Embed Size (px)
Citation preview

The role of adsorption
C. Sanchez and E. Leiva
Volume 2, Part 1, pp 4–13
in
Handbook of Fuel Cells – Fundamentals, Technology and Applications(ISBN: 0-471-49926-9)
Edited by
Wolf VielstichArnold Lamm
Hubert A. Gasteiger
John Wiley & Sons, Ltd, Chichester, 2003

Chapter 2The role of adsorption
C. Sanchez and E. LeivaUnidad de Matematica y Fısica, Facultad de Ciencias Quımicas, INFIQC, Universidad Nacional de Cordoba, Argentina
1 THE ROLE OF ADSORPTION
1.1 Physisorption and chemisorption
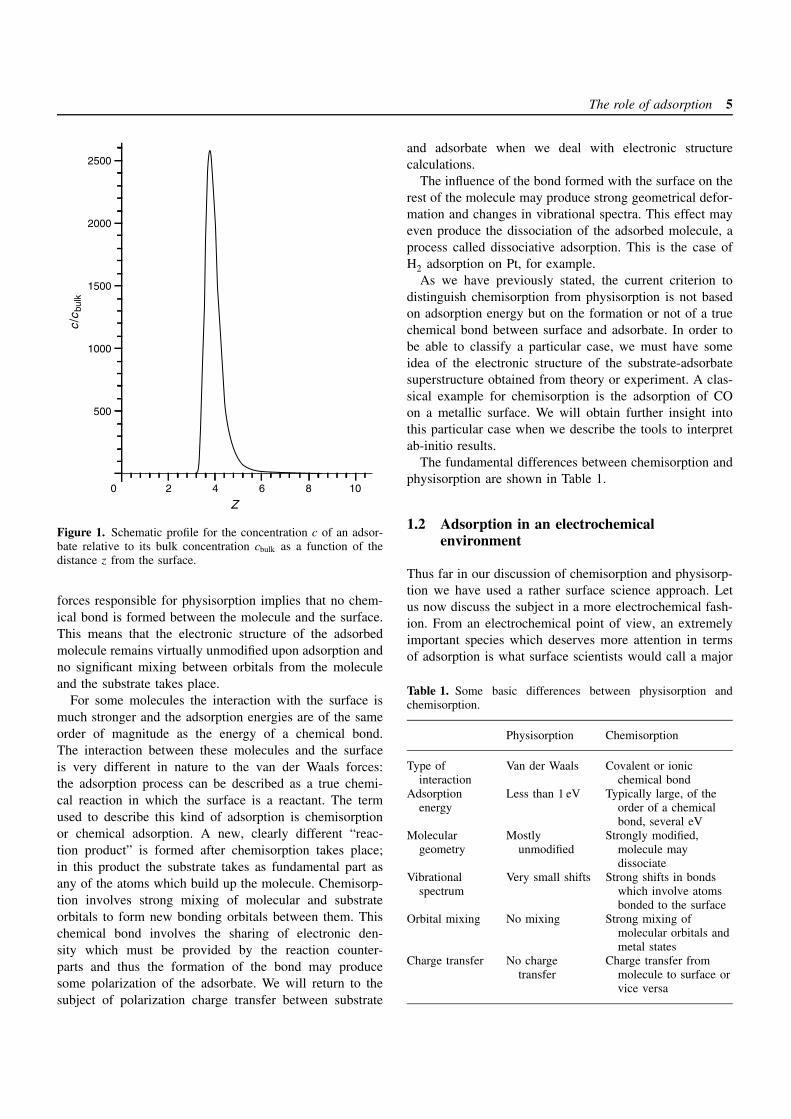
Adsorption is a phenomenon that occurs at interfaces andleads to a concentration of the adsorbed species in theinterfacial region larger than the average concentration inthe bulk fluid (gas or liquid) phase. In Figure 1 the con-centration profile, c(z), of a certain atom or molecule hasbeen plotted as a function of the distance to the surface.Far from the surface, the concentration acquires a con-stant value, the bulk concentration in the fluid phase. Nearthe surface, for small z, the concentration of the speciesdeviates strongly from the bulk concentration; the speciesaccumulate in the neighborhood of the surface; they are thusadsorbed. This increase in concentration within the interfa-cial region is caused by a favorable or attractive interactionbetween the surface and the adsorbed molecule or atom.There is a broad spectrum of interactions which may leadto adsorption, from very weak dispersion forces to differ-ent strengths and types of chemical bonding. The terms“physical” and “chemical” adsorption are a first attempt toclassify the possible interactions within this broad spectrum.These two extreme types of adsorption were originally dis-tinguished in terms of energy or, more specifically, heat ofadsorption.
Physisorption is the result of a weak interaction betweenthe adsorbed molecule (we shall use the term molecule toindicate molecules or atoms) and the surface and hencethe heat of adsorption is rather low. This weak interactionof the Van der Waals type is the result of the electricalforces that appear between polar or neutral molecules and
the surface. In the case of molecules that have a perma-nent dipole moment, the interaction between this dipoleand the permanent and the induced surface dipole resultsin an attractive force. Even for neutral molecules, thisattractive force exists and is called dispersion or Lon-don force. The cause of this is the quantum mechanicalcorrelation between the electronic wave functions of themolecule and the surface. Being quantum mechanical inorigin, this interaction is difficult to explain in classicalterms. Nevertheless, we may try to do so by thinking aboutthe instantaneous fluctuations of a neutral charge distribu-tion which result in transient dipole or multipole moments.When two molecules or a molecule and a surface are suf-ficiently near each other, these fluctuations are no longerindependent but the correlation between them causes anattraction force. This picture aids the interpretation of thephenomenon but must not be taken too literally since thequantum mechanical correlation is not time-dependent inthe same sense as classical fluctuations. The magnitudeof physisorption energy is that of the sublimation energyof molecules; since the forces that keep molecular solidstogether are of the same kind, these energies are below1.0 eV. Since the interaction with the surface is weak, weexpect that a physisorbed molecule may keep its geometricstructure and vibrational spectra upon adsorption more orless unmodified. Extreme and therefore typical candidatesfor physisorption are noble gas atoms adsorbed over anykind of surface.
We must keep in mind that nowadays adsorption energyis not a valid criterion to distinguish between chemisorbedand physisorbed molecules. The weak dispersion or dipolar
Handbook of Fuel Cells – Fundamentals, Technology and Applications, Edited by Wolf Vielstich, Hubert A. Gasteiger, Arnold Lamm.Volume 2: Electrocatalysis. 2003 John Wiley & Sons, Ltd. ISBN: 0-471-49926-9.
The role of adsorption 5
2500
500
1000
1500
2000
c/c
bulk
0 2 8 104 6
Z
Figure 1. Schematic profile for the concentration c of an adsor-bate relative to its bulk concentration cbulk as a function of thedistance z from the surface.
forces responsible for physisorption implies that no chem-ical bond is formed between the molecule and the surface.This means that the electronic structure of the adsorbedmolecule remains virtually unmodified upon adsorption andno significant mixing between orbitals from the moleculeand the substrate takes place.
For some molecules the interaction with the surface ismuch stronger and the adsorption energies are of the sameorder of magnitude as the energy of a chemical bond.The interaction between these molecules and the surfaceis very different in nature to the van der Waals forces:the adsorption process can be described as a true chemi-cal reaction in which the surface is a reactant. The termused to describe this kind of adsorption is chemisorptionor chemical adsorption. A new, clearly different “reac-tion product” is formed after chemisorption takes place;in this product the substrate takes as fundamental part asany of the atoms which build up the molecule. Chemisorp-tion involves strong mixing of molecular and substrateorbitals to form new bonding orbitals between them. Thischemical bond involves the sharing of electronic den-sity which must be provided by the reaction counter-parts and thus the formation of the bond may producesome polarization of the adsorbate. We will return to thesubject of polarization charge transfer between substrate
and adsorbate when we deal with electronic structurecalculations.
The influence of the bond formed with the surface on therest of the molecule may produce strong geometrical defor-mation and changes in vibrational spectra. This effect mayeven produce the dissociation of the adsorbed molecule, aprocess called dissociative adsorption. This is the case ofH2 adsorption on Pt, for example.
As we have previously stated, the current criterion todistinguish chemisorption from physisorption is not basedon adsorption energy but on the formation or not of a truechemical bond between surface and adsorbate. In order tobe able to classify a particular case, we must have someidea of the electronic structure of the substrate-adsorbatesuperstructure obtained from theory or experiment. A clas-sical example for chemisorption is the adsorption of COon a metallic surface. We will obtain further insight intothis particular case when we describe the tools to interpretab-initio results.
The fundamental differences between chemisorption andphysisorption are shown in Table 1.
1.2 Adsorption in an electrochemicalenvironment
Thus far in our discussion of chemisorption and physisorp-tion we have used a rather surface science approach. Letus now discuss the subject in a more electrochemical fash-ion. From an electrochemical point of view, an extremelyimportant species which deserves more attention in termsof adsorption is what surface scientists would call a major
Table 1. Some basic differences between physisorption andchemisorption.
Physisorption Chemisorption
Type ofinteraction
Van der Waals Covalent or ionicchemical bond
Adsorptionenergy
Less than 1 eV Typically large, of theorder of a chemicalbond, several eV
Moleculargeometry
Mostlyunmodified
Strongly modified,molecule maydissociate
Vibrationalspectrum
Very small shifts Strong shifts in bondswhich involve atomsbonded to the surface
Orbital mixing No mixing Strong mixing ofmolecular orbitals andmetal states
Charge transfer No chargetransfer
Charge transfer frommolecule to surface orvice versa

6 Part 1: Introduction
contaminant present in all electrochemical experiments, thesolvent. Water is in most cases the solvent selected to carryout electrochemistry but, in spite of this, relatively littleattention is given to the features of its interaction with thesurface and with other adsorbates. Electrochemistry has anadvantage over surface science to study processes in the sur-face in that the chemical potential of some adsorbed speciescan be easily controlled with a potentiostat. In surface sci-ence, equivalent control of the chemical potential of speciesin the gaseous phase requires precise control of the partialpressure. Clearly the apparatus needed for either case differsdrastically: potentiostatic experiments can be carried out farmore easily. Furthermore, electrochemical potential of elec-trons depends linearly on potential, but chemical potentialin the gaseous phase is proportional to the logarithm ofthe pressure. In order to change significantly the chemicalpotential of a gaseous species, pressure must change severalorders of magnitude while in electrochemical experimentsjust a fraction of a volt is enough. Another great advan-tage of electrochemistry over vacuum surface science is thatsurface contamination from unwanted species can be easilyreduced by protecting the surface with the solution. A majordrawback is that this protection introduces, as we said, anew contaminant in enormous concentration: water. Theelectrochemical double layer owes its complex structure toa variety of adsorption phenomena of water and electrolytespecies; furthermore this structure changes drastically withthe applied potential difference.
The state-of-the-art knowledge of water adsorption inUHV up to the late 80s has been reviewed in a verydetailed form by Thiel and Madey.[1] The main conclu-sions of this review work, which come from theoreticaland experimental sources, are that water bonds throughthe oxygen atom to the surface. This bonding occurs withcharge transfer to the surface lowering the work function ofthe metal, while the internal molecular structure and vibra-tional properties of water are only slightly perturbed. Theadsorption energy of water varies between 0.4 and 0.7 eV.Thus, water is a weakly chemisorbed species when com-pared to other adsorbates such as CO. The low adsorptionenergy means that adsorbed water molecules tend to makehydrogen bonds between each other even at low coverages,thus lowering their energy. Because of this, it is experimen-tally very difficult to study isolated adsorbed molecules.Water clustering is favored by the fact that the diffusionbarrier seems to be quite low allowing for a large mobil-ity of the monomers. The proposed structure, based ondiffraction experiments for high coverage layers and lowcoverage clusters over hexagonal surfaces, is the bilayerstructure shown in Figure 2. This structure consists of anice-like hydrogen bonded network in which the first layeris bound to the metallic surface through oxygen atoms and
Firstlayer
Secondlayer
Figure 2. Schematic diagram of an adsorbed water bilayer overan hexagonal surface. (Reprinted from Thiel and Madey (1987)[1]
with permission from Elsevier Science). Large empty circlesdenote surface atoms, small black-filled circles represent hydrogenatoms in water molecules. The remaining circles denote oxy-gen atoms belonging to adsorbed water molecules. The (
√3 ×√
3)R30◦ unit cell is outlined.
the second layer is held by hydrogen bonding to the firstlayer. The stability of this structure is related to the latticemismatch between ice and the underlying surface.
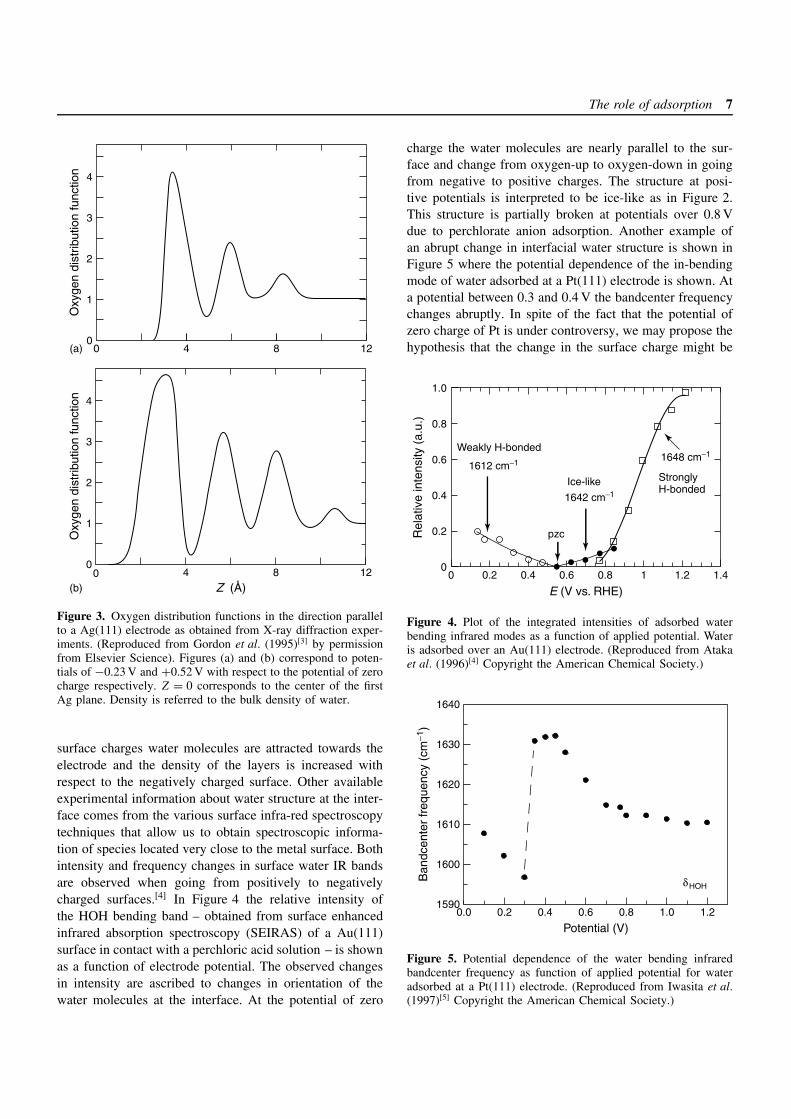
The structure of water at the electrochemical interface isfurther complicated by the presence of an external electricfield. We may clearly distinguish three different circum-stances: the surface at the potential of zero charge, thesurface at a potential more positive than the potential ofzero charge bearing a positive charge, and the surface atmore negative potentials than the potential of zero chargebearing a negative charge. Due to the strong dipole momentof water and the high electric fields present at the interface,we expect that the structure of the water–metal interface inthe region close to the metal will present significant differ-ences between these three situations. The most completeinformation to date of the microscopic structure of thisinterface comes from X-ray diffraction experiments as car-ried out by Toney’s group.[2, 3] The information obtainedfrom these experiments is presented in terms of the oxygendistribution function in the neighborhood of the electrode.In Figure 3 this is plotted for more positive and more neg-ative potentials than the potential of zero charge for aAg(111) surface. These distribution functions represent thelocal increase or decrease of oxygen density at a certaindistance from the surface with respect to the bulk aver-age value. These plots show water adsorbed on the surfacein a similar fashion to the qualitative plot in Figure 1. Aswe may clearly see from the three peaks next to the sur-face in both graphs, the influence of the surface producesa layering of water molecules that extends up to three lay-ers towards the bulk. The differences between positivelyand negatively charged surfaces are important; at positive
The role of adsorption 7
Oxy
gen
dist
ribut
ion
func
tion
00
4
2
1
3
4 8 12(a)
Oxy
gen
dist
ribut
ion
func
tion
00
4
2
1
3
4 8 12
Z (Å)(b)
Figure 3. Oxygen distribution functions in the direction parallelto a Ag(111) electrode as obtained from X-ray diffraction exper-iments. (Reproduced from Gordon et al. (1995)[3] by permissionfrom Elsevier Science). Figures (a) and (b) correspond to poten-tials of −0.23 V and +0.52 V with respect to the potential of zerocharge respectively. Z = 0 corresponds to the center of the firstAg plane. Density is referred to the bulk density of water.
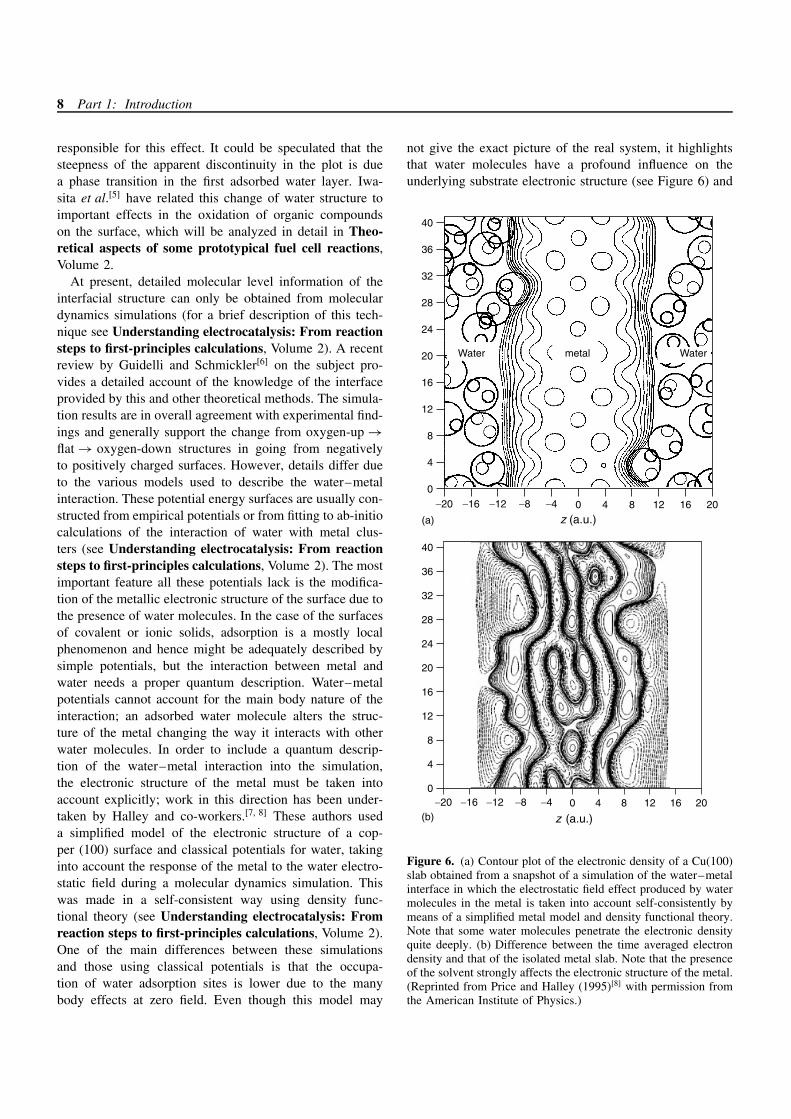
surface charges water molecules are attracted towards theelectrode and the density of the layers is increased withrespect to the negatively charged surface. Other availableexperimental information about water structure at the inter-face comes from the various surface infra-red spectroscopytechniques that allow us to obtain spectroscopic informa-tion of species located very close to the metal surface. Bothintensity and frequency changes in surface water IR bandsare observed when going from positively to negativelycharged surfaces.[4] In Figure 4 the relative intensity ofthe HOH bending band – obtained from surface enhancedinfrared absorption spectroscopy (SEIRAS) of a Au(111)surface in contact with a perchloric acid solution – is shownas a function of electrode potential. The observed changesin intensity are ascribed to changes in orientation of thewater molecules at the interface. At the potential of zero
charge the water molecules are nearly parallel to the sur-face and change from oxygen-up to oxygen-down in goingfrom negative to positive charges. The structure at posi-tive potentials is interpreted to be ice-like as in Figure 2.This structure is partially broken at potentials over 0.8 Vdue to perchlorate anion adsorption. Another example ofan abrupt change in interfacial water structure is shown inFigure 5 where the potential dependence of the in-bendingmode of water adsorbed at a Pt(111) electrode is shown. Ata potential between 0.3 and 0.4 V the bandcenter frequencychanges abruptly. In spite of the fact that the potential ofzero charge of Pt is under controversy, we may propose thehypothesis that the change in the surface charge might be
1.0
0.8
0.6
0.4
0.2
00 0.2 0.4 0.6 0.8 1 1.2 1.4
E (V vs. RHE)
Weakly H-bonded
1612 cm−1
1642 cm−1
1648 cm−1
Ice-like
pzc
StronglyH-bonded
Rel
ativ
e in
tens
ity (
a.u.
)
Figure 4. Plot of the integrated intensities of adsorbed waterbending infrared modes as a function of applied potential. Wateris adsorbed over an Au(111) electrode. (Reproduced from Atakaet al. (1996)[4] Copyright the American Chemical Society.)
1640
1630
1620
1610
1600
15900.0 0.2 0.4 0.6 0.8 1.0 1.2
Potential (V)
Ban
dcen
ter
freq
uenc
y (c
m−1
)
δHOH
Figure 5. Potential dependence of the water bending infraredbandcenter frequency as function of applied potential for wateradsorbed at a Pt(111) electrode. (Reproduced from Iwasita et al.(1997)[5] Copyright the American Chemical Society.)
8 Part 1: Introduction
responsible for this effect. It could be speculated that thesteepness of the apparent discontinuity in the plot is duea phase transition in the first adsorbed water layer. Iwa-sita et al.[5] have related this change of water structure toimportant effects in the oxidation of organic compoundson the surface, which will be analyzed in detail in Theo-retical aspects of some prototypical fuel cell reactions,Volume 2.
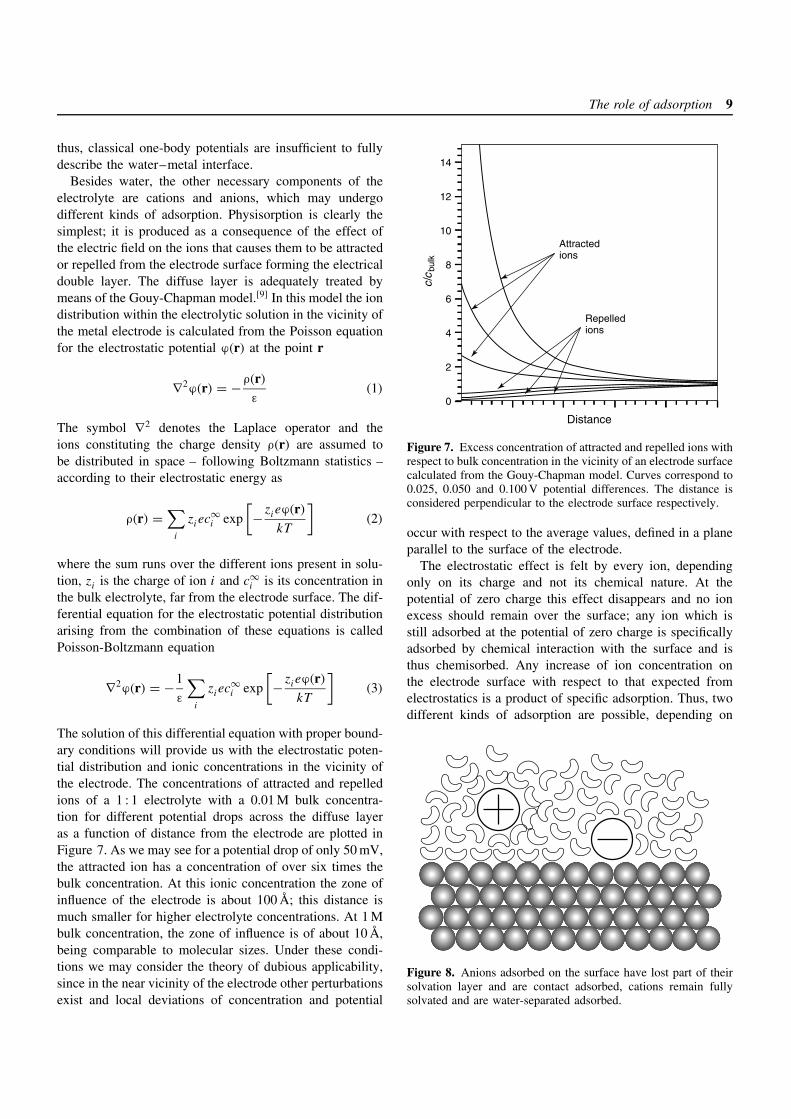
At present, detailed molecular level information of theinterfacial structure can only be obtained from moleculardynamics simulations (for a brief description of this tech-nique see Understanding electrocatalysis: From reactionsteps to first-principles calculations, Volume 2). A recentreview by Guidelli and Schmickler[6] on the subject pro-vides a detailed account of the knowledge of the interfaceprovided by this and other theoretical methods. The simula-tion results are in overall agreement with experimental find-ings and generally support the change from oxygen-up →flat → oxygen-down structures in going from negativelyto positively charged surfaces. However, details differ dueto the various models used to describe the water–metalinteraction. These potential energy surfaces are usually con-structed from empirical potentials or from fitting to ab-initiocalculations of the interaction of water with metal clus-ters (see Understanding electrocatalysis: From reactionsteps to first-principles calculations, Volume 2). The mostimportant feature all these potentials lack is the modifica-tion of the metallic electronic structure of the surface due tothe presence of water molecules. In the case of the surfacesof covalent or ionic solids, adsorption is a mostly localphenomenon and hence might be adequately described bysimple potentials, but the interaction between metal andwater needs a proper quantum description. Water–metalpotentials cannot account for the main body nature of theinteraction; an adsorbed water molecule alters the struc-ture of the metal changing the way it interacts with otherwater molecules. In order to include a quantum descrip-tion of the water–metal interaction into the simulation,the electronic structure of the metal must be taken intoaccount explicitly; work in this direction has been under-taken by Halley and co-workers.[7, 8] These authors useda simplified model of the electronic structure of a cop-per (100) surface and classical potentials for water, takinginto account the response of the metal to the water electro-static field during a molecular dynamics simulation. Thiswas made in a self-consistent way using density func-tional theory (see Understanding electrocatalysis: Fromreaction steps to first-principles calculations, Volume 2).One of the main differences between these simulationsand those using classical potentials is that the occupa-tion of water adsorption sites is lower due to the manybody effects at zero field. Even though this model may
not give the exact picture of the real system, it highlightsthat water molecules have a profound influence on theunderlying substrate electronic structure (see Figure 6) and
−20 −16 −12 −8 −4 0 4 8 12 16 20
40
36
32
28
24
20
16
12
8
4
0
z (a.u.)(b)
−20 −16 −12 −8 −4 0 4 8 12 16 20
0
4
8
12
16
20
24
28
32
36
40
z (a.u.)
metal WaterWater
(a)
Figure 6. (a) Contour plot of the electronic density of a Cu(100)slab obtained from a snapshot of a simulation of the water–metalinterface in which the electrostatic field effect produced by watermolecules in the metal is taken into account self-consistently bymeans of a simplified metal model and density functional theory.Note that some water molecules penetrate the electronic densityquite deeply. (b) Difference between the time averaged electrondensity and that of the isolated metal slab. Note that the presenceof the solvent strongly affects the electronic structure of the metal.(Reprinted from Price and Halley (1995)[8] with permission fromthe American Institute of Physics.)
The role of adsorption 9
thus, classical one-body potentials are insufficient to fullydescribe the water–metal interface.
Besides water, the other necessary components of theelectrolyte are cations and anions, which may undergodifferent kinds of adsorption. Physisorption is clearly thesimplest; it is produced as a consequence of the effect ofthe electric field on the ions that causes them to be attractedor repelled from the electrode surface forming the electricaldouble layer. The diffuse layer is adequately treated bymeans of the Gouy-Chapman model.[9] In this model the iondistribution within the electrolytic solution in the vicinity ofthe metal electrode is calculated from the Poisson equationfor the electrostatic potential ϕ(r) at the point r
∇2ϕ(r) = −ρ(r)ε
(1)
The symbol ∇2 denotes the Laplace operator and theions constituting the charge density ρ(r) are assumed tobe distributed in space – following Boltzmann statistics –according to their electrostatic energy as
ρ(r) =∑
i
ziec∞i exp
[−zieϕ(r)
kT
](2)
where the sum runs over the different ions present in solu-tion, zi is the charge of ion i and c∞
i is its concentration inthe bulk electrolyte, far from the electrode surface. The dif-ferential equation for the electrostatic potential distributionarising from the combination of these equations is calledPoisson-Boltzmann equation
∇2ϕ(r) = −1
ε
∑i
ziec∞i exp
[−zieϕ(r)
kT
](3)
The solution of this differential equation with proper bound-ary conditions will provide us with the electrostatic poten-tial distribution and ionic concentrations in the vicinity ofthe electrode. The concentrations of attracted and repelledions of a 1 : 1 electrolyte with a 0.01 M bulk concentra-tion for different potential drops across the diffuse layeras a function of distance from the electrode are plotted inFigure 7. As we may see for a potential drop of only 50 mV,the attracted ion has a concentration of over six times thebulk concentration. At this ionic concentration the zone ofinfluence of the electrode is about 100 A; this distance ismuch smaller for higher electrolyte concentrations. At 1 Mbulk concentration, the zone of influence is of about 10 A,being comparable to molecular sizes. Under these condi-tions we may consider the theory of dubious applicability,since in the near vicinity of the electrode other perturbationsexist and local deviations of concentration and potential
Attractedions
Repelledions
Distance
0
2
4
6
8
10
12
14
c/c
bulk
Figure 7. Excess concentration of attracted and repelled ions withrespect to bulk concentration in the vicinity of an electrode surfacecalculated from the Gouy-Chapman model. Curves correspond to0.025, 0.050 and 0.100 V potential differences. The distance isconsidered perpendicular to the electrode surface respectively.
occur with respect to the average values, defined in a planeparallel to the surface of the electrode.
The electrostatic effect is felt by every ion, dependingonly on its charge and not its chemical nature. At thepotential of zero charge this effect disappears and no ionexcess should remain over the surface; any ion which isstill adsorbed at the potential of zero charge is specificallyadsorbed by chemical interaction with the surface and isthus chemisorbed. Any increase of ion concentration onthe electrode surface with respect to that expected fromelectrostatics is a product of specific adsorption. Thus, twodifferent kinds of adsorption are possible, depending on
Figure 8. Anions adsorbed on the surface have lost part of theirsolvation layer and are contact adsorbed, cations remain fullysolvated and are water-separated adsorbed.

10 Part 1: Introduction
whether the ion loses its solvation layer or not. In Figure 8the anions adsorbed on the surface have lost part of theirsolvation layer and are contact adsorbed, while cationsremain fully solvated and are water-separated adsorbed. Theinterplay between solvation energy and interaction with thesurface determines whether an ion does or does not losepart of its solvation layer upon adsorption. Since anionsare more loosely solvated by water, the general belief isthat cations tend to remain solvated and anions are con-tact adsorbed. Theoretical results from molecular dynamicssimulations are not conclusive in this respect,[6] since theresults of the simulations depend strongly on the potentialenergy function used for the interaction between ions andthe surface. However, as a general conclusion, cations seemto remain solvated while for anions results are less clear.
From our discussion up to this point we have made afairly complete account of possible adsorption processesthat involve non-electroactive species, such as water andelectrolyte ions. The solution structure in the near vicinityof the electrode is very different from that in the bulkelectrolyte due to water structuring, specific adsorption, andelectrostatic adsorption. This structure must have profoundchanges depending on the potential difference, particularlywith respect to the potential of zero charge. These processesoccur in all electrochemical interfaces and are in many casesunderestimated even though they are so common. In orderto undergo electrochemical reactions, most electroactivespecies have to reach the electrode surface and be adsorbed;water and ion structure over the surface must have profoundeffects on this. So we conclude that detailed knowledge ofthe structure in the near vicinity of the electrode is crucialfor understanding electrocatalytic processes.
1.3 Adsorption and electrocatalysis
In chemical kinetics it is common to decompose a moreor less complicated chemical reaction in a number ofevents, usually called elementary steps or simply reac-tion steps. Although some simple electrochemical reactionsexist where the main event may be described as a singleelectronic transfer, most of them involve some of the fol-lowing processes.
• Mass transport. This may affect reactants, intermediatesof products, and may occur via diffusion, convectionor migration. The first concept refers to the randommotion of particles, the second to the flow of the sol-vent and the third to the action of an electric field.Diffusion may occur in the neighborhood of the elec-trode, due to the depletion or accumulation of a species,or on the surface.
• Electronic transfer reactions. Nowadays it is acceptedthat the simultaneous transfer of two or more electronsis highly unlikely, so that each of the electronic transferof a given reaction can be considered an elementarystep. Every electrochemical reaction will involve atleast one of this type of steps.
• Chemical reactions. This may occur between speciesclose to the surface or adsorbed on the surface.
• Photochemical reactions. This is in the case where thesystem is being illuminated and some of the speciesare light sensitive. In the case of semiconductors,light may also be used to promote electrons from alower state (valence band) to higher energy levels (i.e.conduction band).
These processes are usually sequential, that is, they areassumed to occur one after the other. Thus, it is also usualin electrochemistry to think of a given electrochemicalreaction as a succession of steps, each of which involvessome of the processes mentioned above. For example, inthe case of the oxidation of hydrogen, where the globalreaction is
H2 −−−→ 2H+ + 2e (4)
One of the possible sequences of steps proposed for thisreaction to occur is
H2(aq) −−−→ H2(ads) (diffusion and adsorption)
(5)
H2(ads) −−−→ 2H(ads) (dissociation) (6)
H(ads) + H2O −−−→ H3O+(ads)
+ e(electrooxidation of atoms) (7)
H3O+(ads) −−−→ H3O+(aq) (transport of H+) (8)
This set of reactions is termed reaction mechanism. Depend-ing on the experimental setup employed for the measure-ment, one can measure the system under conditions inwhich the concentration of the different species evolveswith time, in which case one will be speaking about thetransient properties of the system. On the other hand, insome cases the measurement is undertaken under condi-tions in which the concentration of the substances takingpart in the reaction can be assumed to be time indepen-dent, thus the system will be said to be in a steady state.Transient techniques involve some control parameter, likethe speed rate of the potential difference applied to thesystem, which can be varied to probe one of some of thecharacteristic times of the system.
Since the abovementioned processes are of very differentnatures, it usually happens in the steady state that one

The role of adsorption 11
of them is considerably slower than the others. This iscalled rate determining step (RDS), and it can be assumedthat the rate of creation of products is determined by thereaction rate of this step. For example, considering thereaction sequence shown above, if the concentration ofH2 is low and the transport of the dissolved molecules isnot assisted by some convective means (i.e. rotating disk),the production rate of protons will be determined by therate of arrival of H2 molecules at the electrode surfacevia diffusion. This will be the RDS under the conditionsmentioned. On the other hand, if mass transport is grantedby the proper conditions, it may occur that the dissociationreaction (6) (Tafel reaction) is slow on the surface underconsideration as compared to the electrochemical reaction(7). Then, (6) will constitute the RDS. Furthermore, someother alternative reaction mechanism may be proposed. Forexample, electron transfer could take place through thealternative pathway
H2(ads) + H2O −−−→ H(ads) + H3O+
+ e(Heyrovsky reaction) (9)
followed then by (8). Thus, we see that several reactionmechanisms may be proposed for a given global reaction.
Electrochemical reactions for which no adsorbed inter-mediate exists are extremely rare and the electron transferstep or steps almost always involve adsorbed species. Thiskind of electrochemical reaction for which at least someintermediate is specifically adsorbed are called inner spherereactions. Outer sphere reactions are those for which nospecifically adsorbed intermediate exists, no new bonds areformed or broken and surfaces only provide an electronsource. Unless the product clearly forms a new phase asin the case of metallic deposition, inner sphere and outersphere reactions cannot be distinguished by just looking atthe global reaction. Even though the surface in this globalreaction apparently is not involved, it is necessary for thedevelopment of the reaction. In basic chemistry these kindof species that are necessary for a reaction to take place butdo not seem to participate in the global equation, are calledcatalysts. The kind of surface used to carry on the reac-tion is of uppermost importance. For example, the reverseof the reaction just described, the hydrogen evolution reac-tion, occurs easily on platinum but is extremely slow onmercury. Since the metallic surface takes an active rolein the formation of intermediates, the nature and particularstate of the surface is crucial for the success of the reaction.The metallic surface is then an electrocatalyst, a catalyst foran electron transfer reaction. If the rate determining step ofthe reaction involves an adsorbed species, and this is prob-ably the case, then the nature and structure of the metallicsurface will determine the speed of the reaction. Not only
does the structure of the metallic surface change from metalto metal but also the structure of the solution side of theinterface changes, mainly because the change in the poten-tial of zero charge causes a change of the surface charge.This change in the potential of zero charge may be pro-duced upon a specific surface by adsorption of a differentmetal with a different potential of zero charge. The chal-lenge of theoretical electrocatalysis is to understand the wayby which an electrocatalyst works, in order to improve itor to apply this knowledge to other similar systems. As inmany cases the steps in which the metal takes an active partin the electrochemical reaction are adsorption processes, thestudy and understanding of the adsorption phenomena arethe keys to success in this challenge.
1.4 Theoretical approaches to chemisorption
The interface is a highly anisotropic environment in whichdrastic changes of composition occur within a very nar-row spatial extent. The abrupt differences in the molecularstructure of the phases on either side of the interface, whichmake them so interesting and rich in features, turns theminto a nightmare from the theoretical point of view. Theelectrochemical interface is particularly pathologic in thissense when compared to the much simpler metal-gas inter-face. It puts together a handful of problems that are hugescientific problems by themselves, interacting in a non-linear way, namely, an externally polarized metal surface, ahighly complex liquid such as water, electron transfer, sol-vation dynamics and chemisorption, to name only a few.This makes the theoretical description of the electrochemi-cal interface a singular problem, so complex that the subjectstill remains in its very initial steps, in spite of the greateffort dedicated to its study. In what follows, we will givea brief account of what we may call a “modern” theoryof chemisorption. This theory or group of models is nowa-days more or less established to study adsorption in themetal vacuum interphase, but the task to extend the meth-ods to the study of the electrochemical interphase is stillin its early infancy. We intend to follow a perturbativeapproach and after we have established the methods for themetal–vacuum interphase in Understanding electrocatal-ysis: From reaction steps to first-principles calculations,Volume 2, we shall study the main differences that the elec-trochemical interphase poses and how to deal with thesedifferences.
There is no such thing as a single theory of chemisorptionwhich can describe all possible processes and aspects. Thedescription of such a complex phenomenon certainly cannotcome from a single model or theory but from a groupof them which are suited for the description of particular

12 Part 1: Introduction
aspects of the problem. The methods used to describechemisorption from a theoretical point of view may bedivided in three large groups:
1. Methods for the construction of potential energy sur-faces and the study of the electronic structure such asdensity functional theory;
2. Methods to describe equilibrium states and their prop-erties such as thermodynamic Monte Carlo or latticegas approaches;
3. Methods to describe dynamic processes such as transi-tion state theory, stochastic reaction theories, dynamicMonte Carlo, classical trajectory calculations, quan-tum dynamics and molecular dynamics in their vari-ous forms.
Methods in items 2 and 3 are used to describe nuclearmotion and they all need to set up a Hamiltonian operatorfor this motion which must include some kind of potentialenergy surface. So the first group of methods, which allowus to build potential energy surfaces, is of foremost impor-tance. The accuracy of the results provided by methods ingroups 2 and 3 is a priori limited by the accuracy of thepotential energy surface used to build the nuclear Hamil-tonian. This is why special attention must be paid to theconstruction of this potential energy surface.
Implicit in our classification of the methods in these threegroups is the decoupling between electronic and nuclearmotion. This decoupling, also called Born-Oppenheimerapproximation, is what allows us to describe the electronicstructure problem separated from the nuclear motion prob-lem. The approximation is justified by the enormous massdifference between electrons and nuclei. It is supposed thatelectrons in their ground state follow instantaneously thenuclear dynamics, given that nuclear motion is extremelyslow in the electron time scale. This allows us to solveseparately the electronic problem from the nuclear one (aproblem may arise from potential energy surfaces of dif-ferent states crossing for some nuclear configurations; wewill get into more detail about this latter) factoring the sys-tem wave function into a product of nuclear φ({RI }) andelectronic ψ({RI }, {ri}) wave functions
�({RI }, {ri}) = ψ({RI }, {ri})φ({RI }) (10)
where {RI } and {ri} represent the sets of nuclear and elec-tronic coordinates, respectively. Note that the electronicwave function depends parametrically on the nuclear coor-dinates. The solution of the electronic Schrodinger equation
Hel({RI })ψ({RI }, {ri}) = Eel({RI })ψ({RI }, {ri}) (11)
will provide us with the potential energy surface for nuclearmotion from the following function V ({RI }) once the
electronic eigenvalue is added to the coulombic repulsion
V ({RI }) = 1
2
∑I �=J
ZIZJ
|RI − RJ | + Eel({RI }) (12)
There are a great number of methods for the approximatesolution of the electronic Schrodinger equation (equa-tion (11)) with varying degrees of exactitude, from semi-empirical to fully ab-initio methods such as density func-tional theory. Even empirical potentials exist which willprovide potential energy surfaces without the need to solveequation (11), either from fitting of previous ab-initioresults or from fitting experimental data. We repeat anyway,that the choice of potential energy surface for the descrip-tion of nuclear motion is an important decision which willaffect all further results. From an experimental point ofview it might be compared with the use of impure sol-vents; no matter what further sophisticated experimentaltechniques is used in order to study the problem, the resultswill be severely limited by the quality of the stuff you startwith. With the advances in electronics and computer sciencenowadays, it is feasible to obtain fully ab-initio potentialenergy surfaces in large detail and this will be more andmore common in the future. So, we think this will be themethod of choice for the theoretical study of chemisorp-tion problems. In Understanding electrocatalysis: Fromreaction steps to first-principles calculations, Volume 2, adetailed description is given of the state-of-the-art methodto solve the electronic problem, Density Functional The-ory, which is clearly stated as the method of choice withintheoretical surface science.
Our knowledge of the potential energy surface does notnecessarily need to be complete in its full dimensional-ity; the knowledge of critical points, such as minima andsaddle points, or lower dimensionality manifolds of the sur-face may provide us with a wealth of information aboutthe chemical problem. Information about potential energyminima in the surface will give us information on energydifferences between equilibrium states at zero temperature;knowledge of curvature of the surface may allow us tostudy vibrational motion in approximate ways and extrap-olate our results to ambient temperature. If the minima aredeep enough this energy information may be used for theconstruction of a lattice gas Hamiltonian and to predictexperimental results such as thermal desorption spectra. Thelocation and energy of saddle points over the surface willprovide us with transition state characteristics which canbe used to calculate rate constants or decide between mul-tiple possible pathways for a reaction. In Understandingelectrocatalysis: From reaction steps to first-principlescalculations, Volume 2, examples of each of this are givenwhen we deal in detail with the corresponding methods.

The role of adsorption 13
Once we have a sufficiently detailed potential energysurface we must proceed to the application of the methodsin items 2 and 3 for the description of the equilibriumand dynamical properties of nuclear motion. The methodswhich provide the most complete and detailed informationnowadays are simulation methods such as Monte Carloand molecular dynamics in their different variants. Theproblem with these methods is that they require verydetailed potential energy surfaces which are very difficult toobtain ab-initio. This can be solved by the construction ofpotential energy functions fitted to sets of ab-initio resultsor by using ab-initio-tested semi-empirical potentials.
ACKNOWLEDGEMENTS
A fellowship for C. Sanchez from CONICET, financialsupport from CONICET, Agencia Cordoba Ciencia, Secy-tUNC, Program BID 1201/OC-AR PICT No. 06-04505, aswell as language assistance by Pompeya Falcon are grate-fully acknowledged.
REFERENCES
1. P. A. Thiel and T. E. Madey, Surf. Sci. Rep., 7, 211 (1987).
2. M. F. Toney, J. N. Howard, J. Richer, G. L. Borges, J. G. Gor-don, O. R. Melroy, D. G. Wiesler, D. Yee and B. Sorenson,Nature, 368, 444 (1994).
3. J. G. Gordon, O. R. Melroy and M. F. Toney, Electrochim.Acta, 40, 3 (1995).
4. K. Ataka, T. Yotsyanagi and M. Osawa, J. Phys. Chem., 10,10 664 (1996).
5. T. Iwasita, X. H. Xia, H. D. Liess and W. Vielstich, J. Phys.Chem. B, 101, 7542 (1997).
6. R. Guidelli and W. Schmickler, Electrochim. Acta, 45, 2317(2000).
7. J. W. Halley, A. Mazzolo, Y. Zhou and D. Price, J. Elec-troanal. Chem., 450, 273 (1998).
8. D. L. Price and J. W. Halley, J. Chem. Phys., 102, 6603(1995).
9. W. Schmickler, ‘Interfacial Electrochemistry’, Oxford Univer-sity Press, Oxford (1996).





























