Embed Size (px)
Citation preview

In: Recent Developments in Silicone-Based Materials ISBN 978-1-61668-624-6
Editor: Maria Cazacu, pp. 33-75 © 2010 Nova Science Publishers, Inc.
Chapter 2
SOLUTION BEHAVIOR OF POLYSILOXANES
Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi and Maria Cazacu
Petru Poni Institute of Macromolecular Chemistry, 41-A Grigore Ghica Vodǎ Alley,
700487 Iaşi, Romania
ABSTRACT
The thermodynamics of flexible polymers in solution have attracted the interest of
many investigators. Because of its unique properties (low viscosity even at high
molecular weight, high solubility in non-polar solvents, high volatility at low molecular
weight, low surface tension, high refractive index, etc.), poly(dimethylsiloxane) has been
the subject of various studies with respect to its solution properties. Several theories have
been put forward in an effort to explain the various phenomena occurring when very
flexible polymer molecules are dissolved in solvents of different qualities.
The aim of this chapter is to review briefly the background of thermodynamic and
rheological properties for different polysiloxanes in solution and to provide the recent
achievements in this area. The applicability of the theories explaining the various
phenomena that occur when very flexible polymer molecules are dissolved in solvents of
different qualities was critically discussed for the particular case of the polysiloxanes.
The extension of the Flory-Huggins theory by using the new concept of dimensional
relaxation developed recently by Wolf [1,2] enables the modeling of several hitherto
unexplainable anomalous phenomena, as for example the uncommon molecular weight
dependencies of second virial coefficient of poly(dimethylsiloxane) in dilute solutions
[3]. Rheological and optical properties of solutions of polysiloxanes can be regarded in
correlations with their interest for practical applications.
LIST OF SYMBOLS
AP-PDMS -bis(3-aminopropyl)-poly(dimethylsiloxane)
H3C-PDMS -bis(trimethylsilyl)-poly(dimethylsiloxane)
H-PDMS -bis(H)-poly(dimethylsiloxane)

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
HO-PDMS -bis(OH)-poly(dimethylsiloxane) known as
poly(dimethylsiloxane)--diol
MEK methyl ethyl ketone
PDMS poly(dimethylsiloxane)
PDMPS poly(dimethylsiloxane-co-diphenylsiloxane)
2A second virial coefficient
c polymer concentration
*c critical concentration that separate the dilute/semidilute regions
**c critical concentration that separate the semidilute/concentrated regions
c polymer concentration at which the dimensions of the polymer coils
are considered to have shrunk to their unperturbed dimensions
c Flory’s characteristic ratio
d fractal dimension
D , D translational diffusion coefficient in perturbed and unperturbed state
aE flow activation energy
'G storage modulus (elastic)
"G loss modulus (viscous)
Bk Boltzmann constant, 1.380650310-23
m2kg/(s
2 K)
Hk Huggins dimensionless constant
k unperturbed dimensions parameter
l average main chain bond length
m molecular weight of the mer
M molecular weight of the polymer
eM entanglement molecular weight
LM stiff factor
nM number average molecular weight of the polymer
wM weight average molecular weight of the polymer
nw M/M polydispersity index
n relaxation exponent
2n refractive index of the polymer
N number of segments
AN Avogadro’s number, 6.0221023
cn number of clusters
r particle radius in colloidal solutions
R ideal gas constant, 8.314 J/(Kg mol)
cR radius of the clusters
HR hydrodynamic radius
uR molar refractivity

Solution Behavior of Polysiloxanes
35
2ofS hypothetical mean square radius of gyration of a chain in which the
internal rotation around the bonds of the main chain is completely free
2S , 2S mean-square radii of gyration in perturbed and unperturbed states
T absolute temperature
tan loss angle tangent
uV molar volume
z excluded volume parameter
a measure of the effect of contact formation between solvent
molecules and polymer segments at fixed chain conformation
(according to the Wolf’s theory which is based on the dimensional
relaxation concept)
H hydrodynamic expansion factor
S static expansion factor
viscometric expansion factor
binary cluster integral for interactions between repeat units
)( Flory-Huggins interaction parameter dependent on polymer
concentration; frequently it is simply denoted
o Flory-Huggins interaction parameter at infinite dilution
H enthalpy contributions to the Flory-Huggins interaction parameter
S entropy contributions to the Flory-Huggins interaction parameter
o Flory's constant, 2.511021
(for ][ expressed in dL/g)
shear rate
][ , ][ intrinsic viscosity in perturbed and unperturbed state
o zero shear viscosity
s viscosity of the solvent
rel relative viscosity
complex viscosity
shear shear viscosity
sp specific viscosity
c/sp reduced viscosity
volume fraction of the polymer
o constant curvature of the characteristic helix (according to the
Yamakawa’s theory, i.e., the helical wormlike touched-bead model) 1
o characteristic stiffness parameter (according to the Yamakawa’s
theory, i.e., the helical wormlike touched-bead model)
intra-molecular interaction parameter (according to the Wolf’s
concept of the dimensional relaxation)

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
)z( hydrodynamic penetration function
theta condition ( -solvent, -temperature)
coil density
steric factor
long-range relaxation time
o torsion of the characteristic helix (according to the Yamakawa’s
theory, i.e., the helical wormlike touched-bead model)
oscillation frequency
a parameter which quantifies the contributions of the conformational
changes taking place in response to dilution (according to the Wolf’s
concept of the dimensional relaxation)
)z( interpenetration function
1. INTRODUCTION
The increasing interest in polysiloxanes is mainly due to their unique properties, which
are related to their chemical structure and macromolecular architecture, generating a very
important group of products with a wide range of applications.
Poly(dimethylsiloxane) (PDMS) shows some peculiar features in chain construction [4]
because its chain, [Si(CH3)2O]x, has a long SiO bond length, a small van der Waals
radius of the O-atom and a large angle at the O-atom that may reduce the steric repulsion but
does not eliminate the steric conflict for all conformations. The equilibrium flexibility of a
polymer chain, that is the ability of the backbone to rotate, has always been of great interest
for polymer scientists. Differences in physical properties between siloxanes and hydrocarbons
have been attributed to free internal rotation around SiO bond than around SiC bond. The
siloxane chain is flexible in the sense that many configurations are accessible to it. The
inherent rotational barrier around the SiO bond at ordinary temperature is of the order of
RT (where R is the ideal gas constant and T
is the absolute temperature) [5]. Due to their
special properties, the polysiloxanes represent a group of quite unique polymers. They present
very important physical and chemical properties: high chain flexibility, very low glass
transition temperature, very good resistance at temperature, oxidant agents and ultraviolet
radiation, good gas permeability and biocompatibility, but they exhibit rather poor
mechanical properties.
However, the above properties refer mainly to those in the bulk state, so it is not adequate
to conclude that the behavior is similar in the case of isolated polysiloxane chains in dilute
solution, especially regarding the backbone flexibility. Yamakawa and coworkers [6] suggest
that poly(dimethylsiloxane) chains are rather stiff in the static sense. Due to these
considerations, the properties of polysiloxanes in solution are of great interest.
Generally, the classical Flory-Huggins theory [4] was used to describe qualitatively the
behavior of polysiloxanes in solution. However, some inconveniences appear due to several
unrealistic assumptions [2]. In dilute solutions, the segments of the macromolecular coils
cannot distribute among the entire volume of the system due to their chemical bonds (chain
connectivity). Many efforts were carried out in order to find out a better theoretical

Solution Behavior of Polysiloxanes
37
description. Some sophisticated approaches, such as the excluded volume theory [7] or helical
wormlike chain model [8], were applied to explain different behaviors of polysiloxanes [6,9-
14]. However, even these approaches are incapable to accounting some experimental
findings, e.g., the increase of the second virial coefficient (equivalent to a decrease of the
Flory-Huggins interaction parameter, ) with rising the molecular weight of the polymer
[3,15]. Another discrepancy appears in modeling the minima observed in dependence of
on the volume fraction [16]. These difficulties can be overcome by accounting for the
connectivity of monomers in a macromolecular chains and their ability to modify the
conformation in response to the changes in their molecular surrounding resulting from the
mixing process [1-3]. Some observations concerning the application of this concept to
polysiloxanes solutions will be given in this chapter.
Table 1. Polysiloxanes with different terminal groups and their acronyms
CH3
CH3 CH
3
CH3
CH3CH
3
Si SiO O
Si m
HO OH
HO-PDMS
-bis(OH)-
poly(dimethylsiloxane), usually
known as
poly(dimethylsiloxane)--
diol
CH3
CH3 CH
3
CH3
CH3CH
3
Si SiO O
Si m
CH3
CH3
H3C-PDMS
-bis(trimethylsilyl)-
poly(dimethylsiloxane)
CH3
CH3 CH
3
CH3
CH3CH
3
Si SiO O
Si m
H H
H-PDMS
-bis(H)-
poly(dimethylsiloxane)
CH3
CH3 CH
3
CH3
CH3CH
3
Si SiO O
Si HH2
m
H2N
AP-PDMS
-bis(3-aminopropyl)-
poly(dimethylsiloxane)
Si HO O
SiOH
m
n
C6H
5
C6H
5
CH3
CH3
PDMPS
poly(dimethylsiloxane-co-
diphenylsiloxane)

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
Depending on the synthesis procedure, the poly(dimethylsiloxane)s can possess different
terminal groups (Table 1), which influence the properties of their solutions, especially in the
region of low molecular weights.
2. THERMODYNAMIC ASPECTS OF POLYSILOXANES IN SOLUTION
The thermodynamic properties of linear polymer solutions are usually discussed from the
point of view of their dependence on two parameters, polymer concentration, c , and
molecular weight M . Generally, polymer solutions can be divided into three regions: dilute,
semidilute and concentrated, separated by the critical concentrations *c and **c ,
respectively [17]. In dilute solution the concentration is sufficiently low ( c < *c ), so the
macromolecular chains conserve their individuality and the intermolecular interactions are
ignored. The semidilute concentration regime is characterized by intermolecular interactions
and eventually entanglements; segment concentration is still low ( *c < c < **c ) but
polymer coils begin to overlap. In concentrated domain, the segment concentration and the
degree of coil overlapping are high ( c > **c ). The macromolecules lose their individuality
due to the formation of an infinitely large network of overlapping molecules. In view of the
fact that the expressions dilute and concentrated are only qualitative and therefore rather
arbitrary, the state of polymer solutions is defined in terms of the so-called chain overlap
parameter, which is defined as the product of the intrinsic viscosity of the polymer in a given
solvent and its concentration in the solution. A solution is considered as dilute if the chain
overlap parameter remains below 1–2 and as concentrated if it assumes values larger than 6–
10 [3].
According to well established thermodynamic concepts, the influences of chain length on
the thermodynamic properties of polymer solutions should vanish as soon as polymer
concentration exceeds a critical concentration c ( *c <
c < **c ) at which the dimensions
of the polymer coils are considered to have shrunk to their unperturbed dimensions. At higher
concentrations, including **c , the dimensions remain unchanged [18].
Poly(dimethylsiloxane) is one of the most thoroughly studied polymers and there is a
relative abundance of data for various PDMS/solvent systems. However, there is a dispersion
of the parameters calculated from the experimental data even for the same thermodynamic
conditions [19], so it becomes difficult to discuss the basic parameters that characterize its
macromolecular chain in solution.
Cyclic and linear PDMS chains have distinct properties in dilute solutions due to the
differences of the spatial distributions of segments. For linear polymers, the behavior of chain
ends contributes less to overall properties as chain length increases. In concentrated systems,
the individual molecular identity is less important as chain length increases. The
predominance of intermolecular interactions rather than intramolecular ones, even through
individual chains adopt unperturbed conformations, was discussed in terms of segmental
behavior over sections of chains. A comparative study of linear and cyclic PDMS in the bulk
state in the limit of M illustrated the segmental nature. In the two types of chain there
is an increase in segmental flexibility with chain length tending to the same value at infinite
chain length [20].

Solution Behavior of Polysiloxanes
39
The heat of mixing to infinite dilution for PDMS in various solvents and their mixtures
was discussed in different papers [21-25]. Morimoto [23] summarized and discussed
calorimetric results for mixtures of PDMS with a large number of organic solvents. The
thermodynamic properties were also investigated above the usual dilute range of PDMS
solutions [25-28].
Summers et al. [27] have used PDMS as the stationary phase and in gas-liquid
chromatography in order to determine the Flory-Huggins interaction parameter from the
retention volumes at high PDMS concentrations. They used as solvents linear and branched
alkanes as well as aromatic hydrocarbons and the determined values of were in good
agreement with those obtained for the same systems from equilibrium vapor-absorption
experiments.
Flory and Shih [29] analyzed the thermodynamic properties of PDMS solutions and they
found that the entropies of dilution and excess volumes are abnormally low compared with
theoretical calculations. They assume that the discrepancies between experiment and theory
can be explained through the irregularity of the form of PDMS chain having CH3 pendant
groups spaced by comparatively long SiO and SiC bonds. Possibly the irregularity of the
cross section of the chain obstructs efficient packing of the polymer chains in bulk. Effects of
this nature could be ameliorated by using nonpolar solvents which easily fit into irregularities,
determining a decrease of entropy and excess volumes.
The partition function for a solution of PDMS gives rise to an important effect on the
residual free energy of mixing. The thermodynamic functions for the mixing of PDMS
molecules with solvent molecules contain contributions from the exothermic orientation of
the solvent molecules in cooperation with PDMS segments and from an endothermic effect
arising from the penetration of the macromolecular chains into the solvent [30]. During
dissolution of the polymer, a change in dimensions occurs (depending on the solvent quality).
This process is accompanied by a change in intermolecular conformational energy, in
addition to the creation of a certain number of polymer-solvent interactions. The change of
conformational energy in the macromolecular chain influences the heat of mixing of a
polymer-solvent system [31,32]. These results were interpreted with respect the large radius
of the PDMS segment, the existence of the oxygen atom in the PDMS backbone chain and
chain flexibility [4].
The Flory-Huggins polymer-solvent interaction parameter has been traditionally
associated with the heat of mixing of polymer and solvent liquids and with difference in
contact energies. Usually, is mainly entropic and associated with the difference in free
volume between polymer and solvent [33].
A series of papers reported the polymer-solvent interaction parameter for PDMS
solutions [25-28,34-36]. This parameter contains two terms: the first term is due to the
difference in the contact energy between segments of PDMS and the solvent molecules; the
second one is so called structural effect, principally due to the difference in chain length or
bulkiness between the chains [33].
The macromolecular chain of PDMS is able to take a variety of spatial configurations
[29]. It was ascertained [37] that the interaction sites for a PDMS molecule in solution are
assumed to be in peripheral methyl groups and oxygen atoms of the chain, while the terminal
methyl groups may exert the intermolecular forces differing from those of mid-chain

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
repeating units. The proportion of the former to the interaction energy probably decreases
with the molecular weight of PDMS.
2.1. Polysiloxanes in Unperturbed State
In a poor solvent, the polymer-solvent interactions are not favored, and therefore
attraction forces between chains predominate, consequently the random coil adopts a
contracted conformation. The situation when the second virial coefficient, 2A , is zero is
known as - (theta) state and depends on the polymer-solvent system. In this state, the
polymer chains behave as ideal ones, the interactions of monomer units which are far from
each other along the chain are neglected. Any polymer can reach its -state, either choosing
the appropriate solvent (named -solvent) at constant temperature or adjusting the
temperature (named -temperature or Flory temperature) in a given solvent.
The unperturbed dimensions of PDMS chains appear to be affected by the solvent
medium to a much greater degree than for polymethylene chains or other nonpolar polymers.
The polarity of the SiO bond, and perhaps also of SiC, may render this chain more
sensitive to the influence of the medium [38]. The configuration of PDMS chain in
concentrated solutions and in the bulk (in absence of the solvent) was approximated that of
the unperturbed chain in dilute solution [4,39]. For PDMS solutions in bromocyclohexane
there is an uncertainty in temperature: 28C, as determined from viscosity measurements
[40] and 29C [41] or 29.5C, as determined from light scattering measurements [6].
In a series of papers [6,10,11,42], Yamakawa and coworkers investigated the properties
of dilute solutions of PDMS in perturbed and unperturbed states. They explained
quantitatively the dependences of thermodynamic and hydrodynamic parameters on the
molecular weight on the basis of the helical wormlike touched-bead model with a proper set
of values of the model parameters, i.e., the constant curvature, o , and torsion, o , of the
characteristic helix, the characteristic stiffness parameter, 1
o , and the stiff factor, LM .
The parameter 1
o gives a measure of static stiffness of PDMS chains. The high
flexibility of PDMS chains was widely accepted due to the extremely low glass transition
temperature and small bulk modulus of PDMS chains. However, for single isolated PDMS
chain in dilute solution of bromocyclohexane at 29.5C (theta) a value of 31Å was found
for1
o . Thus, it was concluded that PDMSs have the dynamic stiffness of the same order of
magnitude as the atactic polystyrene chains for which 1
o = 23.5Å in cyclohexane at 34.5C
( conditions), although the chemical structure of PDMS is different from those of vinyl
polymers. It appears that PDMS chain is not so flexible in solution as expected from the bulk
properties, e.g., the glass transition temperature and the bulk modulus. According to helical
wormlike touched-bead model [6,11], these quantities reflect the mobility of rather small
portions of polymer chains in the bulk, so that they depend not only on the kinetics of those
portions, but also on the interactions with the surrounding chains. The behavior of a single
PDMS chain can be explained from the correlations between static and dynamic stiffness and
can be due to the so-called draining effect which was found for typical flexible polymers like

Solution Behavior of Polysiloxanes
41
PDMS. In order to explain the smallest values of the intrinsic viscosity reported for siloxane
oligomers, Yamakawa and coworkers [6,43] have considered that the nonslip boundary
conditions on the bead surface may break down for these chains. This suggests that the
intermolecular interactions are rather weak for polysiloxanes, influencing the glass transition
temperature and the bulk modulus.
The transport properties of PDMS in the unperturbed state, i.e., the intrinsic viscosity,
][ , and the translational diffusion coefficient, 1
D
, was considered to be influenced by
the draining effect when ][ becomes negative in the region of oligodimethylsiloxane in
bromocyclohexane [6]. If the draining effect exists in PDMS unperturbed Gaussian chain, the
total friction force exerted by the chain on the surrounding solvent becomes smaller than that
in the nondraining limit. Thus, the values of some parameters, such as: the intrinsic viscosity
][ and the hydrodynamic radius, ,HR (determined from the translational diffusion
coefficient, 1
D
, ,HR1
D
), become smaller than the respective values in the
unperturbed state [44]. The relations 5.0M][ and 5.0
,H MR hold only
asymptotically in the limit of large M (nondraining effect) and the exponents of M become
larger than 0.5. The quantities 5.0M/][ and 5.0M/D for PDMS in bromocyclohexane at
29.5C depend appreciably on the molecular weight, even for large value of M for which the
ratio M/S2 is independent (Figure 7 of the reference [6]), being attributed to the
draining effect.
The intrinsic viscosity becomes negative for PDMS with very small molecular weight in
bromocyclohexane (Figure 4 of reference [6]). Negative viscosity was also reported for
polyisobutylenes in isoamyl isovalerate and in benzene [43]. Such unusual behavior was
regarded by the authors as arising from the specific interactions between polymer and solvent
molecules, being impossible to explain it theoretically within the framework of classical
hydrodynamics. They have converted the negative ][ value to the corrected intrinsic
viscosity which was directly compared with the polymer classical hydrodynamic theories, by
subtracting from the ][ value the negative contribution from the specific interactions
between solute and solvent molecules. For a given polymer, the quantity depends on the
solvent [6, 11], even in the range of large values of wM . This suggests a dependence of the
unperturbed chain dimensions also on the limiting value for the Flory-Fox factor, .
We note that such unusual negative values of the intrinsic viscosity were obtained by
using Huggins and Fous Mead equations [6,9,11]. For many polymer solutions, the Huggins
plots change the slope as a function of polymer concentration, making more difficult the
extrapolation of the experimental data to zero polymer concentration. The turning down of the
specific viscosity in the region of very low concentrations could be responsive for the
negative values of the intrinsic viscosity for oligomers of dimethylsiloxane. It appears now
interesting to evaluate the intrinsic viscosity by using a new method, recently published in the
literature (see section 2.3.2.), which gives linear dependences both for polyelectrolytes [45-
48] and neutral polymers [49].

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
The steric factor, , represents a measure of the hindrance to internal rotation around the
bonds of the backbone of a flexible chain in the unperturbed state. It can be calculated
according to:
2of
2
S
S (1)
where 2ofS is the hypothetical mean square radius of gyration of a chain in which the
internal rotation around the bonds of the main chain is completely free, any type of
interactions are absent ( M/S2of = 3.872 10
-18 [19].
In unperturbed state, the flexibility of macromolecular coils is also determined by the
Flory’s characteristic ratio, c , defined through the equation:
22
nNl/)S6(limc
(2)
where N is the number of bonds, l is the average main chain bond length.
Dilute solutions of poly(dimethylsiloxane)s with different terminal groups were
investigated extensively [8-10,50-53]. It was found that introducing amino end groups the
chains become stiffer as compared to PDMS. One exception was observed for H-PDMS,
when the volume of the terminal group (hydrogen) is lower than the volumes occupied by the
other terminal groups, determining an increase of the flexibility in the unperturbed state
(Table 2).
The unperturbed dimension parameter, k , was determined for polysiloxanes by different
methods (Table 3):
from the Mark-Houwink dependence in theta conditions (theta solvent, theta
temperature):
5.0Mk][ (3)
from the ][ – M dependence in the region of oligomers, when the behavior is
similar with those observed in theta conditions [14,52].
from the relation between k and the unperturbed dimensions 2S through the
average value M/S2 :
2/32o
2/3 M/S6k (4)
where o is Flory's constant, o = 2.511021
(for ][ expressed in dL/g).
Solution Behavior of Polysiloxanes
43
from the viscometric data obtained in good solvent conditions, ][ . The intrinsic
viscosity in unperturbed state, ][ , can be determined according to [18,54]:
*)c/cexp(*c/77.0
*)c/cexp(1][[][
(5)
where *c is the critical concentration at which the polymer coil begin to overlap
each other, is the coil density.
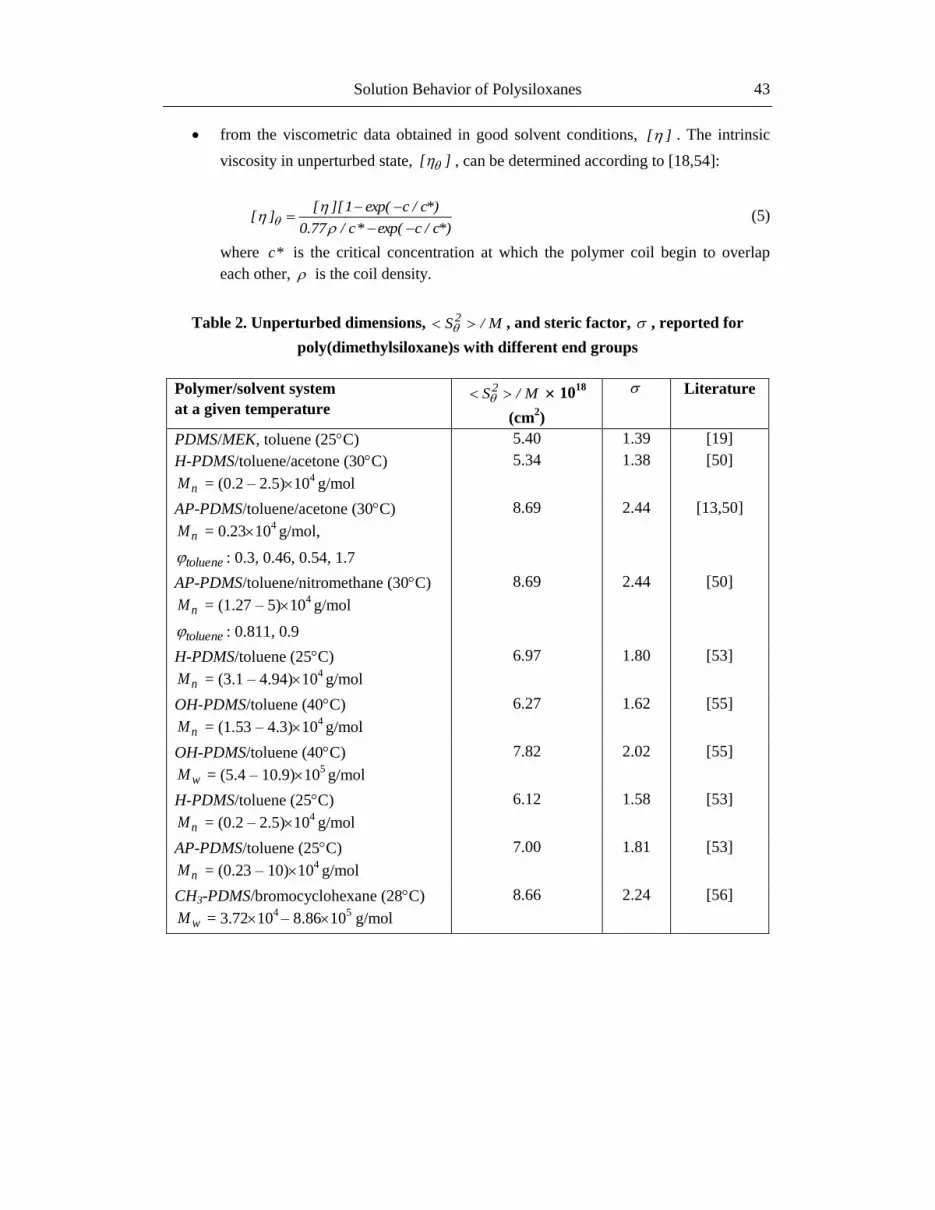
Table 2. Unperturbed dimensions, M/S 2 , and steric factor, , reported for
poly(dimethylsiloxane)s with different end groups
Polymer/solvent system
at a given temperature M/S2 10
18
(cm2)
Literature
PDMS/MEK, toluene (25C) 5.40 1.39 [19]
H-PDMS/toluene/acetone (30C)
nM = (0.2 – 2.5)104 g/mol
5.34 1.38 [50]
AP-PDMS/toluene/acetone (30C)
nM = 0.23104 g/mol,
toluene : 0.3, 0.46, 0.54, 1.7
8.69 2.44 [13,50]
AP-PDMS/toluene/nitromethane (30C)
nM = (1.27 – 5)104 g/mol
toluene : 0.811, 0.9
8.69 2.44 [50]
H-PDMS/toluene (25C)
nM = (3.1 – 4.94)104 g/mol
6.97 1.80 [53]
OH-PDMS/toluene (40C)
nM = (1.53 – 4.3)104 g/mol
6.27 1.62 [55]
OH-PDMS/toluene (40C)
wM = (5.4 – 10.9)105 g/mol
7.82 2.02
[55]
H-PDMS/toluene (25C)
nM = (0.2 – 2.5)104 g/mol
6.12 1.58 [53]
AP-PDMS/toluene (25C)
nM = (0.23 – 10)104 g/mol
7.00 1.81 [53]
CH3-PDMS/bromocyclohexane (28C)
wM = 3.72104 – 8.8610
5 g/mol
8.66 2.24 [56]

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
Table 3. k Values reported for polysiloxanes in different experimental conditions
Solvent, temperature Molecular weight of
PDMS
(g/mol)
k 104
(dL/g)
c Literature
MEK, 20C ( ) 9.3104 – 6.8510
5 7.5
[15]
MEK, 20C ( )
3105 – 1.110
6 8.07 [3]
MEK, 20C ( )
7.5104 – 810
5 7.11 [58]
MEK, 20C ( )
and C8F18/CCl2F
(33/67, w/w) 22.5C ( )
5.5105 – 1.210
6 10.6 7.6 [59]
bromocyclohexane, 28C
( )
> 2105 7.8
6.3 [40]
bromocyclohexane, 28C
( )
4104 – 910
5 7.95 [56]
bromocyclohexane, 29C
( )
3.3104 – 1.0610
6 7.45 [41]
bromocyclohexane, 29.5C
( )
3.3104 – 1.0610
6 8.6 [6,9,11]
bromocyclohexane, 28C
( )
7.5104 – 810
5 6.79 [58]
toluene/nitromethane and
toluene, 25C
(OH-PDMS, CH3-PDMS)
0.23104 – 10
5 5.42 – 9.4 [52]
toluene, 25C 2.8104 – 6.310
4 4.85 [57]
different solvents 7 – 8 [19]
From Tables 2 and 3 one observes that the unperturbed dimensions are influenced by the
solvent and temperature, on the one hand, and by the end group of the polymer, on the other
hand. The amino terminal groups determine the stiffness of the polysiloxane short chains, as it
is illustrated by the increase of the steric factor, , or unperturbed dimension parameter, k .
Also, the method of the determination can influence the values of the unperturbed
dimensions and chain flexibility. As for example, for OH-PDMS and CH3-PDMS samples,
the ][ values obtained by using equation (5) are independent on concentration and
temperature. The resulted value of 4.8510-4
dL/g for k is lower than the values obtained in
theta conditions [57] (Table 3).
The uncertainty observed in the -temperature determination for PDMS in solution of
bromocyclohexane influences the accurate determination of the unperturbed dimensions.

Solution Behavior of Polysiloxanes
45
Thus, the value of 28C for theta temperature, which has been determined from viscosity
measurements [40], is somewhat lower than the value of 29C [41] or 29.5C [6,9,11],
determined from light-scattering measurements.
2.2. Excluded Volume Effects
The behavior of dilute polymer solutions, expressed by different parameters (the second
virial coefficient, the mean dimensions, the intrinsic viscosity) and influenced by temperature,
solvent quality, molecular weight domain, can be discussed through different excluded
volume theories. The theoretical approaches mutually differ in the used mathematical
methods and approximations, but all of them relate the excluded volume effects to measurable
quantities, by considering different possible interactions (polymer-solvent, short- and long-
range, intra- and intermolecular interactions).
The solvent quality plays an important role in determining the type and magnitude of
polymer-solvent interactions. Thus, in a good solvent, the attraction forces between the chain
segments are smaller than the polymer-solvent interactions, so the random coil adopts an
extended conformation. The excluded volume interaction is responsible for the swelling
behavior of the overall chain dimensions in a good solvent, and the polymer is dissolved over
a wide range of temperatures.
Theoretical investigations have established different equations in which the second virial
coefficient, 2A , and the expansion factors ( ) are functions of short-range and long-range
interactions through the interpenetration function, )z( . Thus, for flexible polymers in dilute
solution, some dimensionless parameters, such as the linear expansion factor
22S S/S (where 2S and 2S represent the mean square radii of gyration
in perturbed and unperturbed states, respectively) and the interpenetration function, )z( ,
are universal functions of a single variable, z , called the excluded volume parameter. This
parameter is defined as:
2/12/32o
2/3 M])M/S/(B[)4/1(z (6)
where 2m/B , – the binary cluster integral for interactions between repeat units, m –
molecular weight of the mer.
The interpenetration function, )z( , is defined as:
2/32A
2/3
22
SN4
MA)z(
(7)
2A is the second virial coefficient and AN – the Avogadro’s number.
The hydrodynamic penetration function, )z( , can be expressed as:
][)( 2
MAz (8)

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
For the perturbed Gaussian chain, Douglas and Freed [60-62] have developed the
renormalization group theory (RTP) which predicts that both (the viscometric expansion
factor) and H (hydrodynamic expansion factor) have smaller values in draining limit than in
the nondraining limit for large excluded volume parameter, z . According to the RTP
predictions, and H as a function of z (that takes into account the effects of the
excluded volume and chain stiffness on the basis of the helical wormlike chain [63]) depends
on the degree of draining. Thus, and H plotted against z do not give respective single-
composite curves. The validity of RTP theory was examined for PDMS dilute solutions in
toluene at 25C by comparing the dependences of and H as a function of z with the
simple composite curves obtained for polyisobutylene, atactic polystyrene, atactic and
isotactic poly(methyl methacrylate) [11]. The results showed that there is no draining effect
on and H for PDMS as in the case of others flexible polymers, despite the fact the
draining effect is significant for PDMS in the unperturbed state [6]. The experimental results
were found in a semiquantitatively agreement with the Yamakawa-Yoshizaki theory [64]
which takes into account the possible effect of fluctuating hydrodynamic interactions on H .
The hydrodynamic, static and viscometric expansion factors, H , S and ,
respectively, were investigated as a function of z for PDMS in toluene at 25C [11]. Because
the hydrodynamic radius in toluene is larger than in bromocyclohexane at theta temperature,
even in the region of low molecular weights, the dimensions of the PDMS chains depend
appreciable on the solvent. As it was discussed above (chap. 2.1), due to the specific
interaction between the polymer and solvent molecules, the intrinsic viscosity is also changed
in good and theta solvent [6]. These facts make impossible to evaluate H and for PDMS
in toluene by using the values of ,HR and ][ directly measured in theta conditions.
Horita et al. [11] estimated 0,HR and 0][ for PDMS chains in toluene at 25C by
multiplying the values of ,HR and ][ measured in a theta solvent at theta temperature
(bromocyclohexane at 29.5C) by proper factors that represent the effect of the solvents. By
plotting H as a function of S calculated in this way from the experimental data obtained
for different PDMS samples [11,55], a single composite curve was obtained (Figure 1a). Also,
3 against 3
S form a single composite curve if the values of and S are correctly
evaluated. With increasing the molecular weight (or S ), the draining effect found for PDMS
in the unperturbed state can cause a progressive decrease of 3 from the single composite
curve (Figure 1b).
Yamakawa and coworkers [65,66] predicted a regularity of )z( values in the low
excluded volume region by changing either the solvent quality, either the molecular weight of
the polymer. Starting from the thermodynamic parameters (chain dimensions, second virial
coefficient and intrinsic viscosity data), the excluded volume effect was investigated for
different polysiloxanes [12-14,53]. Negative excluded volume effects ( )z( < 1 and )z( <
0) were obtained for oligomers of dimethylsiloxanes with amino end groups (AP-PDMS) in
toluene and toluene/acetone solvent/non-solvent mixtures at 28C.

Solution Behavior of Polysiloxanes
47
log S
0
0.1
0.2
0.3
0.4
0 0.1 0.2 0.3 0.4
log H
(a)
log S3
0
0.2
0.4
0.6
0 0.2 0.4 0.6 0.8
log 3
(b)
Figure 1. Double logarithmic plots of: (a) H against S and (b) 3 against 3
S for HO-PDMS in
toluene at 25C: (o) [11]; (■) [55].
In contradiction with the behavior observed for long flexible chain oligomers of HO-
PDMS and AP-PDMS, the proportionality 5.0M][ presents some deviations. The
viscosity of AP-PDMS in toluene and toluene/nitromethane mixture (volume fraction of
nitromethane of 0.9) shows upward deviations for nM < 104 [52]. These deviations increase
with increasing the solvent quality and molecular weight. Thus, short chains of PDMS exhibit
anomalous characteristics in dilute solution, the excluded volume effects do not disappear
and 5.0M/][ is not independent on M . This behavior was considered as a consequence of
the non-Gaussian character of flexible chains in good solvents (this state vanishes by
decreasing the solvent quality). Furthermore, in mixtures of solvents, cosolvency and
preferential adsorption phenomena can appear. Preferential adsorption for AP-PDMS in

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
toluene/nitrometane mixture was observed to be relatively independent on the molecular
weight, both components of the solvent mixture having low affinity for the polysiloxanes
[67].
2.3. Thermodynamic Parameters
2.3.1. Hydrodynamic Radius
Dynamic light scattering technique provides the translational diffusion coefficient, D ,
from which HR can be calculated according to:
D6/TkR sBH (9)
where Bk is the Boltzmann constant, T is the absolute temperature, s is the viscosity of the
solvent.
Edwards and coworkers [68] used the classical boundary-spreading technique to
determine the diffusion coefficients of cyclic and linear oligo- and poly(dimethylsiloxane)s
with 444 g/mol < nM < 4.8104 g/mol in toluene at 25C. Mean square radii of gyration,
2S , was calculated from the diffusion data for both cyclic and linear
poly(dimethylsiloxane)s assuming Gaussian statistics, and it was found to be in good
agreement with the corresponding values obtained by neutron scattering. For short PDMS
chains, the HR values were not in agreement with those calculated according the rotational
isomeric state model [69]. This discrepancy was attributed to deviations from Gaussian
behavior, and a better agreement between experiment and theory was achieved by using
appropriate values of the interpenetration function, which relates radii of gyration and
impermeable hydrodynamic diffusion radii.
The hydrodynamic radius ( HR ) in good (toluene, 25C) and theta (bromocyclohexane,
29.5C) solvent for oligo- and poly(dimethylsiloxane)s was determined and discussed by
Yamakawa and coworkers [11]. Figure 2 shows double logarithmic plot of 5.0H M/R ( HR in
Å) as a function of M for PDMS in toluene at 25C [11,57,68] and bromocyclohexane at
29.5C [6].

Solution Behavior of Polysiloxanes
49
log(R H /M w0.5
)
-1.0
-0.9
-0.8
-0.7
-0.6
-0.5
2 3 4 5 6 7log M
TL
BCH
Figure 2. Double logarithmic plot of 5.0H M/R as a function of M for PDMS in toluene (TL) at 25C
(o) [11], (Δ) [68] and () [57] and bromocyclohexane (BCH) at 29.5C (▲) [6] and (■) [11].
For all range of molecular weights, the hydrodynamic radius of PDMS in toluene is
larger than in bromocyclohexane at theta temperature. A significant difference is observed
even in the region of low molecular weights ( M < 103 g/mol) where the excluded volume
effect may be negligible.
2.3.2. Intrinsic Viscosity
Generally, the viscosity of dilute polymer solutions depends on the nature of polymer
and solvent, the concentration of the polymer, its average molecular mass and molecular mass
distribution, the temperature, and the shear rate. Very often, the intrinsic viscosity, ][ , is
determined through the Huggins equation:
c][k][c/ 2HHHsp (10)
Hk is referred as the Huggins dimensionless constant and relates to the size and shape of
polymer segments, as well as to hydrodynamic interactions between different segments of the
same polymer chain. The values of Hk are in the range of 0.3 (for good polymer-solvent
interactions) to 0.5 (for poor polymer-solvent interactions). Other equations and their
applicability limits were recently reviewed [70].
Figure 3 presents a classical way to analyze the viscometric data by following the
dependences of the reduced viscosity ( c/sp ) as a function of concentration ( c ) for
solutions of the five H3C-PDMS and HO-PDMS samples in toluene at 20C, 40C and 60C.
It can be observed a difference of temperature influence on viscometric behavior for the
solutions of H3C-PDMS (samples 2 and 5), on the hand, and HO-PDMS (samples 1, 3 and 4),
on the other hand.

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
c (g/dL)
0.1
0.3
0.5
0 2 4 6 8
sp /c
(dL/g) 1
2
3
4
5
20oC
2
0.1
0.3
0.5
0.7
0.9
0 2 4 6
1
345
sp /c
(dL/g)40
oC
c (g/dL)
60oC
0.0
0.2
0.4
0 2 4 6
sp /c
(dL/g)2
c (g/dL)
1
5
34
Figure 3. c/sp vs. c dependence in toluene at 20C, 40C and 60C, for PDMS samples with
different terminal groups: 1 (HO-PDMS): wM = 6.2104 g/mol, nw M/M = 1.44; 2 (H3C-PDMS):
wM = 8.2104 g/mol, nw M/M = 1.36; 3 (HO-PDMS): wM = 4.710
4 g/mol, nw M/M = 1.52; 4
(HO-PDMS): wM = 2.4104 g/mol, nw M/M = 1.57; 5 (H3C-PDMS): wM = 2.510
4 g/mol,
nw M/M = 1.43 [55,71].

Solution Behavior of Polysiloxanes
51
At 20C, high values of the reduced viscosity can be observed and a change in slope at
very low PDMS concentration for samples 1, 3 and 4 (HO-PDMS) is depicted. According to
different papers, this change in slope corresponds to the transition from extremely dilute to
dilute regimes at a critical concentration, 'c [70,72]. At very low polymer concentration, only
the intramolecular interactions exhibit and the appearance of cyclic structures is possible.
A decrease of the hydrodynamic dimensions of the macromolecular coils occurs above
'c , in the dilute regime, as shown by the deviation of c/sp vs. c dependences. This
deviation is not evident for H3C-PDMS samples at a higher temperature (e.g., 60C). By
increasing the polymer concentration, in the semidilute regime, the reduced viscosity changes
the slope because intra- and intermolecular interactions are in competition. With increasing
the temperature, the c/sp vs. c curves are significantly different. At 40C the intrinsic
viscosity of HO-PDMS samples is lower than at 20C and, at very low concentration, the
change in slope is not evident. One can suppose that possible associations formed at 20C are
destroyed with increasing temperature. For H3C-PDMS samples, viscosity increases with
increasing the temperature. At 60C, the associated structures are destroyed by thermal
motion and the viscosity of HO-PDMS samples decreases.
Acid terminated poly(dimethylsiloxane)s form supramolecular associations in low
polarity solvents due to the hydrogen bonding interactions of chain ends [73,74]. FTIR
spectroscopy and viscometric data obtained in dichloromethane, carbon tetrachloride and
hexane were used to characterize the supramolecular structures. At high concentrations, chain
extension leads to high viscosity values due to intermolecular hydrogen bonding interactions,
whereas at low concentrations, low viscosity cyclic species are formed by intramolecular
hydrogen bonding interactions. The authors used a quantitative model based on Jacobson-
Stockmayer theory in order to describe the competition between the chain extension and
macrocyclization, by taking into account the variation of the free acid fraction with
concentration in non-polar solvents.
According to literature, Hk can be considered as a measure of the solvent quality: values
around 0.3 are usually obtained in good solvents and 0.5 – 1 in theta solvents [19]. For PDMS
in toluene, the values of Huggins constant ( Hk ) are lower than 0.5 and they increase with
increasing the molecular weight in the range 0.31 – 0.41. The temperature influence is not
significant; there is a small decrease of Hk from 20C to 60C [75]. For PDMS in methyl
ethyl ketone (MEK) at 20C the values tend to be higher than 0.5.
As it was observed for many polymer solutions, in the region of very low concentrations,
the Huggins plots change the slope, making more difficult the extrapolation of the
experimental data to zero concentration. Many efforts were made in order to obtain linear
dependences from the viscometric data in the region of very low polymer concentrations. A
new alternative method was developed very recently for the determination of ][ of
polyelectrolytes [45], according to the following equation:
w
w2
wrel
][Bc1
][][Bc][cln
(11)

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
where B represents a system specific constant ( B = 0.5 - Hk ) which holds true for the range
of pair interactions between the solute [46]) and ][ is the characteristic specific
hydrodynamic volume.
The equation (11), successfully verified for different polyelectrolyte solutions [46-48,76],
and neutral polymers in solution [49], thus it can be applied for polysiloxanes in solution. As
for example, from the initial slope of the dependence relln as a function of c at sufficiently
low shear rates and polymer concentrations we can have access to the intrinsic viscosity of
high molecular weight HO-PDMS (Figure 4). For the same polymer sample, the influence of
the solvent quality can be observed as in the case of Huggins plots [3]. n-Octane (n-O), a very
good solvent for PDMS, induces strong polymer-solvent interactions, determining a higher
viscosity as compared with toluene (TL) solutions. Methyl ethyl ketone, which is a theta
solvent for PDMS at 20C, acts as a marginal solvent at 40C, the polymer-polymer
interactions are more favored for this solvent.
c (g/dL)
0.0
1.0
2.0
0 0.4 0.8 1.2 1.6
ln reln-O
TL
MEK
Figure 4. Plot of relln vs. c for HO-PDMS with wM = 1.09106 g/mol, nw M/M = 1.43, in methyl
ethyl ketone, toluene and n-octane at 40C, according to Wolf’s model (eq. (11)).
The viscometric behavior also depends on the chemical structure of siloxane unit. For a
small content of diphenylsiloxane in the copolymer structure, the macromolecular coil is
more extended and the intrinsic viscosity increases (Table 4), despite the fact that the
molecular weight of the copolymer is smaller as compared with HO-PDMS sample [77].
Figure 5 shows clearer the limitation of the Huggins equation in the data evaluation,
especially in the region of very low concentrations.

Solution Behavior of Polysiloxanes
53
Table 4. The number average molecular weight ( nM ), polydispersity index ( nw M/M ),
the intrinsic viscosity values ( H][ , w][ ), and Huggins constants ( Hk ) of HO-PDMS
and two copolymers of dimethylsiloxane with diphenylsiloxane samples in toluene at
25C
Sample
code
Diphenyl
siloxane
units
(%)
nM 10-4
(g/mol)
nw M/M H][a
(dL/g)
Hk a w][ b
(dL/g)
HO-PDMS 0 18.6 1.56 0.90 0.4 0.83
PDMPS1 5.95 14.45 2.28 1.38 0.39 1.20
PDMPS2 47.83 6.88 1.80 0.69 0.25 0.64 aDetermined by using equation (10).
bDetermined by using equation (11).
0.5
1.0
1.5
2.0
2.5
0.0 0.4 0.8 1.2c (g/dL)
sp /c (dL/g)
HO-PDMS
PDMPS1
PDMPS2
Figure 5. Huggins plot (eq. (10)) for HO-PDMS and copolymers of dimethylsiloxane with
diphenylsiloxane (Table 4) in toluene at 25C [77].
Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
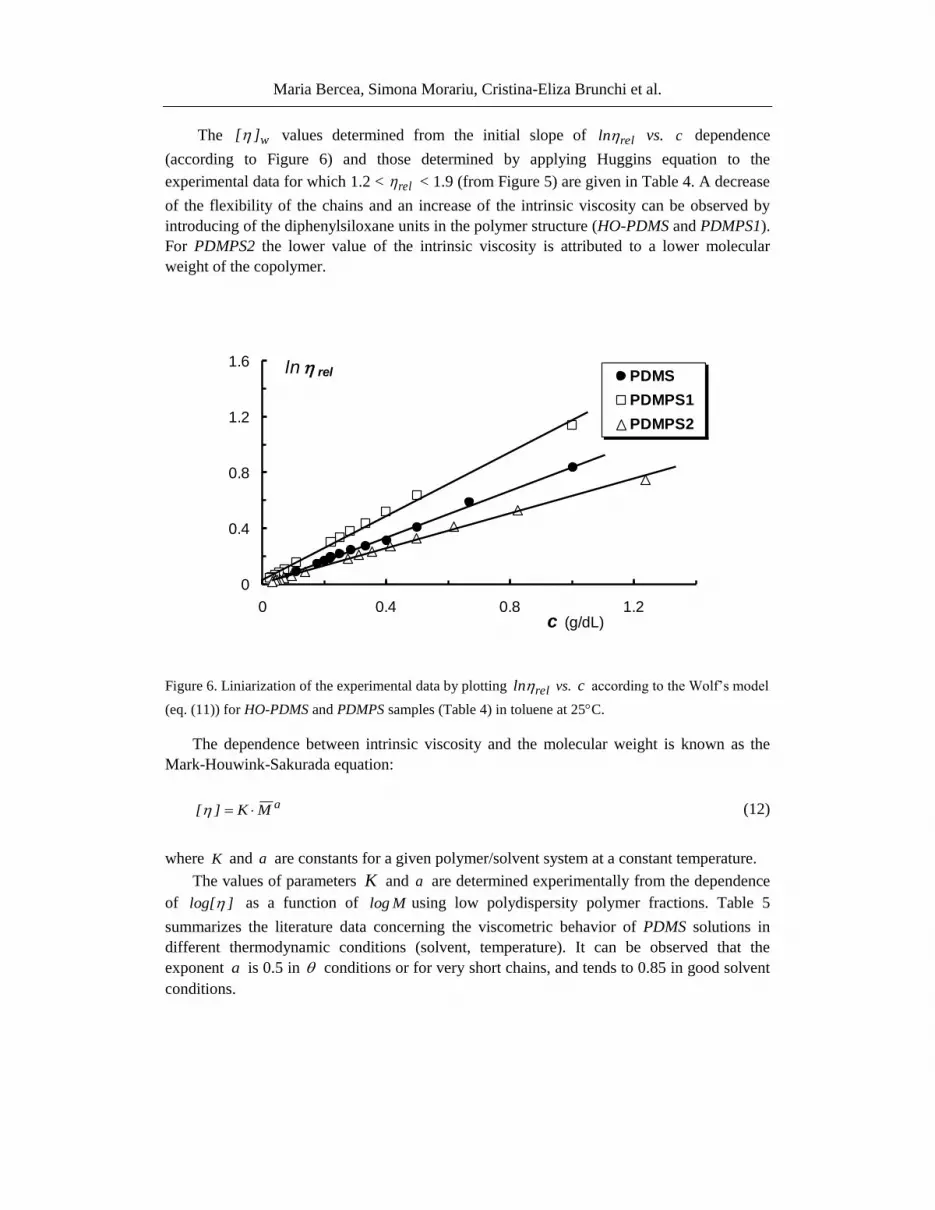
The w][ values determined from the initial slope of relln vs. c dependence
(according to Figure 6) and those determined by applying Huggins equation to the
experimental data for which 1.2 < rel < 1.9 (from Figure 5) are given in Table 4. A decrease
of the flexibility of the chains and an increase of the intrinsic viscosity can be observed by
introducing of the diphenylsiloxane units in the polymer structure (HO-PDMS and PDMPS1).
For PDMPS2 the lower value of the intrinsic viscosity is attributed to a lower molecular
weight of the copolymer.
c (g/dL)
0
0.4
0.8
1.2
1.6
0 0.4 0.8 1.2
ln rel PDMS
PDMPS1
PDMPS2
Figure 6. Liniarization of the experimental data by plotting relln vs. c according to the Wolf’s model
(eq. (11)) for HO-PDMS and PDMPS samples (Table 4) in toluene at 25C.
The dependence between intrinsic viscosity and the molecular weight is known as the
Mark-Houwink-Sakurada equation:
aMK][ (12)
where K and a are constants for a given polymer/solvent system at a constant temperature.
The values of parameters K and a are determined experimentally from the dependence
of ]log[ as a function of Mlog using low polydispersity polymer fractions. Table 5
summarizes the literature data concerning the viscometric behavior of PDMS solutions in
different thermodynamic conditions (solvent, temperature). It can be observed that the
exponent a is 0.5 in conditions or for very short chains, and tends to 0.85 in good solvent
conditions.

Solution Behavior of Polysiloxanes
55
Table 5. The parameters K and a , according to eq. (12), for PDMS solutions in
different thermodynamic conditions
M Solvent, temperature K
(dL/g)
a Literature
< 1.7104
MEK, 20C 0.580 [78]
5104 – 4.710
6 MEK, 30C 8.1510
-4 0.550 [15]
3105 – 1.110
6 MEK, 40C 6.0110
-4 0.565 [3]
7.5104 – 810
5 Bromocyclohexane,
26C (bellow theta
temperature)
1.1110-3
0.430 [58]
2103 - 10
4 Toluene, 25C 0.500 [79]
2103 – 1.3810
6 Toluene, 25C 2.1510
-4 0.650 [80]
> 2105
Toluene, 25C 8.2810-5
0.720 [40]
2.6103 – 6.2510
5 Toluene, 25C 1.3410
-4 0.690 [81]
3103 – 310
5 Toluene, 25C 1.8710
-4 0.658 [82]
1.9104 – 1.310
5 Toluene, 25C 0.840 [83]
0.32104 –
21.73104
Toluene, 25C 2.5110-4
0.623 [52,53]
0.32104 –
21.73104
Toluene/Nitromethane
(90/10), 25C
2.6310-4
0.615 [14,51]
3105 – 1.110
6 Toluene, 25C 4.3310
-4 0.786 [75]
7.5104 – 810
5 Toluene, 30C 1.6510
-4 0.670 [58]
2104 – 910
5 Toluene, 35C 1.2510
-4 0.703 [56]
3105 – 1.110
6 Toluene, 40C 5.3110
-4 0.633 [3]
3105 – 1.110
6 n-Octane, 40C 410
-3 0.787 [3]
1.7104 – 910
5 Cyclohexane, 35C 1.0210
-4 0.735 [19]
The differences observed in Mark-Houwink parameters determined in the same
experimental conditions for a large domain of molecular weights can be due to
measuring/estimating ][ and M , or to differences in chain behavior. For M 104, the
value of ][ in good solvents (as for example toluene) becomes larger that those of ][
determined in theta conditions (as for example bromocyclohexane at 29.5C or MEK at
20C). This suggests that the specific interaction between the polymer and solvent molecules
has a significant effect above this molecular weight.
2.3.3. Polymer-Solvent Interactions
In the polymer solutions, the contributions to the excluded volume depend on the actual
volume of the chain unit as well as on its interaction with the solvent molecules. A measure of
these interactions is the second virial coefficient ( 2A ). In theta conditions, 2A = 0 because the
polymer-solvent interaction vanishes.

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
From the second osmotic virial coefficients the Flory-Huggins interaction parameter at
infinite dilution, o , can be calculated according to:
2212o VA5.0 (13)
o also offers the possibility to quantify the solvent power. From this dependence, it can
be observed that o = 0.5 in theta conditions, o < 0.5 for good solvent conditions and o >
0.5 when the polymer-solvent interactions are unfavorable.
Generally, the thermodynamic particularities of polymer/solvent systems at any
compositions are normally quantified in terms of Flory-Huggins interaction parameter, .
According to experimental evidence and theoretical considerations, does not only depend
on temperature and pressure, but also on the composition of the mixtures and – at least in
dilute solution – on the chain length of the macromolecules. The behavior of dilute solution
properties has been predicted by applying renormalization group methods [7,84,85].
However, these approaches are not able to account the experimental finding that the second
virial coefficients, 2A , may increase with increasing the molar mass of the polymer. In
contrast to the original assumption, the Flory-Huggins interaction parameter ( ) does not
only depend on temperature and pressure, but also on composition and - even in the region of
high polymer concentrations - on the chain length of the polymer. Numerous attempts have
been done to incorporate these features into the theory [6,9-11,25,86].
The experimental information showed that the individuality of macromolecules
survives in thermodynamically good solvents up to almost the pure melt. In order to explain
this behavior, Wolf [1] developed a new concept of dimensional relaxation. This approach
treats the dilution process (the basis for determination of Flory-Huggins interaction
parameters) in two steps: a separation of intersegmental contacts by the insertion of the
solvent, keeping constant the chain conformation; an expansion of the macromolecular coil
(the system rearrange) in order to minimize its Gibbs energy. The following expression was
proposed for the Flory-Huggins interaction parameter at infinite dilution, o :
o (14)
The Wolf concept considers the parameter as a measure of the effect of contact
formation between solvent molecules and polymer segments at fixed chain conformation; this
parameter does not depend on M . is an intra-molecular interaction parameter, which
raises the chemical potential of the solvent in the solution up to the value of the pure solvent.
The parameter quantifies the contributions of the conformational changes taking place in
response to dilution and becomes zero for theta conditions. The sign and extent of the
molecular weight dependence of 2A depends on . For the majority of solutions, 2A
decreases with increasing M and, thus, < 0 explains why solvents are favorable from
thermodynamic point of view. In some rare cases 2A increases with rising M and this is not
in agreement with classical thermodynamic concepts. According to Wolf’s theory, the
increase of 2A with increase of M corresponds to > 0 and expresses the fact that the

Solution Behavior of Polysiloxanes
57
conformational relaxation needs not necessary to be favorable. For sufficiently dilute polymer
solutions, there is the only difference between this new approach and the original Flory-
Huggins theory consists in the second term of eq. (14). According to the theoretical and
experimental considerations, becomes zero under theta conditions (where the coils assume
their unperturbed dimensions) and the conformational relaxation does no longer contribute to
o 2].
By considering the experimental data reported for different polymer-solvent systems,
there is a linear dependence between and .
The influence of the molar mass is included in , a parameter defined as:
)a1(N5.0 (15)
where N is the number of segments and 2NK , a112N ]M)/[(KK , where 1
and
2 represent the density of the solvent and polymer, respectively, and 1M is the molecular
mass of the solvent.
According to the Wolf’s theoretical approach [1,2], the dependence of the second virial
coefficient, 2A , as a function of polymer chain length can be expressed as:
)a1(
122
22 NV
AA
(16)
where:
V2
21A
22
2
(17)
and 1V is the molar volume of the solvent.
For theta solvents 2A = zero and = 0, resulting = 0.5. The sign of
2A depends on
the magnitude of and . An increase of the chain length determines either a decrease
(usually) or an increase of 2A , as a function of the sign of which gives information on the
conformational response of the segments to dilution. and can be evaluated from the
chain length dependence of 2A . We can examine the plot of 2A as a function of )a1(M
and, according to the relation (16), the second virial coefficient is not always zero for infinite
long chains.
Figure 7 is a representation of the experimental data in the traditional way, i.e. in a
double logarithmic plot of the second virial coefficient as a function of number average
molecular weight ( nM ) of PDMS in two thermodynamic situations: good (TL) and poor
(MEK) solvents. [3]. Different qualitative behavior can be depicted in Figure 8: 2A decreases
with increasing the molecular weight for good solvent (TL), when o increases, whereas for
poor solvent (MEK), 2A rises with increasing the molecular weight, thus o decreases for

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
poor solvents. This aspect can not be described theoretically with the well known theories of
thermodynamics [7] according which the second virial coefficient should decrease with
increasing the molecular weight. This discrepancy can be now explained with the new
concepts of dimensional relaxation developed recently by Wolf [1,2].
log A 2
-5
-4
-3
5.4 5.5 5.6 5.7 5.8
log M n
(cm3mol/g
2)
TL
MEK
Figure 7. Double logarithmic plot of 2A as a function of nM for HO-PDMS in TL and MEK at 40C
(data from [1,3]).
104A 2
0
1
2
3
0 2 4 6 8M n
-(1-a)
(cm3mol/g
2) TL
MEK
Figure 8. 2A vs. )a1(
nM
for HO-PDMS in TL and MEK at 40C according to eq. (16) (data from
[1,3]).

Solution Behavior of Polysiloxanes
59
Figure 9 represents the conformational response ( ), as a function of for a fixed
conformation for PDMS [3]. The straight line which is obtained, like for others polymers [1],
shows that, from thermodynamic point of view, the solvents are qualitatively good if the
conformational response is favorable, when the reduction of the Gibbs energy is associated
with the conformational rearrangements, dominated by the entropic contribution.
-1
1
3
5
7
0 1 2 3 4
Figure 9. Parameters of the new Wolf’s approach, as a function of , for a fixed conformation for
HO-PDMS [1,3].
The Gibbs energy of dilution at fixed conformation (dominated by enthalpy) remains low
because the effect must be distributed on the many segments involved in the opening of
intermolecular contacts. The conformational response, , passes zero for 2A = 0,
independent of chain length. Thus, the linear interrelation between the two characteristic
parameters, covering the entire range of solvent quality for PDMS, shows that increases
and decreases as the solvent becomes worse [1,3].
2.3.3.1. Composition Dependence of
The generalization of the Wolf’s approach to arbitrary polymer concentrations yields the
relation:
1(2)q1/()( 2 (18)
where: is the volume fraction of the polymer and q is an additional parameter obtained
from the measured concentration dependence of the interaction parameter.
Thus, only one additional parameter ( q ) is required to incorporate the concentration
dependence in the Flory-Huggins interaction parameter. By using the experimental data

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
obtained for a given polymer-solvent system, for which it is known the o value (limiting
value of for 0 ), the expression can in good approximation be simplified to:
)21(q)q1)(()( 2o (19)
This relation is able to describe the concentration dependence for the Flory-Huggins
interaction parameter for PDMS solutions [2].
2.3.3.2. Enthalpic and Entropic Contributions to
By analyzing how the solvent quality is reflected in concentration and temperature
dependent Flory–Huggins interaction parameter, the enthalpy ( H ) and entropy ( S )
contributions to were evaluated for PDMS in various solvents [25]:
)T/1(T
1H
(20)
HS (21)
where T is the absolute temperature.
Thus, it was possible to obtain information on the enthalpy and entropy parts by plotting
as a function of inverse temperature for different constant concentrations. At infinite
dilution, H is negative for toluene, which is a good solvent (exothermal heat of dilution)
and increases with increasing the polymer concentration, presenting an inversion in the heat
effects. For n-octane, which is a very good solvent, it was also observed a change in sign of
the heat dilution, but, in this case, from an endothermal behavior at low polymer
concentration, to an exothermal one at high polymer concentration. S dependence results as
a mirror image because entropy and enthalpy changes are dependent each other. Thus, the
concentration dependence of the H and S for PDMS in solvents with different
thermodynamic quality is very complex. The noncombinatorial entropy part associated with a
certain heat effect becomes less favorable as the solvent deteriorates. The H values
associated with the combinatorial behavior of the solutions ( S = 0) is largest for the worst
solvent, MEK. These behaviors can result from the arrangements of the solvent molecules and
polymer chains for very different compositions [25].
3. RHEOLOGICAL BEHAVIOR OF
DIFFERENT POLYSILOXANES IN SOLUTIONS
The use of poly(dimethylsiloxane) solutions in many industrial applications (for example
in coating processes) depends on the knowledge of their rheological properties. The
measurements in dynamic regime provide more substantial information than those
Solution Behavior of Polysiloxanes
61
accomplished in stationary regime, especially with regard to the viscoelastic behavior of the
materials, in general, and of multiple phase systems, in particular, in a close connection with
the structure of these systems. The most common way to qualify the fluid properties consists
of the measurements of the complex elasticity modulus components: storage modulus
(elastic) 'G and the loss modulus (viscous) "G , the complex viscosity ( ) and the loss
angle tangent ( tan ) [87,88].
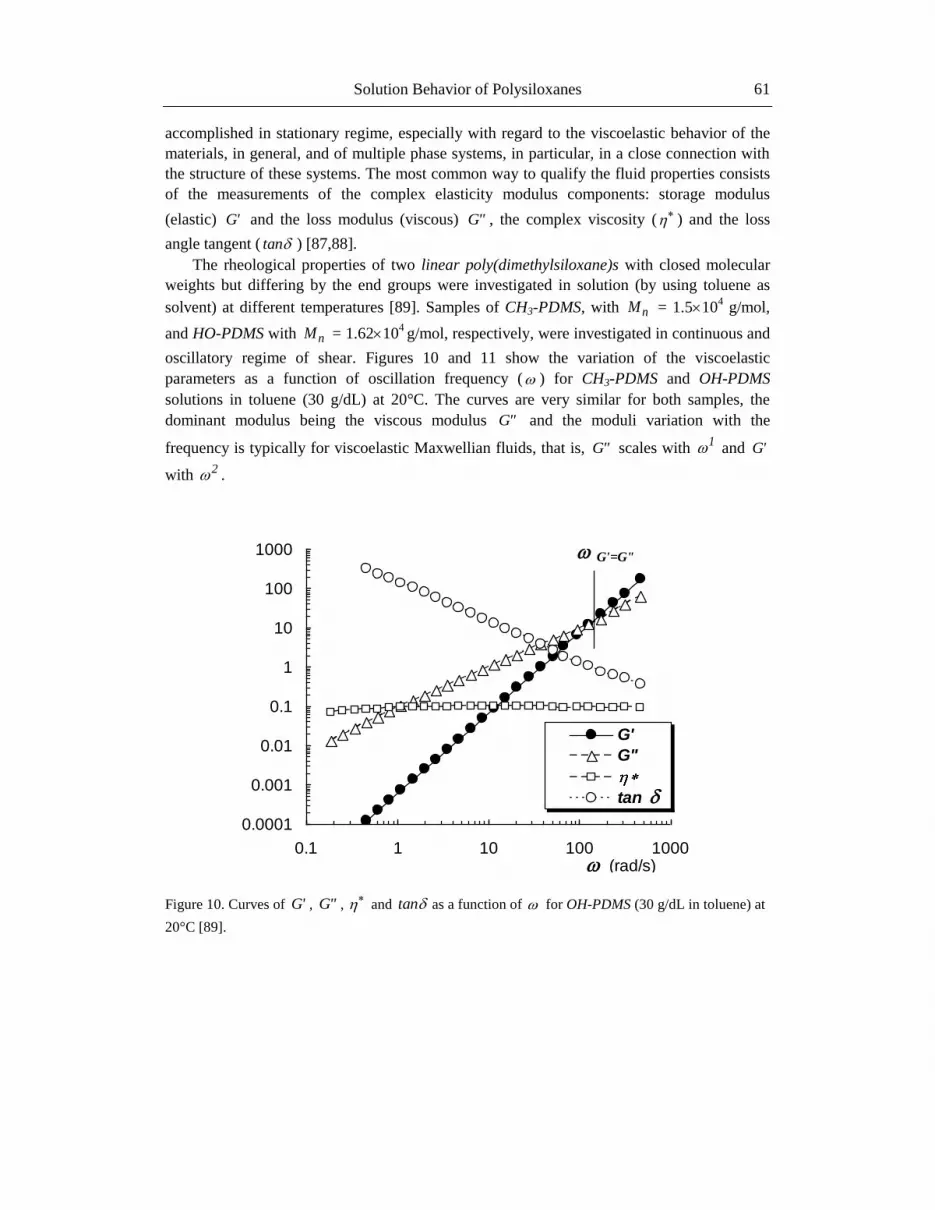
The rheological properties of two linear poly(dimethylsiloxane)s with closed molecular
weights but differing by the end groups were investigated in solution (by using toluene as
solvent) at different temperatures [89]. Samples of CH3-PDMS, with nM = 1.5104 g/mol,
and HO-PDMS with nM = 1.62104 g/mol, respectively, were investigated in continuous and
oscillatory regime of shear. Figures 10 and 11 show the variation of the viscoelastic
parameters as a function of oscillation frequency ( ) for CH3-PDMS and OH-PDMS
solutions in toluene (30 g/dL) at 20°C. The curves are very similar for both samples, the
dominant modulus being the viscous modulus "G and the moduli variation with the
frequency is typically for viscoelastic Maxwellian fluids, that is, "G scales with 1 and 'G
with 2 .
0.0001
0.001
0.01
0.1
1
10
100
1000
0.1 1 10 100 1000 (rad/s)
G'
G"
tan
G'=G"
Figure 10. Curves of 'G , "G , and tan as a function of for OH-PDMS (30 g/dL in toluene) at
20°C [89].

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
0.0001
0.001
0.01
0.1
1
10
100
1000
10000
0.1 1 10 100 1000 (rad/s)
G'
G"
tan
G'=G"
Figure 11. Curves of 'G , "G , and tan as a function of for CH3-PDMS (30 g/dL in toluene)
at 20°C [89].
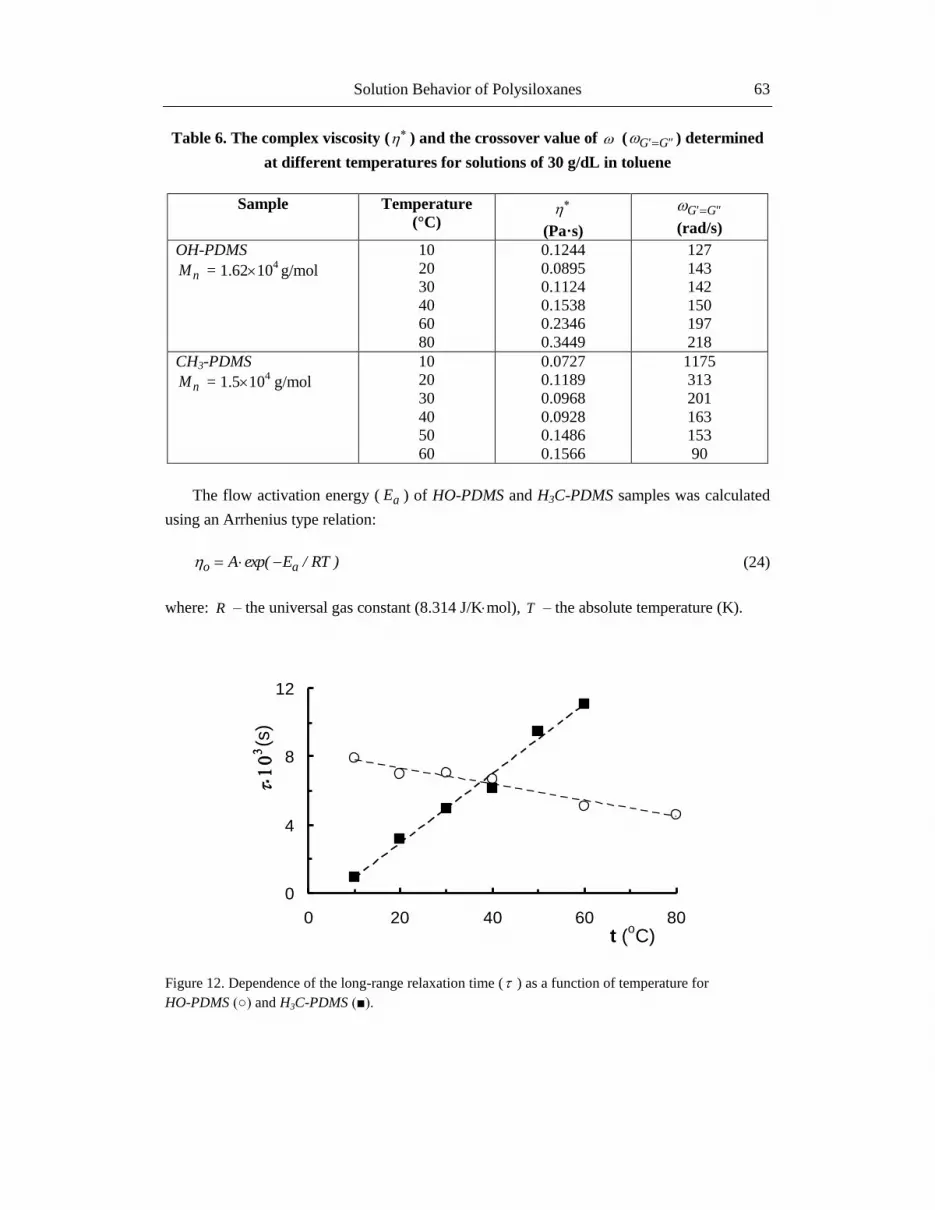
The curves corresponding to elastic and viscous moduli are crossing at a value of
frequency, "G'G , that is inversely proportional with the long-range relaxation time,
(Table 6). The long-range relaxation time for OH-PDMS sample decreases with increasing
the temperature, whereas for H3C-PDMS sample there is a tendency to increase when the
temperature is raised (Figure 12). The higher values obtained below 40°C for HO-PDMS
sample could be explained by the stronger interactions between the polar -OH terminal
groups, comparatively with those corresponding to the -CH3 terminal groups of H3C-PDMS
sample.
The shear viscosity ( shear ) is independent of the shear rate ( ) at all temperatures
showing a Newtonian behavior for both samples. Moreover, the complex viscosity vs.
frequency dependence overlaps with the shear viscosity ( shear ) vs. shear rate ( ) curve
validating the Cox-Merz rule for HO-PDMS and H3C-PDMS samples, according to the
following relationship:
)()(* shear (22)
Thus, the zero shear viscosities, o , were determined by the extrapolation of the complex
viscosity to zero oscillatory frequency, according to the equation:
*)(lim0
o
(23)
Solution Behavior of Polysiloxanes
63
Table 6. The complex viscosity ( ) and the crossover value of ( "G'G ) determined
at different temperatures for solutions of 30 g/dL in toluene
Sample Temperature
(°C)
(Pa·s)
"G'G
(rad/s)
OH-PDMS
nM = 1.62104 g/mol
10
20
30
40
60
80
0.1244
0.0895
0.1124
0.1538
0.2346
0.3449
127
143
142
150
197
218
CH3-PDMS
nM = 1.5104 g/mol
10
20
30
40
50
60
0.0727
0.1189
0.0968
0.0928
0.1486
0.1566
1175
313
201
163
153
90
The flow activation energy ( aE ) of HO-PDMS and H3C-PDMS samples was calculated
using an Arrhenius type relation:
)RT/Eexp(A ao (24)
where: R – the universal gas constant (8.314 J/Kmol), T – the absolute temperature (K).
0
4
8
12
0 20 40 60 80t (
oC)
.103(s
)
Figure 12. Dependence of the long-range relaxation time ( ) as a function of temperature for
HO-PDMS (○) and H3C-PDMS (■).

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
From the plots of *ln on ( T/1 ) (Figure 13) one observes that the dependences are
linear in the temperature range from 20°C to 80°C. The higher value of the activation energy
calculated for HO-PDMS sample (19.53 kJ/mol) as compared with the sample H3C-PDMS
(7.66 KJ/mol) can be attributed to the intermolecular interactions of –OH end groups in a
nonpolar solvent.
-4
-3
-2
-1
0
2.75 3 3.25 3.5
103/T (1/K)
ln(Pa·s)
Figure 13. Dependence of *ln on T/1 for the samples HO-PDMS (●) and H3C-PDMS (▲).
The evolution of linear viscoelasticity during cross-linking of poly(dimethylsiloxane)s
was investigated by Winter and coworkers [90-93] by small amplitude oscillatory shear. The
network junctions were assumed to be due to chemical cross-links and not due to others
association phenomena, crystallization or phase separation.
At the gel point, the relaxation modulus, )t(G , follows a power law which is denoted as
gel equation:
nSt)t(G (25)
where S is the gel stiffness and n is the relaxation exponent.
Stoichiometrically balanced PDMS gels gave the specific exponent value of n = 1/2,
whereas for stoichiometrically imbalanced PDMS samples 1/2 < n < 1. Transformation of
the data from the frequency to time domain required the hypothesis that the power law
behavior extends over the entire frequency range, 0 < < . For the imbalanced gels
)('G)("G and a higher rate of the stress relaxation was observed. Because the gel point
was found to occur before the crossover point between )('G and )("G , the authors firstly
proposed a new method to localize the gel point, i.e., the detection of a loss tangent
independent of the frequency [94,96]. The final modulus of PDMS network scales with

Solution Behavior of Polysiloxanes
65
concentration at a power close to 2. Both gel stiffness and relaxation exponent are strongly
composition dependent. Relaxation exponents between 0.19 and 0.92 and variations of gel
strength over 5 decades were found for PDMS, depending on the entanglement molecular
weight, eM . Thus, prepolymers with the molecular weight below eM produce critical gels
with lower n values, in the range of 0.4 to 0.7, whereas above eM , critical gels with lower
n values (0.2 – 0.4) were obtained, depending on the stoichiometry. Addition of a diluent
affects the relaxation exponent and near the overlap concentration n approaches the limit of
1 [92].
The static and dynamic properties of cross-linked poly(dimethylsiloxane) clusters near
the gelation threshold have been studied also by Adam et al. [94] starting from PDMS
samples with 1.78·105 g/mol < wM <2.06·10
7 g/mol in toluene. In the vicinity of the gelation
point, the mass distribution of the polymer clusters is described by the following power law
[95]:
M
M2.2c fMn (26)
where cn is the number of clusters with mass M and
M
Mf is the cutoff function that
indicates that there is no mass larger than M in the system.
For all clusters of size cR one can write the following relation:
dcRM (27)
where d represents the fractal dimension that depends on the polymer environment: a) in the
reaction medium (without solvent) 5.2d and b) in dilute solution (where the polymer
clusters swell) 2d .
The weight average molecular weight ( wM ), the z-average square radius of gyration
z2S and the z-average diffusion coefficient ( D ) of polymer clusters determined by static
light scattering experiments were found to be linked by the following relation:
d)3(
zw RM
(28)
where zR is either the radius of gyration z2S determined by static light scattering
measurements, or the hydrodynamic radius, HR , deduced from the diffusion coefficient, D
(equation (9)).
The experimental value of d)3( obtained from the wM – zR dependence was 1.71
0.07 in agreement with the theoretical prediction ( 6.1d)3( ) corresponding to the
percolation model.

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
The interaction parameter, wB , was found to depend on the weight average molecular
weight according to the relation:
X
ww M47.0B (29)
where 03.035.0X and wB is determined from the following dependence:
w2/12
H
B
][
S
R
(30)
X exponent obtained by experimental measurements has also a value close to the value
expected by the percolation model ( 375.0X ).
These observations allow concluding that the static properties of cross-linked
poly(dimethylsiloxane) clusters near the gelation threshold are well described by the
percolation model. The rheological data obtained for this chemical system showed that the
local dynamic properties are independent of the distance to the threshold, allowing real
exponents to be evaluated.
Mixtures of PDMS with CO2 were studied in the entire range of compositions at
pressures between 100 bar and 700 bar and temperatures in the range of 30 – 70C [96]. The
volumetric and the viscometric behavior of PDMS solutions in condensed gases have
demonstrated that these systems behave very much like in the ordinary low molecular weight
liquids. Bellow the entanglement point, the viscosity of the mixture is less than those
calculated by additive rule, on the basis of the volume fractions of the components
( 2,1i,i ). If the chains are entangled, the deviations become positive and this effect
increases with increasing the molecular weigh. Large negative excess volumes were obtained
for high volatile solvents. The concentration dependences of the activation energies and
volumes exhibit pronounced sigmoidal shapes. The chain entanglements play an important
role for the occurrence of the inversions in the excess viscosities. This complex behavior was
described quantitatively by means of one ajustable parameter independent of the molecular
weight. The concentration dependence of both excess volume and excess viscosities can be
described quantitatively by a simple relation on the form: 212211 )kk( , in which 1k
and 2k are theoretical parameters, their meaning is explained in ref. [96].
Liquid-crystalline polymer (LCP)/low molar mass nematic (LMMN) composites have
attracted attention due to its application in electrooptic devices. The rheological properties of
LCP solutions are changed dramatically when a small amount of LCP is dissolved in LMMN.
For these composites, both aligning-to-tumbling and tumbling-to-aligning transitions have
been evidenced [97-100]. These changes in the rheological properties are in agreement with
the Brochard theory which affirms that the dissolved polymer can influence differently the
system viscosity, depending on its chain conformation (specifically a prolate vs oblate shape)
[101]. The hydrodynamic theory of Brochard predicts that the dissolution of an oblate side-
chain LCP in a flow-aligning nematogen will produce a transition to tumbling flow and the
dissolution of a prolate main-chain LCP in a flow-tumbling nematogen will produce a
transition to aligning flow. Others investigations concerning of the rheological and

Solution Behavior of Polysiloxanes
67
viscometric properties of dilute solutions of a liquid crystal polymer based on polysiloxane in
a nematic solvent (4’-pentyl-4-cyanobiphenyl and 4,4’-n-octylcyanobiphenyl) were reported
[102,103]. Certain inconsistencies between prediction of the hydrodynamic model of
Brochard and experimental observation of stress transients and electrorheological responses
of side-chain liquid crystal polymer/low molar mass nematic solutions were been observed
and a modification of this theory was been proposed. The modification of Brochard’s theory
supposes that an elastic torque between director rotation and LCP orientation produces
additional viscous dissipation. By application of the modified Brochard’s theory to the
polysiloxane/nematic solvent system a good agreement between theory and experimental data
was obtained.
Tailored poly(dimethylsiloxane) ionomers, which possess highly flexible polysiloxane
backbone, were synthesized and the theoretical predictions of the scaling of viscosity with
polymer volume fraction by both Rubinstein and Semenov theory were compared with the
experimental results [104,105]. This theory predicts a number of regimes of viscosity
dependence on polymer volume fraction based on parameters linked to the solvent quality and
associating polymer structure, for example the length between stickers, overall molecular
mass, strength of interactions and entanglement molar mass of the polymer. The solution
viscosities of poly(dimethylsiloxane) sodium and zinc ionomers with tailored number of
monomers between ions and number of ions per chain in good and in theta solvent conditions
over a broad range of concentrations were reported [106]. For these poly(dimethylsiloxane)
ionomers, the dynamics in both the entangled and unentangled semidilute regimes can be
modeled by intermolecular bonds with renormalized lifetimes.
Aqueous emulsions of linear PDMS have been proposed as a new contrast agent for the
gastro-intestinal nuclear magnetic resonance [107], their use offering some advantages: the
effect of diarrhea and flatulence are reduced; PDMS is an inert photon-rich polymer because
it has two methyl groups in its repeating unit; in 1H-NMR spectrum there is about a 5 ppm
chemical shift from PDMS to water, which is usable for the chemical shift artifact (in
imaging technology, an artifact is a feature appearing on the image which does not
correspond to the properties of the subject in a certain region). Except for this application,
PDMS is widely employed in current researches of new biomedical devices due to its good
hemo- and biocompatibility properties [108]. Crosslinked PDMS colloidal solutions were
proposed as an alternative to emulsions of linear dimethylsiloxane oligomers to avoid
possible absorption by intestine [109]. Emulsions and colloidal PDMS solutions are more
convenient for medical applications because their rheological properties are adjustable. The
rheological properties of colloids are influenced by the volume fraction of the disperse phase
( ), particle radius ( r ), packing effects, the type of surfactant, etc. The rheological
properties changes dramatically above a critical value of the volume fraction, c . For
monodisperse emulsions c 0.635, while for polydisperse emulsions c
0.71 [110]. For
c , the compressed emulsions show predominant elastic behavior and the viscosity
increases abruptly with the increase of the volume fraction. The yield stress appears at low
strain and is proportional to r2/3/1 and it was not observed for c [109].

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
4. OPTICAL PROPERTIES OF POLYSILOXANE SOLUTIONS
The materials based on polysiloxanes are suitable for optical applications due to their
refractivity and transmittance combined with dielectric and mechanic properties. Generally,
the refractive index of a solution depends on the light wavelength, temperature and the
solution concentration. The variation of the refractive index increment of
poly(dimethylsiloxane) in toluene as a function of the temperature and the light wavelength at
different concentrations from 0.005 g/mL to 0.05 g/mL was determined by Nilsson and
Sundelof [111]. The refractive index increment of the poly(dimethylsiloxane) solution slowly
decreases by concentration and temperature increasing. Moreover, the refractive index
depends on the type of the end groups and the structure of polysiloxanes. Brunchi et al. [112]
studied the optical properties of some polymers containing siloxane units. The experimental
refractive index determined at 435 nm and 25°C for HO-PDMS ( nM = 1.62104
g/mol) and
H3C-PDMS ( nM = 1.5104 g/mol) in toluene were 1.401 and 1.403, respectively. The CH3
end groups determine a slight increase of the refractive index in comparison with the OH end
groups. Additional, this parameter was calculated for poly(dimethylsiloxane) with different
end groups by using the following equation, according to the Lorenz-Lorentz approximation
[113]:
)2n/()]1n(V[R 22
22uu (31)
where 2n represents the refractive index of the polymer according to different
approximations, uR is the molar refractivity and uV is the molar volume.
The refractive index was calculated with above equation based on the assumption that the
molar volume and the molar refraction of the chain repeating unit are additive functions of
composition (eqs. (32) and (33) [114]:
ii
iu VaV (32)
ii
iu RaR (33)
where iV and iR are the group contributions, and ia is the number of groups, i , is the
repeating unit.
The experimental values of the refractive index for HO-PDMS and H3C-PDMS in toluene
differed slightly from those calculated with the Lorenz-Lorentz equation, by considering the
corresponding group contributions to the molar refraction and volume. The calculated
refractive indices were 1.402 and 1.400 for HO-PDMS and H3C-PDMS, respectively.
The refractive index can be adjusted by controlling the composition of the polymer. The
influence of polysiloxane composition on the optical properties was studied for PDMPS
(Table 1) with different concentrations of diphenylsiloxane units, in toluene, the results being
presented in Figure 14 [112].

Solution Behavior of Polysiloxanes
69
0 20 40 60 80 1001.35
1.40
1.45
1.50
1.55
1.60
Refr
acti
ve in
dex
Diphenylsiloxane content (%)
Figure 14. Influence of diphenylsiloxane content on the refractive index of PDMPS [112].
The refractive index increases from 1.401 for poly(dimethylsiloxane) to 1.577 for
poly(diphenylsiloxane) by increasing diphenylsiloxane content. The silicone polymers with
the diphenylsiloxane content between 12 % and 21 % mol having the refractive indices
between 1.425 and 1.440 could be used in bio-optic applications. The increase of the
diphenylsiloxane content in the copolymers determines the improvement of the optical
properties but the materials becomes more rigid. It is necessary to change the end groups or to
introduce new substituents in the copolymer structure during the synthesis to remove such
disadvantage.
Some copolymers containing siloxane were developed as materials having optical
properties. Thus, new siloxane-modified methacrylate copolymers were synthesized and
characterized [115] in order to their using in contact lens. An increase of the toughness and a
decrease of the tensile strength of these copolymers by increasing the methyl methacrylate
content in copolymer were observed [116]. The refractive index of vinyl polymers
(poly(methyl methacrylate-allyl methacrylate) and poly(styrene-allyl methacrylate)) decreases
by dimethylsiloxane grafting [117].
The behavior of PDMS and PDMPS with different contents of diphenylsiloxane units in
solvent/nonsolvent (toluene/methanol) mixtures was investigated by turbidimetry [77]. The
characteristics of the samples are listed in Table 4. The turbidity of the polymer containing
siloxane units slowly increases by increasing the polymer concentration (Figure 15). Some
conformational changes were evidenced for PDMS in the entire range of solvent/nonsolvent
binary mixture for polymer concentrations between 0.1 g/dL and 0.2 g/dL (Figure 15a). In the
case of copolymers, these phenomena were not observed (Figure 15b,c).
For all samples, it was observed that the turbidity increases by increasing the nonsolvent
amount in the solvent-nonsolvent mixture (Figure 16).

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
nonsolvent/solvent ratio
0.4
0.6
0.8
1.0
0 0.4 0.8 1.2c (g/dL)
Tu
rbid
ity (N
TU
)
0.11 0.180.250.33
a)
0
1
2
3
4
0 0.4 0.8 1.2c (g/dL)
Tu
rbid
ity (N
TU
)
0.110.180.250.330.43
nonsolvent/solvent ratiob)
0
2
4
6
8
0 0.4 0.8 1.2
c (g/dL)
Tu
rbid
ity (N
TU
)
0.110.18
0.250.330.43
nonsolvent/solvent ratio c)
Figure 15. Variation of turbidity vs. concentration for: (a) PDMS, (b) PDMPS1 and (c) PDMPS2.

Solution Behavior of Polysiloxanes
71
PDMS
0
0.5
1
1.5
2
2.5
3
3.5
0 0.1 0.2 0.3 0.4 0.5 0.6
Nonsolvent/Solvent (vol/vol)
Tu
rbid
ity (N
TU
)
PDMPS1
PDMPS2
Figure 16. Turbidity of PDMS (●), PDMPS1 (□) and PDMPS2 () solutions for 0.550 g/dL as a
function of nonsolvent/solvent ratio at room temperature.
The increase of the turbidity is more accentuated for the copolymer having the higher
diphenylsiloxane unit content. This observation was in accord with experimental data
obtained by Brunchi et al. [112] which showed that by increasing the amount of phenyl
groups, the refractive index boosted to the detriment of transmittance and flexibility.
Moreover, the modification of the wavelength from UV to the visible domain determined the
increase of the transparency which remained constant above 400 nm. The PDMS sample is
insoluble in nonsolvent/solvent mixture at ratios higher than 0.4 unlike
poly(dimethylsiloxane-co-diphenylsiloxane)s that are soluble at higher ratios. The turbidity of
PDMS is lower than the copolymers turbidities at the same concentration and
solvent/nonsolvent ratio; the turbidity of the copolymers is more affected by the addition of
the nonsolvent.
5. CONCLUSION
Based on the literature data and also on own studies, it can be appreciated that the
behavior of the polysiloxanes in solution is different from those in bulk. The high interest in
the study of the polysiloxanes in solution comes in special from the observations that the high
flexible polysiloxane chain behaves rather as stiff one when it is seen as an isolated chain.
Beside the polysiloxane polymer characteristics (cyclic or liniar structure, molecular weight,
nature of the substituent to the silicon atoms or the ending groups nature), both solvent nature
as well as concentration have a significant influence on the polymer behavior in solution.
The data on both thermodynamic (unperturbed state, excluded volume effects,
thermodynamic parameters) and rheological behaviors of the polysiloxanes in solution were
reviewed.
The thermodynamic behavior of polysiloxanes in solution can not be entirely described
by using classical theories of thermodynamics [3]. New theoretical considerations [1-2],

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
which takes into account the chain connectivity and the ability of polymer coils to change the
conformation in response to alterations of their environment, are able to give a better
quantitative description of polysiloxanes in solution.
The results on the investigations of some optical properties (transparence and refractive
index) of the polysiloxanes in solution were also presented and discussed in correlation with
the polymer structure.
ACKNOWLEDGEMENTS
The authors are grateful to Prof. Bernhard A. Wolf from Johannes Gutenberg Universität
Mainz for useful comments and suggestions. The research activity was partially realized in
the framework of an exploratory research project (Contract Number 516/2009, additional
contract nr. 1/2010).
6. REFERENCES
[1] Wolf, B. A. Macromol. Chem. Phys. 2003, 204, 1380-1389.
[2] Wolf, B. A. Adv. Poly. Sci., in press
[3] Bercea, M.; Cazacu, M.; Wolf, B. A. Macromol. Chem. Phys. 2003, 204, 1371-1380.
[4] Flory, P. J. Statistical Mechanics of Chain Molecules; Hanser Publ.: Munich, 1989.
[5] Scott, D. W.; Messerly, J. F.; Todd, S. S.; Guthrie, G. B.; Hossenlopp, I. A.; Moore, R.
T.; Osborn, A.; Berg, W. T.; McCullough, J. P. J. Phys. Chem., 1961, 65, 1320-1326.
[6] Yamada, T.; Koyama, H.; Yoshizaki, T.; Einaga, Y.; Yamakawa, H. Macromolecules
1993, 26, 2566-2574.
[7] Freed, K. F. Renormalization Group Theory of Macromolecules; John Wiley and Sons
Inc.: New York, 1987.
[8] Yamakawa, H. Polym. J. (Tokyo) 1999, 31, 109-119.
[9] Konishi, T.; Yoshizaki, T.; Yamakawa, H. Macromolecules 1991, 24, 5614-5622.
[10] Yamada, T.; Yoshizaki, T.; Yamakawa, H. Macromolecules 1992, 25, 1487-1492.
[11] Horita, K.; Sawatari, N.; Yoshizaki, T.; Einaga, Y.; Yamakawa, H. Macromolecules
1995, 28, 4455-4463.
[12] Grigorescu, G.; Ioan, S.; Harabagiu, V.; Simonescu, B. C. Macromol. Rep. 1996, 138,
163-173.
[13] Grigorescu, G.; Ioan, S.; Simonescu, B. C. Macromol. Symp. 1999, 138, 197-202.
[14] Ioan, S.; Grigorescu, G.; Simonescu, B. C. Polysh J. Appl. Chem. 1999, XLIII, 3-21.
[15] Flory, P. J.; Mandelkern, J. B., Kisinger, J. B., Schurz W. B. J. Am. Chem. Soc. 1952,
74, 3364-3367.
[16] Petri, H. M.; Schuld, N.; Wolf, B. A. Macromolecules 1995, 28, 4975-4980.
[17] Graessley, W. W. Polymer 1980, 21, 258-262.
[18] Qian, J. W.; Rudin A. Eur. Polym. J. 1992, 28, 725-732; 773-787.
[19] Kurata, M.; Tsunashima, Y. Viscosity-Molecular Weight Relationships and
Unperturbed Dimensions of Linear Chain Molecules in Polymer Handbook, 4th Ed.
Brandrup J., Immergut E.H., E. Grulke Eds., John Wiley, New York, 1999.

Solution Behavior of Polysiloxanes
73
[20] Stepto, R. F. T. Theoretical Aspects of Conformation Dependent Properties in Siloxane
Polymers, Eds. S.J. Clarson and J.A. Semlyn, PTR Prentice Hall, Englewood Cliffs,
N.J. 1993.
[21] Delmas, G.; Patterson, D.; Bhattacharyya, S. N. J. Phys. Chem. 1964, 68, 1468-1474.
[22] Patterson, D.; Bhattacharyya, S. N.; Picker, N. Trans Faraday Soc. 1968, 64, 648-661.
[23] Morimoto, S. Makromol. Chem. 1970, 133, 197-204.
[24] Dreifus, D. W.; Patterson, D. Trans. Faraday Soc. 1971, 67, 631-639.
[25] Schuld, N.; Wolf, B. A. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 651-662.
[26] Kuwahara, N.; Okazawa, T.; Kaneko, M. J. Polym. Sci., Part C, 1968, 23, 543-553.
[27] Summers, W. R.; Tewari, Y. T.; Schreiber, H. P. Macromolecules 1972, 5, 12-16.
[28] Schneider, A.; Schuld, N.; Bercea, M.; Wolf, B. A. J. Polym. Sci. Part B Polym. Phys.
2004, 42, 1601-1609.
[29] Flory, P. J.; Shih, H. Macromolecules 1972, 5, 761-766.
[30] Bianchi, U.; Pedemonte, E.; Rossi, C. Makromol. Chem. 1966, 92, 114-121.
[31] Bianchi, U. J. Polym. Sci., B. 1965, 3, 1079-1094.
[32] Morimoto, S. J. Polym. Sci., A-1 1968, 6, 1541-1547.
[33] Patterson, D. Macromolecules 1969, 2, 672-677.
[34] Sugamiya, K.; Kuwahara, N.; Kaneko, M. Macromolecules 1974, 7, 66-72.
[35] Muramoto, A. Polymer 1982, 23, 1311-1316.
[36] Cankurtaran, O.; Yilmaz, F. Polym. Int. 2000, 49, 99-102.
[37] Hamada, F.; Shiomi, T.; Fujishawa, K.; Nakajime, A. Macromolecules 1980, 13, 729-
732.
[38] Flory, P. J.; Crescezi, V.; Mark, J. E. J. Am. Chem. Soc. 1964, 86, 146-152.
[39] Flory, P. J.; Semlyen, J. A. J. Am. Chem. Soc. 1966, 88, 3209-3212.
[40] Haug, A.; Meyerhoff, G. Makromol. Chem. 1962, 53, 91-102.
[41] Schultz, G. V.; Haug, A. Z. Phvs. Chem. (Frankfurt am Main) 1962, 34, 328-345.
[42] Konishi, T.; Yoshizaki, T.; Yamakawa, H. Macromolecules 1991, 24, 5614-5622.
[43] Abe, F.; Einaga Y.; Yamakawa H. Macromolecules 1991, 24, 4423-4428.
[44] Yamakawa, H. Modern Theory of Polymer Solutions Harper & Row, New York, 1971,
Chapter 6.
[45] Wolf, B. A. Macromol. Rapid Commun. 2007, 28, 164-170.
[46] Eckelt, J.; Knopf, A.; Wolf, B. A. Macromolecules 2008, 41, 912-918.
[47] Samadi, F.; Wolf, B. A.; Guo, Y.; Zhang, A.; Dieter Schluter, A. Macromolecules
2008, 41, 8173-8180.
[48] Ghimici, L.; Nichifor, M.; Wolf, B. A. J. Phys. Chem. B 2009, 113, 8020-8025.
[49] Brunchi, C. E.; Bercea, M.; Morariu, S. e-Polymers 2009, no. 065, 1-10.
[50] Grigorescu, G.; Ioan, S.; Harabagiu, V.; Simonescu, B. C. Synth. Polym. J. 1998, V,
213-222.
[51] Grigorescu, G.; Ioan, C.; Ioan, S.; Harabagiu, V.; Simonescu, B. C. Rev. Roum. Chim.
1999, 44, 607-611.
[52] Grigorescu, G.; Ioan, S.; Harabagiu, V.; Simonescu, B. C. Eur. Polym. J. 1998, 34,
827-832.
[53] Grigorescu, G.; Ioan, S.; Pinteala, M.; Simionescu, B. C. J. Serb. Chem. Soc. 1998, 63,
961-966.
[54] Qian, J. W.; Wang, W.; Han, D. L.; Cheng, R. S. Eur. Polym. J. 2003, 37, 1403-1407.
[55] Bercea, M.; Cazacu, M.; unpublished data

Maria Bercea, Simona Morariu, Cristina-Eliza Brunchi et al.
[56] Zilliox, J. G.; Roovers, J. E. L.; Bywater, S. Macromolecules 1975, 8, 573-578.
[57] Brunchi, C. E.; Cazacu, M.; Morariu, S.; Ioan, S. Bull. Transilvania Univ. Brasov
2007, 2, 253-258.
[58] Büyüktanir, E. A.; Küçükyavuz, Z. J. Polym. Sci., Part B: Polym.Phys. 2000, 38,
2678-2686.
[59] Crescenzi, V.; Flory, P. J. J. Am. Chem. Soc. 1964, 86, 141-146.
[60] Douglas, J. F.; Freed, K. F. Macromolecules 1984, 17, 1854-1870; 2344-2352.
[61] Freed, K. F. Renormalization Group Theory of Macromolecules Wily Intersicence:
New York, 1987.
[62] Douglas, J. F.; Freed, K. F. Macromolecules 1994, 27, 6088-6099.
[63] Yamakawa, H. Molecular Conformation and Dynamics of Macromolecules in
Condensed Systems Nagasawa M., Ed.; Elsevier: Amsterdam, 1988.
[64] Yamakawa, H.; Yoshizaki, T. Macromolecules 1995, 28, 3604-3608.
[65] Yamakawa, H. Macromolecules 1992, 25, 1912-1916.
[66] Yamakawa, H.; Abe, F.; Einaga, Y. Macromolecules 1993, 26, 1898-1904.
[67] Grigorescu, G.; Ioan, S.; Cojocaru C.; Simonescu, B. C. Macromol. Sci., Pure Appl.
Chem, 1997, A34, 533-542.
[68] Edwards, C. J. C.; Stepto, R. F. T.; Semlyen, J. A. Polymer 1980, 21, 781-786.
[69] Helfer, C. A.; Mattice, W. L., Chap. The Rotational Isomeric State Model, in Physical
Properties of Polymer Handbook, Part II, pp. 43-57, Springer: New York. 2007.
[70] Morariu, S.; Brunchi, C. E.; Ghimici, L.; Bercea, M. Viscometric Behavior of Polymer
Solutions, in Functional Polymeric Materials Designed for Hi-Tech Applications,
Nechifor M., Ed. SIGNPOST: Kerala (India) 2010, 85-107.
[71] Bercea, M.; Cazacu, M. Proceedings of International Conference Polymeric Materials,
Haale (Saale), Sept. 25th -27
th , 2002, 565-568.
[72] Cheng, R.; Shao, M.; Liu, M.; Qian, R. Eur. Polym. J. 1998, 34, 1613-1619.
[73] Abbed, S.; Boileau, S.; Bouteiller, L. Polymer 2001, 42, 8613-8619.
[74] Abbed, S.; Boileau, S.; Bouteiller, L. Macromolecules 2000, 33, 8479-8487.
[75] Bercea, M.; Cazacu, M. Proceedings of 12th
Romanian International Conference on
Chemistry and Chemical Engineering, Bucharest, Sept. 13-15, 2001, p. 380-385.
[76] Badiger, M. V.; Gupta, N. R.; Eckelt, J.; Wolf, B. A. Macromol. Chem. Phys. 2008,
209, 2087-2093.
[77] Brunchi, C. E.; Alexandru, M.; Morariu, S. 9th
International Balkan Workshop on
Applied Physics, July 7-9, 2008, Constanta, Romania.
[78] Dodgson, K.; Semiyen, J. A. Polymer 1977, 18, 1265-1268.
[79] Bianchi, U.; Dalpiaz, M.; Patrone, E. Makromol. Chem. 1964, 80, 112-119.
[80] Dvornic, P. R.; Jovanovic, J. D.; Govedarcia, M. N. J. Appl. Polym. Sci 1993, 49,
1497-1507.
[81] Lapp, A.; Herz, J.; Strazielle, C. Makromol. Chem., 1985, 186, 1919-1934.
[82] Brezinski, J.; Czlonkowska-Kohutnicka, Z.; Czarnecka, B.; Kornas-Calka, A. Eur.
Polym. J. 1973, 9, 733-738.
[83] Takimoto, H. H.; Forbes, C. T.; Laudenslager, R. K. J. Appl. Polym. Sci. 1961, 5, 153-
156.
[84] des Cloizeaux, J.; Jannink, J. Polymers in Solution, Their Modeling and Structure;
Oxford Science Publishers: New York, 1990.

Solution Behavior of Polysiloxanes
75
[85] Schäfer, L. Excluded Volume Effects in Polymer Solutions; Springer-Verlag: Berlin,
1999.
[86] Wolf, B. A. Macromolecules 2005, 38, 1378-1384.
[87] Barnes, H. A.; Hutton, J. F.; Walters, K. Introduction to Rheology, Amsterdam-
Oxford-New York-Tokyo, 1989.
[88] Barnes, H. A. Handbook of Elementary Rheology, Inst. Of Non -Newtonian Fluid
Mechanics, University of Wales, 2000.
[89] Brunchi, C. E.; Bercea, M.; Cazacu, M.; Ioan, S. The Annals of the Dunarea de Josof
Galati, Mathematics, Physics, Chemistry, Informatics, Fascicle II, Suppl., 2007, 69-76.
[90] Winter, H. H.; Chambon, F. J. Rheol. 1986, 30, 367-382.
[91] Chambon, F.; Winter, H. H. J. Rheol. 1987, 31, 686-697.
[92] Scanlan, J. C.; Winter, H. H. Macromolecules 1991, 24, 47-54.
[93] Winter, H. H.; Mours, M. Adv. Polym Sci. 1997, 134, 165-234.
[94] Adam, M.; Lairez, D.; Karpasas, M.; Gottlieb, M. Macromolecules 1997, 30, 5920-
5929.
[95] Lairez, D.; Adam, M.; Raspaud, E.; Emery, J. R.; Durand, D. Prog. Colloid Polym. Sci.
1992, 90, 37-42.
[96] Mertsch, R.; Wolf, B. A. Macromolecules 1994, 27, 3289-3294.
[97] Jamieson, A. M.; Gu, D. F.; Chen, F. L.; Smith, S. Prog. Polym. Sci. 1996, 21, 981-
1033.
[98] Gu, D.; Jamieson, A. M. Macromolecules 1994, 27, 337-347.
[99] Yao, N.; Jamieson, A. M. J. Rheol. 1998, 42, 603-619.
[100] Yao, N.; Jamieson, A. M. Macromolecules 1998, 31, 5399-5406.
[101] Brochard, F. J. Polym. Sci., Polym. Phys. Ed. 1979, 17, 1367-1374.
[102] Zhao, Y.; Dong, S.; Jamieson, A. M.; Hu, X.; Lal, J.; Nazarenko, S.; Rowan, S. J.
Macromolecules 2005, 38, 5205-5213.
[103] Yao, N.; Jamieson, A. M. Rheol. Acta 2000, 39, 338-345.
[104] Rubinstein, M.; Semenov, A. N. Macromolecules 1998, 31, 1386-1397.
[105] Rubinstein, M.; Semenov, A. N. Macromolecules 2001, 34, 1058-1068.
[106] Batra, A.; Cohen, C.; Duncan, T. M. Macromolecules 2006, 39, 2398-2404.
[107] US patent, 5, 380, 514, 1995.
[108] Tang, L. P.; Sheu, M. S.; Chu, T. M.; Huang, Y. H. Biomaterials 1999, 20, 1365-1370.
[109] Su, F. H.; Shyu S. S.; Chen, Y. C. Eur. Polym. J. 2002, 38, 377-385.
[110] Princen, H. M.; Kiss, A. D. J. Coll. Interf. Sci. 1989, 128, 176-187.
[111] Nilsson, R.; Sundelof, L. O. Die Makromolekulare Chemie 1963, 66, 11-18.
[112] Brunchi, C. E.; Filimon, A.; Cazacu, M.; Ioan, S. High Performance Polymers 2009,
21, 31-47.
[113] van Krevelen, D. W. Properties of Polymers, Elsevier Sci. Pub. Co., Amsterdam, 1976.
[114] Groh, W.; Zimmermann, A. Macromolecules 1991, 24, 6660-6663.
[115] Yilgor, I.; McGrath, J. S. Adv. Polym. Sci. 1988, 86, 1-86.
[116] Bischoff, R.; Cray, S. E. Prog. Polym. Sci. 1999, 24, 185-219.
[117] Ioan, S.; Lungu, V.; Harabagiu, V. Int. J. Polym. Anal. Charact. 2005, 10, 361-372.





























