Embed Size (px)
Citation preview

RESEARCH ARTICLE
Proteomic analysis of p16ink4a-binding proteins
Elielson Souza-Rodrígues1, Josep M. Estanyol2, Erica Friedrich-Heineken3, Eva Olmedo1,Jorge Vera1, Nuria Canela2, Sonia Brun1, Neus Agell1, Ulrich Hübscher3, Oriol Bachs1
and Montserrat Jaumot1*
1 Departament de Biologia Cellular i Anatomia Patològica, Facultat de Medicina, Universitat de Barcelona, Spain2 Unitat de Proteòmica, Serveis Científico-tècnics, Universitat de Barcelona, Spain3 Institute of Veterinary Biochemistry and Molecular Biology, University of Zürich-Irchel, Zürich, Switzerland
The p16ink4a tumor suppressor protein plays a critical role in cell cycle control, tumorogenesisand senescence. The best known activity for p16ink4a is the inhibition of the activity of CDK4 andCDK6 kinases, both playing a key role in cell cycle progression. With the aim to study newp16ink4a functions we used affinity chromatography and MS techniques to identify new p16ink4a-interacting proteins. We generated p16ink4a columns by coupling the protein to activated Sephar-ose 4B. The proteins from MOLT-4 cell line that bind to p16ink4a affinity columns were resolved bySDS-PAGE and identified by MS using a MALDI-TOF. Thirty-one p16ink4a -interacting proteinswere identified and grouped in functional clusters. The identification of two of them, proliferat-ing cell nuclear antigen (PCNA) and minichromosome maintenance protein 6 (MCM6), wasconfirmed by Western blotting and their in vivo interactions with p16ink4a were demonstrated byimmunoprecipitation and immunofluorescence studies. Results also revealed that p16ink4a
interacts directly with the DNA polymerase d accessory protein PCNA and thereby inhibits thepolymerase activity.
Received: February 7, 2007Revised: July 5, 2007
Accepted: August 7, 2007
Keywords:
Affinity chromatography / MALDI-TOF MS / MCM6 / p16ink4a-binding proteins / PCNA
4102 Proteomics 2007, 7, 4102–4111
1 Introduction
In mammals, cell cycle progression is regulated by a familyof protein kinases named cyclin-dependent kinases (CDKs)because its activity depends on the association with reg-ulatory subunits called cyclins [1]. The activity of cyclin-CDKcomplexes is regulated by different mechanisms includingphosphorylation of CDKs and interaction with two families
of inhibitors (CKIs), the Cip-Kip family and the INK4 family[2]. The INK4-class of cell cycle inhibitors includes p15ink4b,p16ink4a, p18ink4c, and p19ink4d. These proteins consist of fouror more highly conserved ankyrin repeats and bind to CDK4and CDK6 and inhibit their kinase activity. The binding ofINK4 proteins prevents the interaction of the CDKs with theD-type cyclins, which are required for catalytic activity [3].The major protein substrate for CDK4 and CDK6 is the reti-noblastoma protein (pRb) which plays a key role in the reg-ulation of the G1 phase of cell cycle. This protein associateswith the transcription factors E2Fs blocking its transcrip-tional activity. During the G1 phase of the cell cycle, whencyclin D-CDK4/6 complexes become activated, pRb is phos-phorylated and as a consequence releases E2Fs that becomefree to transactivate a number of genes involved in cell cycleprogression [4]. The expression of p16ink4a (p16) or other
Correspondence: Professor Oriol Bachs, Departament de Biolo-gia Cellular i Anatomia Patològica, Facultat de Medicina, Univer-sitat de Barcelona, Casanova 143, 08036-Barcelona, SpainE-mail: [email protected]: 134-93-402-19-07
Abbreviations: CDK, cyclin-dependent kinase; EF-2, elongationfactor 2; hnRNP, heterogeneous nuclear ribonucleoprotein;MCM6, minichromosome maintenance protein 6; NMS, normalmouse serum; PCNA, proliferating cell nuclear antigen; pRb, reti-noblastoma protein
* Additional corresponding author: Dr. Montserrat Jaumot,E-mail: [email protected]
DOI 10.1002/pmic.200700133
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 4102–4111 Cell Biology 4103
INK4 members, therefore, produces decreased CDK4/6 ac-tivity and pRb hypophosphorylation, which in turn leads toE2F repression and cell cycle arrest.
The p16 protein is encoded in the INK4a/ARF locus,situated on human chromosome 9p21. This locus containstwo different genes, one named INK4a that encodes for theprotein p16 and the second gene named ARF (for alternativereading frame) encoding the protein p14ARF in the humanand p19ARF in the mouse that is involved in the regulation ofp53 stability [5]. Both, INK4a and ARF genes contain threeexons. Ink4a includes exon 1a, 2, and 3 whereas ARF has itsown promoter and first exon (exon 1b as opposed to exon 1aof p16) and splices into the common second and third exonsshared with p16. Both proteins are encoded in alternativereading frames, and therefore are not isoforms and have noamino acid homology [5].
The INK4a/ARF locus is among the most frequent sitesof genetic loss in human cancer and alterations in this locusare frequently seen in a variety of malignancies [5]. Thelocation of these two different genes at the same locus hasgenerated during the last few years a discussion about therelevance of each one in tumorogenesis. However, by usingspecific knock out mice for each one of these two genes it hasrecently been demonstrated that both genes might be con-sidered as tumor suppressors which is in agreement with thehigh frequency of alterations of this locus in malignancies[6].
Both, p16 and ARF have been also involved in senes-cence [7]. Cells entering senescence cease to respond tomitogenic stimuli, undergo dramatic changes in chroma-tin structure and gene expression, and become enlargedand flattened. This situation is different from other formsof cell cycle arrest such as quiescence in two importantways. First, senescence is generally irreversible and sec-ond it is associated with distinctive molecular and mor-phological alterations as for instance cell flattening.Moreover, the activation of INK4/ARF locus also has beenrelated to aging [8]. Interestingly, although several otherCKIs modulate cell cycle progression, p16 appears amongthese in that its expression dramatically increases withaging.
Thus, p16 is a protein that plays a relevant role in severalimportant processes as tumorogenesis, senescence, andaging. As the only well-known function for p16 is to inhibitthe activity of CDK4 and CDK6 it seems likely that its in-volvement in these mentioned cellular processes might bemediated by the interaction with some other proteins stillunknown. Thus, in this work we aimed to identify new p16-interacting proteins. By affinity chromatography and thesubsequent proteomic analysis, we have identified 31 p16-interacting proteins, among them the proliferating cellnuclear antigen (PCNA) a processivity factor for DNA poly-merases, the key enzymes involved in DNA replication, DNArepair and translesion DNA synthesis [9]. Our results indi-cate that p16 directly interacts with PCNA and thus inhibitsDNA polymerase d.
2 Materials and methods
2.1 Cell culture
MOLT-4 lymphoblastoid cell line and NP18 pancreatic tumorwere cultured in RPMI-1640 medium (Biological Industries).NP29 pancreatic cell line and HeLa cells were cultured inDMEM (Biological Industries). All cell cultures were supple-mented with 10% FCS.
2.1 Sample preparation
MOLT-4 and NP29 cells were lysed in buffer A (50 mMHEPES, pH 7.6, 50 mM KCl, 1 mM MgCl2, 1 mM EGTA)containing 0.5 mg/mL aprotinin, 1 mM PMSF, 10 mg/mLleupeptin, 0.1 mM Na3VO4, and 50 mM NaF for 10 min at47C. After sonication, lysed cells were sedimented at40 000 rpm for 45 min. The supernatant was collected andthe protein content was measured by the method of Brad-ford.
Mice livers were homogenized in STM buffer (250 mMsucrose, 50 mM Tris-HCl pH 7.4, and 5 mM Mg2SO4) con-taining 0.5 mg/mL aprotinin and 1 mM PMSF. Homo-genates were filtered through four sheets of cheesecloth.Nuclei were obtained from homogenates as described byKaufman and Shaper [10]. Nuclei were then resuspended inSTM buffer containing 250 mg/mL DNase I and 250 mg/mLRNase A for 1 h at 47C and sedimented at 8006g for 10 min.The supernatant was collected and named S1 and the proteincontent was measured by the method of Bradford.
2.2 Plasmids
pGEX-KG-p16 and pET-20B(1)-PCNA were provided by Dr.Mazo and Dr. Agell, respectively, from the University of Bar-celona.
2.3 Protein expression and purification
The BL21-DE3 strain of Escherichia coli was transformed withthe vectors pGEX-KG- p16 or pET-20B(1)-PCNA. Therecombinant GST-p16 or His-PCNA proteins were inducedwith 0.4 mM IPTG overnight at 257C. Pellets were frozen at2807C for 30 min and then subjected to three cycles of freezeand defrost of 10 min each. Next, in the case of GST-p16,pellets were lysed in NETN buffer (20 mM Tris-HCl, pH 8.0,100 mM NaCl, 1 mM EDTA, 0.5% IGPAL) containing0.5 mg/mL aprotinin, 10mg/mL leupeptin, and 1 mM PMSF.In the case of PCNA, pellets were lysed in lysis buffer(50 mM NaH2PO4, pH 8.0, 10 mM imidazol, and 300 mMNaCl) containing 0.5 mg/mL aprotinin, 10 mg/mL leupeptin,and 1 mM PMSF. After sonication, cell lysates were cen-trifuged and a supernatant containing GST-p16 or His-PCNA were obtained. For GST-p16, the supernatant wasincubated with glutathione-Sepharose beads (AmershamBiosciences) for 2 h at 47C, spinned and washed twice with
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

4104 E. Souza-Rodrígues et al. Proteomics 2007, 7, 4102–4111
NETN and once with PBS. Next, beads were resuspended in1 mL of PBS and thrombin protease digestion was per-formed to separate GST from p16 according to the manu-facturer’s instructions (Sigma).
For His-PCNA, the supernatant was incubated with NI-NTA-agarose beads (Qiagen) for 4 h at 47C. Afterwards, thebeads were spinned and washed twice with wash buffer(50 mM NaH2PO4, pH 8.0, 20 mM imidazole, and 300 mMNaCl). Next, beads were incubated for 15 min at 47C withelution buffer (50 mM NaH2PO4, pH 8.0, 250 mM imida-zole, and 300 mM NaCl) and eluates containing PCNA wereobtained by centrifugation for 5 min at 15 000 rpm at 47C.
The four subunit recombinant DNA polymerase d waspurified from baculovirus as described by Podust et al. [11]and recombinant PCNA from E. coli according to Schurten-berger et al. [12].
2.4 p16-Sepharose affinity chromatography
To prepare the p16-Sepharose 4B columns, 3–5 mg of puri-fied p16 protein were coupled to 1 mL of CNBr-activatedSepharose 4B (Amersham Biosciencies) following manu-facturer’s instructions. Extracts (20 mg protein) from MOLT-4 or NP29 cell lines or from mice liver nuclei (S1 fraction)were then loaded onto the p16-Sepharose or the Sepharosecontrol columns. After washing in 200 volume of buffer Acontaining 300 mM KCl instead of 50 mM KCl, the boundproteins were eluted with elution buffer (200 mM glycinepH 2.5).
2.5 Gel electrophoresis and Western blotting
Protein samples were analyzed by SDS-PAGE. The gels werestained with CBB or transferred onto Immobilon-P mem-branes (Millipore). The sheets were incubated with TBST(20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20)containing 5% defatted milk powder for 1 h at room tem-perature. Western blots were probed for 1 h at room temper-ature with the following antibodies: anti-p16 (Santa Cruz, sc-468, rabbit polyclonal, 1:200 dilution), anti-p16 (BD Phar-mingen, 554079, mouse monoclonal, 1:500 dilution), anti-PCNA (Santa Cruz, sc-56, mouse monoclonal, 1:200 dilu-tion), anti-PCNA (F-2; Santa Cruz, sc-25280, mouse mono-clonal, 1:200 dilution), anti-minichromosome maintenanceprotein 6 (MCM6; Santa Cruz, sc-9843, goat polyclonal, 1:200dilution), anti-CDK4 (c-22; Santa Cruz, sc-260, rabbit poly-clonal, 1:200 dilution), anti-CDK6 (Santa Cruz, sc-177, rabbitpolyclonal, 1:200 dilution). After washing twice with TBST,sheets were incubated with the corresponding HRP-coupledsecondary antibody (goat antirabbit cat: 1706515, goat anti-mouse cat: 170–6516, BioRad, 1:4000 dilution, rabbit anti-goat cat: A5420, Sigma, 1:5000 dilution) for 45 min at roomtemperature. They were washed twice with TBST, once withTBS (20 mM Tris-HCl, pH 7.5, 150 mM NaCl) and visualizedby enhanced chemiluminiscence (Amersham).
2.6 Identification of proteins by MALDI-TOF MS
The bands were sliced manually from the gel and digested in-dividually with 100–150 ng of trypsin (Promega) at 377C over-night using a Montage In-Gel DigestZP (Millipore) with thestandard manufacturer protocol. The resulting peptides weredried and resuspended in 0.1% TFA. 0.5 mL of them weremixed with the same amount of CHCA. Next they were ana-lyzed by a MALDI-TOF Voyager DE Pro mass spectrometer(Applied Biosystems) operated in delayed extraction reflectormode at 20 kV as accelerating voltage, 90 ns of pulse delaytime, 75% of a grid voltage, and a guide wire voltage of 0.005%.Spectra were accumulated for 100 laser shots and were visual-ized and analyzed using Data Explorer software version 4.2(Applied Biosystems). Monoisotopic peaks were used for PMFusing the Protein Prospector MS-Fit software version 3.2 andthe main databases that describe the human and mouse pro-teomes: Swiss-Prot and the National Center for BiotechnologyInformation (NCBI NIH, Bethesda, MD, USA).
2.7 Immunoprecipitation
Samples (NP-18 and HeLa cells) were lysed in buffer B(50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 5 mM EDTA, 0.1%Triton X-100) containing 50 mM NaF, 1 mM PMSF, 10 mg/mL leupeptin, 0.5 mg/mL aprotinin, and 0.1 mM Na3VO4 for30 min on ice. Lysates were clarified by centrifugation at10 0006g for 10 min at 47C. The supernatants (500–1000 mg)were incubated with 2–3 mg (per 500 mg of total protein) ofanti-PCNA (Santa Cruz, sc-56, mouse monoclonal), anti-p16(BD Pharmingen, 554079, mouse monoclonal), or anti-MCM6 (Santa Cruz, sc-9843, goat polyclonal), overnight at47C, followed by incubation with protein G beads for 1 h at47C. After washing in buffer B, the immunocomplexes weresubjected to Western blotting with the corresponding anti-bodies.
2.8 Immunocytochemistry
To detect p16 and PCNA by immunocytochemistry, cells(NP-18 and HeLa cell lines) were grown in coverslips, fixedin 4% paraformaldehyde/PBS for 30 min at room tempera-ture, washed in PBS, treated with cold methanol for 4 min at2207C, washed in PBS, and blocked with 1% BSA in PBS for1 h. Then, coverslips were then incubated with anti-p16(Santa Cruz, sc-468, rabbit polyclonal, 1:50 dilution) and anti-PCNA (F-2; Santa Cruz, sc-25280, mouse monoclonal, 1:50dilution) antibodies for 1 h at 377C in a humidified atmos-phere. They were then washed five times (5 min each) inPBS and incubated for 45 min at 377C with Alexa-Fluor 594(Invitrogen, Molecular Probes, A11012, goat antirabbit, dilu-tion 1:500) and Alexa-Fluor 488 (Invitrogen, MolecularProbes, A11001, goat antimouse, dilution 1:500). Coverslipswere washed five times (5 min each), mounted on glassslides with Mowiol (Calbiochem) and analyzed by fluores-cence microscopy.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 4102–4111 Cell Biology 4105
2.9 DNA polymerase � assay
The DNA polymerase d assay was essentially performed asdescribed by Jonsson et al. [13]. A final volume of 25 mL con-tained: 50 mM Bis-Tris (pH 6.5), 6.25 mM MgCl2, 20 mM[3H]dTTP (400 cpm/pmol), 0.5 mg polydA/oligo dT12–18 (baseratio 10:1), 1 mM DTT, 250 mg/mL BSA, 10 ng PCNA (ifadded), and DNA polymerase d. The reaction was incubatedfor 15 min at 377C and the TCA precipitable counts deter-mined as described in ref. [11]. One DNA polymerase unit isdefined as the incorporation of 1 nmol dTTP into acid-insol-uble material in 60 min at 377C.
3 Results
3.1 Detection of p16-binding proteins from MOLT-4
cell extracts by affinity chromatography
With the aim of identifying new p16-binding proteins wegenerated affinity chromatography columns of p16-Sephar-ose 4B or Sepharose 4B columns (used as a control) asdescribed in Section 2. The columns were loaded with MOLT-4 cell extracts, washed and the proteins bound to the col-umns were eluted with a buffer containing glycine 200 mM,pH 2.5. The collected proteins were then separated by SDS-PAGE and visualized by CBB staining. As shown in Fig. 1Aapproximately 15 major bands were observed in the eluatesfrom the p16 columns, whereas in contrast, no clear bandswere observed in those from the control columns. Similarexperiments were performed using NP29 cell extracts ornuclear extracts from mice liver. As it may be observed inFigs. 1B and C a significant number of bands were observedin the eluates from the p16 columns, whereas in contrast,only a few nonspecific bands were eluted from the controlcolumns. The patterns of p16-binding proteins from thethree different sources are quite different suggesting thepresence of a distinct population of p16-binding proteins ineach cellular type.
To be sure that the columns were working properly, weanalyzed whether the MOLT-4 eluates from the p16 columncontained CDK4 and CDK6, two-well known p16-bindingproteins. As it can be seen in the Fig. 1D CDK4 and CDK6were detected by Western blotting. The amount of CDK4 inthe eluates was much lower than that of cdk6, probablyreflecting the reduced quantity of CDK4 in this cellular type.
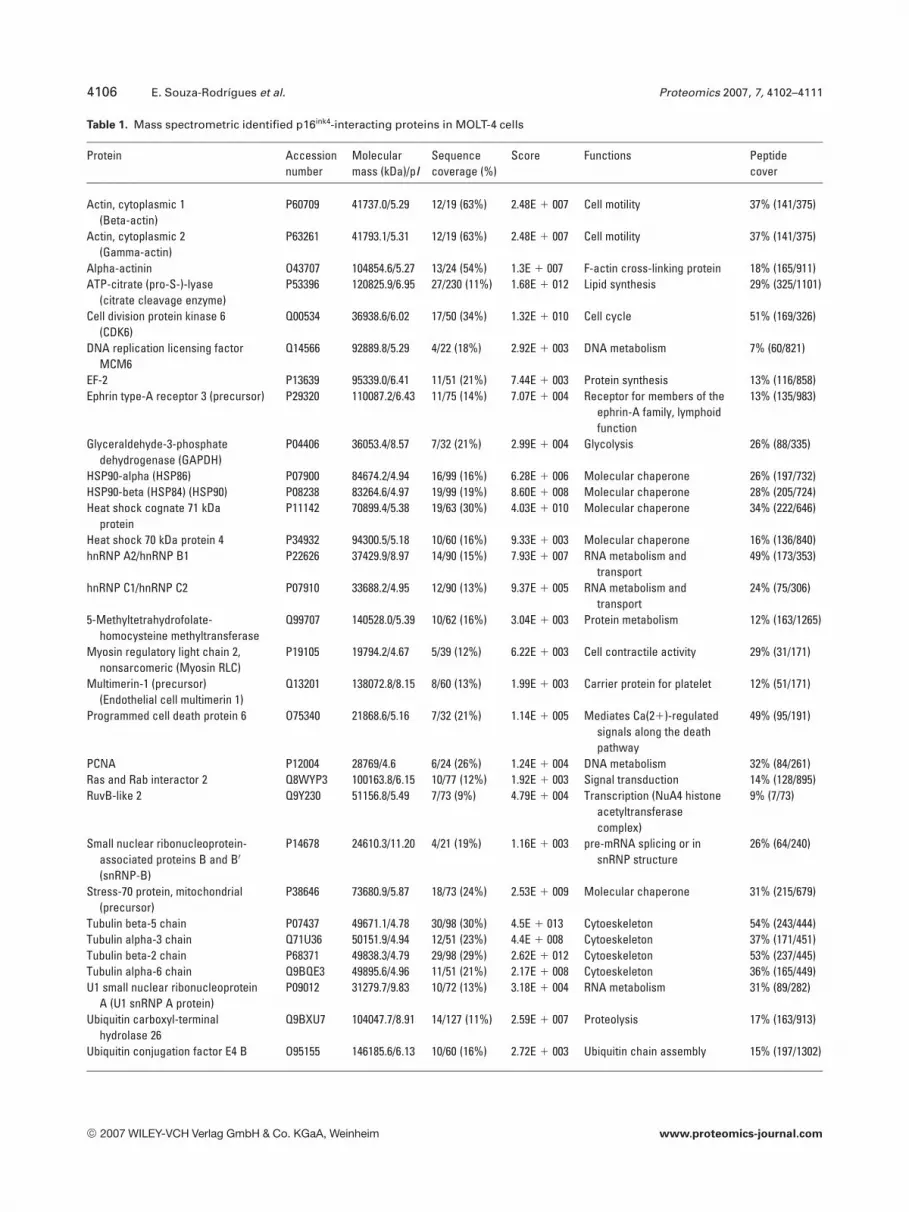
To identify the p16-binding proteins from MOLT-4 cells,the proteins were excised individually from the gels (Fig. 1Ashows a representative gel), trypsinized individually and theresulting peptides analyzed by MS using a MALDI-TOF asdescribed in Section 2. The masses of the monoisotopicpeaks obtained were used to identify each protein by com-paring to theoretical digestions of proteins by trypsin. Thir-tyone proteins were identified by PMF analysis. The list ofthe identified proteins, their functions, and the sequencecoverage is shown in Table 1.
Figure 1. Identification of p16-binding proteins by combining af-finity chromatography, SDS-PAGE, and MS. MOLT-4 (A), NP29(B), and mouse liver nuclei (C) lysates were loaded onto a p16-Sepharose 4B column (p16 col) or onto a Sepharose 4B controlcolumn (C col). After extensive washing with a buffer containing300 mM KCl, the proteins bound to the columns were eluted with200 mM glycine pH 2.5. Samples: precolumn (PC), flow through(FT), wash (W), and eluate (E) were separated in an SDS-PAGEand stained with CBB. (D) Western blot validation of the presenceof CDK4 and CDK6 in the MOLT-4 eluates of the p16 chromatog-raphy columns. Samples were separated by SDS-PAGE, trans-ferred onto membranes and analyzed by Western blotting withCDK6 and CDK4 antibodies.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
4106 E. Souza-Rodrígues et al. Proteomics 2007, 7, 4102–4111
Table 1. Mass spectrometric identified p16ink4-interacting proteins in MOLT-4 cells
Protein Accessionnumber
Molecularmass (kDa)/pI
Sequencecoverage (%)
Score Functions Peptidecover
Actin, cytoplasmic 1(Beta-actin)
P60709 41737.0/5.29 12/19 (63%) 2.48E 1 007 Cell motility 37% (141/375)
Actin, cytoplasmic 2(Gamma-actin)
P63261 41793.1/5.31 12/19 (63%) 2.48E 1 007 Cell motility 37% (141/375)
Alpha-actinin O43707 104854.6/5.27 13/24 (54%) 1.3E 1 007 F-actin cross-linking protein 18% (165/911)ATP-citrate (pro-S-)-lyase
(citrate cleavage enzyme)P53396 120825.9/6.95 27/230 (11%) 1.68E 1 012 Lipid synthesis 29% (325/1101)
Cell division protein kinase 6(CDK6)
Q00534 36938.6/6.02 17/50 (34%) 1.32E 1 010 Cell cycle 51% (169/326)
DNA replication licensing factorMCM6
Q14566 92889.8/5.29 4/22 (18%) 2.92E 1 003 DNA metabolism 7% (60/821)
EF-2 P13639 95339.0/6.41 11/51 (21%) 7.44E 1 003 Protein synthesis 13% (116/858)Ephrin type-A receptor 3 (precursor) P29320 110087.2/6.43 11/75 (14%) 7.07E 1 004 Receptor for members of the
ephrin-A family, lymphoidfunction
13% (135/983)
Glyceraldehyde-3-phosphatedehydrogenase (GAPDH)
P04406 36053.4/8.57 7/32 (21%) 2.99E 1 004 Glycolysis 26% (88/335)
HSP90-alpha (HSP86) P07900 84674.2/4.94 16/99 (16%) 6.28E 1 006 Molecular chaperone 26% (197/732)HSP90-beta (HSP84) (HSP90) P08238 83264.6/4.97 19/99 (19%) 8.60E 1 008 Molecular chaperone 28% (205/724)Heat shock cognate 71 kDa
proteinP11142 70899.4/5.38 19/63 (30%) 4.03E 1 010 Molecular chaperone 34% (222/646)
Heat shock 70 kDa protein 4 P34932 94300.5/5.18 10/60 (16%) 9.33E 1 003 Molecular chaperone 16% (136/840)hnRNP A2/hnRNP B1 P22626 37429.9/8.97 14/90 (15%) 7.93E 1 007 RNA metabolism and
transport49% (173/353)
hnRNP C1/hnRNP C2 P07910 33688.2/4.95 12/90 (13%) 9.37E 1 005 RNA metabolism andtransport
24% (75/306)
5-Methyltetrahydrofolate-homocysteine methyltransferase
Q99707 140528.0/5.39 10/62 (16%) 3.04E 1 003 Protein metabolism 12% (163/1265)
Myosin regulatory light chain 2,nonsarcomeric (Myosin RLC)
P19105 19794.2/4.67 5/39 (12%) 6.22E 1 003 Cell contractile activity 29% (31/171)
Multimerin-1 (precursor)(Endothelial cell multimerin 1)
Q13201 138072.8/8.15 8/60 (13%) 1.99E 1 003 Carrier protein for platelet 12% (51/171)
Programmed cell death protein 6 O75340 21868.6/5.16 7/32 (21%) 1.14E 1 005 Mediates Ca(21)-regulatedsignals along the deathpathway
49% (95/191)
PCNA P12004 28769/4.6 6/24 (26%) 1.24E 1 004 DNA metabolism 32% (84/261)Ras and Rab interactor 2 Q8WYP3 100163.8/6.15 10/77 (12%) 1.92E 1 003 Signal transduction 14% (128/895)RuvB-like 2 Q9Y230 51156.8/5.49 7/73 (9%) 4.79E 1 004 Transcription (NuA4 histone
acetyltransferasecomplex)
9% (7/73)
Small nuclear ribonucleoprotein-associated proteins B and B0
(snRNP-B)
P14678 24610.3/11.20 4/21 (19%) 1.16E 1 003 pre-mRNA splicing or insnRNP structure
26% (64/240)
Stress-70 protein, mitochondrial(precursor)
P38646 73680.9/5.87 18/73 (24%) 2.53E 1 009 Molecular chaperone 31% (215/679)
Tubulin beta-5 chain P07437 49671.1/4.78 30/98 (30%) 4.5E 1 013 Cytoeskeleton 54% (243/444)Tubulin alpha-3 chain Q71U36 50151.9/4.94 12/51 (23%) 4.4E 1 008 Cytoeskeleton 37% (171/451)Tubulin beta-2 chain P68371 49838.3/4.79 29/98 (29%) 2.62E 1 012 Cytoeskeleton 53% (237/445)Tubulin alpha-6 chain Q9BQE3 49895.6/4.96 11/51 (21%) 2.17E 1 008 Cytoeskeleton 36% (165/449)U1 small nuclear ribonucleoprotein
A (U1 snRNP A protein)P09012 31279.7/9.83 10/72 (13%) 3.18E 1 004 RNA metabolism 31% (89/282)
Ubiquitin carboxyl-terminalhydrolase 26
Q9BXU7 104047.7/8.91 14/127 (11%) 2.59E 1 007 Proteolysis 17% (163/913)
Ubiquitin conjugation factor E4 B O95155 146185.6/6.13 10/60 (16%) 2.72E 1 003 Ubiquitin chain assembly 15% (197/1302)
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 4102–4111 Cell Biology 4107
We further aimed to group these proteins in functional clus-ters to investigate new putative cellular processes in whichp16 might participate (Fig. 2).
3.2 Western blot identification of p16-binding
proteins
The identification of two of the novel p16-binding proteinsdiscovered by MS was confirmed by Western blot analysis ofthe eluates from the p16-Sepharose 4B columns. Proteinsamples of each step of the chromatography experimentswere separated by SDS-PAGE and the gels were transferredonto Immobilon-P membranes. Western blot was performedusing anti-PCNA or anti-MCM6 antibodies. Results revealedthat PCNA (Fig. 3A) and MCM6 (Fig. 3B) were present in theeluates from the p16-Sepharose 4B column but not in theeluates from the control column.
3.3 In vivo interaction of p16ink4a with PCNA and
MCM6
To investigate whether p16 interacts in vivo with PCNA andMCM6, immunoprecipitation experiments were per-formed. NP-18 and HeLa cells extracts were immunopre-cipitated with anti-p16 antibodies, with anti-PCNA, or withnormal mouse serum (NMS) as a control. The presence ofPCNA and p16 in the immunoprecipitates was analyzed byWestern blotting using specific antibodies (Fig. 4A). Coim-munoprecipitation of p16 and MCM6 was observed whenNP-18 cell extracts were immunoprecipitated with anti-MCM6, and immunoblotted with anti-p16 antibodies.Results were confirmed in similar experiments immuno-precipitating with anti-p16 antibodies and analyzing byWestern blot the presence of MCM6 in the immunopreci-pitates (Fig. 4B).
3.4 Colocalization and direct interaction of p16 with
PCNA
To further confirm that PCNA and p16 interact in vivo, weexamined the intracellular distribution of PCNA and p16 pro-teins in order to analyze whether they colocalize within the cell.Thus, immunocytochemical experiments were performed inNP-18 and HeLa cells using antibodies against PCNA and p16proteins (Fig. 5A). In these experiments, cells showed bothproteins located in the nucleus, being the nucleoli excluded,and colocalizing in some nuclear regions. Quantification of thenuclear colocalization between both proteins revealed that inNP-18 cells the percentage of nuclear costaining is of 71%whereas in HeLa cells this percentage is of 65%.
Direct interaction between PCNA and p16 was examinedusing affinity chromatography assays. Purified PCNA wasloaded onto a p16-Sepharose 4B column and the bindinganalyzed by CBB staining. Results showed that PCNA wasdirectly bound to the p16 column but not to the control col-umn (Fig. 5B).
3.5 p16 inhibits PCNA-mediated DNA polymerase �activity
Since PCNA is a well-known accessory factor that is involvedin the activation of DNA polymerases, we finally examinedthe ability of p16 to regulate the PCNA-mediated activity ofDNA polymerase d. First we determined the in vitro DNApolymerase d activity in the presence or absence of PCNAand showed that under our experimental conditions PCNAcan activate DNA polymerase d (Fig. 6A). Figure 6B docu-ments that p16 has the ability to inhibit the polymerase ac-tivity up to 50% at a molar ratio of around 100 which is ap-proximately two-fold higher than that observed for p21 (datanot shown), a well-defined inhibitor of PCNA-mediated DNApolymerase d activity.
Figure 2. p16-interacting proteins identified byMS in MOLT-4 cells (shown in Table 1) weregrouped in eight main functional clusters basedon their particular functions. Numbers representthe percentage of the total p16-binding proteinswhich belongs to a specific functional group.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

4108 E. Souza-Rodrígues et al. Proteomics 2007, 7, 4102–4111
Figure 3. Western blot validation of the association of p16 withPCNA and MCM6. MOLT-4 lysates were loaded onto a p16-Sepharose 4B column or onto a Sepharose 4B control column.After extensive washing with a buffer containing 300 mM KCl, theproteins bound to the columns were eluted with 200 mM glycinepH 2.5. Samples: precolumn (PC), flow through (FT), wash (W),and eluate (E) were separated in an SDS-PAGE, transferred ontomembranes, and analyzed by Western blotting with PCNA anti-bodies (A) or with MCM6 antibodies (B).
4 Discussion
The protein p16 is a key regulator of cell cycle progressionand senescence being its major defined function the negativeregulation of CDK4 and CDK6 activity. Despite this protein isencoded by a gene considered to be a tumor suppressor andof this well documented implication in oncogenesis, verylittle is known about the involvement of this protein in theregulation of other cellular processes. Thus, with this aim wehave developed a proteomic approach to identify p16-bindingproteins by combining affinity chromatography and MStechniques which was complemented by Western blot andfunctional analyses.
Results presented here show the identification of 31 p16-binding proteins from MOLT-4 cell extracts. Only one ofthese proteins (CDK6) has been previously identified as ap16-binding protein. Thus, the other 30 proteins can be con-sidered as new p16-interacting proteins. However, it has to beconsidered that not all of these 30 proteins may directly bindto p16 but some of them might be indirectly associatedthrough protein complexes containing p16.
Interestingly, when working with different cell extracts asfor instance NP29 cell extracts or extracts from mice livernuclei, a significant number of p16-binding proteins werealso purified using the same experimental approach. Theseresults indicate that the experimental approach might be ex-tremely useful for the analysis of p16-protein partners indifferent experimental models.
The presence of not only CDK4 and CDK6 but also of twonew p16-binding proteins, MCM6 and PCNA, in the eluatesfrom the p16-Sepharose 4B columns was validated by West-ern blotting using specific antibodies against these proteins.
Figure 4. In vivo interaction of p16 with PCNA and MCM6. (A) NP-18 or HeLa cell lysates were immunoprecipitated with p16 orPCNA antibodies or with NMS as a control. The presence of p16and PCNA in the immunoprecipitates was investigated by West-ern blotting using p16 and PCNA antibodies, respectively. Asample of the respective cell lysates was added as a control (L).(B) NP-18 cell lysates were immunoprecipitated with MCM6 anti-bodies or with normal goat serum (NGS) as a control (upperpanel) or with anti-p16 or NMS as a control (bottom panel). Thepresence of p16 or MCM6 in the immunoprecipitates was inves-tigated by Western blotting using p16 or MCM6 antibodies. Asample of NP-18 cell lysate was added as a control (L).
Moreover, the in vivo interaction between MCM6 and PCNAwith p16 was also demonstrated by immunoprecipitationexperiments. In addition the intracellular colocalization be-tween PCNA and p16 was also demonstrated by immuno-cytochemistry.
The classification of the p16-interacting proteins infunctional clusters revealed that most of the proteins belongto five major functional groups: (i) protein metabolism (nine
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 4102–4111 Cell Biology 4109
Figure 5. (A) Intracellular colocalization of p16 and PCNA. Thepresence of p16 and PCNA in NP18 and HeLa cells was analyzedby immunofluorescence using p16 and PCNA antibodies.Nuclear staining was performed using DAPI. Control was carriedout by incubating cells with secondary antibodies. (B) Directinteraction of PCNA and p16. Purified PCNA was loaded onto ap16-Sepharose 4B column (p16 col) or onto a Sepharose 4B con-trol column (Control col). After extensive washing with a buffercontaining 300 mM KCl, the protein bound to the columns waseluted with 200 mM glycine pH 2.5. Samples: precolumn (PC),flow through (FT), wash (W), and eluate (E) were separated in aSDS-PAGE and stained with CBB.
proteins); (ii) cytoskeleton (eight proteins); (iii) RNA metab-olism (five proteins); (iv) signal transduction (three pro-teins); and (v) cell cycle (three proteins).
The first major group of p16-interacting proteinsincludes a number of chaperones as HSP90-a, HSP90-b,HSPA8 (heat shock cognate 71 kDa protein), HSP70RY (heatshock 70 kDa protein 4), and the stress-70 protein [14–17];two proteins involved in the ubiquitin-dependent pathwaysof protein degradation, USP26 (ubiquitin carboxyl-terminalhydrolase 26) and ubiquitin conjugation factor E4 B; oneprotein which participates in methionine synthesis, 5-methyltetrahydrofolate-homocysteine and one proteinessential in the process of protein synthesis, the elongationfactor 2 (EF-2).
Stress proteins function as molecular chaperones, theyassist protein folding, protein complexes formation, proteindirectioning to target organelles, and prevent protein aggre-gation. When cells are exposed to stress they function asmolecular sensors and react to maintain cell homeostasis byprotecting cells against apoptosis, stabilizing mutations thatcould affect protein folding, etc. Some stress proteins areknown to transiently associate with several cell cycle reg-ulatory proteins, with viral oncoproteins, and with proteinsinvolved in signal transduction cascades. Interestingly, somestress proteins are overexpressed in many human cancersand their inhibition is been currently explored for cancertherapeutics. The chaperones-p16 interactions suggest thatp16 might be participating in some of the chaperone func-tions, or alternatively that the chaperons might participate insome of the p16 pathways [15].
p16 also interacts with USP26 (ubiquitin carboxyl-termi-nal hydrolase 26) and ubiquitin conjugation factor E4.USP26 is a protease included in the family of the deubiqui-tinating enzymes (DUBs) which remove ubiquitin from thepolypeptides. The E4 factors are enzymes responsible for theubiquitin-chain elongation of monoubiquitylated substrateswhich target them for degradation by the proteasome [18].The interaction with these proteins suggests that p16 mightbe degraded via proteasome through ubiquitylation. Al-though p16 is a lysine-less protein a recent paper indicatethat p16 might be targeted for degradation via N-terminalubiquitylation [19]. However, another possibility is that p16might act regulating the activity of these enzymes. In factp14ARF, the other protein encoded by the INK4/ARF locus,interacts with the SUMO E2 conjugating enzyme Ubc9 andenhances the sumoylation of its binding partners [20]. Inter-estingly, ARF also binds and inhibits MDM2, the principalE3-ubiquitin-ligase of p53 [21].
EF-2 mediates the translocation of the tRNAs and themRNA after peptydyl transfer on the ribosome during pro-tein synthesis [22, 23]. The evidence that p16 not onlyassociates with EF-2 but also with 5-methyltetrahydrofolate-homocysteine, (involved in methionine synthesis) [24, 25],suggest a putative role of p16 in the regulation of the proteinsynthesis.
The second group of p16-interacting proteins compriseseight proteins which are components of the cytoeskeleton.Two actin isoforms, four tubulin isoforms, alpha-actinin (a F-actin-crosslinking protein) and myosin regulatory light chain2. Progression through the cell cycle is accompanied of con-tinuous changes in the cytoeskeleton which must be highlyregulated. The finding that p16 associates with several pro-teins members of microfilaments and microtubules net-work, strongly suggest its implication in the cytoeskeletondynamics. This is consistent with the dramatic changes ofcell shape observed during senescence induced by p16.
The third major group of p16-binding proteins is con-stituted by proteins involved in RNA metabolism. The inter-action of p16 with two components (U1 snRNP A and withsnRNP-B proteins) of the pre-mRNA splicing machinery in
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

4110 E. Souza-Rodrígues et al. Proteomics 2007, 7, 4102–4111
Figure 6. p16 inhibits PCNA-mediated DNApolymerase d activity. (A) In vitro DNA poly-merase d assays were performed with increasingamounts of DNA polymerase d (0.5 U/mL) onpoly(dA)/oligo (dT) in the presence or absence of10 ng PCNA as described in Section 2. (B) p16was titrated into a DNA polymerase d assay con-taining (0.4 U DNA polymerase d and 10 ngPCNA) as described in Section 2. The x-axis ofthe graph indicates the molar ratio between p16and PCNA and the y-axis relative DNA synthesiscompared with samples containing no addedp16.
the eukaryotic cells (the spliceosome), strongly suggest theparticipation of p16 in this essential cell process [26, 27].Some proteins of the family of the heterogeneous nuclearribonucleoproteins (hnRNPs) as A2/B1 and C1/C2 alsointeract with p16. HnRNP C1/C2 associate with the RNAcomponent of telomerase and hnRNP A2/B1 binds to thetelomers [28]. It is postulated that hnRNPs may act as dock-ing sites for recruitment of telomerase to the telomers formaintaining telomere length and inhibiting senescence [29].Due to the reported involvement of p16 in aging [30] and inoncogene-induced senescence [31–33], our results suggestthat p16 might participate in this processes by associationwith telomeres or with the telomerase. The interaction of p16with RuvB-like 2 suggest a role in regulation of the genetranscription, since this protein is a component of the NuA4histone acetyltransferase complex required for activation oftranscription of genes involved in growth induction, growtharrest, replicative senescence, apoptosis, and DNA repair[34].
Our results also suggest that p16 can participate in somesignal transduction pathways. Thus, through the interactionwith the ephrin type-A receptor precursor p16 may partici-pate in neuronal communication; it may be also be involvedin the regulation of apoptosis through the association withprogrammed cell death protein 6 (ALG-2) [35]. The associa-
tion of p16 with the Ras and Rab interactor 2, a proteinresponsible for Rab 5 activation it could be involved in endo-cytosis regulation [36, 37].
The last major group of p16-interacting proteins includethose related to cell cycle progression. We observed that inaddition to CDK4 and CDK6 p16 also interacts with PCNAand MCM6, two proteins involved in DNA replication. PCNAis a protein essential for DNA polymerase to advance alongthe template DNA during the process of DNA replication.Specifically, PCNA displaces DNA polymerase a, involved inthe process of priming and recruits DNA polymerase d forfurther DNA synthesis. Several other PCNA partners havebeen previously described [9]. One of them is p21cip1 which inaddition to block PCNA function is also a CDK inhibitor [13,38]. Results shown in Figs. 5B and 6 demonstrate that p16interacts directly with PCNA and inhibits DNA polymerase dactivity in vitro suggesting that p16 might regulate DNAreplication through this mechanism. Interestingly, ourresults suggest that p16 might directly regulate DNA repli-cation by a second mechanism that involves its interactionwith MCM6. This protein belongs to the MCM complexwhich is composed by six members (MCM2 to 7) which arerecruited into the origins of DNA replication through theorigin recognition complex (ORC). MCMs are necessary forthe initiation of DNA replication as DNA helicases that melt
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 4102–4111 Cell Biology 4111
the DNA double helix [39]. Braden et al. [40] have recentlyshown that p16 blocks the assembly of the MCM complexonto chromatin during G1 phase of the cell cycle thus in-hibiting DNA replication. They also observe that p16 expres-sion inhibits the association of MCM7 and PCNA to chro-matin. Thus, these evidences and our results suggest thatp16 might be interfering with DNA replication by directlybinding to PCNA and MCMs.
From our results we conclude that p16 might be partici-pating in a broad spectrum of new functions within the cellthat must be further investigated. The combination of affin-ity chromatography and MS techniques have been confirmedas a very useful approach to study new protein–proteininteractions and thus new protein functions.
This work is supported by grants from the Spanish Ministeriode Educación y Ciencia SAF 2002-00452, SAF 2003-08339, andGEN2003-20243-C08-01. E. S. was supported by the Uni-versidade Estadual de Feira de Santana, BA, Brasil. U. H. andE. F. H. are supported by the Swiss National Science Foundation(grant 3100AO-190312), and by the University of Zürich. Pro-teomic analysis was performed at the Proteomic Unit of the “Ser-veis Científico-tècnics” from the University of Barcelona which isa member of ProteoRed, a Spanish Network of Proteomic facil-ities funded by Genoma Spain.
5 References
[1] Morgan, D. O., Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291.
[2] Sherr, C. J., Roberts, J. M., Genes Dev. 1999, 13, 1501–1512.
[3] Russo, A. A., Tong, L., Lee, J. O., Jeffrey, P. D. et al., Nature1998, 395, 237–243.
[4] Cobrinik, D., Oncogene 2005, 24, 2796–2809.
[5] Sharpless, N. E., Mutat. Res. 2005, 576, 22–38.
[6] Sharpless, N. E., Ramsey, M. R., Balasubramanian, P., Cas-trillon, D. H. et al., Oncogene 2004, 23, 379–385.
[7] Ben Porath, I., Weinberg, R. A., Int. J. Biochem. Cell Biol.2005, 37, 961–976.
[8] Sharpless, N. E., Exp. Gerontol. 2004, 39, 1751–1759.
[9] Maga, G., Hubscher, U., J. Cell Sci. 2003, 116, 3051–3060.
[10] Kaufmann, S. H., Shaper, J. H., Exp. Cell Res. 1984, 155, 477–495.
[11] Podust, V. N., Chang, L. S., Ott, R., Dianov, G. L. et al., J. Biol.Chem. 2002, 277, 3894–3901.
[12] Schurtenberger, P., Egelhaaf, S. U., Hindges, R., Maga, G. etal., J. Mol. Biol. 1998, 275, 123–132.
[13] Jonsson, Z. O., Hindges, R., Hubscher, U., EMBO J. 1998, 17,2412–2425.
[14] Diller, K. R., Annu. Rev. Biomed. Eng. 2006, 8, 403–424.
[15] Helmbrecht, K., Zeise, E., Rensing, L., Cell Prolif. 2000, 33,341–365.
[16] Wegele, H., Muller, L., Buchner, J., Rev. Physiol. Biochem.Pharmacol. 2004, 151, 1–44.
[17] Mosser, D. D., Morimoto, R. I., Oncogene 2004, 23, 2907–2918.
[18] Hoppe, T., Trends Biochem. Sci. 2005, 30, 183–187.
[19] Ben Saadon, R., Fajerman, I., Ziv, T., Hellman, U. et al., J.Biol. Chem. 2004, 279, 41414–41421.
[20] Rizos, H., Woodruff, S., Kefford, R. F., Cell Cycle 2005, 4, 597–603.
[21] Zhang, Y., Xiong, Y., Cell Growth Differ. 2001, 12, 175–186.
[22] Jorgensen, R., Merrill, A. R., Andersen, G. R., Biochem. Soc.Trans. 2006, 34, 1–6.
[23] Browne, G. J., Proud, C. G., Eur. J. Biochem. 2002, 269, 5360–5368.
[24] Brosnan, J. T., Brosnan, M. E., J. Nutr. 2006, 136, 1636S–1640S.
[25] Zou, C. G., Banerjee, R., Antioxid. Redox Signal. 2005, 7,547–559.
[26] Jurica, M. S., Moore, M. J., Mol. Cell 2003, 12, 5–14.
[27] Nilsen, T. W., Bioessays 2003, 25, 1147–1149.
[28] Ford, L. P., Wright, W. E., Shay, J. W., Oncogene 2002, 21,580–583.
[29] Carpenter, B., MacKay, C., Alnabulsi, A., MacKay, M. et al.,Biochim. Biophys. Acta-Rev. Cancer 2006, 1765, 85–100.
[30] Kim, W. Y., Sharpless, N. E., Cell 2006, 127, 265–275.
[31] Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P. et al.,Nature 2006, 444, 633–637.
[32] Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S. et al.,Nature 2006, 444, 638–642.
[33] Jacobs, J. J. L., de Lange, T., Cell Cycle 2005, 4, 1364–1368.
[34] Kanemaki, M., Kurokawa, Y., Matsu-ura, T., Makino, Y. et al.,J. Biol. Chem. 1999, 274, 22437–22444.
[35] Krebs, J., Saremaslani, P., Caduff, R., Biochim. Biophys.Acta-Prot. Proteomics 2002, 1600, 68–73.
[36] Saito, K., Murai, J., Kajiho, H., Kontani, K. et al., J. Biol.Chem. 2002, 277, 3412–3418.
[37] Kimura, T., Sakisaka, T., Baba, T., Yamada, T. et al., J. Biol.Chem. 2006, 281, 10598–10609.
[38] Frouin, I., Maga, G., Denegri, M., Riva, F. et al., J. Biol. Chem.2003, 278, 39265–39268.
[39] Maiorano, D., Lutzmann, M., Mechali, M., Curr. Opin. CellBiol. 2006, 18, 130–136.
[40] Braden, W. A., Lenihan, J. M., Lan, Z., Luce, K. S. et al., Mol.Cell Biol. 2006, 26, 7667–7681.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com





























![[Reliability of the CINtec p16INK4a immunocytochemical test in screening cervical precancerous lesions]](https://img.pdfslide.net/doc/110x75/636144814b9aa63a9e00a9c7/reliability-of-the-cintec-p16ink4a-immunocytochemical-test-in-screening-cervical.jpg)