Embed Size (px)
Citation preview

International Immunology, Vol. 12, No. 6, pp. 851–860 © 2000 The Japanese Society for Immunology
Regulation of TCR-induced IFN-γ releasefrom islet-reactive non-obese diabetic CD8�
T cells by prostaglandin E2 receptorsignaling
Vidya Ganapathy, Tatyana Gurlo, Hilde O. Jarstadmarken andHermann von GrafensteinSchool of Pharmacy, University of Southern California, 1985 Zonal Avenue, Los Angeles, CA 90033, USA
Keywords: cell–cell interactions, cytokine, cytotoxic T lymphocyte, diabetes, rodent
Abstract
Prostaglandins (PG) are released during tissue injury and inflammation, and inhibit immuneresponses at many points. PG may be one of several factors that protect not only against injury-induced, but also spontaneous, organ-specific autoimmune disease. Here we show that theproduction of PGE2, normally produced at a very low rate in islets of Langerhans, is significantlyincreased in inflamed islets of non-obese diabetic (NOD) mice. We investigated a possible role ofPGE2 in controlling TCR-dependent release of IFN-γ from islet-reactive NOD CD8� T cells. PGE2inhibited anti-TCR antibody-triggered release of IFN-γ from CD8� T cell clone 8D8 and frompolyclonal cytotoxic T lymphocytes (CTL). Using receptor subtype selective agonists, we presentevidence that the effect of PGE2 is mediated by EP2 and EP4 receptors, both of which are coupledto an increase in intracellular cAMP production. The cAMP analogs 8-Br-cAMP and Sp-cAMPSmimic the effect of EP2/EP4 receptor agonists, inhibiting TCR-triggered IFN-γ release from NODCD8� T cells in a dose-dependent manner. The inhibitory effect of PGE2 was largely reversed byIL-2 added at the time of culture initiation and decreased with increasing strength of stimulationthrough the TCR. Resting CTL were more sensitive to PGE2 than recently expanded CTL and NODCD8� T cells remained insensitive to PGE2 for a longer time than BALB/c cells. Our studysuggests that PGE2 may be part of a regulatory network that controls local activation of T cells andmay play a role in the balance between the development of islet autoimmunity or maintenance oftolerance.
Introduction
The non-obese diabetic (NOD) mouse is an excellent animal IFN-γ, produced by Th1 CD4� T cells and CD8� T cells, hasbeen implicated as a key disease-enhancing cytokine (4),model for the study of the pathogenesis of insulin-dependent
diabetes mellitus (IDDM) because it shares many features while IL-4 produced by Th2 CD4� T cells as well as systemicIL-10 and transforming growth factor-β are considered to bewith the human disease (1,2). IDDM is an organ-specific
autoimmune disorder characterized by chronic inflammation protective factors (5). Although the Th1–Th2 dichotomy may bean oversimplification, an imbalance of positive and negativeof the islets of Langerhans and by T cell-mediated destruction
of islet β cells (1,3). In NOD mice and in humans, the etiology regulatory signals may influence the development of auto-immune disease (3). It is therefore essential to identify andof IDDM is considered to be multifactorial with disease
development requiring the interactions of a multitude of characterize not only factors and mechanisms that promotedisease, but also those that limit or delay anti-islet immunitycells, which coordinate their function by means of contact-
dependent and soluble factors. Some of these factors and and have the potential to protect against disease devel-opment.the cells that produce them are thought to be disease
promoting while others are considered to be protective. Prostaglandin (PG) E2 is a candidate protective factor
The first two authors contributed equally to this work
Correspondence to: H. von Grafenstein
Transmitting editor: J.-F. Bach Received 8 June 1999, accepted 18 February 2000

852 PGE2 and IFN-γ release from NOD CD8� T cell clones
because it leads to the down-regulation of T cell-dependent uted to the molecular diversity of receptors (20). Four subtypesof pharmacologically active PGE2 receptors differing in theirimmune responses. It has been proposed that one of the
functions of PGE2 is to protect against immunity to self mode of signal transduction have been characterized (21–24). All known PGE2 receptor subtypes, termed EP1–EP4, areantigens when released during tissue injury and inflammation
(6), and PGE2 is thought to mediate some of the immuno- coupled to intracellular signaling via heterotrimeric GTP-binding proteins. EP1 receptors activate phosphatidylinositolsuppressive effects of tumors (7,8). The same reasons may
argue for a protective role of PGE2 against spontaneous turnover and intracellular Ca2� release via a poorly character-ized G-protein-mediated mechanism (25,26). EP2 and EP4autoimmune disease, although this possibility has not been
addressed in detail. In the experimental autoimmune ence- receptors are coupled via Gs protein to an increase inintracellular cAMP production (20,21,27,28). EP3 receptorsphalomyelitis model, local synthesis of PGE2 was found to be
associated with the resolution of brain lesions after the induc- exist in multiple forms that differ in their cytoplasmic tailthat couple the receptor to intracellular different signalingtion of oral tolerance (9). Recent studies have provided
evidence for cytokine-induced synthesis of PGE2 in islets pathways, but have the same extracellular ligand bindingcharacteristics (22,23).(10). Although in that study PGE2 was proposed to mediate
β cell cytotoxic effects of pro-inflammatory cytokines, it is Most studies on intracellular signaling events followingPGE2 receptor activation have focused on EP2/EP4 receptorsequally plausible that PGE2 has a protective effect against
IDDM given its well-known immunosuppressive function. An expressed by CD4� T cells and events downstream ofreceptor-induced cAMP production such as inhibition of IL-2increase in the production of PGE2 in islets was found to be
associated with the protective effect of oral insulin (11). A gene expression (24,29). Although some evidence is availablesuggesting that regulation of IFN-γ synthesis has elements inmore detailed understanding of PGE2’s multiple effects on T
cell subsets and their function is clearly necessary before an common with IL-2 gene expression (30,31), IL-2 and IFN-γare not regulated concordantly in all situations (32). Anergicunderstanding of its role can be reached.
Most of the general information about the effect of PGE2 cells, for example, are characterized by an inability to produceIL-2 and to proliferate (33) while maintaining effector functionon T cell development and function that is available supports
a potential protective role of PGE2 in IDDM. PGE2 has been such as production of IFN-γ (34,35). Little is known about arole of PGE2 in regulating IFN-γ production in CD8� T cellsimplicated in the differentiation of CD4� T cells and the
activation of subsets of CD4� T cells. Thus, it has been shown and any signaling mechanisms that might mediate such PGE2-induced effects.that PGE2, when present at the time of T cell priming, facilitates
the in vitro differentiation of naive CD4� T cells into Th2 rather In the studies reported here we demonstrated that PGE2is produced by inflamed but not healthy NOD islets, andthan Th1 cells (12). Recently, it has been suggested that PGE2
may influence the differentiation of Th subsets indirectly, determined the effect of PGE2 on TCR-dependent release ofIFN-γ from cloned and polyclonal islet-reactive NOD CD8� Tthrough effects on antigen-presenting cells, and that this
mechanism is defective in autoimmune prone animals, thereby cells. Using receptor subtype-specific agonists we identifiedPGE2 receptor subtypes and intracellular signals that mediatefavoring Th1 cell development (13). Subtype-specific effects
of PGE2 were also observed for differentiated Th1 and Th2 the effects PGE2 on IFN-γ release. We also investigated howPGE2 signaling can be overcome and how it is integratedcells. PGE2 was shown to inhibit IL-2 and IFN-γ production in
both short- and long-term clonal Th1 T cells, while IL-4 with other signals that act on T cells. Significant differencesbetween NOD and BALB/c mice in the time-dependent TCR–production by Th2 T cells was not inhibited and IL-5 was
slightly enhanced (14,15). Although this differential effect was PGE2 signal integration may point to a disease-promotingabnormality in NOD mice.complicated by the fact that it was dependent on the presence
of exogenously added IL-2 (15), it implies that Th1 cells aremore sensitive to the inhibitory effect of PGE2 than Th2 cells.
MethodsSince Th1 cells and their cytokines are thought to promotedevelopment of IDDM, whereas Th2 cells are thought to be
Antibodies and reagentsprotective (5) or neutral (4), these data suggest that PGE2may play a role in disease development that has until now PGE2, 8-bromo-cAMP, avidin–horseradish peroxidase, o-
phenylenediamine (OPD), murine rIL-2 and murine rIFN-not been recognized. A similar dichotomy of T cell phenotypesis thought to exist in CD8� T cells (16). We and others have γ were obtained from Sigma (St Louis, MD). Misoprostol,
sulprostone and a PGE2 enzyme immunoassay kit wereproposed that CD8� T cells are an important source of IFN-γduring the development of IDDM (17,18, see also 4). In obtained from Cayman Chemicals (Ann Arbor, MI). Sp-cAMPS
and 19(R)OH-PGE2 were obtained from Biomol (Plymouthcontrast to the considerable amount of information that isavailable on the influence of PGE2 on CD4� T cell function, Meeting, PA). Murine rIL-7 was purchased from Life Tech-
nologies (Gaithersburg, MD). C18 columns were obtainedlittle is known as to how PGE2 affects CD8� T cells. In previousstudies we have shown that IL-2-driven proliferation of CTLL-2 from Waters (Milford, MA). NS-398 (36) was purchased from
Calbiochem (La Jolla, CA). Indo-1-AM was obtained fromcells, a cell line derived from CD8� T cells, could be inhibitedby PGE2 (19). However, the role of PGE2 in the regulation of Molecular Probes (Eugene, OR).
TCX6310 cells were kindly provided by Dr F. MelchersTCR-mediated CD8� T cell responses such as release ofIFN-γ is not known. (Basel Institute for Immunology). Hybridomas H57-597 (anti-
αβTCR), GK1.5 (anti-CD4) and 3.155 (anti-CD8) were pur-PGE2 mediates a broad range of physiological effects indifferent tissues. The diversity of PGE2 effects can be attrib- chased from ATCC (Rockville, MD), and hybridoma YCD3-1

PGE2 and IFN-γ release from NOD CD8� T cell clones 853
(anti-CD3ε) was a kind gift from Dr C. A. Janeway, Jr (Yale with anti-CD3 mAb every 3–4 weeks. T cells (1–5�105) werecultured with mitomycin C-treated spleen cells (5�106) in 5University). Anti-αβTCR mAb was purified from hybridoma
culture supernatant using GammaBind Plus Sepharose ml of TCM in the presence of YCD3-1 (anti-CD3ε mAb) cellculture supernatant diluted 1:20. After 48 h, T cells were(Pharmacia Biotech, Piscataway, NJ) columns. Anti-CD3, anti-
CD4 and anti-CD8 mAb were used in the form of diluted washed and treated with 40 U/ml IL-2 and 10 ng/ml IL-7. Twodays later cells were fed once more with the same medium,hybridoma supernatant. FITC–goat F(ab�)2 anti-rat IgG anti-
body was obtained from Caltag (South San Francisco, CA). and after that every 3–4 days with TCM alone or, during everyother feeding cycle, with TCM supplemented with IL-2 andThe tissue culture medium (TCM) used for cell culture and
for all experiments was based on Click’s medium (Irvine IL-7 (10 U/ml and 10 ng/ml respectively). In most experimentsT cell clones were used 15 days post-stimulation. DeadScientific, Santa Ana, CA) which was supplemented with
4 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin cells were removed using Lymphocyte Separation Medium(Organon Teknika, Durham, NC). In some experiments T cells(Life Technologies), 40 µM β-mercaptoethanol (Sigma) and
10% FBS (Hyclone, Logan, UT). For routine T cell culture we were stimulated with anti-CD3 mAb and splenocytes to studythe influence of full T cell activation versus return to a restingused the supernatant of TCX6310 cells (37) as a source of
IL-2. state on the sensitivity to PGE2. In these experiments the anti-CD3 mAb was removed after 48 h by washing the cells as
Mice above. The stimulatory effect of any remaining anti-TCRantibody bound to Fc receptors of splenocytes does notNOD, B10.BR and BALB/c mice were obtained from the
Jackson Laboratory (Bar Harbor, ME), and were bred and last beyond 48 h, because most antigen-presenting cells insplenocytes die during this time. After this 48 h stimulationmaintained in the USC animal facility under pathogen-free
conditions. The spontaneous incidence of diabetes in our period, T cells were maintained in culture without stimulationfor various time intervals and then used for experiments.NOD colony reaches 65–70% in female mice by 20 weeks of
age and diabetes usually commences by 13 weeks of age.Polyclonal CD8� T cellsFor experiments, 8- to 12-week-old mice were used.Spleen cells (1–2�106) were stimulated with anti-CD3 mAb
PGE2 production in islets and splenocytes, and cultured as above. To purify CD8� Tcells, cultured spleen cells were incubated with anti-CD4Islets were prepared by collagenase digestion as described
elsewhere (38). Islets were cultured for 16 h and the number mAb (supernatant from hybridoma GK1.5, diluted 1 in 3) for30 min on ice, washed and treated with diluted (1:10) pooledof mononuclear cells emanating from them was monitored.
Islets were then divided into two groups: those that did not rabbit complement (ICN, Irvine, CA) for 45 min at 37°C. Theresulting cell preparation was analyzed by flow cytometry andshow any sign of infiltration and those that were severely
infiltrated as indicated by having none or �10 cells around was found to contain �95% of CD8� T cells.them respectively. Between 25 and 50 islets were cultured in
Flow cytometry250 µl of TCM for 40 h and PGE2 release was measured bycompetitive ELISA (Cayman PGE2 enzyme immunoassay kit). For the detection of CD8-expressing cells, polyclonal cells
were stained with anti-CD8 mAb 3.155 and with FITC–F(ab�)2PGE2 was extracted from the culture supernatant as describedby Powell (39). Briefly, two parts (typically 400 µl) of ethanol/ goat anti-rat IgG as second-step antibody. Cells were ana-
lyzed using a FACStar flow cytometer (Becton Dickinson, Sanwater (10/90, v/v) were added to one part (typically 200 µl)of the supernatant to be assayed. The resulting mixture was Jose, CA).adjusted to pH 3.0 (0.1 N HCl) and applied to a pre-activated
Stimulation of IFN-γ release from T cells3 ml C18 column. Column pre-activation and extraction ofPGE2 were performed following the Cayman enzyme immuno- Unless otherwise indicated, IFN-γ release from T cells was
stimulated using anti-αβTCR mAb H57-597 immobilized inassay kit protocol.tissue culture plates (Falcon; 96-well, flat-bottom plate coated
Islet-reactive polyclonal CTL and clones with 1 µg/ml mAb in PBS at 4°C overnight). Clonal T cellswere seeded in antibody-coated plates at a density of 1�104The generation of islet-reactive CTL and cloning of CD8� T
cells is described in detail elsewhere (18). Briefly, spleen cells/well and polyclonal CD8� T cells at 3�104 cells/well.Immediately after seeding the cells, PGE2, PGE2 agonists orcells from newly diabetic female NOD mice were cultured
with NOD islets of Langerhans in the presence of IFN-γ (10 cAMP analogs were added to the culture at the concentrationsindicated in the figure legends. After 24 h, supernatantU/ml), IL-2 (10 U/ml) and IL-7 (10 ng/ml) for 5 days. T cell
clusters surrounding disintegrating islets were picked using was collected and stored at –80°C. PGE2, misoprostol andsulprostone were dissolved in DMSO and dilutions were madea pipette and pooled. Cells were re-stimulated with islets or
the β cell line NIT-1 (40) 4 times at 7–10 day intervals. in TCM. The final concentration of DMSO in culture was lessthen 0.18% and at that concentration did not have anyPolyclonal CTL obtained this way potently destroyed islets
and were used for cloning. For cloning by limiting dilution, T influence on IFN-γ release from T cells (data not shown).cells were stimulated using anti-CD3 mAb and mitomycin
IFN-γ assayC-treated NOD spleen cells as described (18). In this studywe used CD8� T cell clone 8D8, which was one of 18 clones The concentration of IFN-γ in the culture supernatant was
measured by sandwich ELISA using paired anti-cytokine mAbobtained.For routine maintenance, CTL and 8D8 cells were stimulated (PharMingen, San Diego, CA), following protocols recom-

854 PGE2 and IFN-γ release from NOD CD8� T cell clones
mended by the manufacturer. Primary mAb was immobilizedin Immulon-4 plates (Dynatech, Boston, MA). The sensitivityof the assay was 1 U/ml (67 pg/ml).
Measurement of intracellular free calcium in P815 cells
P815 cells, known to express EP3 receptors coupled to anincrease of intracellular free calcium (41), were used as acontrol for the efficacy of the EP3 receptor agonist sulprostone.To load P815 cells with Ca2�-indicator, they were incubatedwith 3 mM of Indo-1-AM for 30 min at 37°C. The cells werethen washed and kept in the form of a pellet at 4°C untilassayed. The cell pellet was resuspended in serum-free tissueculture medium prior to the assay. Indo-1-AM-loaded cells(5�106) were stimulated with PGE2, sulprostone (10–6 M) orincubated with digitonin to determine maximal intracellular
Fig. 1. PGE2 release from islets. Islets from B10.BR mice (B10.BR),free calcium. The fluorescence intensity was recorded onlineNOD islets without any sign of infiltration (NOD) or severely infiltratedwith a fluorescence spectrophotometer.NOD islets (NODinf) (see Methods) were cultured for 40 h in thepresence or absence of lipopolysaccharide (10 µg/ml) as indicated.
Measurement of uterine contractions The specific COX-2 inhibitor NS-398 (10 µM) (36) was added asindicated. Data are the mean � SEM of three measurements.Uteri were excised from NOD mice, cut in half and mounted
in a tension transducer. The bath solution was HBSS (LifeTechnologies), kept at 37°C and bubbled with 95% O2/5%CO2. Various concentrations of PGE2 or sulprostone were
Because PGE2 is produced at sites of inflammation, it is likelyadded and tension development was monitored.that inflamed NOD islets produce this mediator. To address
Curve fitting and statistical analysis this question, islets were isolated from pancreata of NODmice and inflamed islets were separated from non-inflamedTheoretical dose–effect curves were fitted to the data asislets based on the number of mononuclear cells emanatingdescribed earlier (19). Briefly, the following expression wasfrom them during an overnight culture (see Methods andused for the concentration-dependent effect of PGE2:18). Figure 1 shows that inflamed islets produce significantamounts of PGE2, in contrast to islets that do not show signskiL
ln(L) � Σn
i�1
α1 with Σn
i�1
αi � 1 of inflammation. The amount of PGE2 produced by non-1 � kiL inflamed islets was comparable to that produced by B10.BRislets and could be increased somewhat by stimulation withln(L) ranges from 1 (full inhibition) to 0 (no inhibition). L (ligand)lipopolysaccharide, but never reached the high levels pro-refers to the concentration of PGE2, n is the number ofduced by inflamed NOD islets. PGE2 production by thesetheoretical binding sites and ki are individual binding con-islets was catalyzed by the inducible form of cyclooxygenasestants of these sites. αi is the relative contribution of site i toas it was inhibited by NS-398, a specific inhibitor of thisthe total inhibitory effect when it is fully occupied by ligand.enzyme.The sum of all these contributions is 1 (i.e. 100%). For fitting
of ln(L) to the experimental data, the sum of square deviations PGE2 inhibits TCR-dependent IFN-γ releaseof all data points from the theoretical curve was set to a
PGE2 produced by inflamed NOD islets is likely to affect theminimum by varying the binding constants ki, as well as αi.function of cells in the inflammatory infiltrate, including CD8�
For each experiment a scaling factor was used that allowedT cells. The following experiments were conducted to investi-the data to be fitted to a curve ranging from 1 to 0. Statisticalgate the effects of PGE2 on islet-reactive CD8� T cells.significance of a second or third binding constant wasThroughout this study, the NOD CD8� T cell clone 8D8 wasassessed by calculating the F ratio and corresponding Pused which had been described earlier (18). This clonevalues due to the introduction of the additional constant asdestroys islets in vitro and releases IFN-γ in response to islets.compared to a simpler model without it (42). P � 0.001 wasBefore studying PGE2 effects on IFN-γ release, it was ofconsidered to be significant.interest to determine the time course and dose range of anti-TCR antibody for triggering IFN-γ secretion from 8D8 cells.
Results Two weeks after expansion with soluble anti-TCR antibodyand splenocytes, 8D8 cells were stimulated with immobilized
Endogenous production of PGE2 in islets anti-TCR antibody in the absence of splenocytes. To do this,8D8 cells were added to tissue culture plates coated withDuring development of diabetes in the NOD mouse, the islets
of Langerhans become infiltrated with mononuclear cells. This varying concentrations of anti-TCR antibody. In contrast tostimulation with anti-TCR antibody and splenocytes, stimula-phenomenon, also known as islet inflammation or insulitis,
exhibits a large degree of heterogeneity at any one time. tion with anti-TCR antibody alone did not cause proliferationof 8D8 cells (data not shown) but effectively triggered IFN-γHeavily inflamed islets and islets undergoing destruction co-
exist with seemingly healthy islets in the same pancreas. release. A concentration of 1 µg/ml of anti-αβTCR antibody
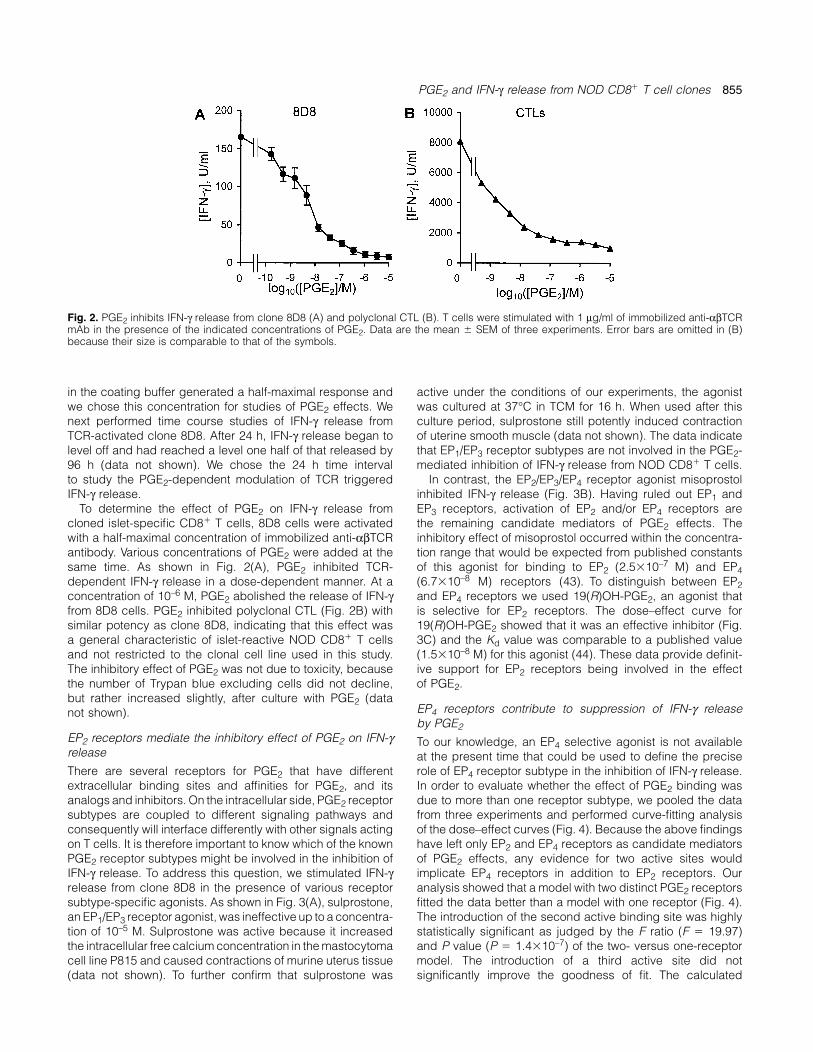
PGE2 and IFN-γ release from NOD CD8� T cell clones 855
Fig. 2. PGE2 inhibits IFN-γ release from clone 8D8 (A) and polyclonal CTL (B). T cells were stimulated with 1 µg/ml of immobilized anti-αβTCRmAb in the presence of the indicated concentrations of PGE2. Data are the mean � SEM of three experiments. Error bars are omitted in (B)because their size is comparable to that of the symbols.
in the coating buffer generated a half-maximal response and active under the conditions of our experiments, the agonistwas cultured at 37°C in TCM for 16 h. When used after thiswe chose this concentration for studies of PGE2 effects. We
next performed time course studies of IFN-γ release from culture period, sulprostone still potently induced contractionof uterine smooth muscle (data not shown). The data indicateTCR-activated clone 8D8. After 24 h, IFN-γ release began to
level off and had reached a level one half of that released by that EP1/EP3 receptor subtypes are not involved in the PGE2-mediated inhibition of IFN-γ release from NOD CD8� T cells.96 h (data not shown). We chose the 24 h time interval
to study the PGE2-dependent modulation of TCR triggered In contrast, the EP2/EP3/EP4 receptor agonist misoprostolinhibited IFN-γ release (Fig. 3B). Having ruled out EP1 andIFN-γ release.
To determine the effect of PGE2 on IFN-γ release from EP3 receptors, activation of EP2 and/or EP4 receptors arethe remaining candidate mediators of PGE2 effects. Thecloned islet-specific CD8� T cells, 8D8 cells were activated
with a half-maximal concentration of immobilized anti-αβTCR inhibitory effect of misoprostol occurred within the concentra-tion range that would be expected from published constantsantibody. Various concentrations of PGE2 were added at the
same time. As shown in Fig. 2(A), PGE2 inhibited TCR- of this agonist for binding to EP2 (2.5�10–7 M) and EP4(6.7�10–8 M) receptors (43). To distinguish between EP2dependent IFN-γ release in a dose-dependent manner. At a
concentration of 10–6 M, PGE2 abolished the release of IFN-γ and EP4 receptors we used 19(R)OH-PGE2, an agonist thatis selective for EP2 receptors. The dose–effect curve forfrom 8D8 cells. PGE2 inhibited polyclonal CTL (Fig. 2B) with
similar potency as clone 8D8, indicating that this effect was 19(R)OH-PGE2 showed that it was an effective inhibitor (Fig.3C) and the Kd value was comparable to a published valuea general characteristic of islet-reactive NOD CD8� T cells
and not restricted to the clonal cell line used in this study. (1.5�10–8 M) for this agonist (44). These data provide definit-ive support for EP2 receptors being involved in the effectThe inhibitory effect of PGE2 was not due to toxicity, because
the number of Trypan blue excluding cells did not decline, of PGE2.but rather increased slightly, after culture with PGE2 (data
EP4 receptors contribute to suppression of IFN-γ releasenot shown).by PGE2
EP2 receptors mediate the inhibitory effect of PGE2 on IFN-γ To our knowledge, an EP4 selective agonist is not availablerelease at the present time that could be used to define the precise
role of EP4 receptor subtype in the inhibition of IFN-γ release.There are several receptors for PGE2 that have differentextracellular binding sites and affinities for PGE2, and its In order to evaluate whether the effect of PGE2 binding was
due to more than one receptor subtype, we pooled the dataanalogs and inhibitors. On the intracellular side, PGE2 receptorsubtypes are coupled to different signaling pathways and from three experiments and performed curve-fitting analysis
of the dose–effect curves (Fig. 4). Because the above findingsconsequently will interface differently with other signals actingon T cells. It is therefore important to know which of the known have left only EP2 and EP4 receptors as candidate mediators
of PGE2 effects, any evidence for two active sites wouldPGE2 receptor subtypes might be involved in the inhibition ofIFN-γ release. To address this question, we stimulated IFN-γ implicate EP4 receptors in addition to EP2 receptors. Our
analysis showed that a model with two distinct PGE2 receptorsrelease from clone 8D8 in the presence of various receptorsubtype-specific agonists. As shown in Fig. 3(A), sulprostone, fitted the data better than a model with one receptor (Fig. 4).
The introduction of the second active binding site was highlyan EP1/EP3 receptor agonist, was ineffective up to a concentra-tion of 10–5 M. Sulprostone was active because it increased statistically significant as judged by the F ratio (F � 19.97)
and P value (P � 1.4�10–7) of the two- versus one-receptorthe intracellular free calcium concentration in the mastocytomacell line P815 and caused contractions of murine uterus tissue model. The introduction of a third active site did not
significantly improve the goodness of fit. The calculated(data not shown). To further confirm that sulprostone was
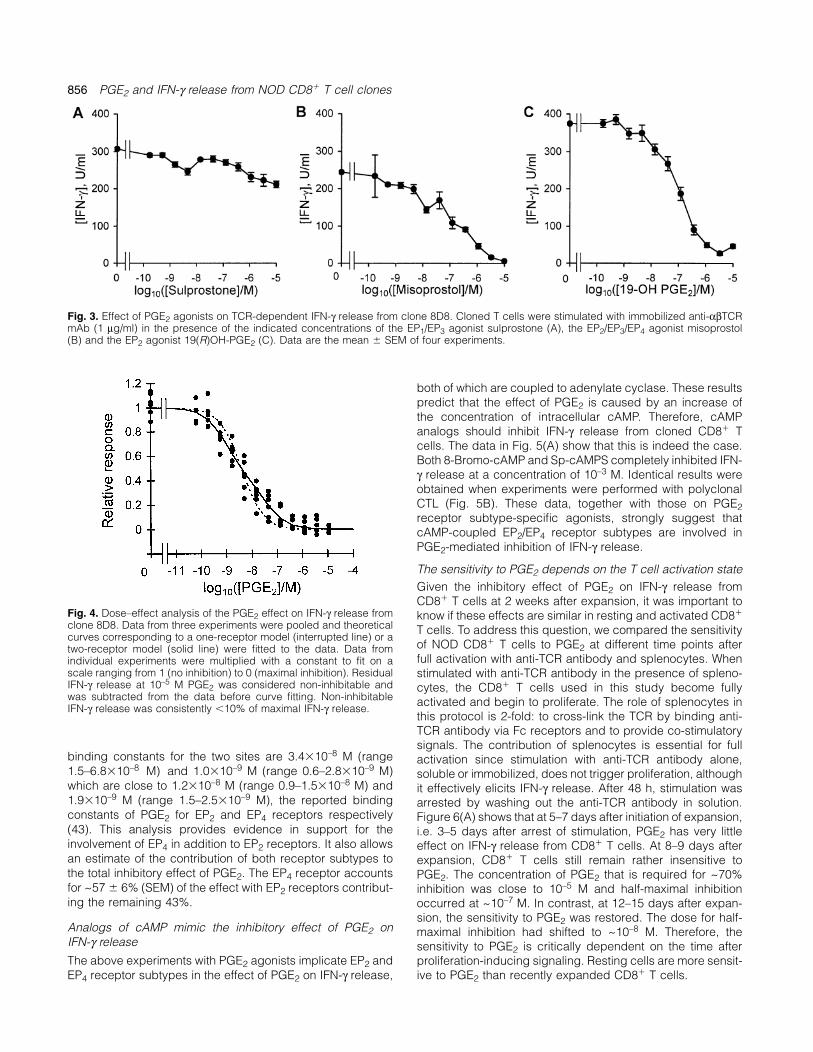
856 PGE2 and IFN-γ release from NOD CD8� T cell clones
Fig. 3. Effect of PGE2 agonists on TCR-dependent IFN-γ release from clone 8D8. Cloned T cells were stimulated with immobilized anti-αβTCRmAb (1 µg/ml) in the presence of the indicated concentrations of the EP1/EP3 agonist sulprostone (A), the EP2/EP3/EP4 agonist misoprostol(B) and the EP2 agonist 19(R)OH-PGE2 (C). Data are the mean � SEM of four experiments.
both of which are coupled to adenylate cyclase. These resultspredict that the effect of PGE2 is caused by an increase ofthe concentration of intracellular cAMP. Therefore, cAMPanalogs should inhibit IFN-γ release from cloned CD8� Tcells. The data in Fig. 5(A) show that this is indeed the case.Both 8-Bromo-cAMP and Sp-cAMPS completely inhibited IFN-γ release at a concentration of 10–3 M. Identical results wereobtained when experiments were performed with polyclonalCTL (Fig. 5B). These data, together with those on PGE2receptor subtype-specific agonists, strongly suggest thatcAMP-coupled EP2/EP4 receptor subtypes are involved inPGE2-mediated inhibition of IFN-γ release.
The sensitivity to PGE2 depends on the T cell activation stateGiven the inhibitory effect of PGE2 on IFN-γ release fromCD8� T cells at 2 weeks after expansion, it was important to
Fig. 4. Dose–effect analysis of the PGE2 effect on IFN-γ release from know if these effects are similar in resting and activated CD8�
clone 8D8. Data from three experiments were pooled and theoretical T cells. To address this question, we compared the sensitivitycurves corresponding to a one-receptor model (interrupted line) or a
of NOD CD8� T cells to PGE2 at different time points aftertwo-receptor model (solid line) were fitted to the data. Data fromfull activation with anti-TCR antibody and splenocytes. Whenindividual experiments were multiplied with a constant to fit on a
scale ranging from 1 (no inhibition) to 0 (maximal inhibition). Residual stimulated with anti-TCR antibody in the presence of spleno-IFN-γ release at 10–5 M PGE2 was considered non-inhibitable and cytes, the CD8� T cells used in this study become fullywas subtracted from the data before curve fitting. Non-inhibitable activated and begin to proliferate. The role of splenocytes inIFN-γ release was consistently �10% of maximal IFN-γ release.
this protocol is 2-fold: to cross-link the TCR by binding anti-TCR antibody via Fc receptors and to provide co-stimulatorysignals. The contribution of splenocytes is essential for full
binding constants for the two sites are 3.4�10–8 M (range activation since stimulation with anti-TCR antibody alone,1.5–6.8�10–8 M) and 1.0�10–9 M (range 0.6–2.8�10–9 M) soluble or immobilized, does not trigger proliferation, althoughwhich are close to 1.2�10–8 M (range 0.9–1.5�10–8 M) and it effectively elicits IFN-γ release. After 48 h, stimulation was1.9�10–9 M (range 1.5–2.5�10–9 M), the reported binding arrested by washing out the anti-TCR antibody in solution.constants of PGE2 for EP2 and EP4 receptors respectively Figure 6(A) shows that at 5–7 days after initiation of expansion,(43). This analysis provides evidence in support for the i.e. 3–5 days after arrest of stimulation, PGE2 has very littleinvolvement of EP4 in addition to EP2 receptors. It also allows effect on IFN-γ release from CD8� T cells. At 8–9 days afteran estimate of the contribution of both receptor subtypes to expansion, CD8� T cells still remain rather insensitive tothe total inhibitory effect of PGE2. The EP4 receptor accounts PGE2. The concentration of PGE2 that is required for ~70%for ~57 � 6% (SEM) of the effect with EP2 receptors contribut- inhibition was close to 10–5 M and half-maximal inhibitioning the remaining 43%. occurred at ~10–7 M. In contrast, at 12–15 days after expan-
sion, the sensitivity to PGE2 was restored. The dose for half-Analogs of cAMP mimic the inhibitory effect of PGE2 on maximal inhibition had shifted to ~10–8 M. Therefore, theIFN-γ release sensitivity to PGE2 is critically dependent on the time afterThe above experiments with PGE2 agonists implicate EP2 and proliferation-inducing signaling. Resting cells are more sensit-
ive to PGE2 than recently expanded CD8� T cells.EP4 receptor subtypes in the effect of PGE2 on IFN-γ release,
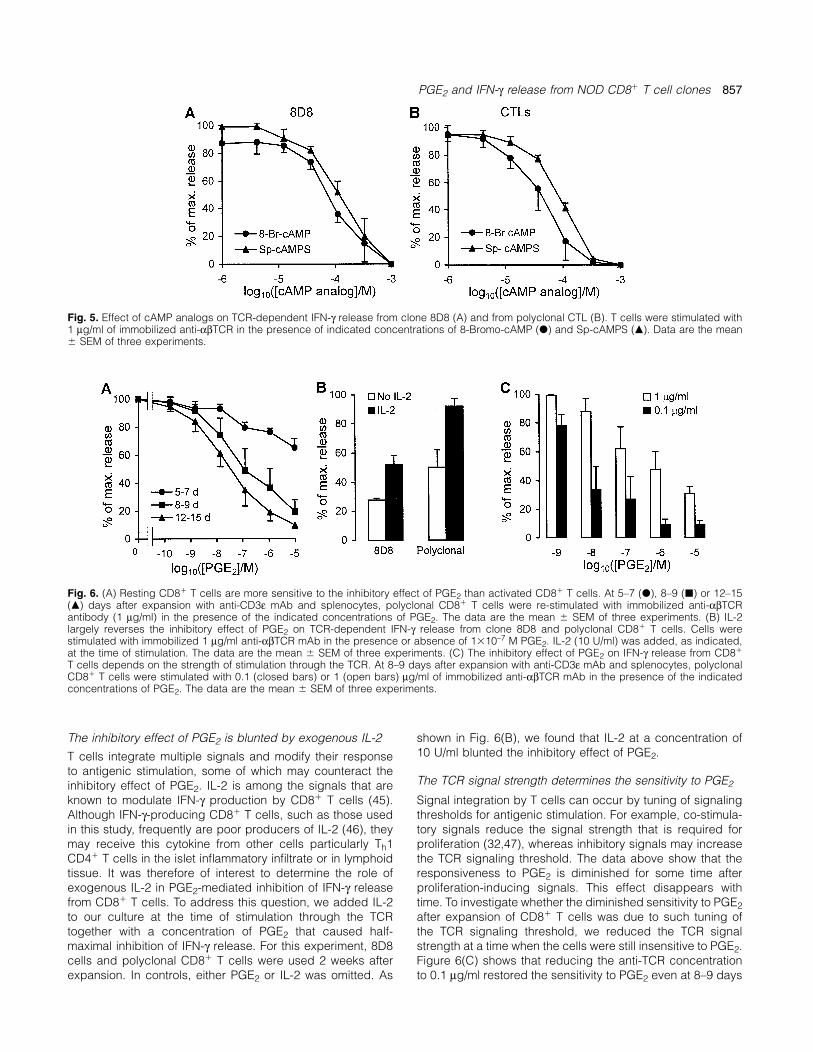
PGE2 and IFN-γ release from NOD CD8� T cell clones 857
Fig. 5. Effect of cAMP analogs on TCR-dependent IFN-γ release from clone 8D8 (A) and from polyclonal CTL (B). T cells were stimulated with1 µg/ml of immobilized anti-αβTCR in the presence of indicated concentrations of 8-Bromo-cAMP (d) and Sp-cAMPS (m). Data are the mean� SEM of three experiments.
Fig. 6. (A) Resting CD8� T cells are more sensitive to the inhibitory effect of PGE2 than activated CD8� T cells. At 5–7 (d), 8–9 (j) or 12–15(m) days after expansion with anti-CD3ε mAb and splenocytes, polyclonal CD8� T cells were re-stimulated with immobilized anti-αβTCRantibody (1 µg/ml) in the presence of the indicated concentrations of PGE2. The data are the mean � SEM of three experiments. (B) IL-2largely reverses the inhibitory effect of PGE2 on TCR-dependent IFN-γ release from clone 8D8 and polyclonal CD8� T cells. Cells werestimulated with immobilized 1 µg/ml anti-αβTCR mAb in the presence or absence of 1�10–7 M PGE2. IL-2 (10 U/ml) was added, as indicated,at the time of stimulation. The data are the mean � SEM of three experiments. (C) The inhibitory effect of PGE2 on IFN-γ release from CD8�
T cells depends on the strength of stimulation through the TCR. At 8–9 days after expansion with anti-CD3ε mAb and splenocytes, polyclonalCD8� T cells were stimulated with 0.1 (closed bars) or 1 (open bars) µg/ml of immobilized anti-αβTCR mAb in the presence of the indicatedconcentrations of PGE2. The data are the mean � SEM of three experiments.
The inhibitory effect of PGE2 is blunted by exogenous IL-2 shown in Fig. 6(B), we found that IL-2 at a concentration of10 U/ml blunted the inhibitory effect of PGE2.T cells integrate multiple signals and modify their response
to antigenic stimulation, some of which may counteract theThe TCR signal strength determines the sensitivity to PGE2inhibitory effect of PGE2. IL-2 is among the signals that are
known to modulate IFN-γ production by CD8� T cells (45). Signal integration by T cells can occur by tuning of signalingthresholds for antigenic stimulation. For example, co-stimula-Although IFN-γ-producing CD8� T cells, such as those used
in this study, frequently are poor producers of IL-2 (46), they tory signals reduce the signal strength that is required forproliferation (32,47), whereas inhibitory signals may increasemay receive this cytokine from other cells particularly Th1
CD4� T cells in the islet inflammatory infiltrate or in lymphoid the TCR signaling threshold. The data above show that theresponsiveness to PGE2 is diminished for some time aftertissue. It was therefore of interest to determine the role of
exogenous IL-2 in PGE2-mediated inhibition of IFN-γ release proliferation-inducing signals. This effect disappears withtime. To investigate whether the diminished sensitivity to PGE2from CD8� T cells. To address this question, we added IL-2
to our culture at the time of stimulation through the TCR after expansion of CD8� T cells was due to such tuning ofthe TCR signaling threshold, we reduced the TCR signaltogether with a concentration of PGE2 that caused half-
maximal inhibition of IFN-γ release. For this experiment, 8D8 strength at a time when the cells were still insensitive to PGE2.Figure 6(C) shows that reducing the anti-TCR concentrationcells and polyclonal CD8� T cells were used 2 weeks after
expansion. In controls, either PGE2 or IL-2 was omitted. As to 0.1 µg/ml restored the sensitivity to PGE2 even at 8–9 days
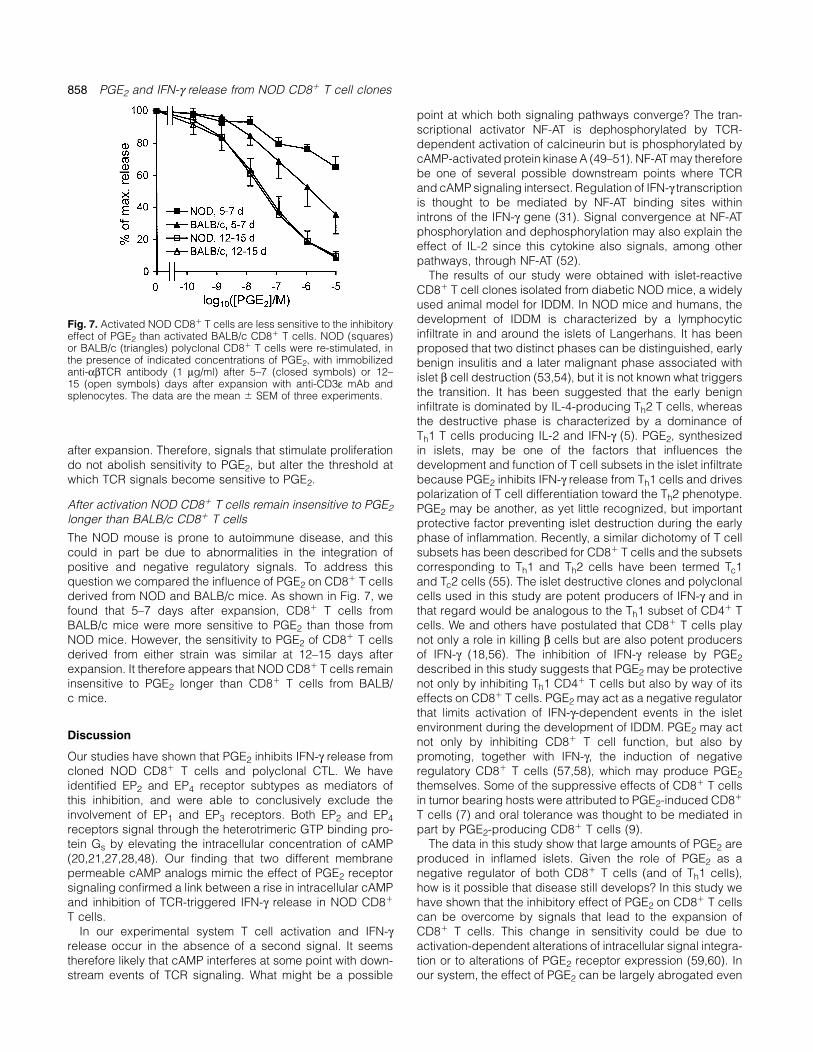
858 PGE2 and IFN-γ release from NOD CD8� T cell clones
point at which both signaling pathways converge? The tran-scriptional activator NF-AT is dephosphorylated by TCR-dependent activation of calcineurin but is phosphorylated bycAMP-activated protein kinase A (49–51). NF-AT may thereforebe one of several possible downstream points where TCRand cAMP signaling intersect. Regulation of IFN-γ transcriptionis thought to be mediated by NF-AT binding sites withinintrons of the IFN-γ gene (31). Signal convergence at NF-ATphosphorylation and dephosphorylation may also explain theeffect of IL-2 since this cytokine also signals, among otherpathways, through NF-AT (52).
The results of our study were obtained with islet-reactiveCD8� T cell clones isolated from diabetic NOD mice, a widelyused animal model for IDDM. In NOD mice and humans, thedevelopment of IDDM is characterized by a lymphocyticFig. 7. Activated NOD CD8� T cells are less sensitive to the inhibitoryinfiltrate in and around the islets of Langerhans. It has beeneffect of PGE2 than activated BALB/c CD8� T cells. NOD (squares)
or BALB/c (triangles) polyclonal CD8� T cells were re-stimulated, in proposed that two distinct phases can be distinguished, earlythe presence of indicated concentrations of PGE2, with immobilized benign insulitis and a later malignant phase associated withanti-αβTCR antibody (1 µg/ml) after 5–7 (closed symbols) or 12– islet β cell destruction (53,54), but it is not known what triggers15 (open symbols) days after expansion with anti-CD3ε mAb and
the transition. It has been suggested that the early benignsplenocytes. The data are the mean � SEM of three experiments.infiltrate is dominated by IL-4-producing Th2 T cells, whereasthe destructive phase is characterized by a dominance ofTh1 T cells producing IL-2 and IFN-γ (5). PGE2, synthesizedin islets, may be one of the factors that influences theafter expansion. Therefore, signals that stimulate proliferation
do not abolish sensitivity to PGE2, but alter the threshold at development and function of T cell subsets in the islet infiltratebecause PGE2 inhibits IFN-γ release from Th1 cells and driveswhich TCR signals become sensitive to PGE2.polarization of T cell differentiation toward the Th2 phenotype.
After activation NOD CD8� T cells remain insensitive to PGE2 PGE2 may be another, as yet little recognized, but importantlonger than BALB/c CD8� T cells protective factor preventing islet destruction during the early
phase of inflammation. Recently, a similar dichotomy of T cellThe NOD mouse is prone to autoimmune disease, and thiscould in part be due to abnormalities in the integration of subsets has been described for CD8� T cells and the subsets
corresponding to Th1 and Th2 cells have been termed Tc1positive and negative regulatory signals. To address thisquestion we compared the influence of PGE2 on CD8� T cells and Tc2 cells (55). The islet destructive clones and polyclonal
cells used in this study are potent producers of IFN-γ and inderived from NOD and BALB/c mice. As shown in Fig. 7, wefound that 5–7 days after expansion, CD8� T cells from that regard would be analogous to the Th1 subset of CD4� T
cells. We and others have postulated that CD8� T cells playBALB/c mice were more sensitive to PGE2 than those fromNOD mice. However, the sensitivity to PGE2 of CD8� T cells not only a role in killing β cells but are also potent producers
of IFN-γ (18,56). The inhibition of IFN-γ release by PGE2derived from either strain was similar at 12–15 days afterexpansion. It therefore appears that NOD CD8� T cells remain described in this study suggests that PGE2 may be protective
not only by inhibiting Th1 CD4� T cells but also by way of itsinsensitive to PGE2 longer than CD8� T cells from BALB/c mice. effects on CD8� T cells. PGE2 may act as a negative regulator
that limits activation of IFN-γ-dependent events in the isletenvironment during the development of IDDM. PGE2 may act
Discussion not only by inhibiting CD8� T cell function, but also bypromoting, together with IFN-γ, the induction of negativeOur studies have shown that PGE2 inhibits IFN-γ release from
cloned NOD CD8� T cells and polyclonal CTL. We have regulatory CD8� T cells (57,58), which may produce PGE2themselves. Some of the suppressive effects of CD8� T cellsidentified EP2 and EP4 receptor subtypes as mediators of
this inhibition, and were able to conclusively exclude the in tumor bearing hosts were attributed to PGE2-induced CD8�
T cells (7) and oral tolerance was thought to be mediated ininvolvement of EP1 and EP3 receptors. Both EP2 and EP4receptors signal through the heterotrimeric GTP binding pro- part by PGE2-producing CD8� T cells (9).
The data in this study show that large amounts of PGE2 aretein Gs by elevating the intracellular concentration of cAMP(20,21,27,28,48). Our finding that two different membrane produced in inflamed islets. Given the role of PGE2 as a
negative regulator of both CD8� T cells (and of Th1 cells),permeable cAMP analogs mimic the effect of PGE2 receptorsignaling confirmed a link between a rise in intracellular cAMP how is it possible that disease still develops? In this study we
have shown that the inhibitory effect of PGE2 on CD8� T cellsand inhibition of TCR-triggered IFN-γ release in NOD CD8�
T cells. can be overcome by signals that lead to the expansion ofCD8� T cells. This change in sensitivity could be due toIn our experimental system T cell activation and IFN-γ
release occur in the absence of a second signal. It seems activation-dependent alterations of intracellular signal integra-tion or to alterations of PGE2 receptor expression (59,60). Intherefore likely that cAMP interferes at some point with down-
stream events of TCR signaling. What might be a possible our system, the effect of PGE2 can be largely abrogated even

PGE2 and IFN-γ release from NOD CD8� T cell clones 859
Benefield, J. and Prechel, M. M. 1996. Mechanisms of immunein resting cells by IL-2, a Th1-derived pro-inflammatory factor.suppression in patients with head and neck cancer: influence onIt is likely that PGE2 acts in concert with other protectivethe immune infiltrate of the cancer. Int. J. Cancer 67:333.
factors such as IL-4 and transforming growth factor-β, and it 9 Khoury, S. J., Hancock, W. W. and Weiner, H. L. 1992. Oralwill be important to uncover the time-dependent interplay tolerance to myelin basic protein and natural recovery from
experimental autoimmune encephalomyelitis are associated withbetween several protective and disease-promoting factors fordownregulation of inflammatory cytokines and differentialan understanding of the transition from benign to malignantupregulation of transforming growth factor beta, interleukin 4, andinsulitis and progression to IDDM. In this study, we report prostaglandin E expression in the brain. J. Exp. Med. 176:1355.
significant differences between the time-dependent sensitivity 10 Rabinovitch, A., Baquerizo, H. and Sumoski, W. 1990. Cytotoxiceffects of cytokines on islet beta-cells: evidence for involvementof NOD and BALB/c CD8� T cells. After full activation,of eicosanoids. Endocrinology 126:67.NOD cells remain insensitive to PGE2 for a longer time
11 Hancock, W. W., Polanski, M., Zhang, J., Blogg, N. and Weiner,than BALB/c CD8� T cells. Whether this difference isH. L. 1995. Suppression of insulitis in non-obese diabetic (NOD)
significant in regard to disease development remains to be mice by oral insulin administration is associated with selectivedetermined. These data do suggest, however, that PGE2 expression of interleukin-4 and -10, transforming growth factor-
beta, and prostaglandin-E. Am. J. Pathol. 147:1193.may be a less effective negative regulator of CD8� T cells12 Katamura, K., Shintaku, N., Yamauchi, Y., Fukui, T., Ohshima, Y.,in NOD mice than in BALB/c mice.
Mayumi, M. and Furusho, K. 1995. Prostaglandin E2 at primingThe putative role of PGE2 has implications with regard to of naive CD4� T cells inhibits acquisition of ability to producethe use of non-steroidal anti-inflammatory drugs in persons IFN-gamma and IL-2, but not IL-4 and IL-5. J. Immunol. 155:4604.at risk of developing type I diabetes. Many of the currently 13 Rothe, H. and Kolb, H. 1998. The APC1 concept of type I diabetes.
Autoimmunity 27:179.used non-steroidal anti-inflammatory drugs block the increase14 Betz, M. and Fox, B. S. 1991. Prostaglandin E2 inhibits productionof PGE2 synthesis during inflammation and may prevent
of Th1 lymphokines but not of Th2 lymphokines. J. Immunol.important immunomodulating actions of PGE2 (61). Since 146:108.PGE2 is inhibitory for CD4� and CD8� T cell effector function, 15 Hilkens, C. M., Vermeulen, H., van Neerven, R. J., Snijdewint,
F. G., Wierenga, E. A. and Kapsenberg, M. L. 1995. Differentialreducing PGE2 levels over a long period of time could leadmodulation of T helper type 1 (Th1) and T helper type 2 (Th2)to an imbalance of positive and negative control of T cellcytokine secretion by prostaglandin E2 critically depends onfunction and adverse effects associated with this lack ofinterleukin-2. Eur. J. Immunol. 25:59.
control. 16 Cerwenka, A., Carter, L. L., Reome, J. B., Swain, S. L. and Dutton,R. W. 1998. In vivo persistence of CD8 polarized T cell subsetsproducing type 1 or type 2 cytokines. J. Immunol. 161:97.
Acknowledgments 17 Rabinovitch, A., Suarez Pinzon, W. L., Sorensen, O., Bleackley,R. C. and Power, R. F. 1995. IFN-gamma gene expressionThis work was supported by National Institutes of Health grant in pancreatic islet-infiltrating mononuclear cells correlates withDK49717. We thank Drs. Gunther Dennert and Sarah Hamm-Alvarez autoimmune diabetes in nonobese diabetic mice. J. Immunol.for critically reading this manuscript, and Donald MacDougall for 154:4874.making the tension transducer available to us. 18 Gurlo, T., Kawamura, K. and von Grafenstein, H. 1999. Role ofinflammatory infiltrate in activation and effector function of clonedislet reactive nonobese diabetic CD8� T cells: involvement of a
Abbreviations nitric oxide-dependent pathway. J. Immunol. 163:5770.19 Gurlo, T., Huang, W. W. and von Grafenstein, H. 1998. PGE2IDDM insulin-dependent diabetes mellitus
inhibits IL-2 and IL-4-dependent proliferation of CTLL-2 and HT2NOD non-obese diabeticcells. Cytokine 10:265.OPD o-phenylenediamine
20 Narumiya, S. 1997. Molecular diversity of prostanoid receptors;PG prostaglandinsubtypes and isoforms of prostaglandin E receptor. Adv. Exp.TCM tissue culture mediumMed. Biol. 400A:207.
21 Nishigaki, N., Negishi, M., Honda, A., Sugimoto, Y., Namba, T.,Narumiya, S. and Ichikawa, A. 1995. Identification of prostaglandinReferences E receptor ‘EP2’ cloned from mastocytoma cells EP4 subtype.FEBS Lett. 364:339.1 Atkinson, M. A. and Maclaren, N. K. 1994. The pathogenesis of
22 Negishi, M., Sugimoto, Y., Namba, T., Irie, A., Narumiya, S. andinsulin-dependent diabetes mellitus. N. Engl J. Med. 331:1428.Ichikawa, A. 1995. Signal transductions of three isoforms of2 Bach, J. F. and Mathis, D. 1997. Organizers, The NOD mouse,mouse prostaglandin E receptor EP3 subtype. Adv. Prostaglandin70th Forum in Immunol. Res. Immunol. 148:285.Thromboxane Leukotriene Res. 23:255.3 Bach, J. F., Chatenoud, L., Herbelin, A., Gombert, J. M. and
23 Negishi, M., Irie, A., Sugimoto, Y., Namba, T. and Ichikawa, A.Carnaud, C. 1997. Autoimmune diabetes: how many steps for1995. Selective coupling of prostaglandin E receptor EP3D to Gione disease? Res. Immunol. 148:332.and Gs through interaction of alpha-carboxylic acid of agonist4 Katz, J. D., Benoist, C. and Mathis, D. 1995. T helper cell subsetsand arginine residue of seventh transmembrane domain. J. Biol.in insulin-dependent diabetes. Science 268:1185.Chem. 270:16122.5 Rabinovitch, A. 1998. An update on cytokines in the pathogenesis
24 Paliogianni, F. and Boumpas, D. T. 1996. Prostaglandin E2 inhibitsof insulin-dependent diabetes mellitus. Diabetes Metab. Rev.the nuclear transcription of the human interleukin 2, but not the14:129.Il-4, gene in human T cells by targeting transcription factors AP-6 Takano, M., Nishimura, H., Kimura, Y., Washizu, J., Mokuno, Y.,1 and NF-AT. Cell. Immunol. 171:95.Nimura, Y. and Yoshikai, Y. 1998. Prostaglandin E2 protects
25 Ichikawa, A., Sugimoto, Y. and Negishi, M. 1996. Molecularagainst liver injury after Escherichia coli infection but hampersaspects of the structures and functions of the prostaglandin Ethe resolution of the infection in mice. J. Immunol. 161:3019.receptors. J. Lipid Mediators Cell Signal. 14:83.7 Walker, T. M., Yurochko, A. D., Burger, C. J. and Elgert, K. D.
26 Katoh, H., Watabe, A., Sugimoto, Y., Ichikawa, A. and Negishi, M.1992. Cytokines and suppressor macrophages cause tumor-1995. Characterization of the signal transduction of prostaglandinbearing host CD8� T cells to suppress recognition of allogeneicE receptor EP1 subtype in cDNA-transfected Chinese hamsterand syngeneic MHC class II molecules. J. Leuk. Biol. 52:661.
8 Young, M. R., Wright, M. A., Lozano, Y., Matthews, J. P., ovary cells. Biochim. Biophys. Acta 1244:41.

860 PGE2 and IFN-γ release from NOD CD8� T cell clones
27 Coleman, R. A., Smith, W. L. and Narumiya, S. 1994. International L. S. 1993. Identification of 19 (R)-OH prostaglandin E2 as aselective prostanoid EP2-receptor agonist. Prostaglandins 46:371.Union of Pharmacology classification of prostanoid receptors:
45 Su, H. C., Cousens, L. P., Fast, L. D., Slifka, M. K., Bungiro, R.properties, distribution, and structure of the receptors and theirD., Ahmed, R. and Biron, C. A. 1998. CD4� and CD8� T cellsubtypes. Pharmacol. Rev. 46:205.interactions in IFN-gamma and IL-4 responses to viral infections:28 Negishi, M., Sugimoto, Y. and Ichikawa, A. 1995. Prostaglandinrequirements for IL-2. J. Immunol. 160:5007.E receptors. J. Lipid Mediators Cell Signal. 12:379.
46 Caruso, A., Licenziati, S., Morelli, D., Fiorentini, S., Ricotta, D.,29 Paliogianni, F., Kincaid, R. L. and Boumpas, D. T. 1993.Malacarne, F., Sfondrini, L. and Balsari, A. 1998. Segregation ofProstaglandin E2 and other cyclic AMP elevating agents inhibittype 1 cytokine production in human peripheral bloodinterleukin 2 gene transcription by counteracting calcineurin-lymphocytes: phenotypic differences between IFN-gamma anddependent pathways. J. Exp. Med. 178:1813.IL-2-producing cells in the CD8� T cell subset. Eur. J. Immunol.30 Campbell, P. M., Pimm, J., Ramassar, V. and Halloran, P. F.28:3630.1996. Identification of a calcium-inducible, cyclosporine sensitive
47 Viola, A. and Lanzavecchia, A. 1996. T cell activation determinedelement in the IFN-gamma promoter that is a potential NFATby T cell receptor number and tunable thresholds [see comments].binding site. Transplantation 61:933.Science 273:104.31 Sica, A., Dorman, L., iggiano, V., Cippitelli, M., Ghosh, P., Rice, N.
48 Narumiya, S. 1994. Prostanoid receptors. Structure, function, andand Young, H. A. 1997. Interaction of NF-kappaB and NFAT withdistribution. Ann. NY Acad. Sci. 744:126.the interferon-gamma promoter. J. Biol. Chem. 272:30412.
49 Li, L., Yee, C. and Beavo, J. A. 1999. CD3- and CD28-dependent32 Dubey, C., Croft, M. and Swain, S. L. 1996. Naive and effectorinduction of PDE7 required for T cell activation. Science 283:848.CD4 T cells differ in their requirements for T cell receptor versus
50 Tsuruta, L., Lee, H. J., Masuda, E. S., Koyano-Nakagawa, N.,costimulatory signals. J. Immunol. 157:3280.Arai, N., Arai, K. and Yokota, T. 1995. Cyclic AMP inhibits33 Powell, J. D., Ragheb, J. A., Kitagawa-Sakakida, S. and Schwartz,expression of the IL-2 gene through the nuclear factor of activatedR. H. 1998. Molecular regulation of interleukin-2 expression byT cells (NF-AT) site, and transfection of NF-AT cDNAs abrogatesCD28 co-stimulation and anergy. Immunol. Rev. 165:287.the sensitivity of EL-4 cells to cyclic AMP. J. Immunol. 154:5255.34 Pawelec, G., Sayers, T. and Busch, F. W. 1989. Selective inhibition
51 Sheridan, C. M. and Gardner, P. 1998. Paper presented at Gordonof interleukin 2 but not of interferon-gamma, tumour necrosisResearch Conference. In Gordon Research Conference. Kimballfactor-alpha or granulocyte/macrophage colony stimulating factorUnion, NH.secretion by human helper T cell clones after antigen-driven
52 Gesbert, F., Delespine-Carmagnat, M. and Bertoglio, J. 1998.tolerisation. Immunol. Lett. 20:187.Recent advances in the understanding of interleukin-2 signal35 O Hehir, R. E., Yssel, H., Verma, S., de Vries, J. E., Spits, H. andtransduction. J. Clin. Immunol. 18:307.Lamb, J. R. 1991. Clonal analysis of differential lymphokine
53 Andre, I., Gonzalez, A., Wang, B., Katz, J., Benoist, C. andproduction in peptide and superantigen induced T cell anergy.Mathis, D. 1996. Checkpoints in the progression of autoimmuneInt. Immunol. 3:819.disease: lessons from diabetes models. Proc. Natl Acad. Sci.36 Futaki, N., Takahashi, S., Yokoyama, M., Arai, I., Higuchi, S.USA 93:2260.and Otomo, S. 1994. NS-398, a new anti-inflammatory agent,
54 Gazda, L. S., Charlton, B. and Lafferty, K. J. 1997. Diabetesselectively inhibits prostaglandin G/H synthase/cyclooxygenaseresults from a late change in the autoimmune response of NOD(COX-2) activity in vitro. Prostaglandins 47:55.mice. J. Autoimmun. 10:261.37 Karasuyama, H. and Melchers, F. 1988. Establishment of mouse
55 Croft, M., Carter, L., Swain, S. L. and Dutton, R. W. 1994.cell lines which constitutively produce large quantities of Generation of polarized antigen-specific CD8 effectorinterleukin 2, 3, 4 or 5, using modified cDNA expression vectors. populations: reciprocal action of interleukin (IL)-4 and IL-12Eur. J. Immunol. 18:97. in promoting type 2 versus type 1 cytokine profiles. J. Exp.38 Agostino, M., Prowse, S. J. and Lafferty, K. J. 1982. Resistance Med. 180:1715.
of established islet allografts to rejection by antibody and 56 Shimizu, J., Kanagawa, O. and Unanue, E. R. 1993. Presentationcomplement. Aust. J. Exp. Biol. Med. Sci. 60:219. of beta-cell antigens to CD4� and CD8� T cells of non-obese
39 Powell, W. S. 1982. Rapid extraction of arachidonic acid diabetic mice. J. Immunol. 151:1723.metabolites from biological samples using octadecylsilyl silica. 57 ElMasry, M. N., Fox, E. J. and Rich, R. R. 1987. Sequential effectsMethods Enzymol. 86:467. of prostaglandins and interferon-gamma on differentiation of
40 Hamaguchi, K., Gaskins, H. R. and Leiter, E. H. 1991. NIT-1, a CD8� suppressor cells. J. Immunol. 139:688.pancreatic beta-cell line established from a transgenic NOD/Lt 58 ElMasry, M. N. and Rich, R. R. 1989. Prostaglandin E2 selectivelymouse. Diabetes 40:842. increases interferon gamma receptor expression on human CD8�
41 Sugimoto, Y., Namba, T., Honda, A., Hayashi, Y., Negishi, M., lymphocytes. J. Clin. Invest. 83:1436.Ichikawa, A. and Narumiya, S. 1992. Cloning and expression of 59 Bloom, D., Jabrane-Ferrat, N., Zeng, L., Wu, A., Li, L., Lo, D.,a cDNA for mouse prostaglandin E receptor EP3 subtype. J. Biol. Turck, C. W., An, S. and Goetzl, E. J. 1999. ProstaglandinChem. 267:6463. E2 enhancement of interferon-gamma production by antigen-
42 Maxwell, S. E. and Delaney, H. D. 1990. Designing Experiments stimulated type 1 helper T cells. Cell. Immunol. 194:21.and Analyzing Data: A Model Comparison Perspective. 60 Holter, W., Spiegel, A. M., Howard, B. H., Weber, S. and Brann,Wadsworth, Belmont, CA. M. R. 1991. Expression of GTP-binding proteins and prostaglandin
43 Kiriyama, M., Ushikubi, F., Kobayashi, T., Hirata, M., Sugimoto, Y. E2 receptors during human T cell activation. Cell. Immunol.and Narumiya, S. 1997. Ligand binding specificities of the eight 134:287.types and subtypes of the mouse prostanoid receptors expressed 61 Roper, R. L. and Phipps, R. P. 1994. Prostaglandin E2 regulationin Chinese hamster ovary cells. Br. J. Pharmacol. 122:217. of the immune response. Adv. Prostaglandin. Thromboxane.
Leukotriene Res. 22:101.44 Woodward, D. F., Protzman, C. E., Krauss, A. H. and Williams,





























