Embed Size (px)
Citation preview

Structural waters define a functional channelmediating activation of the GPCR, rhodopsinThomas E. Angela,1, Sayan Guptab,c,1, Beata Jastrzebskaa, Krzysztof Palczewskia,2, and Mark R. Chanceb,c,d,3
aDepartment of Pharmacology, bCenter for Proteomics and Bioinformatics, cCenter for Synchrotron Biosciences, dDepartment of Physiology and Biophysics,School of Medicine, Case Western Reserve University, Cleveland, OH 44106-4965
Edited by Raymond C. Stevens, The Scripps Research Institute, La Jolla, CA, and accepted by the Editorial Board July 1, 2009 (received for reviewJanuary 30, 2009)
Structural water molecules may act as prosthetic groups indispens-able for proper protein function. In the case of allosteric activationof G protein-coupled receptors (GPCRs), water likely imparts struc-tural plasticity required for agonist-induced signal transmission.Inspection of structures of GPCR superfamily members reveals thepresence of conserved embedded water molecules likely importantto GPCR function. Coupling radiolytic hydroxyl radical labelingwith rapid H2O18 solvent mixing, we observed no exchange ofthese structural waters with bulk solvent in either ground state orfor the Meta II or opsin states. However, the radiolysis approachpermitted labeling of selected side chain residues within thetransmembrane helices and revealed activation-induced changesin local structural constraints likely mediated by dynamics of bothwater and protein. These results suggest both a possible generalmechanism for water-dependent communication in family A GPCRsbased on structural conservation, and a strategy for probingmembrane protein structure.
footprinting � mass spectrometry � signal transduction �membrane proteins � radiolysis
Membrane embedded proteins evolved to facilitate water orion movements both in signaling processes and ion trans-
port across the biological membrane. Many multispanning trans-membrane proteins contain hydrophobic cores with strategicallyplaced ionizable or charged residues that appear critical for theirproper function (1). Structures of many integral membraneproteins, including G protein-coupled receptors (GPCRs), re-veal the presence of ordered water molecules (2–10) that may actlike prosthetic groups with activities distinct from bulk water.Identification of ‘‘ordered’’ waters within a crystalline proteinstructure requires sufficient occupancy of water to enable itsdetection in the protein’s X-ray diffraction pattern, and thus theobserved waters likely represent a subset of the ensemble oftightly bound and functional waters (11). Overall, it is unclear ifthese ordered water molecules provide structural stabilization,mediate conformational changes in signaling, neutralize chargedresidues, or carry out a combination of all these functions.
Radiolytic footprinting of macromolecules in dilute aqueoussolution was developed to analyze the solvent accessibility ofmacromolecular sites in nucleic acids and proteins as a functionof macromolecule conformation, and a detailed understandingof the approach has been developed where radiolysis generatesradicals in bulk solution that freely diffuses to macromolecularsites and covalently labels those sites (12–14). In proteins, thechemistry of modification is well-established, and many types ofside chain can be efficiently labeled and the products easilydetected by mass spectrometry, although the dynamic range oflabeling varies considerably, with aromatic and sulfur containingside chains easily labeled and aliphatic and polar residues labeledless efficiently (15, 16). For soluble proteins, it is well-establishedthat this intrinsic reactivity and the solvent accessibility of theside chains govern their observed reactivity (15, 17, 18). Formembrane proteins, these approaches have not been previouslyexplored, such that it was unclear what factors would govern
labeling, or whether the overall scavenging effects of detergentsor lipids are limiting.
For family A GPCRs, understanding the structure and acti-vation of rhodopsin with its covalently bound ligand, 11-cis-retinylidene, has been greatly facilitated by obtaining X-raycrystal structures of vertebrate and invertebrate rhodopsins (10,19), photo-intermediates with covalently bound all-trans-retinylidene (20–22), and inactive ligand-free end-product opsin(23, 24). For example, crystallographic studies of photoactivatedMeta II-like rhodopsin reveal local structural reorganizationswithout large-scale conformational changes of individual do-mains of this receptor (22). Recent structural studies of otherfamily A GPCRs, including �1 and �2 adrenergic and A2Aadenosine receptors, show conserved membrane topologieswithin this receptor family, indicating the potential for similar ifnot conserved activation mechanisms (2–5). A striking commonfeature of these receptor structures is the presence of conserved,crystallographically ordered clusters of water molecules locatedin the transmembrane core that make contacts with highlyconserved residues in family A GPCRs (11). Water is known tobe critical for the activity of rhodopsin, specifically to achieve theMeta II state (25), and water is also required for the formationof apo-protein opsin via hydrolysis of the chromophore, all-trans-retinal (26). Although, the importance of water has beendemonstrated in another 7-transmembrane protein, bacterio-rhodopsin (27), the molecular details of agonist-induced struc-tural transformations, the changes accompanying chromophorehydrolysis in the signaling process, and the potential role forordered waters in mediating signaling, are not well understood(28).
We used radiolytic protein footprinting to investigate theconformational dynamics of ground state (rhodopsin), photo-activated (Meta II), and inactive ligand-free receptor (opsin) inn-dodecyl-�-maltoside preparations and native membranes. Un-like our previous data on soluble proteins, radiolytic modifica-tions were observed for residues located in both solvent-accessible and solvent-inaccessible regions; in the latter case,residues within the transmembrane domain were labeled, andtheir reactivity varied as a function of rhodopsin activation state.Based on H2O18 exchange studies combined with radiolysis, wedetermined that labeling within the transmembrane region is
Author contributions: T.E.A., S.G., B.J., K.P., and M.R.C. designed research; T.E.A., S.G., andB.J. performed research; T.E.A. contributed new reagents/analytic tools; T.E.A., S.G., andB.J. analyzed data; and T.E.A., S.G., B.J., K.P., and M.R.C. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. R.C.S. is a guest editor invited by the Editorial Board.
1T.E.A. and S.G. contributed equally to this work.
2To whom correspondence may be addressed at: Department of Pharmacology, School ofMedicine, Case Western Reserve University, 10900 Euclid Ave, Cleveland, OH 44106-4965.E-mail: [email protected].
3To whom correspondence may be addressed at: Center for Proteomics and Bioinformatics,School of Medicine, Case Western Reserve University, Cleveland, OH 44106-4988. E-mail:[email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0901074106/DCSupplemental.
www.pnas.org�cgi�doi�10.1073�pnas.0901074106 PNAS � August 25, 2009 � vol. 106 � no. 34 � 14367–14372
BIO
PHYS
ICS
AN
DCO
MPU
TATI
ON
AL
BIO
LOG
Y

partly derived from tightly bound waters and that changes inlabeling reveal regions undergoing local structural alterationsand water reorganization.
Results and DiscussionRadiolytic Labeling of Membrane Embedded Domains of Rhodopsin.Purified rhodopsin was exposed to high flux X-rays for 1- to10-ms time periods. Although the X-ray-irradiated samplesshowed some loss of chromophore absorbance at 500 nm (Fig.S1), this analysis was conducted well after the millisecondirradiation, and gel analysis did not show a significant amount ofamide bond cleavage (Fig. S2). Following exposure to high fluxionizing radiation, protein samples were digested and the re-sulting peptides were analyzed by liquid chromatography cou-pled to mass spectrometry (LC-MS) (Fig. S3A). The extent ofmodification was quantified following extraction of ion currentdata for unmodified and modified peptides of interest. Typicalmass spectrometric sequence coverage of the proteolytic frag-ments was 88% (Fig. 1A). Peptides not detected includedsequences in the solvent exposed N-terminal extracellular re-gion, which has been found not to undergo structural reorgani-zation following photoactivation in crystallographic studies (22),and sequences in the extended cytoplasmic loop III (C-III),which are highly disordered in crystal structures (22), and aportion of helix VI. The remaining regions of rhodopsin weredetected, in some cases with overlapping peptides. Rhodopsinexposed to X-rays either in native disk membranes or detergentexhibited roughly comparable extents of radiolytic labeling, andthe labeling was on the same residues (Fig. S3 B and C).Identities of peptides and sites of modification were confirmedby tandem mass spectrometry (Fig. S3 D and E). Rates ofmodification were determined from X-ray dosage plots as de-scribed (Fig. S3F) (15, 16). The positions of the modifiedresidues are shown in Fig. 1B; surprisingly labeled residues areobserved throughout the transmembrane segment.
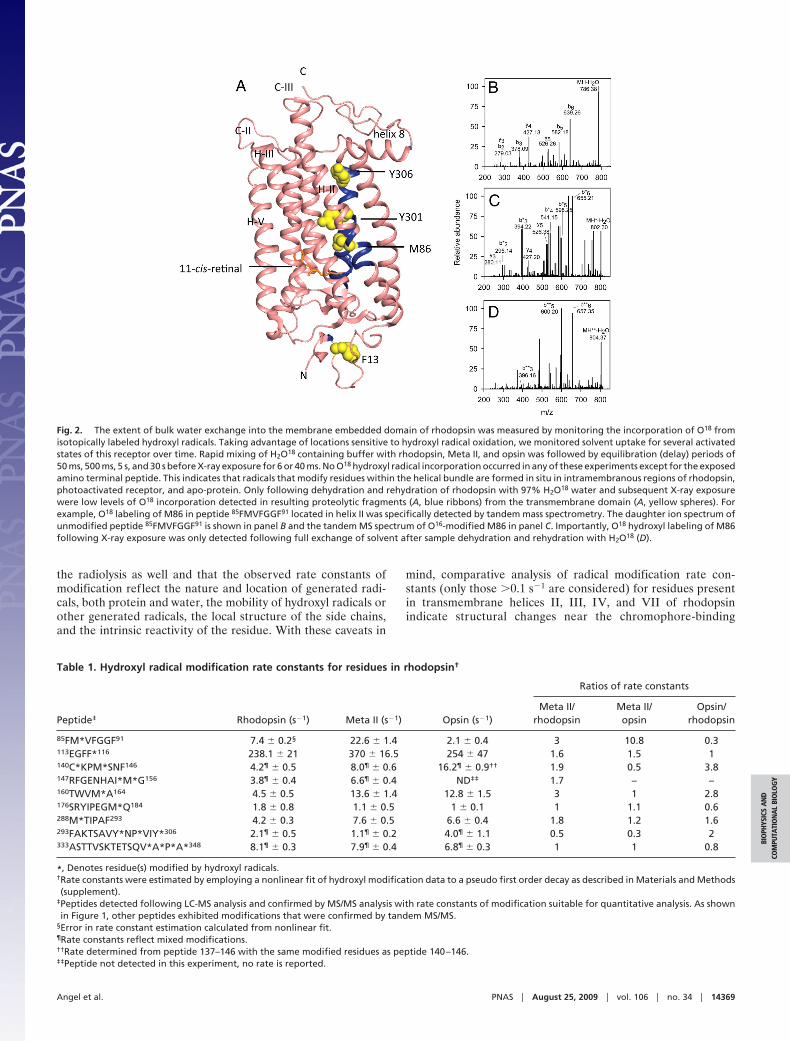
Isotopic Labeling Reveals Limited Exchange of Bulk Solvent withBound Waters Located in the Helical Bundle of Rhodopsin. We usedrapid mixing combined with O18-mediated hydroxyl radicallabeling to determine the potential origins of the transmembranelabeling and to monitor the extent of exchange of bulk solventinto the core of the helical bundle. For most side chains, althoughthe radiolytic labeling under aerobic conditions is mediated byhydroxyl radical, the oxygen atoms are derived from molecularoxygen. The notable exceptions are Phe, Tyr, Cys, and Met,where O18-labeling from radiolysis of H2O18 is entirely derivedfrom radiolysis of water (12, 16). As many of the labeled residuesin the core are Phe, Cys, Met, or Tyr (Fig. 1B), we expected todetect O18-labeling for regions within the core of the receptorthat experience increased bulk solvent exposure as a result of anopening of the helical bundle. Rhodopsin, Meta II, and opsinwere mixed with H2O18 containing buffer (in 1:1 ratio) justbefore X-ray exposure; in these experiments, there was nolabeling from bulk solvent for either of the three receptor statesexcept for the side chain of solvent accessible F13. Trace levelsof O18-labeled Y306, Y301, and M86 were detected only follow-ing dehydration and rehydration with H2O18-containing buffers(Fig. 2). Thus, the labeling of these residues at least is derivedfrom tightly bound waters, including the ordered waters found inrhodopsin crystals. However, the fact that only a few internalresidues are labeled with H2O18 suggests that much of thetransmembrane labeling is derived from a combination of directradiolysis of the protein and radiolysis of nonexchangeablewaters; the numbers of the latter may be substantial as indicatedby the fact that 10% of the weight of a dried protein is becauseof tightly bound water (29). No labeling from bulk solvent isobserved, and major structural rearrangements that would allowinflux of bulk solvent during the time scale of photoactivation
appears unlikely. These results are consistent with recent EPRmeasurements, which suggested that helical bundle reorganiza-tion upon activation is on the order of 6 Å or less, which in ouropinion is not consistent with significant solvent exchange (30).
Water Functions as an Allosteric Modulator in Rhodopsin Activation.In rhodopsin, ordered waters are within hydrogen bondingdistance of highly conserved and functionally important residuessuch as D83, E113, W265, and the NPxxY motif (28). Inspectionof several high-resolution crystal structures of family A GPCRsreveals a set of conserved waters, and those present in rhodopsinare shown in Fig. 1B (11). Interactions mediated by theseordered waters suggest that they are likely to play an importantrole in stabilizing helices I, II, III, and VII. The observed rateconstants of radiolytic modification for residues in rhodopsinsummarized in Table 1 do not show a precise correlation with theproximity to ordered waters revealed by crystallography. Wesuspect that there are ‘‘unobserved’’ waters that are relevant to
Fig. 1. High resolution LC-MS analysis of proteolytic fragments of rhodopsinresults in �88% amino acid sequence coverage. (A) The primary sequence ofrhodopsin with proteolytic peptides detected and confirmed by tandem MSanalysis is indicated by bars below the amino acid sequence. Peptides detectedfollowing digestion with pepsin are underlined by dark gray bars, and thosedetected after cyanogen bromide digestion and LC-MS analysis are underlinedby light gray bars. Residues that make up transmembrane domains of rho-dopsin are colored red. (B) LC-MS/MS analysis reveals radiolytic modificationof many residues (yellow sticks) found in the membrane embedded region ofrhodopsin as shown in this crystallographic model. The shaded rectanglerepresents the membrane; regions detected by LC-MS/MS analysis are coloredgreen and undetected regions are colored gray. The chromophore, 11-cis-retinylidene, is shown as orange sticks. Ordered waters are shown as redspheres in the transmembrane domain.
14368 � www.pnas.org�cgi�doi�10.1073�pnas.0901074106 Angel et al.
the radiolysis as well and that the observed rate constants ofmodification reflect the nature and location of generated radi-cals, both protein and water, the mobility of hydroxyl radicals orother generated radicals, the local structure of the side chains,and the intrinsic reactivity of the residue. With these caveats in
mind, comparative analysis of radical modification rate con-stants (only those �0.1 s�1 are considered) for residues presentin transmembrane helices II, III, IV, and VII of rhodopsinindicate structural changes near the chromophore-binding
Fig. 2. The extent of bulk water exchange into the membrane embedded domain of rhodopsin was measured by monitoring the incorporation of O18 fromisotopically labeled hydroxyl radicals. Taking advantage of locations sensitive to hydroxyl radical oxidation, we monitored solvent uptake for several activatedstates of this receptor over time. Rapid mixing of H2O18 containing buffer with rhodopsin, Meta II, and opsin was followed by equilibration (delay) periods of50 ms, 500 ms, 5 s, and 30 s before X-ray exposure for 6 or 40 ms. No O18 hydroxyl radical incorporation occurred in any of these experiments except for the exposedamino terminal peptide. This indicates that radicals that modify residues within the helical bundle are formed in situ in intramembranous regions of rhodopsin,photoactivated receptor, and apo-protein. Only following dehydration and rehydration of rhodopsin with 97% H2O18 water and subsequent X-ray exposurewere low levels of O18 incorporation detected in resulting proteolytic fragments (A, blue ribbons) from the transmembrane domain (A, yellow spheres). Forexample, O18 labeling of M86 in peptide 85FMVFGGF91 located in helix II was specifically detected by tandem mass spectrometry. The daughter ion spectrum ofunmodified peptide 85FMVFGGF91 is shown in panel B and the tandem MS spectrum of O16-modified M86 in panel C. Importantly, O18 hydroxyl labeling of M86following X-ray exposure was only detected following full exchange of solvent after sample dehydration and rehydration with H2O18 (D).
Table 1. Hydroxyl radical modification rate constants for residues in rhodopsin†
Peptide‡ Rhodopsin (s�1) Meta II (s�1) Opsin (s�1)
Ratios of rate constants
Meta II/rhodopsin
Meta II/opsin
Opsin/rhodopsin
85FM*VFGGF91 7.4 � 0.2§ 22.6 � 1.4 2.1 � 0.4 3 10.8 0.3113EGFF*116 238.1 � 21 370 � 16.5 254 � 47 1.6 1.5 1140C*KPM*SNF146 4.2¶ � 0.5 8.0¶ � 0.6 16.2¶ � 0.9†† 1.9 0.5 3.8147RFGENHAI*M*G156 3.8¶ � 0.4 6.6¶ � 0.4 ND‡‡ 1.7 – –160TWVM*A164 4.5 � 0.5 13.6 � 1.4 12.8 � 1.5 3 1 2.8176SRYIPEGM*Q184 1.8 � 0.8 1.1 � 0.5 1 � 0.1 1 1.1 0.6288M*TIPAF293 4.2 � 0.3 7.6 � 0.5 6.6 � 0.4 1.8 1.2 1.6293FAKTSAVY*NP*VIY*306 2.1¶ � 0.5 1.1¶ � 0.2 4.0¶ � 1.1 0.5 0.3 2333ASTTVSKTETSQV*A*P*A*348 8.1¶ � 0.3 7.9¶ � 0.4 6.8¶ � 0.3 1 1 0.8
*, Denotes residue(s) modified by hydroxyl radicals.†Rate constants were estimated by employing a nonlinear fit of hydroxyl modification data to a pseudo first order decay as described in Materials and Methods(supplement).
‡Peptides detected following LC-MS analysis and confirmed by MS/MS analysis with rate constants of modification suitable for quantitative analysis. As shownin Figure 1, other peptides exhibited modifications that were confirmed by tandem MS/MS.
§Error in rate constant estimation calculated from nonlinear fit.¶Rate constants reflect mixed modifications.††Rate determined from peptide 137–146 with the same modified residues as peptide 140–146.‡‡Peptide not detected in this experiment, no rate is reported.
Angel et al. PNAS � August 25, 2009 � vol. 106 � no. 34 � 14369
BIO
PHYS
ICS
AN
DCO
MPU
TATI
ON
AL
BIO
LOG
Y

pocket and increased labeling of side chains in these helicesfollowing receptor activation.
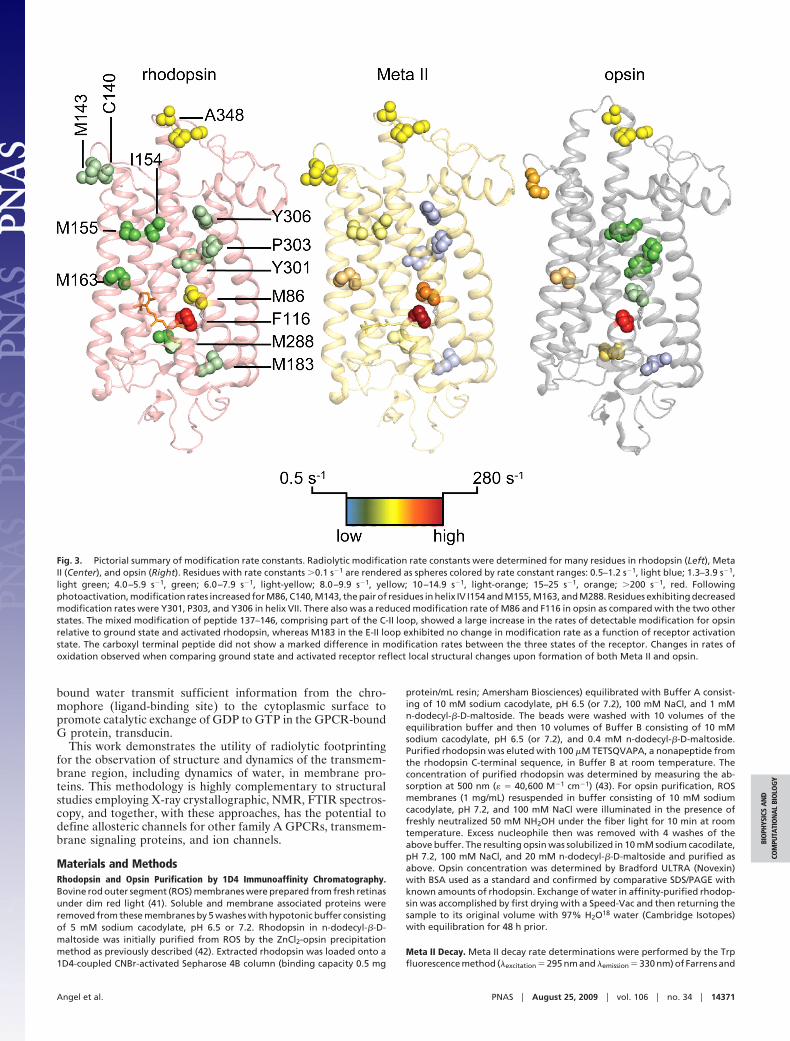
Release of Structural Constraint Resulting from Receptor Activation.In Table 1, a comparison of rates constants of radiolytic labelingfor selected peptides in ground and activated states (Meta II) ofrhodopsin identifies regions of local conformational changeassociated with receptor activation. Following photoactivation ofrhodopsin, a net loss of water upon Meta II formation has beendescribed, and this may be linked to some of the dynamic changesseen here (31, 32). Additionally, rates of radiolytic modificationfor the apo-protein opsin provide insight into the structuraldynamics specific to the deactivated state. These data revealchanges in regions known to undergo structural alterationsfollowing receptor activation. A 2-fold increase in radiolyticlabeling was observed for C140 and M143 in helix III near theconserved ERY residues of the ‘‘ionic lock,’’ and increasedsulfhydryl reactivity following photoactivation has previouslybeen observed (33). Comparison of the rates of modificationbetween Meta II and opsin reveals an even greater increase,suggesting the release of secondary structural constraints anddynamics of water and protein within Meta II following itstransition to opsin.
Residue F116, located in helix III and in the plane of thechromophore, exhibited the highest rate of modification in allstates of the receptor with modification rate constants �10-foldgreater than those found for any other residue. We also note thatthis modification rate is much higher than we have ever previ-ously observed for any Phe residue in a soluble protein. Thisresidue is one turn down (C-terminal) on helix III from the Schiffbase counter ion E113 (28), which was found to be withinhydrogen bonding distance of crystallographically identifiedordered water (atom number 2021 in 1U19.pdb). The very highrate of radiolytic modification of this site in all states of thereceptor is possibly because of proximity of ordered water, andthe increase in labeling upon activation may reflect movement ofwaters and/or side chains favoring the reaction. The reducedmodification rate of F116 found in opsin may be because ofdisorder of residues near F116 upon receptor activation as seenin solid state NMR and crystallographic studies. Thus, in theopsin state, the water may not be positioned as favorably forreaction (22, 34, 35).
The largest relative change in the rate constants of radiolyticmodification was observed for residue M86. This residue liesnear a region of the receptor, namely residues 121–136, thatexhibits an increased disorder following photoactivation (22, 34).We observed a 3-fold increase in the rate of radiolytic modifi-cation of M86 when comparing rate constants for ground stateand activated receptor (Table 1). As for F116, this increase inmodification was specific for the activated state, because theinactive state of the receptor (opsin) evidenced a 10-fold reduc-tion in radiolytic modification as compared with Meta II. M86is located in helix II, 3.9 Å (C-terminal) from the functionallycritical and highly conserved residue D83. Interactions betweenhelix II and VII are mediated by water via hydrogen bondingbetween D83, found in 94% of family A GPCRs, and residuesS298, V300, and N302 located in helix VII, conserved in 75% offamily A GPCRs (36). The functional importance of D83 hasbeen demonstrated by a mutation that led to increased formationof Meta II (37, 38). These findings clearly demonstrate changesin conformational dynamics near the highly conserved andfunctionally important residue D83 differentiate the inverseagonist, agonist bound, and ligand-free states of rhodopsin.
Further away from the ligand-binding pocket toward thecytoplasmic face of the receptor, residue M163 in helix IVshowed a 3-fold increase in modification of the activated versusthe ground state of the receptor. The observed increased rate ofmodification of M163 was common to both Meta II and opsin.
We interpret the increased conformational dynamics of M163 asthe release of constraints mediated by the Y206-H211-M163-E122 hydrogen bond network holding helix III and V togetherin the ground state of the receptor, consistent with studiesemploying solid state NMR spectroscopy (35, 39). Residues154IM155 exhibited a 2-fold increase in the rate of radiolyticlabeling when ground state rates were compared with those ofMeta II. This reflects the dynamics of specific residues at thecytoplasmic face of the receptor following release of the ioniclock holding helix III and helix VI together, likely reflectingmovements of helix VI found to occur as a consequence ofphotoactivaiton (30). Residue M288, located in helix VII on theintradiscal side of the chromophore-binding pocket (28), wasfound in rhodopsin crystal structures near ordered waters (964and 2014 in 1U19 in Protein Data Base). There was a 2-foldincrease in the rate of radiolytic modification for this residue inthe activated receptor and a subsequent modest decrease inmodification rate for the inactive opsin. The observed increasein radiolytic labeling rate of M288 found for the activated statehighlights local structural changes occurring near the ligand-binding pocket and indicate the presence of activated water. Incontrast, the nearby residue M183 in the E-II loop, whosestructure is indicated to be altered upon activation, did not showa similar increase in labeling rate, likely because of movement ofwater away from this site (35). These results demonstrate thatradiolytic labeling of membrane proteins probes both localstructural dynamics and hydration. A slight reduction in the rateof radiolytic modification was observed upon light activation forresidues near the NPxxY domain in helix VII, with the rate foropsin being the highest of the three states; no state-dependentchanges in modification rates were observed for the carboxylterminal tail (Fig. 3 and Movie S1, Movie S2, and Movie S3).
ConclusionThe above results suggest that the structural reorganizationobserved for Meta II does not simply stem from the release ofwater-mediated constraints that allow increased mobility ofregions in helix II and helix VII, because the carboxyl terminaldomain of helix VII exhibited a slight decrease in its observedrates of modification. Rather, we posit a directional increase inthe activation-dependent rate of modification, highest near thechromophore-binding pocket and radiating toward the cytoplas-mic face of the receptor along helix IV as evidenced by increasedmodification rates for residues M163 and 154IM155 (Fig. 3 and-Movie S1, Movie S2, and Movie S3).
Upon light activation, the structural reorganization adjacentto retinal is obvious from many studies; in this work F116, M86,and M288 exhibited clear increases in labeling upon receptoractivation (Fig. 3 and Movie S1, Movie S2, and Movie S3).However, this increased mobility did not extend to M183 or theconserved NPxxY motif as previously speculated (40). Instead,the suggested water reorganization extended to M163, M155,M143, and C140, and, by implication, to loop C-II. This mayrepresent the linkage of the ionic lock on the cytoplasmic faceof the receptor to a secondary internal hydrogen bondingnetwork comprised of residues Y206-H211-M163-E122. Disrup-tion of the secondary interhelical network, analogous to the ioniclock, may in fact be mediated by water reorganization, whichexplains in part the requirement for a hydrated receptor to attainthe activated state. Our radiolytic footprinting of rhodopsin doesdemonstrate the structural activation of bound waters as afunction of receptor signaling status and suggests a possible pathof allosteric communication that may be conserved amongfamily A GPCRs (36). For example, this pathway could beestablished by specific protonation/deprotonation of key resi-dues such as E113 and E134 (28). Overall, we suggest a generalframework where disruption and reorganization of multipleclose packing interactions mediated by both side chains and
14370 � www.pnas.org�cgi�doi�10.1073�pnas.0901074106 Angel et al.
bound water transmit sufficient information from the chro-mophore (ligand-binding site) to the cytoplasmic surface topromote catalytic exchange of GDP to GTP in the GPCR-boundG protein, transducin.
This work demonstrates the utility of radiolytic footprintingfor the observation of structure and dynamics of the transmem-brane region, including dynamics of water, in membrane pro-teins. This methodology is highly complementary to structuralstudies employing X-ray crystallographic, NMR, FTIR spectros-copy, and together, with these approaches, has the potential todefine allosteric channels for other family A GPCRs, transmem-brane signaling proteins, and ion channels.
Materials and MethodsRhodopsin and Opsin Purification by 1D4 Immunoaffinity Chromatography.Bovine rod outer segment (ROS) membranes were prepared from fresh retinasunder dim red light (41). Soluble and membrane associated proteins wereremoved from these membranes by 5 washes with hypotonic buffer consistingof 5 mM sodium cacodylate, pH 6.5 or 7.2. Rhodopsin in n-dodecyl-�-D-maltoside was initially purified from ROS by the ZnCl2-opsin precipitationmethod as previously described (42). Extracted rhodopsin was loaded onto a1D4-coupled CNBr-activated Sepharose 4B column (binding capacity 0.5 mg
protein/mL resin; Amersham Biosciences) equilibrated with Buffer A consist-ing of 10 mM sodium cacodylate, pH 6.5 (or 7.2), 100 mM NaCl, and 1 mMn-dodecyl-�-D-maltoside. The beads were washed with 10 volumes of theequilibration buffer and then 10 volumes of Buffer B consisting of 10 mMsodium cacodylate, pH 6.5 (or 7.2), and 0.4 mM n-dodecyl-�-D-maltoside.Purified rhodopsin was eluted with 100 �M TETSQVAPA, a nonapeptide fromthe rhodopsin C-terminal sequence, in Buffer B at room temperature. Theconcentration of purified rhodopsin was determined by measuring the ab-sorption at 500 nm (� � 40,600 M�1 cm�1) (43). For opsin purification, ROSmembranes (1 mg/mL) resuspended in buffer consisting of 10 mM sodiumcacodylate, pH 7.2, and 100 mM NaCl were illuminated in the presence offreshly neutralized 50 mM NH2OH under the fiber light for 10 min at roomtemperature. Excess nucleophile then was removed with 4 washes of theabove buffer. The resulting opsin was solubilized in 10 mM sodium cacodilate,pH 7.2, 100 mM NaCl, and 20 mM n-dodecyl-�-D-maltoside and purified asabove. Opsin concentration was determined by Bradford ULTRA (Novexin)with BSA used as a standard and confirmed by comparative SDS/PAGE withknown amounts of rhodopsin. Exchange of water in affinity-purified rhodop-sin was accomplished by first drying with a Speed-Vac and then returning thesample to its original volume with 97% H2O18 water (Cambridge Isotopes)with equilibration for 48 h prior.
Meta II Decay. Meta II decay rate determinations were performed by the Trpfluorescence method (�excitation � 295 nm and �emission � 330 nm) of Farrens and
Fig. 3. Pictorial summary of modification rate constants. Radiolytic modification rate constants were determined for many residues in rhodopsin (Left), MetaII (Center), and opsin (Right). Residues with rate constants �0.1 s�1 are rendered as spheres colored by rate constant ranges: 0.5–1.2 s�1, light blue; 1.3–3.9 s�1,light green; 4.0–5.9 s�1, green; 6.0–7.9 s�1, light-yellow; 8.0–9.9 s�1, yellow; 10–14.9 s�1, light-orange; 15–25 s�1, orange; �200 s�1, red. Followingphotoactivation, modification rates increased for M86, C140, M143, the pair of residues in helix IV I154 and M155, M163, and M288. Residues exhibiting decreasedmodification rates were Y301, P303, and Y306 in helix VII. There also was a reduced modification rate of M86 and F116 in opsin as compared with the two otherstates. The mixed modification of peptide 137–146, comprising part of the C-II loop, showed a large increase in the rates of detectable modification for opsinrelative to ground state and activated rhodopsin, whereas M183 in the E-II loop exhibited no change in modification rate as a function of receptor activationstate. The carboxyl terminal peptide did not show a marked difference in modification rates between the three states of the receptor. Changes in rates ofoxidation observed when comparing ground state and activated receptor reflect local structural changes upon formation of both Meta II and opsin.
Angel et al. PNAS � August 25, 2009 � vol. 106 � no. 34 � 14371
BIO
PHYS
ICS
AN
DCO
MPU
TATI
ON
AL
BIO
LOG
Y

Khorana (44). All measurements were performed with 30 nM Rho purified by1D4 affinity chromatography (either exposed or not to radiolysis) dissolved inbuffer consisting of 10 mM BTP, 100 mM NaCl, and 1 mM DDM, pH 6.0, thatfavors formation of Meta II. A Perkin-Elmer LS 55 Luminescence Spectropho-tometer was used to measure the intrinsic fluorescence increase because ofTrp residues, which correlates with the decrease in the protonated Schiff baseconcentration (44–47). Rhodopsin was bleached by a Fiber-Lite illuminatorcovered with a band-pass wavelength filter (480–520 nm) for 15 s immediatelybefore the fluorescence measurements. Bleaching was carried out from adistance of 10 cm to prevent heat accumulation, and a thermostat was appliedto stabilize the temperature of the cuvette at 20 °C. Fluorimeter slit settingswere 5.0 nm at 295 nm for excitation and 10 nm at 330 nm for emission.
Radiolytic Labeling. Protein samples in detergent were exposed to synchrotronX-ray white light at the National Synchrotron Light Source’s (BrookhavenNational Laboratory, Upton, NY) beamline X-28C operating at a ring energyof 2.8 GeV (15). The X-ray beam parameters were optimized by using thestandard fluorophore assay; this assays monitors the loss of intensity of anAlexa fluorophore to determine the effective hydroxyl radical concentration(48, 49). At low flux density, the scavenging effects of detergent requires longexposure times (�100 ms) that permit secondary radical reactions and resultin low signal-to-noise LC-MS data. The high X-ray flux density generated byfocusing the beam with a mirror (mirror angle to 5.5 mrad and the bender
value to 8.0 mm) (17, 49) permits a sufficient dose to be delivered in a fewmilliseconds, reducing chemical noise and enhancing LC-MS data acquisition.Rhodopsin samples were irradiated using time intervals raging from 1–10 mswith a continuous flow method using modified KinTek (KinTek Corporation)apparatus. Solvent exchange experiments were carried out by 1:1 (H2O16
buffer:H2O18 buffer) solvent mixing with 2–3 ms instrumental dead timefollowed by delays of 50, 100, 500, 5,000, and 30,000 ms before synchrotronbeam exposure for 6- or 40-ms time intervals. These experiments were carriedout in the KinTek quench flow mixer (KinTek Corporation) (48). All exposureswere carried out at 4 °C, and to all samples methionine-amide (final concen-tration 10 mM) was added to quench any peroxide-induced or free radical-induced secondary oxidations during the postexposure period (50). Sampleswere frozen in dry ice and stored at �80 °C before proteolytic cleavage andliquid chromatography mass spectrometric (LC-MS) analyses. Details of theselatter procedures are contained in SI Text.
ACKNOWLEDGMENTS. We thank Dr. Leslie T. Webster, Jr. for critical com-ments on the manuscript. This work was supported by National Institutes ofHealth (NIH) Grants EY09339, GM079191, EB09998, EB01979, andT32EY007157. Beamline X28C of the National Synchrotron Light Source (NSLS)is supported by the National Institute of Biomedical Imaging and Bioengi-neering. The National Synchrotron Light Source is financed by the Departmentof Energy.
1. Muller DJ, Wu N, Palczewski K (2008) Vertebrate membrane proteins: Structure, function,and insights from biophysical approaches. Pharmacol Rev 60:43–78.
2. Rosenbaum DM, et al. (2007) GPCR engineering yields high-resolution structural insightsinto beta2-adrenergic receptor function. Science 318:1266–1273.
3. Kobilka B, Schertler GF (2008) New G-protein-coupled receptor crystal structures: Insightsand limitations. Trends Pharmacol Sci 29:79–83.
4. Rasmussen SG, et al. (2007) Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450:383–387.
5. Cherezov V, et al. (2007) High-resolution crystal structure of an engineered humanbeta2-adrenergic G protein-coupled receptor. Science 318:1258–1265.
6. Li J, EdwardsPC,BurghammerM,VillaC, SchertlerGF (2004)Structureofbovine rhodopsinin a trigonal crystal form. J Mol Biol 343:1409–1438.
7. Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y (2002) Functional role ofinternal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl AcadSci USA 99:5982–5987.
8. Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V (2004) The retinal conforma-tion and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol342:571–583.
9. Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE (2001) Advances in determi-nation of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry 40:7761–7772.
10. Palczewski K, et al. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor.Science 289:739–745.
11. Angel TE, Chance MR, Palczewski K (2009) Conserved waters mediate structural andfunctional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc NatlAcad Sci USA 106:8555–8560.
12. Maleknia SD, Brenowitz M, Chance MR (1999) Millisecond radiolytic modification ofpeptidesbysynchrotronX-rays identifiedbymass spectrometry.AnalChem71:3965–3973.
13. Ralston CY, et al. (2000) Time-resolved synchrotron X-ray footprinting and its applicationto RNA folding. Methods Enzymol 317:353–368.
14. Sclavi B, Sullivan M, Chance MR, Brenowitz M, Woodson SA (1998) RNA folding atmillisecondintervalsbysynchrotronhydroxyl radical footprinting.Science279:1940–1943.
15. Takamoto K, Chance MR (2006) Radiolytic protein footprinting with mass spectrometry toprobe the structure of macromolecular complexes. Annu Rev Biophys Biomol Struct35:251–276.
16. Xu G, Chance MR (2007) Hydroxyl radical-mediated modification of proteins as probes forstructural proteomics. Chem Rev 107:3514–3543.
17. Bohon J, Jennings LD, Phillips CM, Licht S, Chance MR (2008) Synchrotron protein foot-printing supports substrate translocation by ClpA via ATP-induced movements of the D2loop. Structure 16:1157–1165.
18. Kiselar JG, Mahaffy R, Pollard TD, Almo SC, Chance MR (2007) Visualizing Arp2/3 complexactivation mediated by binding of ATP and WASp using structural mass spectrometry. ProcNatl Acad Sci USA 104:1552–1557.
19. Murakami M, Kouyama T (2008) Crystal structure of squid rhodopsin. Nature 453:363–367.20. Nakamichi H, Okada T (2006) Crystalographic analysis of primary visual photochemistry.
Angew Chem Int Ed Engl 45:4270–4272.21. Nakamichi H, Okada T (2006) Local peptide movement in the photoreaction intermediate
of rhodopsin. Proc Natl Acad Sci USA 103:12729–12734.22. Salom D, et al. (2006) Crystal structure of a photoactivated deprotonated intermediate of
rhodopsin. Proc Natl Acad Sci USA 103:16123–16128.23. Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP (2008) Crystal structure of the
ligand-free G-protein-coupled receptor opsin. Nature 454:183–187.24. ScheererP,etal. (2008)Crystal structureofopsin in itsG-protein-interactingconformation.
Nature 455:497–502.25. Wald G, Durell J, St George CC (1950) The light reaction in the bleaching of rhodopsin.
Science 111:179–181.
26. Matthews RG, Hubbard R, Brown PK, Wald G (1963) Tautomeric forms of metarhodopsin.J Gen Physiol 47:215–240.
27. Garczarek F, Gerwert K (2006) Functional waters in intraprotein proton transfer moni-tored by FTIR difference spectroscopy. Nature 439:109–112.
28. Palczewski K (2006) G protein-coupled receptor rhodopsin. Annu Rev Biochem 75:743–767.
29. Ball P (2008) Water as an active constituent in cell biology. Chem Rev 108:74–108.30. Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL (2008) High-resolution
distance mapping in rhodopsin reveals the pattern of helix movement because of activa-tion. Proc Natl Acad Sci USA 105:7439–7444.
31. Mitchell DC, Litman BJ (1999) Effect of protein hydration on receptor conformation:Decreased levels of bound water promote metarhodopsin II formation. Biochemistry38:7617–7623.
32. Mitchell DC, Litman BJ (2000) Effect of ethanol and osmotic stress on receptor conforma-tion. Reduced water activity amplifies the effect of ethanol on metarhodopsin II forma-tion. J Biol Chem 275:5355–5360.
33. De Grip WJ, Daemen FJ (1982) Sulfhydryl chemistry of rhodopsin. Methods Enzymol81:223–236.
34. Crocker E, et al. (2006) Location of Trp265 in metarhodopsin II: Implications for theactivation mechanism of the visual receptor rhodopsin. J Mol Biol 357:163–172.
35. Ahuja S, et al. (2009) Helix movement is coupled to displacement of the second extracel-lular loop in rhodopsin activation. Nat Struct Mol Biol 16:168–175.
36. Mirzadegan T, Benko G, Filipek S, Palczewski K (2003) Sequence analyses of G-protein-coupled receptors: Similarities to rhodopsin. Biochemistry 42:2759–2767.
37. Weitz CJ, Nathans J (1993) Rhodopsin activation: Effects on the metarhodopsin I-metarhodopsin II equilibrium of neutralization or introduction of charged amino acidswithin putative transmembrane segments. Biochemistry 32:14176–14182.
38. DeCaluwe GL, Bovee-Geurts PH, Rath P, Rothschild KJ, de Grip WJ (1995) Effect of carboxylmutations on functional properties of bovine rhodopsin. Biophys Chem 56:79–87.
39. Patel AB, et al. (2005) Changes in interhelical hydrogen bonding upon rhodopsin activa-tion. J Mol Biol 347:803–812.
40. Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, Ernst OP (2003) Role of theconserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. ProcNatl Acad Sci USA 100:2290–2295.
41. PapermasterDS(1982)Preparationofantibodies torhodopsinandthe largeproteinofrodouter segments. Methods Enzymol 81:240–246.
42. Okada T, Tsujimoto R, Muraoka M, Funamoto C (2005) Methods and results in X-raycrystallography of bovine rhodopsin, in G Protein-Coupled Receptors: Structure, Function,and Ligand Screening, ed Haga T (CRC Press LLC, Boca Raton, FL), pp 245–261.
43. WaldG,BrownPK(1953)Themolecularexcitationof rhodopsin. JGenPhysiol37:189–200.44. Farrens DL, Khorana HG (1995) Structure and function in rhodopsin. Measurement of the
rate of metarhodopsin II decay by fluorescence spectroscopy. J Biol Chem 270:5073–5076.45. Schadel SA, et al. (2003) Ligand channeling within a G-protein-coupled receptor. The entry
and exit of retinals in native opsin. J Biol Chem 278:24896–24903.46. Heck M, et al. (2003) Signaling states of rhodopsin. Formation of the storage form,
metarhodopsin III, from active metarhodopsin II. J Biol Chem 278:3162–3169.47. Fahmy K, Sakmar TP (1993) Light-dependent transducin activation by an ultraviolet-
absorbing rhodopsin mutant. Biochemistry 32:9165–9171.48. Gupta S, Sullivan M, Toomey J, Kiselar J, Chance MR (2007) The Beamline X28C of the
Center for Synchrotron Biosciences: A national resource for biomolecular structure anddynamics experiments using synchrotron footprinting. J Synchrotron Radiat 14:233–243.
49. Sullivan MR, Rekhi S, Bohon J, Gupta S, Abel D, Toomey J, Chance MR (2008) Installationand testing of a focusing mirror at beamline X28C for high flux X-ray radiolysis ofbiological macromolecules. Rev Sci Instrum 79:025101.
50. Xu G, Kiselar J, He Q, Chance MR (2005) Secondary reactions and strategies to improvequantitative protein footprinting. Anal Chem 77:3029–3037.
14372 � www.pnas.org�cgi�doi�10.1073�pnas.0901074106 Angel et al.





























