Embed Size (px)
Citation preview

The Assessment of Bioequivalence - AStatistical Overview
Thomas Mathew
Department of Mathematics and StatisticsUniversity of Maryland Baltimore County (UMBC)
Baltimore, Maryland 21250

Outline
I Background
I History
I Data for bioequivalence testing
I Experimental designs
I Bioequivalence criteria
I Biosimilars
2 / 52

In an equivalence trial, the aim is to show that two treatments arenot too different in characteristics
Not too different is defined in a clinical manner.
3 / 52

Background
New drug: approved by the Food and Drug Administration (FDA)
Complicated approval process
Company has exclusive rights to sell the brand name drug for anumber of years (patent period)
Brand name drug could be expensive
Once the patent runs out, others can copy the brand name drug
Generic drug: a copy of the brand name drug
Approval process is simple for generic drugs
4 / 52

Enough to show that a generic drug is equivalent to a brand namedrug
Show that the concentration of the active ingredient that entersthe blood is similar for the generic drug and the brand name drug
Called bioequivalence testing
Generic drugs: much cheaper
Bioequivalence testing can also be used to compare different formsof the same drug: tablet, capsule, solution, powder, intra-vascularetc.
5 / 52

Lipitor: Cholesterol lowering medication by Pfizer.
Sales for 2009: $11.4 billion
Canadian and Spanish patents expired in 2010
Sales for 2010: $10.7 billion
Was the top selling drug, worldwide
U.S patent expired on November 30, 2011
Generic versions by several companies
Sales of lipitor plummeted in 2012
www.lipitor.com
6 / 52

Abilify: Drug for treating schizophrenia, from OtzukaPharmaceuticals
Approved by the FDA on November 15, 2002
Approved by the European Medicines Agency on June 4, 2004
Approved by the FDA to treat irritability in children with autismon November 20, 2009.
In 2013, Abilify was the top selling drug in USA, generating$6.3 billion
Patent will expire on April 20, 2015
7 / 52

Biological availability, or bioavailability of a drug: the rate andextent to which the active drug ingredient is absorbed into theblood, and becomes available at the site of drug action.
Two drug products are bioequivalent if they have similar rate andextent of absorption into the blood.
Two drug products are therapeutically equivalent if they providesimilar therapeutic effects.
Fundamental bioequivalence assumption: If two drug productsare bioequivalent, they are also therapeutically equivalent.
8 / 52

History
During the early 1970’s, the U.S. Congress passed the Drug PriceCompetition and Patent Term Restoration Act that authorized theFDA to approve generic drug products.
“The Hatch-Waxman Act” (1984)
The American Statistical Association then formed a BioavailabilityCommittee to establish statistical procedures for establishingbioequivalence.
Under the Drug Price Competition and Patent Term RestorationAct, the FDA was authorized to approve generic drug products in1984.
9 / 52

A Bioequivalence Task Force was formed in 1986 to examine theFDA’s procedures for approving generic drug products.
The Task Force released their report in January 1988, and severalstatistical issues were pointed out.
In 1992, the FDA issued the guidance on statistical procedures forestablishing bioequivalence.
A revised guidance document was issued in 2001.
New Drug Application (NDA)
Abbreviated New Drug Application (ANDA)
10 / 52

Generic drug applications are termed abbreviated because theyare generally not required to include preclinical (animal) andclinical (human) data to establish safety and effectiveness.
Instead, generic applicants must scientifically demonstrate thattheir product is bioequivalent (i.e., performs in a similar manner asthe original drug).
11 / 52

Data for bioequivalence testing
Mysoline: a brand name drug used for treating epilepsy
Primidone: the active ingredient in Mysoline
(www.epilepsyfoundation.org)
A subject receives 250-mg tablet of primidone
Measure the concentration of primidone (micrograms/ml) in theblood several times, over a 32 hour period
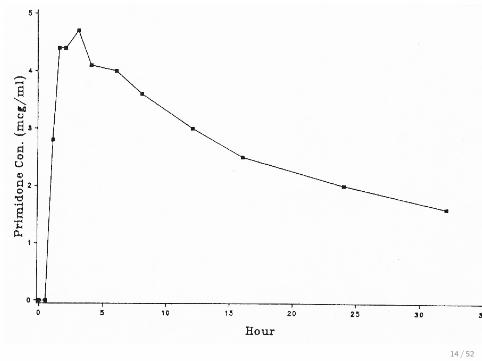
Plot the data against time
12 / 52
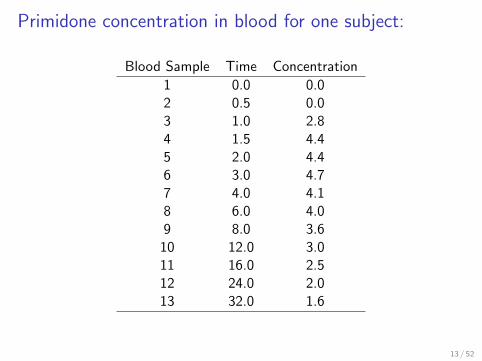
Primidone concentration in blood for one subject:
Blood Sample Time Concentration
1 0.0 0.02 0.5 0.03 1.0 2.84 1.5 4.45 2.0 4.46 3.0 4.77 4.0 4.18 6.0 4.09 8.0 3.610 12.0 3.011 16.0 2.512 24.0 2.013 32.0 1.6
13 / 52
14 / 52

Data obtained from healthy volunteers (male and female)
18-55 years old, BMI = 18-25 kg/m2
Non-smokers/without a history of alcohol or drug abuse
Medical history/Clinical Lab test values must be within normalranges
Other criteria .....
If two drug products perform the same in healthy volunteers, theassumption is made that they will perform the same in patientswith the disease
15 / 52

Data obtained on three variables:
Area under the curve (AUC)
Maximum blood concentration (Cmax)
Time to reach the maximum concentration (Tmax)
AUC: extent of bioavailability
Cmax: rate of bioavailability
Statistical analysis of the data is done to determine if the twodrugs are equivalent.
AUC and Cmax: lognormally distributed.
Is it enough to reduce the data to just three measures?
Pharmacokinetics
16 / 52

Experimental designs
Mysoline: reference drug (R)
Generic drug: test drug (T )
Each subject receives both R and T , separated by a washoutperiod
Crossover designs are used
A two sequence−two period crossover design:
Period
Sequence I II
1 R T2 T R
17 / 52

A two sequence−four period crossover design:
Period
Sequence I II III IV
1 T T R R2 R R T T
A four sequence−four period crossover design:
Period
Sequence I II III IV
1 T T R R2 R R T T3 T R R T4 R T T R
18 / 52

Univariate bioequivalence testing: Based on the separatemodeling and analysis of the univariate responses AUC, Cmax andTmax.
Multivariate bioequivalence testing: Based on the jointmodeling and analysis of all the three responses AUC, Cmax andTmax, or any two of them (typically, AUC and Cmax).
Usually log-transformed data are analyzed
19 / 52

Bioequivalence criteria
Average bioequivalence
µT , µR : average responses among the population of patients whowill take the test drug, and the reference drug, respectively.
The response is usually AUC, after log-transformation (could beCmax or Tmax).
Average bioequivalence holds if µT and µR are equivalent, i.e.,they are “close”
Equivalence is not the same as equality.
20 / 52

µT and µR are considered equivalent if |µT − µR | < ln(1.25).
After obtaining data from a crossover design, perform a statisticaltest to decide if |µT − µR | ≥ ln(1.25) or if |µT − µR | < ln(1.25)
Hypothesis to be tested
H0 : |µT − µR | ≥ ln(1.25) versus H1 : |µT − µR | < ln(1.25)
Conclude average bioequivalence if H0 is rejected after a statisticaltest based on the log-transformed AUC data.
21 / 52

A canonical form
Under an appropriate model for the log-transformed data, acanonical form is
D ∼ N(µT − µR , c2σ2)
vS2
σ2∼ χ2
v
22 / 52

Testing average bioequivalence
D ∼ N(µT − µR , c2σ2), v
S2
σ2∼ χ2
v
H0 : |µT − µR | ≥ ln(1.25), vs H1 : |µT − µR | < ln(1.25).
Rewrite as
H01 : µT − µR ≤ − ln(1.25) vs. H11 : µT − µR > − ln(1.25),
H02 : µT − µR ≥ ln(1.25) vs. H12 : µT − µR < ln(1.25).
Average bioequivalence is concluded if both H01 and H02 arerejected.
23 / 52

Carry out t-tests: conclude average bioequivalence at significancelevel α if
D + ln(1.25)
cS> tv (α) and
D − ln(1.25)
cS< −tv (α)
Equivalently, if
|D| − ln(1.25)
cS< −tv (α)
Two one-sided t-test (TOST)
Schuirmann (1981), Biometrics
Schuirmann (1987), Journal of Pharmacokinetics andBiopharmaceutics.
The TOST is equivalent to computing a 90% confidence intervalfor µT − µR , and verifying if the interval is contained in(− ln(1.25), ln(1.25)).
24 / 52
0.0 0.1 0.2 0.3 0.4
0.00
0.01
0.02
0.03
0.04
0.05
The type I error probability of the TOST
σ0
type
I er
ror

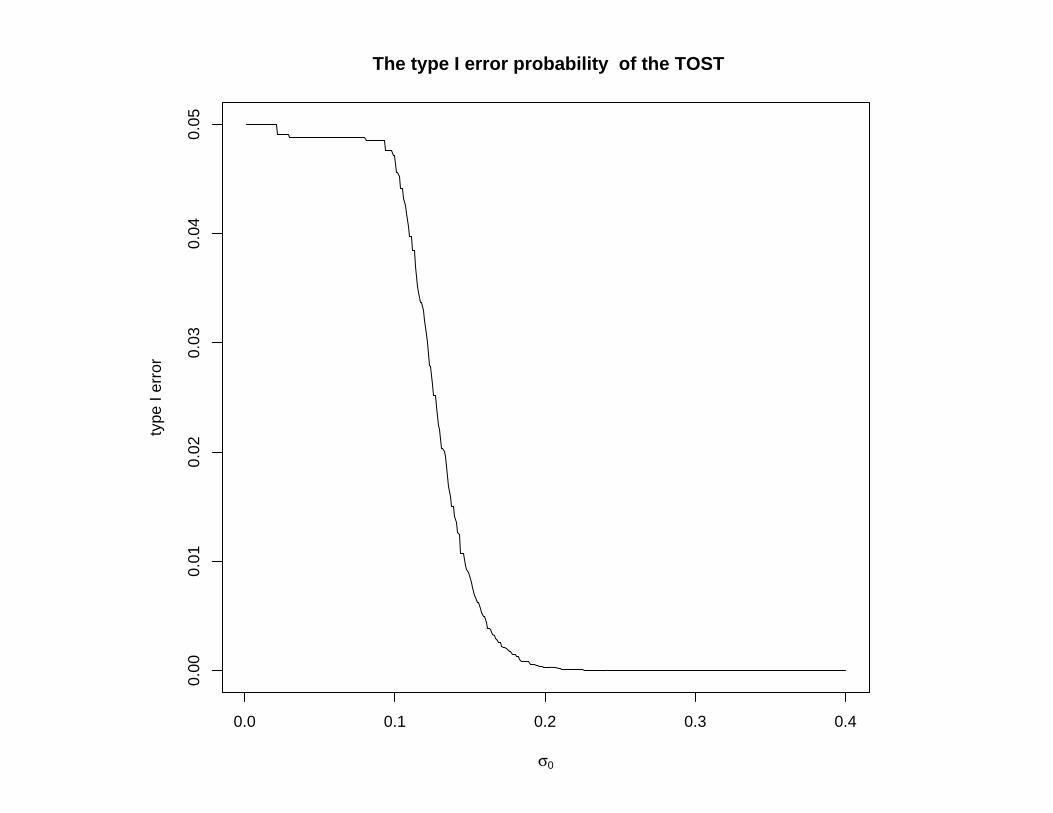
The TOST can be quite conservative as σ gets large
Improved tests due to:
Anderson and Hauck (1983), Communications in Statistics
Munk (1993), Biometrics
Berger and Hsu (1996), Statistical Science
Brown, Hwang and Munk (1997), Annals of Statistics
Munk, Brown and Hwang (2000), Biometrical Journal
Cao and Mathew (2008), Biometrical Journal
Very often, the σ value is not that large.
In cases where it is large, the usual average bioequivalencecriterion, and the TOST are not used.
26 / 52

Highly variable drug products
If the variability is too large, the usual average bioequivalencecriterion is not used in practice
Highly variable drug products: Within subject coefficient ofvariation is at least 0.3
Within subject variance is at least 0.3
27 / 52
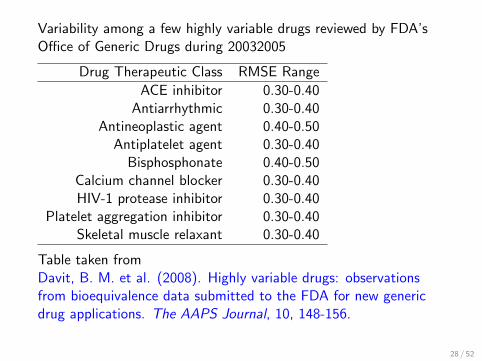
Variability among a few highly variable drugs reviewed by FDA’sOffice of Generic Drugs during 20032005
Drug Therapeutic Class RMSE Range
ACE inhibitor 0.30-0.40Antiarrhythmic 0.30-0.40
Antineoplastic agent 0.40-0.50Antiplatelet agent 0.30-0.40Bisphosphonate 0.40-0.50
Calcium channel blocker 0.30-0.40HIV-1 protease inhibitor 0.30-0.40
Platelet aggregation inhibitor 0.30-0.40Skeletal muscle relaxant 0.30-0.40
Table taken fromDavit, B. M. et al. (2008). Highly variable drugs: observationsfrom bioequivalence data submitted to the FDA for new genericdrug applications. The AAPS Journal, 10, 148-156.
28 / 52

For highly variable drug products, use a Scaled AverageBioequivalence criterion
Instead of the parameter µT − µR , considerµT−µRσWR
Instead of ± ln(1.25), use the limits ± ln(1.25) σ̂WRσW 0
σWR : Within subject variance among those who take the referencedrug
σW 0: a specified constant (σW 0 = 0.25 is a standard choice).
29 / 52

Other concepts of bioequivalance:
Variance bioequivalence (Test if a ratio of variances is around one)
Subject-by-formulation interaction
Population bioequivalence (Drug prescribability)
Individual bioequivalence (Drug switchability)
Probability based criteria
30 / 52

References
Chow, S. C. and Liu, J. P. (2010). Design and Analysis ofBioavailability and Bioequivalence Studies, Third Edition,Chapman and Hall/CRC Press.
Hauschke, H., Steinijans, V. and Pigeot, I. (2007). BioequivalenceStudies in Drug Development: Methods and Applications. Wiley,New York.
Patterson, S. D. and Jones, B. (2006). Bioequivalence and Statisticsin Clinical Pharmacology, Chapman and Hall/CRC Press.
Wellek, S. (2010). Testing Statistical Hypotheses of Equivalence,Second Edition, Chapman and Hall/CRC Press.
31 / 52

Jones, B. and Kenward, M. G. (2003). Design and Analysis ofCross-Over Trials, Second Edition, Chapman and Hall/CRC Press.
Senn, S. (2002). Cross-Over Trials in Clinical Research, SecondEdition, John Wiley.
Journal of Bioequivalence and Bioavailability (online open accessjournal)
32 / 52

FDA guidance documents: www.fda.gov/cder/guidance
FDA Guidance (2001). Guidance for Industry: StatisticalApproaches to Establishing Bioequivalence(www.fda.gov/cder/guidance/3616fnl.htm)
FDA Guidance (2003a). Guidance for Industry: Bioavailability andBioequivalence Studies for Orally Administered Drug Products:General Considerations (www.fda.gov/cder/guidance/5356fnl.htm)
FDA Guidance (2003b). Guidance for Industry: Bioavailability andBioequivalence Studies for Nasal Aerosols and Nasal Sprays forLocal Action (www.fda.gov/cder/guidance/5383DFT.htm)
Data and examples: www.fda.gov/cder/bioequivdata
33 / 52

Biosimilars
A new area of research
Unlike chemical drugs, biological products or medicines(biotechnology drugs or biologics) are therapeutic agents that areproduced using a living system or organism.
Generic drugs are manufactured by chemical synthesis. Biologicsare made of living cells or organisms
Biosimilar: A generic version of a biotechnology drug.
Challenging to establish equivalence or similarity
34 / 52

Unlike the more common chemical drugs, biotechnology drugsgenerally exhibit high molecular complexity.
May be very sensitive to small changes during the manufacturingprocess, which will have an impact on the clinical outcome
The manufacturer of a generic copy does not have access to theoriginator’s molecular clone and original cell bank, nor to the exactfermentation and purification process, nor to the active drugsubstance.
They do have access to the commercialized brand name product.
35 / 52

Differences in impurities and/or breakdown products can haveserious health implications.
Because no two cell lines, developed independently, can beconsidered identical, biotech medicines cannot be fully copied
This has created a concern that copies might perform differentlythan the original branded version of the product.
36 / 52

The first biosimilar product approved by the FDA was the humangrowth hormone Omnitrope
Approval was granted to Sandoz in 2006
Based on an ad hoc review under the Hatch-Waxman Act
FDA stressed that Omnitrope is similar, but not equivalent, toPfizer’s Genotropin
37 / 52

Different terms are used:
European Medicines Agency: Biosimilar
US FDA: Folow-on biologic (FOB)
WHO: Similar biotherapeutic product (SBP)
Health Canada: Subsequent-entered biologics (SEB)
38 / 52

The Biologics Price Competition and Innovation Act of 2009(BPCI Act) was originally sponsored and introduced in 2007 bySenator Kennedy
It was formally passed under the Patient Protection and AffordableCare Act, signed into law by President Obama on March 23, 2010.
The BPCI act is similar to the Hatch-Waxman act, and it createsan abbreviated approval pathway for biosimilars.
39 / 52

The BPCI Act defines a biosimilar product as a biological productthat is highly similar to the reference drug product.
BPCI Act does not explain what is meant by highly similar
The Act does not provide any criteria for assessing biosimilarity
Check similarity of means and similarity of variances?
Separately, or jointly?
Can think of different criteria
40 / 52

The European Medicines Agency (EMEA) has approved biosimilarversions of several drugs
The EMEA has reached the conclusion that there is no standardprocedure that can be applied in order to approve a biosimilar
41 / 52

EMEA Guidelines
The guidelines are product-specific, and stop short of providingprecise equivalence margins
Meeting the standard of clinical similarity to the innovator biologicproduct requires clinical studies that include on the order of 200 to500 subjects, depending on the product in question.
The original product is normally studied in several thousandsubjects in order to secure their range of clinical indications.
There is a considerable reduction in the clinical testing requiredbefore approval of a biosimilar.
42 / 52

Biosimilars should be subject to an approval process which requiressubstantial additional data compared to that required for chemicalgenerics, although not as comprehensive as for the original biotechmedicine.
If comparison is made based on the concept of averagebioequivalence only, the criterion does not take into considerationthe variability, which is known to have a significant impact onclinical performance of biosimilars.
“...... biosimilars have individual characteristics that will needspecific regulatory requirements” (Janet Woodcock, head of FDA’sCenter for Drug Evaluation and Research, May 9, 2011)
43 / 52

The U.S. FDA released three draft guidances on February 9, 2012,related to the establishment of biosimilarity
(1) Scientific considerations in demonstrating biosimilarity to areference product
(2) Quality considerations in demonstrating biosimilarity to areference protein product
(3) Biosimilars: questions and answeres regarding implementationof the BPCI Act of 2009
The guidance documents provide guidelines to sponsors interestedin developing biosimilar products
44 / 52

The documents do not mention any criteria for assessingbiosimilarity
A statistical scientific advisory board (SSAB) was established andsponsored by Amgen in 2009, to look into statistical issues for theassessment of biosimilarity
SSAB members are
Shein-Chung Chow (Duke University School of Medicine)
Laszlo Endrenyi (University of Toronto)
Peter A. Lachenbruch (Oregon State University)
Moment based and probability based criteria
45 / 52

The worldwide market for copies of biotech medicines will grow to$3.7 billion by 2015 from just $243 million in 2010, according to arecent report from market analysis firm Datamonitor
New problems and challenges in the development of criteria, andthe statistical analysis of the data.
46 / 52

References on biosimilars
Chow, S.-C. (2014). Biosimilars: Design and Analysis of Follow-onBiologics, Taylor & Francis/CRC Press.
Journal of Biopharmaceutical Statistics, January 2010, Specialissue on biosimilars
Statistics in Medicine, February 2013, Special issue on biosimilars
Biosimilars, open access journal devoted to research on biosimilars(since 2011).
47 / 52

http://www.emea.europa.eu
European Medicines Agency. (2005). Guideline on SimilarBiological Medicinal Products (CHMP/437/04). EMEA, London,30 October.
European Medicines Agency. (2006). Guideline on SimilarBiological Medicinal Products Containing Biotechnology-DerivedProteins as Active Substance: Non-Clinical and Clinical Issues
European Medicines Agency. (2009). Guideline on non-clinical andclinical development of similar biological medicinal productscontaining low-molecular-weight-heparins.EMEA/CHMP/BMWP/118264/2007. London, 19 March.
48 / 52

FDA Guidance (2012) guidance documents (www.fda.gov)
(1) Scientific considerations in demonstrating biosimilarity to areference product
(2) Quality considerations in demonstrating biosimilarity to areference protein product
(3) Biosimilars: questions and answers regarding implementation ofthe BPCI Act of 2009
49 / 52

www.biosimilars.com: Site with substantive information aboutbiosimilars
Biotechnology Information Institute (www.bioinfo.com)
Generics and Biosimilars Initiative Online (www.gabionline.net)
50 / 52

Senegal to host new company for Generic Drugs in Africa
Sub Saharan Generics (s2generics.com)
To manufacture high-quality generic drugs locally to treat the fivemost common complaints: diabetes, tuberculosis, pain, malariaand hypertension and sell them at ethical prices
Generic drugs exist for all of these ailments and they can bemanufactured cheaply
51 / 52

Sub Saharan Generics was registered as a limited company inSenegal in July 2013
Enjoys the support of Senegal’s Sovereign Wealth Fund and theSenegalese government
The company is in the process of raising capital fromwould-be investors
Drug production is expected to begin in 2015
Article in the magazine Pan African Visions (January 28, 2014).
http://panafricanvisions.com/2014/new-company-works-generic-drugs-africa/
52 / 52





























