Embed Size (px)
Citation preview

This article was downloaded by: [University of Auckland Library]On: 08 September 2011, At: 15:13Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Journal of Environmental Science and Health, Part BPublication details, including instructions for authors and subscription information:http://www.tandfonline.com/loi/lesb20
Simultaneous analysis of free and sulfo-conjugatedsteroid estrogens in artificial urine solution andagricultural soils by high-performance liquidchromatographyFrank F. Scherr a b & Ajit K. Sarmah a ca Soil Chemical & Biological Interactions, Landcare Research New Zealand, Hamilton, NewZealandb Bayer Crop Science, Monheim, Germanyc Department of Civil & Environmental Engineering, University of Auckland, Auckland, NewZealand
Available online: 08 Sep 2011
To cite this article: Frank F. Scherr & Ajit K. Sarmah (2011): Simultaneous analysis of free and sulfo-conjugated steroidestrogens in artificial urine solution and agricultural soils by high-performance liquid chromatography, Journal ofEnvironmental Science and Health, Part B, 46:8, 763-772
To link to this article: http://dx.doi.org/10.1080/03601234.2012.597702
PLEASE SCROLL DOWN FOR ARTICLE
Full terms and conditions of use: http://www.tandfonline.com/page/terms-and-conditions
This article may be used for research, teaching and private study purposes. Any substantial or systematicreproduction, re-distribution, re-selling, loan, sub-licensing, systematic supply or distribution in any form toanyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contentswill be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses shouldbe independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims,proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly inconnection with or arising out of the use of this material.

Journal of Environmental Science and Health, Part B (2011) 46, 763–772Copyright C© Landcare Research New ZealandISSN: 0360-1234 (Print); 1532-4109 (Online)DOI: 10.1080/03601234.2012.597702
Simultaneous analysis of free and sulfo-conjugated steroidestrogens in artificial urine solution and agricultural soils byhigh-performance liquid chromatography
FRANK F. SCHERR1,2 and AJIT K. SARMAH1,3
1Soil Chemical & Biological Interactions, Landcare Research New Zealand, Hamilton, New Zealand2Bayer Crop Science, Monheim, Germany3Department of Civil & Environmental Engineering, University of Auckland, Auckland, New Zealand
A simple and robust analytical method was developed to simultaneously detect and quantify 17β-estradiol (E2), estrone(E1), 17β-estradiol-3-sulphate (E2-3S), and estrone-3-sulphate (E1-3S) in aqueous solutions (calcium chloride and artifi-cial urine solutions) and agricultural soils using high performance liquid chromatography and UV detection. The stan-dards for all four compounds were linear in the range of 0.01 to 1.0 µg mL−1 (n = 6) and 1.0 to 20 µg mL−1
(n = 6), respectively, with correlation coefficients > 0.999. The on-column limits of detection at an injection volume of 50 µLand S/N (signal: noise) ratio of 3 were: 9.0 ng mL−1, 10 ng mL−1, 5.0 ng mL−1, and 7.0 ng mL−1 for E2-3S, E1-3S, E2 and E1,respectively. The limit of detection and quantification in artificial urine solution and CaCl2 solution was 1.0 ng mL−1 for all fourcompounds. Method detection limits for the compounds in the 3 soils ranged from 2 to 2.4 ng g−1 (E2-3S and E1-3S), and 1.0 to 2.9ng g−1 (E2 and E1), respectively.
Keywords: 17β-estradiol; estrone; 17β-estradiol-3-sulphate; estrone-3-sulphate.
Introduction
Animal wastes, in particular animal urine, act as a source ofestrogen sulphates and glucuronides in the environment, es-pecially when the wastes are released into the environmentwithout treatment, e.g., excretion by grazing livestock orland-application of animal wastes.[1] Sulfo-conjugated hor-mones such as estrone-3-sulphate (E1-3S) have been de-tected in various environmental media in recent times.[2,3]
Similarly other estrogen conjugates such as 17β-estradiol-3-sulphate (E2-3S) may also be directly excreted in livestockurine[4] and are likely to act as a precursor to E1-3S found inthe environment. Possible degradation pathways for thesecompounds (Fig. 1) are through hydrolysis of the estrogensulphates’s thio-ester bond resulting in the formation of bi-ologically active free estrogens such as estradiol (E2), whichloses the hydroxyl group through oxidation to form estrone(E1). Furthermore, E2-3S oxidises into E1-3S which even-tually hydrolyses to form E1. Thus, although conjugates are
Address correspondence to Ajit K Sarmah, Department of Civil& Environmental Engineering, Faculty of Engineering, Univer-sity of Auckland, Private Bag 92019, Auckland, New Zealand;E-mail: [email protected] February 3, 2011.
biologically inactive when they are excreted, deconjugationrenders them biologically active in the form of E2 and E1.Therefore estrogen-sulphates could potentially play a smallbut significant indirect role in the endocrine disruption tothe terrestrial and aquatic species in such environments.
Given the potential role of conjugates in the overall fateof their free counterparts in the pasture environment, anal-ysis of these compounds and their corresponding metabo-lites in soil and aqueous media is imperative. To date, therehave been many published studies available to analyse bothfree and conjugated hormones in sewer systems, waste wa-ter treatment plant effluents, river water and sediments.[2,5,6]
However, to date, no analytical method capable of simulta-neously detecting E2-3S, E1-3S, E2 and E1 in agriculturalsoil and aqueous media has been reported in the literature.
High-performance-liquid-chromatography (HPLC) isa reliable and robust analytical approach for separatingorganic contaminants from pre-treated (environmental)samples. Its application in combination with spectropho-tometric (SP) detectors for the quantification of estrogensis commonly reported in the literature, and constitutes acheaper alternative to the more selective mass spectrom-eters (MS).[7] Its application seems especially sufficientin laboratory-scale microcosm experiments that do notnecessarily need the low detection limits often required
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011

764 Scherr and Sarmah
R1=OH, R2=OH:
R1=SO4, R2=OH:
R1=OH, R2= SO4:
R1=SO4, R2= SO4:
estradiol
estradiol-3-sulphate
estradiol-17-sulphate
estradiol-3,17-sulphate
R=OH:
R=SO4:
estrone
estrone-3-sulphate
CH3
R
OCH3
R1
R2
A B
C D1
2
3
45
814
15
16
171312
11
910
A3
6
7
Fig. 1. Molecular structures of estradiol and their sulphate conju-gates (estradiol-17-sulphate and estradiol-3, 17 sulphate are notinvestigated in this study).
for routine environmental residue analysis. The mostcommonly employed SP detectors are diode array detec-tion (DAD), ultraviolet detection (UV), and fluorescencedetection (FD).[8–12] Given these detection units are notable to identify the analytes of interest unambiguously, thecomplete separation within the chromatographic columnis crucial for a successful analysis and quantification oftarget compounds.[7] The most commonly used analyticalcolumns are C18 and ODS-2 silica columns and the domi-nant mobile phase consists of acetonitrile and water, whichis sometimes acidified. The development of monolithicHPLC columns has recently led to a higher sample through-put capacity owing to the higher flow rates that can beemployed on these columns[13] and the implementation forestrogen separation has also been reported.[14] Separationof hormone conjugates employing HPLC in combinationwith SP detection methods has received little attention,though a study by Blom et al.[15] reported the separation ofeight estrogens including five conjugates on a HPLC-UVsystem, suggesting the feasibility of HPLC-UV. The mobilephase in Blom et al’s [15] study consisted of methanol anda 20-mM ammonium sulphate buffer, and separation wasperformed within retention times of ∼40 minutes at aflow rate of 1 mL min−1. Using a conventional HPLC-UV method, and employing sample extraction throughstrong solvent (dichloromethane), cleanup procedure andpre-concentration steps, Lee et al.[16] reported methoddetection limits for E2 and E1 to be in the lower µg L−1
and µg kg−1 range for spiked aqueous and solid samples.There is an extensive body of literature available deal-
ing with various analytical methods capable of analysingfree and conjugated estrogen extraction from aqueous andwater samples. Especially for the purpose of environmen-tal residue analysis the methods are sophisticated and of-ten involve a series of cleanup and separation procedurescomprising filtration, solid-phase-extraction, sonication,accelerated solvent extraction, and microwave assisted sol-vent extraction among others.[6,17–20] The majority of thesemethods are tailored to the respective subsequent analyt-
ical technique and even though methods are validated,reproducing them in a different laboratory environmentoften requires further modifications and adjustments. Inlaboratory-scale experiments the use of less extensive ex-traction methods has been proven suitable to investigateenvironmental fate processes such as sorption and degra-dation. In particular, the use of DCM comprising a subse-quent solvent transfer yielded recoveries in the range of 77to 105 % for free estrogens.[11,16,21] The feasibility of chlo-rinated solvents for the extraction of estrogen sulphatesfrom urine samples was reported earlier and showed excel-lent recoveries when employed with dicyclohexylamine hy-drochloride (patent no 2,711,988, 1955). However, to datethere are no studies available in the literature on the use ofthese solvents and modifiers to extract estrogen sulphatesfrom aqueous as well as soil samples while evaluating themethod performance through demonstration in spiked soilsamples.
In this paper we report on a simple, yet robust HPLCmethod equipped with a conventional UV detector to anal-yse free and conjugated estrogenic steroid hormones andtheir concomitantly formed metabolites in spiked soil andaqueous samples. This method has been evaluated in termsof its recovery, reproducibility, linearity of response, limitof detection, and limit of quantification using three agricul-tural soils. The method has been applied to investigate sorp-tion and degradation studies performed under controlledlaboratory conditions in three agricultural soils collectedfrom a few dairy farming regions of New Zealand.
Materials and methods
Chemicals
The compounds 17β-estradiol (>98 % purity), estrone(>99 % purity), and 17β-estradiol-3-sulphate, andestrone-3-sulphate (all ≥ 95 % purity) were purchased fromSigma-Aldrich Ltd, Australia. Acetonitrile (MallinckrodtChromAR, ≥ 99.8 % purity). Dichloromethane (Mallinck-rodt UltimAR, ≥ 99.9 % purity), methanol (MallinckrodtChromAR, ≥ 99.9 % purity), ammonium sulphate (BDHLaboratory Supplier AnalaR, > 99 % purity), potassiumchloride, potassium sulfate (BDH Laboratory SupplierAnalaR, all >99 % purity), urea (Labserv Analytical Grade,>99 % purity), potassium bicarbonate and glycine (AjaxFinechem Analytical) were obtained from Biolab ScientificLtd, New Zealand. Dicyclohexylamine (Merck, > 99 % pu-rity) was obtained from the University of Waikato Chemi-cal Store, New Zealand, and synthesized with concentratedhydrochloric acid (Ajax Finechem, 36 %) to form solid di-cyclohexylamine hydrochloride (DCH·HCl). HPLC-gradedeionised water was obtained from an onsite arium R© 61316high performance reverse osmosis system (Sartorius Ste-dim Biotech GmbH, Germany). Nitrogen gas oxygen free(gas code 152) was purchased from BOC gas supplier(New Zealand).
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011

Analysis of free and sulfo-conjugated steroid estrogens 765
HPLC-UV system specifications. Analysis of the free andsulphate estrogens was performed on a Dionex SummitHigh Performance Liquid Chromatography (HPLC) sys-tem comprising a Dionex Solvent Rack SOR-100, a DionexThermo-stated Column Compartment TCC-100, a DionexASI-100 Automated Sampler, a Dionex P680 LP-Pump,and a Dionex UVD 170U detector. The UV lamp was aL2D2 deuterium lam (Hamamatsu Photonic, K.K., Japan)and the injection syringe was a Gastight R© from HamiltonBondaduz (#1725, 0.25 mL, Switzerland).
The analytical columns in use were a ChromolithPerformance RP-18 (100 × 4.6 mm, Merck R©, Germany)and an equivalent Onyx R© Monolithic C18 column (100 ×4.6 mm, Phenomenex R©, Australia). Both columns wereoperated with a Security GuardTM column cartridge holderequipped with a C18 cartridge (4 mm, Phenomenex R©,Australia).
HPLC-UV separation and detection. A modified elutionscheme similar to Blom et al.[15] was developed to be ableto separate the estrogens E2 and E1 and their sulphateconjugates E2-3S and E1-3S. The original organic solventmethanol used by Blom et al.[15] was replaced with ace-tonitrile in this study due to the higher auto-absorbance ofmethanol at lower detection wavelengths which were foundto be more sensitive for estrogen detection. The elutionscheme was developed on an Onyx R© Monolithic C18column and all four compounds could be separated within9 minutes using a high sample throughput. Standardlinearity was assessed in the concentration range of 0.1to 1.0 µg mL−1 and 1.0 to 20 µg mL−1. The limits ofdetection were calculated on a signal to noise ratio of 3.
The optimal detection wavelength was determined bypreparing a combined solution of E1, E2, E1-3S, and E2-3S each at 10 µg mL−1 in mobile phase. The UV ab-sorbance of the solution was then measured on a ShimadzuUV 160A UV-Visible Recording Spectrophotometer (Shi-madzu, Japan) in the range from 200 to 300 nm in a full-scanmode.
Extraction from aqueous (artificial urine and CaCl2 solu-tion) matrices and recovery test. Extraction of E1-3S andE1 was performed by spiking the hormones in two aqueousmedia (artificial urine and 5 mM CaCl2 solution). This isto be noted that E2-3S and E2 were not added to aqueousmatrices and therefore no extraction and recovery data areavailable for these two compounds. However, both E2-3Sand E2 extraction from soil during the degradation studiesand associated recoveries are discussed later.
Artificial urine solution consisted of KHCO3 (22.2 gL−1), KCl (3.95 g L−1), K2SO4 (6.7 g L−1), (NH2)2CO (23.5g L−1), and C2H5NO2 (6.2 g L−1) in de-ionised water.[22]
Aqueous solutions of E1-3S, and E1 were prepared at con-centrations of 0.25, 0.5, 0.75, 1.5, 2.5 and 5.0 µg mL−1 byadding appropriate amounts of methanolic stock solutions(200, 400, and 600 µg mL−1) to the respective aqueous solu-
tions of artificial urine and CaCl2 to obtain a total volumeof 25 mL in a glass centrifuge tube. A sub sample of 5 mLof aqueous solution containing the hormones was removedand transferred on to a new glass centrifuge tube, and liq-uid/liquid extraction was performed. Briefly, extraction ofE1-3S was performed by adding 0.25 mL of DCH·HCl (10µg mL−1 in H2O) and 4.9 mL of DCM to the aqueousmatrix. Solvent-added centrifuge tubes were then placedover night on an end-over-end shaker at 22 ± 1◦C, andafter centrifugation at 2200 rpm, an aliquot of 2 mL of theDCM phase was then transferred to an amber glass HPLCvial and evaporated to dryness under a gentle stream of N2at 22 ± 1◦C. An aliquot of 1 mL of 30 % (for E1) and 70% (for E1-3S) methanol was added separately in each vial.Reconstituted samples were then immediately analysed bymeans of HPLC-UV detection without any further storage.
Extraction from soil samples and recovery test. Extractionof the free (E2 and E1) and conjugated hormones (E2-3Sand E1-3S) was performed in a series of sorption and degra-dation experiments in three agricultural soils, and followedby HPLC/UV detection. Three top soils (0–10 cm) fromthree separate dairy farming regions of New Zealand werefreshly collected, sieved (2 mm) and air-dried. These siteswere not previously grazed and therefore were expectedto be devoid of naturally excreted steroid hormones fromlivestock.
For the sorption study, 2–3 g of each soil was placed incentrifuge tubes and equilibrated with 25 mL of 0.005 Mbackground electrolyte solution (CaCl2) for 12 hrs. Afterequilibration, supernatant was decanted and residual soilslurry was extracted for E1 and E1-3S by adding 4.9 mLof DCM to the soil slurry deposited in the tube, sonicatedfor 10 minutes in a sonication bath (Lab Line, Elma, Ger-many) and placed on an end-over-end shaker over night at22 ± 1◦C. Before sonication, an additional volume of 0.25mL DCH·HCl (10 µg mL−1 in H2O) was added in the tubefor E1-3S extraction. After centrifugation at 2200 rpm analiquot of 2 mL of the DCM phase was then transferredto an amber glass HPLC vial and processed as describedabove for the aqueous extraction of the hormones.
During the degradation study, extraction recovery wasevaluated by means of sterile controls. In brief, 150 g of soil,adjusted to 60 % of the maximum water holding capacity(–33 kPa), was autoclaved thrice in preserving jars (122.5◦C,1.13 Bar, Priorclave, Ltd, U.K.). After autoclaving, the wa-ter loss was determined gravimetrically and the lost volumewas reapplied by sterilised deionised water containing ap-propriate amounts of E1-3, E2-3S, E1, and E2, respectively,to yield a nominal soil concentration of 5 µg g−1 soil. Theprocedure was conducted in a laminar flow cabinet andthe aqueous spiking solution was carefully mixed with thesoil with a sterilised spatula. Triplicate sub samples of ap-proximately 2 to 3 g were then removed and extracted by5 mL of DCM and 0.25 mL of DCH·HCl (10 µg mL−1)on an end-over-end shaker at 22 ± 1◦C over night. Further
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011

766 Scherr and Sarmah
Wavelength [nm]
200 220 240 260 280 300
Abs
orba
nce
[-]
0.0
0.5
1.0
1.5
2.0
Fig. 2. UV- absorbance spectrum of a combined estrogen (E2-3S,E1-3S, E2 and E1) standard in mobile phase (10 µg mL−1).
processing and analysis was conducted as described beforefor the aqueous matrices and soil samples during sorptionexperiments.
Results and discussion
HPLC-UV separation and detection
Figure 2 displays the resulting UV absorbance spectrum ofthe combined estrogen standard for the wavelength rangefrom 200 to 300 nm. The graph shows two absorbance op-tima at 201 and 279 nm. The relative absorbance at 201 nmwas about 20 times higher than at 279 nm, indicating thatboth the free and the sulphate conjugated estrogens havean optimum absorbance in the far UV range. A wavelengthof 201 nm was selected for the detection of the hormonesand their sulphate conjugates.
The predominant wavelength for E1 and E2 as well as forestrogen conjugates detection via UV detectors reported inthe literature accounts for 280 nm[10,15, 23, 24] which agreeswith the second optimum found in the present study. Leeet al.[16] and Sarmah et al.[11] reported wavelengths of 205
Fig. 3. Mobile phase gradient system for the separation of E1-3S,E2-3S, E1 and E2 prior to UV detection. Dotted lines indicate achange in mobile phase composition.
and 225 nm, respectively, for the detection of E1 and E2,indicating that lower wavelengths may also be suitable forestrogen detection. The differences may be explained by thediverse UV detectors in use. Older detectors often lack thecapability to run wavelengths in the lower UV range andtherefore the wavelength optimum found in this study maynot have been available in some of the earlier studies.
Compound separation
The aqueous mobile phase for estrogen sulphate separationwas derived from the study by Blom et al.[15] where six estro-gen conjugates, sulphates and glucuronides, were separatedin 40 minutes together with E1 and E2 on a C18 (4.6 ×250 mm) HPLC column employing a gradient systemof ammonium sulphate (20 mM, pH 6.8) and methanol.Methanol has a high auto absorbance at the previouslydetermined optimum detection wavelength, and thereforewas replaced by acetonitrile, which has been employed forthe free estrogen separation on HPLC-UV systems.[8,16]
Furthermore, the ammonium sulphate concentration ofthe buffer solution was reduced to 5 mM and acidified withconcentrated sulphuric acid (H2SO4) to pH 3 in order toprevent the sulphate conjugates to be flushed through theHPLC column without significant retention. Additionally,high purity deionised water was employed as a thirdmobile phase to prevent salt precipitations during gradientrun. The optimized gradient system (Fig. 3) allowedseparation of E2-3S, E1-3S, E2 and E1 within a run timeof 9 minutes (flow rate of 2.0 mL min−1) on a Merc R©Chromolith column, and within 7 minutes (flow rate of 1.5mL min−1) on an Onyx R© column as illustrated in Figures4 and 5. An injection volume of 50 µL was used for bothcolumns, while the column temperature was maintained at22 ± 1◦C. The standard chromatograms (Figs. 4 and 5) in-dicate that the gradient system yielded a distinct separationof the four target compounds and that the matrix influencewas negligible. However, blank matrix runs were alwaysconducted as a matter of quality assurance and to identifypossible additional retention mechanisms that may occuron analytical columns as a result of high sample through-put and/or guard column failure. It can be observed fromthe soil blank run in both sorption and degradation studies(Figs. 4 and 5, bottom plot) that there was no interferencefrom co-eluting peaks reflecting the robustness of theextraction and clean-up steps performed in this study.
The free estrogens E1 and E2 have been successfully sep-arated on a commonly packed C18 analytical column usingmixtures of acetonitrile and water as the mobile phase insome earlier studies. For example, typical runtimes werereported to be in the range of 11–20 minutes to separateE1 and E2 at flow rates of 1.0–1.5 mL min−1 with varyingacetonitrile/water ratios.[16,25,26] The implementation of amonolithic C18 analytical column resulted in a significantreduction of the sample runtime, and separation of E1 andE2 was achieved within 2.5 minutes (chromatograms not
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011
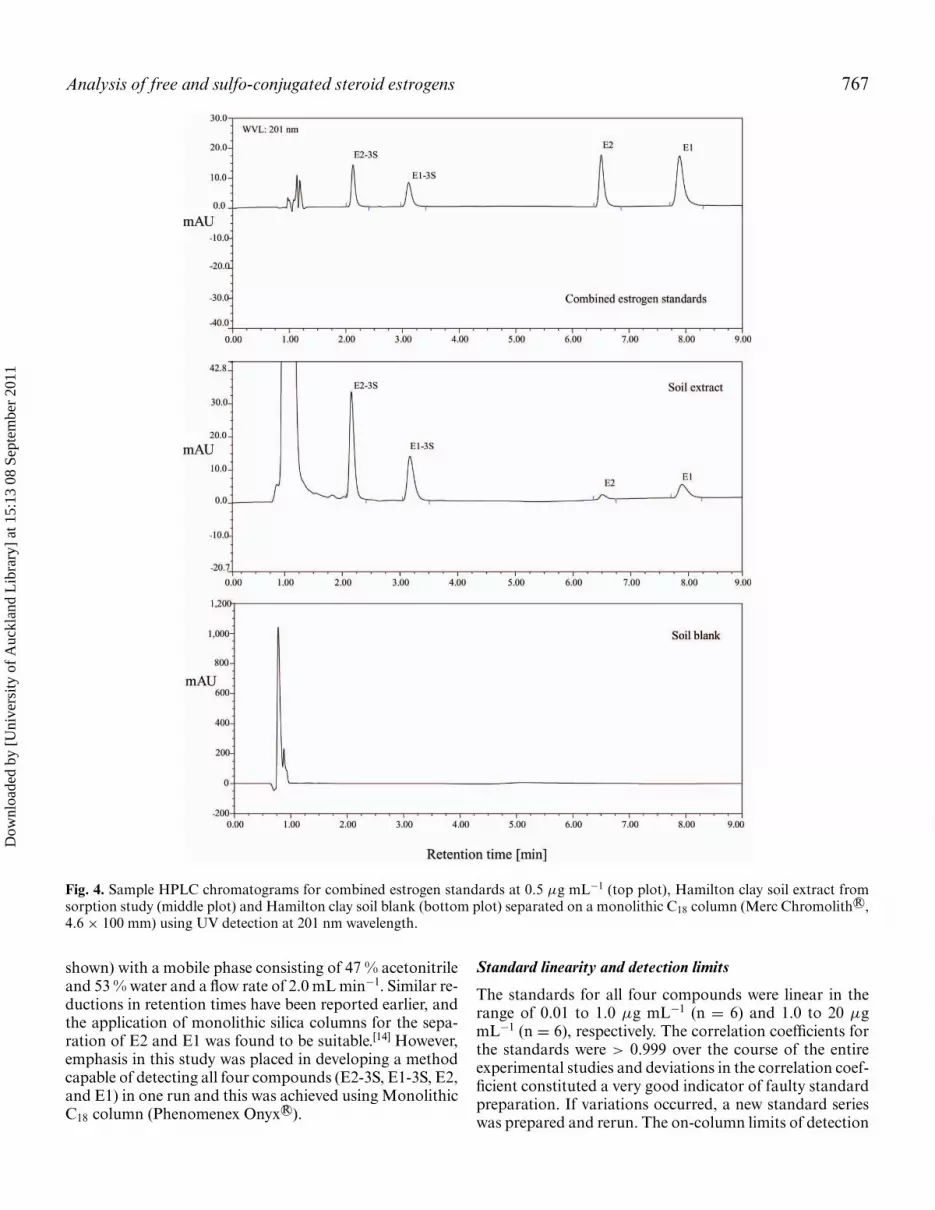
Analysis of free and sulfo-conjugated steroid estrogens 767
Fig. 4. Sample HPLC chromatograms for combined estrogen standards at 0.5 µg mL−1 (top plot), Hamilton clay soil extract fromsorption study (middle plot) and Hamilton clay soil blank (bottom plot) separated on a monolithic C18 column (Merc Chromolith R©,4.6 × 100 mm) using UV detection at 201 nm wavelength.
shown) with a mobile phase consisting of 47 % acetonitrileand 53 % water and a flow rate of 2.0 mL min−1. Similar re-ductions in retention times have been reported earlier, andthe application of monolithic silica columns for the sepa-ration of E2 and E1 was found to be suitable.[14] However,emphasis in this study was placed in developing a methodcapable of detecting all four compounds (E2-3S, E1-3S, E2,and E1) in one run and this was achieved using MonolithicC18 column (Phenomenex Onyx R©).
Standard linearity and detection limits
The standards for all four compounds were linear in therange of 0.01 to 1.0 µg mL−1 (n = 6) and 1.0 to 20 µgmL−1 (n = 6), respectively. The correlation coefficients forthe standards were > 0.999 over the course of the entireexperimental studies and deviations in the correlation coef-ficient constituted a very good indicator of faulty standardpreparation. If variations occurred, a new standard serieswas prepared and rerun. The on-column limits of detection
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011

768 Scherr and Sarmah
Fig. 5. Sample HPLC chromatograms of combined estrogen standards at 0.5 µg mL−1 (top plot), Hamilton clay soil extract fromdegradation study (middle plot) and Hamilton clay soil blank (bottom plot) separated on a monolithic C18 column (PhenomenexOnyx R©, 4.6 × 100 mm) using UV detection at 201 nm wavelength.
at an injection volume of 50 µL and S/N (signal:noise) ra-tio of 3 were: 9.0 ng mL−1, 10 ng mL−1, 5.0 ng mL−1, and7.0 ng mL−1 for E2-3S, E1-3S, E2 and E1, respectively.
Extraction from aqueous matrices
The extraction recovery for E1 and E1-3S from the aqueousmatrices CaCl2 (5 mM) and artificial urine at six concen-trations is displayed in Figure 6. As sorption studies were
not performed for E2-3S and E2, the extraction recoveryfor E2-3S and E2 in the two mediator solutions was notattempted. In general, the recoveries of E1-3S and E1 wereexcellent and ranged from 97 to 107 % across the six con-centrations. A conclusive trend in relation to the aqueousmatrix or the aqueous concentration was not detected. Ma-trix effects that could have been expected from urea andglycine interference in the artificial urine extraction did notoccur. The method detection limits accounted for 1.0 ng
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011

Analysis of free and sulfo-conjugated steroid estrogens 769
Fig. 6. Extraction recoveries of E1 and E1-3S from the aqueousmatrices CaCl2 (5 mM) and artificial urine at six concentrations.Dotted line indicates 100 % mark. Average values and relativestandard deviations for n = 12 samples are shown.
mL−1 for both E1 and E1-3S in both CaCl2 (5 mM) andartificial urine sample.
Dichloromethane has been reported to be suitable forliquid/liquid extraction of estrogens from aqueous sam-ples and the recoveries reached in the present study are inthe range of previously reported values. For instance Sotoet al.[27] achieved a recovery of 102 % for E1 with DCMextraction. However, a lower recovery of 82.5 % for E1was earlier reported by Sarmah et al[11], and Lee et al.[16]
found DCM extractions to be excellent for their indentedpurpose to investigate sorption of estrogens. The lower re-coveries found by Lai et al.[21] may partly be attributed totheir subsequent derivatization for GC-MS analysis duringwhich compound loss could have occurred. The methoddetection limits of the present work are five times lower[21]
and two times higher than those reported earlier.[16] Thiswas mainly attributed to the different DCM/aqueous ma-trix ratios employed amongst the three studies. Lai et al.[21]
used a similar ratio but were not able to achieve a sim-ilar pre-concentration due their lower absolute volumes;
while others have been able to achieve a lower methoddetection limit by using a higher volume of the aqueousmatrix.[16] Information on the extraction of E1-3S fromaqueous samples using DCM is not available in the lit-erature. A patent (patent number 2,711,988, 1955) notedthe successful recovery of E1-3S with ethylene dichlorideand DCH·HCl from which the present extraction schemewas derived. Compared with the method detection limitsyielded from LC-MS methods[5,28,29] the values reportedhere are about an order of magnitude higher and thereforeprobably not sufficient enough to detect estrogen residues inaged environmental field samples. However, for the purposeof laboratory-scale investigations to study sorption, degra-dation and column transport of estrogens the method issuitable to obtain quality data and to infer environmentalfate parameters. Furthermore, the presented methods donot include the usage of expensive solid phase extraction(SPE) procedure and therefore also minimize costs and therisk of compound loss due to extensive solvent transfers.The latter may be overcome by online-SPE techniques,[28]
which are still in the early stages of development and avail-able only at highly specialised laboratories.
Extraction from soil
The standardised extraction of estrogens from natural soilsconstitutes a challenge due to the fact that the free hor-mones and in particular their sulphate conjugates are labilecompounds that undergo rapid decay in natural soils andform metabolites. The study of recoveries is therefore al-ways influenced by degradation processes that can alter soilconcentrations. Sterilization techniques such as autoclav-ing or treatment with sodium azide change soil propertiesand may lead to false conclusions in relation to recoverystudies.[30] Gamma radiation, usually implemented to avoidthose limitations, was, however, not available for this work.
Recoveries during the sorption study
Figure 7 displays the combined recovery from soil andaqueous phase extractions during the sorption of E1 andE1-3S to three different soils. The values for E1-3S includethe recovery for its parent compound E1 which occurredas a metabolite. Except in the case of E1, in the Hamiltonsoil from CaCl2 the recoveries were > 80 % indicating agood extraction yield using this method. It has been earlierreported that a second extraction step could increase therecoveries,[11,25] but in the present study, no significant in-crease in the compound recoveries was observed during asecond extraction. On the other hand, poor soil recoveriesof only 46 % for E1 with DCM extraction were reported byBeck et al.[20] In contrast, excellent recoveries for estrogenextraction from sediments have been reported for a vari-ety of solvents including acetone, acetonitrile/water, ethylacetate and dichloromethane/water.[21]
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011
770 Scherr and Sarmah
Fig. 7. Combined recoveries from soil and aqueous solutionsduring the sorption of E1 and E1-3S to three different soils.Estrone-sulphate recoveries include the metabolite E1. Averagevalues and relative standard deviations for n = 12 samples areshown. ∗indicates n = 10 samples. Dotted line represents 100 %mark.
There is a dearth of published studies on the extractionof estrone-sulphate; however, the preferred solvent is notclearly identified in many past studies. In general it canbe deduced from the literature that either multiple SPEschemes[6] or the implementation of microwave-assisted ex-traction at very high temperatures[7] is necessary to yieldsufficient extraction recoveries for E1-3S. Against this back-ground the use of DCH·HCl in combination with DCM hasbeen proved to be a new development in this study. How-ever, in the present study, occasionally the target peaks wereinterfered with matrix impurities originating from the soil,and in such cases the sample was withdrawn from the sub-sequent data evaluation.
Recoveries during the degradation study
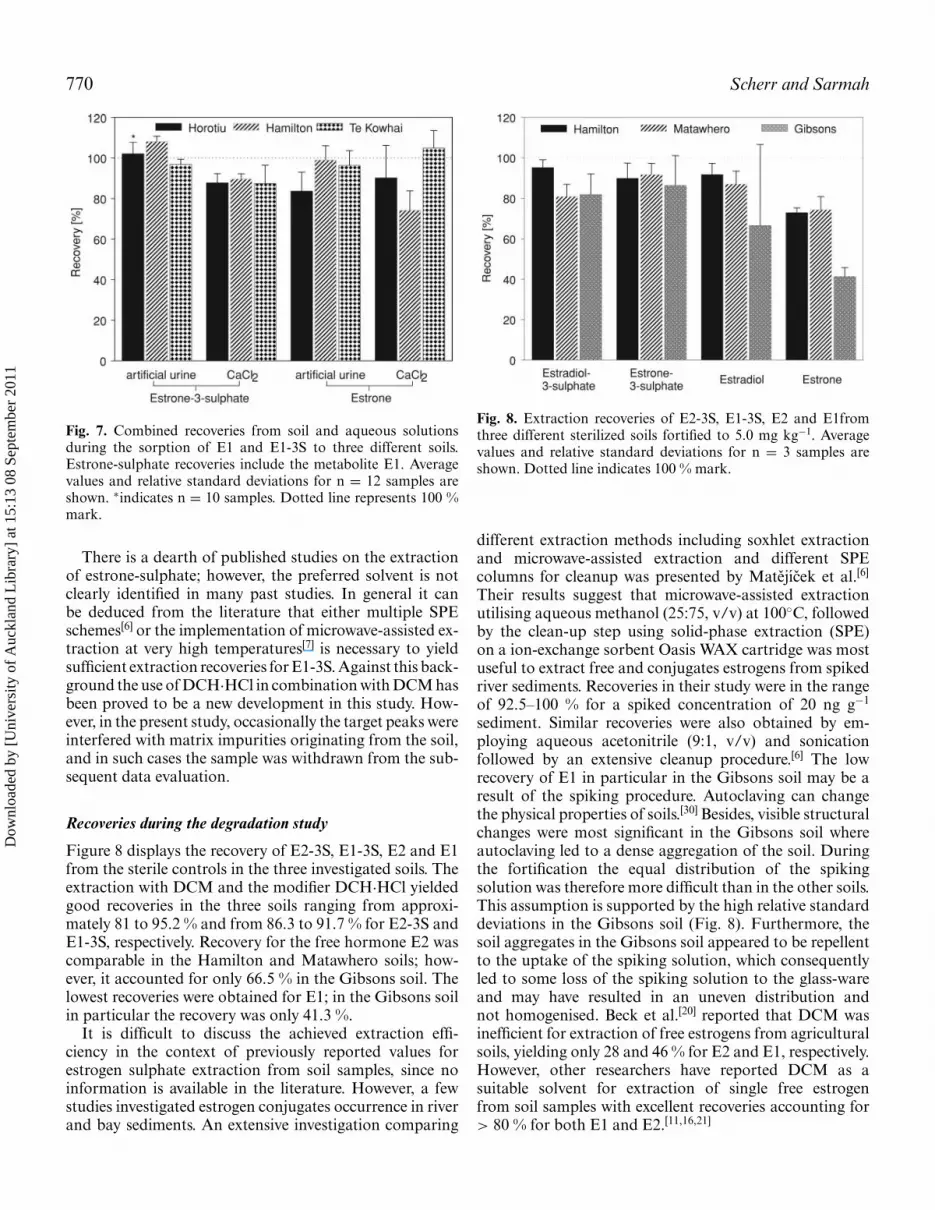
Figure 8 displays the recovery of E2-3S, E1-3S, E2 and E1from the sterile controls in the three investigated soils. Theextraction with DCM and the modifier DCH·HCl yieldedgood recoveries in the three soils ranging from approxi-mately 81 to 95.2 % and from 86.3 to 91.7 % for E2-3S andE1-3S, respectively. Recovery for the free hormone E2 wascomparable in the Hamilton and Matawhero soils; how-ever, it accounted for only 66.5 % in the Gibsons soil. Thelowest recoveries were obtained for E1; in the Gibsons soilin particular the recovery was only 41.3 %.
It is difficult to discuss the achieved extraction effi-ciency in the context of previously reported values forestrogen sulphate extraction from soil samples, since noinformation is available in the literature. However, a fewstudies investigated estrogen conjugates occurrence in riverand bay sediments. An extensive investigation comparing
Fig. 8. Extraction recoveries of E2-3S, E1-3S, E2 and E1fromthree different sterilized soils fortified to 5.0 mg kg−1. Averagevalues and relative standard deviations for n = 3 samples areshown. Dotted line indicates 100 % mark.
different extraction methods including soxhlet extractionand microwave-assisted extraction and different SPEcolumns for cleanup was presented by Matejıcek et al.[6]
Their results suggest that microwave-assisted extractionutilising aqueous methanol (25:75, v/v) at 100◦C, followedby the clean-up step using solid-phase extraction (SPE)on a ion-exchange sorbent Oasis WAX cartridge was mostuseful to extract free and conjugates estrogens from spikedriver sediments. Recoveries in their study were in the rangeof 92.5–100 % for a spiked concentration of 20 ng g−1
sediment. Similar recoveries were also obtained by em-ploying aqueous acetonitrile (9:1, v/v) and sonicationfollowed by an extensive cleanup procedure.[6] The lowrecovery of E1 in particular in the Gibsons soil may be aresult of the spiking procedure. Autoclaving can changethe physical properties of soils.[30] Besides, visible structuralchanges were most significant in the Gibsons soil whereautoclaving led to a dense aggregation of the soil. Duringthe fortification the equal distribution of the spikingsolution was therefore more difficult than in the other soils.This assumption is supported by the high relative standarddeviations in the Gibsons soil (Fig. 8). Furthermore, thesoil aggregates in the Gibsons soil appeared to be repellentto the uptake of the spiking solution, which consequentlyled to some loss of the spiking solution to the glass-wareand may have resulted in an uneven distribution andnot homogenised. Beck et al.[20] reported that DCM wasinefficient for extraction of free estrogens from agriculturalsoils, yielding only 28 and 46 % for E2 and E1, respectively.However, other researchers have reported DCM as asuitable solvent for extraction of single free estrogenfrom soil samples with excellent recoveries accounting for> 80 % for both E1 and E2.[11,16,21]
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011

Analysis of free and sulfo-conjugated steroid estrogens 771
Table 1. Method detection limits (ng g−1) for estrogen and es-trogen sulphates extraction from three agricultural soils.
Soil E2-3S E1-3S E2 E1
Hamilton 2.4 2.0 2.9 1.0Matawhero 2.4 2.0 2.8 1.0Gibsons 1.8 2.0 1.3 1.0
It has to be noted that the high recoveries obtained byMatejıcek et al.[6] and Isobe et al.[2] were based on verysmall sample sizes of 0.5 to 1 g of sediment which reducesthe possibility of compound loss during the fortificationprocedure. In the present study our aim was to investigatethe recovery yield for the intended purpose to study degra-dation of estrogens and estrogen sulphates in microcosmexperiments and therefore the whole experimental processwas investigated involving sub sampling from a fortifiedsoil mass of 150 g.
The method detection limits (based on a signal to noiseratio of 3) are presented in Table 1 and show some variationwith the soil type due to varying matrix interferences fromthe different soils. These method detection limits, however,were only achieved with an increase of the soil mass to5 g and an increase of the DCM volume to 6 mL. Also,the volume of DCM that was evaporated under stream ofN2 was increased to 4 mL.
The presented method detection limits (Table 1) areabout 2-10 times higher than those reported by Matejıceket al.[6] and about 14–100 times higher than those reportedby Isobe et al.[2] and Beck et al.[20] for sediment and soilsamples, respectively. In these studies MS detectors havebeen used that are in general more sensitive than UV de-tectors to analyse very low concentrations.[7] Furthermore,during the extraction and clean-up process these studiesused microwave extractor, accelerated solvent extractor, orgel permeation chromatography clean-up, which were notavailable for the present work. The present study used muchhigher concentrations (at ppb level) that are spiked onto soiland aqueous sample, and therefore HPLC with UV detec-tion seemed to be justifiable rather than LCMS which coulddetect at trace levels (ppt level). The achieved method de-tection limits in the present work were found to be suitableto investigate laboratory batch sorption and degradation ofthe free and sulphate conjugated estrogens in agriculturalsoils.[1,31]
Conclusions
The separation of the free estrogens E1 and E2 and their3-conujated sulphate conjugates has been successfully de-veloped using a mobile-phase gradient system consisting ofacetonitrile, ammonium sulphate (5 mM, pH 3) and waterand UV detection at 201 nm. The implementation of thisgradient system on a monolithic C18 column resulted insignificant runtime reduction which can be regarded as a
crucial achievement for the standard analysis of high sam-ples numbers. A shorter runtime has a number of positiveeffects including shorter sample retention time on the autosampler before analysis, less use of the mobile phase leadingto reduced costs and the decrease of deuterium lamp deadtimes in between target peaks, which consequently increasesthe number of samples that can be analysed by one lamp.
A simple and robust extraction method was developedto extract the estrogen sulphates E1-3S and E2-3S fromaqueous and solid matrices. Dichloromethane in combi-nation with dihydrohexylamine hydrochloride has yieldedexcellent recoveries for the extraction of estrogen sulphatesfrom aqueous matrices and was also found suitable to ex-tract these compounds together with their free counterpartsfrom soil. Poor extraction recoveries for estrone associatedwith one particular soil contributed to the effects originat-ing in the sterilizing procedure, which may have altered thephysical properties of the soil thus hindering the initial soilfortification. The obtained method detection limits wereabout 2–100 times higher than comparable studies involv-ing the analysis of estrogen sulphates in solid samples; how-ever, the extraction and analysis method was found to besuitable for conducting fate-related (sorption and degrada-tion) laboratory studies, especially when highly specialisedextraction equipments and MS detectors are not availablein many laboratories. The method can also be applied toinvestigate sorption experiments involving spiked samplesin soils and sediments, and the soil extraction method canbe employed in laboratory column transport experimentsto extract estrogen soil residues.
Acknowledgments
This study was funded by contracts CO9X0705 (LandcareResearch) from the Foundation for Science, Research andTechnology (New Zealand). Prakash Srinivasan is thankedfor reviewing an earlier version of this manuscript.
References
[1] Scherr, F.F.; Sarmah, A.K.; Di, H. J.; Cameron, K. Sorption of es-trone and estrone-3-sulfate from CaCl2 solution and artificial urinein pastoral soils of New Zealand. Environ. Toxicol. Chem. 2009, 28,2564–2571.
[2] Isobe, T.; Serizawa, S.; Horiguchi, T.; Shibata, Y.; Managaki, S.;Takada, H.; Morita, M.; Shiraishi, H. Horizontal distribution ofsteroid estrogens in surface sediments in Tokyo Bay. Environ. Poll.2006, 144, 632–638.
[3] Komori, K.; Tanaka, H.; Okayasu, Y.; Yasojima, M.S.C. Analy-sis and occurrence of estrogen in wastewater in Japan. Water. Sci.Technol. 2004, 50, 93–100.
[4] Hoffman, B.; Goes di Pinho, T.; Schuler, G. Determination of freeand conjugated oestrogen in peripheral blood plasma, faeces andurine of cattle throughout pregnancy. Exp. Clin. Endocrinol. Diab.1997, 105, 296–303.
[5] D’Ascenzo, G.; Di Corcia, A.; Gentili, A.; Mancini, A.R.; Mas-tropasqua, R.; Nazzari, M.; Samperi, R. Fate of natural estrogenconjugates in municipal sewage transport and treatment facilities.The Sci. Total Environ. 2003, 302, 199–209.
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011

772 Scherr and Sarmah
[6] Matejıcek, D.; Houserova, P.; Kuban, V. Combined isolationand purification procedures prior to the high-performance liquidchromatographic-ion-trap tandem mass spectrometric determina-tion of estrogens and their conjugates in river sediments. J. Chro-matogr. A. 2007, 1171, 80–89.
[7] Ingerslev, F.; Halling-Sorensen, B. Evaluation of analytical chemicalmethods for detection of estrogens in the environment, Report number44; Danish Ministry of the Environment, Copenhagen, Denmark.2003.
[8] Ying, G.G.; Toze, S.; Hanna, J.; Yu, X.Y.; Dillon, P.J.; Kookana,R.S. Decay of endocrine disrupting chemicals in aerobic and anoxicgroundwater. Water Res. 2007, 42, 1133–1141.
[9] Jacobsen, A.M.; Lorenzen, A.; Chapman, R.; Topp, E. Persis-tence of testosterone and 17β-estradiol in soils receiving swinemanure or municipal biosolids. J. Environ. Qual. 2005, 34, 861–871.
[10] Bonin, J.L.; Simpson, M.J. Sorption of steroid estrogens to soiland soil constituents in single- and multi-sorbate systems. Environ.Toxicol. Chem. 2007, 26, 2604–2610.
[11] Sarmah, A.K.; Northcott, G.L.; Scherr, F.F. Retention of estrogenicsteroid hormones by selected New Zealand soils. Environ. Int. 2008,34, 749–755.
[12] Xuan, R.; Blassengale, A.A.; Wang, Q. Degradation of estrogenichormones in a silt loam soil. J. Agric. Food. Chem. 2008, 56,9152–9158.
[13] Cledera-Castro, M.; Santos-Montes, A.; Izquierdo-Hornillos, R.J.Comparison of the performance of conventional microparticulatesand monolithic reversed-phase columns for liquid chromatographyseparation of eleven pollutant phenols. J. Chromatograph. A. 2005,1087, 57–63.
[14] Mizuguchi, T.; Ogasawara, K.; Shimada, K. Application of mono-lithic silica column for HPLC separation of estrogens. Chromatogr.2005, 26, 101–104.
[15] Blom, M.J.; Wassink, M.G.; Kloosterboer, H.J.; Ederveen, A.G.H.;Lambert, J.G.D.; Goos, H.J. Metabolism of estradiol, ethynylestra-diol, and moxestrol in rat uterus, vagina, and aorta: influence of sexsteroid treatment. Drug. Metab. Disp. 2001, 29, 76–81.
[16] Lee, L.S.; Strock, T.J.; Sarmah, A.K.; Rao, P.S.C. Sorption anddissipation of testosterone, estrogens, and their primary transfor-mation products in soils and sediment. Environ. Sci. Technol. 2003,37, 4098–4105.
[17] Lopez de Alda, M.J.; Barcelo, D. Review of analytical methods forthe determination of estrogens and progestogens in waste water.Fresenius J. Anal. Chem. 2001, 371, 437–447.
[18] Dıaz-Cruz, M.S.; Lopez de Alda, M.J.; Barcelo, D.; Environmentalbehaviour and anlysis of veterinary and human drugs in soils, sed-iments and sludge. TrAC Trends Anal. Chem. 2003, 22, 340–351.
[19] Kuster, M.; Lopez de Alda, M.J.; Barcelo, D. Analysis and distri-bution of estrogens and progestogens in sewage sludge, soils andsediments. TrAC Trend. Anal. Chem. 2004, 23, 790–798.
[20] Beck, J.; Totsche, K.U.; Kogel-Knabner, I. A rapid and efficientdetermination of estrogens in soils by pressurised liquid extractionand gas chromatography-mass spectrometry. Chemosphere 2008,71, 954–960.
[21] Lai, K.M.; Johnson, K.L.; Scrimshaw, M.D.; Lester, J.N. Bindingof waterborne steroid estrogens to solid phases in river and estuarinesystems. Environ. Sci. Technol. 2000, 34, 3890–3894.
[22] Early, M.S.; Cameron, K.C.; Fraser, P.M. The fate of potassium,calcium, and magnesium in simulated urine patches on irrigateddairy pasture soil. NZ. J. Agric. Res. 1998, 41, 117–124.
[23] Van Emmerik, T.; Angove, M.J.; Johnson, B.B.; Wells, J.D.; Fernan-des, M.B. Sorption of 17β-estradiol onto selected soil minerals. J.Colloid Inter. Sci. 2003, 266, 33–39.
[24] Yu, Z.; Xiao, B.; Huang, E.; Peng, P. Sorption of steroid estrogensto soils and sediments. Environ. Toxicol. Chem. 2004, 23, 531–539.
[25] Gatti, R.; Gioia, M.G.; Di Pietra, A.M.; Cavrini, V. HPLC fluores-cence determination of unconjugated estrogens in pharmaceuticals.J. Pharma. Biomed. Anal. 1998. 18, 187–192.
[26] Choi, D.W.; Kim, J.Y.; Choi, S.H.; Jung, H.S.; Kim, H.J.; Cho,S.Y.; Kang, C.S.; Chang, S.Y. Identification of steroid hormones inpomegranate (Punica granatum) using HPLC and GC-mass spec-trometry. Food Chem. 2006, 96, 562–571.
[27] Soto, A.M.; Calabro, J.M.; Prechtl, N.V.; Orlando, F.F. Androgenicand estrogenic activity in water bodies receiving cattle feedlot efflu-ent in eastern Nebraska, USA. Environ. Health. Persp. 2004, 112,346–352.
[28] Kuster, M.; Lopez de Alda, M.J.; Hernando, M.D.; Petrovic, M.;Martın-Alonso, J.; Barcelo, D. Analysis and occurrence of phar-maceuticals, estrogens, progestons and polar pesticides in sewagetreatment plant effluents, river water and drinking water in the Llo-bregat river basin (Barcelona, Spain). J. Hydrol. 2008, 358, 112–123.
[29] Rodriguez-Mozaz, S.; Lopez de Alda, M.J.; Barcelo, D. Monitor-ing of estrogens, pesticides and bisphenol A in natural waters anddrinking water treatment plants by solid-phase extraction-liquidchromatography-mass spectrometry. J. Chromatogr. A. 2004, 1045,85–92.
[30] Lotrario, J.B.; Stuart, B.J.; Lam, T.; Arands, R.R.; O’Connor, O.A.;Kosson, D.S. Effects of sterilization methods on the physical char-acteristics of soil: Implications for sorption isotherm analysis. Bull.Environ. Contam. Toxicol. 1995, 54, 668–675.
[31] Scherr, F.F.; Sarmah, A.K.; Di, H.J.; Cameron, K.C. Mod-elling degradation and metabolite formation kinetics of estrone-3-sulphate in agricultural soils. Environ. Sci. Technol. 2008, 42,8388–8394.
Dow
nloa
ded
by [
Uni
vers
ity o
f A
uckl
and
Lib
rary
] at
15:
13 0
8 Se
ptem
ber
2011





























