Embed Size (px)
Citation preview

JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 13 1 OCTOBER 2002
Spin–spin coupling tensors by density-functional linear response theoryPerttu Lanttoa)
NMR Research Group, Department of Physical Sciences, P.O. Box 3000, FIN-90014 University of Oulu,Finland
Juha Vaarab)
Department of Chemistry, P.O. Box 55 (A.I. Virtasen aukio 1), FIN-00014 University of Helsinki, Finland
Trygve Helgakerc)
Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, N-0315 Oslo, Norway
~Received 4 February 2002; accepted 1 July 2002!
Density-functional theory~DFT! calculations of indirect nuclear magnetic resonance spin–spincoupling tensorsJ, with the anisotropic but symmetric parts being the particular concern, are carriedout for a series of molecules with the linear response~LR! method. For the first time, the anisotropiccomponents ofJ are reported for a hybrid functional. Spin–spin tensors calculated using the localdensity approximation~LDA !, the gradient-corrected Becke–Lee–Yang–Parr~BLYP! functional,and the hybrid three-parameter BLYP~B3LYP! functional are compared with previousab initiomulticonfiguration self-consistent-field~MCSCF! LR results and experimental data. In general, theB3LYP functional provides reasonable accuracy not only for the isotropic coupling constants butalso for the anisotropic components ofJ, with the results improving in the sequence LDA→BLYP→B3LYP. Error cancellation often improves the total DFT spin–spin coupling when themagnitude of the paramagnetic spin–orbit contribution is overestimated, or when the spin–dipole~SD! and Fermi-contact~FC! contributions are far from the MCSCF values. For the19F nucleus,known to be difficult for DFT, the anisotropic properties of heteronuclear, in particular19F13Ccouplings are often more accurate than the poorly described isotropic coupling constants. Thishappens since the FC contribution is small at fluorine compared with carbon, leading to a small errorin the total SD/FC term. With the recent implementation of the hybrid B3LYP functional,calculations of predictive quality for theJ tensors are no longer restricted to small model molecules,opening up the possibility of studying the anisotropic components ofJ in large organic andbiomolecules of experimental interest. ©2002 American Institute of Physics.@DOI: 10.1063/1.1502243#
ag
a
linq-r
-o
ag
d
g.thatons in-rgenetic
and
nt-
ster
lesntts,
-
nsnthe
ma
I. INTRODUCTION
The indirect spin–spin coupling tensor between the mnetic nucleiK andL,
JKL5JKL11JKLS 1JKL
A , ~1!
is one of the main parameters determining the nuclear mnetic resonance~NMR! spectrum.1–4 In isotropic liquids andgases, the only spin–spin coupling observable is the coupconstant,J5 1
3Tr J. In anisotropic environments such as liuid crystals and solids, the second-rank symmetric tensopartJS also has an effect on the spectrum.5–7 The role of theantisymmetric, rank-1 partJA has been discussed8,9 but so farnot experimentally observed~see also Ref. 10!.
The spin–spin coupling tensorJ is a second-order molecular property corresponding to the second derivativethe total electronic energy with respect to the nuclear mnetic moments of the coupled nuclei.4 From the point of viewof theoretical calculations, it differs from many other secon
a!Author to whom correspondence should be addressed. [email protected]
b!Electronic mail: [email protected]!Electronic mail: [email protected]
5990021-9606/2002/117(13)/5998/12/$19.00
Downloaded 25 Sep 2002 to 129.240.81.153. Redistribution subject to A
-
g-
g
ial
f-
-
order properties in being more computationally demandin11
First, there are several contributing physical mechanismsare highly dependent on the quality of the wave functinear and at the nuclei; second, some of the mechanismvolve triplet perturbation operators; third, there are a lanumber of response equations to be solved for each magnucleus. Hence, qualitatively correct results call forpost-Hartree–Fock methods that include electron correlationdo not suffer from triplet instability.11
Recent accurateab initio calculations ofJ have beencarried out using the multiconfigurational self-consistefield linear response~MCSCF LR! method,12 the second-order polarization–propagator approach with coupled-clusingles-and-doubles amplitudes, SOPPA~CCSD!,13 and theequation-of-motion coupled-cluster singles-and-doub~EOM-CCSD! method.14–16 Reference 11 provides a recereview of the field. The anisotropic spin–spin componenin particular, have been reported in Refs. 10, 17–26~see alsoRef. 27!. The first successful calculations ofJ using density-functional theory~DFT!28–30have also given rise to an interest in the calculation of the anisotropicJS with DFT.31–33
The first general implementations of all the contributioto the full nonrelativisticJ tensor, using the three maiclasses of exchange–correlation functionals—that is,
il:
8 © 2002 American Institute of Physics
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

--
td
--t
a
ein
n
ichCsth
t oibC
le
rei-acs
t
er
di
Ofther
the, ised
pa-
nsular
g,–-g
or-
al
inre
s inm-
ga,
s.
5999J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Spin–spin coupling tensors
local-density approximation~LDA !,34 the generalized gradient approximation~GGA!, and hybrid functionals—were introduced in Refs. 35 and 36. There, all the contributionsthe spin–spin couplings were calculated using analyticalrivative techniques. The calculatedJ have been found to improve from LDA to GGA and further to the hybrid functionals. The results obtained with the hybrid three-parameBecke–Lee–Yang–Parr~B3LYP! functional37,38 are espe-cially promising: the overal accuracy is almost as highwith that of the much more elaborateab initio calculations.36
Nevertheless, the results for1JFH in the HF molecule havebeen found poor with all these functionals. This has berelated to deficiencies in their long-range behavior, occurrfor elements having lone-pair electrons such as fluorine.28,29
In this work, we examine the zeroth- and second-raparts of J produced by DFT with the LDA,34 BLYP~Becke–Lee–Yang–Parr!,39,40 and B3LYP functionals, re-stricting our attention to molecules and couplings for whrecent MCSCF data exist. In addition, an accurate MCScalculation on FHF2 has been carried out explicitly for thipaper—for details, see Ref. 41. However, our focus is onperformance of DFT for the anisotropic second-rank parspin–spin coupling tensors of several types. Where possthe anisotropic properties calculated using DFT and MCStheory are compared with experimental data.
II. THEORY AND COMPUTATION
A. Spin–spin coupling tensor
The nuclear magnetic momentmK is related to thenuclear spinIK through
mK5gK\ IK , ~2!
wheregK is the nuclear gyromagnetic ratio. TheJ tensor canbe calculated as the second derivative of the perturbed etronic energy with respect to the nuclear spins as~in units ofHz!
JKL51
h
d2E
dIK dILU
IK5IL50
5\
2pgKgLKKL . ~3!
HereK is the reduced spin–spin coupling tensor usuallyported in units of 1019 T2 J21. Since it describes the magntude of the pure electronic coupling without the nuclear ftors, it is particularly useful for comparing couplingbetween different elements and isotopes.
In the nonrelativistic theory of Ramsey,4,5 the nuclearspins are coupled by four distinct interactions, giving risefive different contributions to the coupling tensor:
J5JDSO1JPSO1JSD1JFC1JSD/FC. ~4!
The diamagnetic nuclear-spin–electron-orbit~DSO! contri-bution is a ground-state expectation value of the DSO optor, bilinear inI K and I L :
JKLDSO}^DSO~KL !&. ~5!
The remaining four paramagnetic terms in Eq.~4! can becalculated as linear response functions of the corresponperturbation operators,12 each of which is linear in thenuclear spins:
Downloaded 25 Sep 2002 to 129.240.81.153. Redistribution subject to A
oe-
er
s
ng
k
F
efle,F
c-
-
-
o
a-
ng
JKLPSO}^^PSO~K !; PSO~L !&&0 , ~6!
JKLSD}^^SD~K !; SD~L !&&0 , ~7!
JKLFC}^^FC~K !; FC~L !&&0 , ~8!
JKLSD/FC}^^FC~K !; SD~L !&&0
1^^FC~L !; SD~K !&&0 . ~9!
The purely zeroth-rank Fermi-contact~FC! term is isotropicand usually gives the most important contribution toJ. Like-wise, the purely second-rank spin–dipole/Fermi-contact~SD/FC! cross term usually dominates the anisotropic partJS. Bycontrast, the paramagnetic nuclear-spin–electron-orbit~PSO!and spin–dipole~SD! mechanisms, as well as the DSmechanism, contribute to all the rank-0, -1, and -2 parts oJ.
The rank-0 and -2 contributions can be presented eiin the principal axis system~PAS! or in a molecule-fixedframe common to all tensors. The former way, defined byprincipal values and the directions of the principal axesconventional in solid state NMR, whereas the molecule-fixframe is usually used in liquid–crystal~LC! NMR. There,the observable corresponding toJS is denoted asJaniso.6,7
The anisotropy of the tensor
DJ5Jzz212 ~Jxx1Jyy! ~10!
and, in certain point-group symmetries, the asymmetryrameter
h5Jxx2Jyy
Jzz~11!
enter the expression ofJaniso. Here, thez axis is parallel withthe principal molecular symmetry axis. Other combinatioof tensor elements may also contribute when the molecsymmetry is low.6
The experimentally observable anisotropic couplinDKL
exp5DKL112 J KL
aniso, contains the direct bare-nucleus dipoledipole couplingDKL ~carrying information about the internuclearKL distance! as well as the indirect electronic couplinJKL
aniso.6 To extract structural information fromDKLexp, JKL
aniso
must be known. Accurate calculations of the fullJKL
tensors—not just their isotropic parts—are therefore imptant.
B. DFT calculations
The DFT calculations were carried out by using a locversion of the DALTON quantum chemistry program42 witha recent DFT implementation of NMR spin–spcouplings.36 Three exchange–correlation functionals weused: LDA, GGA~BLYP!, and hybrid~B3LYP!. We haveused the same geometries as in the previous10,17–24,27andpresent41 MCSCF studies. The basis sets are the same athe best calculations of the cited works. Details of the geoetries and basis sets are given in Table I.
In many cases, the segmented basis sets of Huzina43
as contracted and polarized by Kutzelnigget al.,44 wereused. Because of their special contraction scheme,45,46 thesecompact basis sets~denoted HII, HIII, and HIV! have beenfound to be particularly useful for spin–spin calculation
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ell as
F,
6000 J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Lantto, Vaara, and Helgaker
Downloaded 25 Se
TABLE I. Computational details for DFT calculations.a
Molecule Basis set Geometryb Referencec
C2H2 HIVd CC51.207, CH51.060, HCC5180.0 20C2H4 HIVd CC51.338, CH51.088, HCC5121.4 20C2H6 HIII e CC51.535, CH51.094, HCC5111.2 20C6H6 HII f CC51.393, CH51.082 18HCN HIVd CN51.156, CH51.064 17HNC HIVd CN51.165, NH50.994 17CH3CN HIII e CN51.166, CC51.461, CH51.085, HCC5110.0 17CH3NC HIII e CN51.424, CC51.166, CH51.094, HCN5109.1 17HCONH2 HIII e CN51.352, CO51.219, CH151.098, 19
NH2(trans to O)51.001, NH3(cis to O)51.002,OCN5124.7, HCN5112.7, CNH25120.0, CNH35118.5
CH3F HIII e CF51.383, CH51.086, HCF5108.8 23CH2F2 HIII e CF51.354, CH51.092, HCF5108.8, HCH5112.9 23CHF3 HIII e CF51.331, CH51.086, HCF5110.3 23p-C6H4F2
g HIIs3h CF51.368, CH51.093, C7C851.399, C8C951.408, 21C12C7C85122.8, H1C8C95121.4
FHF2 HIVu( t4,d1)i HF51.152, FHF5180.0 41ClF3 cc-pVQZj,k ClFax51.599, ClFeq51.701,FaxClFeq587.5 10OF2 cc-pCVQZl OF51.405, FOF5103.4 10CH3SiH3 HIII e CSi51.864, CH51.095, SiH51.482, HCSi5110.9,
CSiH5110.422
HF HIIIsu4m HF50.918 27HCl HIIIsu4m HCl51.275 27H2O HIIIsu4m HO50.968, HOH575.5 27H2S HIIIsu4m HS51.336, HSH587.9 27NH3 HIIIsu4m HN51.012, HNH5106.7 27PH3 HIIIsu4m HP51.412, HPH593.3 27CH4 HIIIsu4m CH51.086, HCH5109.5 27SiH4 HIIIsu4m SiH51.471, HSiH5109.5 27LiF cc-pV5Zn LiF51.564 24BF cc-pV5Zn BF51.263 24NaF F: cc-pV5Z,n Na:o NaF51.926 24AlF aug-cc-pVQZp AlF51.654 24ClF aug-cc-pVQZp ClF51.628 24KF F: cc-pV5Z,n K:o KF52.171 24LiH cc-pV5Zn LiH51.596 24Na2
o NaNa53.079 24KNa o KNa53.499 24
aBasis sets and geometries common to present DFT as well as present~in the case of FHF2! and previous~seereferences! MCSCF calculations. See the references listed in the last column for complete details as wliterature citations of the basis sets used.
bThe units for the bond lengths and angles are A˚ ngstroms and degrees, respectively.cReference to the earlier MCSCF calculation.dC–F, @11s7p3d1f /8s7p3d1f #; H, @6s3p1d/5s3p1d# in the @primitive/contracted# notation.eSi, @12s8p3d/8s7p3d#; C–F, @11s7p2d/7s6p2d#; H, @6s2p/4s2p#.fC, @9s5p1d/5s4p1d#; H, @5s1p/3s1p#.gThe numbering of nuclei refers to Fig. 1 in Ref. 21.hHII augmented with three sets of tights functions keeping the original contraction for C and F. C–@12s5p1d/8s5p1d#; H, @5s1p/3s1p#.
iHIV fully decontracted and augmented with four sets of tights ~H! andsp ~F! functions and one set of diffusespd ~H! andspd f ~F! functions. F,@16s12p4d2 f #; H, @11s4p2d#.
jF, @12s6p3d2f 1g/5s4p3d2f 1g#; Cl, @16s11p3d2f 1g/6s5p3d2f 1g#.k~ax! denotes axial fluorine in theC2 symmetry axis and~eq! the two equatorial fluorines. The~ax!/~eq!nomenclature has been interchanged in Ref. 10.
lO–F, @15s9p5d3f 1g/8s7p5d3f 1g#.mHIII basis set fully decontracted and augmented with four sets of tights ~H! andsp ~C–Cl! functions. Si–Cl,@16s12p3d#; C–F, @15s11p2d#; H, @10s2p#.
nLi–F, @14s7p4d3f 2g1h/6s5p4d3f 2g1h#.oUncontracted basis set: K,@26s17p1d#; Na, @20s12p4d#.pF, @13s7p4d3f 2g/6s5p4d3f 2g#; Al–Cl, @17s12p4d3f 2g/7s6p4d3f 2g#.
IVhe
ts-
ets
While the small HII set provides qualitatively correctJ ten-sors, quantitative accuracy is reached with the HIII and Hsets augmented with tight primitives. By contrast, tcorrelation-consistent valence sets cc-pVXZ with
p 2002 to 129.240.81.153. Redistribution subject to A
2<X<647,48 and their augmented counterparaug-cc-pVXZ48 converge slowly for spin–spin couplings because of insufficient flexibility in the core region.46 For thecorrelation-consistent core–valence basis s
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

is
xis
gha
ns
cpl
-
d
uth
henc
foly.cne
eavghS
rceala
erndre
pe
xeote
in
dnsn-S
-he
al.erm-
ar-e.
rmnt
to
anthat
f
u-
or
allT
for
ri-ller
e-
m--la-rue
r,O,talg-le
imi-has
6001J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Spin–spin coupling tensors
cc-pCVXZ,47,49the situation is again different as the basiswell-converged already at the quadruple-zeta cc-pCVQZlevel.
In the presentation of the results, the molecular aframe is fixed and the anisotropic properties are defined athe original papers. In most cases, this means that thez axisis along the principal molecular symmetry axis, althouPAS is used for the molecules in Ref. 17. These choicesmade to simplify the comparison of the DFT calculatiowith the previously reported MCSCF results.
C. The quality of the MCSCF reference data
For a complete specification of the MCSCF wave funtions, we refer to the original papers and Table S2 in supmentary material obtainable from EPAPS.50 Here we restrictourselves to some general comments.
The active spaces for HCN, HNC, CH3CN, CH3NC,17
and C6H4F2 ~Ref. 21! are of the multireference restrictedactive-space ~RAS!51 type, with small RAS3 virtual-excitation orbital spaces. For these molecules, therefore,namical correlation is only partly recovered.17,21 This is alsotrue for C6H6 , where the CC coupling tensors were calclated with a small multireference RAS wave function andCH tensors with a complete-active-space~CAS! wavefunction.18 Similarly, the small CAS expansions used for tdiatomic molecules in Ref. 24 and the small single-refereRAS expansion used for OF2 and ClF3 in Ref. 10 can onlybe expected to provide qualitative results.
The single-reference RAS active space usedHCONH2 was quite large but nevertheless allowed onsingle and double excitations into the virtual orbital space19
The lack of higher excitations may compromise the accuraalthough the results are quite well converged in the sequeof calculations presented in Ref. 19. For the singly bondsystems C2H6,20 CH3SiH3 ,22 and CH42nFn (n51,2,3),23
static correlation is unimportant close to the equilibrium gometry. Therefore, the large single-reference RAS wfunctions used for these molecules should provide hiquality tensors. Likewise, the large multireference RAtreatments of the doubly and triply bonded C2H4, and C2H2
should be quite accurate.20 Finally, the large multireferenceRAS functions used for FHF2 in the present work and fothe first-row hydrides27 as well as the large single-referenRAS wave functions correlating also the semicore orbitfor second-row hydrides in Ref. 27 should ensure high quity of the calculatedJ.
The present DFT results are compared also with expmental data, which contain relativistic, rovibrational, asolvent contributions. These contributions are not recoveby the DFT and MCSCF calculations discussed in this pa
III. RESULTS AND DISCUSSION
In this section, we compare the DFT, MCSCF, and eperimental results of the CC, FF, FC, CH, HH, FX, and othspin–spin coupling tensors in turn, and comment finallythe DFT performance in general. For brevity, we have listhe contributions from the different mechanisms to theJ ten-sors only for the anisotropic components. Tables includ
Downloaded 25 Sep 2002 to 129.240.81.153. Redistribution subject to A
sin
re
-e-
y-
-e
e
r
y,ced
-e-
sl-
i-
dr.
-rnd
g
all the individual contributions to the CC, FF, FC, CH, anHH isotropic coupling constants as well as the contributioto the anisotropic components of CH and HH coupling tesors, are obtainable as supplementary material from EPAP50
~see Tables S3–S7!.
A. CC spin–spin coupling tensors
The DFT JCC tensors and their contributions are compared with MCSCF and experimental data in Table II. Tisotropic couplingsJCC are usually well described by BLYPand B3LYP, the latter being the slightly better functionExcept in C2H2 , the totalJCC are either as good as or bettthan the MCSCF values—notably for benzene, where copromises in theab initio work had to be made.18 This is anexcellent result in the sense that, for the simple hydrocbons, the MCSCF wave functions are in fact quite accurat20
The main differences in the totalJ between DFT andMCSCF—in particular, the overestimation by B3LYP foC2H2 and the usual underestimation by LDA—arise froJFC, the other contributions being similar for the differemethods~see Table S3!. For C2H6 , the slightly smallerJFC
obtained with B3LYP gives a total coupling that is closerthe experimental value than with MCSCF. For CH3CN andC6H6, the B3LYP results are much closer to experiment thare the previous small-RAS MCSCF results, suggestingB3LYP is also better for CH3NC.17,18 MCSCF theory over-estimates the magnitude ofJFC in C6H6 , probably because othe small number of active orbitals.18
The results for the anisotropic part of the spin–spin coplings improve in the sequence LDA→BLYP→B3LYP.B3LYP is the only functional that gives the correct sign fD 1JCC
SD/FC in C2H2 . For DJCC in CH3NC and CH3CN, theresults obtained with the B3LYP functional and with a smMCSCF wave function are almost identical. However, DFoverestimates the PSO and SD contributions toD 1JCC and1JCC,zzh in C2H4 and, in particular, toD 1JCC in C2H2 . Werecall that high-quality MCSCF wave functions are usedthe simple hydrocarbons.
For the anisotropic couplings in C6H6 , the situation isnot as clear as for the isotropic couplings. For themetaandpara couplings, the B3LYP values are a bit closer to expement than are the MCSCF values, due to the smaDJSD/FC. For theortho coupling, on the other hand, B3LYPis much further away from experiment than is MCSCF, bcause of a negativeDJSD/FC with B3LYP. However, since theexperimental uncertainties are large for the anisotropic coponents, and sinceDJSD/FC may be overestimated by MCSCF ~due to the insufficient treatment of electron corretion!, we cannot rule out a near-zero or even negative tvalue ofD1JCC
SD/FC.In short, whereas LDA fails to produce reliableJCC,
BLYP works satisfactorily in nearly all cases. HoweveB3LYP is generally the most accurate functional for the PSFC, SD, and SD/FC contributions, resulting in the best toJCC. For all functionals, the description of these paramanetic contributions deteriorates for couplings over multipbonds. Overall, the B3LYP carbon–carbon tensors are slar to the MCSCF tensors whenever the latter approachbeen pursued far enough~e.g., simple hydrocarbons!. How-
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
s in
ge
ata
tes
ri-us
re-ed
an
–A
cel-s
hepic
ted
as ofthe
ct
inl-
A
for
x-be-on-icorare
a
ain
y
6002 J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Lantto, Vaara, and Helgaker
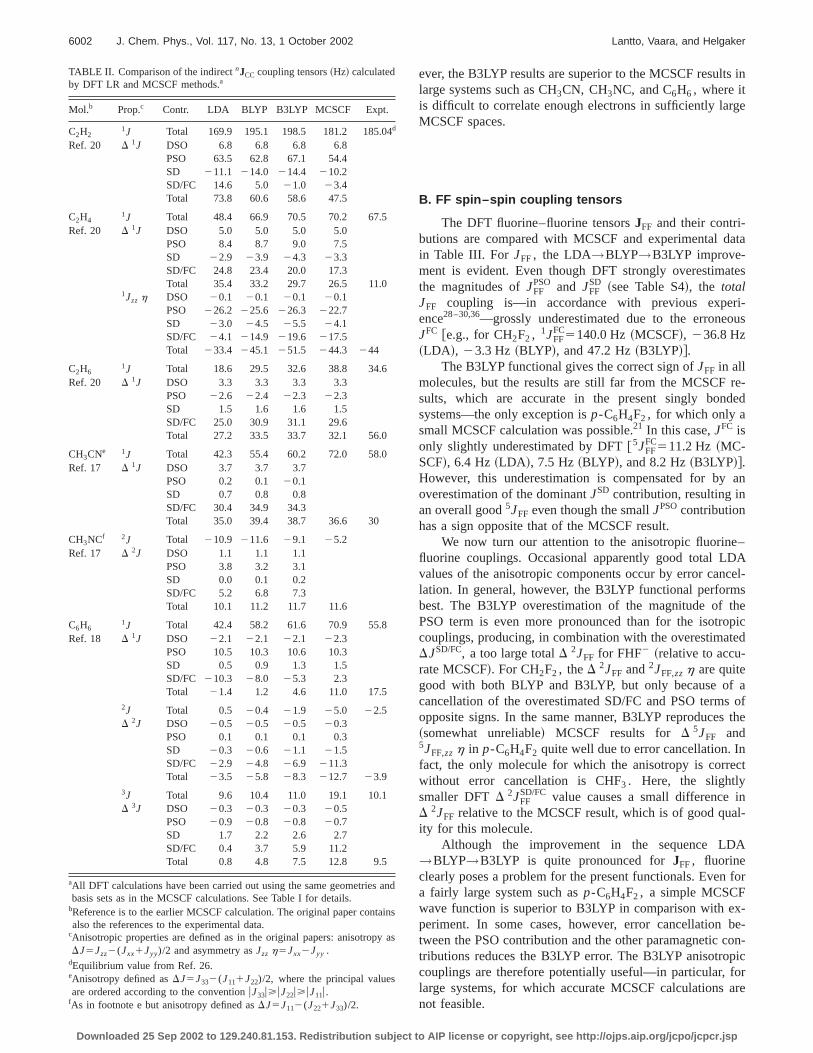
TABLE II. Comparison of the indirectnJCC coupling tensors~Hz! calculatedby DFT LR and MCSCF methods.a
Mol.b Prop.c Contr. LDA BLYP B3LYP MCSCF Expt.
C2H21J Total 169.9 195.1 198.5 181.2 185.04d
Ref. 20 D 1J DSO 6.8 6.8 6.8 6.8PSO 63.5 62.8 67.1 54.4SD 211.1 214.0 214.4 210.2SD/FC 14.6 5.0 21.0 23.4Total 73.8 60.6 58.6 47.5
C2H41J Total 48.4 66.9 70.5 70.2 67.5
Ref. 20 D 1J DSO 5.0 5.0 5.0 5.0PSO 8.4 8.7 9.0 7.5SD 22.9 23.9 24.3 23.3SD/FC 24.8 23.4 20.0 17.3Total 35.4 33.2 29.7 26.5 11.0
1Jzz h DSO 20.1 20.1 20.1 20.1PSO 226.2 225.6 226.3 222.7SD 23.0 24.5 25.5 24.1SD/FC 24.1 214.9 219.6 217.5Total 233.4 245.1 251.5 244.3 244
C2H61J Total 18.6 29.5 32.6 38.8 34.6
Ref. 20 D 1J DSO 3.3 3.3 3.3 3.3PSO 22.6 22.4 22.3 22.3SD 1.5 1.6 1.6 1.5SD/FC 25.0 30.9 31.1 29.6Total 27.2 33.5 33.7 32.1 56.0
CH3CNe 1J Total 42.3 55.4 60.2 72.0 58.0Ref. 17 D 1J DSO 3.7 3.7 3.7
PSO 0.2 0.1 20.1SD 0.7 0.8 0.8SD/FC 30.4 34.9 34.3Total 35.0 39.4 38.7 36.6 30
CH3NCf 2J Total 210.9 211.6 29.1 25.2Ref. 17 D 2J DSO 1.1 1.1 1.1
PSO 3.8 3.2 3.1SD 0.0 0.1 0.2SD/FC 5.2 6.8 7.3Total 10.1 11.2 11.7 11.6
C6H61J Total 42.4 58.2 61.6 70.9 55.8
Ref. 18 D 1J DSO 22.1 22.1 22.1 22.3PSO 10.5 10.3 10.6 10.3SD 0.5 0.9 1.3 1.5SD/FC 210.3 28.0 25.3 2.3Total 21.4 1.2 4.6 11.0 17.5
2J Total 0.5 20.4 21.9 25.0 22.5D 2J DSO 20.5 20.5 20.5 20.3
PSO 0.1 0.1 0.1 0.3SD 20.3 20.6 21.1 21.5SD/FC 22.9 24.8 26.9 211.3Total 23.5 25.8 28.3 212.7 23.9
3J Total 9.6 10.4 11.0 19.1 10.1D 3J DSO 20.3 20.3 20.3 20.5
PSO 20.9 20.8 20.8 20.7SD 1.7 2.2 2.6 2.7SD/FC 0.4 3.7 5.9 11.2Total 0.8 4.8 7.5 12.8 9.5
aAll DFT calculations have been carried out using the same geometriesbasis sets as in the MCSCF calculations. See Table I for details.
bReference is to the earlier MCSCF calculation. The original paper contalso the references to the experimental data.
cAnisotropic properties are defined as in the original papers: anisotropDJ5Jzz2(Jxx1Jyy)/2 and asymmetry asJzz h5Jxx2Jyy .
dEquilibrium value from Ref. 26.eAnisotropy defined asDJ5J332(J111J22)/2, where the principal valuesare ordered according to the conventionuJ33u>uJ22u>uJ11u.
fAs in footnote e but anisotropy defined asDJ5J112(J221J33)/2.
Downloaded 25 Sep 2002 to 129.240.81.153. Redistribution subject to A
ever, the B3LYP results are superior to the MCSCF resultlarge systems such as CH3CN, CH3NC, and C6H6, where itis difficult to correlate enough electrons in sufficiently larMCSCF spaces.
B. FF spin–spin coupling tensors
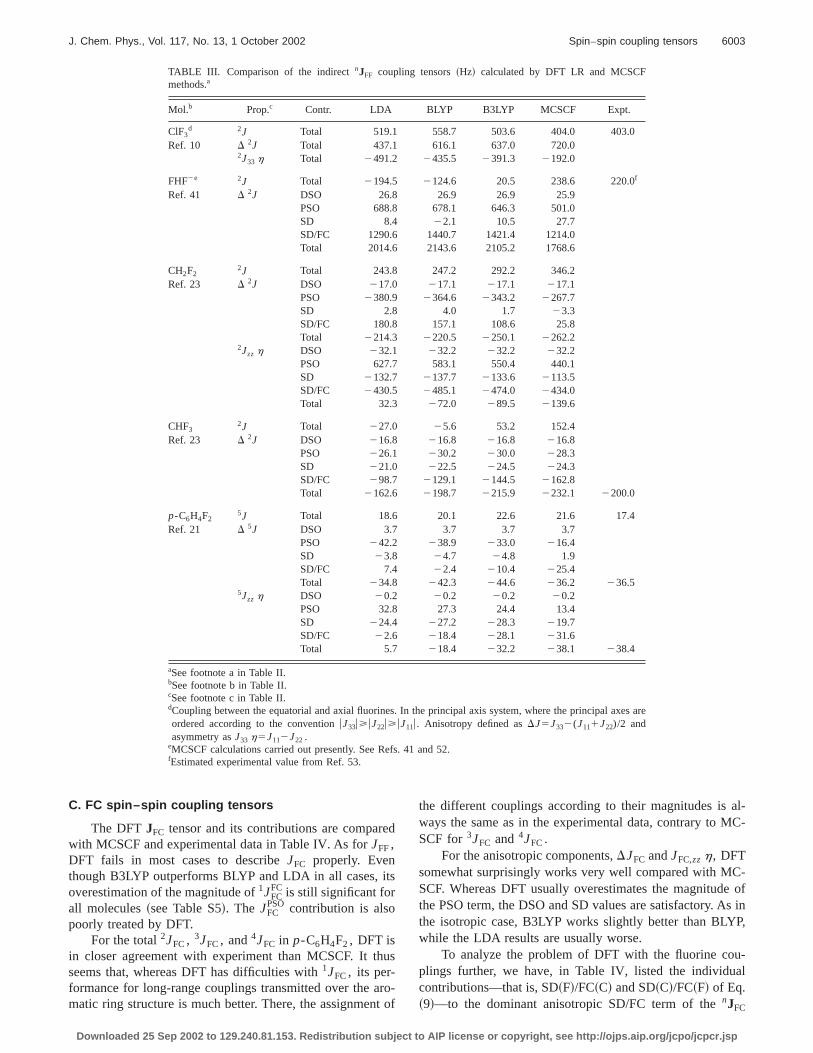
The DFT fluorine–fluorine tensorsJFF and their contri-butions are compared with MCSCF and experimental din Table III. For JFF, the LDA→BLYP→B3LYP improve-ment is evident. Even though DFT strongly overestimathe magnitudes ofJFF
PSO and JFFSD ~see Table S4!, the total
JFF coupling is—in accordance with previous expeence28–30,36—grossly underestimated due to the erroneoJFC @e.g., for CH2F2 , 1JFF
FC5140.0 Hz~MCSCF!, 236.8 Hz~LDA !, 23.3 Hz ~BLYP!, and 47.2 Hz~B3LYP!#.
The B3LYP functional gives the correct sign ofJFF in allmolecules, but the results are still far from the MCSCFsults, which are accurate in the present singly bondsystems—the only exception isp-C6H4F2 , for which only asmall MCSCF calculation was possible.21 In this case,JFC isonly slightly underestimated by DFT@5JFF
FC511.2 Hz ~MC-SCF!, 6.4 Hz~LDA !, 7.5 Hz~BLYP!, and 8.2 Hz~B3LYP!#.However, this underestimation is compensated for byoverestimation of the dominantJSD contribution, resulting inan overall good5JFF even though the smallJPSOcontributionhas a sign opposite that of the MCSCF result.
We now turn our attention to the anisotropic fluorinefluorine couplings. Occasional apparently good total LDvalues of the anisotropic components occur by error canlation. In general, however, the B3LYP functional performbest. The B3LYP overestimation of the magnitude of tPSO term is even more pronounced than for the isotrocouplings, producing, in combination with the overestimaDJSD/FC, a too large totalD 2JFF for FHF2 ~relative to accu-rate MCSCF!. For CH2F2 , theD 2JFF and2JFF,zzh are quitegood with both BLYP and B3LYP, but only because ofcancellation of the overestimated SD/FC and PSO termopposite signs. In the same manner, B3LYP reproduces~somewhat unreliable! MCSCF results for D 5JFF and5JFF,zzh in p-C6H4F2 quite well due to error cancellation. Infact, the only molecule for which the anisotropy is correwithout error cancellation is CHF3. Here, the slightlysmaller DFTD 2JFF
SD/FC value causes a small differenceD 2JFF relative to the MCSCF result, which is of good quaity for this molecule.
Although the improvement in the sequence LD→BLYP→B3LYP is quite pronounced forJFF, fluorineclearly poses a problem for the present functionals. Evena fairly large system such asp-C6H4F2 , a simple MCSCFwave function is superior to B3LYP in comparison with eperiment. In some cases, however, error cancellationtween the PSO contribution and the other paramagnetic ctributions reduces the B3LYP error. The B3LYP anisotropcouplings are therefore potentially useful—in particular, flarge systems, for which accurate MCSCF calculationsnot feasible.
nd
s
as
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
xes are
6003J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Spin–spin coupling tensors
Downloaded 25 Se
TABLE III. Comparison of the indirectnJFF coupling tensors~Hz! calculated by DFT LR and MCSCFmethods.a
Mol.b Prop.c Contr. LDA BLYP B3LYP MCSCF Expt.
ClF3d 2J Total 519.1 558.7 503.6 404.0 403.0
Ref. 10 D 2J Total 437.1 616.1 637.0 720.02J33 h Total 2491.2 2435.5 2391.3 2192.0
FHF2e 2J Total 2194.5 2124.6 20.5 238.6 220.0f
Ref. 41 D 2J DSO 26.8 26.9 26.9 25.9PSO 688.8 678.1 646.3 501.0SD 8.4 22.1 10.5 27.7SD/FC 1290.6 1440.7 1421.4 1214.0Total 2014.6 2143.6 2105.2 1768.6
CH2F22J Total 243.8 247.2 292.2 346.2
Ref. 23 D 2J DSO 217.0 217.1 217.1 217.1PSO 2380.9 2364.6 2343.2 2267.7SD 2.8 4.0 1.7 23.3SD/FC 180.8 157.1 108.6 25.8Total 2214.3 2220.5 2250.1 2262.2
2Jzz h DSO 232.1 232.2 232.2 232.2PSO 627.7 583.1 550.4 440.1SD 2132.7 2137.7 2133.6 2113.5SD/FC 2430.5 2485.1 2474.0 2434.0Total 32.3 272.0 289.5 2139.6
CHF32J Total 227.0 25.6 53.2 152.4
Ref. 23 D 2J DSO 216.8 216.8 216.8 216.8PSO 226.1 230.2 230.0 228.3SD 221.0 222.5 224.5 224.3SD/FC 298.7 2129.1 2144.5 2162.8Total 2162.6 2198.7 2215.9 2232.1 2200.0
p-C6H4F25J Total 18.6 20.1 22.6 21.6 17.4
Ref. 21 D 5J DSO 3.7 3.7 3.7 3.7PSO 242.2 238.9 233.0 216.4SD 23.8 24.7 24.8 1.9SD/FC 7.4 22.4 210.4 225.4Total 234.8 242.3 244.6 236.2 236.5
5Jzz h DSO 20.2 20.2 20.2 20.2PSO 32.8 27.3 24.4 13.4SD 224.4 227.2 228.3 219.7SD/FC 22.6 218.4 228.1 231.6Total 5.7 218.4 232.2 238.1 238.4
aSee footnote a in Table II.bSee footnote b in Table II.cSee footnote c in Table II.dCoupling between the equatorial and axial fluorines. In the principal axis system, where the principal aordered according to the conventionuJ33u>uJ22u>uJ11u. Anisotropy defined asDJ5J332(J111J22)/2 andasymmetry asJ33 h5J112J22 .
eMCSCF calculations carried out presently. See Refs. 41 and 52.fEstimated experimental value from Ref. 53.
ed
ts
u
rot
al-C-
-e ofs inP,
u-al
C. FC spin–spin coupling tensors
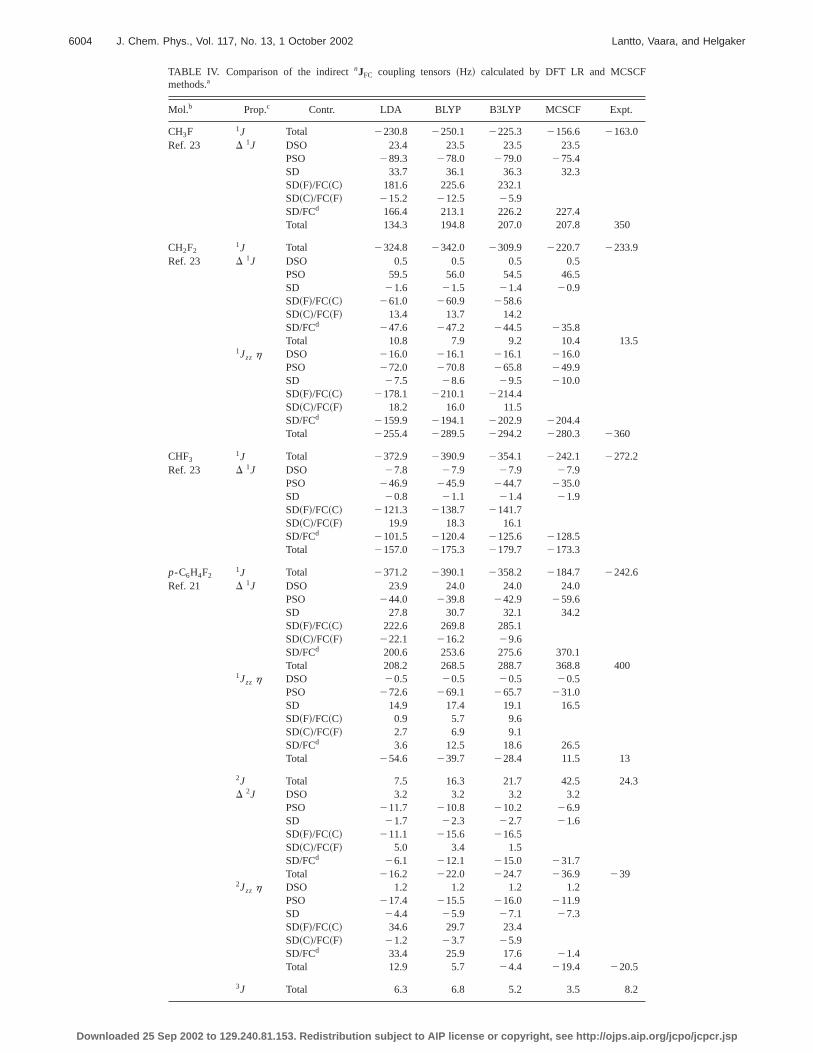
The DFTJFC tensor and its contributions are comparwith MCSCF and experimental data in Table IV. As forJFF,DFT fails in most cases to describeJFC properly. Eventhough B3LYP outperforms BLYP and LDA in all cases, ioverestimation of the magnitude of1JFC
FC is still significant forall molecules~see Table S5!. The JFC
PSO contribution is alsopoorly treated by DFT.
For the total2JFC, 3JFC, and4JFC in p-C6H4F2 , DFT isin closer agreement with experiment than MCSCF. It thseems that, whereas DFT has difficulties with1JFC, its per-formance for long-range couplings transmitted over the amatic ring structure is much better. There, the assignmen
p 2002 to 129.240.81.153. Redistribution subject to A
s
-of
the different couplings according to their magnitudes isways the same as in the experimental data, contrary to MSCF for3JFC and4JFC.
For the anisotropic components,DJFC andJFC,zzh, DFTsomewhat surprisingly works very well compared with MCSCF. Whereas DFT usually overestimates the magnitudthe PSO term, the DSO and SD values are satisfactory. Athe isotropic case, B3LYP works slightly better than BLYwhile the LDA results are usually worse.
To analyze the problem of DFT with the fluorine coplings further, we have, in Table IV, listed the individucontributions—that is, SD~F!/FC~C! and SD~C!/FC~F! of Eq.~9!—to the dominant anisotropic SD/FC term of thenJFC
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
6004 J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Lantto, Vaara, and Helgaker
Downloaded 25 Se
TABLE IV. Comparison of the indirectnJFC coupling tensors~Hz! calculated by DFT LR and MCSCFmethods.a
Mol.b Prop.c Contr. LDA BLYP B3LYP MCSCF Expt.
CH3F 1J Total 2230.8 2250.1 2225.3 2156.6 2163.0Ref. 23 D 1J DSO 23.4 23.5 23.5 23.5
PSO 289.3 278.0 279.0 275.4SD 33.7 36.1 36.3 32.3SD~F!/FC~C! 181.6 225.6 232.1SD~C!/FC~F! 215.2 212.5 25.9SD/FCd 166.4 213.1 226.2 227.4Total 134.3 194.8 207.0 207.8 350
CH2F21J Total 2324.8 2342.0 2309.9 2220.7 2233.9
Ref. 23 D 1J DSO 0.5 0.5 0.5 0.5PSO 59.5 56.0 54.5 46.5SD 21.6 21.5 21.4 20.9SD~F!/FC~C! 261.0 260.9 258.6SD~C!/FC~F! 13.4 13.7 14.2SD/FCd 247.6 247.2 244.5 235.8Total 10.8 7.9 9.2 10.4 13.5
1Jzz h DSO 216.0 216.1 216.1 216.0PSO 272.0 270.8 265.8 249.9SD 27.5 28.6 29.5 210.0SD~F!/FC~C! 2178.1 2210.1 2214.4SD~C!/FC~F! 18.2 16.0 11.5SD/FCd 2159.9 2194.1 2202.9 2204.4Total 2255.4 2289.5 2294.2 2280.3 2360
CHF31J Total 2372.9 2390.9 2354.1 2242.1 2272.2
Ref. 23 D 1J DSO 27.8 27.9 27.9 27.9PSO 246.9 245.9 244.7 235.0SD 20.8 21.1 21.4 21.9SD~F!/FC~C! 2121.3 2138.7 2141.7SD~C!/FC~F! 19.9 18.3 16.1SD/FCd 2101.5 2120.4 2125.6 2128.5Total 2157.0 2175.3 2179.7 2173.3
p-C6H4F21J Total 2371.2 2390.1 2358.2 2184.7 2242.6
Ref. 21 D 1J DSO 23.9 24.0 24.0 24.0PSO 244.0 239.8 242.9 259.6SD 27.8 30.7 32.1 34.2SD~F!/FC~C! 222.6 269.8 285.1SD~C!/FC~F! 222.1 216.2 29.6SD/FCd 200.6 253.6 275.6 370.1Total 208.2 268.5 288.7 368.8 400
1Jzz h DSO 20.5 20.5 20.5 20.5PSO 272.6 269.1 265.7 231.0SD 14.9 17.4 19.1 16.5SD~F!/FC~C! 0.9 5.7 9.6SD~C!/FC~F! 2.7 6.9 9.1SD/FCd 3.6 12.5 18.6 26.5Total 254.6 239.7 228.4 11.5 13
2J Total 7.5 16.3 21.7 42.5 24.3D 2J DSO 3.2 3.2 3.2 3.2
PSO 211.7 210.8 210.2 26.9SD 21.7 22.3 22.7 21.6SD~F!/FC~C! 211.1 215.6 216.5SD~C!/FC~F! 5.0 3.4 1.5SD/FCd 26.1 212.1 215.0 231.7Total 216.2 222.0 224.7 236.9 239
2Jzz h DSO 1.2 1.2 1.2 1.2PSO 217.4 215.5 216.0 211.9SD 24.4 25.9 27.1 27.3SD~F!/FC~C! 34.6 29.7 23.4SD~C!/FC~F! 21.2 23.7 25.9SD/FCd 33.4 25.9 17.6 21.4Total 12.9 5.7 24.4 219.4 220.5
3J Total 6.3 6.8 5.2 3.5 8.2
p 2002 to 129.240.81.153. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
6005J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Spin–spin coupling tensors
Downloaded 25 Se
TABLE IV. ~Continued.!
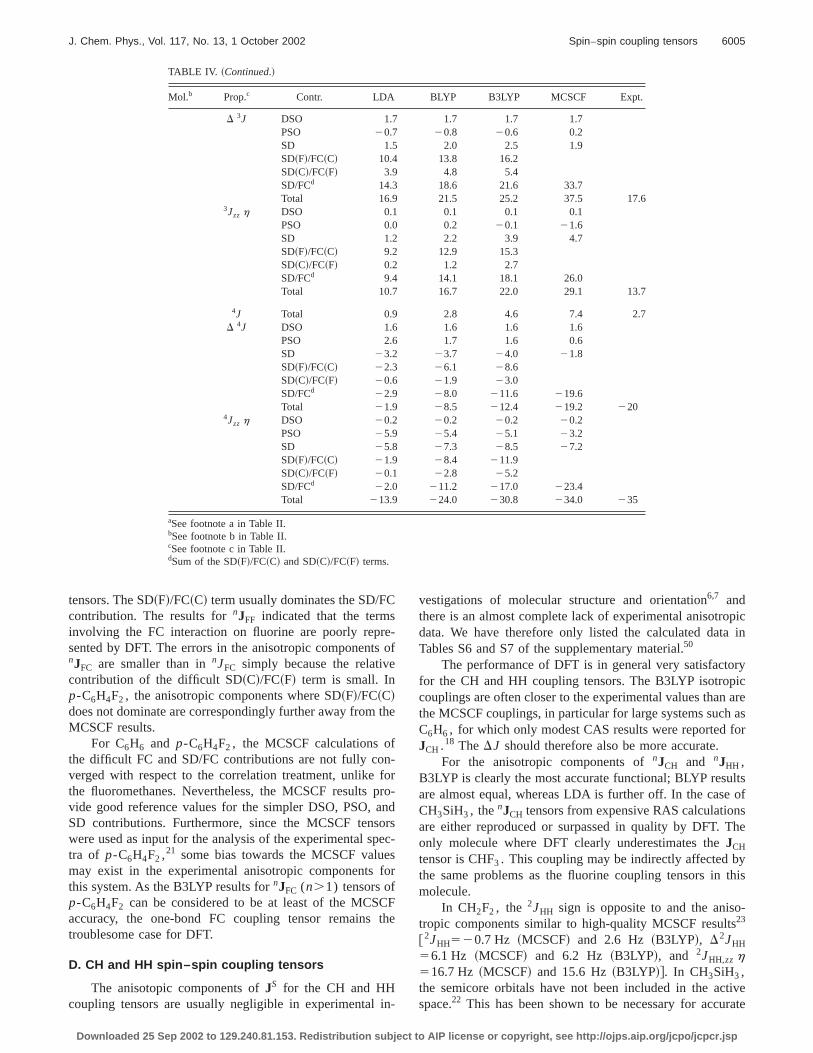
Mol.b Prop.c Contr. LDA BLYP B3LYP MCSCF Expt.
D 3J DSO 1.7 1.7 1.7 1.7PSO 20.7 20.8 20.6 0.2SD 1.5 2.0 2.5 1.9SD~F!/FC~C! 10.4 13.8 16.2SD~C!/FC~F! 3.9 4.8 5.4SD/FCd 14.3 18.6 21.6 33.7Total 16.9 21.5 25.2 37.5 17.6
3Jzz h DSO 0.1 0.1 0.1 0.1PSO 0.0 0.2 20.1 21.6SD 1.2 2.2 3.9 4.7SD~F!/FC~C! 9.2 12.9 15.3SD~C!/FC~F! 0.2 1.2 2.7SD/FCd 9.4 14.1 18.1 26.0Total 10.7 16.7 22.0 29.1 13.7
4J Total 0.9 2.8 4.6 7.4 2.7D 4J DSO 1.6 1.6 1.6 1.6
PSO 2.6 1.7 1.6 0.6SD 23.2 23.7 24.0 21.8SD~F!/FC~C! 22.3 26.1 28.6SD~C!/FC~F! 20.6 21.9 23.0SD/FCd 22.9 28.0 211.6 219.6Total 21.9 28.5 212.4 219.2 220
4Jzz h DSO 20.2 20.2 20.2 20.2PSO 25.9 25.4 25.1 23.2SD 25.8 27.3 28.5 27.2SD~F!/FC~C! 21.9 28.4 211.9SD~C!/FC~F! 20.1 22.8 25.2SD/FCd 22.0 211.2 217.0 223.4Total 213.9 224.0 230.8 234.0 235
aSee footnote a in Table II.bSee footnote b in Table II.cSee footnote c in Table II.dSum of the SD~F!/FC~C! and SD~C!/FC~F! terms.
C
e-se
th
fn-fopranopeesfo
Cth
in
picin
ryicareas
or
ltsofshe
ythis
-
tiverate
tensors. The SD~F!/FC~C! term usually dominates the SD/Fcontribution. The results fornJFF indicated that the termsinvolving the FC interaction on fluorine are poorly reprsented by DFT. The errors in the anisotropic componentnJFC are smaller than innJFC simply because the relativcontribution of the difficult SD~C!/FC~F! term is small. Inp-C6H4F2 , the anisotropic components where SD~F!/FC~C!does not dominate are correspondingly further away fromMCSCF results.
For C6H6 and p-C6H4F2 , the MCSCF calculations othe difficult FC and SD/FC contributions are not fully coverged with respect to the correlation treatment, unlikethe fluoromethanes. Nevertheless, the MCSCF resultsvide good reference values for the simpler DSO, PSO,SD contributions. Furthermore, since the MCSCF tenswere used as input for the analysis of the experimental stra of p-C6H4F2 ,21 some bias towards the MCSCF valumay exist in the experimental anisotropic componentsthis system. As the B3LYP results fornJFC (n.1) tensors ofp-C6H4F2 can be considered to be at least of the MCSaccuracy, the one-bond FC coupling tensor remainstroublesome case for DFT.
D. CH and HH spin–spin coupling tensors
The anisotopic components ofJS for the CH and HHcoupling tensors are usually negligible in experimental
p 2002 to 129.240.81.153. Redistribution subject to A
of
e
ro-d
rsc-
r
Fe
-
vestigations of molecular structure and orientation6,7 andthere is an almost complete lack of experimental anisotrodata. We have therefore only listed the calculated dataTables S6 and S7 of the supplementary material.50
The performance of DFT is in general very satisfactofor the CH and HH coupling tensors. The B3LYP isotropcouplings are often closer to the experimental values thanthe MCSCF couplings, in particular for large systems suchC6H6, for which only modest CAS results were reported fJCH.18 The DJ should therefore also be more accurate.
For the anisotropic components ofnJCH and nJHH ,B3LYP is clearly the most accurate functional; BLYP resuare almost equal, whereas LDA is further off. In the caseCH3SiH3 , thenJCH tensors from expensive RAS calculationare either reproduced or surpassed in quality by DFT. Tonly molecule where DFT clearly underestimates theJCH
tensor is CHF3. This coupling may be indirectly affected bthe same problems as the fluorine coupling tensors inmolecule.
In CH2F2 , the 2JHH sign is opposite to and the anisotropic components similar to high-quality MCSCF results23
@2JHH520.7 Hz ~MCSCF! and 2.6 Hz ~B3LYP!, D2JHH
56.1 Hz ~MCSCF! and 6.2 Hz ~B3LYP!, and 2JHH,zzh516.7 Hz ~MCSCF! and 15.6 Hz~B3LYP!#. In CH3SiH3 ,the semicore orbitals have not been included in the acspace.22 This has been shown to be necessary for accu
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
6006 J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Lantto, Vaara, and Helgaker
Downloaded 25 Se
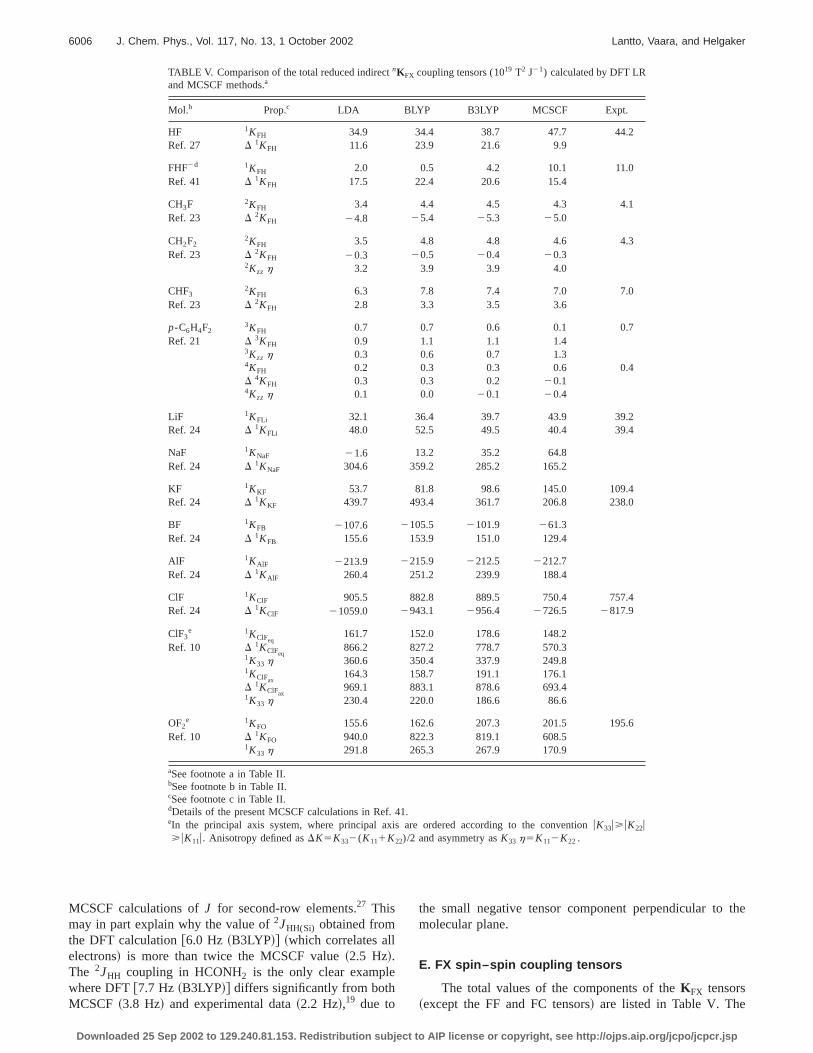
TABLE V. Comparison of the total reduced indirectnKFX coupling tensors (1019 T2 J21) calculated by DFT LRand MCSCF methods.a
Mol.b Prop.c LDA BLYP B3LYP MCSCF Expt.
HF 1KFH 34.9 34.4 38.7 47.7 44.2Ref. 27 D 1KFH 11.6 23.9 21.6 9.9
FHF2d 1KFH 2.0 0.5 4.2 10.1 11.0Ref. 41 D 1KFH 17.5 22.4 20.6 15.4
CH3F 2KFH 3.4 4.4 4.5 4.3 4.1Ref. 23 D 2KFH 24.8 25.4 25.3 25.0
CH2F22KFH 3.5 4.8 4.8 4.6 4.3
Ref. 23 D 2KFH 20.3 20.5 20.4 20.32Kzz h 3.2 3.9 3.9 4.0
CHF32KFH 6.3 7.8 7.4 7.0 7.0
Ref. 23 D 2KFH 2.8 3.3 3.5 3.6
p-C6H4F23KFH 0.7 0.7 0.6 0.1 0.7
Ref. 21 D 3KFH 0.9 1.1 1.1 1.43Kzz h 0.3 0.6 0.7 1.34KFH 0.2 0.3 0.3 0.6 0.4D 4KFH 0.3 0.3 0.2 20.14Kzz h 0.1 0.0 20.1 20.4
LiF 1KFLi 32.1 36.4 39.7 43.9 39.2Ref. 24 D 1KFLi 48.0 52.5 49.5 40.4 39.4
NaF 1KNaF 21.6 13.2 35.2 64.8Ref. 24 D 1KNaF 304.6 359.2 285.2 165.2
KF 1KKF 53.7 81.8 98.6 145.0 109.4Ref. 24 D 1KKF 439.7 493.4 361.7 206.8 238.0
BF 1KFB 2107.6 2105.5 2101.9 261.3Ref. 24 D 1KFB 155.6 153.9 151.0 129.4
AlF 1KAlF 2213.9 2215.9 2212.5 2212.7Ref. 24 D 1KAlF 260.4 251.2 239.9 188.4
ClF 1KClF 905.5 882.8 889.5 750.4 757.4Ref. 24 D 1KClF 21059.0 2943.1 2956.4 2726.5 2817.9
ClF3e 1KClFeq
161.7 152.0 178.6 148.2Ref. 10 D 1KClFeq
866.2 827.2 778.7 570.31K33 h 360.6 350.4 337.9 249.81KClFax
164.3 158.7 191.1 176.1D 1KClFax
969.1 883.1 878.6 693.41K33 h 230.4 220.0 186.6 86.6
OF2e 1KFO 155.6 162.6 207.3 201.5 195.6
Ref. 10 D 1KFO 940.0 822.3 819.1 608.51K33 h 291.8 265.3 267.9 170.9
aSee footnote a in Table II.bSee footnote b in Table II.cSee footnote c in Table II.dDetails of the present MCSCF calculations in Ref. 41.eIn the principal axis system, where principal axis are ordered according to the conventionuK33u>uK22u>uK11u. Anisotropy defined asDK5K332(K111K22)/2 and asymmetry asK33 h5K112K22 .
the
MCSCF calculations ofJ for second-row elements.27 Thismay in part explain why the value of2JHH(Si) obtained fromthe DFT [email protected] Hz ~B3LYP!# ~which correlates allelectrons! is more than twice the MCSCF value~2.5 Hz!.The 2JHH coupling in HCONH2 is the only clear examplewhere [email protected] Hz~B3LYP!# differs significantly from bothMCSCF ~3.8 Hz! and experimental data~2.2 Hz!,19 due top 2002 to 129.240.81.153. Redistribution subject to A
the small negative tensor component perpendicular tomolecular plane.
E. FX spin–spin coupling tensors
The total values of the components of theKFX tensors~except the FF and FC tensors! are listed in Table V. The
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
nt
r-t-
e
e.ol
uaneFTng
s
as
ua
a
teel
g
th
s
.st
n
at
2
6007J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Spin–spin coupling tensors
reduced coupling tensorsK are here used in place ofJ sincecouplings to different nuclei X are compared.
The reduced DFT isotropic couplingsKFX show verydiverse behavior with respect to MCSCF and experimevalues. For fluoromethanes, B3LYP producesKFH that arevery close to the good-quality RAS results.23 For p-C6H4F2 ,the experimental isotropicKFH are reproduced almost pefectly by B3LYP, better than by RAS. In view of the shorcomings of the MCSCF calculation21 and the fact that theMCSCF values converged towards smaller magnitude~datain the original paper!, the B3LYP results are probably morreliable. In HF and FHF2, DFT underestimatesKFH andoverestimates DKFH relative to accurate MCSCFcalculations.27,41 In this case, the PSO error is not largenough to cancel the FC and SD/FC errors, respectively
The reference MCSCF values for the diatomic XF mecules~other than HF! are calculated at the CAS level,10 thusnecessarily with an active space that does not recover mdynamical correlation. Still, the agreement between DFTMCSCF is very satisfactory—in many cases, there is noperiment to compare with. Although the performance of Drelative to experiment or MCSCF is good for the coupliconstants in LiF, AlF, and KF,DK is still quite far off. Thisbehavior is different from1JFC, whereDJ is more accuratethanJ. However, for all diatomics,DK has the same sign ain MCSCF theory.10
In ClF, both atoms carry lone pairs, as reflected inoverestimation of theKClF
PSOterms. Since the FC contributionare also quite small, DFT fails to reproducenKClF , as wewould expect for all halogen–halogen couplings. The sittion is not much better for1KFO, although the isotropic1KFO
is quite close to the MCSCF result obtained with a smactive space.24 The anisotropic components are too large.
Calculations of isotropic couplings have been reporby Patchkovskii, Autschbach, and Ziegler, using the sinteraction correction~SIC! at the LDA level.54 These au-thors demonstrated thatJPSO ~as well as nuclear shieldinconstants, which are theoretically similar toJPSO) can beimproved by the SIC. Hence, the SIC could also improvepresent DFT results when the PSO term is large, as inJFC
and JFF. The JFC term was not improved by the SIC aimplemented in Ref. 54.
F. Other spin–spin coupling tensors
The total values of the properties of a selection ofKXY
coupling tensors are listed in Table VI. WhereasDK is muchbetter in HCl than in HF, the isotropicK exhibits the sameproblems as in HF. ForK andDK between alkali atoms, theagreement with the CAS values10 is qualitative. However, theexperimental values are quite far away from all methods
For KNC and KNH , the agreement with a modeRASSCF wave function is satisfactory.17 For K, B3LYP ismostly closer to experiment than MCSCF; forD 1KNC, DFTgives slightly larger values than does MCSCF. Both DFT aMCSCF underestimateD 2KNC in CH3CN, perhaps indicat-ing a need to revisit the experiment for this molecule.
The B3LYP result for1KSiH is at least as accurate as thof a demanding RAS calculation,22 while B3LYP is clearly
Downloaded 25 Sep 2002 to 129.240.81.153. Redistribution subject to A
al
-
chd
x-
n
-
ll
df-
e
d
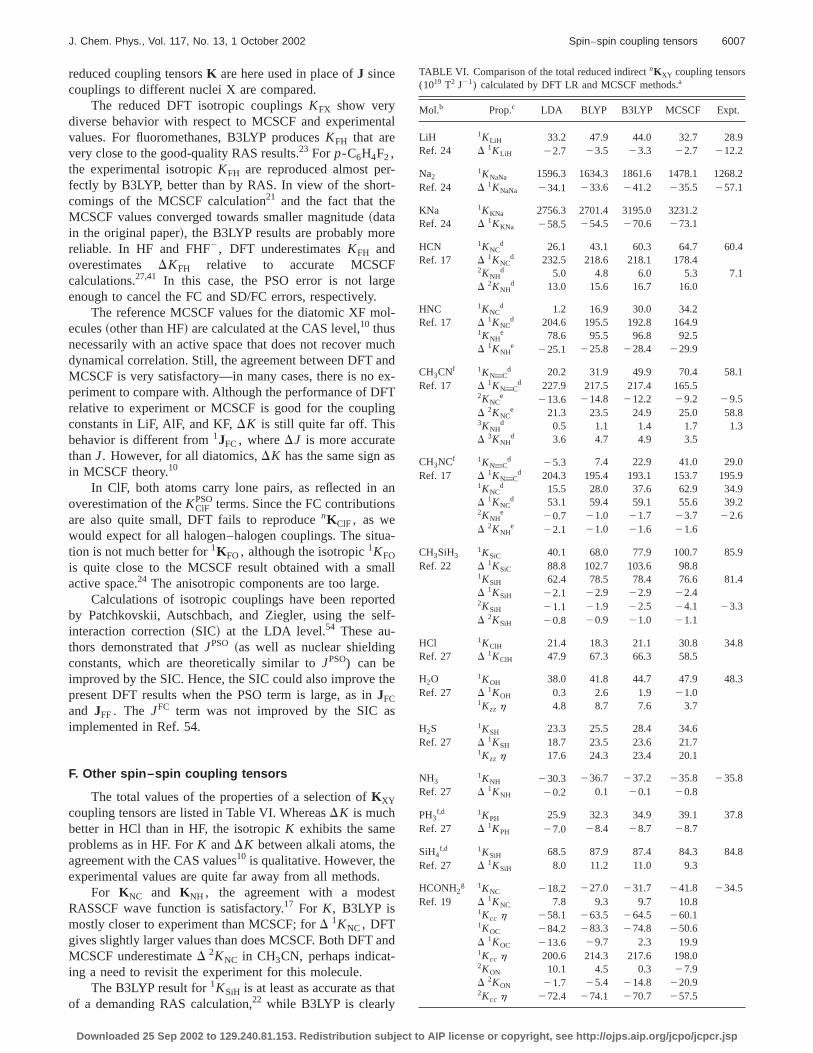
TABLE VI. Comparison of the total reduced indirectnKXY coupling tensors(1019 T2 J21) calculated by DFT LR and MCSCF methods.a
Mol.b Prop.c LDA BLYP B3LYP MCSCF Expt.
LiH 1KLiH 33.2 47.9 44.0 32.7 28.9Ref. 24 D 1KLiH 22.7 23.5 23.3 22.7 212.2
Na21KNaNa 1596.3 1634.3 1861.6 1478.1 1268.
Ref. 24 D 1KNaNa 234.1 233.6 241.2 235.5 257.1
KNa 1KKNa 2756.3 2701.4 3195.0 3231.2Ref. 24 D 1KKNa 258.5 254.5 270.6 273.1
HCN 1KNCd 26.1 43.1 60.3 64.7 60.4
Ref. 17 D 1KNCd 232.5 218.6 218.1 178.4
2KNHd 5.0 4.8 6.0 5.3 7.1
D 2KNHd 13.0 15.6 16.7 16.0
HNC 1KNCd 1.2 16.9 30.0 34.2
Ref. 17 D 1KNCd 204.6 195.5 192.8 164.9
1KNHe 78.6 95.5 96.8 92.5
D 1KNHe 225.1 225.8 228.4 229.9
CH3CNf 1KNwCd 20.2 31.9 49.9 70.4 58.1
Ref. 17 D 1KNwCd 227.9 217.5 217.4 165.5
2KNCe 213.6 214.8 212.2 29.2 29.5
D 2KNCe 21.3 23.5 24.9 25.0 58.8
3KNHd 0.5 1.1 1.4 1.7 1.3
D 3KNHd 3.6 4.7 4.9 3.5
CH3NCf 1KNwCd 25.3 7.4 22.9 41.0 29.0
Ref. 17 D 1KNwCd 204.3 195.4 193.1 153.7 195.9
1KNCd 15.5 28.0 37.6 62.9 34.9
D 1KNCd 53.1 59.4 59.1 55.6 39.2
2KNHe 20.7 21.0 21.7 23.7 22.6
D 2KNHe 22.1 21.0 21.6 21.6
CH3SiH31KSiC 40.1 68.0 77.9 100.7 85.9
Ref. 22 D 1KSiC 88.8 102.7 103.6 98.81KSiH 62.4 78.5 78.4 76.6 81.4D 1KSiH 22.1 22.9 22.9 22.42KSiH 21.1 21.9 22.5 24.1 23.3D 2KSiH 20.8 20.9 21.0 21.1
HCl 1KClH 21.4 18.3 21.1 30.8 34.8Ref. 27 D 1KClH 47.9 67.3 66.3 58.5
H2O 1KOH 38.0 41.8 44.7 47.9 48.3Ref. 27 D 1KOH 0.3 2.6 1.9 21.0
1Kzz h 4.8 8.7 7.6 3.7
H2S 1KSH 23.3 25.5 28.4 34.6Ref. 27 D 1KSH 18.7 23.5 23.6 21.7
1Kzz h 17.6 24.3 23.4 20.1
NH31KNH 230.3 236.7 237.2 235.8 235.8
Ref. 27 D 1KNH 20.2 0.1 20.1 20.8
PH3f,d 1KPH 25.9 32.3 34.9 39.1 37.8
Ref. 27 D 1KPH 27.0 28.4 28.7 28.7
SiH4f,d 1KSiH 68.5 87.9 87.4 84.3 84.8
Ref. 27 D 1KSiH 8.0 11.2 11.0 9.3
HCONH2g 1KNC 218.2 227.0 231.7 241.8 234.5
Ref. 19 D 1KNC 7.8 9.3 9.7 10.81Kcc h 258.1 263.5 264.5 260.11KOC 284.2 283.3 274.8 250.6D 1KOC 213.6 29.7 2.3 19.91Kcc h 200.6 214.3 217.6 198.02KON 10.1 4.5 0.3 27.9D 2KON 21.7 25.4 214.8 220.92Kcc h 272.4 274.1 270.7 257.5
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

to
t
nc-
u
nr tionCw
eneld
hel i
tSOInm
SO-
ity-
ri-of
ereLRPand
-ar-icor
er-
fIn-withlts.ehethe
-s.at
red. In
elyyn-
-ntslso
enesR
em-thetionat-
unc-
y
6008 J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Lantto, Vaara, and Helgaker
better for1KSiC. The B3LYP anisotropies are very closethe MCSCF values.
For HCONH2, the nKNH and nKOH tensors are almosidentical for B3LYP and RAS.19 In the case of1KNC, DFT iscloser to experiment. The largest differences are in1KOC and2KON. As usual, all parameters improve in the sequeLDA→BLYP→B3LYP. In simple first- and second-row hydrides, the 1KXH coupling is reproduced fairly well byB3LYP when compared with the results of large RAS calclations with the semicore correlated.27
G. Comment on the DFT performance
In the present comparison of DFT and MCSCF, oshould keep in mind that the DFT results are often closethe experimental values than the MCSCF results. In additthere are uncertainties related to the neglect of rovibratioand solvent contributions. Hence, a comparison with MSCF does not directly address the accuracy of DFT. Hoever, an advantage ofab initio theory is the ability—in prin-ciple, at least—to monitor the convergence of the differspin–spin contributions with respect to basis-set and corrtion treatment. From such a convergence study, one maytermine whether or not a given DFT result is right for tright reason. Most of the present MCSCF data is usefuthis way.
In most cases, the errors of DFT arise from a failuredescribe the dominant FC contribution. In addition, the Pcontribution is quite sensitive to the choice of functional.such cases, an overall good value of the property someti
TABLE VI. ~Continued.!
Mol.b Prop.c LDA BLYP B3LYP MCSCF Expt.
2KNH1 210.9 213.3 211.8 29.6 212.6D 2KNH1 0.6 0.7 0.7 0.72Kcc h 20.5 20.9 20.8 20.81KNH2 243.0 252.2 253.1 254.4 247.9D 1KNH2 24.7 24.5 25.1 26.61Kcc h 14.5 14.0 15.0 16.81KNH3 242.6 252.2 252.9 254.0 247.9D 1KNH3 24.6 24.4 25.0 26.41Kcc h 14.1 13.6 14.5 16.42KOH1 2.9 3.8 3.9 3.8D 2KOH1 20.1 21.2 21.8 23.32Kcc h 24.2 25.2 25.2 24.63KOH2 0.2 0.4 0.5 0.6D 3KOH2 21.2 21.9 21.9 22.13Kcc h 4.4 4.9 4.5 3.43KOH3 20.7 20.8 21.0 21.0D 3KOH3 0.3 20.1 20.3 20.93Kcc h 20.9 20.8 20.8 20.6
aSee footnote a in Table II.bSee footnote b in Table II.cSee footnote c in Table II.dSee footnote e in Table II.eSee footnote f in Table II.fIn the principal axis system.gIn the principal axis system, where principal values are denoted asKaa ,Kbb , and Kcc according to the definitions in original paper. Anisotropdefined asKcc2(Kaa1Kbb)/2 and asymmetry asKcc h5Kaa2Kbb .
Downloaded 25 Sep 2002 to 129.240.81.153. Redistribution subject to A
e
-
eon,al--
ta-e-
n
o
es
arises fortuitously from error cancellation between the Pand FC contributions~or SD/FC for the anisotropic components!.
IV. CONCLUSIONS
We have compared the performance of the densfunctional linear response~DFT LR! andab initio multicon-figuration self-consistent-field linear response~MCSCF LR!methods for the calculation of all the nonrelativistic contbutions to the diagonal and symmetric off-diagonal partsindirect NMR spin–spin coupling tensors. Comparisons walso made with the existing experimental data. The DFTmethod was tested using the LDA, BLYP, and B3LYexchange–correlation functionals at the same geometriesusing the same basis sets as in the present (FHF2) and pre-vious ~other molecules! MCSCF LR calculations. The purpose of the work was to evaluate the DFT LR and, in pticular, hybrid DFT performance for the anisotropproperties of theJ tensors. Hybrid functionals were tested fthe first time in this sense.
For the investigated main-group systems, B3LYP pforms better than BLYP and in particular LDA. ForJCC,JCH, andJHH , the accuracy of B3LYP is similar to that othe more computationally demanding MCSCF method.deed, for these couplings, one has to make a great effortMCSCF theory to surpass the quality of the B3LYP resuAlso, for couplings to N or Si, DFT provides quantitativresults for both isotropic and anisotropic couplings. Tgreat advantage of DFT over MCSCF is that it providessame accuracy at a lower cost.
Couplings to fluorine are difficult for DFT with the currently available functionals, including hybrid functionalFor this particular atom, the description of the spin densitythe nucleus causes problems for the Fermi-contact~FC! per-turbation. Occasionally, the paramagnetic spin–orbit~PSO!contribution is also inaccurate.
For the anisotropic FC couplings, we have consideseparately the two contributions to the SD/FC interactionheteronuclear couplings such asJFC, the inaccurate SD~C!/FC~F! term ~with FC at fluorine! gives only a small contri-bution and the total coupling is dominated by the accuratcalculated SD~F!/FC~C! term. Consequently, an accuracbetter than for the isotropic coupling is attained. Error cacellation between the PSO, SD, and FC~SD/FC for the an-isotropic components! terms can fortuitously give rise to accurate results—for example, for the anisotropic componeof JFF. Couplings to Cl indicate that other halogens may apresent a challenge to DFT.
To summarize, DFT with the B3LYP functional has beshown to be a very efficient method, which often providsufficient accuracy for the anisotropic components of NMspin–spin coupling tensors. However, as there is no systatic way to improve the correlation treatment beyondcurrent exchange–correlation functionals, some caushould be exercised. In particular, for systems containingoms with many lone pairs such as halogens, the present ftionals are far from satisfactory.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

-h
isn
nnac
th
eb
uc
nd
J.
ari
ed.
S.
ho
hys.
e
f
alitewas
al
e FCer
ation
taryage
Phys.s.
6009J. Chem. Phys., Vol. 117, No. 13, 1 October 2002 Spin–spin coupling tensors
ACKNOWLEDGMENTS
Two of the authors~P.L. and J.V.! are grateful to Professor Jukka Jokisaari for support. P.L. wants to expressgratitude to the Finnish Cultural Foundation, PohjoPohjanmaa Fund of the Finnish Cultural Foundation, TauTonning Fund and the Magnus Ehrnrooth Fund of the Fiish Society of Sciences and Letters for grants. J.V. is on lefrom the NMR Research Group, Department of Physical Sences, University of Oulu, Finland, and is supported byAcademy of Finland~Grant No. 48578! and the MagnusEhrnrooth Fund of the Finnish Society of Sciences and Lters. The computational resources were partially providedthe Center for Scientific Computing, Espoo, Finland.
1E. Hahn, Phys. Rev.80, 580 ~1950!.2W. G. Proctor and F. C. Yu, Phys. Rev.81, 20 ~1951!.3H. S. Gutowsky and D. W. McCall, Phys. Rev.82, 748 ~1951!.4N. F. Ramsey, Phys. Rev.91, 303 ~1953!.5A. D. Buckingham and I. Love, J. Magn. Reson.~1969-1992! 2, 338~1970!.
6J. Lounila and J. Jokisaari, Prog. Nucl. Magn. Reson. Spectrosc.15, 249~1982!.
7J. Vaara, J. Jokisaari, R. E. Wasylishen, and D. L. Bryce Prog. NMagn. Reson. Spectrosc.~in press!.
8E. R. Andrew and L. F. Farnell, Mol. Phys.15, 157 ~1968!.9R. F. Schneider, J. Chem. Phys.48, 4905~1968!.
10D. L. Bryce and R. E. Wasylishen, J. Am. Chem. Soc.122, 11236~2000!.11T. Helgaker, M. Jaszun´ski, and K. Ruud, Chem. Rev.99, 293 ~1999!.12O. Vahtras, H. Ågren, P. Jørgensen, H. J. Aa. Jensen, T. Helgaker, a
Olsen, J. Chem. Phys.96, 2118~1992!; 97, 9178~1992!.13S. P. A. Sauer, J. Phys. B30, 3773~1997!.14H. Sekino and R. J. Bartlett, Chem. Phys. Lett.225, 486 ~1994!.15S. A. Perera, H. Sekino, and R. J. Bartlett, J. Chem. Phys.101, 2186
~1994!.16S. A. Perera, M. Nooijen, and R. J. Bartlett, J. Chem. Phys.104, 3290
~1996!.17A. Barszczewicz, T. Helgaker, M. Jaszun´ski, P. Jørgensen, and K. Ruud,
Magn. Reson., Ser. A114, 212 ~1995!.18J. Kaski, J. Vaara, and J. Jokisaari, J. Am. Chem. Soc.118, 8879~1996!.19J. Vaara, J. Kaski, J. Jokisaari, and P. Diehl, J. Phys. Chem.101, 5069
~1997!; 101, 9185~E! ~1997!.20J. Kaski, P. Lantto, J. Vaara, and J. Jokisaari, J. Am. Chem. Soc.120, 3993
~1998!.21J. Vaara, J. Kaski, and J. Jokisaari, J. Phys. Chem. A103, 5675~1999!.22J. Kaski, P. Lantto, T. Rantala, J. Schroderus, J. Vaara, and J. Jokisa
Phys. Chem. A103, 9669~1999!.23P. Lantto, J. Kaski, J. Vaara, and J. Jokisaari, Chem.-Eur. J.6, 1395
~2000!.24D. L. Bryce and R. E. Wasylishen, J. Am. Chem. Soc.122, 3197~2000!.25R. D. Wigglesworth, W. T. Raynes, S. P. A. Sauer, and J. Oddersh
Mol. Phys. 94, 851 ~1998!; S. P. A. Sauer, W. T. Raynes, and R. ANicholls, J. Chem. Phys.115, 5994~2001!.
26R. D. Wigglesworth, W. T. Raynes, S. Kirpekar, J. Oddershede, andA. Sauer, J. Chem. Phys.112, 3735~2000!; 114, 9192~E! ~2001!.
27P. Lantto and J. Vaara, J. Chem. Phys.114, 5482 ~2001!. Results for theanisotropic properties are unpublished and available from the autupon request.
28V. G. Malkin, O. L. Malkina, and D. R. Salahub, Chem. Phys. Lett.221,91 ~1994!.
29O. L. Malkina, D. R. Salahub, and V. G. Malkin, J. Chem. Phys.105, 8793~1996!.
Downloaded 25 Sep 2002 to 129.240.81.153. Redistribution subject to A
is-o-
vei-e
t-y
l.
J.
, J.
e,
P.
rs
30R. M. Dickson and T. Ziegler, J. Phys. Chem.100, 5286~1996!.31G. Grossmann, M. J. Potrzebowski, U. Fleischer, K. Kru¨ger, O. L.
Malkina, and W. Ciesielski, Solid State Nucl. Magn. Reson.13, 71 ~1998!.32J. Autschbach and T. Ziegler, J. Chem. Phys.113, 936 ~2000!.33J. Autschbach and T. Ziegler, J. Chem. Phys.113, 9410~2000!.34S. J. Vosko, L. Wilk, and M. Nusair, Can. J. Phys.58, 1200~1980!.35V. Sychrovsky, J. Gra¨fenstein, and D. Cremer, J. Chem. Phys.113, 3530
~2000!.36T. Helgaker, M. Watson, and N. C. Handy, J. Chem. Phys.113, 9402
~2000!.37A. D. Becke, J. Chem. Phys.98, 5648~1993!.38P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch, J. P
Chem.98, 11623~1994!.39A. D. Becke, Phys. Rev. A38, 3098~1988!.40C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B37, 785 ~1988!.41The MCSCF calculations for FHF2 were carried out in this work by using
the DALTON software~Ref. 42!. The spin–spin coupling tensors havbeen calculated using the converged HIVu(t4,d1) basis set~H,@11s4p2d#; F, @16s12p4d2f #! that is fully uncontracted from theHuzinaga/Kutzelnigg HIV set~Refs. 43 and 44! and augmented with foursets of tights-(H) or both s- and p-(F) type primitives and one set odiffusesp-(H) or spd-(F) type primitives together with one set ofd-(H)or f -(F) type polarization functions. The exponents for the tight~diffuse/polarization! functions were obtained by multiplying~dividing! the largest~smallest! exponent in a given shell by the factor of 3. The theoreticequilibrium geometry is taken from Ref. 52. The SD contribution is quinsensitive to the dynamical correlation treatment and therefore itcalculated using a multireference type active space1000 1000RAS3221 4221
3110 2110~inthe inactiveRASRAS3
RAS2 notation! chosen on the basis of MP2 natural orbitoccupation numbers. The very large multireference1000 1000RAS9663 10,663
3110 2110
active space was used for the other second-order contributions. Thand SD/FC terms for both1JFH and 2JFF tensors depend most on thcorrelation treatment. The2JFF
PSO is also quite sensitive. According to oucalculations ~not shown here!, all physical contributions are well-converged with respect to both the one-electron basis set and correltreatment.
42T. Helgaker, H. J. Aa. Jensen, P. Jørgensenet al., Dalton release 1.2~2001!, an electronic structure program. Seehttp://www.kjemi.uio.no/software/dalton/dalton.html
43S. Huzinaga,Approximate Atomic Functions~University of Alberta, Edm-onton, 1971!.
44W. Kutzelnigg, U. Fleischer, and M. Schindler, inNMR Basic Principlesand Progress, edited by P. Diehl, E. Fluck, H. Gu¨nther, R. Kosfeld, and J.Seelig~Springer-Verlag, Berlin, 1990!, Vol. 23.
45J. Guilleme and J. San Fabia´n, J. Chem. Phys.109, 8168~1998!.46T. Helgaker, M. Jaszun´ski, K. Ruud, and A. Go´rska, Theor. Chem. Acc.
99, 175 ~1998!.47T. H. Dunning, Jr., J. Chem. Phys.90, 1007~1989!.48D. E. Woon and T. H. Dunning, Jr., J. Chem. Phys.98, 1358~1993!.49D. E. Woon and T. H. Dunning, Jr., J. Chem. Phys.103, 4572~1995!.50See EPAPS Document No. E-JCPSA6-117-306236 for supplemen
tables. This document may be retrieved via the EPAPS homep~http://www.aip.org/pubservs/epaps.html! or from ftp.aip.org in the direc-tory /epaps/. See the EPAPS homepage for more information.
51J. Olsen, B. O. Roos, P. Jørgensen, and H. J. Aa. Jensen, J. Chem.89, 2185 ~1988!; P.-Å. Malmqvist, A. Rendell, and B. O. Roos, J. PhyChem.94, 5477~1990!.
52S. A. Perera and R. J. Bartlett, J. Am. Chem. Soc.122, 1231~2000!.53I. G. Shenderovich, S. N. Smirnov, G. S. Denisovet al., Ber. Bunsenges.
Phys. Chem.102, 422 ~1998!.54S. Patchkovskii, J. Autschbach, and T. Ziegler, J. Chem. Phys.115, 26
~2001!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp














![Measurement of Δ1J(199Hg, 31P) in [HgPCy3(OAc)2]2 and relativistic ZORA DFT investigations of mercury–phosphorus coupling tensors](https://img.pdfslide.net/doc/110x75/63606a090f34b5e422068c90/measurement-of-d1j199hg-31p-in-hgpcy3oac22-and-relativistic-zora-dft-investigations.jpg)














