Embed Size (px)
Citation preview

Structural Insights Into the GTPase Domainof Escherichia coli MnmE Protein
Daniel Monleon,1,2 Marta Martınez-Vicente,3 Vicent Esteve,1,2 Lucia Yim,3 Silvia Prado,3
Maria-Eugenia Armengod,3* and Bernardo Celda1,2*1Department of Physical Chemistry, University of Valencia, C/Dr. Moliner, 50, Burjassot 46100 Valencia, Spain2Central Service for Support of Experimental Sciences (SCSIE), University of Valencia, C/Dr. Moliner, 50,Burjassot 46100 Valencia, Spain3Laboratory of Molecular Genetics, Centro de Investigacion Prıncipe Felipe, Avda. Autopista del Saler 16-3,46013 Valencia, Spain
ABSTRACT The Escherichia coli MnmE pro-tein is a 50-kDa multidomain GTPase involved intRNA modification. Its homologues in eukaryotesare crucial for mitochondrial respiration and, thus,it is thought that the human protein might beinvolved in mitochondrial diseases. Unlike Ras,MnmE shows a high intrinsic GTPase activity andrequires effective GTP hydrolysis, and not simplyGTP binding, to be functionally active. The isolatedMnmE G-domain (165 residues) conserves the GTPaseactivity of the entire protein, suggesting that itcontains the catalytic residues for GTP hydrolysis.To explore the GTP hydrolysis mechanism ofMnmE, we analyzed the effect of low pH on bind-ing and hydrolysis of GTP, as well as on the forma-tion of a MnmE transition state mimic. GTP hy-drolysis by MnmE, but not GTP binding or forma-tion of a complex with mant-GDP and aluminiumfluoride, is impaired at acidic pH, suggesting thatthe chemistry of the transition state mimic is dif-ferent to that of the true transition state, and thatsome residue(s), critical for GTP hydrolysis, isseverely affected by low pH. We use a nuclearmagnetic resonance (NMR)-based approach to getinsights into the MnmE structure and properties.The combined use of NMR restraints and homol-ogy structural information allowed the determina-tion of the MnmE G-domain structure in its freeform. Chemical shift structure-based predictionprovided a good basis for structure refinementand validation. Our data support that MnmE,unlike other GTPases, does not use an argininefinger to drive catalysis, although Arg252 mayplay a role in stabilization of the transition state.Proteins 2007;66:726–739. VVC 2006 Wiley-Liss, Inc.
Key words: GTPase; MnmE; TrmE; aluminium flu-oride; homology modeling; NMR; Era;Ras; Rap2A
INTRODUCTION
GTPases control a variety of cellular processes, includ-ing protein synthesis and translocation, membrane traf-
ficking, signal transduction, and cell cycle control.1,2 Thecommon property shared by these proteins is the pres-ence of a structural module, the G-domain, which isinvolved in the switching of the protein between a GTP-bound and a GDP-bound conformation. This conforma-tional switch determines the active state of the proteinand, therefore, is crucial for its function.
The G-domain typically comprises a central b-sheetflanked by a-helices. Five polypeptide loops form theguanine nucleotide-binding fold and contain the mosthighly conserved motifs in this domain that define the Gprotein superfamily. These motifs (G1–G5) are invaria-bly involved in binding of guanine nucleotides and theimportant cofactor Mg2þ, hydrolysis of GTP, or control-ling the conformational change, which is primarily con-fined to two highly flexible regions called switches I andII that include the G2 and G3 motifs, respectively.
Each GTPase specifically binds and hydrolyses GTP ina cyclic mechanism that activates and deactivates theGTPase protein. Ras-related proteins are in active statewhen GTP-bound; the binding of GTP causes a confor-mational change in these proteins that allows interac-tion with a target molecule or effector; this interaction is
Abbreviations: EcMnmE, Escherichia coli MnmE; G-domain,GTPase domain; GAP, GTPase activating protein; GEF, guanine-nu-cleotide exchange factor; HSQC, heteronuclear single quantum co-herence; NMR, nuclear magnetic resonance; NOE, nuclear over-hauser effect; NOESY, NOE spectroscopy; PDB, Protein Data Bank;RGS, regulators of G protein signaling; RMDSA, restrained molecu-lar dynamics simulated annealing; TmMnmE, Termotoga maritimaMnmE.
Grant sponsor: European Union; Grant number: HPRI-CT-1999-00005 or HPRI-CT-2001-00172; Grant sponsor: Generalidad Valenci-ana; Grant numbers: BMC2001-1555, BFU2004-05819; Grant sponsor:Ministerio de Sanidad y Consumo; Grant numbers: GRUPOS04/07,G03/203, G03/011, and SAF2004-06297.
*Correspondence to: Prof. Bernardo Celda Munoz, Departamentode Quımica Fısica, Universitat de Valencia, Edifici d’Investigacio, C/Dr. Moliner, 50, Burjassot 46100 Valencia, Spain. E-mail: [email protected] or Dra. Marıa-Eugenia Armengod, Laboratory of Molecu-lar Genetics, Centro de Investigacion Prıncipe Felipe, Avda. Autopistadel Saler 16-3, 46013 Valencia, Spain. E-mail:[email protected].
Received 10 November 2005; Revised 31 May 2006; Accepted 17July 2006
Published online 1 December 2006 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/prot.21186
VVC 2006 WILEY-LISS, INC.
PROTEINS: Structure, Function, and Bioinformatics 66:726–739 (2007)

finished by hydrolysis of protein-bound GTP to GDP,which should be released from the inactive GTPase toallow new entry of GTP. The GTPase cycle of Rasrequires participation of GTPase activating proteins(GAPs) and guanine-nucleotide exchange factors (whichcatalyze the hydrolysis step and the release of boundGDP, respectively), since these GTPases typically show avery low intrinsic hydrolase activity and a very high af-finity for guanine nucleotides. In most members of Rasfamily, GAPs stabilize the transition state of the reactionby supplying a so-called arginine finger and enhancingthe stability of amino acids located in switches I andII.3–6 A glutamine adjacent to G3 is also crucial to stabi-lize the transition state and orient the attacking watermolecule. In Ga subunits of trimeric G proteins, the cat-alytic invariant arginine is provided in cis from a helicaldomain of the GTPase polypeptide, inserted just beforethe G2 motif, meanwhile the corresponding GAPs (calledregulators of G protein signaling) do not contribute anyresidue to the catalytic machinery but stabilize theactive conformation.6
Escherichia coli mnmE gene product (EcMnmE) is a50-kDa multidomain protein involved in tRNA modifica-tion, whose middle G-domain is about 165 residues.Unlike proteins from Ras and translation factor families,MnmE exhibits a high intrinsic GTPase hydrolysis rateand low affinity for GTP and GDP; these peculiar bio-chemical properties determine that the GTPase cycle ofMnmE proceeds efficiently, in vitro, without auxiliaryfactors.7 Mutational analyses have indicated that theGTPase domain is essential for the MnmE tRNA modify-ing function, which requires effective hydrolysis of GTP,and not simply GTP binding.8,9
The mitochondrial MSS1 and GTPBP3 proteins arethe MnmE homologues in yeast and human, respec-tively. The MSS1 mutants are defective in mitochondrialtRNA modification, which associates with respiratorydefects.10,11 Several recent works strongly suggest thatlack of modification is a major causative factor of humanmitochondrial diseases associated with specific mito-chondrial tRNA mutations,12–15 which has led to the hy-pothesis that defects in tRNA-modifying proteins mayplay some role in human mitochondrial diseases.9
Strikingly, the isolated G-domain of E. coli MnmEroughly conserves the guanine nucleotide binding andGTPase activities of the intact MnmE molecule,7 whichsuggests that removal of the N- and C-terminal regionsshould not substantially affect its tertiary structure, andthat the MnmE G-domain contains the catalytic residuesthat explain for the high intrinsic GTPase activity of theentire protein. At present, the molecular bases of the pe-culiar biochemical properties of MnmE or its G-domainare unknown. In this respect, the investigation of the pHdependence of the intrinsic GTPase activity of MnmEmay provide some clues on the role of protonation in itscatalytic mechanism. Experimental three-dimensional struc-ture of either E. coli MnmE or its G-domain may also help tounderstand the basis of its intrinsic catalytic activity.Unfortunately, E. coli MnmE did not provide crystals of
the required quality for X-ray diffraction structure deter-mination16 and whole length protein exceeds the size suit-able for structure determination by nuclear magnetic res-onance (NMR) techniques. The solubility of the truncated17 kDa segment of the E. coli MnmE G-domain isextremely reduced and its stability with respect to pH andtemperature conditions is very limited with respect toNMR spectroscopy needs. All these adverse factors makehigh-resolution experimental structure determinationextremely challenging.
A common alternative to experimental procedures forstructure determination is homology modeling three-dimensional structure prediction. Sequence identityscores with respect to other GTPase domains withknown three-dimensional structure are in the validrange for structural homology prediction methods.17 Sev-eral structures have been reported for GTPase proteinsin the Ras family. Therefore, structural information forconserved regions in the amino acid sequence is avail-able and can be translated to the MnmE G-domainamino acid sequence to generate a homology model. Toimprove overall quality and reliability of such a model,NMR experimental data can be included at differentstages of the calculations.18,19 Moreover, the obtainedstructures can be validated by chemical shift predictionalgorithms. Finally, chemical shift prediction may beused for the selection of best models from different tem-plates as an experimental criterion to detect higherstructural homology levels.
In this work, we present a model for the G-domain ofEcMnmE obtained by NMR data and translation ofstructural information from several templates. Both ex-perimental and theoretically derived restraints havebeen used in restrained molecular dynamics simulatedannealing for obtaining and refinement of the model.Overall, our structural studies allow the enunciation ofnew hypotheses concerning the peculiar biochemicalproperties of EcMnm. The possible role of Arg252 andArg275, which might work as arginine fingers for theintrinsic catalytic activity, has also been investigated.
MATERIALS AND METHODSProtein Purification and GTPase Assay
Overproduction and purification of GST fusion pro-teins were done in strain DEV16 transformed with plas-mids pIC684 and pIC758 as previously described.7 GTPhydrolysis was measured by the malachite green assayfor the determination of Pi release.8
Determination of Binding Constants for GTPand GDP
MnmE and MnmE G-domain were titrated againstmant nucleotides (Jena Bioscience) until saturation wasreached. The mant nucleotides (2 lM) were excited at360 nm, and the fluorescence was monitored at 440 nm(Perkin Elmer LS 50 B spectrophotometer, Perkin Elmer,Buckinghamshire, UK). Unless otherwise indicated, allassays were performed at 258C in GTPase buffer (50 mM
727STRUCTURAL INSIGHTS INTO MNME G-DOMAIN
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

Tris-HCl, pH 7.5, 50 mM KCl, 2 mM MgCl2, 5% glycerol).The binding constants (Kd values) were calculated by fit-ting the curves with nonlinear regression, using Graph-Pad Prism software. In these experiments, we used theMnmE mutant G228A, which is impaired for GTP bind-ing,8 as a negative control.
NMR Spectroscopy
NMR sample preparation, resonance assignments pro-cedure, and solution secondary structure determinationfor MnmE G-domain have been previously described byMonleon et al.20 In this previous work, the combined useof automated and manual analysis of triple resonance3D data, along with the distance connectivities extractedfrom a 15N-heteronuclear single quantum coherence(HSQC)-nuclear overhauser effect spectroscopy (NOESY)spectrum recorded at 750 MHz, provided assignmentsfor �95% of assignable backbone atoms of the EcMnmEG-domain. Overall, secondary structure for this G-do-main, based on these chemical shifts and HN-Ha andHN-HN NOE connectivities, is very similar to thatreported for other GTPases such as human Ras or Rac1,or E. coli Era protein. Interestingly, a detailed compari-son of NMR secondary structure shows that the b-strands of MnmE G-domain are more similar to thosereported for Ras than those reported for the other tem-plates, including TmMnmE (the nearest homologue toEcMnmE with known structure) where helix a2 displaysa significant sequence displacement. Structural re-straints derived from NMR experimental data are shownin Table I. Unfortunately, the solubility of MnmE G-do-main NMR sample is extremely reduced and its stabilitywith respect to pH and temperature conditions is verylimited. These adverse factors made the obtaining ofadditional structural information for a high-resolutionexperimental structure determination extremely chal-lenging.
Sequence Analysis
Initial statistics on amino-acid sequence of E. coliMnmE G-domain were performed using ExPASY web
site (http://www.expasy.org). A PSI-BLAST search21 wasperformed over the Protein Data Bank (PDB) databaseto find homologues to G-domain with known three-dimensional structure. Four very well-resolved X-ray orNMR structures of proteins homologous to EcMnmE G-domain were used as templates. These structures werechosen based on a high structural quality and properlevels of sequence identity (TmMnmE, 40%; ERA, 36%;Ras, 30%; and Rap2A, 32%). Alignments between the E.coli MnmE G-domain and amino-acidic sequence of tem-plates were initially achieved using CLUSTALW,22 asshown in Figure 1. A numbering for the isolated G-do-main amino acid sequence was used,20 followed, wherenecessary, by the corresponding positions in the full-length E. coli MnmE amino acid sequence betweenparentheses. Secondary structure for E. coli MnmE G-domain was predicted using PREDATOR23 and PhDsec24
with DSSP database algorithm.25 A consensus predictionwas calculated and then compared with that obtained byNMR backbone assignments (data not shown). Second-ary structure elements and solvent exposure in templatestructures were identified using DSSP method. Pairwise sequence alignments obtained by CLUSTALW weremanually corrected to match maximum number of sec-ondary structure elements and guanine nucleotide bind-ing site motifs between templates and protein target.Lower sequential identity scores were obtained in loopsand turns for increased flexibility and structural vari-ability respect to templates.
Homology Modeling
A structural homology model for EcMnmE G-domainwas built using experimental NMR structural informa-tion and translation of distance restraints from templatestructures to target amino-acid sequence.26,27 Thesewere achieved by using two different types of restraints.First, experimental NMR restraints were obtained fromthe NMR data collected on EcMnmE G-domain. Second,homologous atoms were identified in each template struc-ture and the amino-acid sequence of the target protein,EcMnmE. Then, homology-derived distance restraintswere obtained accordingly as explained elsewhere.27
These two type of restraints were weighed differentlyalong the calculations. Experimental NMR restraintswere initially included in a very conservative fashion.Upper and lower limits for dihedral angle constraintswere obtained by addition and subtraction, respectively, of308 to dihedral angle value reported by TALOS3,28 from13C chemical shift values. Additionally, HN-Ha and HN-HN
NOE cross-peaks identified in 15N-HSQC-NOESY spec-trum were included as 5.0 A upper limit distancerestraints. The final sets of upper and lower bounds wereintroduced in the molecular dynamics program DYANAfor simulated annealing calculations.29 A relative forceconstant of 10 was applied to experimental NMRrestraints over the homology-derived restraints. Highlyviolated homology-derived distance restraints were identi-fied and marked as suspicious. In the initial stages of cal-
TABLE I. Summary of Meaningful ExperimentalStructural Restraints
Total NOE-derived restraints 605Intra-residual 74Sequential 152Medium range (less than 4 residues apart) 332Long range 47
Dihedral angle restraintsC 61/ 61
Chemical shit restraintsCa 167Cb 111Ha 148
Final set of additional homology derived restraints included 615 dis-tance restraints between homologous heavy side chain atoms.
728 D. MONLEON ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

culations, experimental dihedral angle and distancerestraints were heavily weighed with a factor of 20, mean-while suspicious theoretical homology distance restraintswere underweighed with a factor of 0.2. Therefore, the rel-ative weighting factor of experimental NMR restraintsover homology-derived distance restraints varied from 10to 100 depending on the compatibility of the homologywith the NMR data. This procedure was applied to directfinal structures toward better fitting of available experi-mental information. Finally, homologous theoreticalrestraints inconsistent with experimental informationwere detected and analyzed for deletion in final stage ofrefinement. The complete protocol allowed to obtain me-dium resolution structures mostly driven by the experi-mental data.One hundred structures were calculated for each homol-
ogy model and the 20 with lowest target function wereselected for further CNS simulated annealing refine-ment.30 Refinement included all structural restraints ofprevious stages plus Ca/Cb chemical shift values.31 Cus-tomized CNS protocols for simulated annealing, includingstages of weight annealing and temperature annealing,were used for structure refinement. A final ‘‘conjugate gra-dients’’ energy minimization including only experimentalrestraints was performed to fix possible artifacts.The complete protocol was applied to four different pro-
tein structure templates with high sequence similarity toE. coli MnmE G-domain. Initially, close homologous pro-tein Era was chosen as the first template32 (PDB ID1EGA, Era from E. coli). The crystal structure availablein the PDB database was found only in its free form, as
the MnmE G-domain sample studied here. The canonicalGTPase protein Ras was also selected as a template. TheNMR structure of Ras in solution was retrieved in itsGDP-bound form33 (PDB ID 1CRR, Ras from H. sapiens),as no high resolution structure of its free form was found.The NMR solution structure of another mono-domainGTPase protein, Rap2A34 (PDB ID 1KAO from H. sapi-ens), was chosen from the PDB database in its GDP-bound form for similar reasons. In addition, after therecent release of the PDB coordinates for free TmMnmE16
(PDB ID 1XZP), the closest homologue to E. coli MnmE,its crystal structure was included in the set of templates.Homology theoretical distance restraints were derivedfrom the four different template structures separately. Asa consequence, we were able to identify the most structur-ally homologous protein among the templates by compari-son of results with respect to the experimental data. Thecomplete protocol was first tested using Ras, whose struc-ture is well known, as the target molecule. The structureof human Ras was calculated using Rap2A and Era struc-tures as templates. NOE restraints were derived from thecalculated average HN-Hadistances for human Ras. Anumber of distances equivalent to those used in the calcu-lations of EcMnmE G-domain structure was randomlyselected from all HN-Ha distances shorter than 4.5 A.Upper limit for NOE restraints was always set to 5 A.Chemical shift information for human Ras calculation wasintroduced also in the refinement stage.
Stereo chemical quality of final models was measuredwith program PROCHECK. An additional stage of struc-ture validation was performed by back calculation of
Fig. 1. Sequence alignment of EcMnmE G-domain with respect to its closest homologues with known structure (TrmE from T. maritime, Era fromE.coli, human Ras and Rap2A) used for obtaining homology structural information. A bar chart shows the degree of sequence similarity among pro-teins. The secondary structure of the EcMnmE G-domain (as calculated by Molscript, based on our NMR-driven homology model with respect toTmMnmE) has been plotted on the bottom of the graph. Grey boxes indicate hypothetical location for switches I and II based on alignment withTmMnmE. The numbers at the right of the sequences indicate the number of amino acids of each protein. Star indicates conserved residue; a dotor a colon indicates positions with partially conserved residues (dot means minor conservation and colon means higher conservation).
729STRUCTURAL INSIGHTS INTO MNME G-DOMAIN
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
chemical shift values from the homology model PDBstructure with the SHIFTS v4.1 software package.35 Dif-ferences between back-calculated and experimentalchemical shifts were then evaluated. The differenceswere calculated by averaging the deviation between cal-culated and experimental values for Ca and Cb chemicalshifts. These differences together with the violations ofthe experimental structural restraints were used forranking homology models. Structure analysis and ribbondiagrams have been made using MOLMOL.36
Electrostatic Potential Calculations
Best overall model from MnmE G-domain structurecalculations was selected for electrostatic potential dis-tribution plot on the protein surface using the corre-sponding modules of MOLMOL36 program. Solventaccessibility was calculated with PROCHECK37 and res-idues with accessibility values larger than 0.5 were cho-sen as solvent exposed residues. ‘‘Contact’’ was themethod selected for protein surface determination,which was colored using a GRASP standard criterion.Electrostatic potential calculation was performed usingthe standard MOLMOL macro for this purpose, which isbased on simple coulomb interaction. Atom charges andprotonation state of amino acids were derived with the‘‘pdb_charge’’ MOLMOL macro with adequate correc-tions for secondary structure conformations by interpola-tion for a pH of 6.
RESULTS AND DISCUSSIONMnmE G-domain Biochemical Properties
The isolated GTPase domain of MnmE roughly con-serves the guanine nucleotide binding and the GTPaseactivities of the intact protein, i.e. low affinity for nucle-otides and a relatively high intrinsic GTPase activity.7
However, affinity for nucleotides was determined byusing a filter binding assay, which is not the best proce-dure to precisely calculate the Kd values, especiallywhen proteins bind nucleotides with low affinity. In fact,this assay did not allow to accurately measure the Kd
for GDP of MnmE and its G-domain. To gain proper in-formation on the guanine nucleotide binding propertiesof these molecules, we performed binding studies in so-lution with fluorescent nucleotides. Titration experi-ments allowed us to determine that the Kd values formant-GTPgS and mant-GDP of MnmE and its G-domainare in the micromolar range (Table II). It should benoted that affinity for mant-GDP of both the entire pro-tein and its G-domain is somewhat lower than for mant-GTPgS, and that the MnmE G-domain binds mant-GTPgS 2-fold more efficiently than the entire protein.Both proteins, MnmE and its G-domain, have a similarcatalytic efficiency (kcat/Km), although the last one exhib-its smaller values of kcat and Km. The differences in nu-cleotide affinity and catalytic parameters between bothproteins may be due to the higher freedom degree of theN- and C-terminal ends of the MnmE G-domain in rela-tion to the entire protein.
TABLE II. Kinetic and Binding Parameters of MnmE and Its G-domain
Protein
Kd in lM GTP hydrolysis
GTPgSbinding
GDPbinding
Vmax
(nmol/min�mg)Kcat
(min�1)Km
(lM)Kcat/Km
(min�1/lM)
MnmE 1.5 4.1 203 � 8 10.8 754 � 79 0.0143G-domain 0.6 5.6 225 � 9 4.1 252 � 37 0.0162
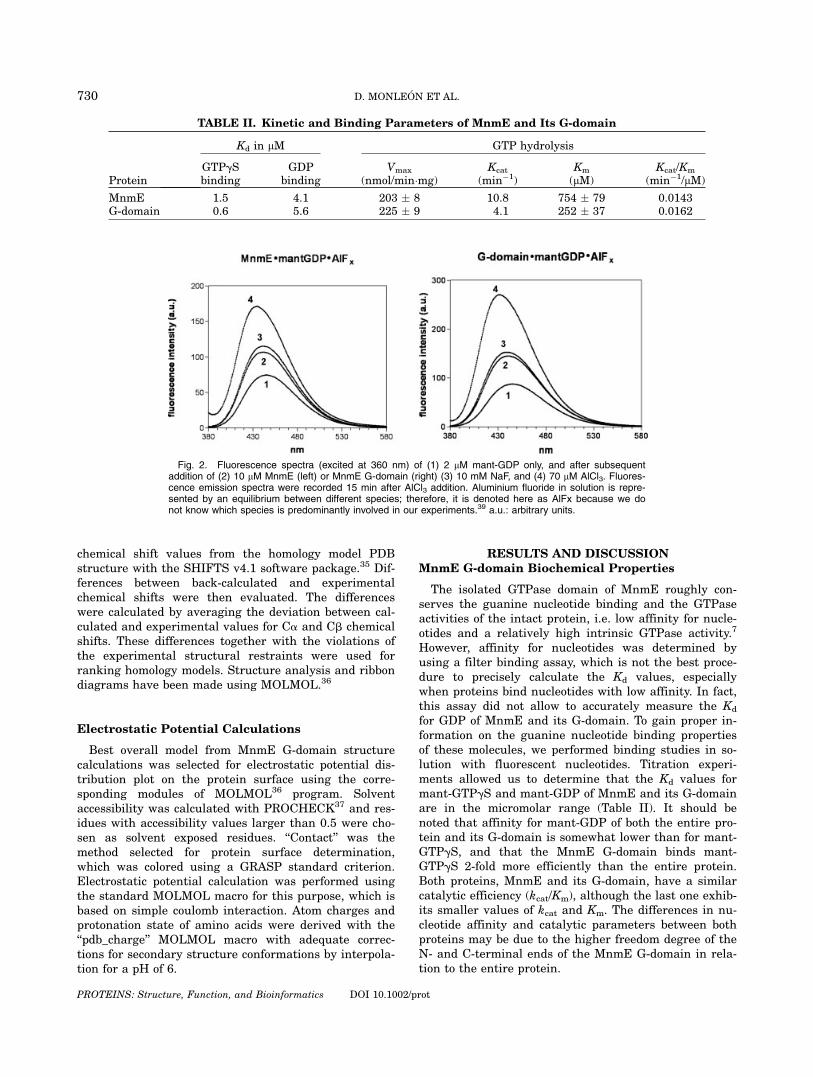
Fig. 2. Fluorescence spectra (excited at 360 nm) of (1) 2 lM mant-GDP only, and after subsequentaddition of (2) 10 lM MnmE (left) or MnmE G-domain (right) (3) 10 mM NaF, and (4) 70 lM AlCl3. Fluores-cence emission spectra were recorded 15 min after AlCl3 addition. Aluminium fluoride in solution is repre-sented by an equilibrium between different species; therefore, it is denoted here as AlFx because we donot know which species is predominantly involved in our experiments.39 a.u.: arbitrary units.
730 D. MONLEON ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
For Ga subunits of heterotrimeric G proteins, the for-mation of a complex was shown among the protein, GDP(including Mg2þ) and aluminium fluoride (for a shortreview, see Ref. 6). This complex is believed to mimicthe transition state of GTP hydrolysis, because it is sta-bilized by the helical domain of Ga (which contains thecatalytic arginine), and the aluminium fluoride complexhas a planar configuration, as supposed for the g-phos-phate group in the transition state. Small GTPases arenot able to form such a complex; however, in the pres-
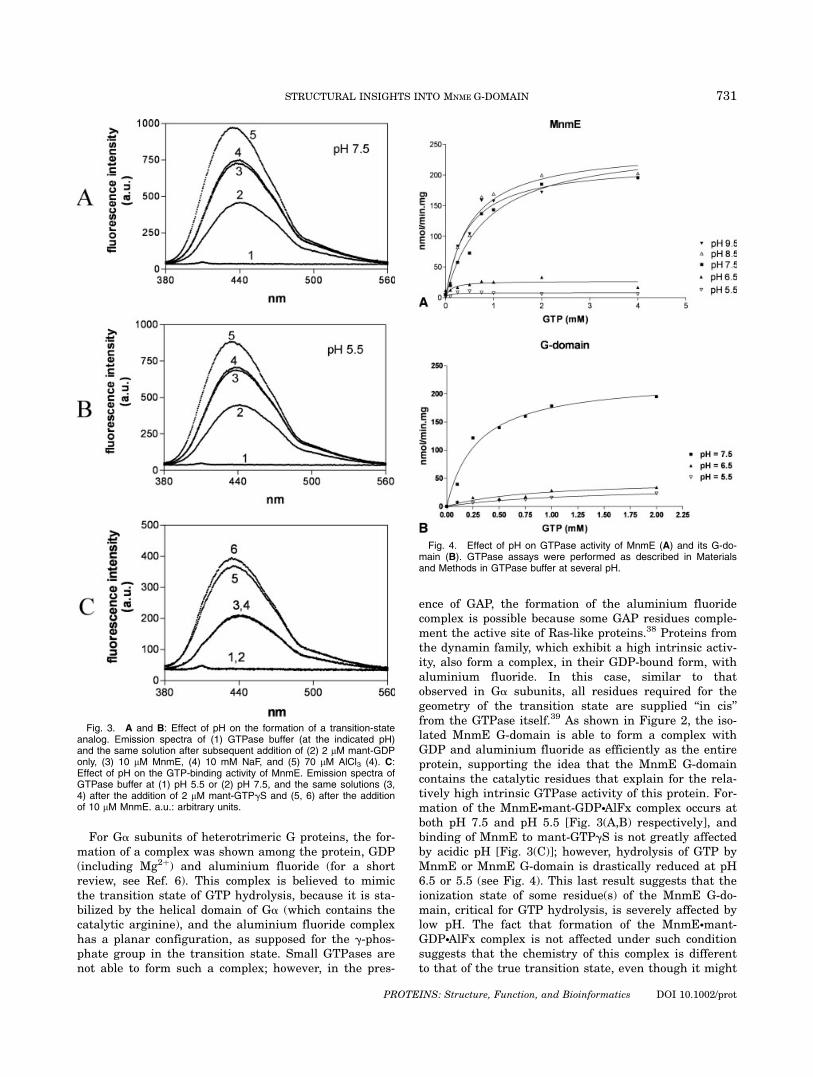
ence of GAP, the formation of the aluminium fluoridecomplex is possible because some GAP residues comple-ment the active site of Ras-like proteins.38 Proteins fromthe dynamin family, which exhibit a high intrinsic activ-ity, also form a complex, in their GDP-bound form, withaluminium fluoride. In this case, similar to thatobserved in Ga subunits, all residues required for thegeometry of the transition state are supplied ‘‘in cis’’from the GTPase itself.39 As shown in Figure 2, the iso-lated MnmE G-domain is able to form a complex withGDP and aluminium fluoride as efficiently as the entireprotein, supporting the idea that the MnmE G-domaincontains the catalytic residues that explain for the rela-tively high intrinsic GTPase activity of this protein. For-mation of the MnmE�mant-GDP�AlFx complex occurs atboth pH 7.5 and pH 5.5 [Fig. 3(A,B) respectively], andbinding of MnmE to mant-GTPgS is not greatly affectedby acidic pH [Fig. 3(C)]; however, hydrolysis of GTP byMnmE or MnmE G-domain is drastically reduced at pH6.5 or 5.5 (see Fig. 4). This last result suggests that theionization state of some residue(s) of the MnmE G-do-main, critical for GTP hydrolysis, is severely affected bylow pH. The fact that formation of the MnmE�mant-GDP�AlFx complex is not affected under such conditionsuggests that the chemistry of this complex is differentto that of the true transition state, even though it might
Fig. 3. A and B: Effect of pH on the formation of a transition-stateanalog. Emission spectra of (1) GTPase buffer (at the indicated pH)and the same solution after subsequent addition of (2) 2 lM mant-GDPonly, (3) 10 lM MnmE, (4) 10 mM NaF, and (5) 70 lM AlCl3 (4). C:Effect of pH on the GTP-binding activity of MnmE. Emission spectra ofGTPase buffer at (1) pH 5.5 or (2) pH 7.5, and the same solutions (3,4) after the addition of 2 lM mant-GTPgS and (5, 6) after the additionof 10 lM MnmE. a.u.: arbitrary units.
Fig. 4. Effect of pH on GTPase activity of MnmE (A) and its G-do-main (B). GTPase assays were performed as described in Materialsand Methods in GTPase buffer at several pH.
731STRUCTURAL INSIGHTS INTO MNME G-DOMAIN
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
well be a very close structural approximation, and thatthe residue(s) critical for the precise geometry of thetransition state is not required for stabilization ofAlFx.40,41
Structure Calculation
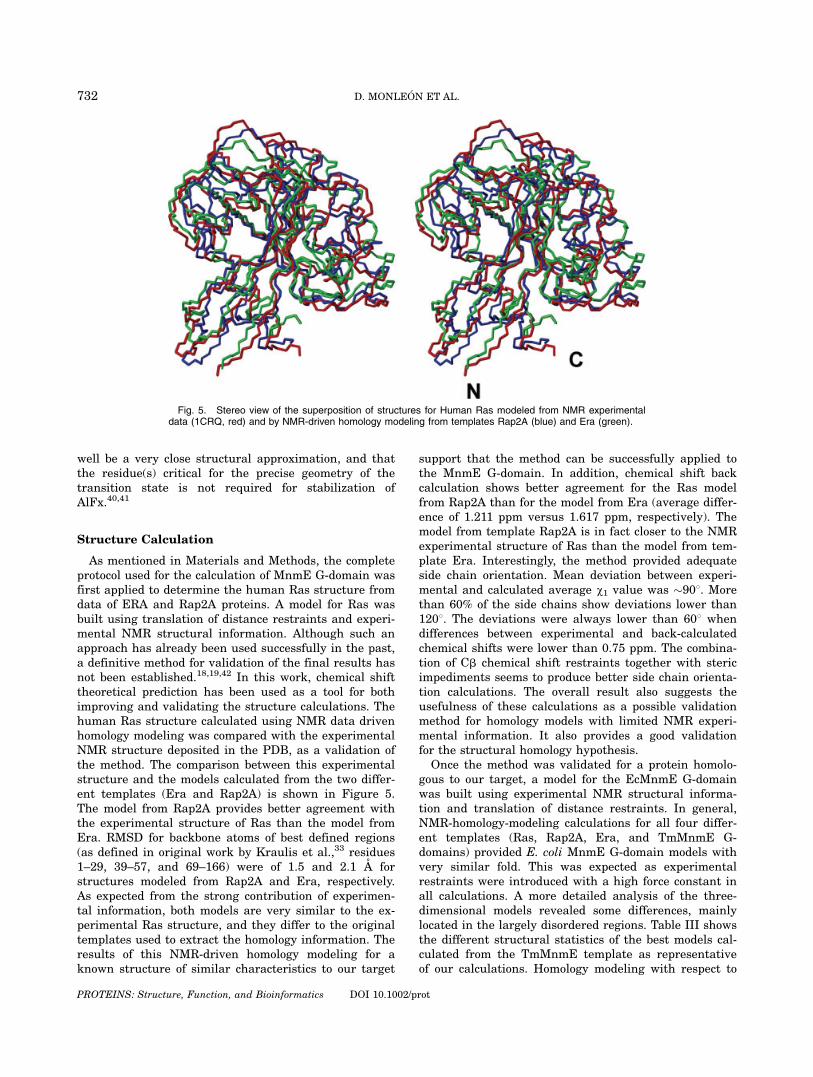
As mentioned in Materials and Methods, the completeprotocol used for the calculation of MnmE G-domain wasfirst applied to determine the human Ras structure fromdata of ERA and Rap2A proteins. A model for Ras wasbuilt using translation of distance restraints and experi-mental NMR structural information. Although such anapproach has already been used successfully in the past,a definitive method for validation of the final results hasnot been established.18,19,42 In this work, chemical shifttheoretical prediction has been used as a tool for bothimproving and validating the structure calculations. Thehuman Ras structure calculated using NMR data drivenhomology modeling was compared with the experimentalNMR structure deposited in the PDB, as a validation ofthe method. The comparison between this experimentalstructure and the models calculated from the two differ-ent templates (Era and Rap2A) is shown in Figure 5.The model from Rap2A provides better agreement withthe experimental structure of Ras than the model fromEra. RMSD for backbone atoms of best defined regions(as defined in original work by Kraulis et al.,33 residues1–29, 39–57, and 69–166) were of 1.5 and 2.1 A forstructures modeled from Rap2A and Era, respectively.As expected from the strong contribution of experimen-tal information, both models are very similar to the ex-perimental Ras structure, and they differ to the originaltemplates used to extract the homology information. Theresults of this NMR-driven homology modeling for aknown structure of similar characteristics to our target
support that the method can be successfully applied tothe MnmE G-domain. In addition, chemical shift backcalculation shows better agreement for the Ras modelfrom Rap2A than for the model from Era (average differ-ence of 1.211 ppm versus 1.617 ppm, respectively). Themodel from template Rap2A is in fact closer to the NMRexperimental structure of Ras than the model from tem-plate Era. Interestingly, the method provided adequateside chain orientation. Mean deviation between experi-mental and calculated average v1 value was �908. Morethan 60% of the side chains show deviations lower than1208. The deviations were always lower than 608 whendifferences between experimental and back-calculatedchemical shifts were lower than 0.75 ppm. The combina-tion of Cb chemical shift restraints together with stericimpediments seems to produce better side chain orienta-tion calculations. The overall result also suggests theusefulness of these calculations as a possible validationmethod for homology models with limited NMR experi-mental information. It also provides a good validationfor the structural homology hypothesis.
Once the method was validated for a protein homolo-gous to our target, a model for the EcMnmE G-domainwas built using experimental NMR structural informa-tion and translation of distance restraints. In general,NMR-homology-modeling calculations for all four differ-ent templates (Ras, Rap2A, Era, and TmMnmE G-domains) provided E. coli MnmE G-domain models withvery similar fold. This was expected as experimentalrestraints were introduced with a high force constant inall calculations. A more detailed analysis of the three-dimensional models revealed some differences, mainlylocated in the largely disordered regions. Table III showsthe different structural statistics of the best models cal-culated from the TmMnmE template as representativeof our calculations. Homology modeling with respect to
Fig. 5. Stereo view of the superposition of structures for Human Ras modeled from NMR experimentaldata (1CRQ, red) and by NMR-driven homology modeling from templates Rap2A (blue) and Era (green).
732 D. MONLEON ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

1XZP (TmMnmE) provides the best structural statisticsin our calculations. Ramachandran plot percentages aswell as amounts of close contacts indicate that homologymodels calculated with respect to 1XZP template showbetter stereo-chemical quality than the other models.Moreover, back-calculated chemical shift values for thismodel show higher degree of consistency with respect toexperimental values than for the other models (see Fig.6). Average chemical shift differences between experi-mental and back-calculated values were 1.176, 1.347,1.488, and 1.679 ppm for structures modeled from tem-plates TmMnmE, Era, Ras, and Rap2A, respectively.Therefore, NMR-driven homology-model obtained from1XZP (TmMnmE) template was selected as G-domainthree-dimensional structure for further analysis. Coordi-nates for final selected model of E. coli MnmE G-domainprotein have been deposited at the PDB (http://www.pdb.org, PDB accession code 1RFL).
MnmE G-domain Structure
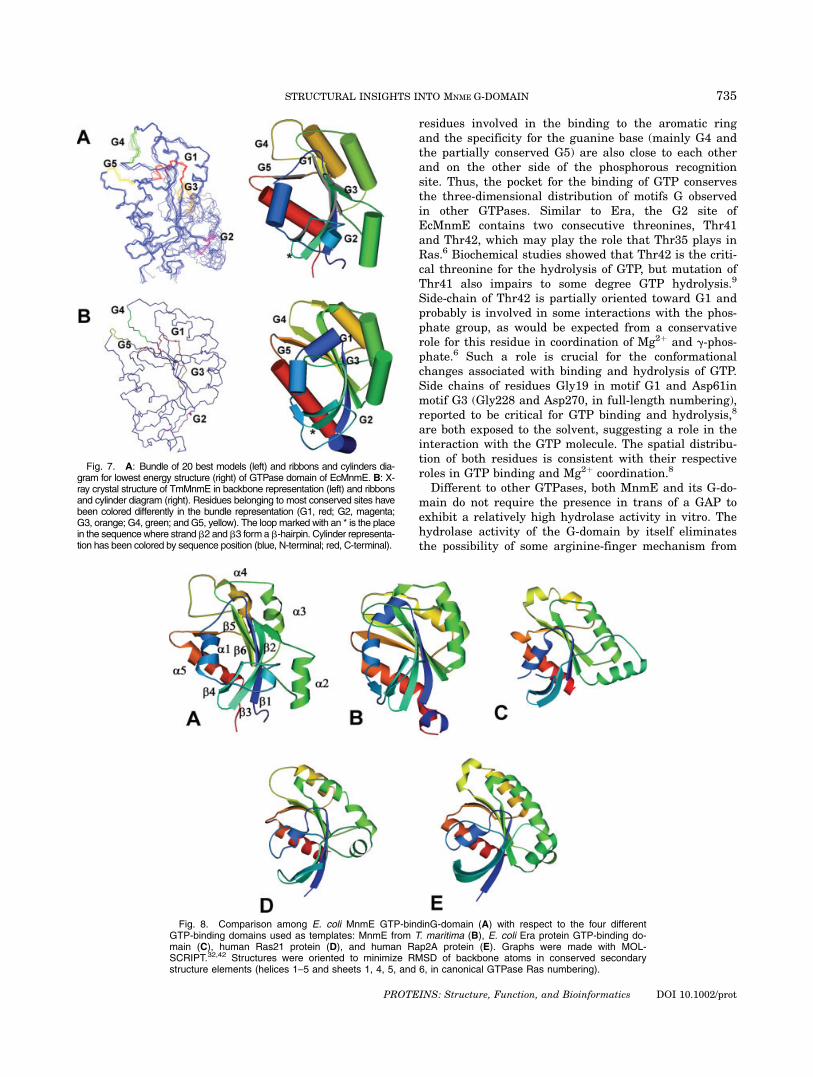
The structure of the GTPase domain of EcMnmE con-sists of a central four-stranded b-sheet flanked by five a-helices, as it is shown in Figure 7(A). This pattern isalso supported by the secondary structure determinedfrom the backbone resonance assignments.20 Residues inhelical conformation include amino acids 22–31, 72–82,102–112, 134–143, and 154–166 (231–240, 281–291, 311–321, 343–352, and 363–375, in full-length numbering).Residues in central b-sheet include 7–14, 89–94, 116–121, and 146–149 (216–223, 298–303, 325–330, and 355–358, in full-length numbering). Additionally, residues48–52 and 55–63 seems to be in b-strand conformationin some of the models, forming a b-hairpin (marked witha * in Fig. 7) detected in most of the other GTPase struc-tures with a similar orientation. Helix a2, although it isconserved in all the calculated models, shows more dis-
persion as a result of the low number of experimentalrestraints. This high dispersion together with somebroad peaks in the NMR spectra suggests some flexibil-ity for this structural element. Interestingly, the orienta-tion of this helix is one of the structural features lessconserved across GTPases, as shown in Figure 8,although it could be due to that the structures here com-pared are in their free or GDP-bound forms.6
In general, the G-domain of EcMnmE resembles otherG-domains although some peculiar features deserve spe-cial attention (see Fig. 8). Differently to mono-domainGTPase proteins as Ras21 and Rap2A, and similarly toother multi-domain proteins as Era and TmMnmE, thecentral b-sheet in EcMnmE G-domain maintains a flatshape. Although TmMnmE and EcMnmE G-domainsshare many common structural features, some signifi-cant differences can be detected. As mentioned earlier,the second helix of the EcMnmE G-domain (south-eastpart of the protein in Fig. 7) displays an orientation dif-ferent to that observed for TmMnmE. Although the loca-tion of this helix is fairly conserved across all GTPases,its orientation seems to be slightly different in eachcase. Helix a2 does not show many interactions with therest of the G-domain in most of the GTPase structuresreported to date. Interestingly, this helix seems to showmore interactions with the core of the GTPase domainin TmMnmE than in the remaining proteins, includingthe EcMnmE G-domain, a fact that could be related tothe higher thermal stability of the T. maritime protein.44
On the other hand, due to the proximity of this helix tothe hypothetical switch II, a change in its orientationmight occur upon GTP binding.
Similar to that observed in TmMnmE and Era, theloop where the G1 site is located (the P-loop) is shorterin EcMnmE than in Ras21 (as determined by MOL-SCRIPT, disordered residues 10–16 in Ras21 and disor-
TABLE III. Structural Statistics for Best Modelsof MnmE G-domain (PDB ID 1RFL) as Calculated
by PROCHECK36
Residual NOE distance constraint violations>0.2 A 21 � 11Maximum (A) 0.77 � 0.40
Residual van der Waals close contacts violations>0.2 A 12 � 7Maximum (A) 0.11 � 0.08
RMSD from ideal geometryBond length (A) 0.0104 � 0.0008Bond angles (deg.) 1.54 � 0.57
RMSD N, Ca, C0 to mean coordinates (A)a 0.614 � 0.172
RMSD heavy atoms to mean coordinates (A)a 1.011 � 0.412Residues distribution on Ramachandran plotIn allowed regions 3079 (92.3%)In unfavorable regions 204 (6.0%)In disallowed regions 121 (3.4%)
Only experimental restraints have been considered.a112 best defined residues in the structure with angular order parameter greaterthan 0.85.
733STRUCTURAL INSIGHTS INTO MNME G-DOMAIN
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
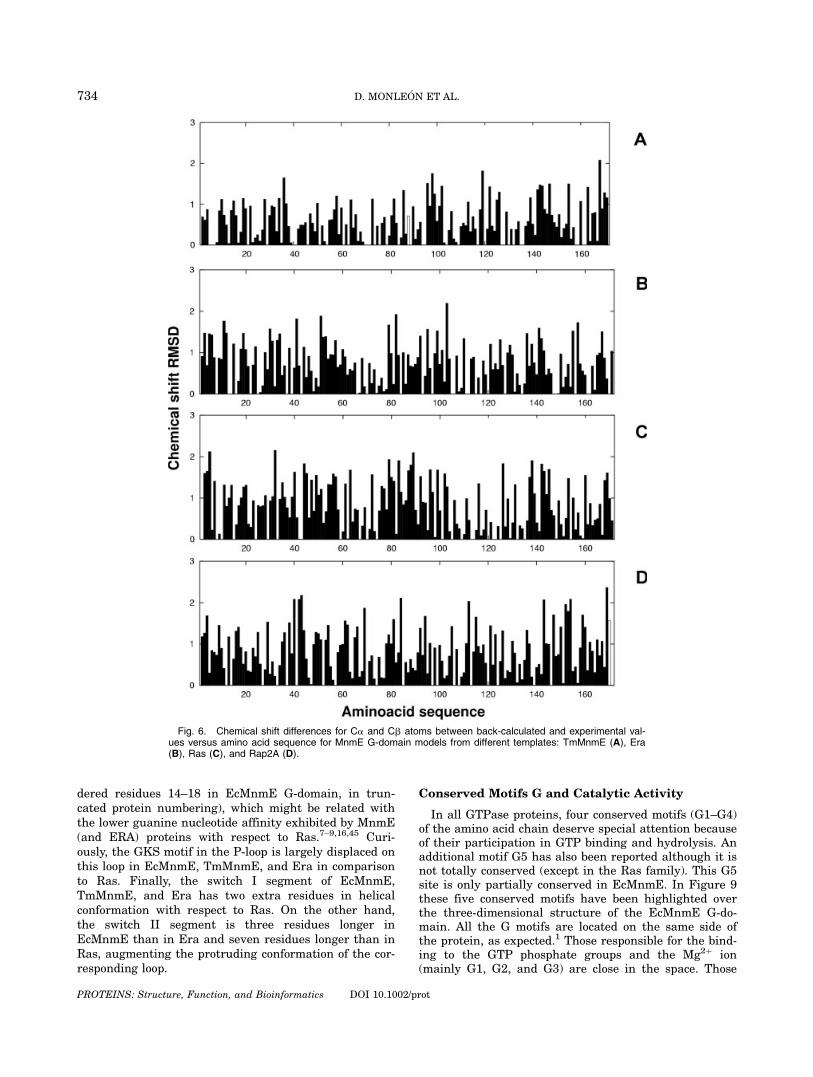
dered residues 14–18 in EcMnmE G-domain, in trun-cated protein numbering), which might be related withthe lower guanine nucleotide affinity exhibited by MnmE(and ERA) proteins with respect to Ras.7–9,16,45 Curi-ously, the GKS motif in the P-loop is largely displaced onthis loop in EcMnmE, TmMnmE, and Era in comparisonto Ras. Finally, the switch I segment of EcMnmE,TmMnmE, and Era has two extra residues in helicalconformation with respect to Ras. On the other hand,the switch II segment is three residues longer inEcMnmE than in Era and seven residues longer than inRas, augmenting the protruding conformation of the cor-responding loop.
Conserved Motifs G and Catalytic Activity
In all GTPase proteins, four conserved motifs (G1–G4)of the amino acid chain deserve special attention becauseof their participation in GTP binding and hydrolysis. Anadditional motif G5 has also been reported although it isnot totally conserved (except in the Ras family). This G5site is only partially conserved in EcMnmE. In Figure 9these five conserved motifs have been highlighted overthe three-dimensional structure of the EcMnmE G-do-main. All the G motifs are located on the same side ofthe protein, as expected.1 Those responsible for the bind-ing to the GTP phosphate groups and the Mg2þ ion(mainly G1, G2, and G3) are close in the space. Those
Fig. 6. Chemical shift differences for Ca and Cb atoms between back-calculated and experimental val-ues versus amino acid sequence for MnmE G-domain models from different templates: TmMnmE (A), Era(B), Ras (C), and Rap2A (D).
734 D. MONLEON ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
residues involved in the binding to the aromatic ringand the specificity for the guanine base (mainly G4 andthe partially conserved G5) are also close to each otherand on the other side of the phosphorous recognitionsite. Thus, the pocket for the binding of GTP conservesthe three-dimensional distribution of motifs G observedin other GTPases. Similar to Era, the G2 site ofEcMnmE contains two consecutive threonines, Thr41and Thr42, which may play the role that Thr35 plays inRas.6 Biochemical studies showed that Thr42 is the criti-cal threonine for the hydrolysis of GTP, but mutation ofThr41 also impairs to some degree GTP hydrolysis.9
Side-chain of Thr42 is partially oriented toward G1 andprobably is involved in some interactions with the phos-phate group, as would be expected from a conservativerole for this residue in coordination of Mg2þ and g-phos-phate.6 Such a role is crucial for the conformationalchanges associated with binding and hydrolysis of GTP.Side chains of residues Gly19 in motif G1 and Asp61inmotif G3 (Gly228 and Asp270, in full-length numbering),reported to be critical for GTP binding and hydrolysis,8
are both exposed to the solvent, suggesting a role in theinteraction with the GTP molecule. The spatial distribu-tion of both residues is consistent with their respectiveroles in GTP binding and Mg2þ coordination.8
Different to other GTPases, both MnmE and its G-do-main do not require the presence in trans of a GAP toexhibit a relatively high hydrolase activity in vitro. Thehydrolase activity of the G-domain by itself eliminatesthe possibility of some arginine-finger mechanism from
Fig. 7. A: Bundle of 20 best models (left) and ribbons and cylinders dia-gram for lowest energy structure (right) of GTPase domain of EcMnmE. B: X-ray crystal structure of TmMnmE in backbone representation (left) and ribbonsand cylinder diagram (right). Residues belonging to most conserved sites havebeen colored differently in the bundle representation (G1, red; G2, magenta;G3, orange; G4, green; and G5, yellow). The loop marked with an * is the placein the sequencewhere strand b2 and b3 form a b-hairpin. Cylinder representa-tion has been colored by sequence position (blue, N-terminal; red, C-terminal).
Fig. 8. Comparison among E. coli MnmE GTP-bindinG-domain (A) with respect to the four differentGTP-binding domains used as templates: MnmE from T. maritima (B), E. coli Era protein GTP-binding do-main (C), human Ras21 protein (D), and human Rap2A protein (E). Graphs were made with MOL-SCRIPT.32,42 Structures were oriented to minimize RMSD of backbone atoms in conserved secondarystructure elements (helices 1–5 and sheets 1, 4, 5, and 6, in canonical GTPase Ras numbering).
735STRUCTURAL INSIGHTS INTO MNME G-DOMAIN
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot
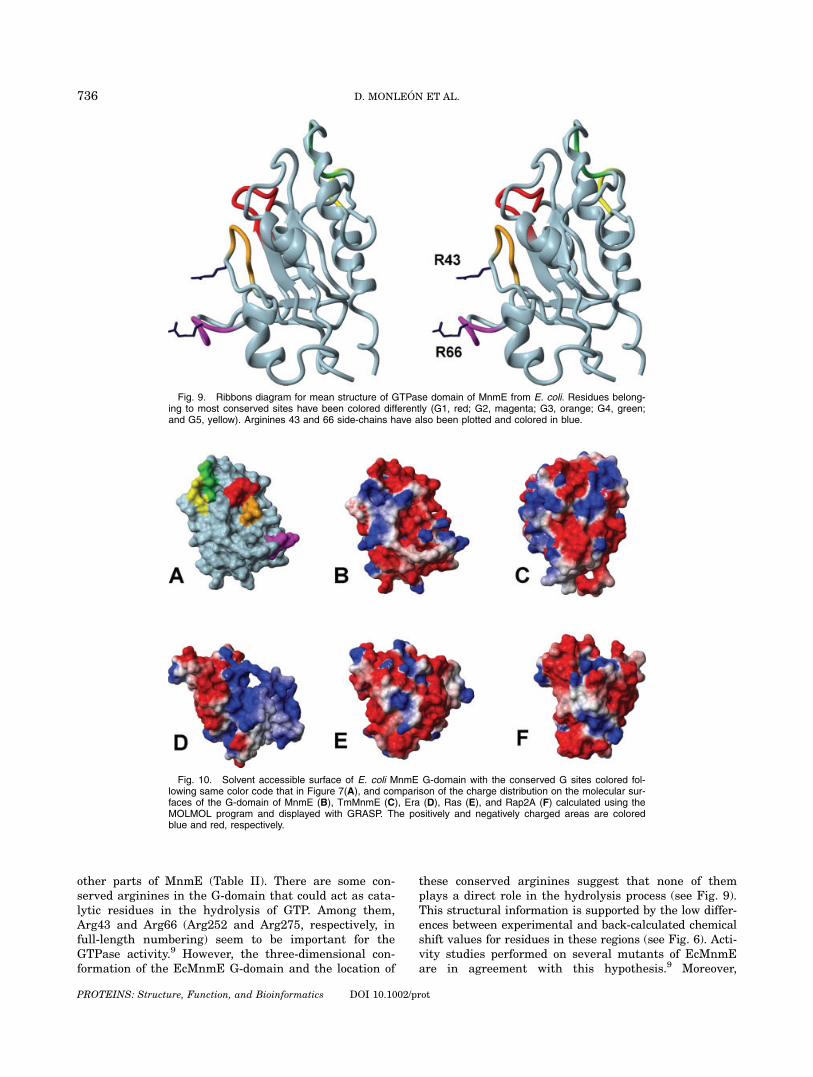
other parts of MnmE (Table II). There are some con-served arginines in the G-domain that could act as cata-lytic residues in the hydrolysis of GTP. Among them,Arg43 and Arg66 (Arg252 and Arg275, respectively, infull-length numbering) seem to be important for theGTPase activity.9 However, the three-dimensional con-formation of the EcMnmE G-domain and the location of
these conserved arginines suggest that none of themplays a direct role in the hydrolysis process (see Fig. 9).This structural information is supported by the low differ-ences between experimental and back-calculated chemicalshift values for residues in these regions (see Fig. 6). Acti-vity studies performed on several mutants of EcMnmEare in agreement with this hypothesis.9 Moreover,
Fig. 9. Ribbons diagram for mean structure of GTPase domain of MnmE from E. coli. Residues belong-ing to most conserved sites have been colored differently (G1, red; G2, magenta; G3, orange; G4, green;and G5, yellow). Arginines 43 and 66 side-chains have also been plotted and colored in blue.
Fig. 10. Solvent accessible surface of E. coli MnmE G-domain with the conserved G sites colored fol-lowing same color code that in Figure 7(A), and comparison of the charge distribution on the molecular sur-faces of the G-domain of MnmE (B), TmMnmE (C), Era (D), Ras (E), and Rap2A (F) calculated using theMOLMOL program and displayed with GRASP. The positively and negatively charged areas are coloredblue and red, respectively.
736 D. MONLEON ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

EcMnmE has a leucine just behind the G3 motif in placeof the catalytic glutamine present in Ras proteins6 (seeFig. 1). Thus, it can be proposed that hydrolysis of GTPby MnmE takes place through a mechanism different tothat involving an intra-molecular arginine-finger and aglutamine to orient the attacking water molecule. Inter-estingly, Arg43 side chain is oriented toward the car-bonyl group of Gly40 and it may form some hydrogenbonding under certain circumstances. The orientation ofthis side chain in our models is partially supported bythe small difference found between experimental andback-calculated values for the Cb chemical shift (only0.58 ppm). This potential hydrogen bonding might exp-lain why when Arg43 is mutated to Ala, the MnmEGTPase activity is reduced in a greater extent thanwhen it is mutated to Lys.9 In addition, the fact thatmutation of Gly40 to Ala in the entire protein (G249A)reduces drastically the GTPase activity and greatly per-turbs formation of a transition-state mimic9 suggeststhat Gly40 lies close to the catalytic center of the MnmEG-domain and that even the smallest possible aminoacid change (to alanine) would sterically interfere withthe geometry of the transition state. The structure ofthe MnmE G-domain here presented supports that theinteraction between Gly40 and Arg43 might play somestabilization role in the intermediate state of hydrolysis.
Electrostatic Potential Distribution at theProtein Surface
To achieve a better understanding of the possibleinteractions of the EcMnmE G-domain with the remain-ing domains and the GTP molecule, the electrostaticpotential at the protein surface has been calculated andcompared with its closest homologues (see Fig. 10). Obvi-ously, a detailed analysis of the electrostatic potentialdistribution at the protein surface of the EcMnmE G-do-main requires a precise determination of the side-chainorientation of all residues. However, a general analysisof the medium-resolution structure presented here alsoprovides useful information on the global electrostaticsof the molecule. The electrostatic potential of the MnmEG-domain resembles that exhibited by mono-domainGTPases and seems fairly equilibrated, in contrast tothat observed for Era. As in the case of Ras and Rap2A,negative regions are predominant on the active-site sideof the protein. However, the areas where the GTP mole-cule is supposed to bind maintain a slightly positive orneutral character. The regions surrounding G2 (whichincludes Arg43) display a fairly equilibrated and neutralpotential, supporting the absence of an intra-moleculararginine finger mechanism. Interestingly, although thestructure of EcMnmE G-domain is very similar to thatof its homologue Era, which is also a multi-domain pro-tein, the electrostatic potential distribution at the sur-face is more resemblance to that reported for mono-do-main GTPases, as mentioned earlier. This feature is alsoshared by the TmMnmE protein, which is the closesthomologue of EcMnmE. This may contribute to the little
or no loss of activity shown by the truncated G-domainof MnmE proteins with respect to the full-length protein(Table II). The G-domain of MnmE is a electrostaticallyequilibrated unit by its own in contrast to the Era G-do-main, which seems to need the effect of its remainingdomains to achieve electrostatic equilibrium.
CONCLUDING REMARKS
MnmE is a multidomain GTPase with peculiar bio-chemical properties. Its G-domain, when isolated, main-tains the GTPase features of the entire protein and,thus, it should contain the catalytic residues responsiblefor the relatively high MnmE GTPase activity. Ourresults suggest that some of these residues suffer pH-sensitive chemical shifts during GTP hydrolysis. Toexplain the GTPase and nucleotide binding properties ofMnmE, structure resolution of free and nucleotide-boundforms of MnmE or its G-domain is crucial. As a firststep, a three-dimensional medium-resolution structurefor the G-domain of E. coli MnmE has been solved. Acombination of experimental NMR restraints and homol-ogy structural information allowed the determination ofthis structure otherwise extremely challenging. The useof chemical shift structure-based prediction provided agood basis for the improvement of the structure calcula-tion process and for the validation of the final results. Ingeneral, the structure of MnmE G-domain resemblesthat of other GTPases. The overall folding consists in acentral b-strand with six extended elements surroundedby five a-helices. The flexible region of the P-loop ofMnmE G-domain is shorter than that observed in canon-ical GTPases, as confirmed by most of our homologymodels and by the X-ray structure of T. maritimeTrmE.16 In contrast, disordered segments of the switch Iand II regions are longer than those observed in otherGTPases, suggesting increased flexibility and strongerconformational changes upon hydrolysis, which appearto be essential for MnmE function.8,9 Our structure andprevious investigations rule out an internal arginine fin-ger mechanism as the reason for the high intrinsic activ-ity for MnmE and its G-domain. The structure of MnmEpresented here provided some insight into the roleplayed by Arg43, which seems to be important for cata-lytic activity. The side chain of this arginine may formunder certain circumstances some hydrogen bond withGly40, stabilizing the conformation of the theoretical in-termediate state. Overall, our structural study allows toanswer some questions raised in previous studies andsupplies a base to approach unsolved questions in thenext future.
ACKNOWLEDGMENTS
We thank the SCSIE of the University of Valencia forproviding access to the NMR facility and high perform-ance computing facilities. The 750 MHz spectra were
737STRUCTURAL INSIGHTS INTO MNME G-DOMAIN
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

recorded at the SON NMR Large Scale Facility inUtrecht. We are also very grateful to Dr. V. Rubio forhelpful discussions and Dr. E. Perez-Paya for help withfluorescence measurements. M. Martınez-Vicente was apredoctoral fellow of the Fundacion Valenciana de Inves-tigaciones Biomedicas. We also thank Bruker EspanolaS.A. economic support as part of the agreement with theUniversity of Valencia for the development of new tech-niques in biomolecular NMR.
REFERENCES
1. Bourne HR, Sanders DA, McCormick F. The GTPase superfam-ily: conserved structure and molecular mechanism. Nature 1991;349:117–127.
2. Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification andevolution of P-loop GTPases and related ATPases. J Mol Biol2002;317:41–72.
3. Bourne HR. G proteins. The arginine finger strikes again. Nature1997;389:673,674.
4. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, LautweinA, Schmitz F, Wittinghofer A. The Ras-RasGAP complex: struc-tural basis for GTPase activation and its loss in oncogenic Rasmutants. Science 1997;277:333–338.
5. Geyer M, Wittinghofer A. GEFs, GAPs, GDIs and effectors: tak-ing a closer (3D) look at the regulation of Ras-related GTP-bind-ing proteins. Curr Opin Struct Biol 1997;7:786–792.
6. Vetter IR, Wittinghofer A. The guanine nucleotide-bindingswitch in three dimensions. Science 2001;294:1299–1304.
7. Cabedo H, Macian F, Villarroya M, Escudero JC, Martinez-Vice-nte M, Knecht E, Armengod ME. The Escherichia coli trmE(mnmE) gene, involved in tRNA modification, codes for an evo-lutionarily conserved GTPase with unusual biochemical proper-ties. EMBO J 1999;18:7063–7076.
8. Yim L, Martinez-Vicente M, Villarroya M, Aguado C, Knecht E,Armengod ME. The GTPase activity and C-terminal cysteine ofthe Escherichia coli MnmE protein are essential for its tRNAmodifying function. J Biol Chem 2003;278:28378–28387.
9. Martinez-Vicente M, Yim L, Villarroya M, Mellado M, Perez-Paya E, Bjork GR, Armengod ME. Effects of mutagenesis in theswitch I region and conserved arginines of Escherichia coliMnmE protein, a GTPase involved in tRNA modification. J BiolChem 2005;280:30660–30670.
10. Colby G, Wu M, Tzagoloff A. MTO1 codes for a mitochondrialprotein required for respiration in paromomycin-resistantmutants of Saccharomyces cerevisiae. J Biol Chem 1998;273:27945–27952.
11. Umeda N, Suzuki T, Yukawa M, Ohya Y, Shindo H, WatanabeK, Suzuki T. Mitochondria-specific RNA-modifying enzymes re-sponsible for the biosynthesis of the wobble base in mitochon-drial tRNAs. J Biol Chem 2005;280:1613–1624.
12. Yasukawa T, Suzuki T, Ohta S, Watanabe K. Wobble modifica-tion defect suppresses translational activity of tRNAs withMERRF and MELAS mutations. Mitochondrion 2002;2:129–141.
13. Yasukawa T, Kirino Y, Ishii N, Holt IJ, Jacobs HT, Makifuchi T,Fukuhara N, Ohta S, Suzuki T, Watanabe K. Wobble modifica-tion deficiency in mutant tRNAs in patients with mitochondrialdiseases. FEBS Lett 2005;579:2948–2952.
14. Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K,Suzuki T. Codon-specific translational defect caused by a wobblemodification deficiency in mutant tRNA from a human mitochon-drial disease. Proc Natl Acad Sci USA 2004;101:15070–15075.
15. Kirino Y, Goto Y, Campos Y, Arenas J, Tsutomu S. Specific cor-relation between the wobble modification deficiency in mutanttRNAs and the clinical features of a human mitochondrial dis-ease. Proc Natl Acad Sci USA 2005;102:7127–7132.
16. Scrima A, Vetter IR, Armengod ME, Wittinghoffer A. The struc-ture of the TrmE GTP-binding protein and its implications fortRNA modification. EMBO J 2005;24:23–33.
17. Fiser A, Sali A. Modeller: generation and refinement of homol-ogy-based protein structure models. Methods Enzymol 2003;374:461–491.
18. Kitchen D, Hoffman RC, Moy FJ, Powers R. Homology modelfor oncostatin M based on NMR structural data. Biochemistry1998;37:10581–10588.
19. Randazzo A, Acklin C, Schafer BW, Heizmann CW, Chazin WJ.Structural insight into human Zn(2þ)-bound S100A2 from NMRand homology modeling. Biochem Biophys Res Commun2001;288:462–467.
20. Monleon D, Yim L, Martinez-Vicente M, Armengod ME, CeldaB. Backbone 1H, 13C and 15N resonance assignments for the18.7 kDa GTPase domain of Escherichia coli MnmE protein.J Biomol NMR 2004;28:307,308.
21. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, MillerW, Lipman DJ. Gapped BLAST and PSI-BLAST: a new genera-tion of protein database search programs. Nucleic Acids Res1997;25:3389–3402.
22. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improvingthe sensitivity of progressive multiple sequence alignmentthrough sequence weighting, position-specific gap penalties andweight matrix choice. Nucleic Acids Res 1994;22:4673–4680.
23. Frishman D, Argos P. Seventy-five percent accuracy in proteinsecondary structure prediction. Proteins 1997;27:329–335.
24. B Rost PHD: predicting one-dimensional protein structure by profilebased neural networks. Methods Enzymol 1996;266:525–539.
25. Kabsch W, Sander C. Dictionary of protein secondary structure:pattern recognition of hydrogen-bonded and geometrical fea-tures. Biopolymers 1983;22:2577–2637.
26. Sali A, Blundell TL. Comparative protein modelling by satisfac-tion of spatial restraints. J Mol Biol 1993;234:779–815.
27. Li H, Tejero R, Monleon D, Bassolino-Klimas D, Abate-Shen C,Bruccoleri RE, Montelione GT. Homology modeling using simu-lated annealing of restrained molecular dynamics and conforma-tional search calculations with CONGEN: application in predict-ing the three-dimensional structure of murine homeodomainMsx-1. Protein Sci 1997;6:956–970.
28. Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraintsfrom searching a database for chemical shift and sequence homol-ogy. J Biomol NMR 1999;13:289–302.
29. Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynam-ics for NMR structure calculation with the new programDYANA. J Mol Biol 1997;273:283–298.
30. Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS,Read RJ, Rice LM, Simonson T, Warren GL. Crystallographyand NMR system: a new software suite for macromolecularstructure determination. Acta Crystallogr D Biol Crystallogr1998;54:905–921.
31. Celda B, Biamonti C, Arnau MJ, Tejero R, Montelione GT. Com-bined use of 13C chemical shift and 1H a-13C a heteronuclearNOE data in monitoring a protein NMR structure refinement. JBiomol NMR 1995;5:161–172.
32. Chen X, Court DL, Ji X. Crystal structure of Era: a GTPase-de-pendent cell cycle regulator containing an RNA binding motif.Proc Natl Acad Sci USA 1999;96:8396–8401.
33. Kraulis PJ, Domaille PJ, Campbell-Burk SL, Van Aken T, LaueED. Solution structure and dynamics of ras p21.GDP deter-mined by heteronuclear three- and four-dimensional NMR spec-troscopy. Biochemistry 1994;33:3515–3531.
34. Cherfils J, Menetrey J, Le Bras G, Janoueix-Lerosey I, de Gunz-burg J, Garel JR, Auzat I. Crystal structures of the small G pro-tein Rap2A in complex with its substrate GTP, with GDP andwith GTPgS. EMBO J 1997;16:5582–5591.
35. Xu XP, Case DA. Automated prediction of 15N, 13Ca, 13Cb and13C0 chemical shifts in proteins using a density functional data-base. J Biomol NMR 2001;21:321–333.
36. Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for dis-play and analysis of macromolecular structures. J Mol Graph1996;14:29–32.
37. Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R,Thornton JM. AQUA and PROCHECK-NMR: programs forchecking the quality of protein structures solved by NMR. J Bio-mol NMR 1996;8:477–486.
38. Ahmadian MR, Stege P, Scheffzek K, Wittinghofer A. Confirma-tion of the arginine-finger hypothesis for the GAP-stimulatedGTP-hydrolysis reaction of Ras. Nat Struct Biol 1997;4:686–689.
738 D. MONLEON ET AL.
PROTEINS: Structure, Function, and Bioinformatics DOI 10.1002/prot

39. Praefcke GJ, Geyer M, Schwemmle M, Robert Kalbitzer H,Herrmann C. Nucleotide-binding characteristics of human gua-nylate-binding protein 1 (hGBP1) and identification of the thirdGTP-binding motif. J Mol Biol 1999;292:321–332.
40. Graham DL, Eccleston JF, Lowe PN. The conserved argi-nine in Rho-GTPase-activating protein is essential for effi-cient catalysis but not for complex formation withRho�GDP and aluminium fluoride. Biochemistry 1999;38:985–991.
41. Schlichting I, Reinstein J. pH influences fluoride coordinationnumber of the AlFx phosphoryl transfer transition state analog.Nat Struct Biol 1999;6:721–723.
42. Sidote DJ, Hoffman DW. NMR structure of an archaeal homo-logue of ribonuclease P protein Rpp29. Biochemistry 2003;42:13541–13550.
43. Esnouf RM. An extensively modified version of MolScript thatincludes greatly enhanced coloring capabilities. J Mol GraphModel 1997;15:132–134.
44. Yamanaka K, Hwang J, Inouye M. Characterization of GTPaseactivity of TrmE, a member of a novel GTPase superfamily, fromThermotoga maritima. J Bacteriol 2000;182:7078–7082.
45. Sullivan SM, Mishra R, Neubig RR, Maddock JR. Analysis ofguanine nucleotide binding and exchange kinetics of the Esche-richia coli GTPase Era. J Bacteriol 2000;182:3460–3466.
739STRUCTURAL INSIGHTS INTO MNME G-DOMAIN





























