Embed Size (px)
Citation preview

The aggregation potential of human amylin determines itscytotoxicity towards islet b-cellsBarbara Konarkowska1,2, Jacqueline F. Aitken1,2, Joerg Kistler1, Shaoping Zhang1,2 andGarth J. S. Cooper1,2,3
1 School of Biological Sciences, Faculty of Science, University of Auckland, New Zealand
2 Centre for Research Excellence in Molecular Biodiscovery, Faculty of Science, University of Auckland, New Zealand
3 Department of Medicine, Faculty of Medical and Health Sciences, University of Auckland, New Zealand
Human amylin (hA) is a small protein cosecreted with
insulin from pancreatic islet b-cells upon stimulation
by glucose or other chemical signals [1,2]. Wild-type
hA, also known as insulinoma amyloid peptide, insuli-
noma amyloid polypeptide (IAPP) or islet amyloid
polypeptide [3], demonstrates a strong in vitro tendency
to aggregate into fibrils [4,5], which is dependent on
specific residues in different regions of the molecule,
notably, the amyloidogenic region of amino acids 20–
29 [6]. It is the main protein in the amyloid aggregates
that are frequently present in the islets of human sub-
jects with type 2 diabetes mellitus (T2DM) [7,8], as
well as in diabetic cats and primates [9]. It has been
reported that aggregated hA might contribute to the
loss of insulin-producing pancreatic b-cells cells in
T2DM [10,11]. The following lines of evidence have
associated hA with the pancreatic pathology of
T2DM: (a) amyloid is often concentrated near areas of
islet b-cell degeneration in humans or primates with
T2DM [10,12–14]; (b) synthetic hA is toxic to pancre-
atic b-cells in vitro [11,15–17]; (c) spontaneous loss of
b-cells has been reported in a line of hA-transgenic
mice [18]; and (d) toxicity of hA towards cultured cells
correlates with its ability to form fibrils - for example,
rat amylin does not form fibrils and nor does it evoke
b-cell apoptosis in vitro [11,17,19].
Keywords
amylin; amyloid formation; pancreatic islet
b-cells; protein aggregation; type 2 diabetes
Correspondence
G. J. S. Cooper, Level 4, Thomas Building,
School of Biological Sciences, University of
Auckland, Private Bag 92019, Auckland,
New Zealand
Fax: +64 9 373 7045
Tel: +64 9 373 7599, ext. 87394
E-mail: [email protected]
(Received 5 April 2006, revised 6 June
2006, accepted 8 June 2006)
doi:10.1111/j.1742-4658.2006.05367.x
Human amylin is a small fibrillogenic protein that is the major constituent
of pancreatic islet amyloid, which occurs in most subjects with type 2 dia-
betes. There is evidence that it can elicit in vitro apoptosis in islet b-cells,but the physical properties that underpin its cytotoxicity have not been
clearly elucidated. Here we employed electron microscopy, thioflavin T
fluorescence and CD spectroscopy to analyze amylin preparations whose
cytotoxic potential was established by live–dead assay in cultured b-cells.Highly toxic amylin contained few preformed fibrils and initially showed
little b-sheet content, but underwent marked time-dependent aggregation
and b-conformer formation following dissolution. By contrast, low-toxicity
amylin contained abundant preformed fibrils, and demonstrated high initial
b-sheet content but little propensity to aggregate further once dissolved.
Thus, mature amylin fibrils are not toxic to b-cells, and aggregates of fibrils
such as occur in pancreatic islet amyloid in vivo are unlikely to contribute
to b-cell loss. Rather, the toxic molecular species is likely to comprise sol-
uble oligomers with significant b-sheet content. Attempts to find ways of
protecting b-cells from amylin-mediated death might profitably focus on
preventing the conformational change from random coil to b-sheet.
Abbreviations
AM, acetomethoxyl ester; EthD, ethidium homodimer; hA, human amylin; HFIP, hexafluoroisopropanol; IAPP, insulinoma amyloid
polypeptide; JNK1, Jun NH2-terminal kinase 1; KRB, Krebs-Ringer bicarbonate buffer; RINm5F, rat insulinoma m5F; T2DM, type 2 diabetes
mellitus; TEM, transmission electron microscopy; ThT, thioflavin T.
3614 FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS

Preventing hA-induced loss of pancreatic b-cells has
been proposed as a route to attenuate the gradual
decline in endogenous production of insulin in T2DM
[20]. However, at this time our knowledge of the mech-
anism by which hA evokes pancreatic b-cell death is
incomplete. The small oligomeric structures formed
very early in the process of hA aggregation [4,21]
could be toxic to pancreatic b-cells, but whether such
structures form in vivo and whether mature fibrils are
cytotoxic remains unresolved.
It was initially assumed that mature amyloid fibrils
were the most likely toxic species, as these are the form
of aggregates that have been commonly detected adja-
cent to islet cells [11,22,23]. However, a growing body
of experimental data suggests that in the case of many
amyloid diseases, the most toxic form of protein to cells
are the prefibrillar aggregates [24–29]. Protofibrillar sol-
uble intermediates are known to exist in the early stages
of fibril formation of various amyloidogenic proteins,
including hA, b-amyloid and a-synuclein [21,30–32].
Prefibrillar aggregates of proteins not associated with
amyloid diseases have also been shown to cause cyto-
toxicity in a similar manner and extent to those of dis-
ease-associated proteins [33]. Production of an antibody
that recognizes micellar Ab, but not its soluble or fibril-lar forms, has been reported [31]. This antibody also
recognized and inhibited the cytotoxicity of soluble
oligomers of structurally unrelated proteins such as
a-synuclein, insulin, amylin and polyglutamine, suggest-
ing that these proteins may have a common structure
and may share a common pathogenic mechanism.
Clearly, the nature of the cytotoxic species of amylin has
implications that may be applicable to other amyloid
pathologies, making its investigation generally relevant.
Kayed et al. [34] and Tenidis et al. [35] previously
observed that a conformational transition of soluble
amylin into b-sheet precedes formation of insoluble
amylin aggregates, and in addition, the kinetics of this
amyloid formation are consistent with a nucleation-
dependent polymerization mechanism.
Wogulis et al. [36] suggested that the neuronal cell
death associated with b-amyloid and human amylin
required the presence of both fibrillar and soluble pep-
tide. They postulated that nucleation-dependent poly-
merization was a common feature of amyloid-mediated
cell death.
While studying their cytotoxicity in a b-cell culturemodel, we noted that different synthetic hA prepara-
tions vary markedly in their levels of intrinsic bioactivity
and hypothesized that this variation might be directly
related to differences in the rate of formation of soluble
cytotoxic intermediates, resulting from the initial aggre-
gation state of the synthetic hA preparation.
To test this hypothesis, we employed three independ-
ent techniques: transmission electron microscopy
(TEM), thioflavin T (ThT) fluoroscopy and CD spec-
troscopy, to characterize relevant physical properties
of hA preparations whose toxicity was known, as
determined by live–dead assay in rat insulinoma m5F
(RINm5F) islet b-cells. In particular, we characterized
their aggregation state with respect to the presence or
absence of preformed fibrils, aggregate content and
b-sheet content following initial dissolution. We also
measured time-dependent rates of aggregation and
b-sheet formation in solution.
We have previously described the RINm5F model of
amylin-mediated b-cell apoptosis [15–17,20]. Treatment
of these cells with fibrillogenic amylin preparations
(i.e. preparations that are able form fibrils from mono-
meric and oligomeric species) evokes apoptosis charac-
terized by typical changes in the cell membrane and
nucleus, formation of apoptotic bodies and internucle-
osomal cleavage of genomic DNA [37]. Fibrillogenic
amylin evokes cytotoxicity via a molecular signaling
pathway that incorporates sequential activation of Jun
NH2-terminal kinase 1 (JNK1), c-Jun, caspase 8, and
caspases 1 and 3 [16,17], as well as increased expres-
sion of p53 and p21WAF1 ⁄CIP1 [19]. Significantly, these
cellular and molecular events are only elicited by inter-
action between cells and fibrillogenic amylin variants,
whereas, by contrast, nonfibrillogenic amylin molecules
are nontoxic. We have also shown that responses
evoked by fibrillogenic amylin molecules in RINm5F
cells are echoed by those in the human islet b-cell line,CM [16,17].
Here we show that the cytotoxicity of preparations
of two fibrillogenic proteins, wild-type hA and 8)37hA,
correlates with their initial aggregation states.8)37hA is a fragment of hA that lacks the NH2-
terminal seven amino acids of the wild-type protein
(necessary for receptor binding and peripheral biologi-
cal activity [38]), but that retains its ability to elicit
cytotoxicity in b-cells [17,39]. Although the physiologic
function of 8)37hA is unknown, it is nevertheless a
second closely related amyloidogenic member of the
amylin family, and was therefore used to generate a
set of independent data to complement those obtained
with the wild-type molecule (Table 1).
Two different preparations of each of these proteins
were studied: one containing a high proportion of
mature fibrils and the other containing an abundance
of small globular structures. These preparations
showed remarkable differences in cytotoxicity, differ-
ences in their secondary structure, and differences in
their potential to form new aggregates. Our results
demonstrate an inverse relationship between the extent
B. Konarkowska et al. Aggregation potential of human amylin
FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS 3615
of aggregation and the cytotoxic potential of hA. They
show that mature hA fibrils are not cytotoxic in vitro,
and instead implicate a much earlier precursor stage,
namely, the transition from random coil to b-sheetcoupled with de novo aggregation, as necessary proper-
ties for the induction of b-cell death.
Results and Discussion
Cytotoxic potential varies between different
hA preparations
We have previously noted major differences in cyto-
toxic potential among different commercial prepara-
tions of hA. Here, we employed a well-characterized
live–dead assay to measure the extent of cell death [37]
following 24 h incubations with various preparations
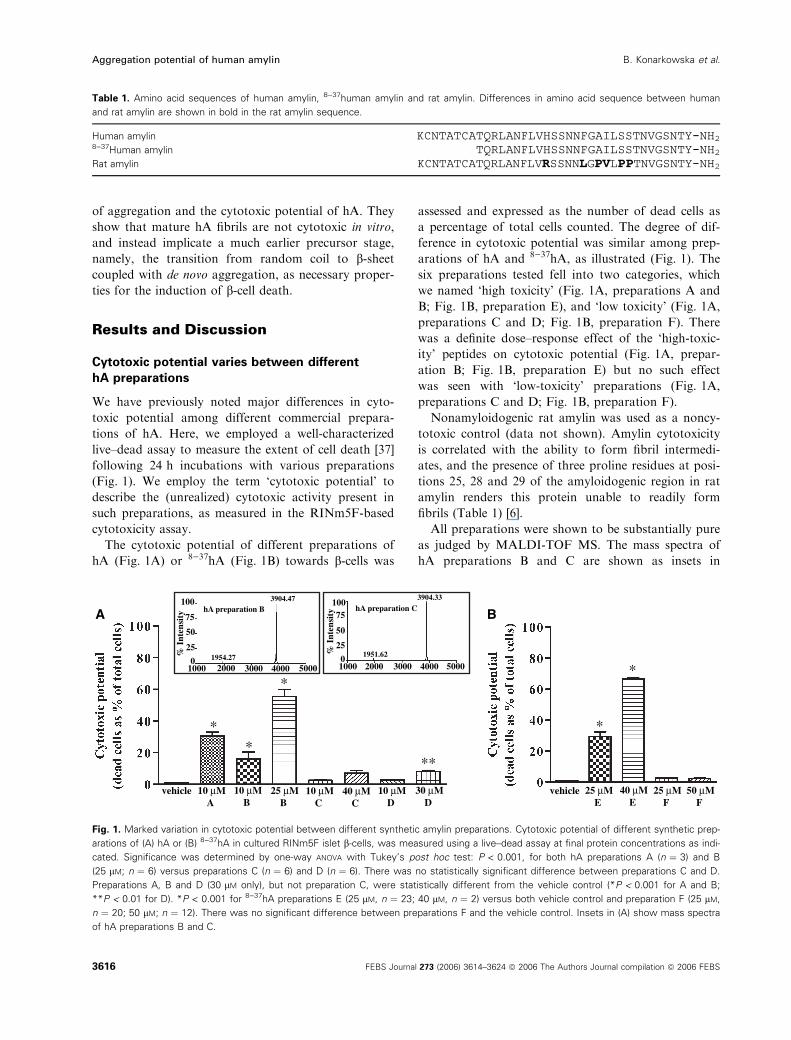
(Fig. 1). We employ the term ‘cytotoxic potential’ to
describe the (unrealized) cytotoxic activity present in
such preparations, as measured in the RINm5F-based
cytotoxicity assay.
The cytotoxic potential of different preparations of
hA (Fig. 1A) or 8)37hA (Fig. 1B) towards b-cells was
assessed and expressed as the number of dead cells as
a percentage of total cells counted. The degree of dif-
ference in cytotoxic potential was similar among prep-
arations of hA and 8)37hA, as illustrated (Fig. 1). The
six preparations tested fell into two categories, which
we named ‘high toxicity’ (Fig. 1A, preparations A and
B; Fig. 1B, preparation E), and ‘low toxicity’ (Fig. 1A,
preparations C and D; Fig. 1B, preparation F). There
was a definite dose–response effect of the ‘high-toxic-
ity’ peptides on cytotoxic potential (Fig. 1A, prepar-
ation B; Fig. 1B, preparation E) but no such effect
was seen with ‘low-toxicity’ preparations (Fig. 1A,
preparations C and D; Fig. 1B, preparation F).
Nonamyloidogenic rat amylin was used as a noncy-
totoxic control (data not shown). Amylin cytotoxicity
is correlated with the ability to form fibril intermedi-
ates, and the presence of three proline residues at posi-
tions 25, 28 and 29 of the amyloidogenic region in rat
amylin renders this protein unable to readily form
fibrils (Table 1) [6].
All preparations were shown to be substantially pure
as judged by MALDI-TOF MS. The mass spectra of
hA preparations B and C are shown as insets in
Table 1. Amino acid sequences of human amylin, 8)37human amylin and rat amylin. Differences in amino acid sequence between human
and rat amylin are shown in bold in the rat amylin sequence.
Human amylin KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY-NH28)37Human amylin TQRLANFLVHSSNNFGAILSSTNVGSNTY-NH2
Rat amylin KCNTATCATQRLANFLVRSSNNLGPVLPPTNVGSNTY-NH2
vehicle 10 µMA
10 µMB
25 µMB
10 µMC
40 µMC
10 µMD
30 µMD
vehicle 25 µME
40 µME
25 µMF
50 µMF
A B100
75
50
25
01000 2000 3000 4000 5000
ytisnetnI %
hA preparation B
1954.27
3904.47 10075
50
25
01000 2000 3000 4000 5000
ytisnetnI %
hA preparation C
1951.62
3904.33
**
*
*
*
**
Fig. 1. Marked variation in cytotoxic potential between different synthetic amylin preparations. Cytotoxic potential of different synthetic prep-
arations of (A) hA or (B) 8)37hA in cultured RINm5F islet b-cells, was measured using a live–dead assay at final protein concentrations as indi-
cated. Significance was determined by one-way ANOVA with Tukey’s post hoc test: P < 0.001, for both hA preparations A (n ¼ 3) and B
(25 lM; n ¼ 6) versus preparations C (n ¼ 6) and D (n ¼ 6). There was no statistically significant difference between preparations C and D.
Preparations A, B and D (30 lM only), but not preparation C, were statistically different from the vehicle control (*P < 0.001 for A and B;
**P < 0.01 for D). *P < 0.001 for 8)37hA preparations E (25 lM, n ¼ 23; 40 lM, n ¼ 2) versus both vehicle control and preparation F (25 lM,
n ¼ 20; 50 lM; n ¼ 12). There was no significant difference between preparations F and the vehicle control. Insets in (A) show mass spectra
of hA preparations B and C.
Aggregation potential of human amylin B. Konarkowska et al.
3616 FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS
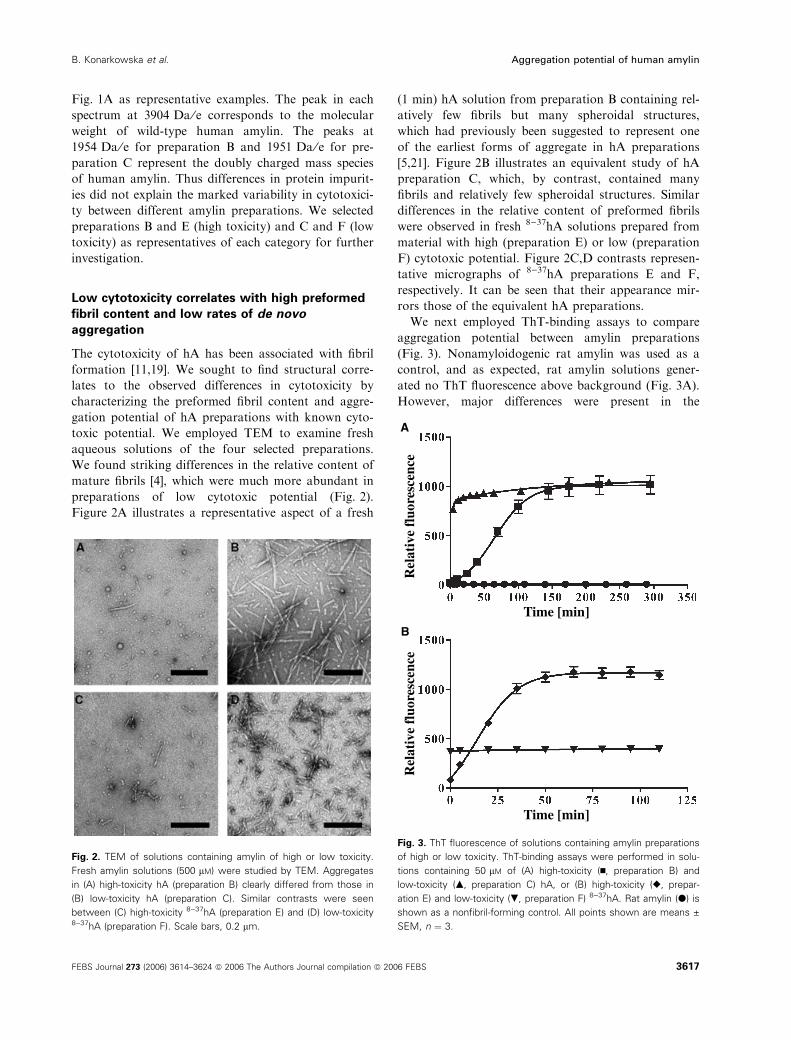
Fig. 1A as representative examples. The peak in each
spectrum at 3904 Da ⁄ e corresponds to the molecular
weight of wild-type human amylin. The peaks at
1954 Da ⁄ e for preparation B and 1951 Da ⁄ e for pre-
paration C represent the doubly charged mass species
of human amylin. Thus differences in protein impurit-
ies did not explain the marked variability in cytotoxici-
ty between different amylin preparations. We selected
preparations B and E (high toxicity) and C and F (low
toxicity) as representatives of each category for further
investigation.
Low cytotoxicity correlates with high preformed
fibril content and low rates of de novo
aggregation
The cytotoxicity of hA has been associated with fibril
formation [11,19]. We sought to find structural corre-
lates to the observed differences in cytotoxicity by
characterizing the preformed fibril content and aggre-
gation potential of hA preparations with known cyto-
toxic potential. We employed TEM to examine fresh
aqueous solutions of the four selected preparations.
We found striking differences in the relative content of
mature fibrils [4], which were much more abundant in
preparations of low cytotoxic potential (Fig. 2).
Figure 2A illustrates a representative aspect of a fresh
(1 min) hA solution from preparation B containing rel-
atively few fibrils but many spheroidal structures,
which had previously been suggested to represent one
of the earliest forms of aggregate in hA preparations
[5,21]. Figure 2B illustrates an equivalent study of hA
preparation C, which, by contrast, contained many
fibrils and relatively few spheroidal structures. Similar
differences in the relative content of preformed fibrils
were observed in fresh 8)37hA solutions prepared from
material with high (preparation E) or low (preparation
F) cytotoxic potential. Figure 2C,D contrasts represen-
tative micrographs of 8)37hA preparations E and F,
respectively. It can be seen that their appearance mir-
rors those of the equivalent hA preparations.
We next employed ThT-binding assays to compare
aggregation potential between amylin preparations
(Fig. 3). Nonamyloidogenic rat amylin was used as a
control, and as expected, rat amylin solutions gener-
ated no ThT fluorescence above background (Fig. 3A).
However, major differences were present in the
Fig. 2. TEM of solutions containing amylin of high or low toxicity.
Fresh amylin solutions (500 lM) were studied by TEM. Aggregates
in (A) high-toxicity hA (preparation B) clearly differed from those in
(B) low-toxicity hA (preparation C). Similar contrasts were seen
between (C) high-toxicity 8)37hA (preparation E) and (D) low-toxicity8)37hA (preparation F). Scale bars, 0.2 lm.
ecnecseroulf evitaleR
Time [min]
Aecnecseroulf evitale
R
B
Time [min]
Fig. 3. ThT fluorescence of solutions containing amylin preparations
of high or low toxicity. ThT-binding assays were performed in solu-
tions containing 50 lM of (A) high-toxicity (n, preparation B) and
low-toxicity (m, preparation C) hA, or (B) high-toxicity (r, prepar-
ation E) and low-toxicity (., preparation F) 8)37hA. Rat amylin (d) is
shown as a nonfibril-forming control. All points shown are means ±
SEM, n ¼ 3.
B. Konarkowska et al. Aggregation potential of human amylin
FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS 3617

ThT-binding profiles of hA preparations with differing
cytotoxic potential. First, the hA preparation of low
cytotoxic potential (preparation C) demonstrated high
levels of ThT-detectable aggregates immediately after
dissolution into ThT-containing buffer, and initial
fluorescence values comprised more than 80% of maxi-
mal values obtained during the time of the assay
(Fig. 3A). By contrast, the preparation of high cyto-
toxic potential (preparation B) contained few pre-
formed aggregates, and initial fluorescence values
comprised less than 5% of the maximal values that
developed during the time course (Fig. 3A). These data
are consistent with our TEM findings, both demonstra-
ting major differences in the aggregation state immedi-
ately after dissolution.
Prolonged incubation of these preparations with
ThT revealed very different time-dependent increases
in fluorescence. In the case of low-toxicity hA prepar-
ation C, this increase was small compared to that of
high-cytotoxicity preparation B (Fig. 3A). Moreover,
the ThT-binding profile of the highly toxic preparation
displayed a sigmoidal-shaped fluorescence time curve,
consistent with the presence of three phases: lag, elon-
gation and saturation. These findings are consistent
with those previously reported for hA [40]. By con-
trast, the best fit to the time-dependent ThT fluores-
cence data from the weakly toxic hA preparation was
a two-phase association curve with a relatively short
elongation phase followed by saturation. The presence
of ThT itself in the assay did not appear to affect the
kinetics of aggregation, as a control experiment where
samples were incubated in the absence of ThT and
aliquots removed at different times showed similar
results to those described above (data not shown).
This difference between the shapes of the fluores-
cence–time curves suggests that the initial aggregation
state differs markedly between high-toxicity and
low-toxicity hA preparations, consistent with the dif-
ferences observed by electron microscopy. In the
high-toxicity preparation, hA was initially mostly non-
aggregated, but underwent progressive aggregation
during the first 150 min in solution (Fig. 3A). By con-
trast, the low-toxicity hA was largely aggregated at the
beginning of the experiment, and aggregation was
essentially complete within 30 min.
The results obtained with hA were paralleled by sim-
ilar time-dependent fluorescence data for high-toxicity
and low-toxicity preparations of 8)37hA (Fig. 3B).
High-toxicity 8)37hA (preparation E) displayed a ThT-
binding profile similar to that of the high-toxicity pre-
paration of full-length protein, with the exception that
the lag phase was shorter. Initial fluorescence values
were low (they comprised less than 15% of maximal
values obtained during the time course of the assay),
indicating a low level of ThT-detectable aggregates at
the beginning. In the case of low-cytotoxicity 8)37hA
(preparation F), the initial fluorescence values were at
least three-fold higher than those observed for highly
toxic protein. Moreover, they remained within a narrow
range over the duration of the experiment, and there
was no statistically significant difference between the
initial and maximal values obtained during the time
course of the assay. The paucity of de novo formation of
ThT-detectable aggregates by the low-toxicity prepar-
ation is reflected in the observation that the data were
best fitted to a one-phase exponential decay model.
In summary, the results of the ThT-binding study
indicate that (a) the initial amount of ThT-detectable
aggregates in freshly prepared aqueous solutions of
hA, and (b) the extent of subsequent de novo forma-
tion of ThT-detectable aggregates, differed markedly
between the high-toxicity and low-toxicity preparations
of full-length or truncated amylin.
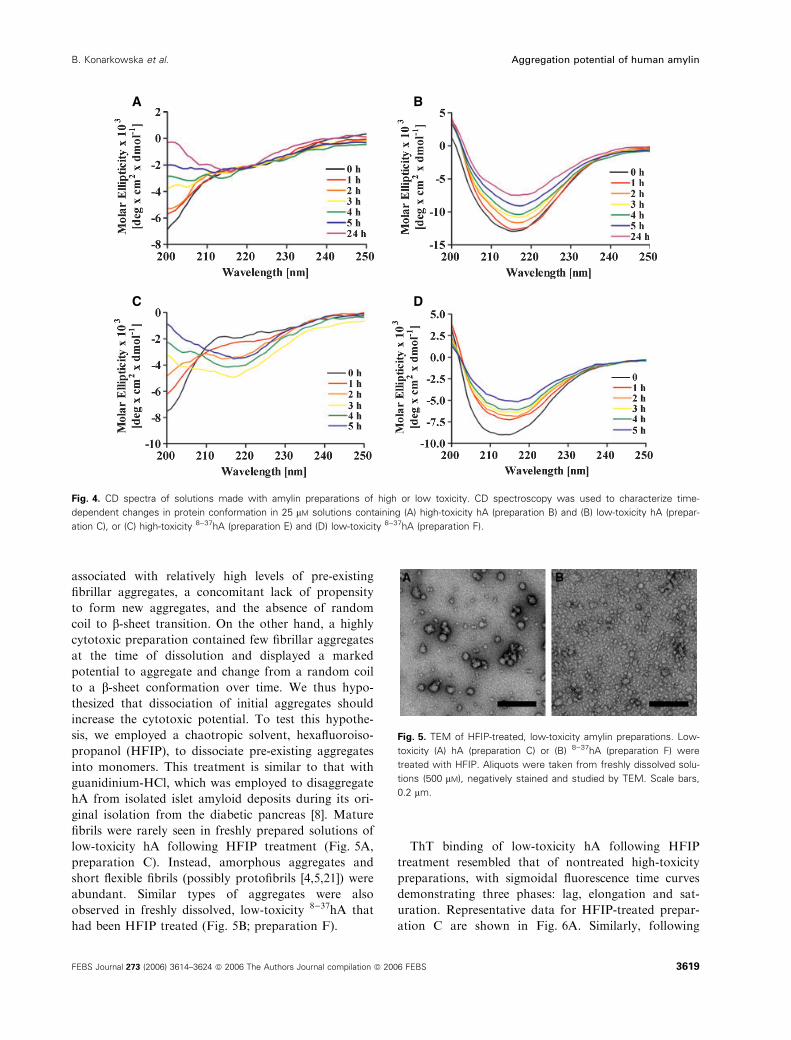
We next employed CD spectroscopy to probe the ini-
tial conformational status of hA from preparations
with different cytotoxic potential, and extended these
studies to characterize time-dependent conformational
changes of the proteins in solution. CD spectra from
the high-toxicity hA (preparation B) demonstrated a
time-dependent conversion from random coil to b-sheetconformation (Fig. 4A), as shown by a loss of signal at
205 nm and a subsequent appearance of a minimum at
217 nm (24 h), indicating the random coil to b-sheettransition. By contrast, low-toxicity hA already showed
predominantly b-sheet conformation immediately upon
dissolution in water (strong minimum at 217 nm), and
then underwent little conformational change (Fig. 4B;
preparation C).
Since the b-sheet conformer is reportedly character-
istic of hA in its fibrillar form [40], these CD data are
consistent with both the electron microscopy and ThT-
binding studies, which also showed an advanced aggre-
gation state immediately upon dissolution. Equivalent
differences of CD spectral behavior were found to exist
between preparations of 8)37hA of high (preparation
E) and low (preparation F) cytotoxic potential
(Fig. 4C,D).
The CD spectrum of rat amylin (a nonfibril-forming
control), although not shown, was similar to that of
hA (Fig. 4A) at 0 h.
Disaggregation restores cytotoxic potential to
low-toxicity hA
Based on the data presented so far, we observed
that the low cytotoxicity of an hA preparation was
Aggregation potential of human amylin B. Konarkowska et al.
3618 FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS
associated with relatively high levels of pre-existing
fibrillar aggregates, a concomitant lack of propensity
to form new aggregates, and the absence of random
coil to b-sheet transition. On the other hand, a highly
cytotoxic preparation contained few fibrillar aggregates
at the time of dissolution and displayed a marked
potential to aggregate and change from a random coil
to a b-sheet conformation over time. We thus hypo-
thesized that dissociation of initial aggregates should
increase the cytotoxic potential. To test this hypothe-
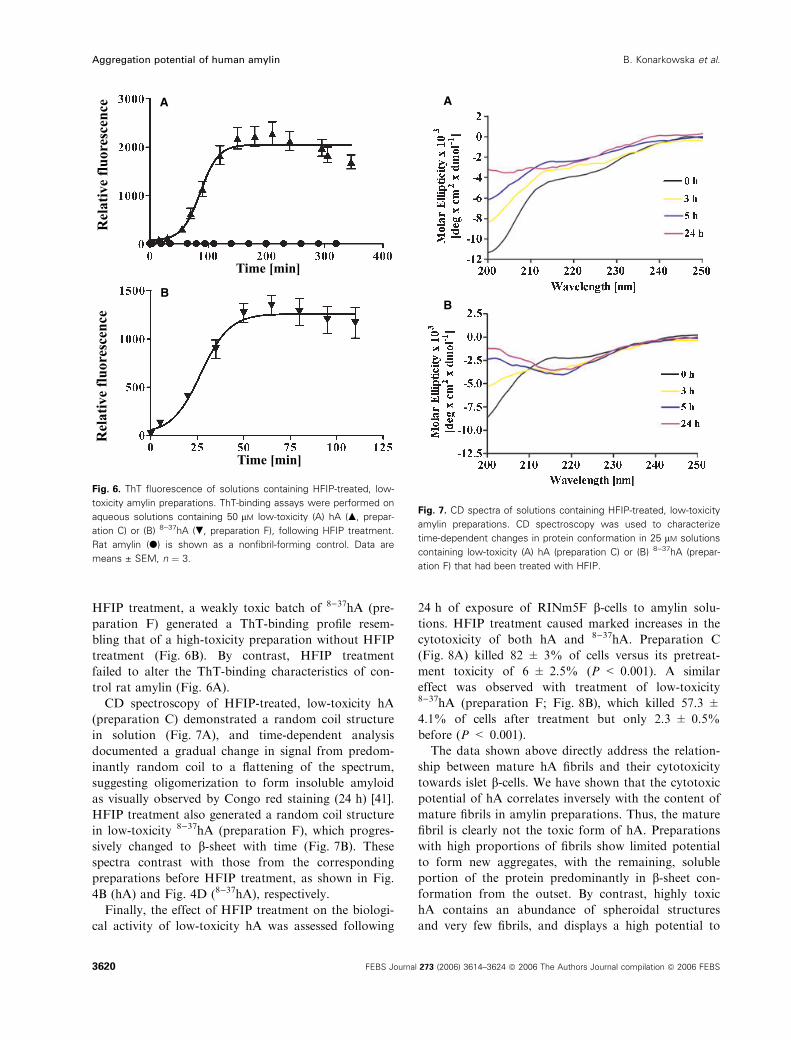
sis, we employed a chaotropic solvent, hexafluoroiso-
propanol (HFIP), to dissociate pre-existing aggregates
into monomers. This treatment is similar to that with
guanidinium-HCl, which was employed to disaggregate
hA from isolated islet amyloid deposits during its ori-
ginal isolation from the diabetic pancreas [8]. Mature
fibrils were rarely seen in freshly prepared solutions of
low-toxicity hA following HFIP treatment (Fig. 5A,
preparation C). Instead, amorphous aggregates and
short flexible fibrils (possibly protofibrils [4,5,21]) were
abundant. Similar types of aggregates were also
observed in freshly dissolved, low-toxicity 8)37hA that
had been HFIP treated (Fig. 5B; preparation F).
ThT binding of low-toxicity hA following HFIP
treatment resembled that of nontreated high-toxicity
preparations, with sigmoidal fluorescence time curves
demonstrating three phases: lag, elongation and sat-
uration. Representative data for HFIP-treated prepar-
ation C are shown in Fig. 6A. Similarly, following
A B
C D
Fig. 4. CD spectra of solutions made with amylin preparations of high or low toxicity. CD spectroscopy was used to characterize time-
dependent changes in protein conformation in 25 lM solutions containing (A) high-toxicity hA (preparation B) and (B) low-toxicity hA (prepar-
ation C), or (C) high-toxicity 8)37hA (preparation E) and (D) low-toxicity 8)37hA (preparation F).
Fig. 5. TEM of HFIP-treated, low-toxicity amylin preparations. Low-
toxicity (A) hA (preparation C) or (B) 8)37hA (preparation F) were
treated with HFIP. Aliquots were taken from freshly dissolved solu-
tions (500 lM), negatively stained and studied by TEM. Scale bars,
0.2 lm.
B. Konarkowska et al. Aggregation potential of human amylin
FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS 3619
HFIP treatment, a weakly toxic batch of 8)37hA (pre-
paration F) generated a ThT-binding profile resem-
bling that of a high-toxicity preparation without HFIP
treatment (Fig. 6B). By contrast, HFIP treatment
failed to alter the ThT-binding characteristics of con-
trol rat amylin (Fig. 6A).
CD spectroscopy of HFIP-treated, low-toxicity hA
(preparation C) demonstrated a random coil structure
in solution (Fig. 7A), and time-dependent analysis
documented a gradual change in signal from predom-
inantly random coil to a flattening of the spectrum,
suggesting oligomerization to form insoluble amyloid
as visually observed by Congo red staining (24 h) [41].
HFIP treatment also generated a random coil structure
in low-toxicity 8)37hA (preparation F), which progres-
sively changed to b-sheet with time (Fig. 7B). These
spectra contrast with those from the corresponding
preparations before HFIP treatment, as shown in Fig.
4B (hA) and Fig. 4D (8)37hA), respectively.
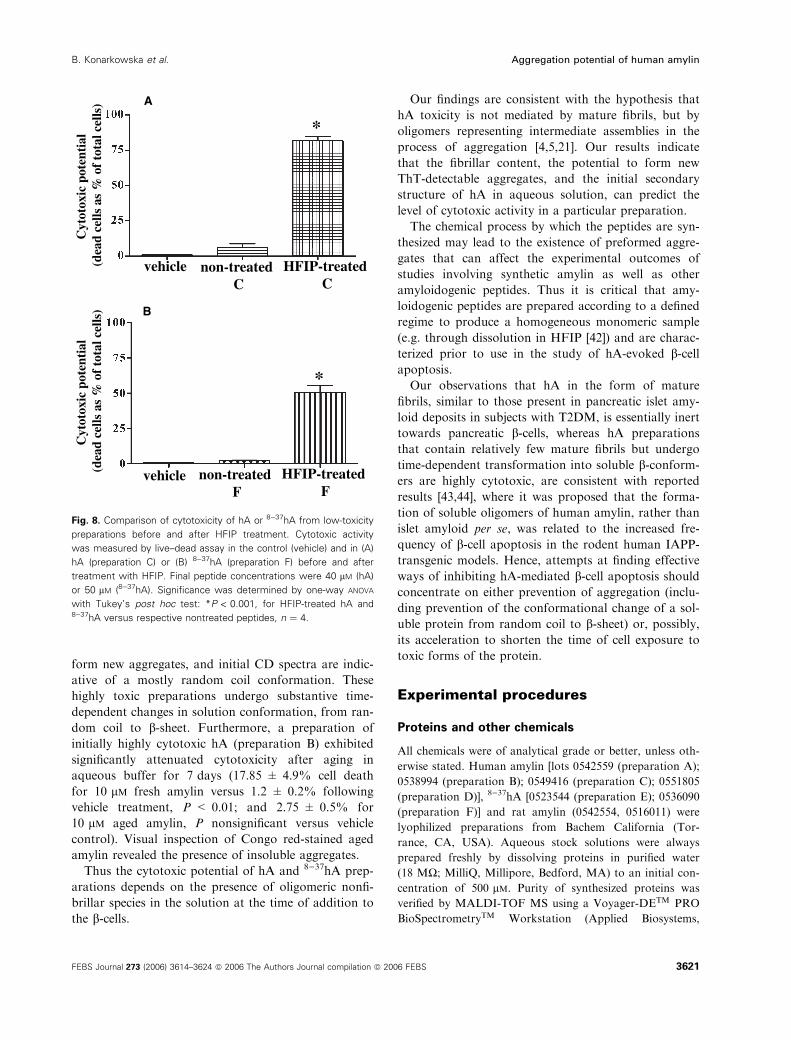
Finally, the effect of HFIP treatment on the biologi-
cal activity of low-toxicity hA was assessed following
24 h of exposure of RINm5F b-cells to amylin solu-
tions. HFIP treatment caused marked increases in the
cytotoxicity of both hA and 8)37hA. Preparation C
(Fig. 8A) killed 82 ± 3% of cells versus its pretreat-
ment toxicity of 6 ± 2.5% (P < 0.001). A similar
effect was observed with treatment of low-toxicity8)37hA (preparation F; Fig. 8B), which killed 57.3 ±
4.1% of cells after treatment but only 2.3 ± 0.5%
before (P < 0.001).
The data shown above directly address the relation-
ship between mature hA fibrils and their cytotoxicity
towards islet b-cells. We have shown that the cytotoxic
potential of hA correlates inversely with the content of
mature fibrils in amylin preparations. Thus, the mature
fibril is clearly not the toxic form of hA. Preparations
with high proportions of fibrils show limited potential
to form new aggregates, with the remaining, soluble
portion of the protein predominantly in b-sheet con-
formation from the outset. By contrast, highly toxic
hA contains an abundance of spheroidal structures
and very few fibrils, and displays a high potential to
B
ro
ulf e
vital
eR
ec
ne
cse
ro
ulf e
vital
eR
ec
ne
cse
A
Time [min]
Time [min]
Fig. 6. ThT fluorescence of solutions containing HFIP-treated, low-
toxicity amylin preparations. ThT-binding assays were performed on
aqueous solutions containing 50 lM low-toxicity (A) hA (m, prepar-
ation C) or (B) 8)37hA (., preparation F), following HFIP treatment.
Rat amylin (d) is shown as a nonfibril-forming control. Data are
means ± SEM, n ¼ 3.
B
A
Fig. 7. CD spectra of solutions containing HFIP-treated, low-toxicity
amylin preparations. CD spectroscopy was used to characterize
time-dependent changes in protein conformation in 25 lM solutions
containing low-toxicity (A) hA (preparation C) or (B) 8)37hA (prepar-
ation F) that had been treated with HFIP.
Aggregation potential of human amylin B. Konarkowska et al.
3620 FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS
form new aggregates, and initial CD spectra are indic-
ative of a mostly random coil conformation. These
highly toxic preparations undergo substantive time-
dependent changes in solution conformation, from ran-
dom coil to b-sheet. Furthermore, a preparation of
initially highly cytotoxic hA (preparation B) exhibited
significantly attenuated cytotoxicity after aging in
aqueous buffer for 7 days (17.85 ± 4.9% cell death
for 10 lm fresh amylin versus 1.2 ± 0.2% following
vehicle treatment, P < 0.01; and 2.75 ± 0.5% for
10 lm aged amylin, P nonsignificant versus vehicle
control). Visual inspection of Congo red-stained aged
amylin revealed the presence of insoluble aggregates.
Thus the cytotoxic potential of hA and 8)37hA prep-
arations depends on the presence of oligomeric nonfi-
brillar species in the solution at the time of addition to
the b-cells.
Our findings are consistent with the hypothesis that
hA toxicity is not mediated by mature fibrils, but by
oligomers representing intermediate assemblies in the
process of aggregation [4,5,21]. Our results indicate
that the fibrillar content, the potential to form new
ThT-detectable aggregates, and the initial secondary
structure of hA in aqueous solution, can predict the
level of cytotoxic activity in a particular preparation.
The chemical process by which the peptides are syn-
thesized may lead to the existence of preformed aggre-
gates that can affect the experimental outcomes of
studies involving synthetic amylin as well as other
amyloidogenic peptides. Thus it is critical that amy-
loidogenic peptides are prepared according to a defined
regime to produce a homogeneous monomeric sample
(e.g. through dissolution in HFIP [42]) and are charac-
terized prior to use in the study of hA-evoked b-cellapoptosis.
Our observations that hA in the form of mature
fibrils, similar to those present in pancreatic islet amy-
loid deposits in subjects with T2DM, is essentially inert
towards pancreatic b-cells, whereas hA preparations
that contain relatively few mature fibrils but undergo
time-dependent transformation into soluble b-conform-
ers are highly cytotoxic, are consistent with reported
results [43,44], where it was proposed that the forma-
tion of soluble oligomers of human amylin, rather than
islet amyloid per se, was related to the increased fre-
quency of b-cell apoptosis in the rodent human IAPP-
transgenic models. Hence, attempts at finding effective
ways of inhibiting hA-mediated b-cell apoptosis shouldconcentrate on either prevention of aggregation (inclu-
ding prevention of the conformational change of a sol-
uble protein from random coil to b-sheet) or, possibly,its acceleration to shorten the time of cell exposure to
toxic forms of the protein.
Experimental procedures
Proteins and other chemicals
All chemicals were of analytical grade or better, unless oth-
erwise stated. Human amylin [lots 0542559 (preparation A);
0538994 (preparation B); 0549416 (preparation C); 0551805
(preparation D)], 8)37hA [0523544 (preparation E); 0536090
(preparation F)] and rat amylin (0542554, 0516011) were
lyophilized preparations from Bachem California (Tor-
rance, CA, USA). Aqueous stock solutions were always
prepared freshly by dissolving proteins in purified water
(18 MW; MilliQ, Millipore, Bedford, MA) to an initial con-
centration of 500 lm. Purity of synthesized proteins was
verified by MALDI-TOF MS using a Voyager-DETM PRO
BioSpectrometryTM Workstation (Applied Biosystems,
laitnetop cixototyC
)sllec latot fo % sa sllec daed( vehicle non-treated
CHFIP-treated
C
A
laitnetop cixototyC
)sllec latot fo % sa sllec daed( non-treated
FHFIP-treated
F
B
*
*
vehicle
Fig. 8. Comparison of cytotoxicity of hA or 8)37hA from low-toxicity
preparations before and after HFIP treatment. Cytotoxic activity
was measured by live–dead assay in the control (vehicle) and in (A)
hA (preparation C) or (B) 8)37hA (preparation F) before and after
treatment with HFIP. Final peptide concentrations were 40 lM (hA)
or 50 lM (8)37hA). Significance was determined by one-way ANOVA
with Tukey’s post hoc test: *P < 0.001, for HFIP-treated hA and8)37hA versus respective nontreated peptides, n ¼ 4.
B. Konarkowska et al. Aggregation potential of human amylin
FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS 3621

Foster City, CA, USA) in positive-ion mode with a matrix
of saturated a-cyano-4-hydroxycinnamic acid.
Cell culture and treatments
RINm5F cells were a gift from H. K. Oie (National Institutes
of Health, Bethesda, MD, USA). Cells were cultured in
RPMI 1640 medium (Roswell Park Memorial Institute
Media 1640; Invitrogen, Carlsbad, CA, USA) supplemented
with 10% (v ⁄ v) fetal bovine serum (Invitrogen), 290 lgÆmL)1
l-glutamine, 100 IUÆmL)1 penicillin, 100 lgÆmL)1 strepto-
mycin and 2.5 mgÆmL)1 NaHCO3 at 37 �C in a humidified
incubator with 5% CO2, as previously described [15,19]. All
experiments were performed using cells between passages 28
and 39, which were plated in 24-well culture plates at a
density of 1.2 · 105 per well.
Assessment of cell viability
Cell viability following hA treatment was assessed by live–
dead assay [20,37]. After plating, cells were grown for
approximately 48 h. Following a single rinse with NaCl ⁄Pi,
fresh medium was added to each well, and aliquots of
aqueous amylin or vehicle added to a final volume of
200 lL. Following a 24 h incubation, cell viability was
determined by double-staining with calcein–acetomethoxyl
ester (AM) and ethidium homodimer-1 (EthD-1; Molecular
Probes, Eugene, OR, USA). Cells were gently rinsed with
Krebs-Ringer bicarbonate buffer (KRB) and incubated
with 1 lm calcein–AM and 4 lm EthD-1 in KRB in the
dark at 37 �C for 10 min. Green fluorescence of live cells
and red fluorescence marking dead cells were simulta-
neously visualized using a Zeiss Axiovert S100 microscope
(Zeiss filter set no. 09, Carl Zeiss International, Oberko-
chen, Germany). Live and dead cells were counted in two
to four fields per well (two wells per experiment) and pho-
tographed at 400· magnification using a digital camera
(Zeiss Axiocam). Statistical analysis of data was by one-
way ANOVA with Tukey’s post hoc test, and significance
was determined at P < 0.05.
HFIP treatment
Lyophilized amylin aliquots were weighed into microcentri-
fuge tubes, and HFIP added to a final protein concentra-
tion of 250 lm. Lids were sealed (Parafilm; American Can
Co., Greenwich, CT, USA) and protein dissolution was
aided by gentle agitation, with recovery of contents by
pulse-spinning. Tubes were stored at room temperature
(approximately 22 �C) in the dark for 5–24 h. Contents
were mixed and again pulse-spun. HFIP was removed by
evaporation under N2, leaving a thin transparent film of
peptide on the internal surface of the tube, which was
then dissolved in sterile water by trituration followed by
pulse-spinning. HFIP-treated amylin was dissolved in water
to a final concentration of 500 lm. Vehicle controls were
constructed exactly as described above, but with the omis-
sion of protein.
TEM
Aliquots (5 lL) of freshly prepared aqueous amylin
(500 lm) were adsorbed onto glow-discharged carbon-coa-
ted collodion film on 400-mesh copper grids for 30 s. Next,
grids were blotted, washed three times in water droplets,
negatively stained for 20 s with 2% (w ⁄ v) uranyl acetate,
reblotted and left to air-dry inside a glass Petri dish [40].
Grids were examined within 1–2 days following their pre-
paration, using a Tecnai Transmission Electron Microscope
(FEI, Hillsboro, OR, USA) operated at 120 kV. Images
were recorded at a nominal magnification of 6 · 104 using
a Bioscan digital camera (Gatan, Pleasanton, CA, USA).
All experiments were performed in duplicate or triplicate,
and the images shown are representative of at least six pho-
tomicrographs.
ThT-binding assay
The aggregation potential of amylin was assessed using the
ThT-binding assay. Experiments were performed in tripli-
cate using black plastic microtiter plates (Nunc, Roskilde,
Denmark). Assays were initiated by adding 89 lL of
10 mm Tris ⁄HCl, pH 7.2, buffer and 1 lL of a 1 mm aque-
ous solution of ThT per well. Next, an aliquot of a fresh
aqueous solution of amylin or vehicle was added to each
well to a final volume of 100 lL, giving final concentrations
of amylin and ThT of 50 lm and 10 lm, respectively.
Fluorescence was measured at 510 nm with an excitation of
450 nm and a cutoff filter at 495 nm using a Spectra MAX
Gemini XS fluorescence spectrophotometer (Molecular
Devices, Sunnyvale, CA, USA). Fluorescence measurements
were performed immediately after addition of amylin, and
subsequent measurements were made at intervals over the
next 2–6 h. Data analysis was performed by nonlinear
regression using graphpad prism v.4.00 for Windows
(GraphPad Software Inc., San Diego, CA, USA).
CD spectroscopy
Samples were prepared by diluting freshly prepared aque-
ous amylin (500 lm) into 10 mm Tris ⁄HCl, pH 7.2, buffer,
yielding a final protein concentration of 25 lm. Blanks wereprepared by adding water (vehicle) instead of amylin to the
buffer. Spectra were recorded at room temperature (p*-180Spectrometer; Applied Photophysics, Leatherhead, UK),
using a 1 mm pathlength quartz cuvette with 250 lLsample aliquots, at 1.0-nm intervals between k ¼ 200 and
250 nm, with automatic subtraction of buffer-blank spectra.
Aggregation potential of human amylin B. Konarkowska et al.
3622 FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS

For presentation of the results, the data were expressed
as molar ellipticity (h). Interpretation was performed
according to generally accepted guidelines for the prediction
of protein secondary structure from CD spectra [40].
Acknowledgements
We thank J. D. Green, K. M. Loomes, J. Z. Bai, A.
Turner, M. S. Cameron-Cooper and C. A. Tse for dis-
cussions and criticism.
References
1 Moore CX & Cooper GJ (1991) Co-secretion of amylin
and insulin from cultured islet b-cells: modulation by
nutrient secretagogues, islet hormones and hypoglycemic
agents. Biochem Biophys Res Commun 179, 1–9.
2 Cooper GJ (1994) Amylin compared with calcitonin
gene-related peptide: structure, biology, and relevance
to metabolic disease. Endocr Rev 15, 163–201.
3 Westermark P, Wernstedt C, Wilander E, Hayden DW,
O’Brien TD & Johnson KH (1987) Amyloid fibrils in
human insulinoma and islets of Langerhans of the dia-
betic cat are derived from a neuropeptide-like protein
also present in normal islet cells. Proc Natl Acad Sci
USA 84, 3881–3885.
4 Goldsbury CS, Cooper GJ, Goldie KN, Muller SA,
Saafi EL, Gruijters WT, Misur MP, Engel A, Aebi U &
Kistler J (1997) Polymorphic fibrillar assembly of
human amylin. J Struct Biol 119, 17–27.
5 Goldsbury C, Kistler J, Aebi U, Arvinte T & Cooper
GJ (1999) Watching amyloid fibrils grow by time-lapse
atomic force microscopy. J Mol Biol 285, 33–39.
6 Green J, Goldsbury C, Mini T, Sunderji S, Frey P, Kis-
tler J, Cooper G & Aebi U (2003) Full-length rat amylin
forms fibrils following substitution of single residues
from human amylin. J Mol Biol 326, 1147–1156.
7 Bell ET (1959) Hyalinization of the islet of Langerhans
in non-diabetic individuals. Am J Pathol 35, 801–805.
8 Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB &
Reid KB (1987) Purification and characterization of a
peptide from amyloid-rich pancreases of type 2 diabetic
patients. Proc Natl Acad Sci USA 84, 8628–8632.
9 Hoppener JW, Ahren B & Lips CJ (2000) Islet amyloid
and type 2 diabetes mellitus. N Engl J Med 343, 411–
419.
10 Clark A, Cooper GJ, Lewis CE, Morris JF, Willis AC,
Reid KB & Turner RC (1987) Islet amyloid formed
from diabetes-associated peptide may be pathogenic in
type-2 diabetes. Lancet 2, 231–234.
11 Lorenzo A, Razzaboni B, Weir GC & Yankner BA
(1994) Pancreatic islet cell toxicity of amylin associated
with type-2 diabetes mellitus. Nature (Lond) 368, 756–
760.
12 Opie EL (1901) The relation of diabetes mellitus to
lesions of the pancreas. Hyaline degeneration of the
islands of Langerhans. J Exp Med 5, 527–540.
13 Ehrlich JC & Ratner IM (1961) Amyloidosis of the
islets of Langerhans. Am J Pathol 38, 49–59.
14 de Koning EJ, Bodkin NL, Hansen BC & Clark A
(1993) Diabetes mellitus in Macaca mulatta monkeys is
characterised by islet amyloidosis and reduction in beta-
cell population. Diabetologia 36, 378–384.
15 Bai JZ, Saafi EL, Zhang S & Cooper GJ (1999) Role of
Ca2+ in apoptosis evoked by human amylin in pancre-
atic islet b-cells. Biochem J 343, 53–61.
16 Zhang S, Liu J, MacGibbon G, Dragunow M &
Cooper GJ (2002) Increased expression and activation
of c-Jun contributes to human amylin-induced apoptosis
in pancreatic islet b-cells. J Mol Biol 324, 271–285.
17 Zhang S, Liu J, Dragunow M & Cooper GJ (2003)
Fibrillogenic amylin evokes islet b-cell apoptosisthrough linked activation of a caspase cascade and
JNK1. J Biol Chem 278, 52810–52819.
18 Janson J, Soeller WC, Roche PC, Nelson RT, Torchia
AJ, Kreutter DK & Butler PC (1996) Spontaneous dia-
betes mellitus in transgenic mice expressing human islet
amyloid polypeptide. Proc Natl Acad Sci USA 93,
7283–7288.
19 Zhang S, Liu J, Saafi EL & Cooper GJ (1999) Induction
of apoptosis by human amylin in RINm5F islet b-cellsis associated with enhanced expression of p53 and
p21WAF1 ⁄ CIP1. FEBS Lett 455, 315–320.
20 Aitken JF, Loomes KM, Konarkowska B & Cooper GJ
(2003) Suppression by polycyclic compounds of the
conversion of human amylin into insoluble amyloid.
Biochem J 374, 779–784.
21 Green JD, Goldsbury C, Kistler J, Cooper GJ & Aebi
U (2004) Human amylin oligomer growth and fibril
elongation define two distinct phases in amyloid forma-
tion. J Biol Chem 279, 12206–12212.
22 Jaikaran ET & Clark A (2001) Islet amyloid and type 2
diabetes: from molecular misfolding to islet pathophy-
siology. Biochim Biophys Acta 1537, 179–203.
23 Janciauskiene S & Ahren B (1998) Different sensitivity
to the cytotoxic action of IAPP fibrils in two insulin-
producing cell lines, HIT-T15 and RINm5F cells.
Biochem Biophys Res Commun 251, 888–893.
24 Janson J, Ashley RH, Harrison D, McIntyre S & Butler
PC (1999) The mechanism of islet amyloid polypeptide
toxicity is membrane disruption by intermediate-sized
toxic amyloid particles. Diabetes 48, 491–498.
25 Ritzel RA & Butler PC (2003) Replication increases
b-cell vulnerability to human islet amyloid polypeptide-
induced apoptosis. Diabetes 52, 1701–1708.
26 Hardy J & Selkoe DJ (2002) The amyloid hypothesis of
Alzheimer’s disease: progress and problems on the road
to therapeutics. Science 297, 353–356.
B. Konarkowska et al. Aggregation potential of human amylin
FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS 3623

27 Lambert MP, Barlow AK, Chromy BA, Edwards C,
Freed R, Liosatos M, Morgan TE, Rozovsky I,
Trommer B, Viola KL et al. (1998) Diffusible, nonfibril-
lar ligands derived from Abeta1-42 are potent central
nervous system neurotoxins. Proc Natl Acad Sci USA
95, 6448–6453.
28 Goldberg MS & Lansbury PT Jr (2000) Is there a
cause-and-effect relationship between alpha-synuclein
fibrillization and Parkinson’s disease? Nat Cell Biol 2,
E115–E119.
29 Sousa MM, Cardoso I, Fernandes R, Guimaraes A &
Saraiva MJ (2001) Deposition of transthyretin in early
stages of familial amyloidotic polyneuropathy: evidence
for toxicity of nonfibrillar aggregates. Am J Pathol 159,
1993–2000.
30 Lashuel HA, Hartley D, Petre BM, Walz T & Lansbury
PT Jr (2002) Neurodegenerative disease: amyloid pores
from pathogenic mutations. Nature 418, 291.
31 Kayed R, Head E, Thompson JL, McIntire TM, Milton
SC, Cotman CW & Glabe CG (2003) Common struc-
ture of soluble amyloid oligomers implies common
mechanism of pathogenesis. Science 300, 486–489.
32 Kayed RK, Sokolov Y, Edmonds B, McIntire TM, Mil-
ton SC, Hall JE & Glabe CG (2004) Permeabilization
of lipid bilayers is a common conformation-dependent
activity of soluble amyloid oligomers in protein misfold-
ing diseases. J Biol Chem 279, 46363–46366.
33 Bucciantini M, Calloni G, Chiti F, Formigli L, Nosi D,
Dobson CM & Stefani M (2004) Prefibrillar amyloid
protein aggregates share common features of cytotoxi-
city. J Biol Chem 279, 31374–31382.
34 Kayed R, Bernhagen J, Greenfield N, Sweimeh K,
Brunner H, Voelter W & Kapurniotu A (1999) Confor-
mational transitions of islet amyloid polypeptide (IAPP)
in amyloid formation in vitro. J Mol Biol 287, 781–796.
35 Tenidis K, Waldner M, Bernhagen J, Fischle W, Berg-
mann M, Weber M, Merkle M, Voelter W, Brunner H
& Kapurniotu A (2000) Identification of a penta- and
hexapeptide of islet amyloid polypeptide (IAPP) with
amyloidogenic and cytotoxic properties. J Mol Biol 295,
1055–1071.
36 Wogulis M, Wright S, Cunningham D, Chilcote T,
Powell K & Rydel R (2005) Nucleation-dependent
polymerization is an essential component of amyloid-
mediated neuronal cell death. J Neurosci 25, 1071–1080.
37 Saafi EL, Konarkowska B, Zhang S, Kistler J & Cooper
GJ (2001) Ultrastructural evidence that apoptosis is the
mechanism by which human amylin evokes death in
RINm5F pancreatic islet b-cells. Cell Biol Int 25, 339–350.
38 Hettiarachchi M, Chalkley S, Furler SM, Choong YS,
Heller M, Cooper GJ & Kraegen EW (1997) Rat amy-
lin-(8–37) enhances insulin action and alters lipid meta-
bolism in normal and insulin-resistant rats. Am J
Physiol 273, E859–E867.
39 Konarkowska B, Aitken J, Kistler J, Zhang S &
Cooper G (2005) Thiol reducing compounds prevent
human amylin-evoked cytotoxicity. FEBS J 272,
4949–4959.
40 Goldsbury C, Goldie K, Pellaud J, Seelig J, Frey P,
Muller SA, Kistler J, Cooper GJ & Aebi U (2000)
Amyloid fibril formation from full-length and
fragments of amylin. J Struct Biol 130, 352–362.
41 Kapurniotu A, Bernhagen J, Greenfield N, Al-Abed Y,
Teichberg S, Frank RW, Voelter W & Bucala R (1998)
Contribution of advanced glycosylation to the amyloi-
dogenicity of islet amyloid polypeptide. Eur J Biochem
251, 208–216.
42 Higham CE, Jaikaran ET, Fraser PE, Gross M & Clark
A (2000) Preparation of synthetic human islet amyloid
polypeptide (IAPP) in a stable conformation to enable
study of conversion to amyloid-like fibrils. FEBS Lett
470, 55–60.
43 Butler AEJJ, Soeller WC & Butler PC (2003)
Increased beta-cell apoptosis prevents adaptive
increase in beta-cell mass in mouse model of type 2
diabetes: evidence for role of islet amyloid formation
rather than direct action of amyloid. Diabetes 52,
2304–2314.
44 Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC &
Butler PC (2004) Diabetes due to a progressive defect in
beta-cell mass in rats transgenic for human islet amyloid
polypeptide (HIP Rat): a new model for type 2 diabetes.
Diabetes 53, 1509–1516.
Aggregation potential of human amylin B. Konarkowska et al.
3624 FEBS Journal 273 (2006) 3614–3624 ª 2006 The Authors Journal compilation ª 2006 FEBS





























