Embed Size (px)
Citation preview

Materials Science and Engineering C 29 (2009) 1387–1398
Contents lists available at ScienceDirect
Materials Science and Engineering C
j ourna l homepage: www.e lsev ie r.com/ locate /msec
The comparison of powder characteristics and physicochemical, mechanical andbiological properties between nanostructure ceramics of hydroxyapatite andfluoridated hydroxyapatite
Hossein Eslami, Mehran Solati-Hashjin, Mohammadreza Tahriri ⁎Biomaterial Group, Faculty of Biomedical Engineering (Center of Excellence), Amirkabir University of Technology, P. O. Box: 15875-4413, Tehran, Iran
⁎ Corresponding author. Tel.: +98 21 44218562; fax:E-mail address: [email protected] (M. Tahriri).
0928-4931/$ – see front matter © 2008 Elsevier B.V. Adoi:10.1016/j.msec.2008.10.033
a b s t r a c t
a r t i c l e i n f oArticle history:
In this study, several fluori Received 12 August 2008Received in revised form 8 October 2008Accepted 26 October 2008Available online 6 November 2008Keywords:FluorideHydroxyapatiteFluorhydroxyapatiteFluorapatiteSinterabilityXRD
ne-substituted hydroxyapatite ceramics with the general chemical formula Ca5(PO4)3(OH)1− xFx (0≤x≤1), where x=0.0 (hydroxyapatite; HA), x=0.68 (fluorhydroxyapatite; FHA) andx=0.97 (fluorapatite; FA) were prepared. The powders were characterized using scanning electronmicroscopy (SEM), transmission electron microscopy (TEM), Fourier transform infra-red (FTIR), X-raydiffraction (XRD), F-selective electrode, atomic absorption spectroscopy (AAS) and EDTA titration analyses.The powders were uniaxially pressed and were formed as a disc shape. Subsequently, sinterability andthermal stability of synthesized powders were compared together. Also the effect simultaneously of fluoridecontent and temperature were examined on the lattice parameters and crystallites size of the obtainedpowders. Mechanical properties including hardness, elastic modulus and fracture toughness were measuredusing indentation. The in vitro dissolution studies of the samples were carried out at osteoclastic resorptionconditions. Finally, the biocompatibility and cytotoxicity of the samples were carried out using osteoblast-like cells and L929 cell line, respectively. The obtained results showed that the thermal stability substantiallyis increased with increase incorporated fluoride into HA structure. Also it was found that the fluoride reducedthe lattice parameters and crystallites size of HA. Finally, the in vitro dissolution studies results suggest thatthe fluoride substitutions in HA offer the ability to prepare HAs with different degrees of solubility.
© 2008 Elsevier B.V. All rights reserved.
1. Introduction
Hydroxyapatite (HA; Ca5(PO4)3OH)), the mineral component ofbones and hard tissues in mammals, has been the subject of muchresearch over the years, particularly in the field of biomaterials science.This is due to its importance in clinical applications involving medicaldevices and implants and more recently in the broad field of tissueengineering. Substitutions within the HA lattice are observed both fornaturally occurring and synthetic HA. The most common are substitu-tions involving carbonate,fluoride and chloride ions forhydroxyl ions [1].However, bone mineral hydroxyaptite exceptionally is containing smallamounts of fluoride ions, sodium, magnesium, and other tracecomponents [2]. The mineral phase of tooth enamel consists of apatitecontaining 0.04 wt.% to 0.07 wt.% of fluoride, and constitutes about 95 to97% of the dry mass. Fluoride ions (F−) present in salvia and bloodplasma, are required for normal dental and skeletal development. It hasbeen suggested that a fluoride intake of 1.5–4 mg/day significantlyreduces the risk of dental caries [3]. The fluoride ion has attractedattention due to its therapeutic ability of osteoporosis healing since thebonemass is increasedby F− administration [4]. F− is known to stimulateosteoblast activity both in vitro and in vivo. Sodium fluoride (NaF)
+98 21 33235707.
ll rights reserved.
directly increases the proliferation rate and the alkaline phosphataseactivity of osteoblastic cells, resulting in the enhancement of new bonetissue formation [5]. The mechanism by which F− may influence theproliferation anddifferentiation of orthoclastic cells strongly suggests thealteration of one or several G-protein-dependent tyrosine phosphoryla-tionprocesses, activation of the extracellular signal-regulated kinase, andpossibly other signaling pathways [6,7]. Hence, sodium fluoride therapyhas been studied as one of the treatments for osteoporosis [8–10].
When F− substitutes for the hydroxyl (OH−) group, solubility ofthe bone mineral decreases because partially F-substituted hydro-xyapatite or fluorhydroxyapatite (FHA; Ca5(PO4)3(OH)1− xFx 0≤x≤1)and fluorapatite (FA; Ca5(PO4)3F) are less soluble than pure hydro-xyapatite at pH 5–7 [11]. However, it has been reported that if all of theOH groups in HA are replaced by F to form FA, the resultingmaterials isnot osteo-conductive [12]. Moreover, the high F content might lead tosevere adverse effects such as osteomalacia. As a result, variousmethods have been developed in an attempt to tailor the fluoridecontent of FHA to achieve the best biological properties.
Synthetic HA, presents poor thermal stability as indicated by thedecomposition into other phases such as tricalcium phosphate (TCP;Ca3(PO4)2) at sintering temperatures higher than 900 °C. In contrast, itis expected that FA or FHAmight have superior mechanical propertieswhen sintered at high temperatures because of their higher thermalstability than HA [13].

1388 H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
FHA can be either prepared using a solid-state reaction or a wet-chemical process, but the latter is used more commonly. There areseveral methods of synthesizing fluoridated hydroxyapatite with variesfluorine contents, such as by sol–gel [14] a solid state reaction [15], andpyrolysis methods [16]. The pH-cycling method as the modified wetchemical process was first introduced by Ramsey et al. [17] to avoid ahigh temperature operation and the use of volatilized alcohol (fluorinecontaining reagent). These different approaches have been character-ized using X-ray diffraction (XRD), Fourier transform intra-red spectro-scopy (FTIR), Scanning electron microscopy (SEM) [18–20]. In thismanner, Okazaki et al. [18] demonstrates that a laminated structure isformed in micro sized FHA precipitates produced via a multi-stepfluorine supply process.
In this investigation, we reported synthesis of nanocrystalline hy-droxyapatite, fluorhydroxyapatite and fluorapatite powders via a wetchemical technique, and the resulting powders were characterized bycommonly used bulk techniques. Subsequently, the thermal stability,mechanical properties, solubility and biocompatibility of them wereevaluated.
2. Experimental procedure
2.1. Powders preparation
2.1.1. Synthesis of HACalcium nitrate 4-hydrate (36.15 g; 98%, Merck PROLABO) dissolved
in water (525 ml) was prepared. Diammonium hydrogen phosphate(12 g; 99%, Merck Company) dissolved in water (375 ml) was slowlyadded to the calciumnitrate solutionwith vigorous stirring. The solutionwas brought to pH 11 by addition of concentrated sodium hydroxidesolution (99%,Merck Company). In consequence, obtained precipitationwas HA. The precipitation of HA can be described by Eq. (1) and/or (2)[21]:
10Ca2þ þ 6HPO
−4 þ 2OH
−→Ca10ðPO4Þ6ðOHÞ2 þ 6Hþ ð1Þ
10Caþ2 þ 6H2PO
−4 þ 2OH
−→Ca10ðPO4Þ6ðOHÞ2 þ 12Hþ ð2Þ
The precipitate was aged overnight at room temperature andthoroughly was centrifuged and washed with de-ionized water. Theprocesses of centrifuging and washing were carried out three times.The resulting powder was dried in a freeze-drier system (Alpha 1–2LD, Germany) for 10 h. Finally, the dried powder was calcined in anelectrical box furnace at 1000 °C for 1 h after heating at the rate of5 °C/min in air.
2.1.2. Synthesis of FHA and FAFor the preparation of FHA fine powder, 5 g of HA prepared by the
above method was suspended in 500 ml of 0.02 M sodium fluoride(98.5%, Merck company) solution. This systemwas equilibrated to pH7 for overnight. The pH of the solution was decreased to 4 by slowlyadding 1 M nitric acid (68%, Merck Company). After 30 min, the pH ofthe solution brought back to 7 by the addition of 1 M sodiumhydroxide. The reaction of the FHA can be expressed by the reaction(3) [21]:
Ca10ðPO4Þ6ðOHÞ2 þ 2xNaF→Ca10ðPO4Þ6ðOHÞ2−2xF2x þ xNa2O þ xH2O
ð3Þ
The cycle of pH fluctuation was repeated three times. The solutionwas centrifuged and then washed with de-ionized water. It is notablethat, the processes of centrifuging and washing were carried out threetimes. The wet powders were dried in an oven at 80 °C for 24 h. Theresulting FHA powder was manually milled by an agate mortar. Finally,the dried powderwas calcined in an electrical box furnace at 1000 °C for1 h after heating at the rate of 5 °C/min in air.
Theoretically, if sodium fluoride concentration is 0.02 M, allhydroxyl groups can be substituted by fluoride. However, practically,pure FA is not synthesized. For synthesizing the FA, the amount offluoride content entrance to the solution is considered constant andeffective parameters on the diffusion of fluoride ions in the structureof apatite are modified. Consequently, below process for the synthesisof FA is suggested:
(1) The pH fluctuation from 7 to 4 changed into 7 to 3 which in thisstate, HA powder dissolved some more in the sodium fluoridesolution.
(2) The residence solution time in each pH, increased from 30 to45 min.
(3) The number of cycles increased from 3 to 4.
After the above process, the solution was aged at room tempera-ture for 24 h, centrifuged and washed by de-ionized water for threetimes consecutively. Then, the wet powders were dried in an oven at80 °C for 24 h. The resulting FA powders were thenmanually milled byan agate mortar. Finally, the dried powder was calcined in an electricalbox furnace at 1000 °C for 1 h then the cooling took place at the rate of5 °C/min in the air.
2.2. Samples characterization
2.2.1. X-ray diffractionXRD of all calcined powders was carried out using a Siemens-
Brucker D5000 diffractometer, with voltage and current setting of40 kV and 40 mA, respectively and uses Cu-Kα radiation (1.5406 Å).For qualitative analysis, XRD diagrams were recorded in the interval5°≤2θ≤70° at a scan speed of 2°/min giving a step size 0.02° and thestep time 1 s.
2.2.2. Transform infra-red spectroscopyThe powder samples were examined by Fourier transform infra-
red spectroscopy with a BomemMB 100 spectrometer. For IR analysis,first 1 mg of the powder sample was carefully mixed with 300 mg ofKBr (infrared grade) and palletized under vacuum. Then the pelletwas analyzed in the range of 400 to 4000 cm−1 at a scan speed of23 scan/min with 4 cm−1 resolution.
2.2.3. AAS and EDTA titration techniquesIn order to calculate the Ca/P molar ratio of the precipitated pow-
ders, the content of Ca and Pwere chemically analyzed by quantitativechemical analysis via an EDTA titration technique and atomic ab-sorption spectroscopy (AAS) with a Shimadzu UV-31005 instrument.It is notable that three replicate samples were processed for each sam-ple composition.
2.2.4. F-selective electrodeFluorine concentration measurements were conducted using a F-
selective electrode in citrate/hydrochloride acid (TISAB) buffer. 10 mgof the powder sample was dissolved in 0.2 M HCl (20 mg) to whichwas added 10mgwater and 0.2 M trisodium citrate (40 mg). Standardsolution made from NaF was used to calibrate the measurements inthe same buffer solution. It is worth mentioning that three replicatesamples were processed for each sample composition.
2.2.5. Scanning electron microscopyThe powder sample was coated with a thin layer of Gold (Au) by
sputtering (EMITECHK450X, England) and then themicrostructureof thepowder sample was observed in a scanning electron microscope (SEM;Tescan Vega 2XMU) that operated at an acceleration voltage of 15 kV.
2.2.6. Transmission electron microscopyThe morphology of precipitates was observed by transmission elec-
tron microscopy (TEM; CM200-FEG-Philips). Carbon-coated 200 mesh

1389H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
copper grids were dipped in a dilute suspension of the precipitate. Theparticles were deposited onto the support grids by deposition from adilute suspension in acetone or ethanol. The particle shapes and sizeswere characterized by diffraction (amplitude) contrast and, for crystal-line materials, by high resolution (phase contrast) imaging.
2.2.7. Sinterability and lattice parametersAn amount of 0.2 g of each milled powder was uniaxially pressed
into discs using a pressure of 80 MPa. All the discs were then sinteredat three different temperatures, 1200, 1300 and 1400 °C at a heatingrate of 5 °C/min. They were soaked at each temperature for 1 h beforecooling with the furnace. Subsequently, the sinterability of thementioned discs is evaluated by using the XRD. Also the lattice pa-rameters such as a and c of all powders at different temperatures werecalculated by using the Eqs. (4) and (5), respectively:
a = d ×
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi43
× h2 + hk + k2� �r
For h k0ð Þ planes½ � ð4Þ
c = l × d For 0 0 lð Þ planes½ � ð5Þ
Where d is the spacing between the planes in the atomic lattice.The volume V of the hexagonal unit cell was determined for each HAformulation from the relation (6):
V = 2:589 × a2 × c ð6Þ
Crystallographic identification of the phases of synthesized apa-tites was accomplished by comparing the experimental XRD patternsto standards compiled by the International Center for Diffraction Data(ICDD), which were card # 09-0432 for HA and # 15-0876 for FA. Theaverage size of the individual HA crystallites were calculated fromXRD data using the Scherrer approximation (7):
Dhkl =kλ
β1=2 cos θð7Þ
Where D(hkl) is the crystallite size, as calculated for the (h k l)reflection, λ is the wavelength of Cu-Kα radiation (1.5406 Å), β1/2 isfull width at half maximum intensity and k is the broading constantwith crystal habit and chosen as 0.9 for elongated apatite crystallites.
2.2.8. Contact angleA dynamic contact angle analyzer system (FTA200, First Ten
Angstroms, Portsmouth, VA) was used tomeasure the contact angle ofthe FHA discs with different fluorine contents. Cell culture mediumwas placed on the surface of each FHA disc, and the contact angle wasmeasured to determine the wettability of the sample. The sampleswere cleaned with ethanol in an ultrasonic bath for 10 min and thenair dried between each measurement. The images were continuouslycaptured by the grabber and transferred to the user screen. From theimage, the contact angle was measured and determined. Five drops ofeach liquid were recorded to determine the measurements.
2.2.9. Mechanical propertiesThe mechanical properties of the discs were determined using
indentation with Digital Microhardness Tester (Model MXT70,Matsuzawa Seiki Co., Ltd, Tokyo, Japan). The 10 mm diameter discswere subjected to a load of 200 g for 15 s with a Vickers and Knoopindenter. A total of 5 indentations were made and the resultinghardness values were averaged to determine the standard deviation ofthe hardness of each of the sintered and polished discs. All resultswere calculated to provide a mean with a variability of two standard
deviations. Hardness was calculated according to the procedure inASTM E-384 using the Eq. (8):
HV = 1:8544P = d2 ð8Þ
where HV is the Vickers hardness number (kg/mm2), P is the appliedload (Newton) and d is the diagonal of indentation (meter). In thespecific situation of the Vickers indent, the two diagonals on eachindentationweremeasuredmanually and averaged. The elastic modu-lus was calculated according to the procedure described by Marshallet al. [22] using the Eq. (9):
E =0:45HV
a = b − a= b Vð9Þ
where a is the length of the shorter diagonal, b is the length of thelonger diagonal and b′ is the crack length. The variables a and b are incorrespondence to the Knoop indenter.
The fracture toughness was calculated with using the Eq. (10):
K1C = E=HVð Þ2=3 P = c3=2� �
c=að Þ−1½ �−1=2Þ ð10Þ
where E is the Young's modulus (GPa) and c is the crack lengthindentation (meter) and a is the mean length of the diagonal, both asobserved in the Vickers indentation.
2.2.10. SolubilityThe in vitro dissolution studies of HA, FHA and FA were carried out
with the conditions of osteoclastic resorption, i.e. at a pH of about 4.5in order to simulate general remodelling of the skeletal system. Forthis purpose, the compacted samples were immersed into water(100 ml each) at pH 4.5 and temperature, 37 °C. The pH was checkedand adjusted at regular intervals (3 h). If the pH was increased due toneutralization of the basic calcium phosphate, 0.001 N HNO3 wasadded in order to maintain an average pH of 4.5. The samples weretaken out after 3 days and weighed after drying.
Also the compacted samples were immersed in covered containerswith TRIS solution buffered at pH 7.25, and then the containers weretransferred into water bath held at 37 °C for up to 2 weeks. Theconcentration of Ca2+ ions in the soaked solution was measured byEDTA titration technique.
2.2.11. Biological evaluation
2.2.11.1. Biocompatibility. The osteoblast-like cells were extractedfrom adult rabbit back shank. The cells were washed twice inphosphate buffer solution (PBS), and then collected via centrifugation.In the standard incubation condition (5%CO2, 37 °C), the cells wereincubated for about one week in different cultural media containingDulbecco's Modified Eagle's Medium (DMEM), dexamethasone,vitamin C and β-sodium glycerol-phosphate.
The samples were sterilized in 120 °C water steam for 20 min. Thesamples were then placed in 24-well plates for osteoblast-like cellsimplantation at a set density of 1×104 cells/cm2. The osteoblast-likecells were further incubated in DMEM solution supplemented with10% FBS for 1, 3 and 7 days in the standard culture condition. Incharacterization of cell attachment, the samples were taken out everyday for digestion with 0.25% trypsin EDTA solution, and then the cellnumbers were evaluated by haemacytometer. Eight replicate sampleswere processed for each sample composition.
2.2.11.2. Cytotoxicity. L929 mouse fibroblast cell line (ATCC) was usedfor cytotoxicity. The cells were seeded in polystyrene plates enrichedwith Minimal Essential Medium supplemented with 10% fetal bovineserum,100 U/ml of penicillin and 100 μg/ml streptomycin, respectivelyand incubated at 37 °C in humid atmosphere and 5% CO2. When the

Fig. 1. XRD patterns of HA, FHA and FA calcined powders at 1000 °C for 1 h.
1390 H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
cells attained confluency, the sterilized discs were placed in directcontact with the cells and incubated for 48 h under the same condition.Negative (Ultra High molecular weight Poly Ethylene) and positive(copper) controls were used. After 48 h, the cells were observed underoptical microscopy (Nicon E200). Cellular responses were scored as 0,1,2 and 3 according to non-cytotoxic, mildly cytotoxic and severelycytotoxic as per ISO 10993-5.
3. Results and discussion
3.1. The characteristic of powder
Broad range X-ray diffractometer pattern (Fig. 1) from the calc-HAsample was consistent a pure HA. The pattern from calc-FHA wasqualitatively similar to that of calc-HA. Both HA and FHA showedbroader peaks than their calcined forms, while the pattern from FAdisplayed sharp peaks. No tricalcium phosphate was detected by XRDin any of the three powders. However, it was not possible to dis-tinguish a FA phase in the presence of HA from such as long-range XRDpatterns as in Fig. 1. It is worth mentioning that the XRD resultssuggest that calc-FHA has a structure intermediate between the HAand FA forms. However, no structures for partially F-substitutedcarbonate-free apatites are recorded in the ICDD database.
The Ca/P molar ratio of HA, FHA and FA powders are summarizedin Table 1.
The measured Ca/P ratio for this synthesized powders was higherthan stoichiometric ratio (1.667) that can arise from:(a) local presenceof carbonate apatite in which the Ca/P molar ratio can be as high as3.33 [23], or (b) the presence of impurities such as CaO. With respectto the FTIR spectra, the first explanation is much more reasonable.
The fluorine concentration of all powder samples that wasmeasured by F-selective electrode is listed in Table 2.
With respect to the obtained amount from F-selective electrode,chemical formula of FHA and FA was calculated as Ca5(PO4)3(OH)0.32F0.68 and Ca5(PO4)3(OH)0.03F0.97, respectively. Therefore, theefficiency of F− incorporation into the HA was calculated as 68% forFHA and 0.97% for FA. For describing these obtained efficiencies, it is a
Table 1Ca/P molar ratio of the HA, FHA and FA samples.
Sample Ca/P molar ratio
HA 1.69±0.01FHA 1.71±0.02FA 1.70±0.02
requirement that we know ion exchange between F and OH in theapatite structure [24]. It was suggested that when the pH of thefluoride solution decreased from 7 to 4, more HA dissolved into thesolution (in fact, the Ca2+ ions dissociate from the surface of hy-droxyapatite particles) and then Ca2+ ions rapidly reacted with F ions,and formed calcium fluoride (CaF2). The CaF2 then reacted with theother ions in the solution and formed precalcination fluorhydrox-yapatite as the pH increased to 7. It is notable that, during the cal-cinations process, the hydroxyapatite containing fluoride (FHA)becomes homogenous [15]. The reaction for this phenomenon isinterdiffusion of F− and OH− ions [21]. These ion exchange processescan be expressed by reactions (11) and (12):
Ca5ðPO4Þ3OH þ 810F− þ ð7−mÞHþ→5CaF2 þ H2O
þ ð3−mÞH2PO−4 þmHPO
24 ð11Þ
5CaF2 þ ð3−mÞH2PO−4 þmHPO
2−4
þ ð1−xÞH2O→Ca5ðPO4Þ3ðOHÞ1−xFx þ ð10−xÞF−þ ð7−x−mÞH−ð0bxb1; 0bmb3Þ ð12Þ
Combining Eqs. (11) and (12), the chemical reaction at each pHcycle can be written as:
Ca5ðPO4Þ3ðOHÞ þ xF− þ xHþ→Ca5ðPO4Þ3ðOHÞ1−xFx þ xH2O ð13ÞAs a result, identical XRD patterns were observed for FHA and FA
samples, as these samples were a mixture of HA and F-rich apatite (orFA). However, they were substantially different from HA that wasabsent of fluorine ions.
Also the rate of this F:OH exchange process is very slow. Ramseyet al. [17] reported that it took 10 days, five pH cycled, and with slightexcess of fluorine ions the F− totally replaces the OH− group in HA.But, the sample in the current study was held only for 30 min in eachpH cycle while three pH cycles were applied only. As a result, theefficiency of fluorine incorporation in this paper (68% for FHA powdersample) was much lower than the value of 95% reported by Ramsey[17].
Table 2Measured F value using F-selective electrode and amount of X in FHAs chemical formula.
Sample Measured content (wt.%) Amount of X in Ca5(PO4)3(OH)1− xFx
HA 0 0FHA 2.51±0.05 0.68FA 3.57±0.03 0.97
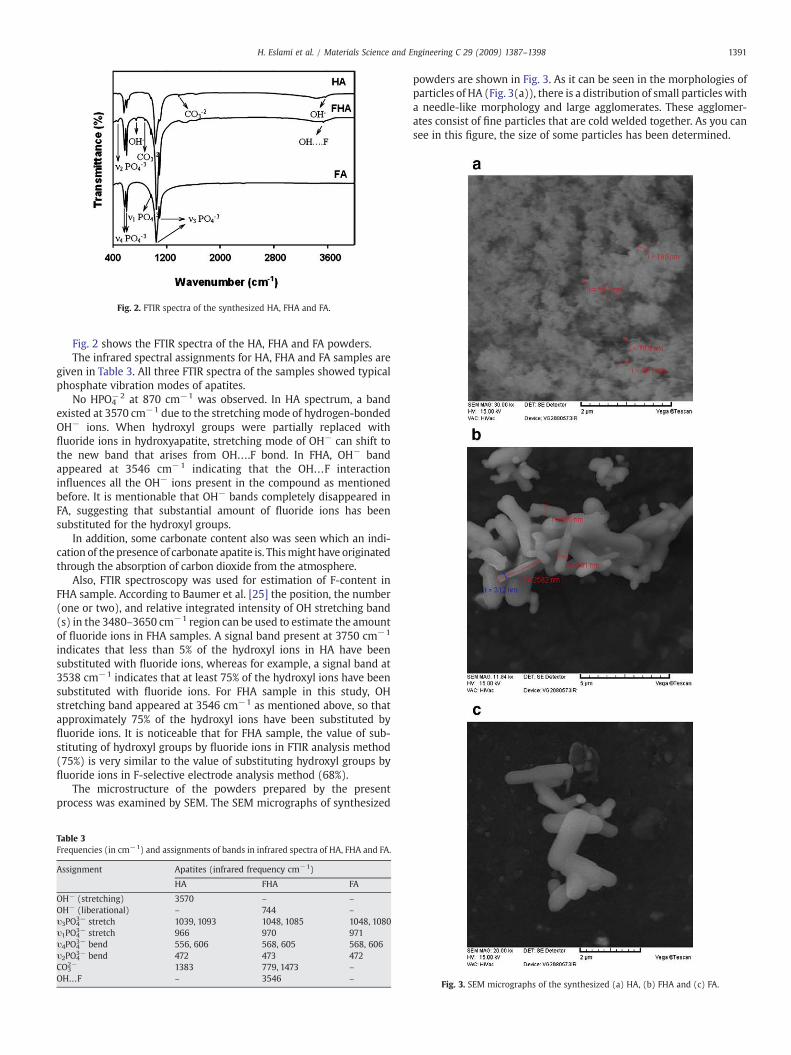
Fig. 2. FTIR spectra of the synthesized HA, FHA and FA.
1391H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
Fig. 2 shows the FTIR spectra of the HA, FHA and FA powders.The infrared spectral assignments for HA, FHA and FA samples are
given in Table 3. All three FTIR spectra of the samples showed typicalphosphate vibration modes of apatites.
No HPO4−2 at 870 cm−1 was observed. In HA spectrum, a band
existed at 3570 cm−1 due to the stretching mode of hydrogen-bondedOH− ions. When hydroxyl groups were partially replaced withfluoride ions in hydroxyapatite, stretching mode of OH− can shift tothe new band that arises from OH….F bond. In FHA, OH− bandappeared at 3546 cm−1 indicating that the OH…F interactioninfluences all the OH− ions present in the compound as mentionedbefore. It is mentionable that OH− bands completely disappeared inFA, suggesting that substantial amount of fluoride ions has beensubstituted for the hydroxyl groups.
In addition, some carbonate content also was seen which an indi-cation of the presence of carbonate apatite is. Thismight have originatedthrough the absorption of carbon dioxide from the atmosphere.
Also, FTIR spectroscopy was used for estimation of F-content inFHA sample. According to Baumer et al. [25] the position, the number(one or two), and relative integrated intensity of OH stretching band(s) in the 3480–3650 cm−1 region can be used to estimate the amountof fluoride ions in FHA samples. A signal band present at 3750 cm−1
indicates that less than 5% of the hydroxyl ions in HA have beensubstituted with fluoride ions, whereas for example, a signal band at3538 cm−1 indicates that at least 75% of the hydroxyl ions have beensubstituted with fluoride ions. For FHA sample in this study, OHstretching band appeared at 3546 cm−1 as mentioned above, so thatapproximately 75% of the hydroxyl ions have been substituted byfluoride ions. It is noticeable that for FHA sample, the value of sub-stituting of hydroxyl groups by fluoride ions in FTIR analysis method(75%) is very similar to the value of substituting hydroxyl groups byfluoride ions in F-selective electrode analysis method (68%).
The microstructure of the powders prepared by the presentprocess was examined by SEM. The SEM micrographs of synthesized
Table 3Frequencies (in cm−1) and assignments of bands in infrared spectra of HA, FHA and FA.
Assignment Apatites (infrared frequency cm−1)
HA FHA FA
OH− (stretching) 3570 – –
OH− (liberational) – 744 –
υ3PO43− stretch 1039, 1093 1048, 1085 1048, 1080
υ1PO43− stretch 966 970 971
υ4PO43− bend 556, 606 568, 605 568, 606
υ2PO43− bend 472 473 472
CO32− 1383 779, 1473 –
OH…F – 3546 –
powders are shown in Fig. 3. As it can be seen in the morphologies ofparticles of HA (Fig. 3(a)), there is a distribution of small particles witha needle-like morphology and large agglomerates. These agglomer-ates consist of fine particles that are cold welded together. As you cansee in this figure, the size of some particles has been determined.
Fig. 3. SEM micrographs of the synthesized (a) HA, (b) FHA and (c) FA.
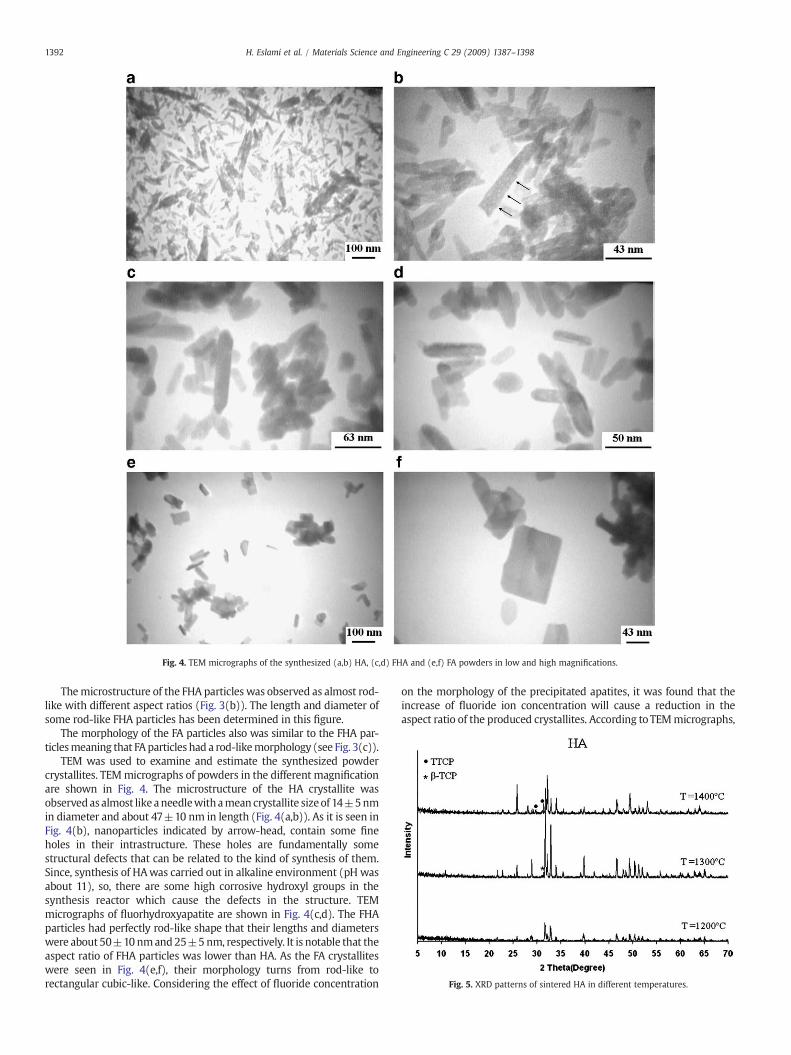
Fig. 4. TEM micrographs of the synthesized (a,b) HA, (c,d) FHA and (e,f) FA powders in low and high magnifications.
Fig. 5. XRD patterns of sintered HA in different temperatures.
1392 H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
Themicrostructure of the FHA particles was observed as almost rod-like with different aspect ratios (Fig. 3(b)). The length and diameter ofsome rod-like FHA particles has been determined in this figure.
The morphology of the FA particles also was similar to the FHA par-ticlesmeaning that FA particles had a rod-likemorphology (see Fig. 3(c)).
TEM was used to examine and estimate the synthesized powdercrystallites. TEMmicrographs of powders in the different magnificationare shown in Fig. 4. The microstructure of the HA crystallite wasobservedasalmost like aneedlewithamean crystallite sizeof 14±5nmin diameter and about 47±10 nm in length (Fig. 4(a,b)). As it is seen inFig. 4(b), nanoparticles indicated by arrow-head, contain some fineholes in their intrastructure. These holes are fundamentally somestructural defects that can be related to the kind of synthesis of them.Since, synthesis of HAwas carried out in alkaline environment (pH wasabout 11), so, there are some high corrosive hydroxyl groups in thesynthesis reactor which cause the defects in the structure. TEMmicrographs of fluorhydroxyapatite are shown in Fig. 4(c,d). The FHAparticles had perfectly rod-like shape that their lengths and diameterswere about 50±10nmand25±5nm, respectively. It is notable that theaspect ratio of FHA particles was lower than HA. As the FA crystalliteswere seen in Fig. 4(e,f), their morphology turns from rod-like torectangular cubic-like. Considering the effect of fluoride concentration
on the morphology of the precipitated apatites, it was found that theincrease of fluoride ion concentration will cause a reduction in theaspect ratio of the produced crystallites. According toTEMmicrographs,
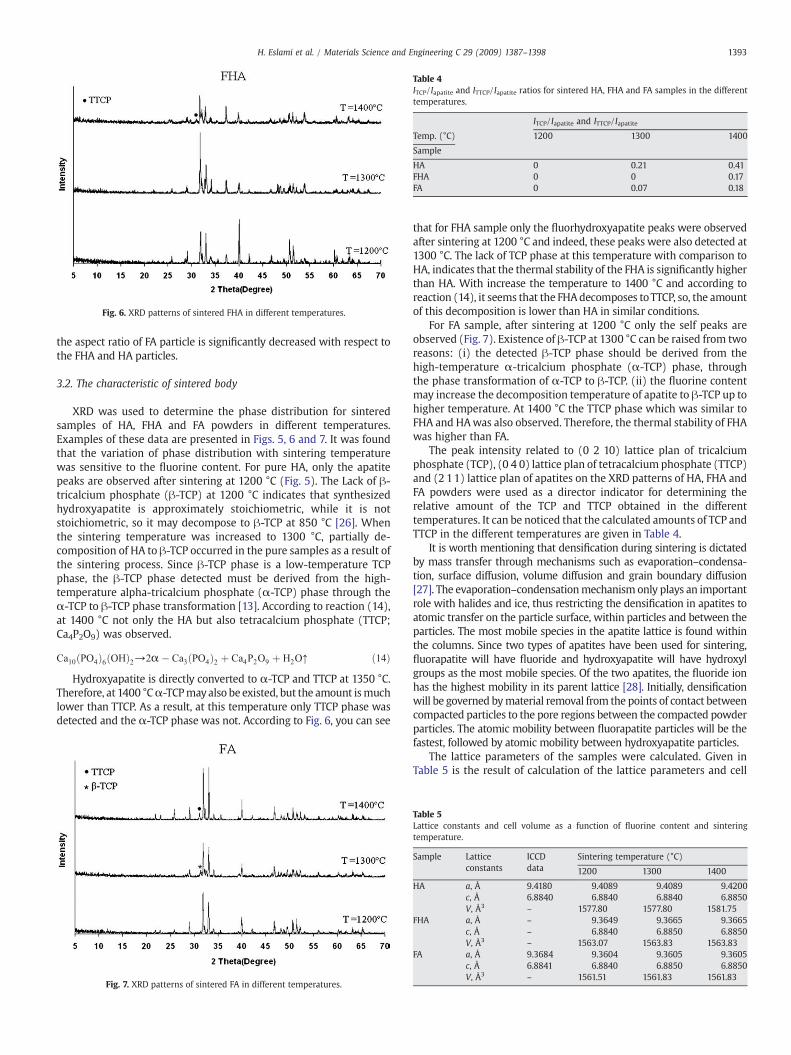
Fig. 6. XRD patterns of sintered FHA in different temperatures.
Table 4ITCP/Iapatite and ITTCP/Iapatite ratios for sintered HA, FHA and FA samples in the differenttemperatures.
ITCP/Iapatite and ITTCP/Iapatite
Temp. (°C) 1200 1300 1400
Sample
HA 0 0.21 0.41FHA 0 0 0.17FA 0 0.07 0.18
1393H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
the aspect ratio of FA particle is significantly decreased with respect tothe FHA and HA particles.
3.2. The characteristic of sintered body
XRD was used to determine the phase distribution for sinteredsamples of HA, FHA and FA powders in different temperatures.Examples of these data are presented in Figs. 5, 6 and 7. It was foundthat the variation of phase distribution with sintering temperaturewas sensitive to the fluorine content. For pure HA, only the apatitepeaks are observed after sintering at 1200 °C (Fig. 5). The Lack of β-tricalcium phosphate (β-TCP) at 1200 °C indicates that synthesizedhydroxyapatite is approximately stoichiometric, while it is notstoichiometric, so it may decompose to β-TCP at 850 °C [26]. Whenthe sintering temperature was increased to 1300 °C, partially de-composition of HA to β-TCP occurred in the pure samples as a result ofthe sintering process. Since β-TCP phase is a low-temperature TCPphase, the β-TCP phase detected must be derived from the high-temperature alpha-tricalcium phosphate (α-TCP) phase through theα-TCP to β-TCP phase transformation [13]. According to reaction (14),at 1400 °C not only the HA but also tetracalcium phosphate (TTCP;Ca4P2O9) was observed.
Ca10ðPO4Þ6ðOHÞ2→2α� Ca3ðPO4Þ2 þ Ca4P2O9 þ H2O↑ ð14ÞHydroxyapatite is directly converted to α-TCP and TTCP at 1350 °C.
Therefore, at 1400 °Cα-TCPmayalso be existed, but the amount ismuchlower than TTCP. As a result, at this temperature only TTCP phase wasdetected and the α-TCP phase was not. According to Fig. 6, you can see
Fig. 7. XRD patterns of sintered FA in different temperatures.
that for FHA sample only the fluorhydroxyapatite peaks were observedafter sintering at 1200 °C and indeed, these peaks were also detected at1300 °C. The lack of TCP phase at this temperature with comparison toHA, indicates that the thermal stability of the FHA is significantly higherthan HA. With increase the temperature to 1400 °C and according toreaction (14), it seems that the FHAdecomposes toTTCP, so, the amountof this decomposition is lower than HA in similar conditions.
For FA sample, after sintering at 1200 °C only the self peaks areobserved (Fig. 7). Existence of β-TCP at 1300 °C can be raised from tworeasons: (i) the detected β-TCP phase should be derived from thehigh-temperature α-tricalcium phosphate (α-TCP) phase, throughthe phase transformation of α-TCP to β-TCP. (ii) the fluorine contentmay increase the decomposition temperature of apatite to β-TCP up tohigher temperature. At 1400 °C the TTCP phase which was similar toFHA and HAwas also observed. Therefore, the thermal stability of FHAwas higher than FA.
The peak intensity related to (0 2 10) lattice plan of tricalciumphosphate (TCP), (0 4 0) lattice plan of tetracalcium phosphate (TTCP)and (2 11) lattice plan of apatites on the XRD patterns of HA, FHA andFA powders were used as a director indicator for determining therelative amount of the TCP and TTCP obtained in the differenttemperatures. It can be noticed that the calculated amounts of TCP andTTCP in the different temperatures are given in Table 4.
It is worth mentioning that densification during sintering is dictatedby mass transfer through mechanisms such as evaporation–condensa-tion, surface diffusion, volume diffusion and grain boundary diffusion[27]. The evaporation–condensationmechanism only plays an importantrole with halides and ice, thus restricting the densification in apatites toatomic transfer on the particle surface, within particles and between theparticles. The most mobile species in the apatite lattice is found withinthe columns. Since two types of apatites have been used for sintering,fluorapatite will have fluoride and hydroxyapatite will have hydroxylgroups as the most mobile species. Of the two apatites, the fluoride ionhas the highest mobility in its parent lattice [28]. Initially, densificationwill be governed bymaterial removal from the points of contact betweencompacted particles to the pore regions between the compacted powderparticles. The atomic mobility between fluorapatite particles will be thefastest, followed by atomic mobility between hydroxyapatite particles.
The lattice parameters of the samples were calculated. Given inTable 5 is the result of calculation of the lattice parameters and cell
Table 5Lattice constants and cell volume as a function of fluorine content and sinteringtemperature.
Sample Latticeconstants
ICCDdata
Sintering temperature (°C)
1200 1300 1400
HA a, Å 9.4180 9.4089 9.4089 9.4200c, Å 6.8840 6.8840 6.8840 6.8850V, Å3 – 1577.80 1577.80 1581.75
FHA a, Å – 9.3649 9.3665 9.3665c, Å – 6.8840 6.8850 6.8850V, Å3 – 1563.07 1563.83 1563.83
FA a, Å 9.3684 9.3604 9.3605 9.3605c, Å 6.8841 6.8840 6.8850 6.8850V, Å3 – 1561.51 1561.83 1561.83
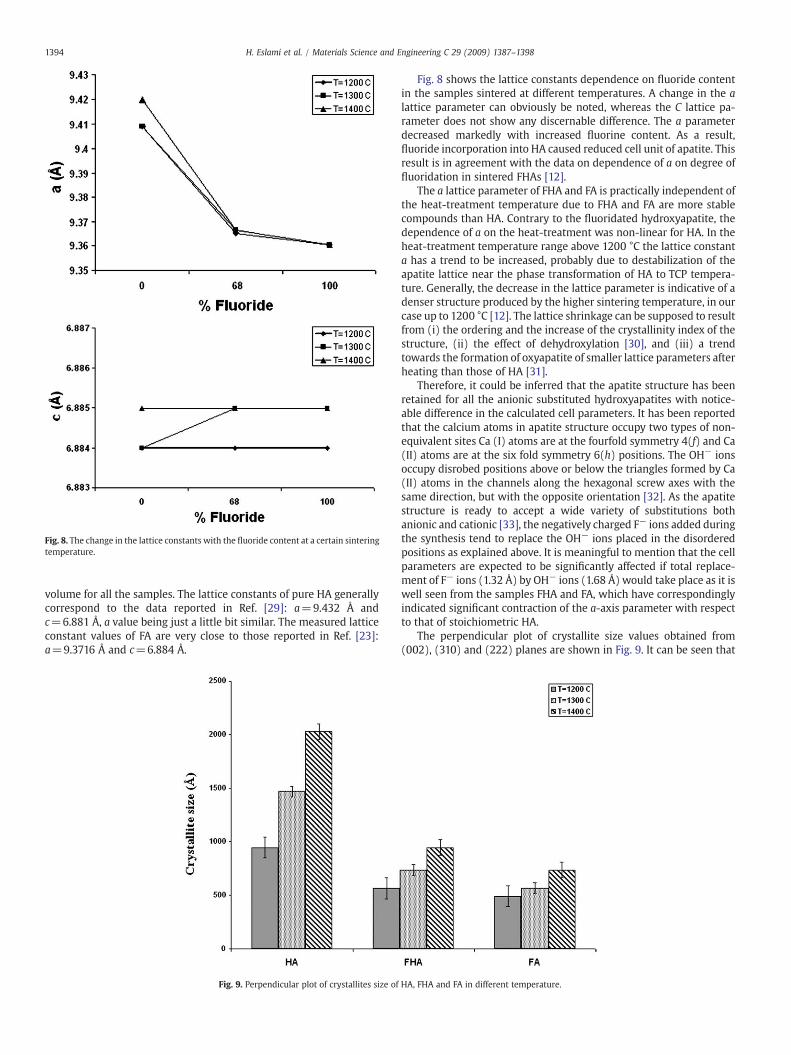
Fig. 8. The change in the lattice constants with the fluoride content at a certain sinteringtemperature.
1394 H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
volume for all the samples. The lattice constants of pure HA generallycorrespond to the data reported in Ref. [29]: a=9.432 Å andc=6.881 Å, a value being just a little bit similar. The measured latticeconstant values of FA are very close to those reported in Ref. [23]:a=9.3716 Å and c=6.884 Å.
Fig. 9. Perpendicular plot of crystallites size of
Fig. 8 shows the lattice constants dependence on fluoride contentin the samples sintered at different temperatures. A change in the alattice parameter can obviously be noted, whereas the C lattice pa-rameter does not show any discernable difference. The a parameterdecreased markedly with increased fluorine content. As a result,fluoride incorporation into HA caused reduced cell unit of apatite. Thisresult is in agreement with the data on dependence of a on degree offluoridation in sintered FHAs [12].
The a lattice parameter of FHA and FA is practically independent ofthe heat-treatment temperature due to FHA and FA are more stablecompounds than HA. Contrary to the fluoridated hydroxyapatite, thedependence of a on the heat-treatment was non-linear for HA. In theheat-treatment temperature range above 1200 °C the lattice constanta has a trend to be increased, probably due to destabilization of theapatite lattice near the phase transformation of HA to TCP tempera-ture. Generally, the decrease in the lattice parameter is indicative of adenser structure produced by the higher sintering temperature, in ourcase up to 1200 °C [12]. The lattice shrinkage can be supposed to resultfrom (i) the ordering and the increase of the crystallinity index of thestructure, (ii) the effect of dehydroxylation [30], and (iii) a trendtowards the formation of oxyapatite of smaller lattice parameters afterheating than those of HA [31].
Therefore, it could be inferred that the apatite structure has beenretained for all the anionic substituted hydroxyapatites with notice-able difference in the calculated cell parameters. It has been reportedthat the calcium atoms in apatite structure occupy two types of non-equivalent sites Ca (I) atoms are at the fourfold symmetry 4(f) and Ca(II) atoms are at the six fold symmetry 6(h) positions. The OH− ionsoccupy disrobed positions above or below the triangles formed by Ca(II) atoms in the channels along the hexagonal screw axes with thesame direction, but with the opposite orientation [32]. As the apatitestructure is ready to accept a wide variety of substitutions bothanionic and cationic [33], the negatively charged F− ions added duringthe synthesis tend to replace the OH− ions placed in the disorderedpositions as explained above. It is meaningful to mention that the cellparameters are expected to be significantly affected if total replace-ment of F− ions (1.32 Å) by OH− ions (1.68 Å) would take place as it iswell seen from the samples FHA and FA, which have correspondinglyindicated significant contraction of the a-axis parameter with respectto that of stoichiometric HA.
The perpendicular plot of crystallite size values obtained from(002), (310) and (222) planes are shown in Fig. 9. It can be seen that
HA, FHA and FA in different temperature.
1395H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
with incorporation of fluoride into hydroxyapatite structure, crystal-lites size markedly has been decreased. It is meaningful to the fluoridewhich significantly prevented growth of grains in HA structure.
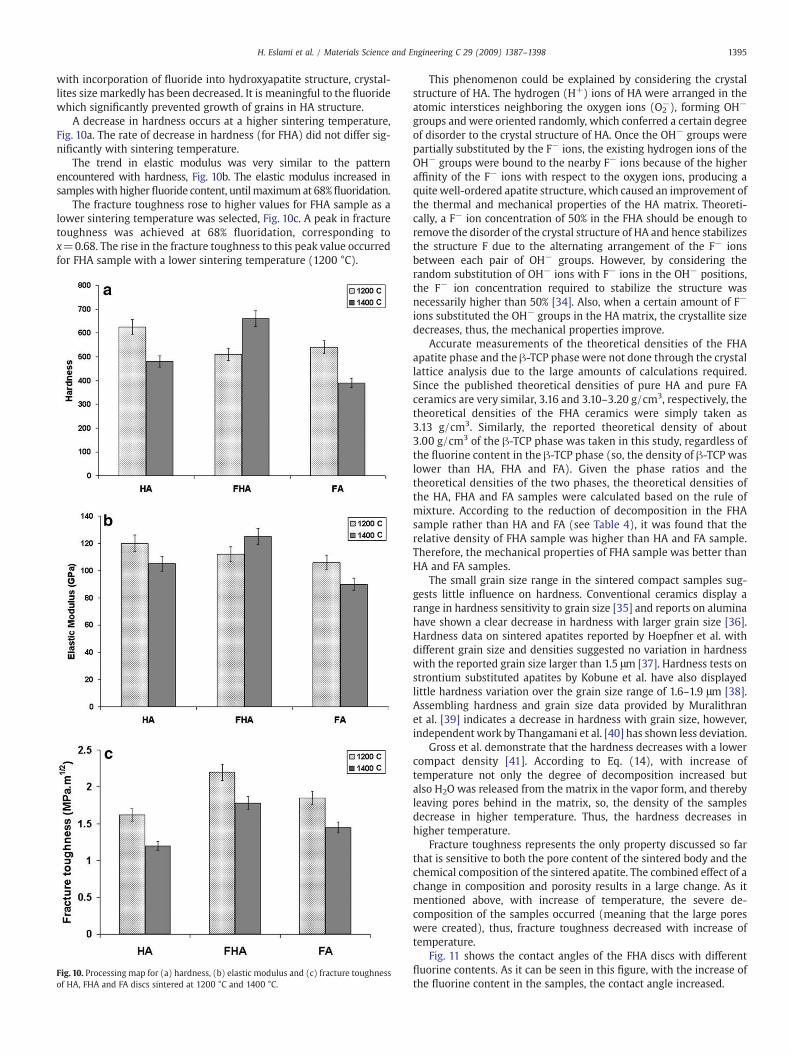
A decrease in hardness occurs at a higher sintering temperature,Fig. 10a. The rate of decrease in hardness (for FHA) did not differ sig-nificantly with sintering temperature.
The trend in elastic modulus was very similar to the patternencountered with hardness, Fig. 10b. The elastic modulus increased insampleswith higher fluoride content, untilmaximumat 68% fluoridation.
The fracture toughness rose to higher values for FHA sample as alower sintering temperature was selected, Fig. 10c. A peak in fracturetoughness was achieved at 68% fluoridation, corresponding tox=0.68. The rise in the fracture toughness to this peak value occurredfor FHA sample with a lower sintering temperature (1200 °C).
Fig. 10. Processing map for (a) hardness, (b) elastic modulus and (c) fracture toughnessof HA, FHA and FA discs sintered at 1200 °C and 1400 °C.
This phenomenon could be explained by considering the crystalstructure of HA. The hydrogen (H+) ions of HA were arranged in theatomic interstices neighboring the oxygen ions (O2
−), forming OH−
groups and were oriented randomly, which conferred a certain degreeof disorder to the crystal structure of HA. Once the OH− groups werepartially substituted by the F− ions, the existing hydrogen ions of theOH− groups were bound to the nearby F− ions because of the higheraffinity of the F− ions with respect to the oxygen ions, producing aquite well-ordered apatite structure, which caused an improvement ofthe thermal and mechanical properties of the HA matrix. Theoreti-cally, a F− ion concentration of 50% in the FHA should be enough toremove the disorder of the crystal structure of HA and hence stabilizesthe structure F due to the alternating arrangement of the F− ionsbetween each pair of OH− groups. However, by considering therandom substitution of OH− ions with F− ions in the OH− positions,the F− ion concentration required to stabilize the structure wasnecessarily higher than 50% [34]. Also, when a certain amount of F−
ions substituted the OH− groups in the HA matrix, the crystallite sizedecreases, thus, the mechanical properties improve.
Accurate measurements of the theoretical densities of the FHAapatite phase and the β-TCP phase were not done through the crystallattice analysis due to the large amounts of calculations required.Since the published theoretical densities of pure HA and pure FAceramics are very similar, 3.16 and 3.10–3.20 g/cm3, respectively, thetheoretical densities of the FHA ceramics were simply taken as3.13 g/cm3. Similarly, the reported theoretical density of about3.00 g/cm3 of the β-TCP phase was taken in this study, regardless ofthe fluorine content in the β-TCP phase (so, the density of β-TCP waslower than HA, FHA and FA). Given the phase ratios and thetheoretical densities of the two phases, the theoretical densities ofthe HA, FHA and FA samples were calculated based on the rule ofmixture. According to the reduction of decomposition in the FHAsample rather than HA and FA (see Table 4), it was found that therelative density of FHA sample was higher than HA and FA sample.Therefore, the mechanical properties of FHA sample was better thanHA and FA samples.
The small grain size range in the sintered compact samples sug-gests little influence on hardness. Conventional ceramics display arange in hardness sensitivity to grain size [35] and reports on aluminahave shown a clear decrease in hardness with larger grain size [36].Hardness data on sintered apatites reported by Hoepfner et al. withdifferent grain size and densities suggested no variation in hardnesswith the reported grain size larger than 1.5 μm [37]. Hardness tests onstrontium substituted apatites by Kobune et al. have also displayedlittle hardness variation over the grain size range of 1.6–1.9 μm [38].Assembling hardness and grain size data provided by Muralithranet al. [39] indicates a decrease in hardness with grain size, however,independentwork by Thangamani et al. [40] has shown less deviation.
Gross et al. demonstrate that the hardness decreases with a lowercompact density [41]. According to Eq. (14), with increase oftemperature not only the degree of decomposition increased butalso H2O was released from the matrix in the vapor form, and therebyleaving pores behind in the matrix, so, the density of the samplesdecrease in higher temperature. Thus, the hardness decreases inhigher temperature.
Fracture toughness represents the only property discussed so farthat is sensitive to both the pore content of the sintered body and thechemical composition of the sintered apatite. The combined effect of achange in composition and porosity results in a large change. As itmentioned above, with increase of temperature, the severe de-composition of the samples occurred (meaning that the large poreswere created), thus, fracture toughness decreased with increase oftemperature.
Fig. 11 shows the contact angles of the FHA discs with differentfluorine contents. As it can be seen in this figure, with the increase ofthe fluorine content in the samples, the contact angle increased.
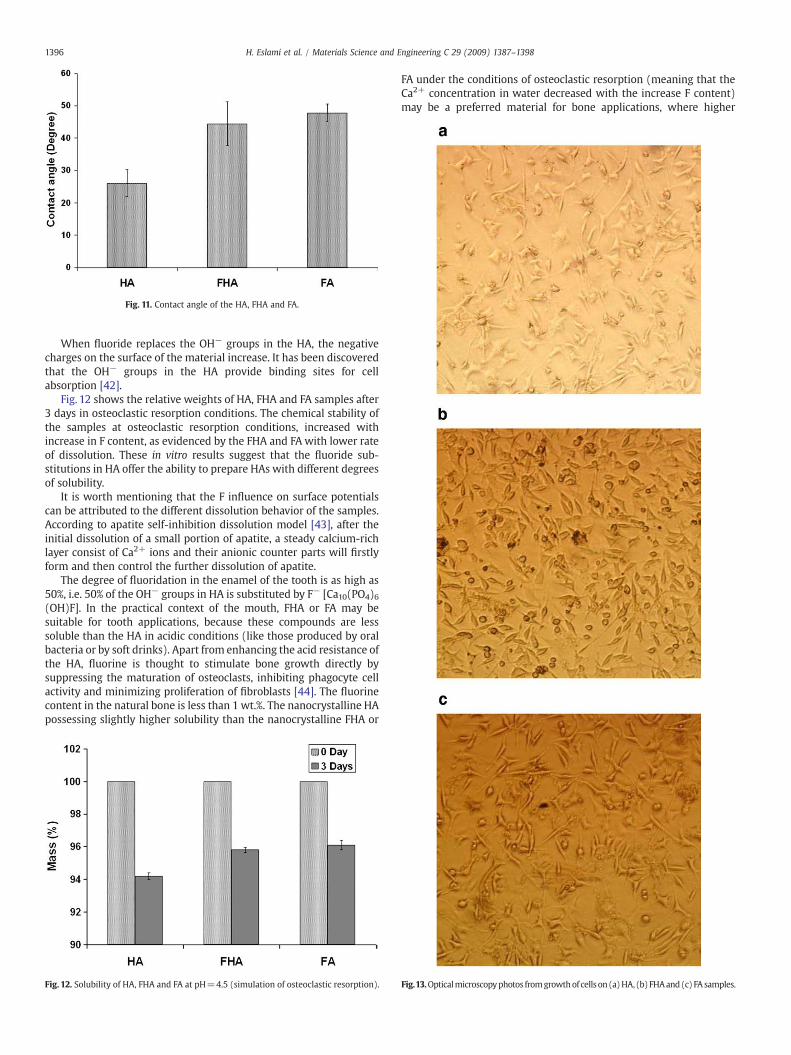
Fig. 11. Contact angle of the HA, FHA and FA.
1396 H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
When fluoride replaces the OH− groups in the HA, the negativecharges on the surface of the material increase. It has been discoveredthat the OH− groups in the HA provide binding sites for cellabsorption [42].
Fig. 12 shows the relative weights of HA, FHA and FA samples after3 days in osteoclastic resorption conditions. The chemical stability ofthe samples at osteoclastic resorption conditions, increased withincrease in F content, as evidenced by the FHA and FA with lower rateof dissolution. These in vitro results suggest that the fluoride sub-stitutions in HA offer the ability to prepare HAs with different degreesof solubility.
It is worth mentioning that the F influence on surface potentialscan be attributed to the different dissolution behavior of the samples.According to apatite self-inhibition dissolution model [43], after theinitial dissolution of a small portion of apatite, a steady calcium-richlayer consist of Ca2+ ions and their anionic counter parts will firstlyform and then control the further dissolution of apatite.
The degree of fluoridation in the enamel of the tooth is as high as50%, i.e. 50% of the OH− groups in HA is substituted by F− [Ca10(PO4)6(OH)F]. In the practical context of the mouth, FHA or FA may besuitable for tooth applications, because these compounds are lesssoluble than the HA in acidic conditions (like those produced by oralbacteria or by soft drinks). Apart from enhancing the acid resistance ofthe HA, fluorine is thought to stimulate bone growth directly bysuppressing the maturation of osteoclasts, inhibiting phagocyte cellactivity and minimizing proliferation of fibroblasts [44]. The fluorinecontent in the natural bone is less than 1 wt.%. The nanocrystalline HApossessing slightly higher solubility than the nanocrystalline FHA or
Fig. 12. Solubility of HA, FHA and FA at pH=4.5 (simulation of osteoclastic resorption).
FA under the conditions of osteoclastic resorption (meaning that theCa2+ concentration in water decreased with the increase F content)may be a preferred material for bone applications, where higher
Fig.13.Opticalmicroscopyphotos fromgrowthof cells on(a)HA,(b)FHAand(c) FA samples.
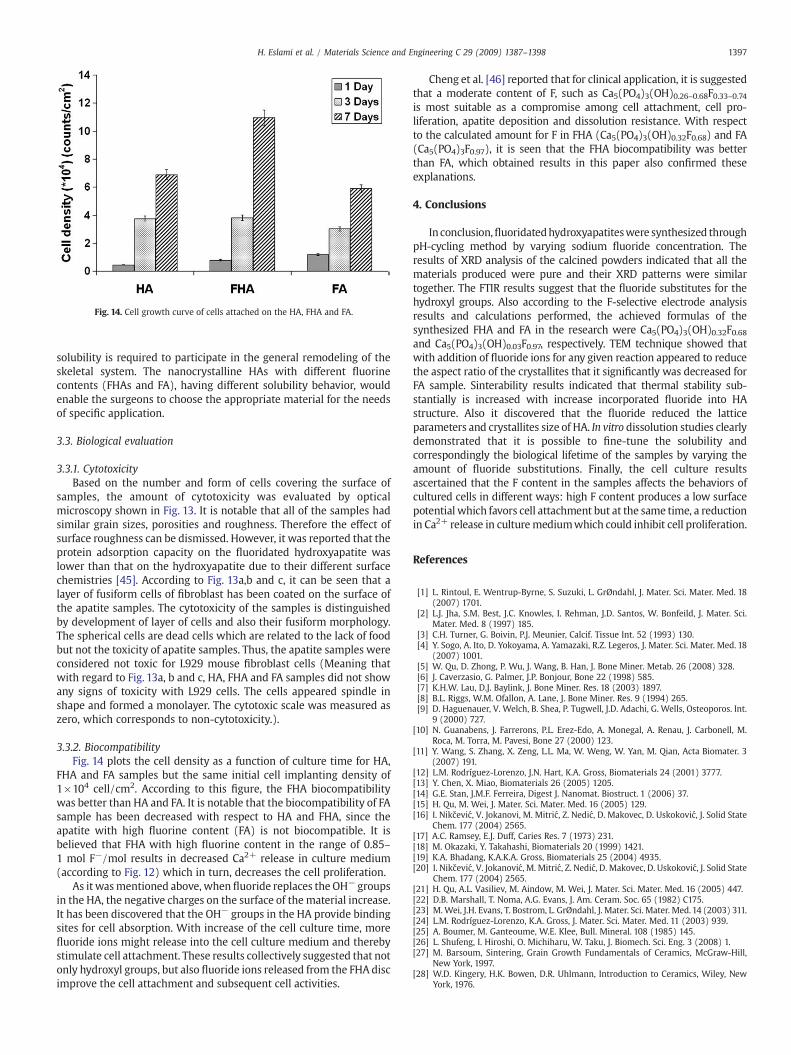
Fig. 14. Cell growth curve of cells attached on the HA, FHA and FA.
1397H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
solubility is required to participate in the general remodeling of theskeletal system. The nanocrystalline HAs with different fluorinecontents (FHAs and FA), having different solubility behavior, wouldenable the surgeons to choose the appropriate material for the needsof specific application.
3.3. Biological evaluation
3.3.1. CytotoxicityBased on the number and form of cells covering the surface of
samples, the amount of cytotoxicity was evaluated by opticalmicroscopy shown in Fig. 13. It is notable that all of the samples hadsimilar grain sizes, porosities and roughness. Therefore the effect ofsurface roughness can be dismissed. However, it was reported that theprotein adsorption capacity on the fluoridated hydroxyapatite waslower than that on the hydroxyapatite due to their different surfacechemistries [45]. According to Fig. 13a,b and c, it can be seen that alayer of fusiform cells of fibroblast has been coated on the surface ofthe apatite samples. The cytotoxicity of the samples is distinguishedby development of layer of cells and also their fusiform morphology.The spherical cells are dead cells which are related to the lack of foodbut not the toxicity of apatite samples. Thus, the apatite samples wereconsidered not toxic for L929 mouse fibroblast cells (Meaning thatwith regard to Fig. 13a, b and c, HA, FHA and FA samples did not showany signs of toxicity with L929 cells. The cells appeared spindle inshape and formed a monolayer. The cytotoxic scale was measured aszero, which corresponds to non-cytotoxicity.).
3.3.2. BiocompatibilityFig. 14 plots the cell density as a function of culture time for HA,
FHA and FA samples but the same initial cell implanting density of1×104 cell/cm2. According to this figure, the FHA biocompatibilitywas better than HA and FA. It is notable that the biocompatibility of FAsample has been decreased with respect to HA and FHA, since theapatite with high fluorine content (FA) is not biocompatible. It isbelieved that FHA with high fluorine content in the range of 0.85–1 mol F−/mol results in decreased Ca2+ release in culture medium(according to Fig. 12) which in turn, decreases the cell proliferation.
As it wasmentioned above, when fluoride replaces the OH− groupsin the HA, the negative charges on the surface of the material increase.It has been discovered that the OH− groups in the HA provide bindingsites for cell absorption. With increase of the cell culture time, morefluoride ions might release into the cell culture medium and therebystimulate cell attachment. These results collectively suggested that notonly hydroxyl groups, but also fluoride ions released from the FHA discimprove the cell attachment and subsequent cell activities.
Cheng et al. [46] reported that for clinical application, it is suggestedthat a moderate content of F, such as Ca5(PO4)3(OH)0.26–0.68F0.33–0.74is most suitable as a compromise among cell attachment, cell pro-liferation, apatite deposition and dissolution resistance. With respectto the calculated amount for F in FHA (Ca5(PO4)3(OH)0.32F0.68) and FA(Ca5(PO4)3F0.97), it is seen that the FHA biocompatibility was betterthan FA, which obtained results in this paper also confirmed theseexplanations.
4. Conclusions
In conclusion,fluoridatedhydroxyapatiteswere synthesized throughpH-cycling method by varying sodium fluoride concentration. Theresults of XRD analysis of the calcined powders indicated that all thematerials produced were pure and their XRD patterns were similartogether. The FTIR results suggest that the fluoride substitutes for thehydroxyl groups. Also according to the F-selective electrode analysisresults and calculations performed, the achieved formulas of thesynthesized FHA and FA in the research were Ca5(PO4)3(OH)0.32F0.68and Ca5(PO4)3(OH)0.03F0.97, respectively. TEM technique showed thatwith addition of fluoride ions for any given reaction appeared to reducethe aspect ratio of the crystallites that it significantly was decreased forFA sample. Sinterability results indicated that thermal stability sub-stantially is increased with increase incorporated fluoride into HAstructure. Also it discovered that the fluoride reduced the latticeparameters and crystallites size of HA. In vitro dissolution studies clearlydemonstrated that it is possible to fine-tune the solubility andcorrespondingly the biological lifetime of the samples by varying theamount of fluoride substitutions. Finally, the cell culture resultsascertained that the F content in the samples affects the behaviors ofcultured cells in different ways: high F content produces a low surfacepotential which favors cell attachment but at the same time, a reductionin Ca2+ release in culturemediumwhich could inhibit cell proliferation.
References
[1] L. Rintoul, E. Wentrup-Byrne, S. Suzuki, L. GrØndahl, J. Mater. Sci. Mater. Med. 18(2007) 1701.
[2] L.J. Jha, S.M. Best, J.C. Knowles, I. Rehman, J.D. Santos, W. Bonfeild, J. Mater. Sci.Mater. Med. 8 (1997) 185.
[3] C.H. Turner, G. Boivin, P.J. Meunier, Calcif. Tissue Int. 52 (1993) 130.[4] Y. Sogo, A. Ito, D. Yokoyama, A. Yamazaki, R.Z. Legeros, J. Mater. Sci. Mater. Med. 18
(2007) 1001.[5] W. Qu, D. Zhong, P. Wu, J. Wang, B. Han, J. Bone Miner. Metab. 26 (2008) 328.[6] J. Caverzasio, G. Palmer, J.P. Bonjour, Bone 22 (1998) 585.[7] K.H.W. Lau, D.J. Baylink, J. Bone Miner. Res. 18 (2003) 1897.[8] B.L. Riggs, W.M. Ofallon, A. Lane, J. Bone Miner. Res. 9 (1994) 265.[9] D. Haguenauer, V. Welch, B. Shea, P. Tugwell, J.D. Adachi, G. Wells, Osteoporos. Int.
9 (2000) 727.[10] N. Guanabens, J. Farrerons, P.L. Erez-Edo, A. Monegal, A. Renau, J. Carbonell, M.
Roca, M. Torra, M. Pavesi, Bone 27 (2000) 123.[11] Y. Wang, S. Zhang, X. Zeng, L.L. Ma, W. Weng, W. Yan, M. Qian, Acta Biomater. 3
(2007) 191.[12] L.M. Rodríguez-Lorenzo, J.N. Hart, K.A. Gross, Biomaterials 24 (2001) 3777.[13] Y. Chen, X. Miao, Biomaterials 26 (2005) 1205.[14] G.E. Stan, J.M.F. Ferreira, Digest J. Nanomat. Biostruct. 1 (2006) 37.[15] H. Qu, M. Wei, J. Mater. Sci. Mater. Med. 16 (2005) 129.[16] I. Nikčević, V. Jokanovi, M. Mitrić, Z. Nedić, D. Makovec, D. Uskoković, J. Solid State
Chem. 177 (2004) 2565.[17] A.C. Ramsey, E.J. Duff, Caries Res. 7 (1973) 231.[18] M. Okazaki, Y. Takahashi, Biomaterials 20 (1999) 1421.[19] K.A. Bhadang, K.A.K.A. Gross, Biomaterials 25 (2004) 4935.[20] I. Nikčević, V. Jokanović, M. Mitrić, Z. Nedić, D. Makovec, D. Uskoković, J. Solid State
Chem. 177 (2004) 2565.[21] H. Qu, A.L. Vasiliev, M. Aindow, M. Wei, J. Mater. Sci. Mater. Med. 16 (2005) 447.[22] D.B. Marshall, T. Noma, A.G. Evans, J. Am. Ceram. Soc. 65 (1982) C175.[23] M.Wei, J.H. Evans, T. Bostrom, L. GrØndahl, J. Mater. Sci. Mater. Med.14 (2003) 311.[24] L.M. Rodríguez-Lorenzo, K.A. Gross, J. Mater. Sci. Mater. Med. 11 (2003) 939.[25] A. Boumer, M. Ganteoume, W.E. Klee, Bull. Mineral. 108 (1985) 145.[26] L. Shufeng, I. Hiroshi, O. Michiharu, W. Taku, J. Biomech. Sci. Eng. 3 (2008) 1.[27] M. Barsoum, Sintering, Grain Growth Fundamentals of Ceramics, McGraw-Hill,
New York, 1997.[28] W.D. Kingery, H.K. Bowen, D.R. Uhlmann, Introduction to Ceramics, Wiley, New
York, 1976.

1398 H. Eslami et al. / Materials Science and Engineering C 29 (2009) 1387–1398
[29] M. Kay, R.A. Young, A.S. Posner, Nature 204 (1964) 1050.[30] S.M. Barinov, L.I. Shvorneva, D. Ferro, I.V. Fadeeva, S.V. Tumanov, Sci. Tech. Adv.
Mater. 5 (2004) 537.[31] M. Kikuchi, A. Yamazaki, M. Akao, H. Akao, Mineral. J. 18 (1996) 79.[32] A. Hadrich, A. Lautic, T.J. Mhiri, J. Raman Spectrosc. 32 (2001) 33.[33] S. Cazalbou, C. Combes, D. Eichert, C. Rey, J. Mater. Chem. 14 (2004) 2148.[34] H. Eslami, M. Solati-Hashjin, M. Tahriri, J. Ceram Process. Res. 9 (2008) 224.[35] R.W. Rice, C.C. Wu, F. Borchelt, J. Am. Ceram. Soc. 77 (1994) 2539.[36] A. Krell, P. Blank, J. Am. Ceram. Soc. 78 (1995) 1118.[37] T.P. Hoepfner, E.D. Case, Ceram. Int. 29 (2003) 699.
[38] M. Kobune, A. Mineshige, S. Fujii, H. Iida, J. Ceram. Soc. Jpn. 105 (1997) 210.[39] G. Muralithran, S. Ramesh, Ceram. Int. 26 (2000) 221.[40] N. Thangamani, K. Chinnakali, F.D. Gnanam, Ceram. Int. 28 (2002) 355.[41] K.A. Gross, K.A. Bhadang, Biomaterials 25 (2004) 1395.[42] T.M. Lee, E. Cheng, C.Y. Yang, Biomaterials 25 (2004) 23.[43] S.V. Dorozhkin, Prog. Cryst. Growth Charact. Mater. 44 (2002) 45.[44] L.J. Pullen, K.A. Gross, J. Mater. Sci. Mater. Med. 16 (2005) 399.[45] K.H. Eggen, G. Rolla, Scand. J. Dent. Res. 91 (1983) 347.[46] K. Cheng, W. Weng, H. Wang, S. Zhang, Biomaterials 26 (2005) 6288.





























