Embed Size (px)
Citation preview

ARTICLES
Self-assembly of metal–polymer analoguesof amphiphilic triblock copolymers
ZHIHONG NIE1, DANIELE FAVA1, EUGENIA KUMACHEVA1,2,3*, SHAN ZOU1, GILBERT C. WALKER1
AND MICHAEL RUBINSTEIN4
1Department of Chemistry, University of Toronto, 80 St George Street, Toronto, Ontario M5S 3H6, Canada2Department of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College Street, Toronto, Ontario M5S 3E5, Canada3Institute of Biomaterials & Biomedical Engineering, University of Toronto, 4 Taddle Creek Road, Toronto, Ontario M5S 3G9, Canada4Department of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599-3290, USA*e-mail: [email protected]
Published online: 8 July 2007; doi:10.1038/nmat1954
Organized arrays of anisotropic nanoparticles show electronic and optical properties that originate from the coupling ofshape-dependent properties of the individual nanorods. The organization of nanorods in a controllable and predictable way providesa route to the fabrication of new materials and functional devices. So far, significant progress has been achieved in the self-assemblyof nanorod arrays, yet the realization of a range of different structures requires changing the surface chemistry of the nanoparticles.We organized metal nanorods in structures with varying geometries by using a striking analogy between amphiphilic ABA triblockcopolymers and the hydrophilic nanorods tethered with hydrophobic polymer chains at both ends. The self-assembly was tunable andreversible and it was achieved solely by changing the solvent quality for the constituent blocks. This approach provides a new route tothe organization of anisotropic nanoparticles by using the strategies that are established for the self-assembly of block copolymers.
One-dimensional nanoparticles have a range of shape-dependentproperties, for example, absorption, photoluminescence, surface-enhanced Raman cross-sections and conductivity1–4. Theseproperties determine potential applications of discrete anisotropicnanostructures in optical and electronic devices1–3,5,6, sensingand imaging7,8, biodiagnostics8 and drug and gene delivery9,10.Self-assembly or assisted assembly of nanorods into organizedarrays allows the realization of their collective properties thatarise from the coupling of the optical and electronic properties ofthe neighbouring individual nanoparticles. Further scientific andtechnological advances in the application of nanorods in functionaldevices depend on the ability to organize them in complex one- ormulti-dimensional functional architectures.
So far, a few techniques exist that allow the organizationof nanorods in two orientation modes, namely, end-to-end andside-to-side nanorod assembly. These techniques use chemicalor physical binding of ligands that stabilize nanoparticles11,12,biorecognition7,13, the self-assembly of nanorods using DNA,carbon nanotube or block copolymer templates14–16 or externalfields17,18 and interactions between segmented polymer–metal rodsin selective solvents19. Most of these methods suffer from limitedcontrol over the geometry and complexity of the assemblies.Generally, to obtain varying self-assembled structures of nanorods,their surface chemistry has to be changed by replacing ormodifying ligands.
Here, we report a new ‘block copolymer’ paradigm forthe self-assembly of nanorods. As an exemplary system, weused gold nanorods whose self-assembly allows for the controlof the plasmonic properties of the nanoparticles1,2,4,12. Weconsidered a hydrophilic gold nanorod tethered with a hydrophobic
homopolymer at both ends as an amphiphile resembling a‘pom-pom’ ABA triblock copolymer and used the ability of blockcopolymers to organize in a variety of thermodynamically andkinetically controlled structures20,21 owing to the segregation ofthe constituent blocks. We assumed that the different architecture,flexibility and dimensions of the nanorods (comparable withthe size of block copolymers) would lead to their assemblyin structures not observed previously for the micrometre-longmetal–polymer rods19.
By using one of the major strategies used for the self-assemblyof block copolymers—the selective variation of the quality of thesolvent for the constituent blocks—we achieved reversible andtunable self-assembly of the nanorods in dilute solutions andproduced a range of nanorod structures without changing theligands. A ‘block copolymer’ paradigm allowed us to conceptualizeand predict the organization of the nanorods in specific structures.We expect that this general and straightforward approach canbe applied to the assembly of other anisotropic nanoparticles,leading to unique optical and electronic properties of the self-organized structures, for example, tunable plasmonic wavelengthsand resonating effects1,3.
Figure 1 shows the architecture of the model ‘block copolymer’.The central rigid hydrophilic block is formed by a gold nanorodcovered with a double layer of cetyl trimethylammonium bromide1
(CTAB). The flexible hydrophobic side blocks are formed by thiol-terminated polystyrene molecules strongly anchored to the {111}facets at the ends of the nanorods1,13. The ‘block copolymer’structural unit in Fig. 1 is best described as a ‘pom-pom’ ABA blockcopolymer22 with at least several polystyrene molecules graftedto each end of the nanorods. By selectively changing the quality
nature materials VOL 6 AUGUST 2007 www.nature.com/naturematerials 609
© 2007 Nature Publishing Group

ARTICLES
CTAB-coated gold nanorod
Polystyrene
Polystyrene
a
b
c
de
Figure 1 Self-assembly of polymer-tethered gold nanorods in selective solvents. An amphiphilic gold nanorod carrying a double layer of CTAB along the longitudinal side(the {100} facet) and polystyrene molecules grafted to both ends. a–e, SEM images of the self-assembled nanorod structures: rings (a) and chains (b) self-assembled in thedimethyl formamide/water mixture at water contents of 6 and 20 wt%, respectively, side-to-side aggregated bundles of nanorods (c) and nanospheres (d) self-organized inthe tetrahydrofuran/water mixture at water contents of 6 and 20 wt%, respectively, and bundled nanorod chains obtained in the ternary dimethyl formamide/tetrahydrofuran/water mixture at a weight ratio of liquids 42.5:42.5:15 (e). The scale bars are 100 nm. The insets show corresponding schematic diagrams of thenanorod assemblies.
of the solvent for the stabilizing molecules—polystyrene andCTAB—we assembled the nanorods in rings, nanochains, bundles,nanospheres and bundled nanochains, which are all shown inFig. 1, along with schematic diagrams of the nanorod assemblyshown in the corresponding insets.
Gold nanorods with a mean diameter of 8.0 nm and a lengthof 42 nm were synthesized using the procedure reported byNikoobakht and El-Sayed23. Preferential binding of CTAB alongthe {100} facet of the longitudinal side of the nanorods left theirends (the {111} faces) deprived of CTAB and allowed for thebinding of thiol-terminated polystyrene (Mn = 12,000) to the endsof the nanorods1,13. From now on, these amphiphilic nanorodsare referred to as ‘triblocks’. Polystyrene molecules grafted to theends of the nanorods formed two brush ‘crowns’ surroundingthe ends of the nanorods and a part of the longitudinal side ofthe nanorods24 and stabilized the nanorods in organic solvents.Here, we used solutions of triblocks in dimethyl formamide (DMF)and tetrahydrofuran (THF). These solvents were chosen for thefollowing reasons. Both DMF and THF are good solvents for thepolystyrene blocks: the values of the second virial coefficient, A2,are 3.5 × 10−4 mol cm3 g−2 and 9.0 × 10−4 mol cm3 g−2 for DMFand THF, respectively (equivalent to Flory–Huggins interactionparameters of 0.46 and 0.4, respectively)25–27. However, the CTAB-coated nanorods without polystyrene grafting exhibited a goodsolubility in DMF and poor solubility in THF.
The self-assembly process was triggered by adding water toa solution of triblocks in an organic solvent, thereby reducingthe solubility of the polystyrene blocks. The self-organizationof triblocks in DMF/water and THF/water mixtures wasmonitored with ultraviolet–visible spectroscopy and in dynamiclight-scattering experiments (see Supplementary Information,Figs S1,S3) and examined by imaging the self-assembled structuresof triblocks on mica, gold-coated borosilicate glass and carbon-coated transmission electron microscopy (TEM) grids. We did
not observe a notable effect of the substrate on the self-assemblyprocess. Control experiments conducted at different solventevaporation rates did not significantly change the self-assemblyof pom-pom species. The self-assembled structures were stablefor at least several weeks in water/organic mixtures and inwater (following the dialysis of solutions of triblocks againstdeionized water).
The scanning electron microscopy (SEM) images in Fig. 1a,bshow typical mesoscopic architectures obtained by adding a varyingamount of water, CW, to the solution of amphiphilic species inDMF. With the addition of water, the mixture became a poorsolvent for the polystyrene blocks but remained a good solventfor the hydrophilic CTAB-stabilized metal blocks. As a result, thetriblocks organized in chains following the end-to-end binding ofthe polystyrene-tethered ends. We followed the evolution of chainsin the DMF/water mixture by tracing the shift in the nanorodlongitudinal plasmonic band: the formation of chains resulted ina red-shift of the plasmonic band, in comparison with individualnanorods4 (see Supplementary Information, Fig. S3). The assemblyof the nanorods into nanochains started immediately after theaddition of water, and it was completed in several hours.
At equilibrium, the aggregation number of triblocks in thelinear chains was in the range from 10 to 100, increasing with theamount of water added to the solution. The average aggregationnumber, 〈N〉, was determined by the surface energy recovered whenpairs of polystyrene blocks of different triblocks associated witheach other to form chains as 〈N〉 = 2∗(c∗ exp(α))1/2, where c is thedimensionless concentration of triblocks and α is the ‘bond’ energyin units of kT (ref. 28). As the quality of the solvent became worse,the energy gain on attachment of two polystyrene ends increasedand so did the number of ‘triblocks’ per chain.
In contrast to DMF, THF did not favourably interact withCTAB-coated metal blocks. With the addition of water, aTHF/water mixture became a better solvent for CTAB-coated
610 nature materials VOL 6 AUGUST 2007 www.nature.com/naturematerials
© 2007 Nature Publishing Group
ARTICLES
10
20
30
DR (nm)
Yiel
d of
ring
s (%
)
CW (%)
L / Lp = 3.6
L / Lp = 3.8
L / Lp = 9.8L / Lp ≈ 1.0
0
40
%
0 200 400 600 8000
15
30
45
0 5 10 15 20 25
a b c
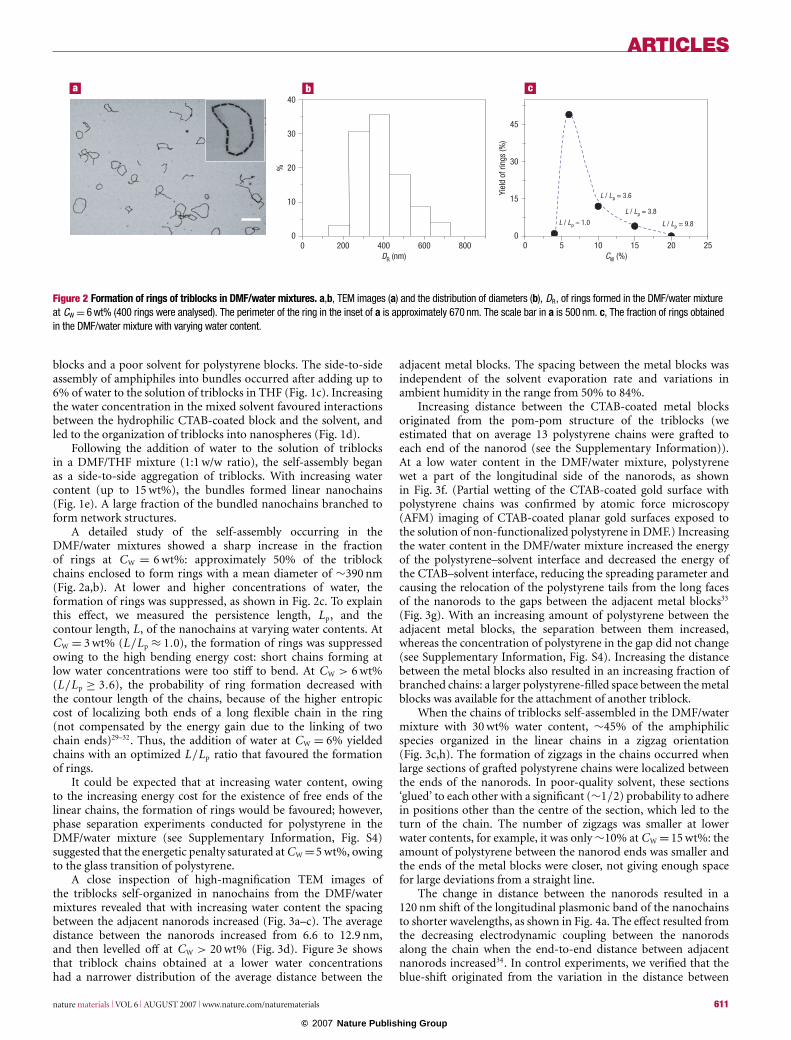
Figure 2 Formation of rings of triblocks in DMF/water mixtures. a,b, TEM images (a) and the distribution of diameters (b), DR, of rings formed in the DMF/water mixtureat CW = 6 wt% (400 rings were analysed). The perimeter of the ring in the inset of a is approximately 670 nm. The scale bar in a is 500 nm. c, The fraction of rings obtainedin the DMF/water mixture with varying water content.
blocks and a poor solvent for polystyrene blocks. The side-to-sideassembly of amphiphiles into bundles occurred after adding up to6% of water to the solution of triblocks in THF (Fig. 1c). Increasingthe water concentration in the mixed solvent favoured interactionsbetween the hydrophilic CTAB-coated block and the solvent, andled to the organization of triblocks into nanospheres (Fig. 1d).
Following the addition of water to the solution of triblocksin a DMF/THF mixture (1:1 w/w ratio), the self-assembly beganas a side-to-side aggregation of triblocks. With increasing watercontent (up to 15 wt%), the bundles formed linear nanochains(Fig. 1e). A large fraction of the bundled nanochains branched toform network structures.
A detailed study of the self-assembly occurring in theDMF/water mixtures showed a sharp increase in the fractionof rings at CW = 6 wt%: approximately 50% of the triblockchains enclosed to form rings with a mean diameter of ∼390 nm(Fig. 2a,b). At lower and higher concentrations of water, theformation of rings was suppressed, as shown in Fig. 2c. To explainthis effect, we measured the persistence length, Lp, and thecontour length, L, of the nanochains at varying water contents. AtCW = 3 wt% (L/Lp ≈ 1.0), the formation of rings was suppressedowing to the high bending energy cost: short chains forming atlow water concentrations were too stiff to bend. At CW > 6 wt%(L/Lp ≥ 3.6), the probability of ring formation decreased withthe contour length of the chains, because of the higher entropiccost of localizing both ends of a long flexible chain in the ring(not compensated by the energy gain due to the linking of twochain ends)29–32. Thus, the addition of water at CW = 6% yieldedchains with an optimized L/Lp ratio that favoured the formationof rings.
It could be expected that at increasing water content, owingto the increasing energy cost for the existence of free ends of thelinear chains, the formation of rings would be favoured; however,phase separation experiments conducted for polystyrene in theDMF/water mixture (see Supplementary Information, Fig. S4)suggested that the energetic penalty saturated at CW =5 wt%, owingto the glass transition of polystyrene.
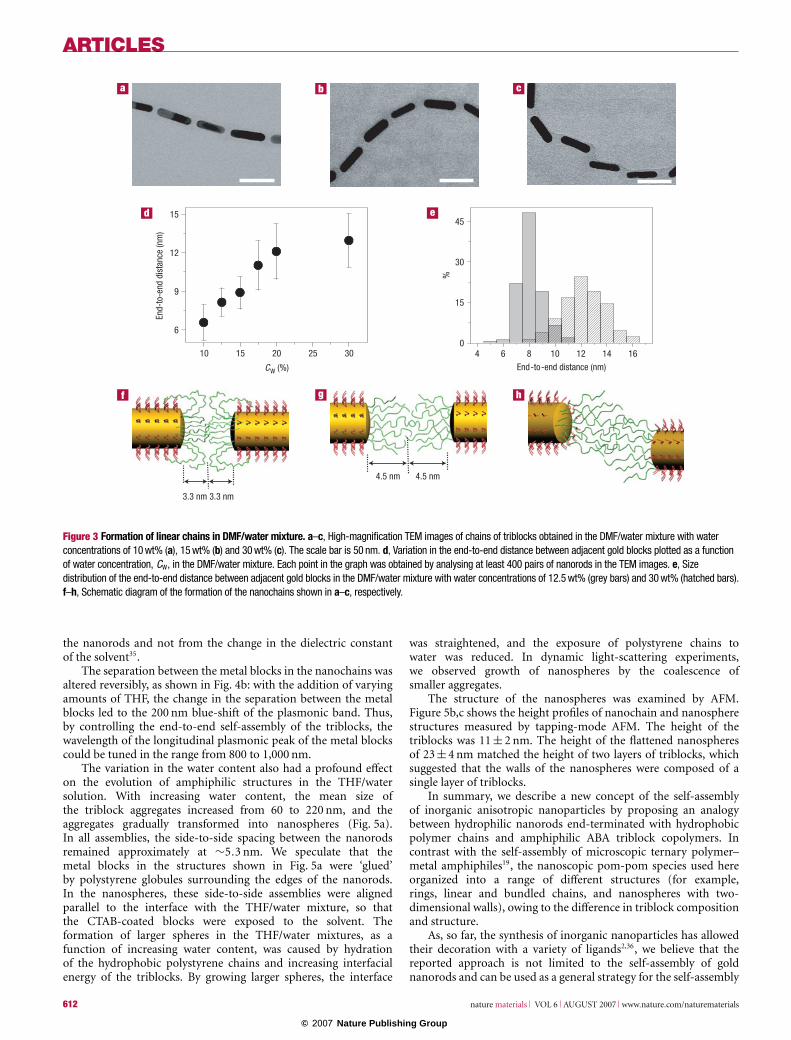
A close inspection of high-magnification TEM images ofthe triblocks self-organized in nanochains from the DMF/watermixtures revealed that with increasing water content the spacingbetween the adjacent nanorods increased (Fig. 3a–c). The averagedistance between the nanorods increased from 6.6 to 12.9 nm,and then levelled off at CW > 20 wt% (Fig. 3d). Figure 3e showsthat triblock chains obtained at a lower water concentrationshad a narrower distribution of the average distance between the
adjacent metal blocks. The spacing between the metal blocks wasindependent of the solvent evaporation rate and variations inambient humidity in the range from 50% to 84%.
Increasing distance between the CTAB-coated metal blocksoriginated from the pom-pom structure of the triblocks (weestimated that on average 13 polystyrene chains were grafted toeach end of the nanorod (see the Supplementary Information)).At a low water content in the DMF/water mixture, polystyrenewet a part of the longitudinal side of the nanorods, as shownin Fig. 3f. (Partial wetting of the CTAB-coated gold surface withpolystyrene chains was confirmed by atomic force microscopy(AFM) imaging of CTAB-coated planar gold surfaces exposed tothe solution of non-functionalized polystyrene in DMF.) Increasingthe water content in the DMF/water mixture increased the energyof the polystyrene–solvent interface and decreased the energy ofthe CTAB–solvent interface, reducing the spreading parameter andcausing the relocation of the polystyrene tails from the long facesof the nanorods to the gaps between the adjacent metal blocks33
(Fig. 3g). With an increasing amount of polystyrene between theadjacent metal blocks, the separation between them increased,whereas the concentration of polystyrene in the gap did not change(see Supplementary Information, Fig. S4). Increasing the distancebetween the metal blocks also resulted in an increasing fraction ofbranched chains: a larger polystyrene-filled space between the metalblocks was available for the attachment of another triblock.
When the chains of triblocks self-assembled in the DMF/watermixture with 30 wt% water content, ∼45% of the amphiphilicspecies organized in the linear chains in a zigzag orientation(Fig. 3c,h). The formation of zigzags in the chains occurred whenlarge sections of grafted polystyrene chains were localized betweenthe ends of the nanorods. In poor-quality solvent, these sections‘glued’ to each other with a significant (∼1/2) probability to adherein positions other than the centre of the section, which led to theturn of the chain. The number of zigzags was smaller at lowerwater contents, for example, it was only ∼10% at CW = 15 wt%: theamount of polystyrene between the nanorod ends was smaller andthe ends of the metal blocks were closer, not giving enough spacefor large deviations from a straight line.
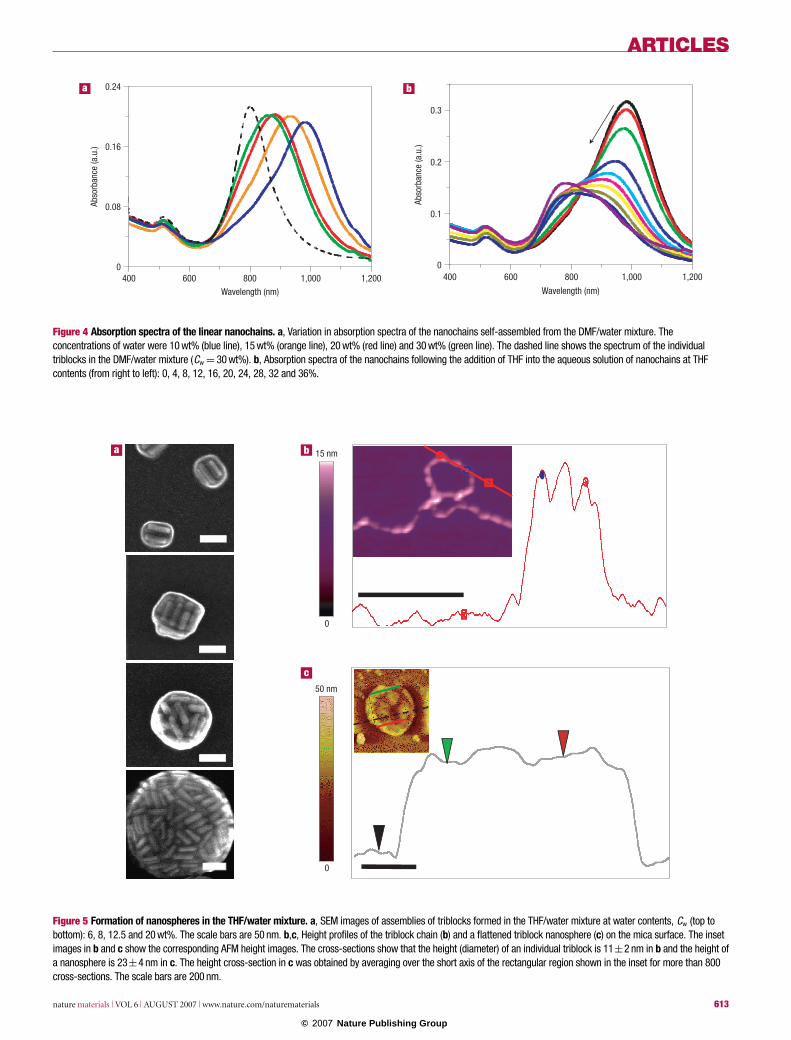
The change in distance between the nanorods resulted in a120 nm shift of the longitudinal plasmonic band of the nanochainsto shorter wavelengths, as shown in Fig. 4a. The effect resulted fromthe decreasing electrodynamic coupling between the nanorodsalong the chain when the end-to-end distance between adjacentnanorods increased34. In control experiments, we verified that theblue-shift originated from the variation in the distance between
nature materials VOL 6 AUGUST 2007 www.nature.com/naturematerials 611
© 2007 Nature Publishing Group
ARTICLES
3.3 nm 3.3 nm
End-
to-e
nd d
ista
nce
(nm
)
4.5 nm 4.5 nm
%
End-to-end distance (nm)
6
9
12
15
15 20 25
CW (%)
30100
15
30
45
4 6 8 10 12 14 16
a b
d e
c
f g h
Figure 3 Formation of linear chains in DMF/water mixture. a–c, High-magnification TEM images of chains of triblocks obtained in the DMF/water mixture with waterconcentrations of 10 wt% (a), 15 wt% (b) and 30 wt% (c). The scale bar is 50 nm. d, Variation in the end-to-end distance between adjacent gold blocks plotted as a functionof water concentration, CW, in the DMF/water mixture. Each point in the graph was obtained by analysing at least 400 pairs of nanorods in the TEM images. e, Sizedistribution of the end-to-end distance between adjacent gold blocks in the DMF/water mixture with water concentrations of 12.5 wt% (grey bars) and 30 wt% (hatched bars).f–h, Schematic diagram of the formation of the nanochains shown in a–c, respectively.
the nanorods and not from the change in the dielectric constantof the solvent35.
The separation between the metal blocks in the nanochains wasaltered reversibly, as shown in Fig. 4b: with the addition of varyingamounts of THF, the change in the separation between the metalblocks led to the 200 nm blue-shift of the plasmonic band. Thus,by controlling the end-to-end self-assembly of the triblocks, thewavelength of the longitudinal plasmonic peak of the metal blockscould be tuned in the range from 800 to 1,000 nm.
The variation in the water content also had a profound effecton the evolution of amphiphilic structures in the THF/watersolution. With increasing water content, the mean size ofthe triblock aggregates increased from 60 to 220 nm, and theaggregates gradually transformed into nanospheres (Fig. 5a).In all assemblies, the side-to-side spacing between the nanorodsremained approximately at ∼5.3 nm. We speculate that themetal blocks in the structures shown in Fig. 5a were ‘glued’by polystyrene globules surrounding the edges of the nanorods.In the nanospheres, these side-to-side assemblies were alignedparallel to the interface with the THF/water mixture, so thatthe CTAB-coated blocks were exposed to the solvent. Theformation of larger spheres in the THF/water mixtures, as afunction of increasing water content, was caused by hydrationof the hydrophobic polystyrene chains and increasing interfacialenergy of the triblocks. By growing larger spheres, the interface
was straightened, and the exposure of polystyrene chains towater was reduced. In dynamic light-scattering experiments,we observed growth of nanospheres by the coalescence ofsmaller aggregates.
The structure of the nanospheres was examined by AFM.Figure 5b,c shows the height profiles of nanochain and nanospherestructures measured by tapping-mode AFM. The height of thetriblocks was 11 ± 2 nm. The height of the flattened nanospheresof 23 ± 4 nm matched the height of two layers of triblocks, whichsuggested that the walls of the nanospheres were composed of asingle layer of triblocks.
In summary, we describe a new concept of the self-assemblyof inorganic anisotropic nanoparticles by proposing an analogybetween hydrophilic nanorods end-terminated with hydrophobicpolymer chains and amphiphilic ABA triblock copolymers. Incontrast with the self-assembly of microscopic ternary polymer–metal amphiphiles19, the nanoscopic pom-pom species used hereorganized into a range of different structures (for example,rings, linear and bundled chains, and nanospheres with two-dimensional walls), owing to the difference in triblock compositionand structure.
As, so far, the synthesis of inorganic nanoparticles has allowedtheir decoration with a variety of ligands2,36, we believe that thereported approach is not limited to the self-assembly of goldnanorods and can be used as a general strategy for the self-assembly
612 nature materials VOL 6 AUGUST 2007 www.nature.com/naturematerials
© 2007 Nature Publishing Group
ARTICLES
600 800 1,000
Wavelength (nm)
400 1,200
Abso
rban
ce (a
.u.)
600 800 1,000
Wavelength (nm)
400 1,200
Abso
rban
ce (a
.u.)
a b
0.08
0.16
0
0.24
0
0.2
0.1
0.3
Figure 4 Absorption spectra of the linear nanochains. a, Variation in absorption spectra of the nanochains self-assembled from the DMF/water mixture. Theconcentrations of water were 10 wt% (blue line), 15 wt% (orange line), 20 wt% (red line) and 30 wt% (green line). The dashed line shows the spectrum of the individualtriblocks in the DMF/water mixture (Cw = 30 wt%). b, Absorption spectra of the nanochains following the addition of THF into the aqueous solution of nanochains at THFcontents (from right to left): 0, 4, 8, 12, 16, 20, 24, 28, 32 and 36%.
15 nm
50 nm
0
b
c
a
0
Figure 5 Formation of nanospheres in the THF/water mixture. a, SEM images of assemblies of triblocks formed in the THF/water mixture at water contents, Cw (top tobottom): 6, 8, 12.5 and 20 wt%. The scale bars are 50 nm. b,c, Height profiles of the triblock chain (b) and a flattened triblock nanosphere (c) on the mica surface. The insetimages in b and c show the corresponding AFM height images. The cross-sections show that the height (diameter) of an individual triblock is 11±2 nm in b and the height ofa nanosphere is 23±4 nm in c. The height cross-section in c was obtained by averaging over the short axis of the rectangular region shown in the inset for more than 800cross-sections. The scale bars are 200 nm.
nature materials VOL 6 AUGUST 2007 www.nature.com/naturematerials 613
© 2007 Nature Publishing Group

ARTICLES
of one-dimensional nanoparticles. Furthermore, we believe thatgrafting of block copolymers37,38 or multiple homopolymers36—tothe ends of nanorods—would lead to a new phenomenology inthe self-assembly of nanorods. The strategy described here wouldalso be useful for the assembly of other amphiphilic species, if theloss in entropy of the system owing to their self-organization iscompensated by the gain in enthalpy owing to the self-assembly.
METHODS
Gold nanorods were synthesized following the procedure developed byNikoobakht and El-Sayed23. The synthesis was scaled up to obtain a 100 mldispersion of the nanorods. Seed nanoparticles were prepared by reducingHAuCl4 (0.12 ml, 5 mM) mixed with 2.5 ml of an aqueous 0.2 M solution ofCTAB and sodium borohydride (0.5 ml, 10 mM) in ice-cold water. For thepreparation of a growth solution, 50 ml of a 0.2 M CTAB solution was mixedwith 5 ml of an aqueous 5 mM solution of HAuCl4, 2.8 ml of an aqueous 4 mMAgNO3 solution and 40 ml of water. Following the addition of 1 ml of anaqueous 0.8 M solution of ascorbic acid, the dark yellow solution turnedcolourless. Finally, we added 0.8 ml of a 5-min-aged seed solution ofnanoparticles to the growth solution. The nanorods were purified using three30-min-long centrifugation cycles at 8,000–10,000 r.p.m. (Eppendorf centrifuge5417R). At the end of each centrifugation cycle, the supernatant was removedand the precipitated nanorods were redispersed in deionized water.
Approximately 0.5 ml of the concentrated (∼1.0 mg ml−1) aqueoussolution of nanorods was added to 10 ml of a 0.2 wt% solution ofthiol-terminated polystyrene in THF. The solution was sonicated for 30 minand incubated for at least 24 h. The modified nanorods were purified using four30-min-long centrifugation cycles at 10,000 r.p.m. The supernatant wasremoved and the precipitated nanorods were redispersed in THF or DMF.
Self-assembly of triblocks was carried out at room temperature by addingwater (in a mixture with the corresponding organic solvent) to the solution oftriblocks in THF, DMF or a DMF/THF mixture. The concentration ofnanorods in the original organic solvent was approximately 0.015 mg ml−1. Forexample, to trigger the assembly of triblocks in the DMF solution, we slowlyadded a mixture of water with DMF to this solution. Following the addition ofwater, the sample was incubated at room temperature for at least 24 h. BeforeSEM and TEM imaging, water was added to a concentration of 80 wt%, and/orthe system was dialysed against deionized water, using a 6,000–8,000molecular-weight-cutoff dialysis membrane tubing (Spectrum Laboratories)for 3 days. Before these experiments, we verified that the values of the glasstransition temperature, Tg, of polystyrene in water–organic solvent mixturesare above room temperature and thus the self-assembled structures werevitrified before dilution or dialysis.
SEM, TEM and high-resolution TEM images were obtained using a HitachiHD-2000 STEM. Samples for TEM analysis were prepared by depositing a dropof dilute nanorod solution on a 400 mesh carbon-coated copper grid andallowing the solvent to evaporate. The values of L and Lp for the nanochains oftriblocks were obtained by image analysis of the TEM images.
In the AFM experiments, we dropped ∼0.5 ml of the solution ofself-assembled triblocks onto a freshly cleaved mica surface. The samples wereincubated for about 1 h, then the surface was rinsed with water and dried undervacuum. AFM imaging was carried out in air with the tapping mode using aNanoscope III Dimension 5000 microscope (Veeco Digital Instruments) and anMFP-3D AFM (Asylum Research). The silicon probe cantilevers (MikroMasch,resonance frequencies in the range of 135–190 kHz, free amplitude: 20–25 nm)were used with nominal spring constants of between 3.5 and 12.5 N m−1.
Received 20 March 2007; accepted 5 June 2007; published 8 July 2007.
References1. Murphy, C. J. et al. Anisotropic metal nanoparticles: Synthesis, assembly, and optical applications.
J. Phys. Chem. B 109, 13857–13870 (2005).2. Murphy, C. J., Gole, A. M., Hunyadi, S. E. & Orendorff, C. J. One-dimensional colloidal gold and
silver nanostructures. Inorg. Chem. 45, 7544–7554 (2006).3. Xia, Y. N. et al. One-dimensional nanostructures: Synthesis, characterization, and applications. Adv.
Mater. 15, 353–389 (2003).4. Jain, P. K., Eustis, S. & El-Sayed, M. A. Plasmon coupling in nanorod assemblies: Optical absorption,
discrete dipole approximation simulation, and exciton-coupling model. J. Phys. Chem. B 110,18243–18253 (2006).
5. Hu, J. T. et al. Linearly polarized emission from colloidal semiconductor quantum rods. Science 292,2060–2063 (2001).
6. Huynh, W. U., Dittmer, J. J. & Alivisatos, A. P. Hybrid nanorod-polymer solar cells. Science 295,2425–2427 (2002).
7. Sudeep, P. K., Joseph, S. T. S. & Thomas, K. G. Selective detection of cysteine and glutathione usinggold nanorods. J. Am. Chem. Soc. 127, 6516–6517 (2005).
8. Huang, X. H., El-Sayed, I. H., Qian, W. & El-Sayed, M. A. Cancer cell imaging and photothermaltherapy in the near-infrared region by using gold nanorods. J. Am. Chem. Soc. 128, 2115–2120 (2006).
9. Gorelikov, I., Field, L. M. & Kumacheva, E. Hybrid microgels photoresponsive in the near-infraredspectral range. J. Am. Chem. Soc. 126, 15938–15939 (2004).
10. Salem, A. K., Searson, P. C. & Leong, K. W. Multifunctional nanorods for gene delivery. Nature Mater.2, 668–671 (2003).
11. Thomas, K. G., Barazzouk, S., Ipe, B. I., Joseph, S. T. S. & Kamat, P. V. Uniaxial plasmon couplingthrough longitudinal self-assembly of gold nanorods. J. Phys. Chem. B 108, 13066–13068 (2004).
12. Joseph, S. T. S., Ipe, B. I., Pramod, P. & Thomas, K. G. Gold nanorods to nanochains: Mechanisticinvestigations on their longitudinal assembly using alpha, omega-alkanedithiols and interplasmoncoupling. J. Phys. Chem. B 110, 150–157 (2006).
13. Caswell, K. K., Wilson, J. N., Bunz, U. H. F. & Murphy, C. J. Preferential end-to-end assembly of goldnanorods by biotin-streptavidin connectors. J. Am. Chem. Soc. 125, 13914–13915 (2003).
14. Dujardin, E., Hsin, L. B., Wang, C. R. C. & Mann, S. DNA-driven self-assembly of gold nanorods.Chem. Commun. 14, 1264–1265 (2001).
15. Zhang, Q. L., Gupta, S., Emrick, T. & Russell, T. P. Surface-functionalized CdSe nanorods forassembly in diblock copolymer templates. J. Am. Chem. Soc. 128, 3898–3899 (2006).
16. Correa-Duarte, M. A., Perez-Juste, J., Sanchez-Iglesias, A., Giersig, M. & Liz-Marzan, L. M. AligningAu nanorods by using carbon nanotubes as templates. Angew. Chem. Int. Edn 44, 4375–4378 (2005).
17. Gupta, S., Zhang, Q. L., Emrick, T. & Russell, T. P. “Self-corralling” nanorods under an appliedelectric field. Nano Lett. 6, 2066–2069 (2006).
18. Ryan, K. M., Mastroianni, A., Stancil, K. A., Liu, H. T. & Alivisatos, A. P. Electric-field-assistedassembly of perpendicularly oriented nanorod superlattices. Nano Lett. 6, 1479–1482 (2006).
19. Park, S., Lim, J. H., Chung, S. W. & Mirkin, C. A. Self-assembly of mesoscopic metal-polymeramphiphiles. Science 303, 348–351 (2004).
20. Discher, D. E. & Eisenberg, A. Polymer vesicles. Science 297, 967–973 (2002).21. Yan, X. H., Liu, G. J. & Li, Z. Preparation and phase segregation of block copolymer nanotube
multiblocks. J. Am. Chem. Soc. 126, 10059–10066 (2004).22. Knauss, D. M. & Huang, T. Z. Star-block-linear-block-star triblock (pom-pom) polystyrene by
convergent living anionic polymerization. Macromolecules 35, 2055–2062 (2002).23. Nikoobakht, B. & El-Sayed, M. A. Preparation and growth mechanism of gold nanorods (NRs) using
seed-mediated growth method. Chem. Mater. 15, 1957–1962 (2003).24. Zhulina, E. B., Birshtein, T. M. & Borisov, O. V. Curved polymer and polyelectrolyte brushes beyond
the Daoud-Cotton model. Eur. Phys. J. E 20, 243–256 (2006).25. Wolf, B. A. & Willms, M. M. Measured and calculated solubility of polymers in
mixed-solvents-Co-non-solvency. Makromol. Chem. 179, 2265–2277 (1978).26. Schulz, G. V. & Baumann, H. Thermodynamic properties expansion coefficient and viscosity value of
polystyrene in tetrahydrofuran. Makromol. Chem. 114, 122–138 (1968).27. Rubinstein, M. & Colby, R. H. Polymer Physics (Oxford Univ. Press, Oxford, 2003).28. Israelachvili, J. N. Intermolecular and Surface Forces (Academic, London, 1992).29. Lairez, D., Adam, M., Carton, J. P. & Raspaud, E. Aggregation of telechelic triblock copolymers: From
animals to flowers. Macromolecules 30, 6798–6809 (1997).30. Raspaud, E., Lairez, D., Adam, M. & Carton, J. P. Triblock copolymers in a selective solvent. 1.
Aggregation process in dilute-solution. Macromolecules 27, 2956–2964 (1994).31. Tenbrinke, G. & Hadziioannou, G. Topological constraints and their influence on the properties of
synthetic macromolecular systems. 2. Micelle formation of triblock copolymers. Macromolecules 20,486–489 (1987).
32. Wang, Y. M., Mattice, W. L. & Napper, D. H. Simulation of the self-assembly of symmetrical triblockcopolymers in dilute-solution. Macromolecules 25, 4073–4077 (1992).
33. De Gennes, P. G., Brochard-Wyart, F. & Quere, D. Capillarity and Wetting Phenomena: Drops,Bubbles, Pearls, Waves (Springer, New York, 2004).
34. Su, K. H. et al. Interparticle coupling effects on plasmon resonances of nanogold particles. Nano Lett.3, 1087–1090 (2003).
35. Link, S., Mohamed, M. B. & El-Sayed, M. A. Simulation of the optical absorption spectra of goldnanorods as a function of their aspect ratio and the effect of the medium dielectric constant. J. Phys.Chem. B 103, 3073–3077 (1999).
36. Shan, J. et al. Amphiphilic gold nanoparticles grafted with poly(N-isopropylacrylamide) andpolystyrene. Macromolecules 38, 2918–2926 (2005).
37. Zubarev, E. R., Xu, J., Sayyad, A. & Gibson, J. D. Amphiphilicity-driven organization of nanoparticlesinto discrete assemblies. J. Am. Chem. Soc. 128, 15098–15099 (2006).
38. Zubarev, E. R., Xu, J., Sayyad, A. & Gibson, J. D. Amphiphilic gold nanoparticles with V-shaped arms.J. Am. Chem. Soc. 128, 4958–4959 (2006).
AcknowledgementsE.K. is grateful for Canada Research Chair financial support (NSERC Canada). G.C.W. gratefullyacknowledges NSERC Canada (grant 312497), NSF (grant CHE-0404579), ONR (grantN00014-05-10765), ARO (grant W911NF-04-1-0191) and NIH (grant 1 R21 EB003101-01) forfinancially supporting this work. M.R. acknowledges financial support from NSF (grantsCHE-0616925 and CTS-0609087), NIH (grant 1-R01-HL0775486A) and NASA (agreementNCC-1-02037). The authors thank Y. Wang for assistance in the synthesis of thiol-terminatedpolystyrene and D. Shirvanyants for assistance in image analysis.Correspondence and requests for materials should be addressed to E.K.Supplementary Information accompanies this paper on www.nature.com/naturematerials.
Author contributionsZ.N. was responsible for project planning, data analysis and experimental work, D.F. was responsiblefor experimental work, S.Z. was responsible for experimental work and data analysis, G.C.W. and M.R.were responsible for data analysis and interpretation and E.K. was responsible for project planning anddata analysis and interpretation.
Competing financial interestsThe authors declare no competing financial interests.
Reprints and permission information is available online at http://npg.nature.com/reprintsandpermissions/
614 nature materials VOL 6 AUGUST 2007 www.nature.com/naturematerials
© 2007 Nature Publishing Group





























