0.6mg ratio. Cells were assayed 40 h posttransfection forluciferase (Promega) and galactosidase (Applied Biosystems, FosterCity, CA) activity using a Packard TopCount luminometer or a 2450Microbeta Counter (Perkin-Elmer, Waltham, MA).
Cell Cycle Analysis
U2OS osteosarcoma cells (2.5� 105) were transfected with 3 mgwild-type p16INK4a-FLAG or mutant p16INK4a-FLAG plasmid with1 mg EGFP-spectrin. Cells were maintained at 371C or transferredto 401C 24 h posttransfection. Approximately 48 h posttransfec-tion cells were harvested, fixed, and stained as previouslydescribed [Rizos et al., 2001b]. DNA content from at least 5,000cells was analyzed using ModFIT software (Verity Software,Topsham, ME).
HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010 693

Western Blotting
Total cellular proteins were extracted at 41C using RIPA lysisbuffer containing 6 M urea (Sigma) and CompleteTM proteaseinhibitors (Roche, Basel, Switzerland). Proteins (30–50 mg) wereresolved on 12% SDS-polyacrylamide gels and transferred toImmobilon-P membranes (Millipore, Bedford, MA). Westernblots were probed with antibodies against p16INK4a (N20; SantaCruz), FLAG (Sigma), GFP (7.1 and 13.1; Roche), total pRb (G3-245; Becton Dickinson, Franklin Lakes, NJ), phosphorylatedp-pRb807/811 (9308; Cell Signalling, Danvers, MA), E2F-1 (C20;Santa Cruz), cyclin A (BF683; Becton Dickinson), and b-actin(AC-74; Sigma).
Results
Selection and In Silico Analyses of p16INK4a Variants
A total of 28 p16INK4a variants were selected for this study.These include 26 missense mutations located throughout theprotein, a 24-bp amino terminal duplication (p.M1_S8dup, alsoknown as 24 bp duplication), a 19-bp deletion (c.225_243del19,also known as p16INK4a-Leiden), the silent p.A73A mutant andtwo population polymorphisms (p.A148T, p.R144C) [Yang et al.,1995]. Of these 28 mutations, 24 have been identified as inheritedmutations in the germline of multiple affected members ofmelanoma families from countries such as Australia, France, andthe United Kingdom. To predict the clinical consequences ofmissense variants we applied six missense substitution algorithms;four used evolutionary sequence conservation (BLOSUM62pairwise, SIFT, PolyPhen, and Align-GVGD) and two methodsonly considered the amino acid substitution (Grantham scale andBLOSUM62 matrix score). According to Chan et al. [2007], thefour evolutionary based methods predict 73–82% of all variantsand this value improves to over 90% when three of the fourmethods predict deleterious effects [Chan et al., 2007]. Thus, wecalculated the ‘‘prediction score’’ from the number of conserva-tion-based methods that defined an alteration as deleterious. Only12 of the 25 missense mutations had a prediction score Z3, andseven of these were classified deleterious by all four methods(Table 1). Of the 12 mutations with a prediction score Z3, onlyeight were classified as deleterious by the Grantham andBLOSUM62 scores, although Grantham and BLOSUM62 bothpredicted an additional seven missense mutations were deleter-ious. Importantly, only BLOSUM62 classified the commonp.A148T polymorphism as deleterious, while Grantham, BLO-SUM62 and Polyphen all tagged the p.R144C polymorphism ashaving deleterious effects (Table 1).
Measuring p16INK4a Interactions In Vivo
The in vivo binding activity of all p16INK4a variants was assessedusing a mammalian two-hybrid assay. Each p16INK4a mutantconstruct was cloned in frame with the GAL4 nuclear localizationsequence and transiently cotransfected with either a CDK4 or aCDK6 expression plasmid into Saos-2 cells. These pRb-null cellswere selected because they do not undergo p16INK4a-mediated cellcycle arrest [Rizos et al., 2001a].
Previous results have shown that a 15% decrease in cell cyclearrest correlates well with clinical disease [Chan et al., 2007], andwe used the same cutoff to assess p16INK4a binding affinity toCDK4 and CDK6. We classified any variant with o85% of wild-type CDK4 or CDK6 binding affinity as deleterious. Using these
criteria p16INK4a variants could be divided into three functionalgroups based on their binding affinity for CDK4 and CDK6. In thefirst group, 20 p16INK4a variants were impaired in their ability tobind both CDK4 and CDK6 (Table 2). The second group consistedof four p16INK4a variants (24 bp duplication, p.R24P, p.M53I,p.S56I) that bound CDK6 at least as well as the wild-type proteinbut showed diminished affinity for CDK4. Notably, we found thatp.S56I consistently bound CDK6 with an affinity greater thantwofold that of the wild-type protein (Fig. 1). The third groupincluded the p.A148T, p.R144C polymorphisms, which bound toboth CDK4 and CDK6 at least as effectively as the wild-typep16INK4a protein (Fig. 1). Notably, no mutation showed wild-typebinding for CDK4 and diminished CDK6 affinity.
Measuring Cell Cycle Inhibitory Activity of p16INK4a
Variants
The predictive value of cell cycle inhibitory assays was initiallyinvestigated using eight p16INK4a variants. In this assay the effectof p16INK4a proteins on cell cycle progression was analyzed aspreviously reported [Parry and Peters, 1996] by transientlytransfecting FLAG-tagged constructs into U2OS osteosarcomacells. As the cell cycle inhibitory activity of melanoma-associatedp16INK4a variants may be temperature sensitive [Parry and Peters,1996], the assays were performed using cells transfected in parallel,and then maintained at 371C or 401C. Forty-eight hours aftertransfection, DNA content was assayed by flow cytometry todetermine cell cycle distribution. At this time point, the expressionof wild-type p16INK4a in U2OS cells induced potent cell cyclearrest at both temperatures; p16INK4a induced S-phase inhibitionof 4273% and 4973% at 371C and 401C, respectively. Similarly,the 24 bp duplication, p.R24P, p.G67S, p.N71K, p.V126D, andp.A148T remained fully active in mediating cell cycle arrest at371C and 401C (Fig. 2A). As reported previously, the p.G101Wvariant induced S-phase inhibition that was indistinguishablefrom the wild-type protein at 371C, but was significantly impairedat promoting arrest at 401C [Parry and Peters, 1996]. Finally, thep.M53I mutation reproducibly induced only partial cell cycleinhibition at both temperatures when compared with wild-typep16INK4a (Fig. 2A). However, the expression of p.M53I wasreduced compared to the other p16INK4a variants, and this wouldinfluence cell cycle inhibitory activity (Fig. 2A and Supp. Fig. S2).
We had expected that flow cytometry based cell cycle inhibitoryassays would be a useful predictor of p16INK4a function, and yet inour transient assay the highly penetrant p.R24P, p.G101W, andp.V126D mutations retained significant cell cycle inhibitoryactivity. We reevaluated p.R24P and p.G101W using a melanomacell model with IPTG inducible, physiological levels of p16INK4a
expression [McKenzie et al., 2009]. Three cell models werecompared; one inducible for wild-type p16INK4a and two induciblefor either the melanoma-associated p.R24P or p.G101W mutant.
Induction of wild-type p16INK4a-induced cell cycle arrest thatwas evident as early as 16 h postinduction and was maintained48 h after p16INK4a induction (data not shown). In contrast,induced expression of the p.R24P and p.G101W proteins did notpromote effective cell cycle inhibition (Fig. 2B), and thiscorrelated with their diminished ability to inhibit pRb phosphor-ylation, compared to the wild-type protein (Fig. 2C). Importantly,there was also a marked difference in the ability of the p16INK4a
variants to reduce expression of proteins required for S-phaseentry that are downstream of pRb, including E2F-1 and cyclin A.
An alternative approach for measuring p16INK4a-mediated cellcycle inhibition, involved examining the expression of the
694 HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010

Tabl
e1.
InS
ilic
oEv
alua
tion
ofp1
6INK
4aM
isse
nse
Mut
atio
ns
Var
ian
tF
amil
iesa
B62
PW
sco
reb
B62
PW
pre
dic
tio
n
SIF
Tal
ign
ed
sco
re
SIF
Tal
ign
ed
pre
dic
tio
nP
oly
ph
enA
-GV
GD
c
Pre
dic
tio
n
sco
red
BL
OSU
M62
sco
ree
BL
OSU
M62
pre
dic
tio
n
Gra
nth
am
sco
ref
Gra
nth
amsc
ore
pre
dic
tio
n
p.L
16P
34.
00D
elet
erio
us
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
03
�2
Del
eter
iou
s98
Del
eter
iou
s
p.G
23D
26.
00D
elet
erio
us
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
03
�1
Del
eter
iou
s94
Del
eter
iou
s
p.R
24P
90.
54To
lera
ted
0.18
Tole
rate
dP
oss
ibly
Dam
agin
gC
01
�2
Del
eter
iou
s10
3D
elet
erio
us
p.L
32P
64.
00D
elet
erio
us
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
03
�3
Del
eter
iou
s98
Del
eter
iou
s
p.G
35A
34.
62D
elet
erio
us
0.12
Tole
rate
dB
enig
nC
01
0To
lera
ted
60D
elet
erio
us
p.A
36P
11.
24To
lera
ted
0.07
Tole
rate
dP
oss
ibly
Dam
agin
gC
01
�1
Del
eter
iou
s27
To
lera
ted
p.I
49S
13.
38To
lera
ted
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
02
�2
Del
eter
iou
s14
2D
elet
erio
us
p.M
53I
194.
37D
elet
erio
us
0.02
Del
eter
iou
sP
rob
ably
Dam
agin
gC
03
1To
lera
ted
10T
ole
rate
d
p.S
56I
32.
51To
lera
ted
0.01
Del
eter
iou
sP
rob
ably
Dam
agin
gC
152
�2
Del
eter
iou
s14
2D
elet
erio
us
p.A
60R
24.
00D
elet
erio
us
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
654
�1
Del
eter
iou
s11
2D
elet
erio
us
p.G
67S
26.
00D
elet
erio
us
0.00
Del
eter
iou
sP
oss
ibly
Dam
agin
gC
554
0To
lera
ted
56T
ole
rate
d
p.A
68L
14.
00D
elet
erio
us
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
654
�1
Del
eter
iou
s96
Del
eter
iou
s
p.A
68T
gN
A4.
00D
elet
erio
us
0.00
Del
eter
iou
sP
oss
ibly
Dam
agin
gC
554
�1
Del
eter
iou
s58
To
lera
ted
p.E
69G
14.
30D
elet
erio
us
0.01
Del
eter
iou
sB
enig
nC
152
�2
Del
eter
iou
s98
Del
eter
iou
s
p.N
71I
15.
37D
elet
erio
us
0.11
Tole
rate
dP
rob
ably
Dam
agin
gC
152
�3
Del
eter
iou
s14
9D
elet
erio
us
p.N
71K
25.
37D
elet
erio
us
0.11
Tole
rate
dB
enig
nC
01
0To
lera
ted
94D
elet
erio
us
p.N
71S
25.
37D
elet
erio
us
0.11
Tole
rate
dB
enig
nC
01
1To
lera
ted
46T
ole
rate
d
p.P
81T
26.
05D
elet
erio
us
0.04
Del
eter
iou
sP
rob
ably
Dam
agin
gC
03
1To
lera
ted
38T
ole
rate
d
p.L
97R
14.
00D
elet
erio
us
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
654
�2
Del
eter
iou
s10
2D
elet
erio
us
p.R
99P
12.
05To
lera
ted
0.05
Del
eter
iou
sP
oss
ibly
Dam
agin
gC
02
�2
Del
eter
iou
s10
3D
elet
erio
us
p.G
101W
164.
57D
elet
erio
us
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
254
�2
Del
eter
iou
s18
4D
elet
erio
us
p.R
112G
32.
61To
lera
ted
0.07
Tole
rate
dP
oss
ibly
Dam
agin
gC
01
�2
Del
eter
iou
s12
5D
elet
erio
us
p.V
126D
73.
51D
elet
erio
us
0.00
Del
eter
iou
sP
rob
ably
Dam
agin
gC
454
�3
Del
eter
iou
s15
2D
elet
erio
us
p.R
144C
gN
A0.
27To
lera
ted
0.08
Tole
rate
dP
oss
ibly
Dam
agin
gC
01
�3
Del
eter
iou
s18
0D
elet
erio
us
p.A
148T
gN
A1.
58To
lera
ted
0.16
Tole
rate
dB
enig
nC
00
�1
Del
eter
iou
s58
To
lera
ted
aN
um
ber
of
fam
ilie
sre
po
rted
toca
rry
p16
INK
4a
alte
rati
on
bas
edo
n[G
old
stei
net
al.,
2006
a;K
ann
engi
esse
ret
al.,
2009
;M
on
ner
atet
al.,
2007
;P
oll
ock
etal
.,19
98]
and
htt
ps:
//b
iod
eskt
op
.uvm
.ed
u/p
erl/
p16
.bB
62P
WSc
ore
5A
vera
geB
LO
SUM
62M
atri
xSc
ore
for
all
pai
rwis
eco
mp
aris
on
s,va
lueo
3.5
clas
sifi
edas
neu
tral
and4
3.5
asd
elet
erio
us
[Ch
anet
al.,
2007
].c A
-GV
GD
clas
sifi
ers
ran
gefr
om
C65
(mo
stli
kely
del
eter
iou
s)to
C0
(mo
stli
kely
neu
tral
).In
this
rep
ort
anA
-GV
GD
grad
eZ
C25
was
con
sid
ered
like
lyd
elet
erio
us
[Tav
tigi
anet
al.,
2008
a,b
].dP
red
icti
on
sco
reeq
ual
sth
en
um
ber
of
evo
luti
on
ary
pro
gram
s(B
62P
W,
SIF
T,
Po
lyp
hen
,A
-GV
GD
)th
atju
dge
dth
eal
tera
tio
nd
elet
erio
us.
e BL
OSU
M62
sco
reb
ased
on
mat
rix
sco
reo
fth
en
ewam
ino
acid
sub
stit
uti
on
for
the
old
.Sc
ore
sZ
0w
ere
con
sid
ered
del
eter
iou
s[H
enik
off
and
Hen
iko
ff,
1992
].f G
ran
tham
sco
recl
assi
fied
amin
oac
idsu
bst
itu
tio
ns
asei
ther
tole
rate
d(s
core
o60
)o
rd
elet
erio
us
(sco
reZ
60)
[Ch
anet
al.,
2007
;G
ran
tham
,19
74].
gp
.R14
4Can
dp
.A14
8Th
ave
bee
nre
po
rted
tob
ep
oly
mo
rph
ism
s[G
om
bar
tet
al.,
1997
;H
uss
uss
ian
etal
.,19
94;
San
chez
-Ces
ped
eset
al.,
2001
].T
he
p.A
68T
was
fou
nd
insp
ora
dic
mel
ano
ma
case
s[P
icci
nin
etal
.,19
97].
NA
,n
ot
app
lica
ble
.
HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010 695

proliferation marker, Ki67 in transiently transfected NM39 cells.In this assay, no marker of transfection is required as dualimmunofluorescent staining was employed to detect p16INK4a
expression and Ki67 accumulation in transfected NM39 cells. Thisassay also allowed the evaluation of p16INK4a subcellular distribu-tion (see below) (Fig. 3A). As shown in Figure 3B, only 972% oftransfected NM39 cells expressing wild-type p16INK4a werepositive for Ki67, 48 h posttransfection. Of the 28 p16INK4a
variants tested in this assay, 10 melanoma-associated p16INK4a
variants showed at least a twofold higher Ki67 index, ranging from1972% for the p.A60R mutant to 3374% for p16INK4a-Leiden(Fig. 3B). These include nine missense mutations (p.G23D,p.R24P, p.A60R, p.E69G, p.L97R, p.R99P, p.G101W, p.R112G,and p.V126D) that had in silico prediction scores ranging from 1(p.R24P and p.R112G) to 4 (p.A60R, p.L97R, p.G101W, andp.V126D) (Table 1).
Analysis of p16INK4a Subcellular Distribution
The subcellular distribution of each p16INK4a construct was alsoevaluated in transiently transfected NM39 melanoma cells. Aspreviously reported, wild-type p16INK4a-FLAG localized evenlythroughout NM39 cells [McKenzie et al., 2009; Rizos et al., 2001a;Walker et al., 1999]. Of the 28 p16INK4a-FLAG variants tested, onlythe silent p.A73A variant, the two polymorphisms p.A148T, andp.R144C and the melanoma-associated 24-bp duplication showedwild-type localization and were distributed evenly throughouttransiently transfected NM39 cells (Fig. 3A and C). All otherp16INK4a-FLAG mutants showed disrupted localization within
Table 2. Functional Evaluations of p16INK4a Missense Mutations
Variant Familiesa Prediction scoreb % Altered localizationc Ki67 indexd CDK4 bindinge CDK6 bindinge
wild type 2.3 9.3 100.0 100.0
p.M1_S8dup 5 1.5 2.5 80.2 94.5
c.225_243del19 22 92.3 32.9 �1.2 1.0
p.L16P 3 3 40.7 18.6 0.5 2.7
p.G23D 2 3 38.6 20.5 3.6 27.7
p.R24P 9 1 34.2 20.2 54.7 124.1
p.L32P 6 3 29.1 15.7 �1.9 5.4
p.G35A 3 1 11.2 7.1 3.0 8.2
p.A36P 1 1 14.2 5.5 4.1 3.2
p.I49S 1 2 37.5 15.8 14.4 50.6
p.M53I 19 3 23.7 8.0 31.5 137.5
p.S56I 3 2 28.7 12.9 49.5 221.6
p.A60R 2 4 28.7 19.0 �4.2 11.5
p.G67S 2 4 36.1 17.8 31.9 28.0
p.A68L 1 4 32.3 16.2 4.0 16.7
p.A68T NA 4 40.1 12.6 10.3 2.3
p.E69G 1 2 38.2 20.0 1.8 �4.2
p.N71I 1 2 37.8 16.8 2.6 2.0
p.N71K 2 1 32.3 16.8 7.3 11.7
p.N71S 2 1 28.4 11.3 21.4 14.2
p.A73A NA NA 2.2 7.8 112.0f ND
p.P81T 2 3 23.2 14.9 6.2 13.0
p.L97R 1 4 37.2 30.3 �3.0 �4.5
p.R99P 1 2 14.7 21.5 0.4 0.5
p.G101W 16 4 62.8 21.2 19.9 66.3
p.R112G 3 1 46.3 20.9 32.1 ND
p.V126D 7 4 45.9 24.8 0.7 8.7
p.R144C NA 1 3.7 9.3 91.4 108.8
p.A148T NA 0 0.5 8.7 102.3 98.0
aNumber of families reported to carry p16INK4a alteration (see Table 1). The p.A73A was found in sporadic melanoma cases [Piccinin et al., 1997].bPrediction score equals the number of evolutionary programs (B62PW, SIFT, Polyphen, A-GVGD) that judged the alteration deleterious.c% transfected NM39 cells displaying punctate and speckled p16INK4a localization.d% transfected NM39 cells showing Ki67 expression comparable to untransfected cells.ePercentage binding affinities of p16INK4a variants with CDK4 or CDK6 shown relative to wild-type p16INK4a.fData derived from Rizos et al. [2001a].NA, not applicable; ND, not determined; DNA numbering relates to CDKN2A, GenBank accession no. NM_000077.2.
Figure 1. Interaction of melanoma associated p16INK4a variantswith CDK4 and CDK6. Binding activity was determined using the two-hybrid assay system in Saos-2 cells. Binding of mutant p16INK4a
protein to CDK4 and CDK6 was expressed as percentage relative tothe binding activity of the wild-type protein. At least two independenttransfection experiments were performed in duplicate for eachconstruct. The position of the wild-type protein is boxed. Two groupsof p16INK4a variants are circled; mutants with limited affinity for bothCDK4 and CDK6, and mutants with reduced CDK4 binding, but nearwild-type CDK6 affinity.
696 HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010

these cells. The subcellular distribution of the mutant p16INK4a
proteins was no longer homogeneous but showed a speckled(small discrete areas of intense fluorescence, e.g., p.R24P) orpunctate distribution (larger aggregates of intense fluorescence,e.g., p.M53I) (Fig. 3A). Speckling was observed in the nucleus andin the cytoplasm, whereas the punctate aggregates accumulated inthe cytoplasm only.
To determine whether the Ki67 index contributes any distinctinformation to the subcellular distribution data, we correlated thedata generated from both assays. As shown in Figure 4, the Ki67index was closely related to aberrant localization; increasing Ki67index was associated with increasing percentage of transfected cellswith mislocalized p16INK4a (Fig. 4). However, analysis ofsubcellular distribution was the more sensitive assay as it clearlydifferentiated between the wild-type protein and 24 p16INK4a
variants, whereas Ki67 index only distinguished 10 variants asaltered. In fact, the analysis of subcellular distribution only failedto detect one melanoma-associated variant, the 24 bp duplication.This variant was also indistinguishable from wild type p16INK4a inthe Ki67 assay and flow cytometry cell cycle analyses.
Discussion
Mutations affecting the p16INK4a tumor suppressor occurin approximately 40% of multiple-case melanoma families[Goldstein et al., 2006a], and the majority of these mutationshave only been observed in a single family. Genetic counsellingregarding these rare variants would be considerably assisted byfunctional data, particularly in small ‘‘nuclear’’ kindreds in whichcosegregation with disease cannot be determined. However, thevalue of functional analyses has been limited by inconsistent andunreliable assays. For example, the p.R24P mutation, whichcosegregates with disease in a multiple case Australian melanomakindred with 15 affected individuals [Holland et al., 1995] and inanother eight melanoma-prone families (Table 1) displayedinteraction with CDK4 similar to that of the wild-type proteinin an in vitro GST pull-down assay [Becker et al., 2001], but hadno binding affinity for CDK4 in vitro [Harland et al., 1997].Similarly, the p.G101W mutation, originally discovered in familialmelanoma kindreds [Holland et al., 1999; Hussussian et al., 1994;Weaver-Feldhaus et al., 1994], was defective in an in vitro kinaseinhibition assay, but retained its ability to arrest cells in G1 [Kohet al., 1995; Ranade et al., 1995].
In this study we compared the location, binding affinity, andcell cycle inhibitory activity of 28 p16INK4a variants. Of these, 12mutations also altered the p14ARF tumor suppressor. Althoughp14ARF inactivation via nonsense, frameshift, and splice sitevariants have been reported in melanomas and melanoma-pronekindreds [Freedberg et al., 2008; Rizos et al., 2001c], mutationsspecifically targeting p14ARF are rare. Furthermore, of the subsetof missense mutations affecting both p16INK4a and p14ARF,p16INK4a seems to be the primary target of inactivation, asp14ARF is not always functionally impaired [Rizos et al., 2001b].Nevertheless, to evaluate the contribution of p14ARF inmelanoma susceptibility the functional analysis of p14ARFvariants should be included.
In this report we propose that the combination of themammalian two-hybrid assay, to quantitate CDK4 binding affinityand dual immunofluorescence, to determine p16INK4a subcellulardistribution provides a rapid and reliable assessment of p16INK4a
function. Assessment of Ki67 expression can also be included inthe subcellular distribution assay, but the Ki67 data does not
Figure 2. Cell cycle inhibitory activities of melanoma-associatedp16INK4a variants. A: Upper panel: U2OS osteosarcoma cells weretransfected with the indicated p16INK4a-FLAG plasmid and pEGFP-spectrin. The cell cycle distribution of green fluorescent cells wasdetermined 48 h posttransfection using propidium iodide staining. Thepercentage of S-phase inhibition was calculated using the followingformula: (percentage of cells in S-phase in the vector transfectedcells�percentage of cells in S-phase in cells transfected withp16INK4a expression plasmids)/(percentage of cells in S phase in thevector transfected cells)� 100. The results (mean7standard devia-tion [SD]), are derived from at least two independent transfectionexperiments. Lower panel: the expression of p16INK4a-FLAG constructswas determined 40 h after cotransfecting U20S cells with theindicated FLAG plasmid and the pEGFPN1 vector. B: The relative S-phase inhibition induced by the expression of wild-type p16INK4a,p.R24P and p.G101W variants was determined either by transientlyexpressing these proteins in the NM39 melanoma cells, or inducingtheir expression in stable melanoma cell models. Cell cycledistribution was determined 48 h posttransfection or -induction fromat least two independent experiments. The percentage of S-phaseinhibition was calculated as detailed above. C: Expression of theindicated proteins was determined 48 h after inducing the expressionof wild-type p16INK4a, p.R24P or p.G101W proteins in stable melanomacells with 4 mM IPTG (1) or PBS (�).
HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010 697
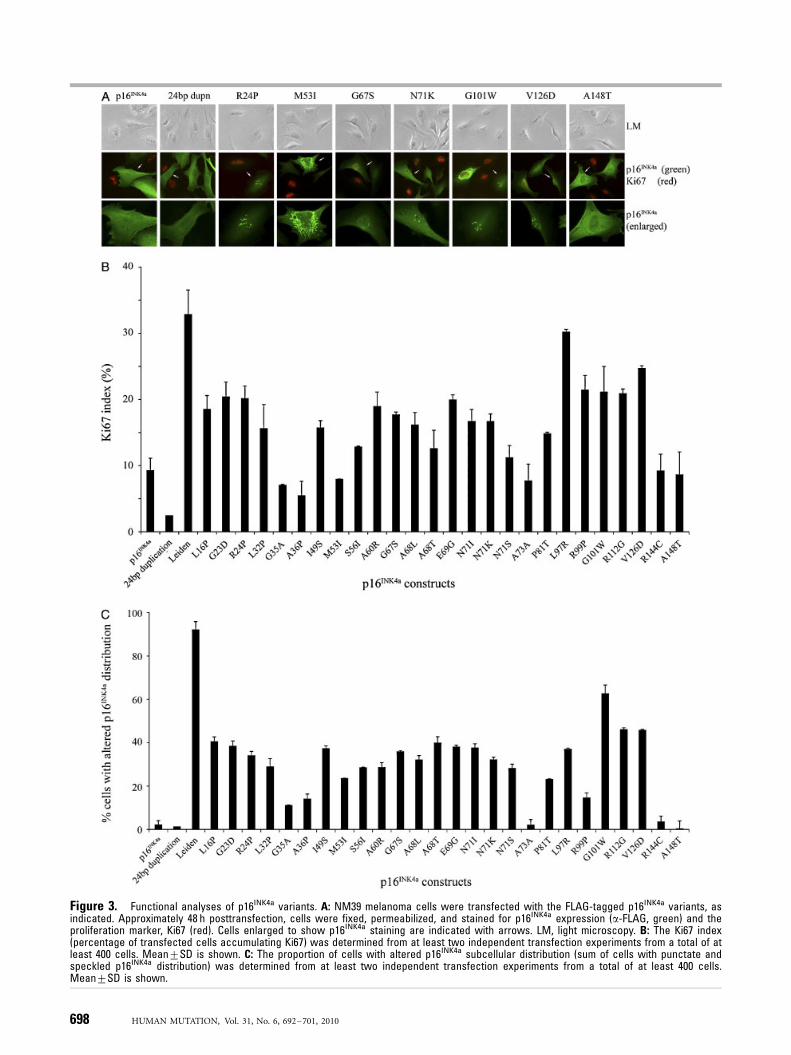
Figure 3. Functional analyses of p16INK4a variants. A: NM39 melanoma cells were transfected with the FLAG-tagged p16INK4a variants, asindicated. Approximately 48 h posttransfection, cells were fixed, permeabilized, and stained for p16INK4a expression (a-FLAG, green) and theproliferation marker, Ki67 (red). Cells enlarged to show p16INK4a staining are indicated with arrows. LM, light microscopy. B: The Ki67 index(percentage of transfected cells accumulating Ki67) was determined from at least two independent transfection experiments from a total of atleast 400 cells. Mean7SD is shown. C: The proportion of cells with altered p16INK4a subcellular distribution (sum of cells with punctate andspeckled p16INK4a distribution) was determined from at least two independent transfection experiments from a total of at least 400 cells.Mean7SD is shown.
698 HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010

contribute new information, and inclusion as a predictive tool isnot justified.
The mammalian two-hybrid assay has the advantage ofexpressing proteins in mammalian cells and thus maintainingappropriate posttranslational synthesis. Of the 28 mutations testedwe found that no mutation had diminished CDK6 affinity whileretaining wild-type binding to CDK4, and considering that such amutation has never been reported it seems reasonable to limit thescreening of p16INK4a interactions to CDK4. In fact, all melanoma-associated variants tested in this study showed diminishedp16INK4a-CDK4 affinity, including a small reduction in CDK4binding for the 24 bp duplication. Although this mutant has beenfound in five melanoma-prone kindreds [Flores et al., 1997;Pollock et al., 1998; Walker et al., 1995] and appears highlypenetrant for melanoma, it shows only minimal loss of cell cycleinhibitory function in stable cell models [Becker et al., 2005;Newton-Bishop et al., 2000], and behaved as wild-type protein intransient colony forming assays [Walker et al., 1999]. It is possiblethat the 24 bp duplication impacts on functions of p16INK4a thatare independent of CDK binding. For instance, p16INK4a alsointeracts with the chromatin remodeling factor BRG1 and thenovel ISOC2 protein [Becker et al., 2009; Huang et al., 2007]. Thecontribution of these interactions on the tumor suppressorfunctions of p16INK4a remains to be defined, and the impact ofmelanoma-associated mutations on these interactions requiresinvestigation. It is also possible that the 24 bp repeat alters thestability, expression, and/or processing of the p16INK4a transcript,and we are currently investigating these possibilities. The fact thatthis variant has arisen independently at least three times showsthat it is not simply in linkage disequilibrium with an undetected,separate pathogenic variant of INK4a/ARF [Pollock et al., 1998].Finally, the 24 bp duplication may represent a rare populationvariant that is either not pathogenic or only weakly so. However,no population-based studies have yet detected this variant incontrols [Orlow et al., 2007] so this possibility remains unlikely.
Although CDK4 and CDK6 kinases are likely to adopt similarstructures, it appears that a small subset of melanoma-associatedp16INK4a variants are specifically defective for binding CDK4. Inparticular, the 24 bp duplication, p.R24P, p.M53I, and p.S56Ivariants showed CDK6 binding similar to or greater than thewild-type protein. p.R24P and p.M53I have been shown
previously to bind CDK6, and not CDK4 [Harland et al., 1997;Jones et al., 2007; Walker et al., 1999] and, to our knowledge, therehave been no reports measuring CDK6 binding activity of the S56Iand 24 bp duplication variants. It will be interesting to comparethe growth inhibitory activity of these mutations, considering thatp.R24P was unable to arrest the proliferation of human fibroblasts[Jones et al., 2007]. These mutations highlight the importance ofCDK4 rather than CDK6 in melanoma development, consistentwith the evidence that CDK4 is a high-risk melanoma suscept-ibility gene [Goldstein et al., 2006a]. These data also underscorethe importance of determining p16INK4a-CDK4, rather thanp16INK4a-CDK6, affinity as a measure of p16INK4a functionalactivity.
The subcellular localization experiments were also effective atdiscriminating between wild-type p16INK4a, the silent p.A73Amutation, the p.A148T, and p.R144C polymorphisms andcausative p16INK4a mutations. Only the 24 bp duplication wasnot identified using this assay as a disease-associated variant, as itlocalized like wild-type protein, throughout the cell. Thelocalization of mutant proteins appears to reflect the supra-physiological expression levels achieved in our transient assays, asp.R24P, p.M53I, and p.G101W mutants did not show abnormaldistribution patterns when expressed at physiological levels instable cell models [McKenzie et al., 2009]. Thus, althoughp16INK4a variant mislocalization presumably reflects the accumu-lation of misfolded, unstable proteins within the endoplasmicreticulum [Malhotra and Kaufman, 2007], it has proven a valuablepredictive tool for p16INK4a function. This method is robust, canbe performed in various cell lines (we have used U20S and NM39),is not affected by ectopic p16INK4a expression levels (compare thelow levels of p16INK4a-Leiden expression [Supp. Fig. S2] with itsaberrant localization [Fig. 3]), and can be performed in a few days.
It is worth mentioning that the p.A148T polymorphismbehaved as the wild-type protein in all the functional assaysdescribed in this work, and indeed this mutation has not beenshown to compromise protein function. Nevertheless, thecommon single-nucleotide polymorphism (SNP) allele thatencodes protein variant p.A148T has been associated with amodest (twofold) elevation of melanoma risk in case–controlstudies [Debniak et al., 2005], although it is unclear whether this ismediated by protein effects or other linked, functionallysignificant SNPs.
Flow cytometry-based cell cycle inhibitory assays are commonlyused to assess p16INK4a activity [Parry and Peters, 1996; Rizoset al., 2001a], but proved ineffective in detecting impairedmelanoma-associated mutations. This was most evident with thep.R24P variant, which was unable to arrest stable cell models whenexpressed at physiologic levels, or to bind CDK4 in themammalian two-hybrid assay, but effectively arrested the U2OScells at 371C and 401C when overexpressed. Certainly stable cellmodels are powerful tools for analyzing many aspects of p16INK4a
function, as they accumulate only physiological levels of p16INK4a
and are clonal cell populations. Unfortunately, the time taken,almost 3 months, and the resources required to generate thesemodels precludes their use as a rapid and general functional assay.
The inclusion of in silico analyses is helpful, but theevolutionary-based algorithms we applied did not offer thesensitivity required to confidently classify most melanoma-associated mutants as deleterious, and the data generated can beinfluenced by the protein alignments used [Chan et al., 2007].Certainly, the predictive value of these algorithms is useful when atleast three of the evolutionary-based methods agree that a variantis deleterious. In this study, 12 missense mutations had an in silico
Figure 4. Comparison of Ki67 and subcellular distribution assays inpredicting deleterious p16INK4a variants. The Ki67 index and percen-tage of transfected NM39 cells showing aberrant p16INK4a distributionare directly compared for each p16INK4a variant. The position of thewild-type protein is boxed.
HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010 699

prediction score of Z3, and these mutations were all functionallycompromised in our CDK4 and localization studies. Therefore, wepropose that a combination of the mammalian two-hybrid assaywith cellular localization studies provides a quick (both assays canbe performed in approximately 96 h) and highly sensitive strategyfor determining the functional activity of p16INK4a variants in anin vivo setting. These functional assays are not limited to missensep16INK4a mutations and can be used to assess any alteration withinthe p16INK4a reading frame that can be cloned. This includes smalldeletions, like the Leiden variant, and small insertions, such as the24 bp duplication. Accumulation of such functional data on alarge proportion of p16INK4a variants, so that they may beadequately classified, is an important translational research goalthat will support effective genetic counselling of individuals athigh risk of melanoma. Such data may also help identify morereliable bioinformatic principles for predicting the pathogenicityof novel variants of this important protein.
Acknowledgments
We thank Paula Torres and Sieu Tran for technical expertise. This work is
supported by Program Grant 402761 of the National Health and Medical
Research Council of Australia (NHMRC), the Cancer Council of NSW, and
an infrastructure grant to Westmead Millennium Institute by the Health
Department of NSW through Sydney West Area Health Service. Westmead
Institute for Cancer Research is the recipient of capital grant funding from
the Australian Cancer Research Foundation. H.R. is a Cancer Institute New
South Wales, Research Fellow. H.M. is a Cancer Institute of NSW Scholar,
and is a recipient of an Australian Postgraduate Award.
References
Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. 1996. Involvement of
the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of
normal human fibroblasts. Proc Natl Acad Sci USA 93:13742–13747.
Becker TM, Ayub AL, Kefford RF, Mann GJ, Rizos H. 2005. The melanoma-
associated 24 base pair duplication in p16INK4a is functionally impaired. Int J
Cancer 117:569–573.
Becker TM, Haferkamp S, Dijkstra MK, Scurr LL, Frausto M, Diefenbach E,
Scolyer RA, Reisman DN, Mann GJ, Kefford RF, Rizos H. 2009. The chromatin
remodelling factor BRG1 is a novel binding partner of the tumor suppressor
p16INK4a. Mol Cancer 8:4.
Becker TM, Rizos H, Kefford RF, Mann GJ. 2001. Functional impairment of
melanoma-associated p16INK4a mutants in melanoma cells despite retention of
cyclin-dependent kinase 4 binding. Clin Cancer Res 7:3282–3288.
Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B,
Chompret A, Ghiorzo P, Gruis N, Hansson J, Harland M, Hayward N, Holland EA,
Mann GJ, Mantelli M, Nancarrow D, Platz A, Tucker MA, Melanoma Genetics
Consortium. 2002. Geographical variation in the penetrance of CDKN2A
mutations for melanoma. J Natl Cancer Inst 94:894–903.
Brenner AJ, Stampfer MR, Aldaz CM. 1998. Increased p16 expression with first
senescence arrest in human mammary epithelial cells and extended growth
capacity with p16 inactivation. Oncogene 17:199–205.
Chan PA, Duraisamy S, Miller PJ, Newell JA, McBride C, Bond JP, Raevaara T,
Ollila S, Nystrom M, Grimm AJ, Christodoulou J, Oetting WS, Greenblatt MS.
2007. Interpreting missense variants: comparing computational methods in
human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR).
Hum Mutat 28:683–693.
Debniak T, Scott RJ, Huzarski T, Byrski T, Rozmiarek A, Debniak B, Zaluga E,
Maleszka R, Kladny J, Gorski B, Cybulski C, Gronwald J, Kurzawski G,
Lubinski J. 2005. CDKN2A common variants and their association with
melanoma risk: a population-based study. Cancer Res 65:835–839.
Flores JF, Pollock PM, Walker GJ, Glendening JM, Lin AHT, Palmer JM, Walters MK,
Hayward NK, Fountain JW. 1997. Analysis of the CDKN2A, CDKN2B and
CDK4 genes in 48 Australian melanoma kindreds. Oncogene 15:2999–3005.
Foulkes WD, Flanders TY, Pollock PM, Hayward NK. 1997. The CDKN2A (p16) gene
and human cancer. Mol Med 3:5–20.
Freedberg DE, Rigas SH, Russak J, Gai W, Kaplow M, Osman I, Turner F,
Randerson-Moor JA, Houghton A, Busam K, Timothy Bishop D, Bastian BC,
Newton-Bishop JA, Polsky D. 2008. Frequent p16-Independent Inactivation of
p14ARF in Human Melanoma. J Natl Cancer Inst 100:784–795.
Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF, Azizi E,
Bianchi-Scarra G, Bishop DT, Bressac-de Paillerets B, Bruno W, Calista D,
Cannon Albright LA, Demenais F, Elder DE, Ghiorzo P, Gruis NA, Hansson J,
Hogg D, Holland EA, Kanetsky PA, Kefford RF, Landi MT, Lang J, Leachman SA,
Mackie RM, Magnusson V, Mann GJ, Niendorf K, Newton Bishop J, Palmer JM,
Puig S, Puig-Butille JA, de Snoo FA, Stark M, Tsao H, Tucker MA, Whitaker L,
Yakobson E Melanoma Genetics Consortium (GenoMEL). 2006a. High-risk
Melanoma Susceptibility Genes and Pancreatic Cancer, Neural System Tumors,
and Uveal Melanoma across GenoMEL. Cancer Res 66:9818–9828.
Goldstein AM, Chan M, Harland M, Hayward NK, Demenais F, Bishop DT, Azizi E,
Bergman W, Bianchi-Scarra G, Bruno W, Calista D, Albright LA, Chaudru V,
Chompret A, Cuellar F, Elder DE, Ghiorzo P, Gillanders EM, Gruis NA,
Hansson J, Hogg D, Holland EA, Kanetsky PA, Kefford RF, Landi MT, Lang J,
Leachman SA, MacKie RM, Magnusson V, Mann GJ, Bishop JN, Palmer JM,
Puig S, Puig-Butille JA, Stark M, Tsao H, Tucker MA, Whitaker L, Yakobson E;
Lund Melanoma Study Group; Melanoma Genetics Consortium (GenoMEL)
2006b. Features associated with germline CDKN2A mutations: a GenoMEL
study of melanoma-prone families from three continents. J Med Genet
44:99–106.
Gombart AF, Yang R, Campbell MJ, Berman JD, Koeffler HP. 1997. Inhibition of
growth of human leukemia cell lines by retrovirally expressed wild-type
p16INK4A. Leukemia 11:1673–1680.
Grantham R. 1974. Amino acid difference formula to help explain protein evolution.
Science 185:862–864.
Greenblatt MS, Beaudet JG, Gump JR, Godin KS, Trombley L, Koh J, Bond JP. 2003.
Detailed computational study of p53 and p16: using evolutionary sequence
analysis and disease-associated mutations to predict the functional conse-
quences of allelic variants. Oncogene 22:1150–1163.
Haferkamp S, Becker TM, Scurr LL, Kefford RF, Rizos H. 2008. p16INK4a-induced
senescence is disabled by melanoma-associated mutations. Aging Cell
7:733–745.
Harland M, Meloni R, Gruis N, Pinney E, Brookes S, Spurr NK, Frischauf A-M,
Bataille V, Peters G, Cuzick J, Selby P, Bishop DT, Bishop JN. 1997. Germline
mutations of the CDKN2 gene in UK melanoma families. Hum Mol Genet
6:2061–2067.
Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein
blocks. Proc Natl Acad Sci USA 89:10915–10919.
Holland EA, Beaton SC, Becker TM, Grulet OM, Peters BA, Rizos H, Kefford RF,
Mann GJ. 1995. Analysis of the p16 gene, CDKN2, in 17 Australian melanoma
kindreds. Oncogene 11:2289–2294.
Holland EA, Schmid H, Kefford RF, Mann GJ. 1999. CDKN2A (p16INK4a) and CDK4
mutation analysis in 131 Australian melanoma probands: effect of family history
and multiple primary melanomas. Genes Chromosomes Cancer 25:339–348.
Huang X, Shi Z, Wang W, Bai J, Chen Z, Xu J, Zhang D, Fu S. 2007. Identification and
characterization of a novel protein ISOC2 that interacts with p16INK4a.
Biochem Biophys Res Commun 361:287–293.
Hussussian CJ, Struewing JP, Goldstein AM, Higgins PAT, Ally DS, Sheahan MD,
ClarkJr WH, Tucker MA, Dracopoli NC. 1994. Germline p16 mutations in
familial melanoma. Nat Genet 8:15–21.
Jones R, Ruas M, Gregory F, Moulin S, Delia D, Manoukian S, Rowe J, Brookes S,
Peters G. 2007. A CDKN2A mutation in familial melanoma that abrogates
binding of p16INK4a to CDK4 but not CDK6. Cancer Res 67:9134–9141.
Kannengiesser C, Brookes S, del Arroyo AG, Pham D, Bombled J, Barrois M,
Mauffret O, Avril MF, Chompret A, Lenoir GM, Sarasin A; French Hereditary
Melanoma Study Group, Peters G, Bressac-de Paillerets B. 2009. Functional,
structural, and genetic evaluation of 20 CDKN2A germ line mutations identified
in melanoma-prone families or patients. Hum Mutat 30:564–574.
Koh J, Enders GH, Dynlacht BD, Harlow E. 1995. Tumour-derived p16 alleles
encoding proteins defective in cell-cycle inhibition. Nature 375:506–510.
Mahony D, Parry DA, Lees E. 1998. Active cdk6 complexes are predominantly nuclear
and represent only a minority of the cdk6 in T cells. Oncogene 16:603–611.
Malhotra JD, Kaufman RJ. 2007. The endoplasmic reticulum and the unfolded
protein response. Semin Cell Dev Biol 18:716–731.
McConnell BB, Gregory FJ, Stott FJ, Hara E, Peters G. 1999. Induced expression of
p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by
reassortment of cyclin-CDK-inhibitor complexes. Mol Cell Biol 19:1981–1989.
McKenzie H, Becker TM, Scurr LL, Kefford RF, Rizos H. 2009. Wild type and
melanoma-associated mutant p16(INK4a) proteins do not oligomerize in vivo.
Pigment Cell Melanoma Res 22:131–133.
Monnerat C, Chompret A, Kannengiesser C, Avril MF, Janin N, Spatz A,
Guinebretiere JM, Marian C, Barrois M, Boitier F, Lenoir GM,
Bressac-de Paillerets B. 2007. BRCA1, BRCA2, TP53, and CDKN2A germline
mutations in patients with breast cancer and cutaneous melanoma. Fam Cancer
6:453–461.
Newton-Bishop JA, Wachsmuth RC, Harland M, Bataille V, Pinney E, Mack P,
Baglietto L, Cuzick J, Bishop DT. 2000. Genotype/phenotype and penetrance
700 HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010

studies in melanoma families with germnline CDKN2A mutations. J Invest
Dermatol 114:28–33.
Orlow I, Begg CB, Cotignola J, Roy P, Hummer AJ, Clas BA, Mujumdar U, Canchola R,
Armstrong BK, Kricker A, Marrett LD, Millikan RC, Gruber SB, Anton-Culver H,
Zanetti R, Gallagher RP, Dwyer T, Rebbeck TR, Kanetsky PA, Wilcox H, Busam K,
From L, Berwick M; GEM Study Group. 2007. CDKN2A germline mutations
in individuals with cutaneous malignant melanoma. J Invest Dermatol
127:1234–1243.
Parry D, Peters G. 1996. Temperature-sensitive mutants of p16CDKN2 associated
with familial melanoma. Mol Cell Biol 16:3844–3852.
Piccinin S, Doglioni C, Maestro R, Vukosavljevic T, Gasparotto D, D’Orazi C,
Boiocchi M. 1997. p16/CDKN2 and CDK4 gene mutations in sporadic
melanoma development and progression. Int J Cancer 74:26–30.
Pollock PM, Spurr N, Bishop T, Newton-Bishop J, Gruis N, van der Velden PA,
Goldstein AM, Tucker MA, Foulkes WD, Barnhill R, Haber D, Fountain J,
Hayward NK. 1998. Haplotype analysis of two recurrent CDKN2a mutations in
10 melanoma families: evidence for common founders and independent
mutations. Hum Mutat 11:424–431.
Ranade K, Hussussian CJ, Sirkosi RS, Varmus HE, Goldstein AM, Tucker MA,
Serrano M, Hannon GJ, Beach D, Dracopoli NC. 1995. Mutations associated
with familial melanoma impair p16INK4 function. Nat Genet 10:114–116.
Reymond A, Brent R. 1995. p16 proteins from melanoma-prone families are deficient
in binding to Cdk4. Oncogene 11:1173–1178.
Rizos H, Darmanian AP, Holland EA, Mann GJ, Kefford RF. 2001a. Mutations in the
INK4a/ARF melanoma susceptibility locus functionally impair p14ARF. J Biol
Chem 276:41424–41434.
Rizos H, Darmanian AP, Holland EA, Mann GJ, Kefford RF. 2001b. Mutations in the
INK4a/ARF melanoma susceptibility locus functionally impair p14ARF. J Biol
Chem 276:41424–41434.
Rizos H, Darmanian AP, Indsto JO, Shannon JA, Kefford RF, Mann GJ. 1999.
Multiple abnormalities of the p16INK4a-pRb regulatory pathway in cultured
melanoma cells. Melanoma Res 9:10–19.
Rizos H, Puig S, Badenas C, Malvehy J, Darmanian AP, Jimenez L, Milà M,
Kefford RF. 2001c. A melanoma-associated germline mutation in exon 1binactivates p14ARF. Oncogene 20:5543–5557.
Sanchez-Cespedes M, Decker PA, Doffek KM, Esteller M, Westra WH, Alawi EA,
Herman JG, Demeure MJ, Sidransky D, Ahrendt SA. 2001. Increased loss of
chromosome 9p21 but not p16 inactivation in primary non-small cell lung
cancer from smokers. Cancer Res 61:2092–2096.
Serrano M, Hannon GJ, Beach D. 1993. A new regulatory motif in cell-cycle
control causing specific inhibition of cyclin D/CDK4. Nature 366:704–707.
Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. 2008a. Classification of rare
missense substitutions, using risk surfaces, with genetic- and molecular-
epidemiology applications. Hum Mutat 29:1342–1354.
Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB. 2008b. In silico analysis of
missense substitutions using sequence-alignment based methods. Hum Mutat
29:1327–1336.
Walker GJ, Gabrielli BG, Castellano M, Hayward NK. 1999. Functional reassessment
of P16 variants using a transfection-based assay. Int J Cancer 82:305–312.
Walker GJ, Hussussian CJ, Flores JF, Glendening JM, Haluska FG, Dracopoli NC,
Hayward NK, Fountain JW. 1995. Mutations of the CDKN2/p16INK4 gene in
Australian melanoma kindreds. Hum Mol Gen 4:1845–1852.
Wallace IM, O’Sullivan O, Higgins DG, Notredame C. 2006. M-Coffee: combining
multiple sequence alignment methods with T-Coffee. Nucleic Acids Res 34:
1692–1699.
Weaver-Feldhaus J, Gruis NA, Neuhausen S, Le Paslier D, Stockert E, Skolnick MH,
Kamb A. 1994. Localization of a putative tumor suppressor gene by using
homozygous deletions in melanomas. Proc Natl Acad Sci USA 91:7563–7567.
Yang R, Gombart AF, Serrano M, Koeffler HP. 1995. Mutational effects on the
p16INK4a tumor suppressor protein. Cancer Res 55:2503–2506.
Zhang B, Peng ZY. 1996. Defective folding of mutant p16(INK4) proteins encoded by
tumor-derived alleles—communication. J Biol Chem 271:28734–28737.
Zhu J, Woods D, McMahon M, Bishop JM. 1998. Senescence of human fibroblasts
induced by oncogenic Raf. Genes Dev 12:2997–3007.
HUMAN MUTATION, Vol. 31, No. 6, 692–701, 2010 701





![[Reliability of the CINtec p16INK4a immunocytochemical test in screening cervical precancerous lesions]](https://img.pdfslide.net/doc/110x75/636144814b9aa63a9e00a9c7/reliability-of-the-cintec-p16ink4a-immunocytochemical-test-in-screening-cervical.jpg)























