Embed Size (px)
Citation preview

University of Ulm
Institute of General Physiology
Head Prof. Dr. Paul Dietl
The role of TRPV4 in membrane barrier integrity and
inhibition in stretch-induced pathological lung cellular
responses during mechanical ventilation
Dissertation
submitted to obtain the doctoral degree of Human Biology
of the Medical Faculty of Ulm University
Nicolas Pairet
Castres, Frankreich
2018

II
Amtierender Dekan: Prof. Dr. Thomas Wirth
Erstgutachter: Prof. Dr. Paul Dietl
Zweitgutachter: PD. Dr. Jürgen Schymeinsky
Tag der Promotion: 01.02.2019

Index
III
Index:
List of abbreviations ....................................................................................................................... V
1 Introduction ............................................................................................................................. 1
1.1 Transient receptor potential (TRP) channels an overview ....................................................... 1
1.2 Transient receptor potential cation channel subfamily V member 4 (TRPV4) ...................... 5
1.2.1 TRPV4 gene and structure ......................................................................................................... 5
1.2.2 Protein interaction and regulation of TRPV4 ............................................................................ 8
1.2.3 Chemical activation and inhibition of TRPV4......................................................................... 11
1.2.4 TRPV4 function and physiological activation ......................................................................... 13
1.3 Ventilator induced lung injury (VILI)...................................................................................... 19
1.4 Acute respiratory distress syndrome (ARDS) .......................................................................... 21
1.5 The role of TRPV4 in ARDS and VILI .................................................................................... 23
1.6 The aim of the thesis ................................................................................................................... 26
2 Methods .................................................................................................................................. 27
2.1 In vitro studies ............................................................................................................................. 27
2.1.1 TER measurement ................................................................................................................... 27
2.1.2 Vascular Permeability Assay ................................................................................................... 28
2.1.3 Calcium 6 assay on the FLIPRTETRA
........................................................................................ 29
2.1.4 TRPV4 agonism effect on LDH release .................................................................................. 30
2.1.5 RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay .................................................. 30
2.1.6 Cell-IQ®
................................................................................................................................... 31
2.1.7 TRPV4 agonism effect on cytokine release ............................................................................. 31
2.1.8 Uniaxial cell strain and microscopy......................................................................................... 32
2.1.9 Equibiaxial cell strain .............................................................................................................. 33
2.1.10 Cells ......................................................................................................................................... 33
2.2 In vivo studies .............................................................................................................................. 35
2.2.1 Effect of TRPV4 activation on vascular permeability ............................................................. 35
2.2.2 Murine mechanical ventilation model ..................................................................................... 36
2.3 Molecular biology assays ........................................................................................................... 37
2.3.1 Pierce™ BCA Protein Assay Kit ............................................................................................. 37
2.3.2 ELISA/MSD ............................................................................................................................ 38
2.3.3 Phospho/Total ERK1/2 assay .................................................................................................. 38
2.3.4 ATP release measurement ....................................................................................................... 39
2.3.5 LDH release ............................................................................................................................. 39
2.3.6 Human cAMP / Calcium Signaling PathwayFinder ................................................................ 40
2.4 Compounds ................................................................................................................................. 44
2.5 Calculations & Statistics ............................................................................................................ 44
2.6 Ethics statement .......................................................................................................................... 45

Index
IV
3 Results ..................................................................................................................................... 46
3.1 Results: Role of TRPV4 in regulating endothelial membrane integrity ................................ 46
3.1.1 TRPV4 mediated calcium influx ............................................................................................. 46
3.1.2 TER measurement in HUVECs ............................................................................................... 47
3.1.3 Effect of TRPV4 agonism on TER .......................................................................................... 48
3.1.4 TRPV4 agonism effect on vascular permeability assay with FITC-Dextran in HUVECs ...... 50
3.1.5 Effect of TRPV4 agonism and antagonism on TER ................................................................ 51
3.1.6 Effect of TRPV4 activation on vascular permeability in vivo ................................................. 53
3.1.7 Effect of TRPV4 activation on lung vascular permeability in Balb/c mice ............................. 54
3.1.8 Effect of TRPV4 activation and inhibition on lung vascular permeability in vivo .................. 56
3.1.9 TRPV4 antagonist reverses the effect of TRPV4 agonism ...................................................... 56
3.1.10 TRPV4 mediated cytotoxicity .................................................................................................. 58
3.1.11 Time point of TRPV4 induced cytotoxicity and calcium dependent TRPV4 induced
LDH release ........................................................................................................................ 61
3.1.12 Life cell imaging of HUVECs exposed to the TRPV4 agonist GSK1016790A ...................... 62
3.1.13 TRPV4 activation in the RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay ........... 64
3.2 Results: Role of TRPV4 in stretch induced pathological cellular response ........................... 66
3.2.1 Effect of TRPV4 agonism on cells Ca2+
influx........................................................................ 66
3.2.2 Effect of stretch on cells Ca2+
influx ........................................................................................ 67
3.2.3 Effect of TRPV4-agonist on cell cytokine release ................................................................... 69
3.2.4 Effect of TRPV4 antagonism on stretch induced cytokine release .......................................... 70
3.2.5 TRPV4 mediated regulation of genes in the Human cAMP / Calcium Signaling
PathwayFinder ......................................................................................................................... 72
3.2.6 Effect of stretch on macrophages cytokine release .................................................................. 76
3.2.7 TRPV4 antagonist effect on mechanical ventilation induced cytokine release
and permeability increase in vivo............................................................................................ 77
4 Discussion ............................................................................................................................... 80
4.1 Role of TRPV4 in regulating endothelial membrane integrity ............................................... 80
4.2 Role of TRPV4 in stretch induced pathological cellular response ......................................... 86
4.3 Summary and clinical relevance ............................................................................................... 92
4.4 Next steps .................................................................................................................................... 95
5 Abstract .................................................................................................................................. 98
6 References ............................................................................................................................ 100
Acknowledgement ........................................................................................................................ 119

List of abbreviations
V
List of abbreviations
4α-PDD 4α-phorbol 12,13 didecanoate
5´,6´-EET 5´,6´-epoxyeicosatrienoic acid
aa amino acid
AA arachidonic acid
AC Voltage
AECC American-European Consensus Conference
AIP4 Ubiquitin ligase Atrophin-1-interacting protein 4
Ag Agonist
AKAP79 A kinase anchoring Protein 79
ALI Acute lung injury
ANK Ankyrin repeats
Ant Antagonist
AQP2 Aquaporin 2
AQP5 Aquaporin 5
ARDS Acute respiratory distress syndrome
ATP Adenosine triphosphate
BAA Bisandrographolide A
BALF Bronchoalveolar lavage fluid
BCA Bicinchoninic acid
BKCa Ca2+
-sensitive large-conductance K+ channels
CaM Calmodulin
CCL Capacitance
CHO Chinese hamster ovary cells
CIRB Calmodulin/inositol 1,4,5-trisphophate receptor binding domain
COPD Chronic obstructive pulmonary disease
CT Computed tomography
Ctrl Control
DMAPP dimethylallyl pyrophosphate
DPBS Dulbecco's Phosphate-Buffered Saline
DRG Dorsal root ganglia
ECMO Extracorporeal membrane oxygenation
EET Epoxyeicosatrienoic acids
ER Endoplasmatic reticulum
ERK Extracellular signal Regulated Kinases

List of abbreviations
VI
EthD-III Ethidium homodimer III
FITC Fluorescein isothiocyanate
GM-CSF Granulocyte-Macrophage Colony Stimulating Factor
HBSS Hank´s Balanced Salt Solution
HCL Hydrochlorid acid
HUVEC Human umbilical vein endothelial cell
ICU Intensive Care Units
IL Interleukin
INT Iodonitrotetrazolium
IP3 Inositol 1,4,5-trisphophate
i.p. Intraperitoneal
i.t. Intratracheal
i.v. Intravenous
KC Keratinocyte chemoattractant chemokine
KCa2.3 Calcium-activated potassium channels
KO / -/- Knockout
LDH Lactate dehydrogenase
LPS Lipopolysaccharide
M1 Macrophage phenotype M1
M2 Macrophage phenotype M2
MACS Magnetic activated cell sorting
MAP7 Microtubule-associated protein 7 domain
MCP-1 Monocyte Chemoattractant Protein-1
M-CSF Macrophage Colony Stimulating Factor
MMP Matrix metalloproteinase
MSOF Multiple-system organ failure
MV Mechanical ventilation
NCI-H292 Human lung epithelial cells
NGF Nerve growth factor
NO Nitric oxide
OS-9 Osteosarcoma amplified 9 protein
OTRPC4 Osmosensitive transient receptor potential channel 4
P Pore domain
PACSIN-3 Protein kinace C and casein kinase substrate in neurons protein 3
PBMC Peripheral blood mononuclear cell
PBS Phosphate-Buffered Saline
PDE5 Phosphodiesterase 5

List of abbreviations
VII
PEEP Positive end-expiratory pressure
PIBS Phosphoinositide-binding site
PIP2 Phosphatidylinositol 4,5-biphosphate
PKA Protein kinases A
PKC Protein kinases C
PLA2 Phospholipase A2
p.o. Per-oral
PRD Proline-rich domain
PS Phosphatidylserine
RANTES Regulated upon activation, normal T cell expressed and secreted
chemokine
RR Ruthenium red
RVD Regulatory volume decreases
S Transmembrane spanning domain
SACs Stretch-activated ion channels
SAEC Small airway epithelial cell
SGK1 Serum glucocorticoid-induced protein kinase 1
SR Sarcoplasmatic reticulum
STIM1 Stromal interaction molecule 1
SU Subunit
TER Transepithelial/transendothelial electrical resistance
TM Transmembrane domain
TNF-α Tumor necrosis factor α
TRP Transient receptor potential channel
TRPA Transient receptor potential ankyrin channel
TRPC Transient receptor potential canonical channel
TRPM Transient receptor potential melastin channel
TRPML Transient receptor potential mucolipin channel
TRPP Transient receptor potential polycystin channel
TRPN / NOMPC No mechanoreceptor potential C channel
TRPV Transient receptor potential vanilloid channel
TRPV4 Transient receptor potential vanilloid type 4 channel
TRP12 Transient receptor potential channel 12
VEGF Vascular endothelial growth factor
Veh Vehicle
VILI Ventilation induced lung injury
VRL-2 Vanilloid receptor-like channel 2

List of abbreviations
VIII
VR-OAC Vanilloid receptor-related osmotically activated channel
WT Wild-type
Z Impedance

Introduction
1
1 Introduction
1.1 Transient receptor potential (TRP) channels an overview
Transient receptor potential (TRP) channels form an ion channel superfamily that is
involved in sensing and transmission of a plethora of external and internal stimuli (Yin and
Kuebler 2010). The first TRP channel was described in the Drosophila photoreceptor,
where a deletion of the trp gene led only to a transient response in the presence of
continuous light instead of a substained retinal depolarization (Minke 1977, Montell et al.
1985). Further investigations identified about 70 TRP channels in both invertebrates and
vertebrates (60 in zebrafish, 24 in nematodes, 16 in fruit flies and one in yeast) (Montell
2005). In mammals, 33 different TRP channels have been found so far (Montell 2001,
Clapham 2003). Based on amino acid homologies, the TRP channel superfamily can be
differentiated into seven main subfamilies: TRPA (ankyrin), TRPC (canonical), TRPV
(vanilloid), TRPM (melastin), TRPML (mucolipin), TRPP (polycystin) and TRPN (no
mechanoreceptor potential C, or NOMPC) (Clapham 2003). In humans and mice 28 TRP
channels have been identified with one member of TRPA, seven members of TRPC (with
TRPC2 as a pseudogene in humans), six members of TRPV, eight members of TRPM,
three members of TRPML and three members of TRPP (Venkatachalam and Montell
2007). TRPN is the only TRP subfamily not represented in mammals and have only been
found in worms, Drosophila and zebra fish (Clapham 2003, Montell 2005).
For all TRP channels the predicted subunit structure consists of six helix transmembrane
(TM) spanning domains (S1-S6) with a loop between the fifth (S5) and sixth (S6) TMs
(Figure 1A) forming a pore domain (P) (Clapham 2003, Hoenderop et al. 2003). The NH2
and COOH termini are located intracellularly in the cytoplasm and differ depending on the
TRP families with N-termini containing a various number of ankyrin repeats, a putative
caveolin-binding site and a predicted coiled coil region. The C-terminal comprises of a
TRP domain of about 23-25 amino acids that is loosely conserved in all TRP mammalian
subfamilies and encompass a highly conserved 6-amino acid TRP box1 (EWKFAR in
TRPCs) and a proline rich domain that has been referred as TRP box2. Depending on the
TRP subfamily the C-terminus can contain a calmodulin/inositol 1,4,5-trisphophate (IP3)
receptor binding (CIRB) domain, a coiled coil region, an enzyme domain and an PDZ
binding domain for protein-protein interactions (Montell 2005, Ramsey et al. 2006,
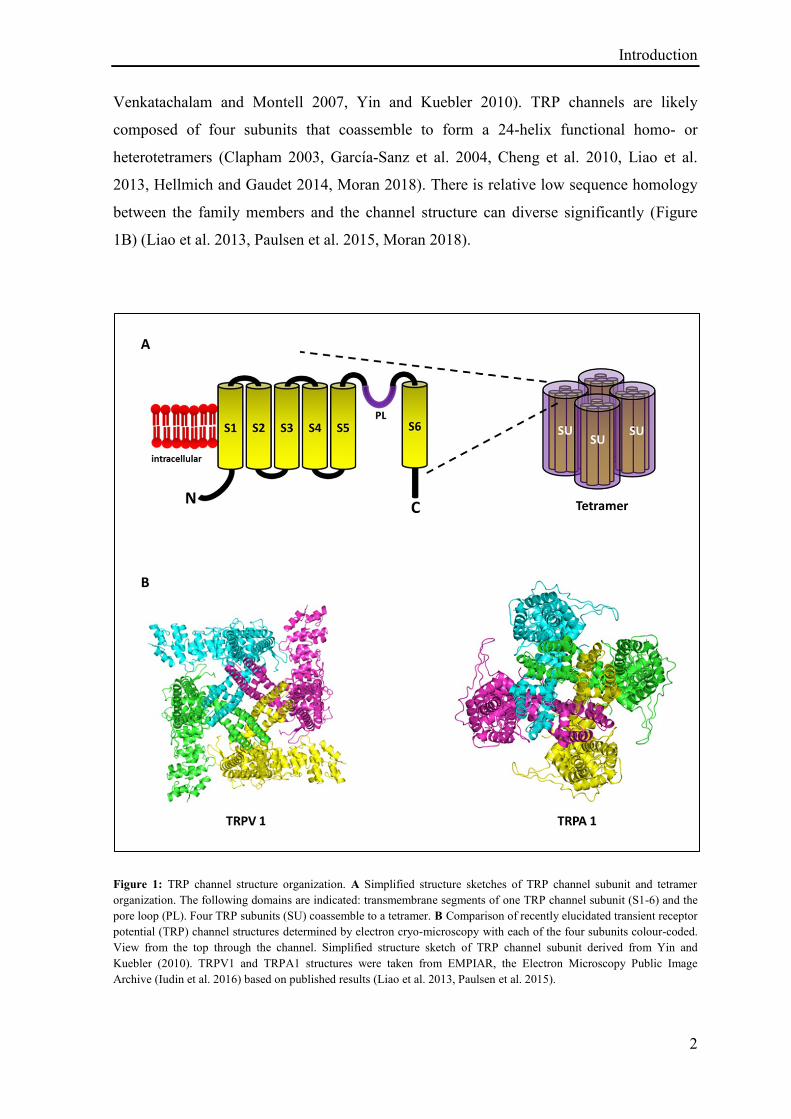
Introduction
2
Venkatachalam and Montell 2007, Yin and Kuebler 2010). TRP channels are likely
composed of four subunits that coassemble to form a 24-helix functional homo- or
heterotetramers (Clapham 2003, García-Sanz et al. 2004, Cheng et al. 2010, Liao et al.
2013, Hellmich and Gaudet 2014, Moran 2018). There is relative low sequence homology
between the family members and the channel structure can diverse significantly (Figure
1B) (Liao et al. 2013, Paulsen et al. 2015, Moran 2018).
Figure 1: TRP channel structure organization. A Simplified structure sketches of TRP channel subunit and tetramer
organization. The following domains are indicated: transmembrane segments of one TRP channel subunit (S1-6) and the
pore loop (PL). Four TRP subunits (SU) coassemble to a tetramer. B Comparison of recently elucidated transient receptor
potential (TRP) channel structures determined by electron cryo-microscopy with each of the four subunits colour-coded.
View from the top through the channel. Simplified structure sketch of TRP channel subunit derived from Yin and
Kuebler (2010). TRPV1 and TRPA1 structures were taken from EMPIAR, the Electron Microscopy Public Image
Archive (Iudin et al. 2016) based on published results (Liao et al. 2013, Paulsen et al. 2015).

Introduction
3
TRP channels are widely distributed throughout the body and are expressed in a large
number of tissues and excitable and nonexcitable cell types, including immune cells. They
are particularly expressed in sensory organs and receptor cells, pointing to their critical role
as cellular sensors for diverse signal sensation and transduction (Clapham 2003). The
majority of TRP channels are Ca2+
permeable non-selective cation channels (Montell 2001,
Owsianik et al. 2006) and are exceptional in the sense that they are polymodal and
activated by many types of different stimuli, such as temperature, pH, osmotic and
mechanical stress, pheromones, chemicals, intra- and extracellular messengers and
probably by the filling state of intracellular Ca2+
stores (Clapham 2003, Pedersen et al.
2005).
TRP channels are permeable for cations and except for only two TRP channels (TRPM4,
TRPM5) that are impermeable for calcium, all other TRP channels are Ca2+
permeable.
The permeability ratios between these channels vary significantly with PCa/PNa selectivity
ranging from 0.3 to ˃ 100 with TRPV5 and TRPV6 showing the highest Ca2+
permeability
(Pedersen et al. 2005, Owsianik et al. 2006). The Ca2+
concentration in extracellular
biologic fluids ranges from 1.6 to 2 mM. In contrast the cytosolic free Ca2+
concentration
is maintained by cells around 100 nM, meaning that for a cell at rest the [Ca2+
] is ~ 20.000
times lower in the cytoplasm than outside the cell, producing a high electrochemical
gradient of about 180 mV between the extracellular and intracellular (cytoplasmic) space
(Berridge et al. 2003, Clapham 2003, Bootman 2012). TRP channels modulate the cations
flux through the plasma membrane down an electrochemical gradient, thereby playing an
important role in raising the free intracellular Ca2+
concentration. In addition to their role
as plasmalemmal Ca2+
channels a number of studies also indicate that in some cases TRP
channels could also function as calcium release channels from organelles acting as
intracellular calcium store such as the endoplasmatic (ER) and sarcoplasmatic reticulum
(SR) (Pedersen et al. 2005, Bootman 2012). Changes in cytosolic free calcium
concentration has a fundamental role in cellular process and calcium entry through plasma
membrane channels is recognized as a cellular signalling event per se. Changes in
transmembrane voltage leads to central cellular events such as neurotransmitter release,
neuronal action potential propagation and muscle contraction, but Ca2+
entry also plays a
crucial role in nonexcitable cells by gating other voltage-dependent channels and affecting
effector proteins sensitive to elevated intracellular calcium concentrations and so

Introduction
4
controlling a plethora of cellular processes such as transcriptional regulation, proliferation,
cell death and migration (Berridge et al. 2003, Ramsey et al. 2006, Bootman 2012).
There is also considerable evidence that TRP channels are regulated by post-translational
mechanisms such as multimerization of TRP subunit to heterometric complexes,
translocation and interaction with membrane proteins may dramatically modulate TRP
channel function (Yin and Kuebler 2010) and that these regulatory processes may be
triggered via chemical or physical stimulation, demonstrated in the mechanical shear stress
induced translocation of TRPV4 and TRPM7 to the plasma membrane (Bezzerides et al.
2004, Oancea et al. 2006, Loot et al. 2008, Yin and Kuebler 2010). The gating mechanism
of TRP channels is poorly understood and it remains unclear whether they are directly
activated by a stimulus or indirectly via second messengers and serve rather as transducers
that are functionally activated by an upstream stimulus (Clapham 2003, Ramsey et al.
2006, Christensen and Corey 2007, Yin and Kuebler 2010).
TRP channels are involved in numerous fundamental cell functions, diverse physiological
processes and act as sensors for external irritants and inflammation products (Nilius et al.
2005). Because of their properties it is not surprising that an increasing number of
pathophysiological conditions and diseases are now been linked to TRP channels
(Pedersen et al. 2005). The importance of these channels is emphasized by the broad
number of genetic diseases caused by aberrant TRP functions leading to skeletal, skin,
sensory, cardiac, ocular and neuronal disturbance (Moran 2018). Other indications for the
role of TRP channels implication in diseases is shown by their correlation between the
level of channel expression and the disease symptoms, e.g. the abundance of TRPV1 is
higher in patients with gastrointestinal diseases such as inflammatory bowel disease,
Crohn´s disease and ulcerative colitis (Yiangou et al. 2001, Geppetti and Trevisani 2004,
Nilius et al. 2005) and TRPV1 expression is considerably increased in the airway nerves of
patients exhibiting chronic cough (Groneberg et al. 2004). Furthermore phenotypes of TRP
knockout mice and other transgenic models also point to the potential role of this channel
in diseases and allow a degree of extrapolation to their impact in human diseases, e.g.
TRPV4 knockout mice display a blunting of inflammation induced thermal hyperalgesia
(Todaka et al. 2004, Nilius et al. 2005, Nilius et al. 2007) and in a mouse model of chronic
itch, scratching evoked by impaired skin barrier was shown to be abolished in TRPA1-
deficient animals (Wilson et al. 2013). Compounds modulating TRP channels have been
studied in preclinical experiments targeting indications such as pain, atopic dermatitis, itch,

Introduction
5
disorders of the central nervous system and cardiovascular disorders with some of those
already entered clinical trials, e.g. a TRPV4 inhibitor from GlaxoSmithKline entered Phase
2 clinical trials as a potential treatment for pulmonary edema and reduced pulmonary gas
transfer in patients with heart failure, as well as TRPA1 antagonist entered Phase 1 trials,
where a significant reduction in pain scores was reported after treatment with the
antagonist GRC 17536 in patients with painful diabetic neuropathy who have intact
neuronal function (Moran 2018).
1.2 Transient receptor potential cation channel subfamily V member 4 (TRPV4)
The transient receptor potential vanilloid 4 (TRPV4) ion channel is a Ca2+
-permeable
nonselective cation channel (Yin and Kuebler 2010). It has a higher permeability to Ca2+
than to Ba2+
, Sr2+
or Mg2+
and in absence of divalent ions it is also permeate by
monovalent cations, such as K+, Cs
+, Rb
+, Na
+ and Li
+ and discriminates poorly between
them (Nilius et al. 2001, Voets et al. 2002). It is distributed widely throughout the body
and participates in the transduction of both chemical stimuli and physical stimuli such as
heat, pH, osmotic and mechanical stimuli (reviewed in Garcia-Elias et al. 2014). TRPV4
was first described in 2000 as a channel with a role in osmosensation and was initially
given different names: osmosensitive transient receptor potential channel 4 (OTRPC4),
vanilloid receptor-related osmotically activated channel (VR-OAC), vanilloid receptor-like
channel 2 (VRL-2), and transient receptor potential channel 12 (TRP12). Finally in 2002
the current nomenclature TRPV4 was accepted (Liedtke et al. 2000, Strotmann et al. 2000,
Wissenbach et al. 2000, Delany et al. 2001, Nilius et al. 2001, Garcia-Elias et al. 2014,
White et al. 2016).
1.2.1 TRPV4 gene and structure
The human TRPV4 gene is found in chromosome 12 at q23-q24.1 and has 15 exons with
five splice variants (TRPV4-A-E) (Arniges et al. 2006, Garcia-Elias et al. 2014).
Progesterone has been shown to reduce expression of TRPV4 in epithelial and vascular
smooth muscle cells (Jung et al. 2009). Other factors have been identified that increase
TRPV4 expression such as interleukin 1β and interleukin 17 in dorsal root ganglia (DRG)
neurons (Segond von Banchet et al. 2013) and nerve growth factor (NGF) in the
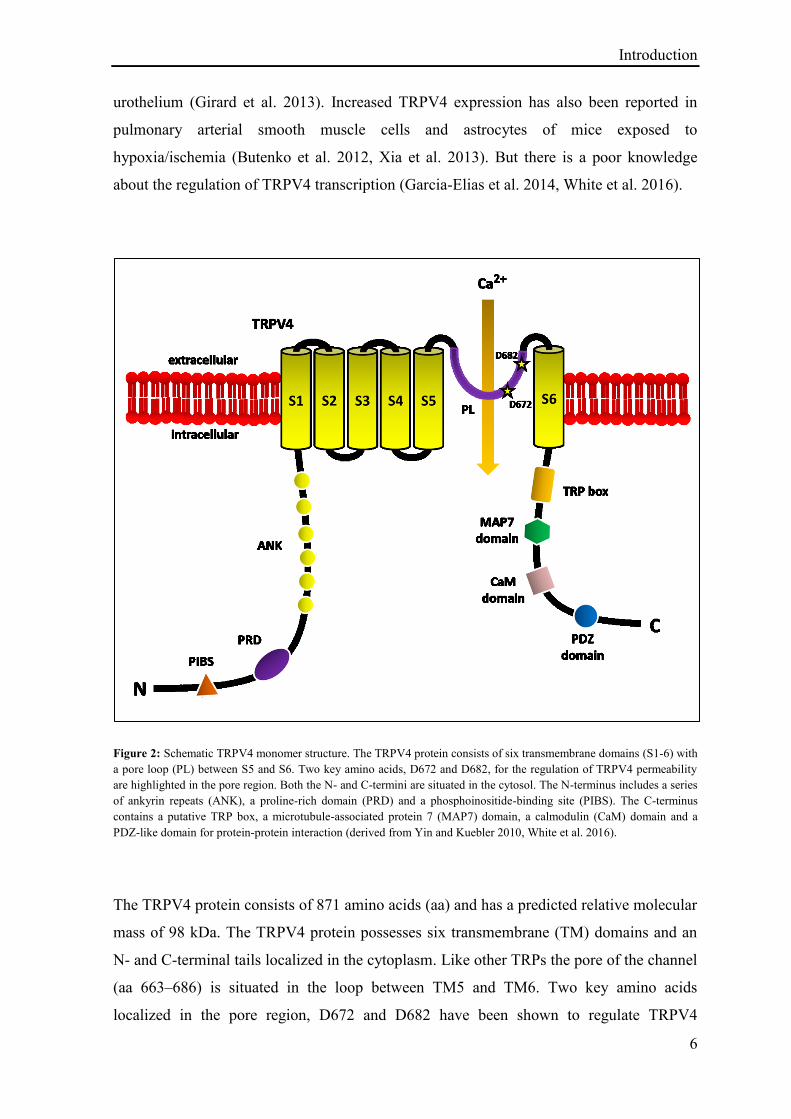
Introduction
6
urothelium (Girard et al. 2013). Increased TRPV4 expression has also been reported in
pulmonary arterial smooth muscle cells and astrocytes of mice exposed to
hypoxia/ischemia (Butenko et al. 2012, Xia et al. 2013). But there is a poor knowledge
about the regulation of TRPV4 transcription (Garcia-Elias et al. 2014, White et al. 2016).
Figure 2: Schematic TRPV4 monomer structure. The TRPV4 protein consists of six transmembrane domains (S1-6) with
a pore loop (PL) between S5 and S6. Two key amino acids, D672 and D682, for the regulation of TRPV4 permeability
are highlighted in the pore region. Both the N- and C-termini are situated in the cytosol. The N-terminus includes a series
of ankyrin repeats (ANK), a proline-rich domain (PRD) and a phosphoinositide-binding site (PIBS). The C-terminus
contains a putative TRP box, a microtubule-associated protein 7 (MAP7) domain, a calmodulin (CaM) domain and a
PDZ-like domain for protein-protein interaction (derived from Yin and Kuebler 2010, White et al. 2016).
The TRPV4 protein consists of 871 amino acids (aa) and has a predicted relative molecular
mass of 98 kDa. The TRPV4 protein possesses six transmembrane (TM) domains and an
N- and C-terminal tails localized in the cytoplasm. Like other TRPs the pore of the channel
(aa 663–686) is situated in the loop between TM5 and TM6. Two key amino acids
localized in the pore region, D672 and D682 have been shown to regulate TRPV4

Introduction
7
permeability (Figure 2). Neutralization of these two negatively charged residues decreases
the permeability for calcium and D682 has also been shown to participate in Ruthenium
red block (Voets et al. 2002, Garcia-Elias et al. 2014).
The N-terminal tail is the longest part of the TRPV4 protein and represents more than 50%
of the total protein and is suggested to play an important role in gating of the channel
(Phelps et al. 2010). The N-terminus houses up to six ankyrin repeats (ANK), depending
on the splice variant of TRPV4, which are involved in protein-protein interactions, channel
oligomerization and are necessary for the channel function (Arniges et al. 2006). The N-
terminal tail also contains a phosphoinositide-binding site (PIBS) which enables to bind to
phosphatidylinositol 4,5-biphosphate in the plasma membrane and that is required for
channel activation by physiological stimuli such as heat and hypotonicity (Garcia-Elias et
al. 2013). In addition a proline-rich domain (PRD) is localized in the NH2 terminus,
playing an important role in the regulation of TRPV4. The PRD is used to bind to kinases
like protein kinace C and casein kinase substrate in neurons protein 3 (PACSIN-3)
(Cuajungco et al. 2006) and is complete deletion renders the channel insensitive to all
stimuli, including the synthetic small molecule agonist 4α-phorbol 12,13 didecanoate (4α-
PDD) (Garcia-Elias et al. 2008, Garcia-Elias et al. 2014).
The C-terminal tail contains a putative TRP box, proposed for TRPV1 and suggested for
TRPV4 because of the similar structure predicted for TRPV1 and TRPV4 (Garcia-Sanz et
al. 2007, Garcia-Elias et al. 2014). This region, localized adjacent to the channel gate is
essential for the tetramerization of the channel subunits into functional channels (Garcia-
Sanz et al. 2007). Channel protein folding, maturation and trafficking are dependent on the
component of TRPV4 COOH-terminal and the COOH terminus interacts with the
microtubule-associated protein 7 (MAP7) (Suzuki, Hirao et al. 2003). MAP7 in the C-
terminal tail of TRPV4 has also been suggested to interact with the cytoskeleton and a
single mutation at E797 in the MAP7 domain results in constitutive opening of the channel
(Watanabe, Vriens et al. 2002). TRPV4 N-terminus houses a calmodulin domain for the
binding of calcium-calmodulin to TRPV4 promote to lead to conformational change
followed by channel opening and calcium influx (Strotmann et al. 2003). In the final four
amino acid residues of the TRPV4 C-terminus a PDZ-like domain is found for further
protein-protein interaction (Garcia-Elias et al. 2008, Garcia-Elias et al. 2014).

Introduction
8
The monomer structure normally coassembles to a homotetrameric functional TRPV4 ion
channel and is suggested to have the same tetramer structure as TRPV1 (Figure 1B)
(Shigematsu et al. 2010). TRPV4 has been reported to heterotetramize with e.g. TRPC1,
TRPP2 and also to form TRPV4-TRPC1-TRPP2 complexes (Stewart et al. 2010, Ma et al.
2011, Du et al. 2014). This heteromerization alter the properties of TRPV4 and give these
channels additional functions to its already large array of tasks (Du et al. 2014).
1.2.2 Protein interaction and regulation of TRPV4
In addition to the activation of TRPV4 by a wide range of stimuli, TRPV4 activity in the
plasma membrane is modulated at different levels: modifying TRPV4 localization and
expression on the plasma membrane, interaction with signal molecules and proteins,
cytoskeletal protein interaction and interaction with other ion channel proteins (reviewed in
Garcia-Elias et al. 2014, White et al. 2016).
Proteins modulating TRPV4 expression and location on the plasma membrane
Like other integral membrane proteins TRPV4 is synthesized in the endoplasmatic
reticulum (ER) and targeted to the plasma membrane. In the ER, TRPV4 has been shown
to interact with osteosarcoma amplified 9 (OS-9), a ubiquitous protein on the cytoplasmic
side of the ER playing a role in selecting substrates for degradation. OS-9 interacts with all
TRPV4 splice variants and this interaction is strongest with those lacking full ankyrin
repeats. It interacts with the N-tail of TRPV4 monomers and so reduces the amount of
channels in the membrane and protects TRPV4 monomers from ubiquitination and
degradation allowing formation of mature tetramers to occur (Wang et al. 2007).
Expression of TRPV4 at the plasma membrane is a net consequence of endocytosis and
exocytosis and expression of TRPV4 at the cell membrane has been shown to be regulated
by Protein kinase C casein kinase substrate in neuron protein 3 (PACSIN-3) (Cuajungco et
al. 2006). Binding of PACSIN 3 to TRPV4 decreases endocytosis resulting in an increased
TRPV4 plasma membrane expression and an overexpression in PASCIN-3 has been
associated with an increase in plasma membrane associated TRPV4. Interestingly, TRPV4
bound to PACSIN 3 is no longer activated by cellular swelling or heat but remain sensitive

Introduction
9
to the TRPV4 agonist 4α-Phorbol 12,13-didecanoate (4α-PDD) (D'Hoedt et al. 2008),
suggesting that TRPV4 is activated by different non-overlapping mechanism.
Conversely, activation of TRPV4 can be associated with its downregulation at the cell
membrane. Ubiquitin ligase Atrophin-1-interacting protein 4 (AIP4) and the protein β-
arrestin, which serves as an adaptor between TRPV4 and AIP4, binds TRPV4 in the
presence of angiotensin and lead to monoubiquitination of TRPV4 and its subsequent
endocytosis (Wegierski et al. 2006, Shukla et al. 2010).
Finally, the precise location of TRPV4 in the plasma membrane also seems to have
important functional consequences and appear to be tightly regulated (Goldenberg et al.
2015). TRPV4 has been shown to associate with caveolin-1, a primary structure
component of the caveolae, which are plasma membrane microdomains rich in proteins as
well as lipids and have several functions in signal transduction, such as mechanosensation
(Cuajungco et al. 2006, Saliez et al. 2008). This placement associates TRPV4 in close
proximity of proteins with critical importance in vascular biology. In the lung, caveolae are
key sites of nitric oxide (NO) production and interestingly other TRP channels have been
shown to translocate to caveolae in response to acute hypoxia (Tabeling et al. 2015),
making such translocation critical to the function of TRP channels. The stromal interaction
molecule 1 (STIM1) has also been proposed to complex with the COOH-terminal tail of
TRPV4 for guiding TRPV4 from the ER to the cell membrane and for its proper function
(Shin et al. 2015).
Cytoskeletal proteins interacting with TRPV4
TRPV4´s C-tail interacts with the microtube-associated protein 7 (MAP7) and it has been
described that TRPV4 interacts with the cytoskeleton via F-actin and tubulin which
compete for the binding to the COOH terminus (Suzuki, Hirao et al. 2003). The interaction
between TRPV4 and F-actin support channel activation following cell swelling (Becker et
al. 2009). Additionally MAP7 has been promoted to enhance TRPV4 presence at the
plasma membrane, thereby indirectly increasing its activity (Suzuki, Hirao et al. 2003).
TRPV4 has also been shown to interact with key molecules that connect the cytoskeleton
with structures that maintain the barrier function in epithelia. β-catenin and E-cadherin,
major components of the tight junctions in keratinocytes, interacts with the N-tail of

Introduction
10
TRPV4 to maintain the integrity of skin barrier (Sokabe et al. 2010, Sokabe and Tominaga
2010). Additionally mechanical forces applied to β1 integrin are activating TRPV4
(Matthews et al. 2010).
Proteins and signal molecules modulating TRPV4
It has been observed that several enzymes affect the activity of TRPV4. Activation of
TRPV4 is enhanced by the phosphorylation of specific sites in the N- and C-tail of TRPV4
by protein kinases C (PKC) (Xu et al. 2003) as well as by protein kinases A (PKA)
(Alessandri-Haber et al. 2006) and TRPV4 phosphorylation by PKA and PKC has been
shown to be dependent on interaction with A kinase anchoring Protein 79 (AKAP79) (Fan
et al. 2009). Phosphorylation of TRPV4 by Serum glucocorticoid-induced protein kinase 1
(SGK1), amplifies the TRPV4 response to appropriate stimuli and enable TRPV4 binding
to F-actin (Shin et al. 2012). An important nonprotein modulator of TRPV4 is the
membrane phospholipid, phosphatidylinositol 4,5-biphosphate (PIP2), localized on the
inner leaflet of the plasma membrane (Garcia-Elias et al. 2013). The TRPV4 N-terminal
proline-rich domain (PRD) has been shown to interact with plasma membrane PIP2 and is
thought to stabilize the intracellular tail of TRPV4 in an open conformation. Depletion of
PIP2 makes the channel unresponsive to heat or osmotic stimuli, but maintain activation by
epoxyeicosatrienoic acids EETs or 4α-PDD (Garcia-Elias et al. 2013). Adenosine
triphosphate (ATP) also interacts with these sites and is a positive modulator of TRPV4
channel activity (Lorenzo et al. 2008). Nitric oxide (NO) has been shown to cause S-
nitrosylation of TRPV4 in a residue of the C-tail and reduces activation of TRPV4 (Lee et
al. 2011). Calmodulin (CaM) has been identified to bind to TRPV4 within the second ANK
domain of the N-tail and at the C-tail (Phelps et al. 2010). The reported effects of CaM
binding on TRPV4 range from a positive modulation (Strotmann et al. 2003) to an
inhibitory effect (Phelps et al. 2010). In a heterologous expression system, increasing
intracellular calcium has been shown to inhibit TRPV4 channel function (Phelps et al.
2010). However another group demonstrated that TRPV4 is activated by increasing
intracellular Ca2+
through direct binding to TRPV4 calmodulin (Strotmann et al. 2003).

Introduction
11
Channel proteins interacting with TRPV4
As already mentioned heteromeric channels are formed by TRPV4 interacting with TRPP2
resulting in a mechano- and thermosensitive sensor in the cilium (Kottgen et al. 2008).
TRPV4-TRPC1-TRPP2 channel complexes found in TRPV4, TRPC1, and TRPP2
cotransfected cells of the vascular endothelium, are activated by flow to mediate calcium
influx (Du et al. 2014). TRPV4 and aquaporin 5 (AQP5) cell membrane expression is
increased by hypotonicity, and in this system AQ5 is essential for gating TRPV4 (Liu et al.
2006). TRPV4 and Aquaporin 2 (AQ2) are suggested to assemble in response to
anisosmotic conditions (Galizia et al. 2012). TRPV4 may also interact indirectly with other
calcium-sensitive proteins and channels located close to TRPV4 channels (White et al.
2016). TRPV4 functions in conjunction with Ca2+
-sensitive large-conductance K+ channels
(BKCa) in the bronchial epithelium and vascular smooth muscle (Earley et al. 2005,
Fernandez-Fernandez et al. 2008). Activation of Ca2+
-sensitive K+ channels via as few as
three TRPV4 channels mediating a localized Ca2+
influx (sparklets) has been shown for
intermediate- and small-conductance K+ channels (Sonkusare et al. 2012). Similar
observations were made for calcium-activated potassium channels (KCa2.3) that have been
shown to interact with TRPV4 inducing vascular relaxation (Ma et al. 2013).
1.2.3 Chemical activation and inhibition of TRPV4
TRPV4 is activated by a wide array of chemicals. The relevant ones will be described and
compared in this section. Furthermore an overview of relevant chemical antagonists of
TRPV4 will be given.
Activators of TRPV4:
Endogenous arachidonic acid (AA) and its metabolites epoxyeicosatrienoic acids (EETs)
and dimethylallyl pyrophosphate (DMAPP) are TRPV4 activators and thought to be
downstream effectors of other stimuli affecting TRPV4, including endocannabinoid
anandamide and cellular swelling (Watanabe et al. 2003, Vriens et al. 2004, Bang et al.
2012). The natural 5´,6´-EET gates TRPV4 by a direct action on a site formed by residues
from the S2-S3, S4 and S4-S5 transmembrane domains (Berna-Erro et al. 2017).

Introduction
12
Plant derived non-selective agonists of TRPV4 are e.g. Bisandrographolide A (BAA) with
an EC50 of about 800 nM (Smith et al. 2006), Apigenin (EC50 ~ 10 µM) (Ma et al. 2012)
and several plant cannabinoids (De Petrocellis et al. 2012). Phorbol is an organic
compound of croton plants and its derivatives are also agonists of TRPV4 (Watanabe,
Davis et al. 2002).
One of the more specific agonists of TRPV4 and a widely used synthetic activator of
TRPV4 is the ester 4α-Phorbol 12,13-didecanoate (4α-PDD) activating TRPV4 in the
micromolar range by binding between the transmembrane domain 3 and 4 (S3, S4), which
is not mediated by PKC enzymes (Vriens et al. 2007, Klausen et al. 2009). Although its
exclusivity for TRPV4 has been put in question, by the fact that it can activates mouse
DRG neurons independently of TRPV4 (Alexander et al. 2013).
Finally a potent and selective small molecule created by GlaxoSmithKline, GSK106790A,
is a useful TRPV4 activator with an EC50 in the low nanomolar range (Thorneloe et al.
2008). Treatment with this compound in vivo was shown to cause serious vascular effects,
leading to disruption of endothelial barrier, particularly in the lung and widespread
vascular leakage (Willette et al. 2008).
Inhibitors of TRPV4:
Lanthanium and gadolinium are non-selective TRP channel blockers, and gadolinium was
one of the earliest inhibitor used to address TRPV4 (Nilius et al. 2004). Gadolinium was
identified as an inhibitor of stretch-activated ion channels and is viewed these days as a
nonselective inhibitor of extracellular Ca2+
entry (Goldenberg et al. 2015).
A commonly used but nonspecific compound for studying TRPV4, is the cationic dye
Ruthenium red (RR). RR is unfortunately nonspecific and blocks most TRPV channels,
and also members of the TRPM and TRPA subfamily (Guler et al. 2002, Goldenberg et al.
2015).
Newer compounds have appeared with more specificity and affinity to TRPV4. One of the
most selective TRPV4 inhibitor used to date is the antagonist HC-067047 with an IC50
ranging from 17 to 133 nM (Everaerts et al. 2010). At doses, that block TRPV4 function, it
also displays no adverse cross signs of sickness in mice, but his clinical safety profile

Introduction
13
remains untested. HC-0670747 is a powerful tool for studying TRPV4, although a study in
pulmonary vasculature in mice lacking TRPV4 shown vasodilatation caused by HC-
0670747 [30 µM] indicating that this inhibitor may have off-target effects at high
concentrations and has also been shown to inhibit TRPM8 at submicromolar
concentrations (Everaerts et al. 2010, Xia et al. 2013, Goldenberg et al. 2015).
One of the largest efforts in TRPV4 inhibitor design has been conducted by
GlaxoSmithKline (Darby et al. 2016). The newer antagonist GSK2193874 displayed
remarkable specificity for rodent and human TRPV4, demonstrated by a screen against
approximately 200 other channel proteins, including other TRPV subfamily members
(Thorneloe et al. 2012). A key advantage of this compound lies also in its oral activity and
can so potentially be dosed repeatedly. GSK2193874 has been shown to prevent
pulmonary edema in a mouse model of heart failure and in isolated human lung tissues and
appeared safe for potential use in human trials (Thorneloe et al. 2012). This compound and
newer version of it, may present an important tool for a variety of pulmonary disease
states.
Finally another strategy for clinical inhibition of TRPV4 is by blocking phosphodiesterase
5 (PDE5) a downstream effector of TRPV4 (Goldenberg et al. 2015). PDE5 inhibitor,
sildenafil has been shown to attenuate TRPV4 mediated endothelial calcium entry and
pulmonary edema formation in ex vivo and in vivo models of congestive heart failure (Yin
et al. 2008) and indicate a potential indirect route for preventing adverse physiologic
effects of TRPV4 activation.
1.2.4 TRPV4 function and physiological activation
TRPV4 is a polymodal ion channel activated by a wide range of diverse stimuli and
simultaneous stimuli of different natures may interact. E.g. TRPV4 activation by 4α-PDD
or hypotonic solutions induces minor channel activation compared to all stimuli at 37°C
(Gao et al. 2003). TRPV4 is a mechano-, osmo- and thermosensitive Ca2+
channel that is
involved in multiple physiological functions such as hearing, renal function, skeletal
development, nociception, vascular tone and blood pressure, endothelial and epithelial
barrier function, and has also been related to several motor sensory neuropathies and has
been shown to play a role in regulatory volume decreases (RVD) of cells (reviewed in

Introduction
14
Darby et al. 2016, White et al. 2016). This section will concentrate on TRPV4 functions
relevant for this thesis.
TRPV4 and heat:
Non-noxious heat was one of the earliest physiological activator of TRPV4 described and
TRPV channels in general are activated by specific non-overlapping temperature ranges.
TRPV4 is activated at temperature between 24 and 38°C, TRPV1 is activated at
temperature greater than 43°C, while TRPV2 is activated by temperature greater than 52°C
(Watanabe, Vriens et al. 2002, Clapham 2003). Therefore TRPV4 has been suggested to
play a role in normal thermoregulation (Guler et al. 2002, Watanabe, Vriens et al. 2002).
However, there is no agreement for TRPV4 in the detection of noxious temperature in vivo
(Garcia-Elias et al. 2014, Darby et al. 2016, White et al. 2016), e.g. TRPV4-/- mice show
normal escape latencies from hot plates and conversely TRPV4-/- exhibit reduced sensory
nerve discharge frequency in response to noxious temperature during electrophysiological
studies (Todaka et al. 2004). Additionally, heat (37°C) increases the efficacy of other
stimuli in activating TRPV4 (Gao et al. 2003).
TRPV4 and pH:
TRPV4 has been reported to be activated by low pH or citrate in Chinese hamster ovary
(CHO) cells expressing TRPV4 in vitro. Mice lacking TRPV4 have been shown to exhibit
a diminished response to acids (Suzuki, Mizuno et al. 2003). Further studies implicate
TRPV4 in acid induced lung injury, where it has been demonstrated to mediate the lung
injury response in mice exposed to hydrochlorid acid (HCL), assessed by lung
permeability increase, inflammatory cell influx and pro-inflammatory cytokine levels
increase (Balakrishna et al. 2014, Yin et al. 2016, Scheraga et al. 2017). Protection from
acute lung injury response to HCL was observed in TRPV4-KO mice or in mice treated
with different small molecule TRPV4 inhibitors (Balakrishna et al. 2014, Yin et al. 2016,
Scheraga et al. 2017), which will be further described below.

Introduction
15
TRPV4 in epithelial and endothelial barrier function:
Endothelial and epithelial barriers are characterized by intercellular cell junctions
consisting of tight junctions and adherens junctions (Mullin et al. 2005, Bazzoni 2006).
Adherent junctions containing VE-cadherins interconnect cells to a width of approximately
3 nm and tight junctions prevent extravasation of much smaller molecule (˂ 1 k Da) (Curry
2005, Mehta and Malik 2006). These junctions play an important role in barrier function
by restricting the paracellular passage of fluid and proteins across tissue membranes.
Pathophysiological states such as inflammation can disrupt barrier integrity and increased
endothelial permeability can be triggered by endothelial Ca2+
influx, resulting in
cytoskeletal reorganization and loss of interendothelial junction proteins (Tiruppathi et al.
2006, Darby et al. 2016). TRPV4 activation has been shown to result in epithelial and
endothelial permeability increase from in vitro and in vivo studies (Darby et al. 2016). For
example, TRPV4 has been shown to function in linking cell-to-cell junctions in skin
keratinocytes with the actin cytoskeleton to ensure the development of a tight barrier
(Sokabe et al. 2010). Activation of TRPV4 was observed to reduce the level of filamentous
actin and to disintegrate cell junctions between epithelial cells of the brain ventricles
(Narita et al. 2015). In the lung TRPV4 activation activates matrix metalloproteinases
(MMPs) MMP2 and MMP9, that contributes to lung injury by degrading components of
the basement membrane as well as non-matrix components such as integrins and
intercellular structure like E-cadherin (Villalta et al. 2014). TRPV4 has been shown to
regulate vascular permeability most notably within the lungs (Willette et al. 2008) and its
activation, whether via physical stimuli such as mechanical ventilation, pulmonary venous
hypertension or with pharmacological tools leads to an increased endothelial permeability
in an intracellular calcium-influx dependent manner (Hamanaka et al. 2007, Jian et al.
2008). TRPV4 regulates the integrity of the alveolar barrier and its activation has been
shown to causes endothelial detachment from the basement membrane, leading to
disruption of the pulmonary endothelial barrier, resulting in pulmonary edema formation
and alveolar flooding (Alvarez et al. 2006, Jian et al. 2008). TRPV4 also has been shown
to initiate the acute endothelial calcium-dependent permeability increase during ventilator-
induced lung injury in isolated mouse lungs (Hamanaka et al. 2007), which will be further
discussed below.

Introduction
16
TRPV4 in osmoregulation and response to mechanical deformation:
In cell-based assays TRPV4 respond to osmotic changes in the cell environment,
decreasing its activity in hypertonic solutions and increasing its activity in hypotonic
solutions and so contributing to cellular homeostasis (Strotmann et al. 2000). TRPV4-KO
mice have been shown impaired osmotic regulation, supporting a role of TRPV4 in
osmosensation (Liedtke and Friedman 2003, Mizuno et al. 2003). Changes in osmolarity
causes cell swelling or shrinkage that deform the plasma membrane and may therefore
involve aspects of mechanosensation (Darby et al. 2016, White et al. 2016). Cell
deformation and lipid bilayer tension can affect further cellular processes (Hoffmann et al.
2009). TRPV4 has been implicated in the control of regulatory volume decrease (RVD), a
regulatory response to cell swelling after exposure to a hypotonic solution that is normally
associated with changes in intracellular calcium concentration (Arniges et al. 2004).
TRPV4 has been shown to provide the Ca2+
signal, required to activate further Ca2+
potassium channel and the subsequent RVD in epithelial cells and also interacts with
aquaporins to control RVD in astrocytes (Arniges et al. 2004, Benfenati et al. 2011, Jo et
al. 2015), an important observation, suggesting that disruption of cell volume regulation
may have crucial consequences for cell signalling, barrier integrity and cell viability
(Benfenati et al. 2011).
Whether mechanical forces are generated by hypotonicity, trauma, pressure, shear stress
evoked by flow or direct cell stretch, it typically results in the deformation of the cell
membrane and it is now clear that TRPV4 responds to the application of mechanical forces
to the cell membrane and therefore it is correct to describe TRPV4 as mechanosensitive
(White et al. 2016). Thus cell stretch evoked increase in intracellular Ca2+
applied to
urothelial cells is significantly reduced in cells from TRPV4-KO mice compared to
wildtype mice (Mochizuki et al. 2009). Furthermore TRPV4 has been shown to be
activated when cyclically stretch in capillary endothelial cells (Thodeti et al. 2009). Flow
evoked shear stress activates TRPV4 leading to an increased intracellular calcium
concentration in vascular endothelial cells and HEK293 cells (Mendoza et al. 2010,
Baratchi et al. 2014). Mechanical activation of TRPV4 has also been reported to trigger
ATP release from different epithelial cells (Seminario-Vidal et al. 2011, Ueda et al. 2011).
Shear stress has also been shown to cause TRPV4 to traffic from cytoplasmic vesicles to
the plasma membrane (Baratchi et al. 2016). TRPV4 is expressed in the bladder urothelium
where it has been show to participate in sensing of intravesical mechanical pressure during

Introduction
17
bladder filling and ATP release (Birder et al. 2007, Everaerts et al. 2010), additionally
TRPV4-/- mice manifest an incontinent phenotype (Gevaert et al. 2007). Furthermore
TRPV4 in the lung has been shown to initiate the acute calcium-dependent permeability
increase during mechanical ventilation with high tidal volumes leading to ventilator-
induced lung injury in isolated mouse lungs (Hamanaka et al. 2007) that will be further
discussed below.
Gating of TRPV4 by mechanical stimuli:
The question remains however, whether TRPV4 is directly or indirectly gated by
mechanical stimuli and there are several possible mechanism of TRPV4 activation in this
purpose (Darby et al. 2016):
First TRPV4 may respond directly to the effect of mechanical deformation of the
membrane, whether secondary to hypotonicity or to a direct mechanical pressure
impinging on the cell membrane. This concept includes the direct gating of TRPV4 by
mechanical forces to the cell membrane, which induces a conformational change within the
ion channel and results in channel gating because of energy differences between the open
and closed conformation (Brohawn et al. 2014) and in this context the lipid-bilayer directly
effects TRPV4 gating (Liedtke 2005). But the direct response of TRPV4 to the effect of
mechanical deformation also includes an alternative theory, by which mechanical forces
applied to cell membrane structures, attached or tethered to the ion channel, leads to its
opening (Christensen and Corey 2007, White et al. 2016). Such structures include
accessory proteins, the cytoskeleton or even the extracellular matrix and mechanical forces
are transmitted via these structures to effect a conformational change of the channel
resulting in gating (Kung 2005, Christensen and Corey 2007, Pedersen and Nilius 2007).
The concept of direct gating of TRPV4 by mechanical forces is supported by a study on rat
TRPV4 expressed in Xenopus oocytes by repeatedly examining excised patches in a simple
buffer (Loukin et al. 2010). In this system TRPV4 could be activated by pipette suction
even in the presence of relevant enzyme inhibitors to eliminate any enzyme effects. The
evidence that TRPV4 interacts with the cytoskeleton also supports the concept that
mechanical deformation of the cell membrane per se is capable of activating TRPV4. This
is also supported by a study showing that forces applied to β1-integrins resulted in ultra-
rapid activation of Ca2+
influx through TRPV4 within 4 msec and that TRPV4 is rather

Introduction
18
activated by mechanical stretch in the cytoskeletal backbone than by deformation of the
lipid bilayer (Matthews et al. 2010).
A second putative mechanism of TRPV4 activation, is consistent with TRPV4 as
mechanosensitive rather than mechanically gated, explaining an indirect gating of TRPV4
by a force-sensing protein, that might be more distant to TRPV4 and communicate with the
channel by generating a secondary signal such as a diffusible second messenger molecule
or activation of a kinase (White et al. 2016). In this view osmotic and mechanical
sensitivity of TRPV4 has been claimed to be dependent of phospholipase A2 (PLA2).
Activation of TRPV4 by cell swelling has been described to depend on formation of
arachidonic acid (AA) and its subsequent metabolization to 5´,6´-epoxyeicosatrienoic acid
(5´,6´-EET) by cytochrome P450 epoxygenase (Watanabe et al. 2003, Vriens et al. 2004,
Fernandes et al. 2008), whereby 5´,6´-EET has then been recently shown to directly bind to
TRPV4 resulting in its gating (Berna-Erro et al. 2017). AA has also been claimed a direct
and potent activation of TRPV4 (Zheng et al. 2013). Both viscous loading and
hypotonicity have been suggested to employ a PLA2 dependent mechanism and the
production of EET to gate TRPV4 in ciliated epithelial cells (Fernandes et al. 2008).
Furthermore when limited activation of PLA2 is possible, these stimuli employ
extracellular ATP-mediated activation of inositol trisphosphate (IP3) to gate TRPV4,
thereby IP3 do not act as an agonist of TRPV4 but sensitise TRPV4 to EET in which an
interaction of TRPV4 with IP3 receptor 3 appears to occur (Fernandes et al. 2008),
requiring the binding of IP3 to a domain in the TRPV4 COOH-terminal and so leading to a
IP3-mediated sensitization of TRPV4 to these stimuli (Garcia-Elias et al. 2008).
Interestingly heat and 4α-PDD are suggested to activate TRPV4 independently of PLA2
and P-450 epoxygenase (Vriens et al. 2004) in turn pointing to the possibility that TRPV4
may be activated by more than one mechanism.
A third possible mechanism is that alterations in extracellular tonicity per se, e.g. induced
by mechanical pressure, activates intracellular proteins independent of plasma membrane
deformation, which in turn gates TRPV4 (White et al. 2016). In doing so, osmotic
stimulation results in activation of various intracellular phosphorylation/dephosphorylation
signalling processes. Given its range of activators it is not unexpected that TRPV4 has, as
already mentioned, more than a single mechanism of activation (Brewster et al. 1993,
Liedtke 2005, White et al. 2016).

Introduction
19
1.3 Ventilator induced lung injury (VILI)
Mechanical ventilation (MV) is an important tool in intensive care units (ICU) for the
treatment of respiratory failure. Despite its lifesaving effects, mechanical ventilation has
been demonstrated to induce lung damage by itself. It may aggravate lung conditions in
previously diseased lungs as well as induce serious tissue damage in previously healthy
lungs, a process named ventilator-induced lung injury (Halbertsma et al. 2005, Sutherasan
et al. 2014, Carrasco Loza et al. 2015). Several experimental studies postulate that a
previous inflammation, also named first inflammatory hit of the lung, is crucial for the
development of VILI (Carrasco Loza et al. 2015).
During mechanical ventilation, lung strain is poorly defined, especially in humans and
difficult to estimate because of the heterogeneous local lung susceptibility during MV
(Protti et al. 2014, Carrasco et al. 2015). During MV injured regions of the lung will
receive smaller fractions of the total tidal volume from the inspired tidal volume, e.g. due
to alveolar collapse and fluid extravasation, therefore other lung areas will receive the
majority of the tidal volume leading to massive overdistension of this areas and local
damage perhaps even with protective ventilation strategies (Carrasco Loza et al. 2015,
Bellani et al. 2016). In turn areas that receive the higher tidal volume, may promote a local
inflammatory response, that might trigger a subsequent generalized inflammatory response
in the lung tissue (Carrasco Loza et al. 2015, Beitler et al. 2016).
Ventilator induced lung injury (VILI) is characterized by a reduction of the alveolar epi-
and endothelial barrier function resulting in pulmonary oedema formation, inflammation
and alveolar flooding (Webb and Tierney 1974). Two main forces act on the lung tissues
and cells during mechanical ventilation, excessive volumes and/or pressures, leading to
volu- or barotrauma that causes rupture of the lung parenchyma (Dreyfuss and Saumon
1993, Dreyfuss and Saumon 1998). Studies revealed that the end-inspiratory volume
responsible for the volutrauma was the main determinant of VILI rather than a barotrauma
induced by an end-inspiratory pressure (Halbertsma et al. 2005). Another process termed
Atelectrauma, describes the cyclical opening and collapse of the alveoli in response to
mechanical ventilation, resulting in increasing stretch and shear forces in other regions
leading to lung damage and surfactant dysfunction. This effect can be attenuated by an
increased positive end-expiratory pressure (PEEP), to prevent the collapse of the alveoli,
but requires elevated inspiratory pressures (Dreyfuss and Saumon 1993, Halbertsma et al.

Introduction
20
2005). In the lungs, cytokines are produced by alveolar macrophages but also by bronchial,
bronchiolar and alveolar epithelial cells (Pugin et al. 1998, Vlahakis et al. 1999, Carrasco
Loza et al. 2015). Previous studies have demonstrated that most alveolar cells are capable
of producing pro-inflammatory mediators such as tumor necrosis factor (TNF)-α,
interleukin (IL) -6, IL-8 and IL-1β when stretched in vitro or when ventilated in ex vivo
experiments (nicely reviewed in Halbertsma et al. 2005). High level of mechanical stretch
is also associated with an increased epithelial cell necrosis and a reduction of apoptosis
(Lionetti et al. 2005, Carrasco Loza et al. 2015). A mechanism of injury, termed biotrauma,
has been elaborated postulating that the stress produced by mechanical ventilation through
overdistension of lung units not only exacerbate, but also initiate an inflammatory response
in form of an upregulation of pulmonary cytokine production due to the MV (Tremblay
and Slutsky 1998, Lionetti et al. 2005). Loss of the alveolar-capillary barrier due to the
mechanical forces may result in losing the compartmentalization of the local pulmonary
response and releasing pro-inflammatory mediators into the systemic circulation leading to
multiple-system organ failure (MSOF) (Slutsky and Tremblay 1998, Frank and Matthay
2002). Ranieri et al. (1999) support this concept by demonstrating that the concentration of
pro-inflammatory cytokines in both bronchoalveolar lavage fluid (BALF) and serum could
be decreased with a lung-protective ventilation strategy. This concept may also explain the
observation that most ARDS patients die from MSOF rather than from respiratory failure
(Montgomery et al. 1985, Halbertsma et al. 2005).
How mechanical stimuli is converted into a biochemical response (mechanotransduction)
such as cytokine release when lung cells are stretched during mechanical ventilation
remains to be clarified. Mechanical ventilation causes the expansion of the plasma
membrane and transmembrane receptors such as integrins, stretch-activated ion channels
and also the cytoskeleton by itself have been identified as key structures in
mechanosensing this physical stimuli, that then induces various cellular processes (Pugin
2003, Vlahakis and Hubmayr 2003, Halbertsma et al. 2005).
The potential involvement of cation channels in mediating the response generated in the
lung after mechanical stress has been demonstrated in isolated rat lungs in which the
increase in microvascular permeability was abolished by gadolinium (inhibitor of stretch-
activated nonselective cation channels) and concluded that stretch-activated cation
channels may initiate the increase in permeability induced by mechanical ventilation
through an increase in intracellular Ca2+
concentration (Parker et al. 1998). From the TRP

Introduction
21
channels known to be implicated in mechanotransduction such as TRPA1, TRPC1,
TRPC3, TRPC6, TRPM4, TRPM7, TRPP2 , TRPV1, TRPV2 and TRPV4 (Yin and
Kuebler 2010), TRPV4 has received specific attention as potential new molecular target
for the treatment of mechanical stress induced pathological conditions of the lung such as
ventilator-induced lung injury (Hamanaka et al. 2007, Yin and Kuebler 2010, Hamanaka et
al. 2010). The force-sensitive ion channel TRPV4 (Yin and Kuebler 2010) that is also
expressed in many cells of the lung (Alvarez et al. 2006, Hamanaka et al. 2010), has been
suggested to initiate the acute calcium-dependent permeability increase during ventilator-
induced lung injury in isolated mouse lungs (Hamanaka et al. 2007).
1.4 Acute respiratory distress syndrome (ARDS)
Acute respiratory distress syndrome (ARDS) is a rapidly progressive form of acute
respiratory failure characterized by severe hypoxemia and noncardiogenic pulmonary
edema (Ashbaugh et al. 2005) contributing to systemic inflammation and frequently
resulting in death (Silversides and Ferguson 2013).
Because ARDS is not a disease, but a syndrome composed of a multifaceted means of
diagnosis and is determined by different causes with many different clinical histories, an
entirely satisfactory definition of ARDS remains an elusive goal (Rezoagli et al. 2017).
The first common definition of ARDS was achieved in 1994 during the American-
European Consensus Conference (AECC) on ARDS (Umbrello, Formenti et al. 2017).
However, it had numerous limitations across the diagnostic criteria and a new definition
emerged in 2012. This most recent revisited definition of ARDS, known as the Berlin
definition of ARDS was proposed by an expert panel endorsed by the European Society of
Intensive Care Medicine (Ranieri et al. 2012, Rezoagli et al. 2017). The Berlin criteria
provided a small but significant improvement in the predictive ability for mortality when
compared to the AECC criteria (Umbrello et al. 2017). The Berlin definition of ARDS is
based on four variables including timing (1), chest imaging (2), origin of edema (3) and
oxygenation (4) and is defined by the following criteria: (1) onset within 1 week of a
known clinical insult or new/worsening respiratory symptoms; (2) presence of bilateral
opacities in radiograph (X-ray) or computed tomography (CT) scan on the chest that are
not fully explained by effusion, lobar/lung collapse or nodules; (3) diagnosis of respiratory
failure not fully explained by cardiac failure or fluid overload; (4) presence of hypoxemia,

Introduction
22
as defined by a specific threshold of the arterial partial pressure of oxygen to fraction of
inspired oxygen ratio (PaO2/FiO2) measured with a minimum of required positive end-
expiratory pressure (PEEP) ≥ 5 cm H2O, thus able to identify three categories of severity
based on the degree of hypoxemia: mild (200 millimeters of mercury (mm) Hg <
PaO2/FiO2 ≤ 300mmHg), moderate (100 mmHg < PaO2/FiO2 ≤ 200 mmHg), severe
(PaO2/FiO2 ≤ 100 mmHg) (Ashbaugh et al. 2005, Ranieri et al. 2012, Umbrello et al.
2017).
Approximately 5% of hospitalized patients with the need for mechanical ventilation meet
the diagnostic criteria for ARDS and it has been shown that only 25% of these patients
have a mild form of ARDS, while the remaining 75% display a moderate to severe form
(Rubenfeld et al. 2005, Esteban et al. 2008, Umbrello et al. 2017). Based on various
population studies the incidence of ARDS varies from about 10 to 80 per 100.000 person
per year with a relevant geographic diversity (in Europe 17.9, in USA 78.8 per 100.000
person per year) (Rezoagli et al. 2017). In the US alone more than 200.000 cases per year
are affected by this clinical syndrome (Rubenfeld et al. 2005) and this number could even
be significantly higher according to the LUNG SAFE study, an international multicentre
prospective cohort study conducted in intensive care units (ICU) in 50 countries based on
the current Berlin definition, showing that clinicians, even trained on ARDS diagnosis,
missed almost 40% of ARDS diagnosis (Bellani et al. 2016, Rezoagli et al. 2017). This
study also pointed to the fact, that ARDS occurrence in intensive care units was estimated
to be 10.4% of the admissions and more than doubled (23.4%) when patients had to be
mechanically ventilated (Rezoagli et al. 2017).
One of the main hallmarks of ARDS is an increased pulmonary capillary permeability
leading to accumulation of protein-rich fluid inside the alveoli. This results in damage to
the capillary endo- and alveolar epithelium, causing the release of cytokines further
producing diffuse alveolar damage (Martin 1999, Umbrello et al. 2017). The pathological
features of ARDS have been described by three overlapping phases: an inflammatory
phase, a proliferative phase and a fibrotic phase. However these sequences may be
complicated by other variables such as ventilator induced lung injury (VILI) (Umbrello et
al. 2017). ARDS remains a syndrome with an elevated incidence and is associated with a
mortality ranging from 40% to 60% and a significant long-term morbidity (Phua et al.
2009, Herridge 2011, Umbrello et al. 2017). A high dead space fraction in the lung,
restricting the proportion of the lungs capable of participating in gas exchange, was

Introduction
23
correlated to an increase in mortality for patients with ARDS (Nuckton et al. 2002,
Rezoagli et al. 2017). The ultimate cause of death is often through multiple-system organ
failure (MSOF) due to systemic inflammation rather than hypoxia and is not fully
understood (Montgomery et al. 1985, Meduri et al. 2009, Umbrello et al. 2017).
ARDS can be caused by several factors like pneumonia, sepsis, gastric content aspiration,
trauma, pancreatitis, inhalation injury, burns, non-cardiogenic shock, drug overdose, near
drowning, acute lung injury, smoking and also by mechanical ventilation (Ferguson et al.
2012, Rezoagli et al. 2017, Umbrello et al. 2017). It is noteworthy that ARDS does not
develop in the majority of patients with clinical risk factors for this syndrome, suggesting
that genetic or epigenetic susceptibility may also play an important role in the pathogenesis
of this disorder. In fact one third of all patients with ARDS have a hyper-inflammatory
subphenotype with elevated plasma concentrations of interleukin-6 (IL-6), interleukin-8
(IL-8), and tumor necrosis factor α (TNF-α) (Thompson et al. 2017).
Currently, no effective pharmacological treatments exist for ARDS (Thompson et al. 2017)
and the primary target for the treatment of ARDS is to ensure gas exchange while
minimizing the risk of VILI. The patient management strategies remain to date largely only
supportive (Umbrello et al. 2017) and consists of prone positioning patients, fluid
management, extracorporeal membrane oxygenation (ECMO), inhaled vasodilators,
corticosteroids and a protective mechanical ventilation with low tidal volumes (Matthay et
al. 2012, Umbrello et al. 2017). Despite the fact that mechanical ventilation is an important
tool for life support of ARDS patients, it also has the potential to exert pathological
mechanical forces on different lung cells leading to Ventilator-Induced Lung Injury (VILI)
(Slutsky and Imai 2003).
1.5 The role of TRPV4 in ARDS and VILI
In the lungs TRPV4 is expressed in different pulmonary cell types, such as bronchiolar and
alveolar epithelial cells, alveolar macrophages, neutrophils, smooth muscle cells and
endothelial cells (Jia et al. 2004, Alvarez et al. 2006, Hamanaka et al. 2010, Nayak et al.
2015, Yin et al. 2016) and has been suggested to play a role in pulmonary diseases and
diseases conditions including pulmonary hypertension, cough, asthma, cystic fibrosis,
edema formation, ciliary beat dysfunction, chronic obstructive pulmonary disease (COPD),

Introduction
24
acute lung injury (ALI) and ARDS (reviewed in Goldenberg et al. 2015, Darby et al. 2016,
Scheraga et al. 2017). ARDS can be induced by acid inhalation or by ventilation with high
tidal volumes leading to ventilator induced lung injury (VILI) (Goldenberg et al. 2015) and
this section will focus on the role of TRPV4 in ARDS and VILI.
In this concept TRPV4 has been shown to mediate the acute lung injury response to a
sterile stimulus in a murine model of acid inhalation. It has been demonstrated to mediate
the lung injury response in mice exposed to hydrochlorid acid (HCL), assessed by lung
vascular permeability increase, inflammatory cell influx and pro-inflammatory cytokine
levels (e.g. IL- IL-6, KC, IL-1β, MCP-1, RANTES) (Balakrishna et al. 2014). Protection
from acute lung injury in response to HCL was observed in TRPV4-KO mice or in mice
treated with different small molecule TRPV4 inhibitors showing significantly lower levels
of chemokines/cytokines and permeability increase compared to wildtype mice
(Balakrishna et al. 2014).
Another sterile cause with the potential to result in lung injury is mechanical ventilation
(Goldenberg et al. 2015). The potential involvement of cation channels in mediating the
response generated in the lung after mechanical stress has been demonstrated in isolated rat
lungs in which the increase in microvascular permeability was abolished by gadolinium
(inhibitor of stretch-activated nonselective cation channels) and concluded that stretch-
activated cation channels may initiated the increase in permeability induced by mechanical
ventilation through an increase in intracellular Ca2+
concentration (Parker et al. 1998).
TRPV4 has been shown to be a particularly promising candidate for the initiation of the
acute calcium-dependent permeability increase during ventilation in isolated mouse lungs
(Hamanaka et al. 2007). In this study pretreatment with inhibitors of TRPV4 (Ruthenium
red), arachidonic acid production (methanandamide), or P-450 epoxygenases (miconazole)
prevented the increases in lung permeability in isolated perfused mice lungs during
mechanical ventilation, an effect that was also absent in TRPV4-KO mice compared to
untreated WT mice. Furthermore lung distention caused calcium entry in the isolated mice
lungs which was absent in TRPV4-KO and Ruthenium red treated lungs (Hamanaka et al.
2007). Pharmacological activation of TRPV4 with 4α-PDD also showed an increase in
endothelial permeability in isolated rat lungs and this effect was reversed by Ruthenium
red administration (Alvarez et al. 2006). Prevention of ventilator induced-lung edema
formation was also demonstrated by inhalation of nanoparticles releasing Ruthenium red in
an murine isolated perfused lung model of ventilation (Jurek et al. 2014).

Introduction
25
TRPV4 has also been suggested to play a prominent role in mediating the mechanical
activation of macrophages suggesting to initiate this pathological response during
ventilation (Hamanaka et al. 2010). An important role for alveolar macrophages in
mechanical ventilation models has been demonstrated by depletion of macrophages in rat
lungs using clodronate-filled liposomes resulting in an attenuation of ventilator-induced
lung injury, where high volume ventilation resulted not only in an activation-associated
adhesion of alveolar macrophages but also in an increased alveolar protein leakage and
lung edema formation that was attenuate by depletion of macrophages (Frank et al. 2006,
Eyal et al. 2007). A more recent investigation linked TRPV4 channels and macrophages in
the role of modulating VILI. In this study the ventilator-induced lung injury was markedly
attenuated in TRPV4-KO mice, whereas reintroduction of TRPV4-WT macrophages in
TRPV4-KO mice reconstituted the lung injury response to mechanical ventilation, showing
that TRPV4 activation in macrophages plays a crucial role in initiating this injury
(Hamanaka et al. 2010). Additionally macrophages isolated from WT mice exhibited an
increase in intracellular Ca2+
and produced reactive oxygen species in response to 4α-PDD
that was not seen in TRPV4-KO cells (Hamanaka et al. 2010). Macrophages TRPV4 has
also been shown to regulate cytokine secretion (Scheraga et al. 2016, Scheraga et al. 2017).
Previous studies have demonstrated that most alveolar cells are capable of producing pro-
inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL) -6, IL-8
and IL-1β when stretched in vitro or when ventilated in ex vivo experiments (nicely
reviewed in Halbertsma et al. 2005). TRPV4 activation has also been promoted to induce
inflammatory pathways in immune cells and to induce pro-inflammatory
cytokines/chemokines secretion in response to lipopolysaccharide (LPS) in epithelial cells
(Henry et al. 2016, Scheraga et al. 2017). A recent study in fetal mouse distal lung
epithelial cells linked cell stretch and an inflammatory response to TRPV4 in vitro and
demonstrated that TRPV4 may also play an important role in the transduction of
mechanical signals in the lung epithelium during ventilation by modulating the stretch-
induced release of pro-inflammatory cytokines (Nayak et al. 2015). Taken together, these
data demonstrated the potential of TRPV4 inhibition for the prevention of ARDS in
response to mechanical ventilation.

Introduction
26
1.6 The aim of the thesis
To better understand TRPV4 biology and its role in the regulation of membrane barrier
integrity, the purpose was firstly to establish in vitro and in vivo models of permeability
and to investigate on the role of TRPV4 in modulating membrane barrier integrity with
pharmacological tools. We used two reported selective activators of TRPV4,
GSK1016790A and 4α-PDD and the potent and selective TRPV4 blocker GSK2193874,
promoted as an excellent in vitro and in vivo tool for the study of TRPV4. We also
questioned the link between pharmacological activation of TRPV4 and the corresponding
functional observations on calcium influx and barrier integrity when there is no classical
signal transduction pathway affirmed that can be followed to substantiate such a link. We
therefore hypothesized that such effects may also be caused by selective TRPV4 mediated
cytotoxicity.
The second part of this thesis focusses on the effect of lung cell stretch due to over-
distention of lung region during mechanical ventilation and the role of TRPV4 in
mediating a pathological cellular response to these physical stimuli. Therefore the purpose
was to establish cell stretch experiments in vitro to investigate firstly the cellular calcium
response of lung cells to a mechanical stimulus and secondly the effect of TRPV4
inhibition in this system. The next aim was to find other possible stretch induced cellular
responses in lung cells that might play a role in VILI and ARDS such as inflammatory
mediators and the potential of TRPV4 inhibition in such a system. The final aim was to
establish a disease related murine model of ventilation hypothesizing that a selective orally
active inhibitor of TRPV4 could improve cell stretch induced pathological cellular
response during mechanical ventilation also in vivo, such as lung permeability increase and
inflammatory mediator release.

Methods
27
2 Methods
2.1 In vitro studies
2.1.1 TER measurement
The CellZscope Automated Cell Monitoring System (nanoAnalytics GmbH; Münster,
Germany) was used for continuous measurement of transepithelial/transendothelial
electrical resistance (TER). A direct correlation between the permeability of a cell layer
and its transepithelial/-endothelial electric resistance exists. Therefore a cell layer cultured
till confluence on a permeable membrane forms the interface between two medium-filled
compartments while a voltage (AC) is applied across the electrodes and TER and
capacitance (CCL) of the layer is measured over time by recording the frequency-dependent
impedance (Z) and using an electrical equivalent circuit to analyze the data. Human
umbilical vein endothelial cells (HUVECs, EndoGRO™, SCCE001, Merck Millipore,
USA) were seeded at a density of 3 x 104 cells per transwell filters (Corning #3470; 0.4 μm
Pores; Polystyrene; 24 wp) in 100 µl EndoGRO-LS Complete Culture Media Kit
(SCME001, Millipore, Billerica, MA, USA) and incubated at 37°C in humidified air for 24
h. Afterwards cells on Transwell filters were incubated in humidified air for another 24 h
in an Invivo2 300 Hypoxia Chamber (Ruskinn Technology, Pencoed, UK) at 1% O2, 5%
CO2 at 37°C. The 24 wells in the CellZscope were filled with 810 µL of medium and
warmed up in the incubator. Afterwards cells on transwell filters were transferred in the
machine and another 160 µl of medium was given on the apical side of the transwell filters.
To maintain optimal culture conditions, the CellZscope was placed in a tissue culture
incubator (37°C, 5% CO2) and TER measurement was initiated. Cells were preincubated in
the presence or absence of different concentrations of the TRPV4 antagonist GSK2193874
for 1h or more and afterwards different concentrations of the TRPV4 agonists 4α-PDD or
GSK1016790A were added from a 10-fold concentrate in medium on the apical side of the
transwell filters and TER was measured continually for up to 24 h. In this system the effect
of the cytokines IL-1β and TNF-α on TER were also investigated. For better illustration,
TER was normalized (0% was defined as the TER level of the no cell control and 100%
was defined as the largest mean in each data set) with GraphPad Prism Software and
group mean only was shown as percentage.
To investigate whether the effect of TRPV4 activation can not only be prevented but also
reversed by TRPV4 inhibition, HUVECs were treated firstly with the TRPV4 agonist

Methods
28
GSK1016790A and afterward with the TRPV4 antagonist GSK2193874 during TER
measurement.
TER measurement was also performed in small airway epithelial cells (SAEC)
differentiated on transwells and cultured in air-liquid interface (ALI), as described in
section 2.1.10. Therefore ALI cultures on transwell filters were placed in the cellZscope
and medium was added basolaterally and apically to enable impedance measurement as
previously described. The cellZscope was placed in an incubator at 37°C, 5% CO2 in
humidified air and TER measurement was initiated. Cells were preincubated in presence or
absence of different concentrations of the TRPV4 antagonist GSK2193874 for 1 h or more
and afterwards were treated with the TRPV4 agonist 4α-PDD or GSK1016790A and TER
measurement was performed continuously for up to 24 h.
2.1.2 Vascular Permeability Assay
HUVECs were seeded on HTS-Transwell-96 Well Plates (Corning # 3391; 0.4 µm Pores;
Polycarbonate Membrane, NY, USA) with a density of 25 x 103 cells/well in 100 µl
medium. The reservoir plate was filled with 25 ml medium and cells were incubated at
37°C and 5% CO2 in humidified air for 24 h. Afterwards cells on Transwell filters were
incubated in humidified air for another 24 h in an Invivo2 300 Hypoxia Chamber (Ruskinn
Technology, Pencoed, UK) at 1% O2, 5% CO2 at 37°C. The HTS-Transwell-96 well plate
was then transferred on a receiver plate which was filled on the basal side with 225 µl
medium/well and different concentrations of the stimuli. The apical medium was
exchanged by 100 µl medium with the same concentration of the stimuli as the basal
medium and cells were incubated at 37°C and 5% CO2 in humidified air up to 20 h.
Fluorescein isothiocyanate-dextran (FITC-Dextran, 2000 kDa, #FD2000S, Merck KGaA,
Darmstadt, Germany) solved in H2O (25 mg/ml) was diluted 1:100 in medium. Afterwards
20 µl of the FITC-Dextran dilution was given on the apical side of each transwells and the
transwell plate was incubated protected from light at RT for 1 h. Then 100 µl medium from
each well of the basal side of the receiver plate was transferred to a black 96-well plate
(96F Nunclon Delta Black Microwell SI, Nunc, Langenselbold, Germany) and
fluorescence was measured in a SpectrMax M5 plate reader (Molecular Devices,
Sunnyvale, CA). In this system the effect of the cytokines IL-1β and TNF-α on membrane
permeability were also investigated.

Methods
29
2.1.3 Calcium 6 assay on the FLIPRTETRA
Pharmacological activation and inhibition of TRPV4 was analyzed using the FLIPR
Calcium 6 Assay kit (molecular devices #R8191 bulk kit) and was performed according to
the manufacturer’s instructions. Briefly cells were seeded with a density of 1 x 104
(HUVECs) or 3 x 104 (NCI-H292) cells/well in appropriated medium with 25 μL
medium/well on assay plates (384 well Poly-D-Lysin black/clear bottom, Biocoat #4663)
and incubated for 24 h. Cells were incubated for 2 h with the calcium 6 dye solution
(calcium 6 dye in assay buffer (HBSS [+ CaCl2/MgCl2] + 20 mM Hepes + 0.1% BSA; pH
7,4) according to the manufacturer´s instruction at 37°C in 5% CO2, humidified air. For
fluorescence measurement, cells were transferred to the FLIPR and buffer or compounds
were given in 10 μl/well with different concentrations and cells were preincubated in the
presence or absence of different concentrations of the TRPV4 antagonist GSK2193874 for
15 min during measurement (FLIPRTETRA
, Molecular Devices, excitation 470-495 nm,
emission 515-575 nm, with 2 read intervals, first read interval with 1 read per second for
10 s before compound dispersion and 1 read per second after first compound addition for
50 s and a second read interval with 1 read every 10 s for 84 times) before second
compound addition. For EC 50 or IC 50 measurement cells were afterward stimulated with
10 μl/well of different concentrations of the TRPV4 agonist 4α-PDD or GSK1016790A
during read out (FLIPRTETRA
, Molecular Devices, excitation 470-495 nm, emission 515-
575 nm, with 2 read intervals, first read interval with 1 read per second for 10 s with 1 read
before and 9 reads after agonist dispersion and a second read interval with 1 read every 3 s
for 210 times) and the concentration-dependent inhibition or activation of calcium influx
was determined. To investigate the different effects of the TRPV4 agonist 4α-PDD or
GSK1016790A on calcium-influx over time the readout after the second compound
addition was also extended up to ~ 5 h (FLIPRTETRA
, Molecular Devices, excitation 470-
495 nm, emission 515-575 nm, with 2 read intervals, first read interval with 1 read per
second for 10 s with 1 read before and 59 reads after second compound dispersion and a
second read interval with 1 read every 30 s for 596 times). To investigate whether the
effect of TRPV4 activation can also be reversed by TRPV4 inhibition, cells were also
treated firstly with the TRPV4 agonist GSK1016790A and afterward with the TRPV4
antagonist GSK2193874 in intracellular calcium concentration measurements.

Methods
30
2.1.4 TRPV4 agonism effect on LDH release
Cells were seeded (25 x 103 cells/well for HUVECs; 5 x 10
4 cells/well for NCI-H292) in
appropriate medium on 96 well culture plates (NunclonTM Delta Surface, Thermo
scientific) and incubated for 24 h. Afterwards cells were preincubated for 1 h in the
presence or absence of the TRPV4 antagonist GSK2193874 in 100 μL medium. Medium
was removed one more time and cells were incubated at 37°C in 5% CO2, humidified air
for up to 12 h in 100 μL medium in presence or absence of different concentrations of the
TRPV4 agonist GSK1016790A or 4α-Phorbol 12, 13-didecanoate (4α-PDD). Then
supernatant was collected at different time points and lactate dehydrogenase (LDH) release
was detected using a CytoTox96® Non-Radioactive Cytotoxicity Assay kit (Promega,
Madison, WI) following manufacturers instruction (see section 2.3.5).
Experiments were also performed with Hank´s Balanced Salt Solution (HBSS, Gibco, Life
technologies, Grand Island, NY) or Dulbecco's Phosphate-Buffered Saline (DPBS, Gibco,
Life technologies, Grand Island, NY) in the presence or absence of CaCl2 and MgCl2.
2.1.5 RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay
The RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay (Promega, Madison,
USA) is a live-cell real-time assay that measures the exposure of phosphatidylserine (PS)
on the outer leaflet of the cell membrane during the apoptotic process and is detected by
annexin V binding with a simple luminescence signal. The assay also includes a cell-
impermeant, profluorescent DNA dye, which detects necrosis. In the assay, time-dependent
increases in luminescence that occur before increases in fluorescence reflect the apoptotic
process. A significant time delay between the emergences of PS, indicated by Annexin V
binding, leading to a luminescence signal and the loss of membrane integrity visualized by
fluorescence signal, indicate an apoptotic phenotype leading to secondary necrosis.
Increases in fluorescence or increase in both luminescence and fluorescence concurrently
consist with necrosis or other non-apoptotic mechanisms.
The RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay were performed as
prescribed by the manufacturer. Briefly HUVECs were seeded with a density of 25 x 103
cells/well on 96 well white plates (96F Nunclon™ Delta White Microwell SI, Thermo
Fisher Scientific, Roskilde, Denmark) in 50 µl medium (SCME001, Millipore, Billerica,

Methods
31
MA, USA) and incubated at 37°C in 5% CO2, humidified air for 24 h. Afterwards cells
were preincubated for 1 h in the presence or absence of the TRPV4 antagonist
GSK2193874. Cells were then treated with different concentration of the TRPV4 agonists
GSK1016790A or 4α-PDD and directly equal volume of detection reagent was added, the
plate was covered with an imaging seal (4titude 4ti-0516/96, LabSource, Switzerland) and
a kinetic mode (1 read every 30 sec for up to 20 h) using a multimode instrument with
temperature control was initiated for assay signal detection.
2.1.6 Cell-IQ®
The Cell-IQ® (Chip-Man Technologies, Tampere, Finland) is a fully integrated live cell
imaging and comprises a temperature controlled incubator, a tailored gaseous environment
and the use of light emitting diodes for both phase and fluorescence imaging allowing
biological cellular responses to be monitored in real time.
HUVECs were seeded (25 x 103 cells/well) in medium (SCME001, Millipore, Billerica,
MA, USA) on 96 well culture plates (NunclonTM
Delta Surface, Thermo scientific) and
incubated at 37°C in 5% CO2, humidified air for 24 h. Afterwards cells were preincubated
for 1 h in the presence or absence of the TRPV4 antagonist GSK2193874. Cells were then
incubated at 37°C in 5% CO2, humidified air for up to 4 h in 100 μL medium in presence
or absence of different concentrations of the TRPV4 agonist GSK1016790A and live cell
imaging was recorded in a Cell-IQ®
.
2.1.7 TRPV4 agonism effect on cytokine release
NCI-H292 cells were seeded with a density of 5 x 104 cells/well with 200 μL RPMI-1640
medium (Gibco, Grand Island, N.Y.) containing 10% heat-inactivated fetal bovine serum
(FBS) on 96 well culture plates (NunclonTM Delta Surface, Thermo scientific) and
incubated at 37° C in 5% CO2, humidified air for 24 h. Afterwards medium was removed
and cells were preincubated for 1 h in the presence or absence of the TRPV4 antagonist
GSK2193874 [1 µM] in 200 μL medium. Medium was removed one more time and cells
were incubated at 37°C in 5% CO2, humidified air for 24 h in 200 μL medium in presence
or absence the TRPV4 agonist GSK1016790A or 4α-PDD. Then supernatant was collected

Methods
32
and stored at −80°C for later analyses or cytokine measurement was performed using
multiplexing technology from Meso Scale Discovery (see section 2.3.2).
2.1.8 Uniaxial cell strain and microscopy
Uniaxial cell strain was performed on the Stretch/compression device (University Ulm,
Ulm, Germany), a device for simultaneous live cell imaging during uniaxial mechanical
strain or compression (Gerstmair et al. 2009). An elastic silicon membrane (Specialty
Manufacturing, Saginaw, MI 48603-3440 USA) was cut into a rectangular piece (9 x 2 cm)
and clamped into the membrane holders that shape the membrane into a chamber
(Gerstmair et al. 2009), autoclaved and coated overnight at 4°C with fibronectin (5 µg/ml
in PBS, both from Sigma-Aldrich, Steinheim, Germany). Human lung epithelial cells
(NCI-H292) were seeded in the elastic silicon chamber (4 x 105 cells/membrane) and
cultivated in medium at 37°C in 5% CO2, humidified air for 24h. Prior to imaging, the cells
were pre-incubated in medium at 37°C, 5% CO2 with 2 µM of the fluorescent Ca2+
dye
fluo-4 and 0.2% Pluronic F127 (Molecular Probes, Karlsruhe, Germany), protected from
light with or without the TRPV4 antagonist GSK2193874 [1 μM] for 30 min and another
30 min at RT. For cell stretch, the medium was replaced with bath solution (pH 7.4; 140
mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM glucose, and 10 mM HEPES; all
from Sigma-Aldrich). Then the membranes were fastened onto the stretch apparatus
(Gerstmair et al. 2009), mounted on a Zeiss Axiovert 200 (Carl Zeiss, Oberkochen,
Germany) with a 20X plan Neofluar Zeiss objective. Images were acquired with a
CoolSnap EZ CCD camera and Metamorph software (exposure time of 30 ms and an
acquisition rate of 0.5 frames per second) and an EGFPfilter cube (excitation 470/20 nm,
emission 525/25 nm, dichroic 490 nm). The membranes were stretched at RT with a
triangular waveform one single time from 0% to 80% length increase and back to 0%
within 800 ms. Prior to this stretch protocol other stretch protocols with different stretch
amplitudes and frequencies were performed.
The average grey values in the image sequence were determined with ImageJ (Abràmoff et
al. 2004) by drawing a region of interest that comprised the adherence area of a single cell.
To compensate for the slight sideward shift of the cell after the strain, the region of interest
was manually repositioned. Data were transferred to MS-excel and after background
subtraction, the average fluorescence values of each cell before and 10 s after the strain

Methods
33
were determined. The strain-induced change after stimulation was expressed as the percent
change in intensity compared to the baseline signal before stretch.
2.1.9 Equibiaxial cell strain
The Flexcell FX-5000 Tension System (FX5K®; Flexcell International Corp,
Hillsborough, NC) was used to apply mechanical cyclic tensile stretch on lung epithelial
cells (NCI-H292) and human monocyte derived macrophages (see section 2.1.10). The
FX5K® is a computer-based system that uses a vacuum to strain cells adhered to flexible
silicon membranes (BioFlex® plates; Flexcell International Corp) arranged in a format of
six wells per plate with a total growth area of 9.62 cm2 per well. The deformation of the
flexible membrane also causes the attached cells to deform. NCI-H292 cells were seeded
onto Collagen Type I-coated BioFlex® plates at a density of 1 × 106 cells/well,
macrophages at a density of 2 x 106 cells/well. Cells remained untreated or were pretreated
with compounds and exposed to continuous mechanical stimulation with an equibiaxial
half sinusoidal waveform with an elongation from 8% to 30% and a frequency of 1.25 Hz
for up to 48 h at 37°C in 5% CO2, humidified air. Control cultures were grown under the
same conditions but without the strain protocol. Then supernatant was collected and stored
at −80°C for later analyses. Prior to this stretch protocol other stretch protocols with
different stretch amplitudes and frequencies had to be performed.
2.1.10 Cells
HUVECs
Human umbilical vein endothelial cells (HUVECs, EndoGRO™, SCCE001, Merck
Millipore, USA) were cultured in EndoGRO-LS Complete Culture Media Kit (SCME001,
Millipore, Billerica, MA, USA). Washing steps were performed with Dulbecco's
Phosphate-Buffered Saline (DPBS, Gibco, Life technologies, Grand Island, NY) and
TrypLE Express (GIBCO, Life technologies, Grand Island, NY) was used for cell
detachment.

Methods
34
NCI-H292
Lung epithelial cells NCI-H292 (Cat. No. CRL-1848TM
from the American Type Culture
Collection ATCC, Manassas, VA) were cultured in RPMI-1640 medium (Gibco, Grand
Island, N.Y., containing 10% heat-inactivated fetal bovine serum). Washing steps were
performed with DPBS (Gibco, Life technologies, Grand Island, NY) and TrypLE Express
(GIBCO, Life technologies, Grand Island, NY) was used for cell detachment.
SAEC
Small airway epithelial cells (SAECs, Lonza, Donor # 408031, Verviers, Belgium) were
cultured and differentiated following the Lonza CloneticsTM
S-ALITM
air-liquid interface
medium protocol. Briefly SAECs were seeded into cell culture flask (T175 NUNC flask,
178883, Thermo Fischer) on day - 8 in Clonetics S-ALI growth medium. On day - 4 cells
were trypsinised and seeded with a density of 22 x 103 cells/well on Corning Transwell
filters (Corning #3470; 0.4 μm Poren; Polystyrene; 24 wp). On day 0 airlift of the cells was
performed by removing the apical medium and substituting the basolateral growth medium
with S-ALI Differentiation Medium (Clonetics S-ALI differentiation medium). On the
apical side cells were washed to remove growth factors. Afterwards SAECs were
differentiated in air-liquid interface (ALI) for at least 4 weeks with basolateral medium
changes 3 times a week with apical washing step ones a week.
Human Monocyte Derived Macrophages
Human whole blood was obtained from anonymised healthy volunteers. Blood was
donated by internal donors at the centre for occupational health at Boehringer Ingelheim in
Biberach. The donors provided signed informed consent that allows use for scientific
purposes. Peripheral blood mononuclear cells (PBMCs) were isolated by means of density
gradient centrifugation using Ficoll-Paque™ and a Leucosep Tube (Greiner Bio-One
GmbH) according to manufacturer’s instructions. CD14 positive monocyte purification
was performed by magnetic activated cell sorting (MACS) according to the manufacturer’s
instructions (Monocyte Isolation Kit II, Miltenyi Biotec) and seeded 2 x 106 cells/well in
XVIVO-10 medium (Lonza) on Collagen Type I-coated BioFlex®
plates (BioFlex® plates;

Methods
35
Flexcell International Corp). Medium was supplemented with either 100 ng/mL
Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) to induce an M1
phenotype or Macrophage Colony Stimulating Factor (M-CSF) to induce an M2 phenotype
for 7 days.
2.2 In vivo studies
2.2.1 Effect of TRPV4 activation on vascular permeability
TRPV4 induced vascular permeability increase was investigated in male Balb/c or
C57BL/6J mice (Charles River, Sulzfeld, Germany) with weights ranging from 20 - 25 g.
Mice were held at 55% relative humidity, 22°C with a day-night-cycle of 12 h. For
experimental procedure the dye Evans blue was used to monitor vascular leakage by
measurement of Evans blue in formamide extracts of mice tissue after protein leakage
induction.
Experimental procedure:
time point - 2 h : application of TRPV4 antagonist (p.o.)
time point - 10 min: narcosis (i.p.)
time point - 1 min: Evans blue application (i.v.)
time point 0 h: application of TRPV4 agonist (i.v. or i.t.)
time point + x min: euthanasia and samples collection
Two hours before agonist addition mice were pretreated with different concentrations of
the TRPV4 antagonist GSK2193874 (solved in 0.5% Natrosol, Merck, # 8.22068.0500)
given orally (p.o.) with a volume of 10 ml/kg. Afterwards mice were anesthetized with
Medetomidin (Zoetis, # 07725752) /Midazolam (Roche, # 03085793) /Fentanyl (Janssen, #
4795545) (0.5 mg/kg + 5 mg/kg + 0.05 mg/kg; application volume 150 μl/animal) given
intraperitoneal (i.p.) at time point -10 min and were placed on thermostatically-controlled
heat mats to preserve body temperature during experimental procedure. At time point -1
min, 2% Evans blue (Sigma; # E2129) and 33 U/ml Heparin (Ratiopharm; # 3029843)
solved in 0.9% NaCl was applicated with a volume of 100 µl per mouse intravenously (i.v.)
on the tail vein. At time point 0 h different concentrations of the TRPV4 agonist

Methods
36
GSK1016790A (solved in 0.9% NaCl, Fresenius) were given intratracheally (i.t.; 50
µl/mouse) or i.v. (100 µl/mouse) through the tail vein. At the desired time points
experiment was stopped. Mice were euthanized by application of an overdose of narcoren
(Merial; # 6088986.00.00) given i.p. (400 mg/ml; ~ 400 µl/mouse). Afterwards mice were
perfused with 10 ml buffer/animal (25 000 IE Heparin, Ratiopharm, # 196621, solved in
500 ml NaCl). Therefor mice chest were opened and a small cut was performed in the left
heart ventricle, where a cannula (23G, 0.6 x 25 mm, Luer Lock, Braun, # 105107) was
inserted to inject the perfusion-buffer. Afterwards organs were excised and washed in PBS
(Lonza, # BE17-516F). Lungs were further prepared and separated in bronchus and
parenchyma. Organs were then added to 750 µl formamide (Sigma, # F7503) in Safe-Lock
reaction tubes (2 ml, Eppendorf, # 0030120094) and incubated overnight at 65°C. On the
next day samples were collected and 250 µl formamide supernatant were given to a 96-
well plate and absorbance was measured at 620 nm in a spectrometer.
2.2.2 Murine mechanical ventilation model
Experiments were performed in female Balb/c mice (n=82; Charles River, Sulzfeld,
Germany) aged 10 - 12 weeks, with weights ranging from 23 to 28 g and were held in the
same conditions as previously describe. Mice were mechanically ventilated and the effect
of ventilation on lung permeability increase, function and inflammatory response was
investigated with or without pretreatment with the orally active TRPV4 inhibitor
GSK2193874.
Experimental procedure:
time point – 1 h: application of the TRPV4 antagonist (p.o.)
time point 0 h: narcosis (i.p.)
time point + 1 h: start of mechanical ventilation
time point + 4 h: euthanasia and samples collection
The TRPV4 antagonist GSK2193874 [90 mg/kg] or the solvent (0.5% Natrosol with
0.015% Tween80) were administered orally by gavage 2 h before ventilation (time point -1
h). At time point 0 h mice received intraperitoneal narcoren [60 mg/kg] and rompun [2.5

Methods
37
mg/kg] solved in 0.9% NaCl (Fresenius) with additional dosing as needed to maintain
appropriate anesthesia. A cannula (Fa. Harvard, USA, Art.-Nr.: NP73-2836) was inserted
into the trachea, sutured, and coupled to a flexiVent (SCIREQ, Montreal, Quebec, Canada)
small animal ventilator and controlled with the Software FlexiWare (Fa. EMKA
Technologies, Paris, France). Mechanical ventilation was performed with different
ventilation protocols, with tidal volumes of 20 ml/kg (control n=5; treated n=4), 30 ml/kg
(control and treated n=8) and 40 ml/kg (control and treated n=4) with a frequency of
75/min and 2 cm H2O PEEP and a control group ventilated with a normal tidal volume of
6.5 ml/kg (n=4) and a frequency of 150/min and 3 cm H2O PEEP for 3 h and an additional
unventilated control group (n=6). During the experiments mice were placed on
thermostatically-controlled heat mats to preserve body temperature in a supine position.
During the 3 h of ventilation on the FlexiVent, parameters such as lung resistance,
compliance and elastance were recorded every 15 min. With the ending of the ventilation
protocol, mice were euthanized with an overdose of narcoren (500 mg/ml, ~ 0.5 ml/ animal
given i.p.) remaining therefor on the ventilator until cardiac arrest. Bronchoalveolar lavage
(BAL) was performed using 2 times 0.8 ml Hanks Salt Solution (Fa. Biochrom AG) and
0.6 mM EDTA (Fa.: Promega). The bronchoalveolar lavage fluid (BALF) was centrifuged
at 1500 rpm at 4°C for 10 minutes and the supernatant was stored at -80°C for later
analyses such as BCA protein assay and cytokine measurements.
2.3 Molecular biology assays
2.3.1 Pierce™ BCA Protein Assay Kit
For total protein concentration measurement in BALF supernatant the Pierce™
BCA Protein Assay Kit (Thermo Scientific, Rockford, USA) was used. The assay is a
formulation based on bicinchoninic acid (BCA) for colorimetric detection and quantitation
of total protein and combines the reduction of Cu+2
to Cu1+
by protein in an alkaline
medium with a selective colorimetric detection of Cu1+
using a reagent containing BCA.
Assays were performed according to the manufacturer’s instructions and absorbance at 562
nm was measured in a spectrophotometer (SpectrMax M5 plate reader, Molecular Devices,
Sunnyvale, CA).

Methods
38
2.3.2 ELISA/MSD
For cytokine measurement in human cell culture supernatants and murine BALF
supernatants the Meso Scale Discovery V- and U-PLEX multiplexing technology (MESO
SCALE DISCOVERY®, Rockville, USA) was used. The assays are sandwich
immunoassays for measuring the levels of protein targets within a single sample. Samples
and detection antibodies conjugated with electrochemiluminescent labels (MSD SULFO-
TAGTM
) are added to plates coated with capture antibodies on independent and well-
defined spots. Analytes in the samples bind to the capture antibodies on the working
electrode surface and later bounding of the detection antibodies to the analytes completes
the sandwich. Plates were loaded in the MSD instrument (MESOTM
SECTOR S 600,
MESO SCALE DISCOVERY, Rockville, USA), where a voltage applied to the plates
electrodes causes the captured labels to emit light, which intensity is measured to provide a
quantitative measure of analytes in the sample. V-PLEX plates were used in the human
Chemokine Panel 1 (Eotaxin, MIP-1β, Eotaxin-3, TARC, IP-10, MIP-1α, IL-8, MCP-1,
MDC and MCP-4), Cytokine Panel 1 (GM-CSF, IL-1α, IL-5, IL-7, IL-12/IL-23p40, IL-15,
IL-16, IL-17A, TNF-β and VEGF) and Pro-inflammatory Panel 1 (IFN-γ, IL-1β, IL-2, IL-
4, IL-6, IL-8, IL-10, IL-12p70, IL-13 and TNF-α) configuration or U-PLEX plates were
individually spotted with the antibody pairs against the desired analytes. For cytokine
measurement in mouse BALF the V-PLEX Pro-inflammatory Panel 1 Mouse Kit (IFN-γ,
IL-1β, IL-2, IL-4, IL-5, IL-6, KC/GRO, IL-10, IL-12p70, and TNF-α.) was used. Assays
were performed according to the manufacturer’s instructions.
2.3.3 Phospho/Total ERK1/2 assay
For the measurement of Extracellular signal Regulated Kinases (ERK) 1 and 2 in cell
lysate the Meso Scale Discovery phosphoprotein assay was used. The Phospho
(Thr202/Tyr204; Thr185/Tyr187)/Total ERK1/2 Assay (MESO SCALE DISCOVERY®,
Gaithersburg, USA) is a sandwich immunoassay and provides a plate pre-coated with
capture antibodies for phosphorylated ERK1/2 ((Thr202/Tyr204; Thr185/Tyr187) and total
ERK1/2 on spatial distinct spots. As for the cytokine measurement, analytes in the samples
binds to the capture antibody on the working electrode and afterwards conjugated detection
antibody bound to the analytes completing the sandwich. Plates were loaded in the MSD

Methods
39
instrument for analysis. Assays were performed according to the manufacturer’s
instructions.
2.3.4 ATP release measurement
For quantifying cell ATP release in supernatant, the ATP-GloTM
Bioluminometric Cell
Viability Assay Kit (Biotium, Hayward, CA) was performed. ATP is an indicator of
metabolically active cells and therefore the number of viable cells can be assessed based on
the amount of ATP available. Furthermore the assay was used for the detection of ATP
release from cells exposed to mechanical stretch. The ATP detection kit takes advantage of
firefly luciferase’s use of ATP to oxidize D-Luciferin and the resulting production of light
to detect the amount of ATP available and was measured in a luminometer. Assays were
performed according to the manufacturer’s instructions.
2.3.5 LDH release
Lactate dehydrogenase (LDH) release was detected using a CytoTox96® Non-Radioactive
Cytotoxicity Assay kit (Promega, Madison, WI) following manufacturers instruction.
Briefly released LDH in culture supernatants is measured with a 30-minute coupled
enzymatic assay, which results in the conversion of a tetrazolium salt (iodonitrotetrazolium
violet; INT) into a red formazan product. The amount of color formed is proportional to the
number of lysed cells. Briefly the release of LDH from damaged cells is measured by
supplying lactate, NAD+ and INT as substrates in the presence of diaphorase. Generation
of a red formazan product in the presence of LDH is proportional to the amount of LDH
released from lysed cells. The conversion of tetrazolium salt to a red formazan product in
the presence of LDH is detectable by measurement of the absorbance at 490 nm using a
SpectrMax M5 plate reader (Molecular Devices, Sunnyvale, CA). Percent cytotoxicity is
calculated by the following formula: 100 x Experimental LDH release divided by the
maximum LDH release (= lysate).

Methods
40
2.3.6 Human cAMP / Calcium Signaling PathwayFinder
NCI-H292 seeded in 1 ml medium with a density of 3 x 105 cells/well on a 12 well plate
(Thermo Fisher Scientific, NunclonTM
Delta Surface, #150628, Roskilde, Denmark), were
incubated at 37°C in 5% CO2, humidified air for 24 h. Afterwards cells were preincubated
for 2 h in the presence or absence of the TRPV4 antagonist GSK2193874 [1 µM]. Medium
was removed and cells were stimulated either with 0.1 % DMSO, 1 µM GSK2193874, 10
µM 4α-PDD or 10 µM 4α-PDD and 1 µM GSK2193874 in 1 ml medium and incubated at
37°C in 5% CO2, humidified air for 24 h. For purification of total RNA from cells the
RNeasy®
Mini Kit (Qiagen, #7410, Hilden, Germany) was used.
Cells were washed twice with Dulbecco's Phosphate-Buffered Saline (DPBS, Gibco, Life
technologies, Grand Island, NY) without Ca2+
and Mg2+
and 350 μL RLT buffer
(QIAGEN, #79216) + 1% β-Mercaptoethanol was given to each well and incubated for 5
min on ice. Samples were pooled from 3 wells each group to 1 sample. Afterwards samples
were transferred on a QIAshredder tubes (QUIAGEN, #79656) and centrifuge at 10.000
rpm for 2 min. Afterwards 350 µl 70% cold EtOH was given to each flow-through and
samples were transferred to an RNeasy spin column placed in a 2 ml collection tube and
centrifuge for 15 s at 10.000 rpm. The flow-through was discarded and the collection tube
was used for the next step. 350 μL Buffer RW1 (QIAGEN, Hilden, Germany) was given to
the RNeasy spin column and centrifuge for 15 s at 10.000 rpm to wash the spin column
membrane. The flow-through was discarded and the collection tube was used for the next
step. Afterwards 80 µl of DNase I incubation mix (10 µl DNase I stock solution and 70 µl
buffer RDD) was added to each column and incubated at RT for 15 min. Afterwards 350
μL Buffer RW1 were given to the RNeasy spin column and centrifuge for 15 s at 10.000
rpm to wash the spin column membrane. Flow-through was discarded and collection tubes
were used in the next step. 500 µl Buffer RPE (QIAGEN, Hilden, Germany) was given to
the RNeasy spin column and centrifuge for 15 s at 10.000 rpm. Flow-through was
discarded and another 500 µl Buffer RPE was given to each RNeasy spin column and
centrifuge for 2 min at 10.000 rpm. Afterwards the RNeasy spin column was removed
carefully from the collection tube and placed in a new 2 mL collection tube and centrifuge
at ≥ 10.000 rpm for 1 min. The RNeasy spin column was then placed in a new 1.5 mL
collection tube and 50 µl RNase-free water (QIAGEN, Hilden, Germany) was added
directly to the spin columns membrane and centrifuge for 1 min at 10.000 rpm to elute the

Methods
41
RNA. RNA concentration of each sample was determined by measuring the absorbance
(260 nm) in a NanoDrop 8000 (Thermo Fisher Scientific).
For cDNA synthesis from purified RNA in prior to real-time PCR the RT2 First Strand Kit
12 (Qiagen, #330401, Hilden, Germany) was used and performed according to the
manufacturer’s instructions. Briefly the RNA samples were treated with DNA elimination
mix according to the RNA concentration of each samples (25 ng - 5 µg RNA sample + 2 µl
Buffer GE + x µl RNase-free water to a total volume of 10 µl) and incubated for 5 min at
42°C and then placed immediately on ice for at least 1 min. 10 µl of reverse-transcription
mix was added to each tube containing 10 µl DNA elimination mix and incubated at 42°C
for exactly 15 min. The reaction was then immediately stopped by incubating at 95°C for 5
min. 91 µl RNase-free water was added to each reaction and cDNA samples were placed
on ice or stored at -20°C before proceeding with PCR.
Afterwards the Human cAMP / Calcium Signaling PathwayFinder RT2 Profiler PCR Array
(Qiagen, # 330231, Hilden, Germany) for 384-well (4 x 96 PCR arrays) was performed
according to the manufacturer’s instructions. Briefly the PCR components mix was
prepared by adding 548 µl of RNase-free water and 650 µl RT2 SYBR Green ROX qPCR
Mastermix 2 (SABiosciences, Qiagen, #330520, Hilden, Germany) to 102 µl of each
cDNA samples. Afterwards the PCR components mix from the 4 samples was dispensed
into the cAMP / Calcium Signaling PathwayFinder RT2 Profiler PCR Array. The array was
sealed with an optical adhesive film and placed in the real-time PCR cycler system ViiA 7
(Applied Biosystems by Life Technologies, Carlsbad, USA) with 1 cycle at 95°C for 10
min to HotStart DNA Taq Polymerase and further 40 cycles (1 min each) at 60°C
performing fluorescence data collection. Afterwards the CT values for all wells were
exported to a blank Excel® spreadsheet for use with the SABiosciences PCR Array Data
Analysis Template Excel and Web-based software
(www.SABiosciences.com/pcrarraydataanalysis.php.).
Table 1: Gene table of the Human cAMP / Calcium Signaling PathwayFinder RT2 Profiler PCR Array
Position Unigene Refseq Symbol Description
A01 Hs.99913 NM_000684 ADRB1 Adrenergic, beta-1-, receptor
A02 Hs.171189 NM_001621 AHR Aryl hydrocarbon receptor
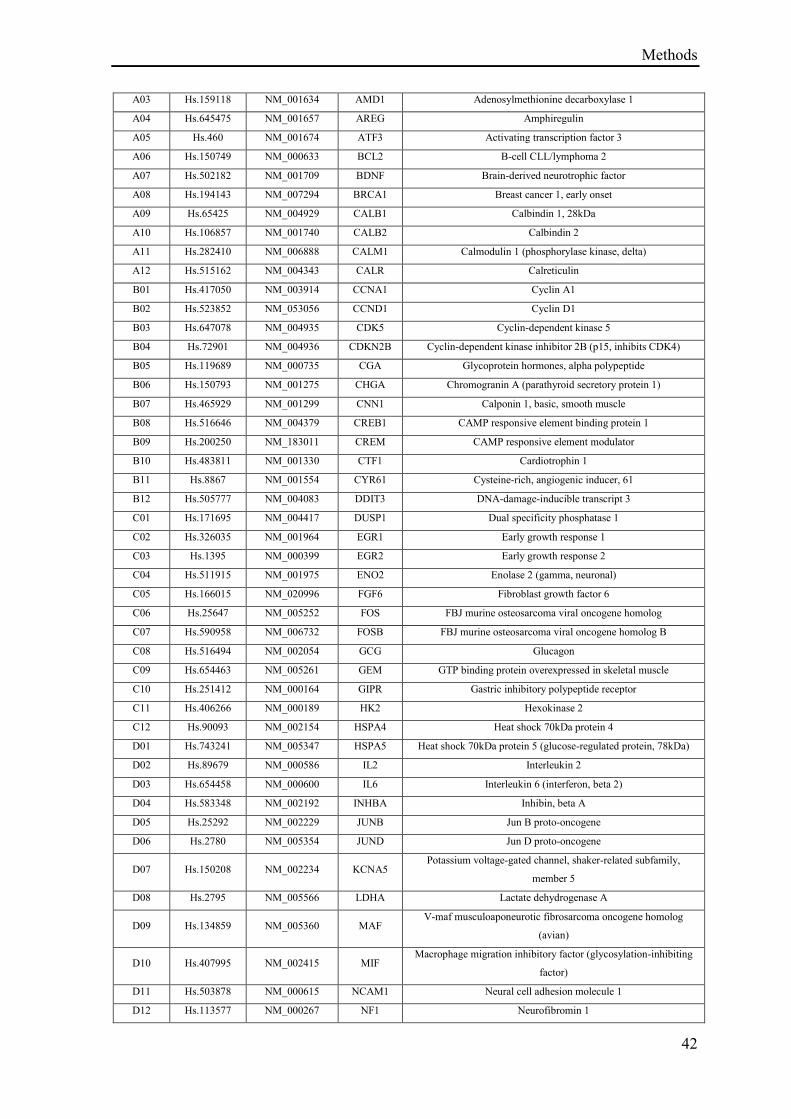
Methods
42
A03 Hs.159118 NM_001634 AMD1 Adenosylmethionine decarboxylase 1
A04 Hs.645475 NM_001657 AREG Amphiregulin
A05 Hs.460 NM_001674 ATF3 Activating transcription factor 3
A06 Hs.150749 NM_000633 BCL2 B-cell CLL/lymphoma 2
A07 Hs.502182 NM_001709 BDNF Brain-derived neurotrophic factor
A08 Hs.194143 NM_007294 BRCA1 Breast cancer 1, early onset
A09 Hs.65425 NM_004929 CALB1 Calbindin 1, 28kDa
A10 Hs.106857 NM_001740 CALB2 Calbindin 2
A11 Hs.282410 NM_006888 CALM1 Calmodulin 1 (phosphorylase kinase, delta)
A12 Hs.515162 NM_004343 CALR Calreticulin
B01 Hs.417050 NM_003914 CCNA1 Cyclin A1
B02 Hs.523852 NM_053056 CCND1 Cyclin D1
B03 Hs.647078 NM_004935 CDK5 Cyclin-dependent kinase 5
B04 Hs.72901 NM_004936 CDKN2B Cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4)
B05 Hs.119689 NM_000735 CGA Glycoprotein hormones, alpha polypeptide
B06 Hs.150793 NM_001275 CHGA Chromogranin A (parathyroid secretory protein 1)
B07 Hs.465929 NM_001299 CNN1 Calponin 1, basic, smooth muscle
B08 Hs.516646 NM_004379 CREB1 CAMP responsive element binding protein 1
B09 Hs.200250 NM_183011 CREM CAMP responsive element modulator
B10 Hs.483811 NM_001330 CTF1 Cardiotrophin 1
B11 Hs.8867 NM_001554 CYR61 Cysteine-rich, angiogenic inducer, 61
B12 Hs.505777 NM_004083 DDIT3 DNA-damage-inducible transcript 3
C01 Hs.171695 NM_004417 DUSP1 Dual specificity phosphatase 1
C02 Hs.326035 NM_001964 EGR1 Early growth response 1
C03 Hs.1395 NM_000399 EGR2 Early growth response 2
C04 Hs.511915 NM_001975 ENO2 Enolase 2 (gamma, neuronal)
C05 Hs.166015 NM_020996 FGF6 Fibroblast growth factor 6
C06 Hs.25647 NM_005252 FOS FBJ murine osteosarcoma viral oncogene homolog
C07 Hs.590958 NM_006732 FOSB FBJ murine osteosarcoma viral oncogene homolog B
C08 Hs.516494 NM_002054 GCG Glucagon
C09 Hs.654463 NM_005261 GEM GTP binding protein overexpressed in skeletal muscle
C10 Hs.251412 NM_000164 GIPR Gastric inhibitory polypeptide receptor
C11 Hs.406266 NM_000189 HK2 Hexokinase 2
C12 Hs.90093 NM_002154 HSPA4 Heat shock 70kDa protein 4
D01 Hs.743241 NM_005347 HSPA5 Heat shock 70kDa protein 5 (glucose-regulated protein, 78kDa)
D02 Hs.89679 NM_000586 IL2 Interleukin 2
D03 Hs.654458 NM_000600 IL6 Interleukin 6 (interferon, beta 2)
D04 Hs.583348 NM_002192 INHBA Inhibin, beta A
D05 Hs.25292 NM_002229 JUNB Jun B proto-oncogene
D06 Hs.2780 NM_005354 JUND Jun D proto-oncogene
D07 Hs.150208 NM_002234 KCNA5 Potassium voltage-gated channel, shaker-related subfamily,
member 5
D08 Hs.2795 NM_005566 LDHA Lactate dehydrogenase A
D09 Hs.134859 NM_005360 MAF V-maf musculoaponeurotic fibrosarcoma oncogene homolog
(avian)
D10 Hs.407995 NM_002415 MIF Macrophage migration inhibitory factor (glycosylation-inhibiting
factor)
D11 Hs.503878 NM_000615 NCAM1 Neural cell adhesion molecule 1
D12 Hs.113577 NM_000267 NF1 Neurofibromin 1
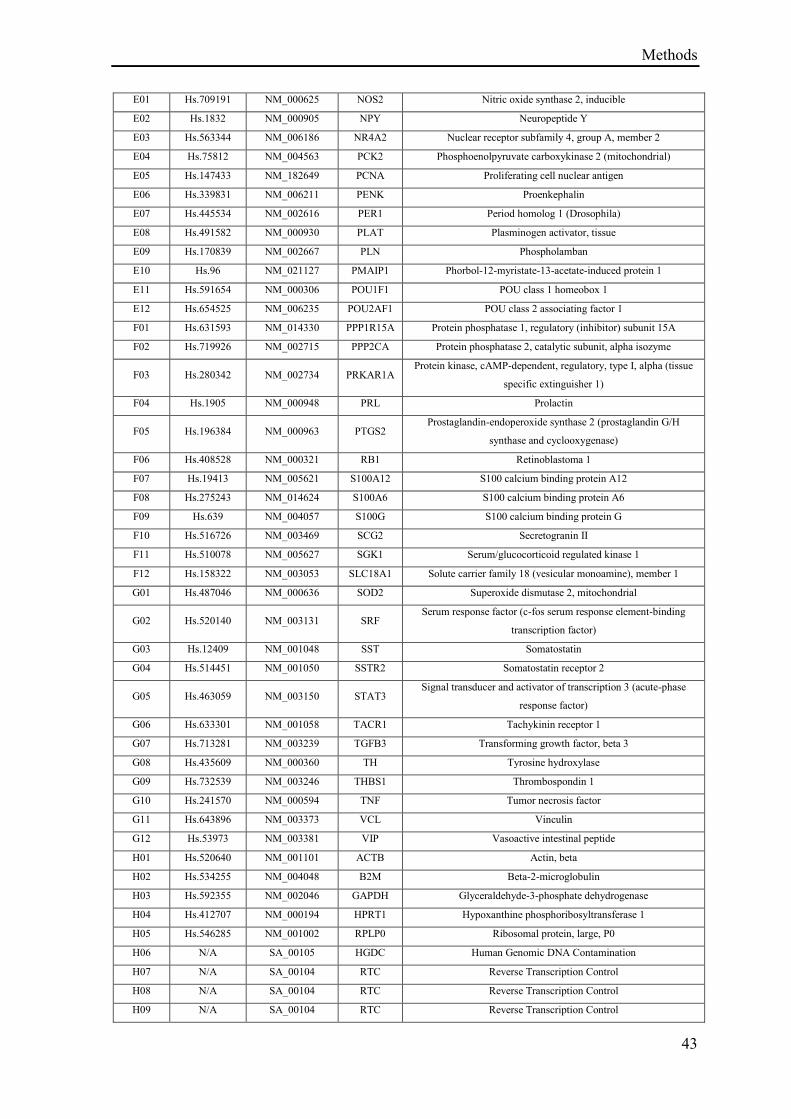
Methods
43
E01 Hs.709191 NM_000625 NOS2 Nitric oxide synthase 2, inducible
E02 Hs.1832 NM_000905 NPY Neuropeptide Y
E03 Hs.563344 NM_006186 NR4A2 Nuclear receptor subfamily 4, group A, member 2
E04 Hs.75812 NM_004563 PCK2 Phosphoenolpyruvate carboxykinase 2 (mitochondrial)
E05 Hs.147433 NM_182649 PCNA Proliferating cell nuclear antigen
E06 Hs.339831 NM_006211 PENK Proenkephalin
E07 Hs.445534 NM_002616 PER1 Period homolog 1 (Drosophila)
E08 Hs.491582 NM_000930 PLAT Plasminogen activator, tissue
E09 Hs.170839 NM_002667 PLN Phospholamban
E10 Hs.96 NM_021127 PMAIP1 Phorbol-12-myristate-13-acetate-induced protein 1
E11 Hs.591654 NM_000306 POU1F1 POU class 1 homeobox 1
E12 Hs.654525 NM_006235 POU2AF1 POU class 2 associating factor 1
F01 Hs.631593 NM_014330 PPP1R15A Protein phosphatase 1, regulatory (inhibitor) subunit 15A
F02 Hs.719926 NM_002715 PPP2CA Protein phosphatase 2, catalytic subunit, alpha isozyme
F03 Hs.280342 NM_002734 PRKAR1A Protein kinase, cAMP-dependent, regulatory, type I, alpha (tissue
specific extinguisher 1)
F04 Hs.1905 NM_000948 PRL Prolactin
F05 Hs.196384 NM_000963 PTGS2 Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H
synthase and cyclooxygenase)
F06 Hs.408528 NM_000321 RB1 Retinoblastoma 1
F07 Hs.19413 NM_005621 S100A12 S100 calcium binding protein A12
F08 Hs.275243 NM_014624 S100A6 S100 calcium binding protein A6
F09 Hs.639 NM_004057 S100G S100 calcium binding protein G
F10 Hs.516726 NM_003469 SCG2 Secretogranin II
F11 Hs.510078 NM_005627 SGK1 Serum/glucocorticoid regulated kinase 1
F12 Hs.158322 NM_003053 SLC18A1 Solute carrier family 18 (vesicular monoamine), member 1
G01 Hs.487046 NM_000636 SOD2 Superoxide dismutase 2, mitochondrial
G02 Hs.520140 NM_003131 SRF Serum response factor (c-fos serum response element-binding
transcription factor)
G03 Hs.12409 NM_001048 SST Somatostatin
G04 Hs.514451 NM_001050 SSTR2 Somatostatin receptor 2
G05 Hs.463059 NM_003150 STAT3 Signal transducer and activator of transcription 3 (acute-phase
response factor)
G06 Hs.633301 NM_001058 TACR1 Tachykinin receptor 1
G07 Hs.713281 NM_003239 TGFB3 Transforming growth factor, beta 3
G08 Hs.435609 NM_000360 TH Tyrosine hydroxylase
G09 Hs.732539 NM_003246 THBS1 Thrombospondin 1
G10 Hs.241570 NM_000594 TNF Tumor necrosis factor
G11 Hs.643896 NM_003373 VCL Vinculin
G12 Hs.53973 NM_003381 VIP Vasoactive intestinal peptide
H01 Hs.520640 NM_001101 ACTB Actin, beta
H02 Hs.534255 NM_004048 B2M Beta-2-microglobulin
H03 Hs.592355 NM_002046 GAPDH Glyceraldehyde-3-phosphate dehydrogenase
H04 Hs.412707 NM_000194 HPRT1 Hypoxanthine phosphoribosyltransferase 1
H05 Hs.546285 NM_001002 RPLP0 Ribosomal protein, large, P0
H06 N/A SA_00105 HGDC Human Genomic DNA Contamination
H07 N/A SA_00104 RTC Reverse Transcription Control
H08 N/A SA_00104 RTC Reverse Transcription Control
H09 N/A SA_00104 RTC Reverse Transcription Control
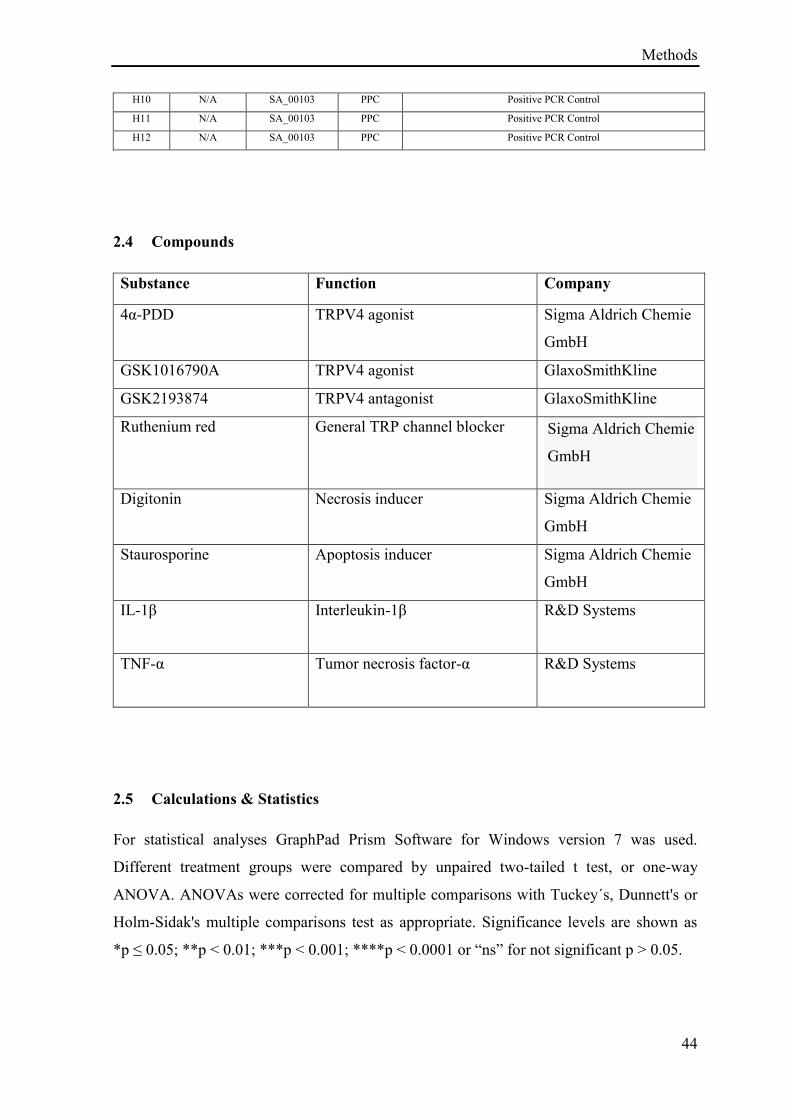
Methods
44
H10 N/A SA_00103 PPC Positive PCR Control
H11 N/A SA_00103 PPC Positive PCR Control
H12 N/A SA_00103 PPC Positive PCR Control
2.4 Compounds
Substance Function Company
4α-PDD TRPV4 agonist Sigma Aldrich Chemie
GmbH
GSK1016790A TRPV4 agonist GlaxoSmithKline
GSK2193874 TRPV4 antagonist GlaxoSmithKline
Ruthenium red General TRP channel blocker
Sigma Aldrich Chemie
GmbH
Digitonin Necrosis inducer Sigma Aldrich Chemie
GmbH
Staurosporine Apoptosis inducer Sigma Aldrich Chemie
GmbH
IL-1β Interleukin-1β R&D Systems
TNF-α Tumor necrosis factor-α R&D Systems
2.5 Calculations & Statistics
For statistical analyses GraphPad Prism Software for Windows version 7 was used.
Different treatment groups were compared by unpaired two-tailed t test, or one-way
ANOVA. ANOVAs were corrected for multiple comparisons with Tuckey´s, Dunnett's or
Holm-Sidak's multiple comparisons test as appropriate. Significance levels are shown as
*p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 or “ns” for not significant p > 0.05.

Methods
45
2.6 Ethics statement
All human blood samples were obtained from volunteers with prior written informed
consent. The blood donation was voluntary and in compliance with data protection
standards. Please note that the current process of the donation service at Boehringer
Ingelheim in Biberach is approved by the appropriate external review board of the federal
state of Baden-Württemberg, Germany named Ethik-Kommission der Landesärztekammer
Baden-Württemberg. Blood donation from healthy subjects is voluntary. Ensure data
protection for the participants and prior written informed consent is obtained from each
blood donor. The same principles was applied to the blood donation at the time of blood
sampling for the current manuscript. Please note that we have obtained blood from less
than 5 healthy volunteers for the purpose of our investigation and the blood draw was not
part of a clinical trial but for basic research purposes only. Therefore, no prospective ethics
approval was necessary for the blood donation. In any case, the principles of voluntary
donation, data protection and prior written informed consent were applied.
Human cells sourced from commercial vendors were verified to have associated signed
informed consents in place. This process was endorsed by a panel of senior company
scientists and physicians.
All animal experimental procedures were performed in accordance with European and
local animal welfare regulations and approved by the Regierungspräsidium Tübingen in
Germany with the approved animal experimental licenses TVV 13-014-02 (approval date:
10.03.2013) and TVV 15-001-02 (approval date: 22.05.2015).

Results
46
3 Results
3.1 Results: Role of TRPV4 in regulating endothelial membrane integrity
For better understanding TRPV4 biology and its role in the regulation of membrane barrier
integrity, its activation and inhibition was investigated in different in vitro and in vivo
models focusing on permeability using two reported selective activators of TRPV4,
GSK1016790A and 4α-PDD (Thorneloe et al. 2008, Willette et al. 2008) and the potent
and selective TRPV4 blocker GSK2193874 (Thorneloe et al. 2012).
3.1.1 TRPV4 mediated calcium influx
TRPV4 mediated calcium influx has been identified as key regulator of endothelial
permeability in previous study (Hamanaka et al. 2007, Yin et al. 2008). To investigate the
different mode of action of GSK1016790A and 4α-PDD, TRPV4 mediated calcium influx
was studied with these TRPV4 agonists (described in section 2.1.3). Pharmacological
activation and inhibition of TRPV4 on calcium influx was analysed in the FLIPRTETRA
using the FLIPR Calcium 6 Assay kit in human umbilical vein endothelial cells
(HUVECs). TRPV4 activation with GSK1016790A resulted in a direct, significant and
strong increase in intracellular calcium concentration in a dose-dependent manner (Figure
3A, 3C), that could be nearly completely block by preincubation for 15 min with 1 µM of
the TRPV4 antagonist GSK2193874 (Figure 3C). Activation of TRPV4 with 4α-PDD led
to a very small but significant increase of intracellular calcium concentration, that occurred
hours after agonist addition (Figure 3B, 3D) and that was blocked by inhibition with
GSK2193874 (Figure 3D).
Results
47
Figure 3: TRPV4 mediated Calcium influx with the FLIPR Calcium 6 Assay in HUVECs. (A) Calcium influx
measured in the FLIPRTETRA before and after addition of different concentrations of the TRPV4 agonist GSK1016790A
(Ag). (B) Calcium influx measurement after addition of different concentrations of the TRPV4 agonist 4α-PDD. (C)
Calcium influx maxima in cells preincubated for 15 min in presence or absence of a TRPV4 antagonist GSK2193874 [1
µM] (Ant) and treated afterwards with different concentrations of the TRPV4 agonist GSK1016790A (Ag). (D) Calcium
influx in cells preincubated for 15 min in presence or absence of a TRPV4 antagonist GSK2193874 [1 µM] (Ant) and
treated afterwards with different concentrations of the TRPV4 agonist 4α-PDD. Data are shown as mean ± SEM; (n=3;
*p < 0.05; ****p < 0.0001 vs control or as indicated above graphs; one-way ANOVA Tukey's multiple comparisons
test). Grid lines labelled with Ag showing time points of TRPV4 agonist addition in the graphs.
3.1.2 TER measurement in HUVECs
Transient receptor potential vanilloid 4 (TRPV4) has been suggested to be a critical
regulator of endothelial barrier integrity. Pharmacological activation of TRPV4 was
therefor studied in a cellZscope allowing continuous measurement of
transepithelial/transendothelial electrical resistance (TER) (described in section 2.1.1).
During method establishment a TER improvement was observed when endothelial cells
were cultured for 24 hours under the more physiological hypoxic conditions (1% O2, 5%
CO2 at 37°C) prior to TER measurement. Under these conditions it was possible to
establish a model with an assay window high enough to test compound effect on TER in
HUVECs. An initial TER improvement was observed when cells were cultured 24 h under
hypoxic conditions (Figure 4B) compared to cells cultured under normoxia (Figure 4A).
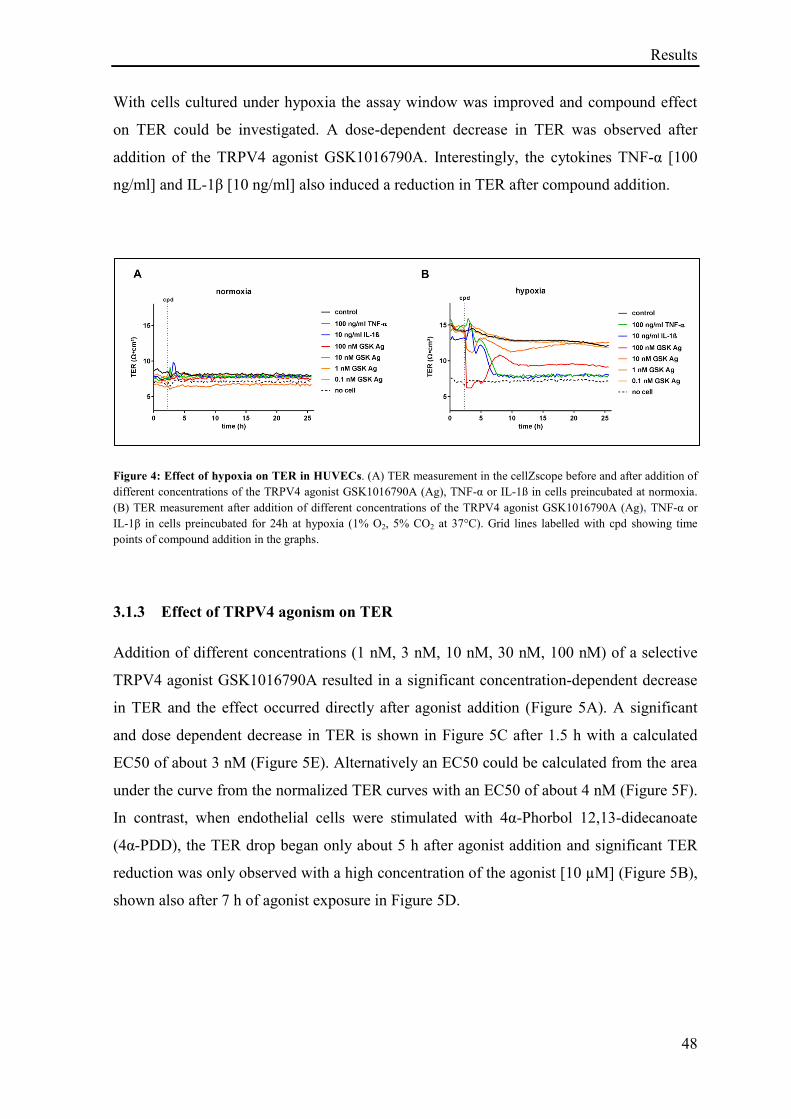
Results
48
With cells cultured under hypoxia the assay window was improved and compound effect
on TER could be investigated. A dose-dependent decrease in TER was observed after
addition of the TRPV4 agonist GSK1016790A. Interestingly, the cytokines TNF-α [100
ng/ml] and IL-1β [10 ng/ml] also induced a reduction in TER after compound addition.
Figure 4: Effect of hypoxia on TER in HUVECs. (A) TER measurement in the cellZscope before and after addition of
different concentrations of the TRPV4 agonist GSK1016790A (Ag), TNF-α or IL-1ß in cells preincubated at normoxia.
(B) TER measurement after addition of different concentrations of the TRPV4 agonist GSK1016790A (Ag), TNF-α or
IL-1β in cells preincubated for 24h at hypoxia (1% O2, 5% CO2 at 37°C). Grid lines labelled with cpd showing time
points of compound addition in the graphs.
3.1.3 Effect of TRPV4 agonism on TER
Addition of different concentrations (1 nM, 3 nM, 10 nM, 30 nM, 100 nM) of a selective
TRPV4 agonist GSK1016790A resulted in a significant concentration-dependent decrease
in TER and the effect occurred directly after agonist addition (Figure 5A). A significant
and dose dependent decrease in TER is shown in Figure 5C after 1.5 h with a calculated
EC50 of about 3 nM (Figure 5E). Alternatively an EC50 could be calculated from the area
under the curve from the normalized TER curves with an EC50 of about 4 nM (Figure 5F).
In contrast, when endothelial cells were stimulated with 4α-Phorbol 12,13-didecanoate
(4α-PDD), the TER drop began only about 5 h after agonist addition and significant TER
reduction was only observed with a high concentration of the agonist [10 µM] (Figure 5B),
shown also after 7 h of agonist exposure in Figure 5D.
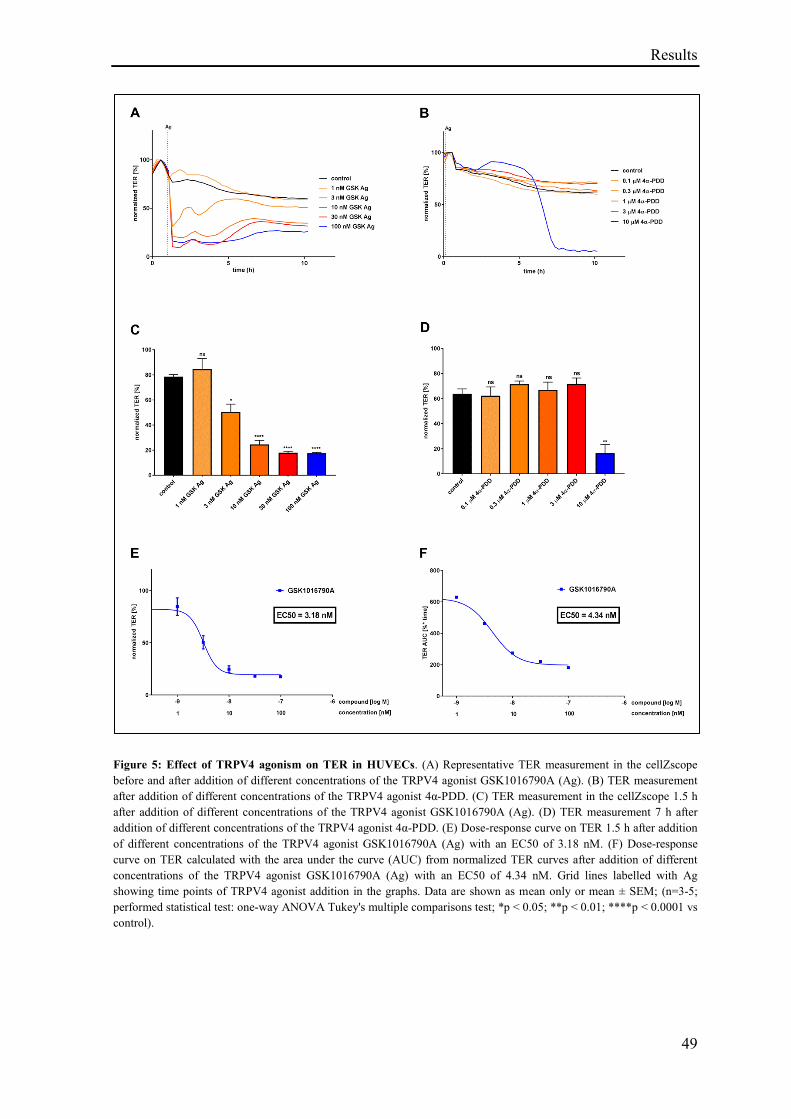
Results
49
Figure 5: Effect of TRPV4 agonism on TER in HUVECs. (A) Representative TER measurement in the cellZscope
before and after addition of different concentrations of the TRPV4 agonist GSK1016790A (Ag). (B) TER measurement
after addition of different concentrations of the TRPV4 agonist 4α-PDD. (C) TER measurement in the cellZscope 1.5 h
after addition of different concentrations of the TRPV4 agonist GSK1016790A (Ag). (D) TER measurement 7 h after
addition of different concentrations of the TRPV4 agonist 4α-PDD. (E) Dose-response curve on TER 1.5 h after addition
of different concentrations of the TRPV4 agonist GSK1016790A (Ag) with an EC50 of 3.18 nM. (F) Dose-response
curve on TER calculated with the area under the curve (AUC) from normalized TER curves after addition of different
concentrations of the TRPV4 agonist GSK1016790A (Ag) with an EC50 of 4.34 nM. Grid lines labelled with Ag
showing time points of TRPV4 agonist addition in the graphs. Data are shown as mean only or mean ± SEM; (n=3-5;
performed statistical test: one-way ANOVA Tukey's multiple comparisons test; *p < 0.05; **p < 0.01; ****p < 0.0001 vs
control).
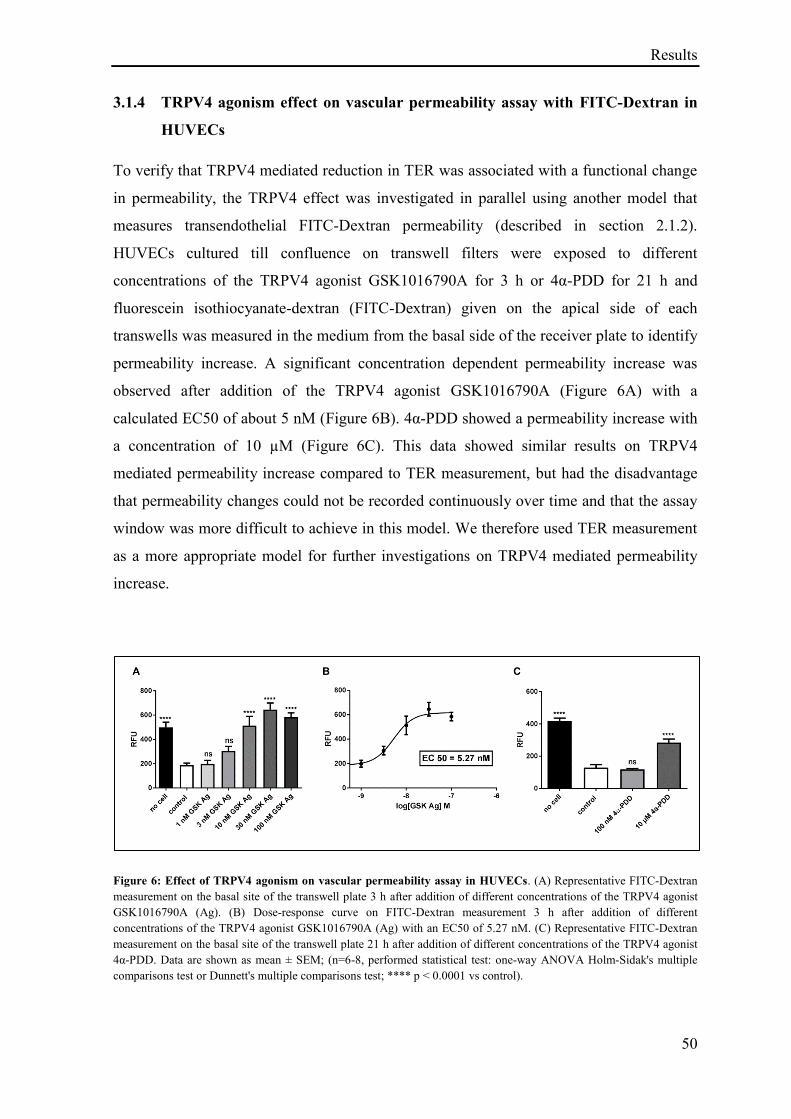
Results
50
3.1.4 TRPV4 agonism effect on vascular permeability assay with FITC-Dextran in
HUVECs
To verify that TRPV4 mediated reduction in TER was associated with a functional change
in permeability, the TRPV4 effect was investigated in parallel using another model that
measures transendothelial FITC-Dextran permeability (described in section 2.1.2).
HUVECs cultured till confluence on transwell filters were exposed to different
concentrations of the TRPV4 agonist GSK1016790A for 3 h or 4α-PDD for 21 h and
fluorescein isothiocyanate-dextran (FITC-Dextran) given on the apical side of each
transwells was measured in the medium from the basal side of the receiver plate to identify
permeability increase. A significant concentration dependent permeability increase was
observed after addition of the TRPV4 agonist GSK1016790A (Figure 6A) with a
calculated EC50 of about 5 nM (Figure 6B). 4α-PDD showed a permeability increase with
a concentration of 10 µM (Figure 6C). This data showed similar results on TRPV4
mediated permeability increase compared to TER measurement, but had the disadvantage
that permeability changes could not be recorded continuously over time and that the assay
window was more difficult to achieve in this model. We therefore used TER measurement
as a more appropriate model for further investigations on TRPV4 mediated permeability
increase.
Figure 6: Effect of TRPV4 agonism on vascular permeability assay in HUVECs. (A) Representative FITC-Dextran
measurement on the basal site of the transwell plate 3 h after addition of different concentrations of the TRPV4 agonist
GSK1016790A (Ag). (B) Dose-response curve on FITC-Dextran measurement 3 h after addition of different
concentrations of the TRPV4 agonist GSK1016790A (Ag) with an EC50 of 5.27 nM. (C) Representative FITC-Dextran
measurement on the basal site of the transwell plate 21 h after addition of different concentrations of the TRPV4 agonist
4α-PDD. Data are shown as mean ± SEM; (n=6-8, performed statistical test: one-way ANOVA Holm-Sidak's multiple
comparisons test or Dunnett's multiple comparisons test; **** p < 0.0001 vs control).

Results
51
3.1.5 Effect of TRPV4 agonism and antagonism on TER
Preincubation with different doses of the TRPV4 antagonist GSK2193874 resulted in a
significant concentration-dependent inhibition of the TRPV4 agonist GSK1016790A (30
nM) effect on TER 1.5 h after agonist addition (Figure 7A, 7C) with a calculated IC50 of
about 200 nM (Figure 7E). In contrast the TRPV4 antagonist GSK2193874 was not able to
inhibit the effect of the TRPV4 agonist 4α-PDD (Figure 7B, 7D) under this assay
procedure. Additionally TRPV4 agonism and antagonism effect with GSK1016790A and
GSK2193874 were also investigated in human small airway epithelial cells (SAECs) by
our group (cultured as described in section 2.1.10) in TER measurement (described in
section 2.1.1). SAECs showed a strong initial TER without the need of preincubation in
hypoxia. Similar to the observation in HUVECs, TRPV4 agonism with GSK1016790A
resulted in a direct and significant reduction in TER that could be inhibited by
preincubation with the TRPV4 antagonist GSK2193874 in SAECs.
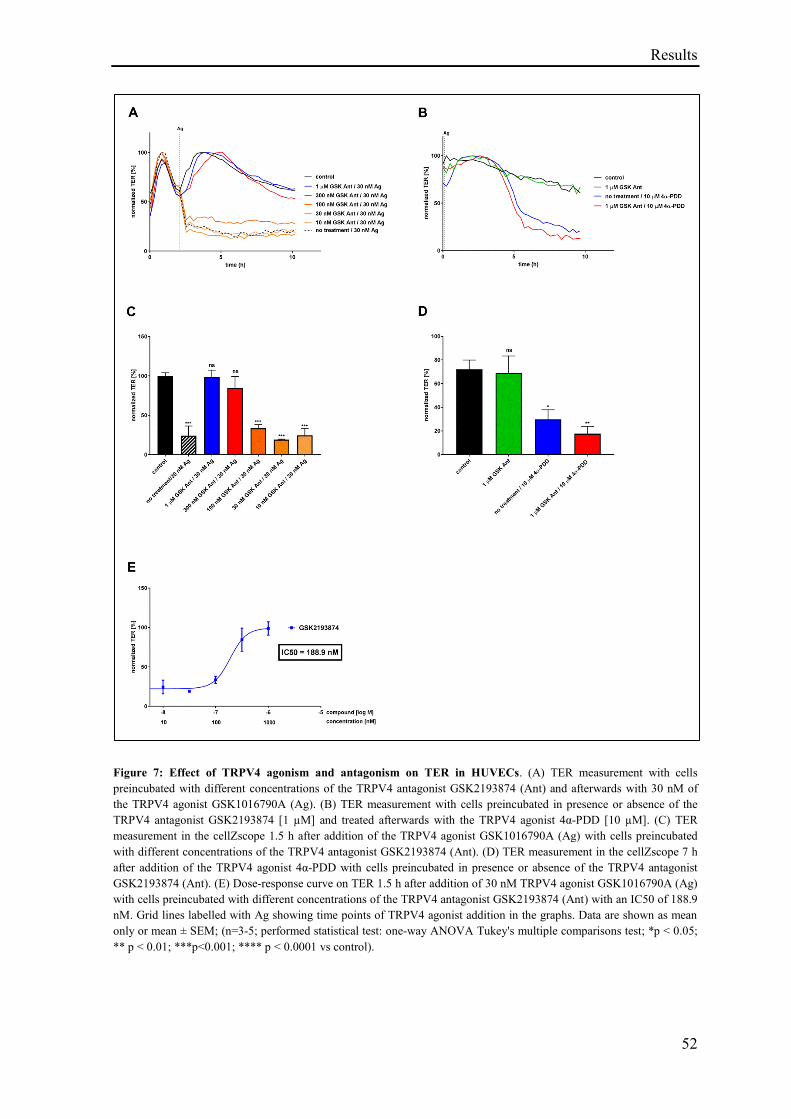
Results
52
Figure 7: Effect of TRPV4 agonism and antagonism on TER in HUVECs. (A) TER measurement with cells
preincubated with different concentrations of the TRPV4 antagonist GSK2193874 (Ant) and afterwards with 30 nM of
the TRPV4 agonist GSK1016790A (Ag). (B) TER measurement with cells preincubated in presence or absence of the
TRPV4 antagonist GSK2193874 [1 µM] and treated afterwards with the TRPV4 agonist 4α-PDD [10 µM]. (C) TER
measurement in the cellZscope 1.5 h after addition of the TRPV4 agonist GSK1016790A (Ag) with cells preincubated
with different concentrations of the TRPV4 antagonist GSK2193874 (Ant). (D) TER measurement in the cellZscope 7 h
after addition of the TRPV4 agonist 4α-PDD with cells preincubated in presence or absence of the TRPV4 antagonist
GSK2193874 (Ant). (E) Dose-response curve on TER 1.5 h after addition of 30 nM TRPV4 agonist GSK1016790A (Ag)
with cells preincubated with different concentrations of the TRPV4 antagonist GSK2193874 (Ant) with an IC50 of 188.9
nM. Grid lines labelled with Ag showing time points of TRPV4 agonist addition in the graphs. Data are shown as mean
only or mean ± SEM; (n=3-5; performed statistical test: one-way ANOVA Tukey's multiple comparisons test; *p < 0.05;
** p < 0.01; ***p<0.001; **** p < 0.0001 vs control).

Results
53
3.1.6 Effect of TRPV4 activation on vascular permeability in vivo
After having demonstrated that TRPV4 activation with the agonist GSK1016790A led to a
permeability increase in vitro and that this effect can be inhibited by the TRPV4 antagonist
GSK2193874 in endothelial cells, further investigations on TRPV4 activation were
performed in a murine vascular permeability model (described in section 2.2.1). Evans
blue given intravenously (i.v.) was used to monitor vascular leakage in different organs
after 15 min exposure with the TRPV4 agonist GSK1016790A [300 µg/kg] given
intravenously. Vascular leakage could not be observed in the different tissues (lung
parenchyma, bronchus, kidney, liver and colon) of C57BL/6J mice (Figure 8A) and
suggest that Balb/c mice may be the more appropriate animal model in this permeability
assay, an observation also made by others showing that mannitol permeability is greater in
Balb/c gut compared to C57BL/6J by assessing lipopolysaccharide (LPS) in portal vein
plasma (Volynets et al. 2016). In the tissues of kidney, colon and liver of Balb/c mice
permeability increase could not be observed. In contrast in lung tissues of Balb/c mice a
significant vascular leakage was observed in the lung parenchyma and an elevated but not
significant Evans blue signal was observed in the bronchus (Figure 8B) and may be
explained by an higher TRPV4 protein expression in the mice lungs compared to the colon,
liver and kidney like the situation seen in humans
(https://www.proteinatlas.org/ENSG00000111199-TRPV4/tissue, in the Human Protein
Atlas, available from www.proteinatlas.org (Uhlén et al. 2015)).
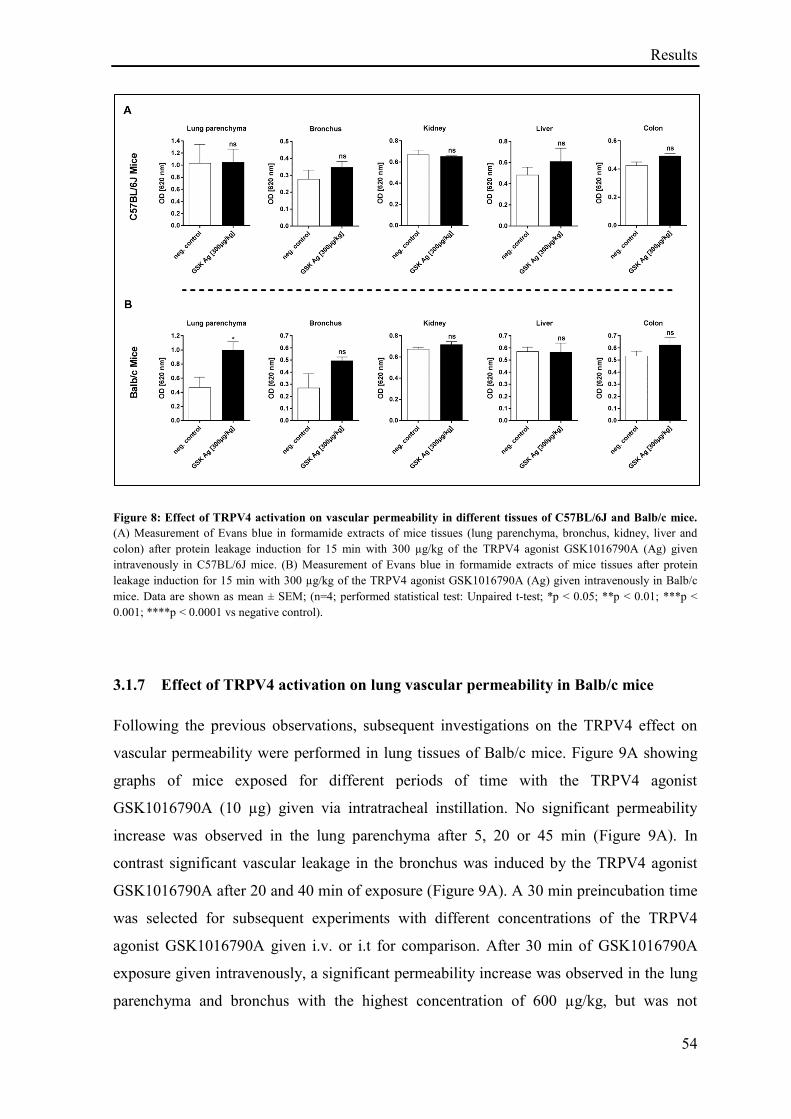
Results
54
Figure 8: Effect of TRPV4 activation on vascular permeability in different tissues of C57BL/6J and Balb/c mice.
(A) Measurement of Evans blue in formamide extracts of mice tissues (lung parenchyma, bronchus, kidney, liver and
colon) after protein leakage induction for 15 min with 300 µg/kg of the TRPV4 agonist GSK1016790A (Ag) given
intravenously in C57BL/6J mice. (B) Measurement of Evans blue in formamide extracts of mice tissues after protein
leakage induction for 15 min with 300 µg/kg of the TRPV4 agonist GSK1016790A (Ag) given intravenously in Balb/c
mice. Data are shown as mean ± SEM; (n=4; performed statistical test: Unpaired t-test; *p < 0.05; **p < 0.01; ***p <
0.001; ****p < 0.0001 vs negative control).
3.1.7 Effect of TRPV4 activation on lung vascular permeability in Balb/c mice
Following the previous observations, subsequent investigations on the TRPV4 effect on
vascular permeability were performed in lung tissues of Balb/c mice. Figure 9A showing
graphs of mice exposed for different periods of time with the TRPV4 agonist
GSK1016790A (10 µg) given via intratracheal instillation. No significant permeability
increase was observed in the lung parenchyma after 5, 20 or 45 min (Figure 9A). In
contrast significant vascular leakage in the bronchus was induced by the TRPV4 agonist
GSK1016790A after 20 and 40 min of exposure (Figure 9A). A 30 min preincubation time
was selected for subsequent experiments with different concentrations of the TRPV4
agonist GSK1016790A given i.v. or i.t for comparison. After 30 min of GSK1016790A
exposure given intravenously, a significant permeability increase was observed in the lung
parenchyma and bronchus with the highest concentration of 600 µg/kg, but was not
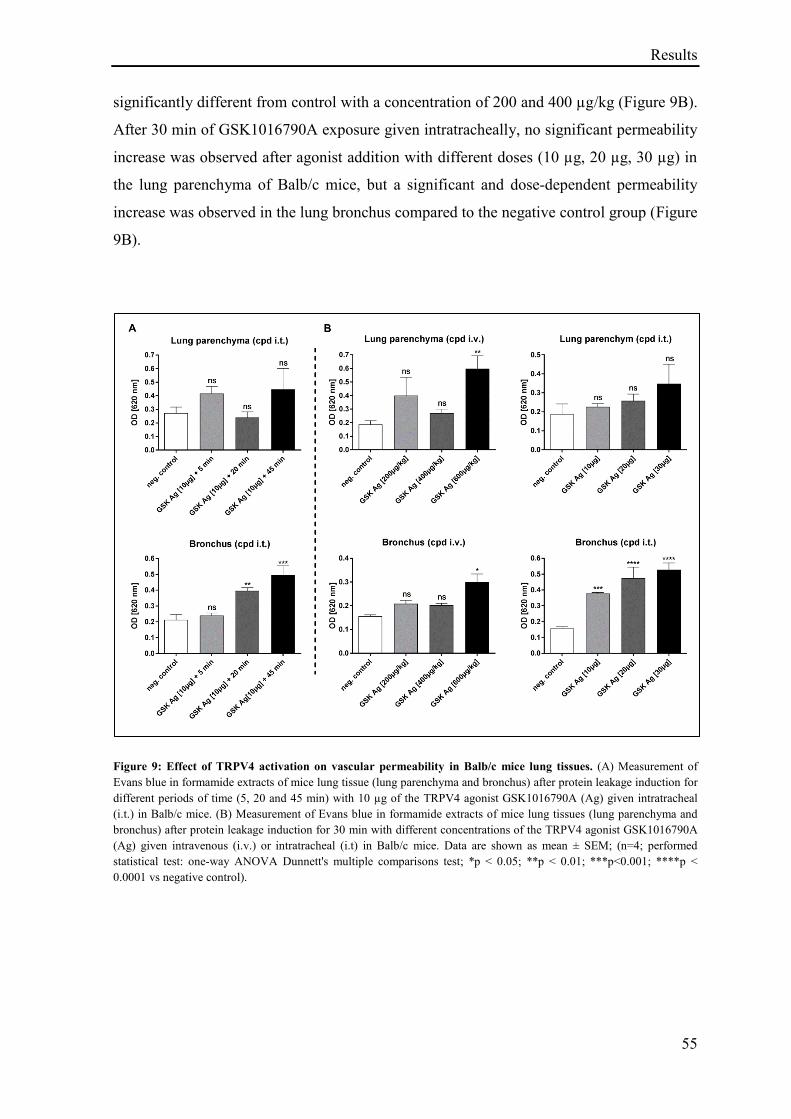
Results
55
significantly different from control with a concentration of 200 and 400 µg/kg (Figure 9B).
After 30 min of GSK1016790A exposure given intratracheally, no significant permeability
increase was observed after agonist addition with different doses (10 µg, 20 µg, 30 µg) in
the lung parenchyma of Balb/c mice, but a significant and dose-dependent permeability
increase was observed in the lung bronchus compared to the negative control group (Figure
9B).
Figure 9: Effect of TRPV4 activation on vascular permeability in Balb/c mice lung tissues. (A) Measurement of
Evans blue in formamide extracts of mice lung tissue (lung parenchyma and bronchus) after protein leakage induction for
different periods of time (5, 20 and 45 min) with 10 µg of the TRPV4 agonist GSK1016790A (Ag) given intratracheal
(i.t.) in Balb/c mice. (B) Measurement of Evans blue in formamide extracts of mice lung tissues (lung parenchyma and
bronchus) after protein leakage induction for 30 min with different concentrations of the TRPV4 agonist GSK1016790A
(Ag) given intravenous (i.v.) or intratracheal (i.t) in Balb/c mice. Data are shown as mean ± SEM; (n=4; performed
statistical test: one-way ANOVA Dunnett's multiple comparisons test; *p < 0.05; **p < 0.01; ***p<0.001; ****p <
0.0001 vs negative control).
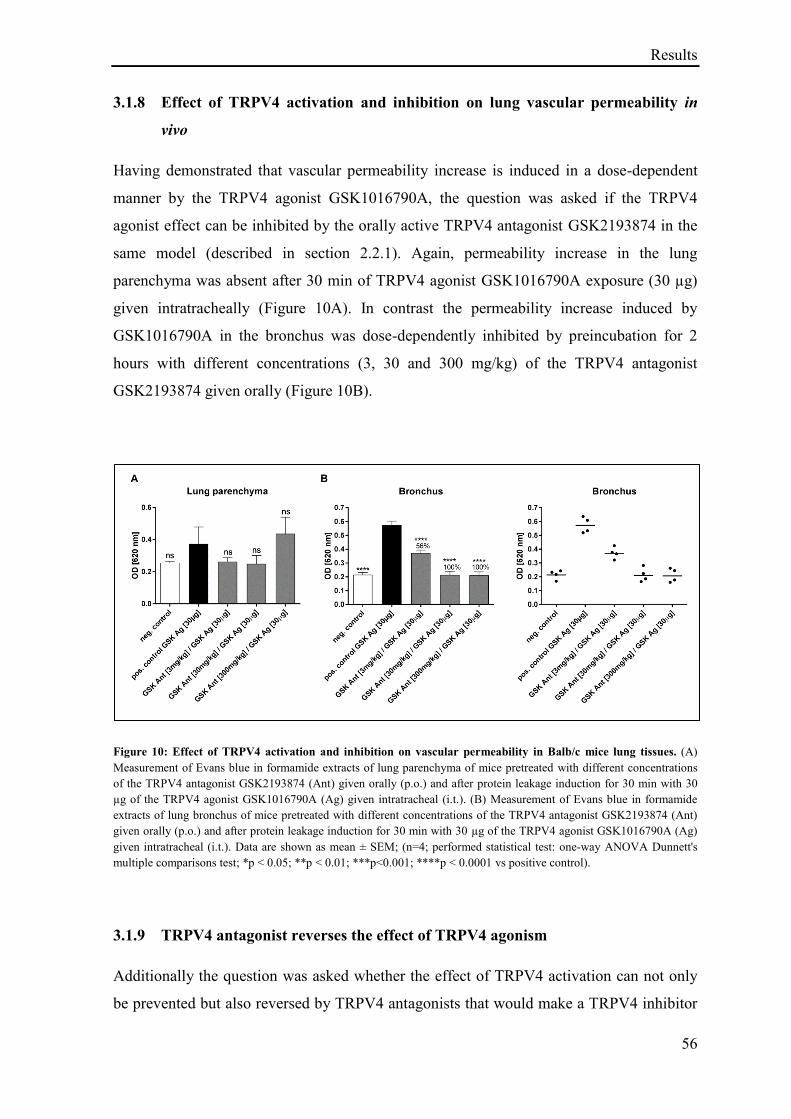
Results
56
3.1.8 Effect of TRPV4 activation and inhibition on lung vascular permeability in
vivo
Having demonstrated that vascular permeability increase is induced in a dose-dependent
manner by the TRPV4 agonist GSK1016790A, the question was asked if the TRPV4
agonist effect can be inhibited by the orally active TRPV4 antagonist GSK2193874 in the
same model (described in section 2.2.1). Again, permeability increase in the lung
parenchyma was absent after 30 min of TRPV4 agonist GSK1016790A exposure (30 µg)
given intratracheally (Figure 10A). In contrast the permeability increase induced by
GSK1016790A in the bronchus was dose-dependently inhibited by preincubation for 2
hours with different concentrations (3, 30 and 300 mg/kg) of the TRPV4 antagonist
GSK2193874 given orally (Figure 10B).
Figure 10: Effect of TRPV4 activation and inhibition on vascular permeability in Balb/c mice lung tissues. (A)
Measurement of Evans blue in formamide extracts of lung parenchyma of mice pretreated with different concentrations
of the TRPV4 antagonist GSK2193874 (Ant) given orally (p.o.) and after protein leakage induction for 30 min with 30
µg of the TRPV4 agonist GSK1016790A (Ag) given intratracheal (i.t.). (B) Measurement of Evans blue in formamide
extracts of lung bronchus of mice pretreated with different concentrations of the TRPV4 antagonist GSK2193874 (Ant)
given orally (p.o.) and after protein leakage induction for 30 min with 30 µg of the TRPV4 agonist GSK1016790A (Ag)
given intratracheal (i.t.). Data are shown as mean ± SEM; (n=4; performed statistical test: one-way ANOVA Dunnett's
multiple comparisons test; *p < 0.05; **p < 0.01; ***p<0.001; ****p < 0.0001 vs positive control).
3.1.9 TRPV4 antagonist reverses the effect of TRPV4 agonism
Additionally the question was asked whether the effect of TRPV4 activation can not only
be prevented but also reversed by TRPV4 antagonists that would make a TRPV4 inhibitor

Results
57
even more attractive as a drug candidate. HUVECs were treated firstly with the TRPV4
agonist GSK1016790A and afterward with the TRPV4 antagonist GSK2193874 in TER
and intracellular calcium concentration measurement (described in section 2.1.1 and
section 2.1.3). In TER measurement HUVECs that were first stimulated with 15 nM of the
TRPV4 agonist GSK1016790A and that were subsequently left untreated for 30 min after
agonist exposure, showed a drop in TER that remained until the end of the experiment. In
contrast HUVECs treated within the first 30 min of agonism with 1 µM of the TRPV4
antagonist GSK2193874 showed a recovery in TER too that of the control group, treated
only with 0.1% DMSO (Figure 11C). The same picture was observed in the intracellular
calcium influx measurement with the FLIPRTETRA
. HUVECs first treated with the TRPV4
agonist GSK1016790A [300 nM] and 15 min later with the TRPV4 antagonist
GSK2193874 [1 µM], showed a large and significant increase in intracellular calcium
concentration directly after agonist addition, that could be significantly reduced from the
time point of the antagonist addition compared to the group, that remained treated with the
agonist only (Figure 11A, 11B). In the calcium measurement a too low concentration of the
TRPV4 agonist GSK1016790A resulted in a assay window not high enough to be reversed
and the effect of a too high concentration of GSK1016790A could not be reversed by
TRPV4 antagonism with GSK2193874 (Figure 11B).
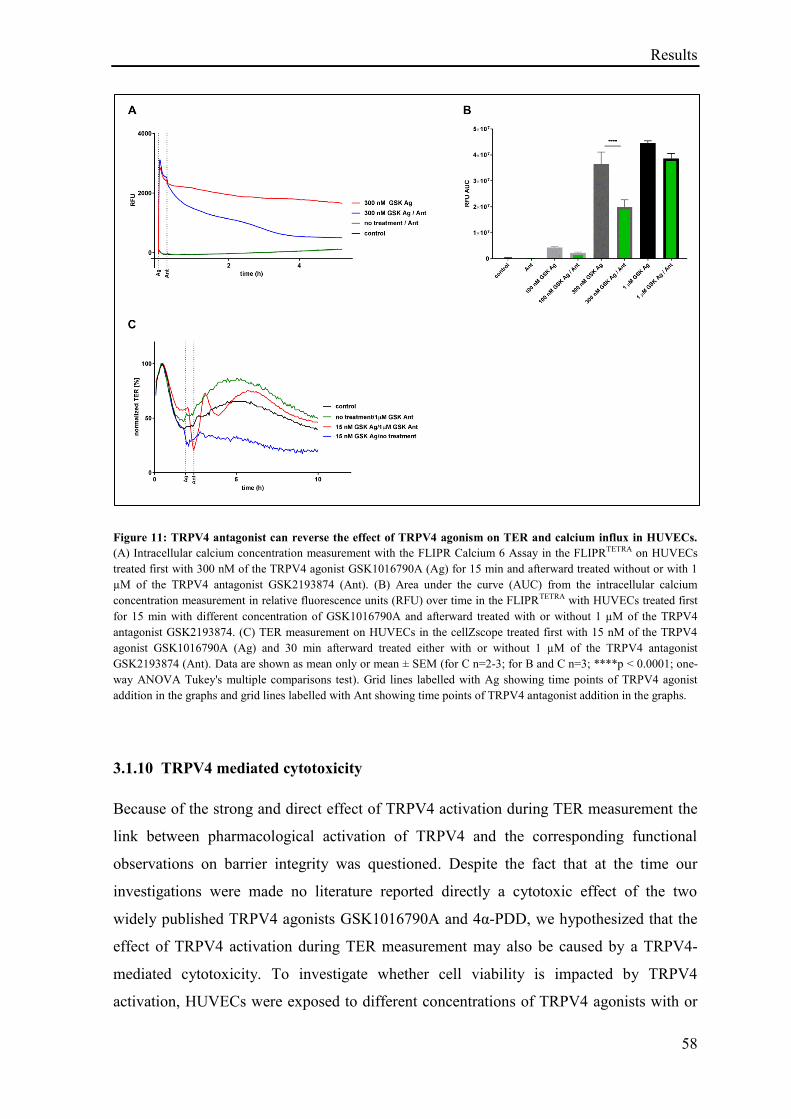
Results
58
Figure 11: TRPV4 antagonist can reverse the effect of TRPV4 agonism on TER and calcium influx in HUVECs.
(A) Intracellular calcium concentration measurement with the FLIPR Calcium 6 Assay in the FLIPRTETRA on HUVECs
treated first with 300 nM of the TRPV4 agonist GSK1016790A (Ag) for 15 min and afterward treated without or with 1
µM of the TRPV4 antagonist GSK2193874 (Ant). (B) Area under the curve (AUC) from the intracellular calcium
concentration measurement in relative fluorescence units (RFU) over time in the FLIPRTETRA with HUVECs treated first
for 15 min with different concentration of GSK1016790A and afterward treated with or without 1 µM of the TRPV4
antagonist GSK2193874. (C) TER measurement on HUVECs in the cellZscope treated first with 15 nM of the TRPV4
agonist GSK1016790A (Ag) and 30 min afterward treated either with or without 1 µM of the TRPV4 antagonist
GSK2193874 (Ant). Data are shown as mean only or mean ± SEM (for C n=2-3; for B and C n=3; ****p < 0.0001; one-
way ANOVA Tukey's multiple comparisons test). Grid lines labelled with Ag showing time points of TRPV4 agonist
addition in the graphs and grid lines labelled with Ant showing time points of TRPV4 antagonist addition in the graphs.
3.1.10 TRPV4 mediated cytotoxicity
Because of the strong and direct effect of TRPV4 activation during TER measurement the
link between pharmacological activation of TRPV4 and the corresponding functional
observations on barrier integrity was questioned. Despite the fact that at the time our
investigations were made no literature reported directly a cytotoxic effect of the two
widely published TRPV4 agonists GSK1016790A and 4α-PDD, we hypothesized that the
effect of TRPV4 activation during TER measurement may also be caused by a TRPV4-
mediated cytotoxicity. To investigate whether cell viability is impacted by TRPV4
activation, HUVECs were exposed to different concentrations of TRPV4 agonists with or

Results
59
without preincubation with 1 µM of the TRPV4 antagonist GSK2193874 for 1 h and
lactate dehydrogenase (LDH) release was recorded to monitor cytotoxic effects (described
in section 2.1.4 and 2.3.5). Cells exposed up to 3 µM 4α-PDD showed no significant LDH
release compared to the control groups after 12 h (Figure 12B), whereas cells exposed to
10 µM 4α-PDD showed no increase in LDH release after 3 h, but LDH release was
observed to begin after 8 h reaching a maximum after 12 h (Figure 12A, 12B). This effect
could not be blocked when cells were preincubated with the TRPV4 antagonist
GSK2193874 (Figure 12A, 12B). In contrast activation of TRPV4 with the agonist
GSK1016790A led to a rapid concentration-dependent increase in cytotoxicity as measured
by LDH release (Figure 12C), even at low concentrations of the agonist (Figure 12D). In
contrast to 4α-PDD, the effect of TRPV4 activation with GSK1016790A could completely
be blocked by preincubation with the TRPV4 antagonist GSK2193874 (Figure 12E) and
was also inhibited in a dose-dependent manner by the TRPV4 antagonist GSK2193874,
when challenged against 30 nM of the TRPV4 agonist GSK1016790A (Figure 12F).
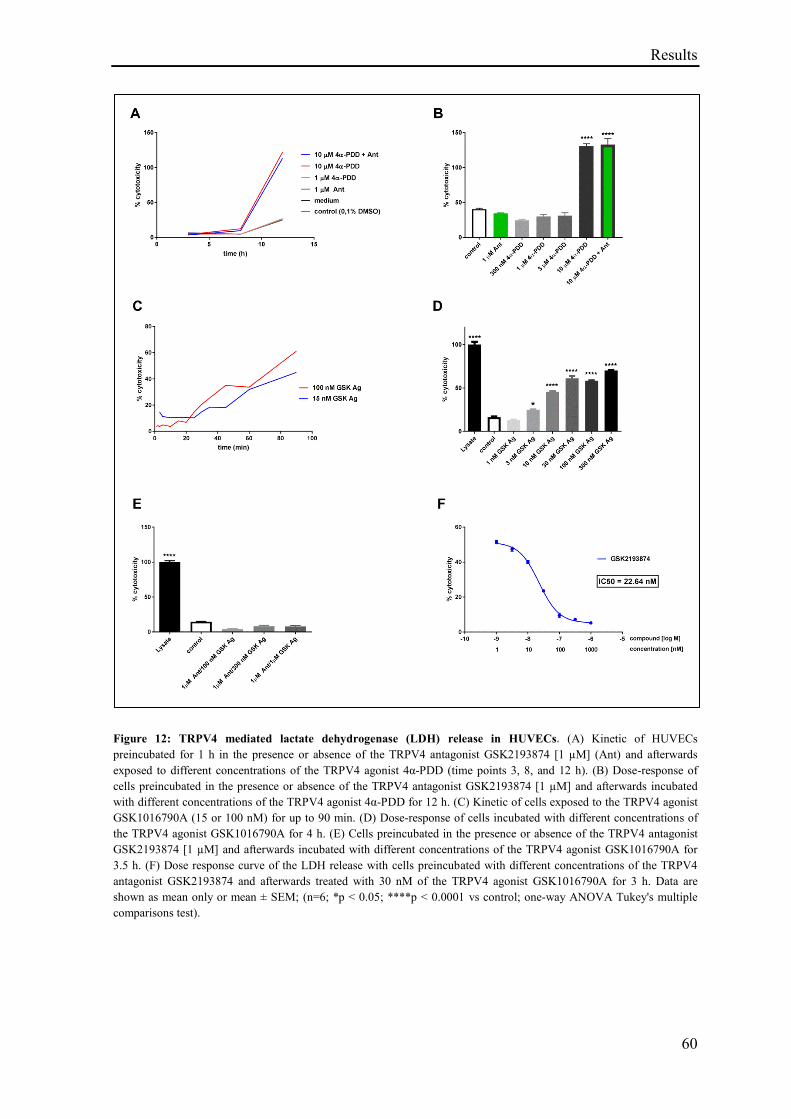
Results
60
Figure 12: TRPV4 mediated lactate dehydrogenase (LDH) release in HUVECs. (A) Kinetic of HUVECs
preincubated for 1 h in the presence or absence of the TRPV4 antagonist GSK2193874 [1 µM] (Ant) and afterwards
exposed to different concentrations of the TRPV4 agonist 4α-PDD (time points 3, 8, and 12 h). (B) Dose-response of
cells preincubated in the presence or absence of the TRPV4 antagonist GSK2193874 [1 µM] and afterwards incubated
with different concentrations of the TRPV4 agonist 4α-PDD for 12 h. (C) Kinetic of cells exposed to the TRPV4 agonist
GSK1016790A (15 or 100 nM) for up to 90 min. (D) Dose-response of cells incubated with different concentrations of
the TRPV4 agonist GSK1016790A for 4 h. (E) Cells preincubated in the presence or absence of the TRPV4 antagonist
GSK2193874 [1 µM] and afterwards incubated with different concentrations of the TRPV4 agonist GSK1016790A for
3.5 h. (F) Dose response curve of the LDH release with cells preincubated with different concentrations of the TRPV4
antagonist GSK2193874 and afterwards treated with 30 nM of the TRPV4 agonist GSK1016790A for 3 h. Data are
shown as mean only or mean ± SEM; (n=6; *p < 0.05; ****p < 0.0001 vs control; one-way ANOVA Tukey's multiple
comparisons test).

Results
61
3.1.11 Time point of TRPV4 induced cytotoxicity and calcium dependent TRPV4
induced LDH release
Additionally the time points, when a significant cytotoxic effect occurred was investigated
when HUVECs were treated with either 15 nM or 100 nM of the TRPV4 agonist
GSK1016790A monitored by LDH release (described in section 2.1.4 and 2.3.5). Cells
exposed to 15 nM of the TRPV4 agonist GSK1016790A showed no significant LDH
release for up to 45 min and a significant cytotoxic effect was observed after 1 h. When
cells were treated with 100 nM GSK1016790A, a cytotoxic effect was not observed for up
to 20 min and LDH release was significantly increased after 25 min of agonist exposure
compared to control group (Figure 13A, 13B).
After having shown that the TRPV4 agonist GSK1016790A induces a large increase in
intracellular calcium concentration in HUVECs and also induces cytotoxicity, further
investigations were made on the question whether the cytotoxic effect induced by the
TRPV4 activator GSK1016790A is dependent on extracellular calcium influx. Therefor
HUVECs were incubated for 1 h in absence or presence of calcium with 100 nM
GSK1016790A in HBSS and LDH release was recorded (described in section 2.1.4 and
2.3.5). The TRPV4 agonist GSK1016790A showed, in cells incubated in HBSS with
calcium, the same cytotoxic effect as in cells treated with the agonist in medium. 100 nM
of the agonist induced a strong and significant increase in LDH release, that could be
blocked by preincubation with the TRPV4 antagonist. In contrast the TRPV4 agonist
GSK1016790A [100 nM] caused no cytotoxic effect compared to control, when incubated
in HBSS without calcium (Figure 13C).
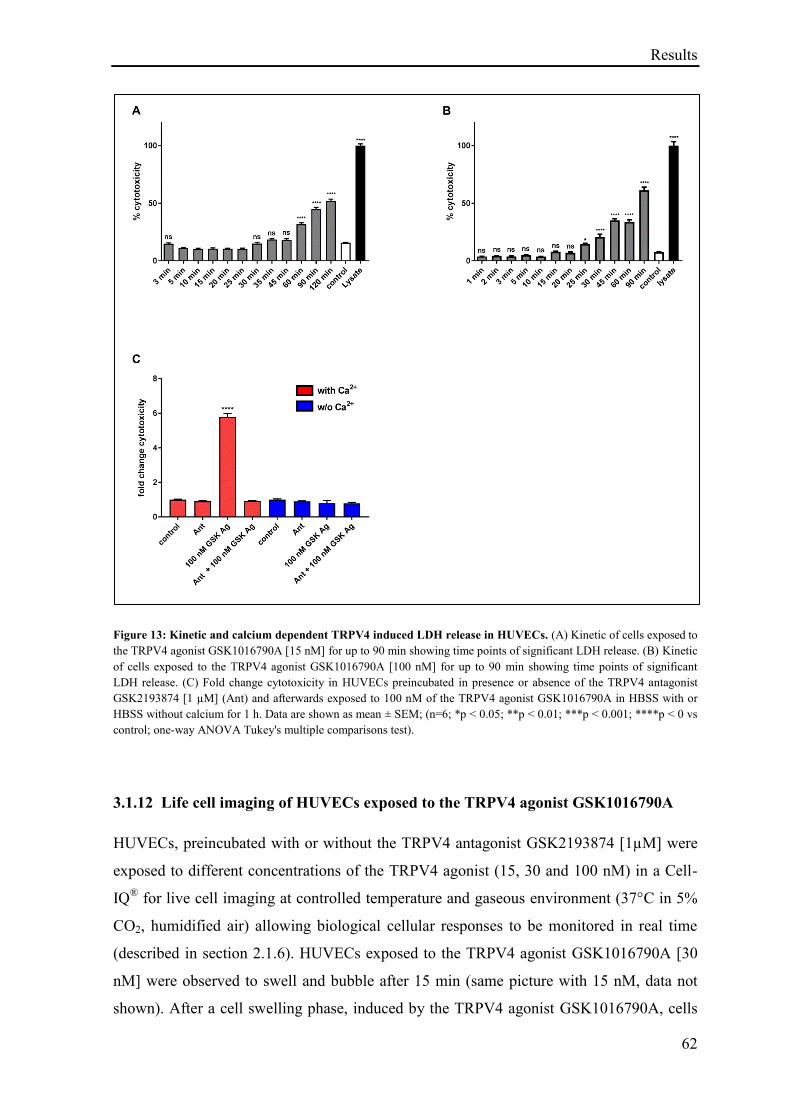
Results
62
Figure 13: Kinetic and calcium dependent TRPV4 induced LDH release in HUVECs. (A) Kinetic of cells exposed to
the TRPV4 agonist GSK1016790A [15 nM] for up to 90 min showing time points of significant LDH release. (B) Kinetic
of cells exposed to the TRPV4 agonist GSK1016790A [100 nM] for up to 90 min showing time points of significant
LDH release. (C) Fold change cytotoxicity in HUVECs preincubated in presence or absence of the TRPV4 antagonist
GSK2193874 [1 µM] (Ant) and afterwards exposed to 100 nM of the TRPV4 agonist GSK1016790A in HBSS with or
HBSS without calcium for 1 h. Data are shown as mean ± SEM; (n=6; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0 vs
control; one-way ANOVA Tukey's multiple comparisons test).
3.1.12 Life cell imaging of HUVECs exposed to the TRPV4 agonist GSK1016790A
HUVECs, preincubated with or without the TRPV4 antagonist GSK2193874 [1µM] were
exposed to different concentrations of the TRPV4 agonist (15, 30 and 100 nM) in a Cell-
IQ® for live cell imaging at controlled temperature and gaseous environment (37°C in 5%
CO2, humidified air) allowing biological cellular responses to be monitored in real time
(described in section 2.1.6). HUVECs exposed to the TRPV4 agonist GSK1016790A [30
nM] were observed to swell and bubble after 15 min (same picture with 15 nM, data not
shown). After a cell swelling phase, induced by the TRPV4 agonist GSK1016790A, cells
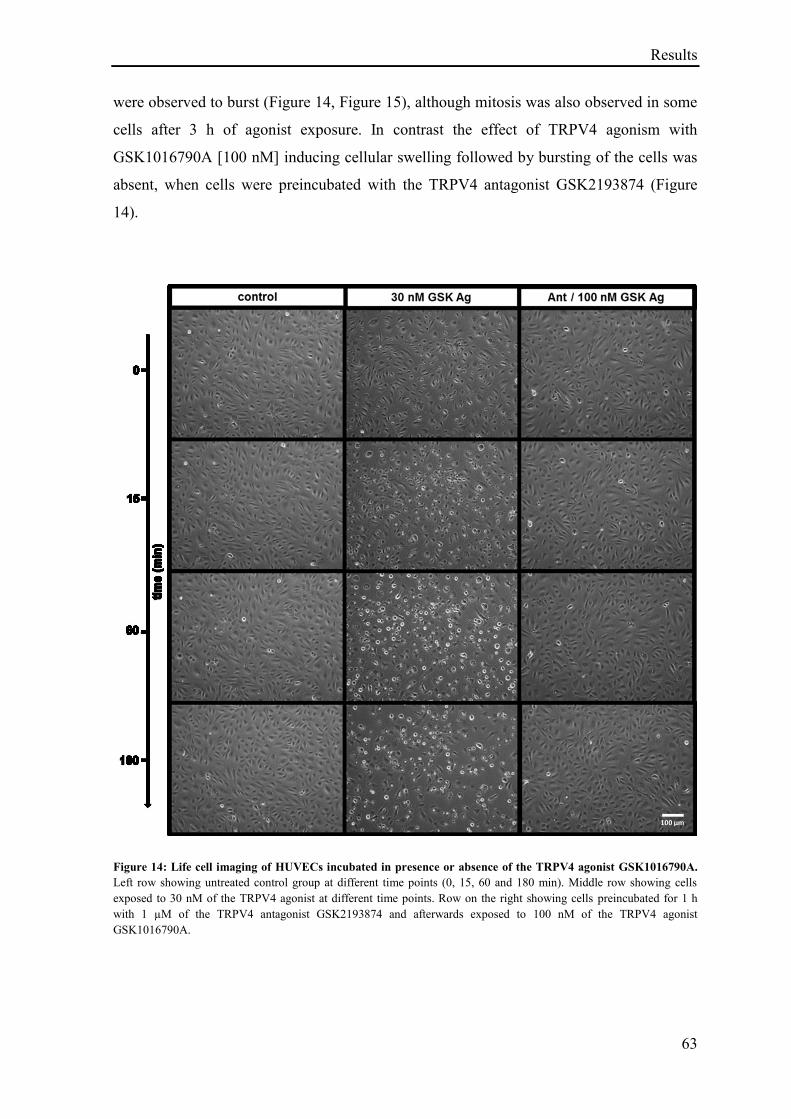
Results
63
were observed to burst (Figure 14, Figure 15), although mitosis was also observed in some
cells after 3 h of agonist exposure. In contrast the effect of TRPV4 agonism with
GSK1016790A [100 nM] inducing cellular swelling followed by bursting of the cells was
absent, when cells were preincubated with the TRPV4 antagonist GSK2193874 (Figure
14).
Figure 14: Life cell imaging of HUVECs incubated in presence or absence of the TRPV4 agonist GSK1016790A.
Left row showing untreated control group at different time points (0, 15, 60 and 180 min). Middle row showing cells
exposed to 30 nM of the TRPV4 agonist at different time points. Row on the right showing cells preincubated for 1 h
with 1 µM of the TRPV4 antagonist GSK2193874 and afterwards exposed to 100 nM of the TRPV4 agonist
GSK1016790A.

Results
64
Figure 15: Life cell imaging of HUVECs incubated in presence of the TRPV4 agonist GSK1016790A. Cells exposed
to 30 nM of the TRPV4 agonist GSK1016790A for 10, 23, 24 and 25 min. Arrows pointing to cell that swell and then
burst.
3.1.13 TRPV4 activation in the RealTime-Glo™ Annexin V Apoptosis and Necrosis
Assay
In TER measurement and LDH release assays different modes of action were observed
with the two different TRPV4 agonists GSK1016790A and 4α-PDD. Therefore the
question was made whether the observed effects could be explained by different cytotoxic
processes. To investigate whether the TRPV4 induced cytotoxicity is an apoptotic or
necrotic process, HUVECs were exposed to different concentrations of the TRPV4
agonists GSK1016790A or 4α-PDD after being preincubated in presence or absence of the
TRPV4 antagonist GSK2193874 [1 µM] and necrotic or apoptotic process was recorded in
real time using the RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay (described
in section 2.1.5). The TRPV4 agonist GSK1016790A showed a direct, strong and
concentration-dependent increase in the fluorescence signal from the DNA-intercolating
dye, similar to the necrosis inducer digitonin, that could be significantly blocked, when
pretreated with the TRPV4 antagonist GSK2193874 (Figure 16A, 16B). In contrast the
agonist 4α-PDD [10 µM] led to an significant increase in the fluorescence signal, that
began only after 8 h reaching the maximum signal after more than 15 hours and that could
not be blocked by the TRPV4 antagonist GSK2193874 [1 µM] (Figure 16A, 16B).
Luminescence detection of phosphatidylserine (PS) on the outer leaflet of the cell was not
increased by the TRPV4 agonist GSK1016790A compared with the control group. The
agonist 4α-PDD [10 µM] in contrast led to an increase in the luminescence signal
beginning after 5 h and reaching its maximum signal at a time point of 8 h. The effect of
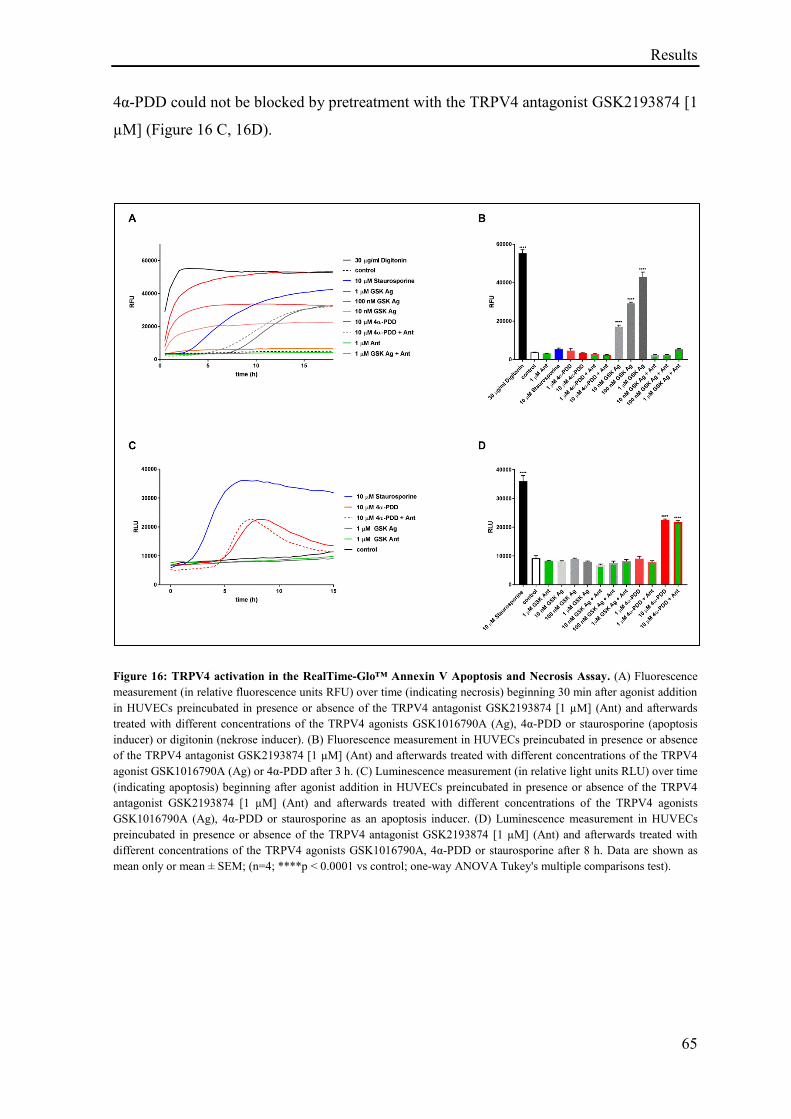
Results
65
4α-PDD could not be blocked by pretreatment with the TRPV4 antagonist GSK2193874 [1
µM] (Figure 16 C, 16D).
Figure 16: TRPV4 activation in the RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay. (A) Fluorescence
measurement (in relative fluorescence units RFU) over time (indicating necrosis) beginning 30 min after agonist addition
in HUVECs preincubated in presence or absence of the TRPV4 antagonist GSK2193874 [1 µM] (Ant) and afterwards
treated with different concentrations of the TRPV4 agonists GSK1016790A (Ag), 4α-PDD or staurosporine (apoptosis
inducer) or digitonin (nekrose inducer). (B) Fluorescence measurement in HUVECs preincubated in presence or absence
of the TRPV4 antagonist GSK2193874 [1 µM] (Ant) and afterwards treated with different concentrations of the TRPV4
agonist GSK1016790A (Ag) or 4α-PDD after 3 h. (C) Luminescence measurement (in relative light units RLU) over time
(indicating apoptosis) beginning after agonist addition in HUVECs preincubated in presence or absence of the TRPV4
antagonist GSK2193874 [1 µM] (Ant) and afterwards treated with different concentrations of the TRPV4 agonists
GSK1016790A (Ag), 4α-PDD or staurosporine as an apoptosis inducer. (D) Luminescence measurement in HUVECs
preincubated in presence or absence of the TRPV4 antagonist GSK2193874 [1 µM] (Ant) and afterwards treated with
different concentrations of the TRPV4 agonists GSK1016790A, 4α-PDD or staurosporine after 8 h. Data are shown as
mean only or mean ± SEM; (n=4; ****p < 0.0001 vs control; one-way ANOVA Tukey's multiple comparisons test).

Results
66
3.2 Results: Role of TRPV4 in stretch induced pathological cellular response
Major content in this part of the thesis are published in PLOS ONE with the title “TRPV4
inhibition attenuates stretch-induced inflammatory cellular responses and lung barrier
dysfunction during mechanical ventilation” (Pairet et al. 2018).
After having shown that pharmacological activation of TRPV4 leads to a disruption of
membrane integrity and permeability increase in vitro and in vivo and that this effect could
be inhibited with the orally active TRPV4 antagonist GSK2193874, the second part of this
thesis focussed on the effect of a more physiologically relevant mean to activate TRPV4,
namely lung cell stretch, due to over-distention of lung region during mechanical
ventilation and the role of TRPV4 in modulating a cellular response to this physical
stimuli.
3.2.1 Effect of TRPV4 agonism on cells Ca2+
influx
To investigate the effect of lung cell stretch, epithelial cells were chosen as a more
appropriate model to investigate cell stretch induced cellular response rather than
endothelial cells, who would be more suitable to study the effect of shear stress. Because
of this consideration and the observed permeability increase in the bronchus of Balb/c mice
after TRPV4 activation in our murine vascular permeability model, human bronchial
epithelial cells NCI-H292, shown to express TRPV4 by our group (RT-PCR, CT value ≈
26), were selected to study the effect of cell stretch. In a first step, to confirm the presence
of functional TRPV4 activity, human lung epithelial cells (NCI-H292) were incubated with
the TRPV4 agonist GSK1016790A and intracellular calcium influx was measured. A
significant increase in intracellular calcium concentration was observed after TRPV4
agonist addition with EC50s ranging from 1-2 nM, and the significant increase in calcium
after agonist addition [2 nM] was concentration-dependently inhibited by the TRPV4
antagonist GSK2193874 with an IC50 of approximately 50 nM and an IC95 of
approximately 1 µM (Figure 17A, 17B). The TRPV4 agonist 4α-PDD led also to a
intracellular calcium concentration increase in a dose-dependent manner in NCI-H292 with
an EC50 of about 5 µM and the effect of 4α-PDD [4 µM] could be also inhibited by the
TRPV4 antagonist GSK2193874 concentration-dependently with an IC50 of about 45 nM
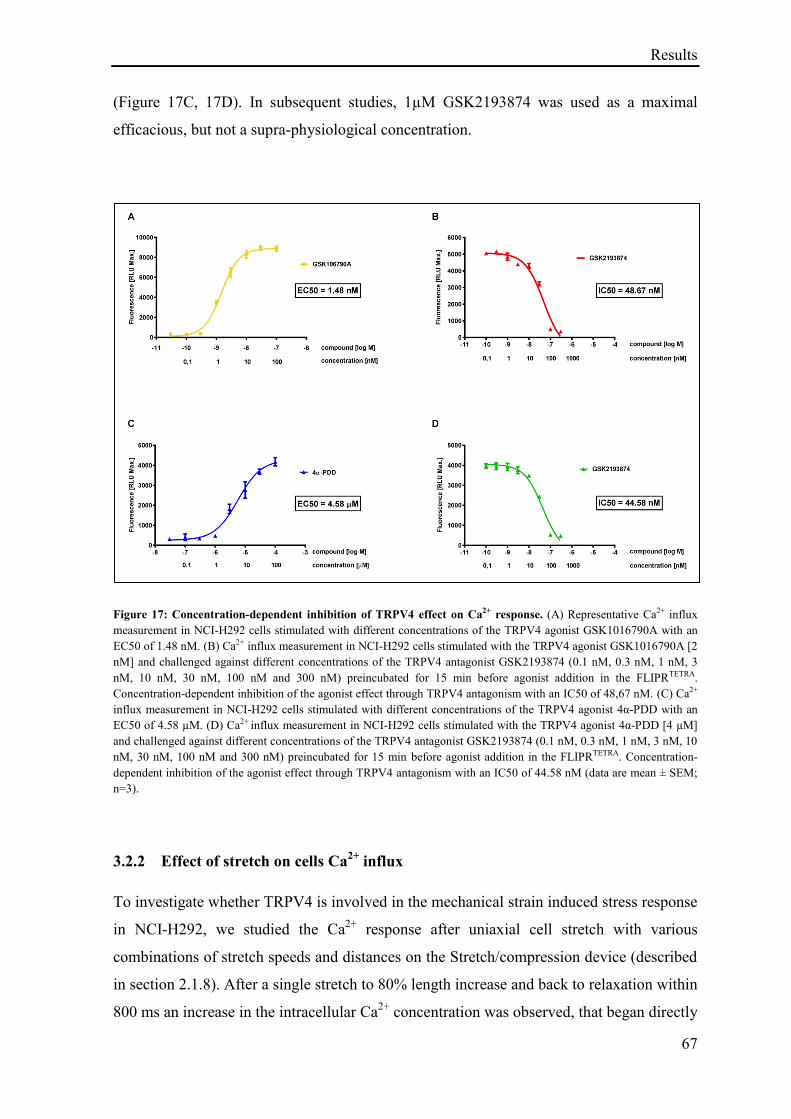
Results
67
(Figure 17C, 17D). In subsequent studies, 1µM GSK2193874 was used as a maximal
efficacious, but not a supra-physiological concentration.
Figure 17: Concentration-dependent inhibition of TRPV4 effect on Ca2+ response. (A) Representative Ca2+ influx
measurement in NCI-H292 cells stimulated with different concentrations of the TRPV4 agonist GSK1016790A with an
EC50 of 1.48 nM. (B) Ca2+ influx measurement in NCI-H292 cells stimulated with the TRPV4 agonist GSK1016790A [2
nM] and challenged against different concentrations of the TRPV4 antagonist GSK2193874 (0.1 nM, 0.3 nM, 1 nM, 3
nM, 10 nM, 30 nM, 100 nM and 300 nM) preincubated for 15 min before agonist addition in the FLIPRTETRA.
Concentration-dependent inhibition of the agonist effect through TRPV4 antagonism with an IC50 of 48,67 nM. (C) Ca2+
influx measurement in NCI-H292 cells stimulated with different concentrations of the TRPV4 agonist 4α-PDD with an
EC50 of 4.58 µM. (D) Ca2+ influx measurement in NCI-H292 cells stimulated with the TRPV4 agonist 4α-PDD [4 µM]
and challenged against different concentrations of the TRPV4 antagonist GSK2193874 (0.1 nM, 0.3 nM, 1 nM, 3 nM, 10
nM, 30 nM, 100 nM and 300 nM) preincubated for 15 min before agonist addition in the FLIPRTETRA. Concentration-
dependent inhibition of the agonist effect through TRPV4 antagonism with an IC50 of 44.58 nM (data are mean ± SEM;
n=3).
3.2.2 Effect of stretch on cells Ca2+
influx
To investigate whether TRPV4 is involved in the mechanical strain induced stress response
in NCI-H292, we studied the Ca2+
response after uniaxial cell stretch with various
combinations of stretch speeds and distances on the Stretch/compression device (described
in section 2.1.8). After a single stretch to 80% length increase and back to relaxation within
800 ms an increase in the intracellular Ca2+
concentration was observed, that began directly
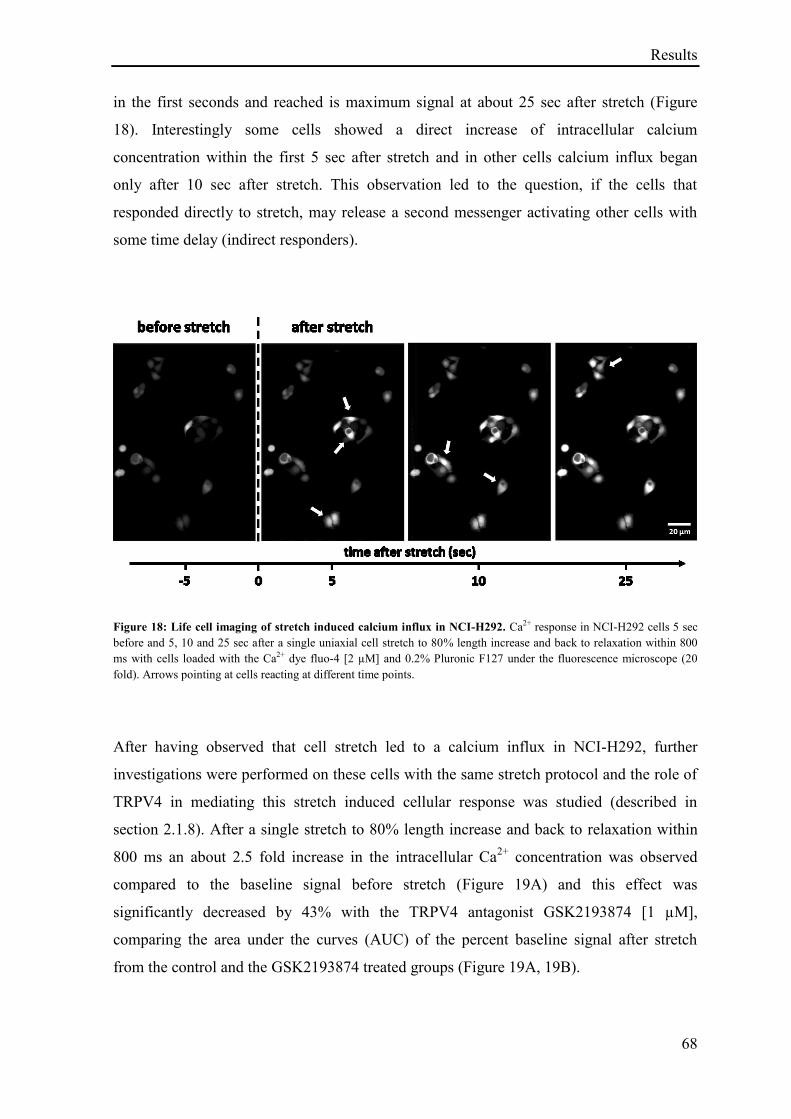
Results
68
in the first seconds and reached is maximum signal at about 25 sec after stretch (Figure
18). Interestingly some cells showed a direct increase of intracellular calcium
concentration within the first 5 sec after stretch and in other cells calcium influx began
only after 10 sec after stretch. This observation led to the question, if the cells that
responded directly to stretch, may release a second messenger activating other cells with
some time delay (indirect responders).
Figure 18: Life cell imaging of stretch induced calcium influx in NCI-H292. Ca2+ response in NCI-H292 cells 5 sec
before and 5, 10 and 25 sec after a single uniaxial cell stretch to 80% length increase and back to relaxation within 800
ms with cells loaded with the Ca2+ dye fluo-4 [2 µM] and 0.2% Pluronic F127 under the fluorescence microscope (20
fold). Arrows pointing at cells reacting at different time points.
After having observed that cell stretch led to a calcium influx in NCI-H292, further
investigations were performed on these cells with the same stretch protocol and the role of
TRPV4 in mediating this stretch induced cellular response was studied (described in
section 2.1.8). After a single stretch to 80% length increase and back to relaxation within
800 ms an about 2.5 fold increase in the intracellular Ca2+
concentration was observed
compared to the baseline signal before stretch (Figure 19A) and this effect was
significantly decreased by 43% with the TRPV4 antagonist GSK2193874 [1 µM],
comparing the area under the curves (AUC) of the percent baseline signal after stretch
from the control and the GSK2193874 treated groups (Figure 19A, 19B).
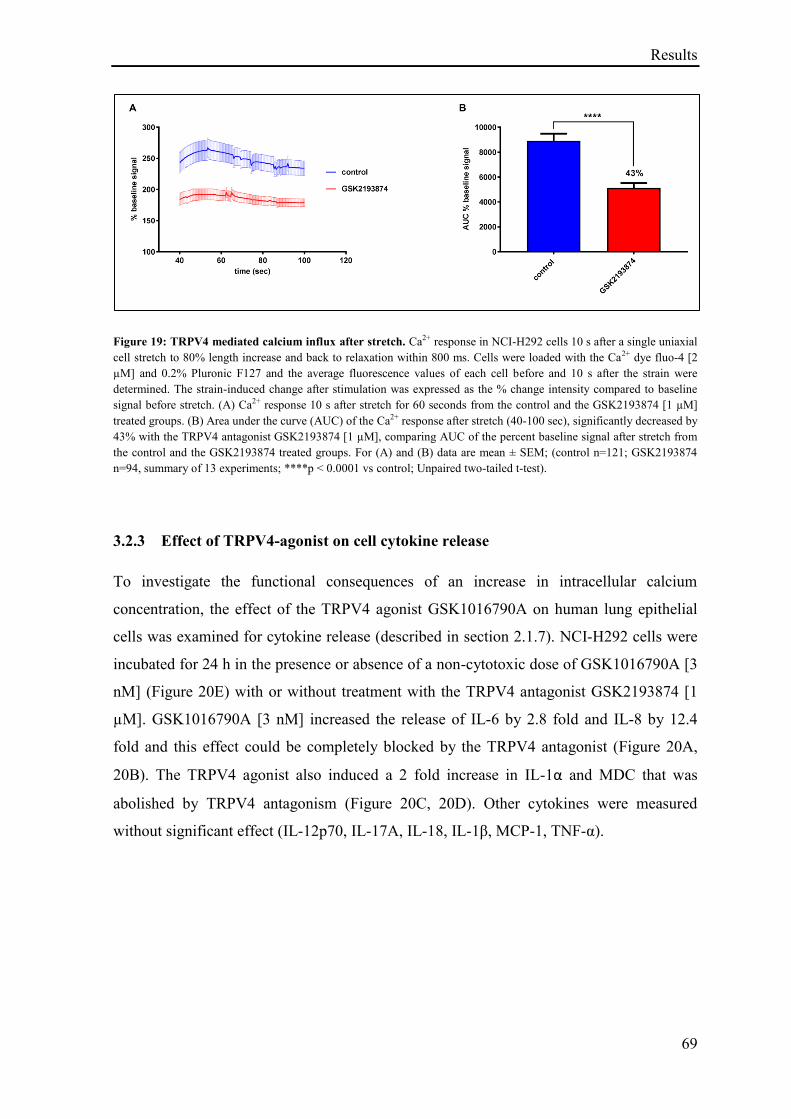
Results
69
Figure 19: TRPV4 mediated calcium influx after stretch. Ca2+ response in NCI-H292 cells 10 s after a single uniaxial
cell stretch to 80% length increase and back to relaxation within 800 ms. Cells were loaded with the Ca2+ dye fluo-4 [2
µM] and 0.2% Pluronic F127 and the average fluorescence values of each cell before and 10 s after the strain were
determined. The strain-induced change after stimulation was expressed as the % change intensity compared to baseline
signal before stretch. (A) Ca2+ response 10 s after stretch for 60 seconds from the control and the GSK2193874 [1 µM]
treated groups. (B) Area under the curve (AUC) of the Ca2+ response after stretch (40-100 sec), significantly decreased by
43% with the TRPV4 antagonist GSK2193874 [1 µM], comparing AUC of the percent baseline signal after stretch from
the control and the GSK2193874 treated groups. For (A) and (B) data are mean ± SEM; (control n=121; GSK2193874
n=94, summary of 13 experiments; ****p ˂ 0.0001 vs control; Unpaired two-tailed t-test).
3.2.3 Effect of TRPV4-agonist on cell cytokine release
To investigate the functional consequences of an increase in intracellular calcium
concentration, the effect of the TRPV4 agonist GSK1016790A on human lung epithelial
cells was examined for cytokine release (described in section 2.1.7). NCI-H292 cells were
incubated for 24 h in the presence or absence of a non-cytotoxic dose of GSK1016790A [3
nM] (Figure 20E) with or without treatment with the TRPV4 antagonist GSK2193874 [1
µM]. GSK1016790A [3 nM] increased the release of IL-6 by 2.8 fold and IL-8 by 12.4
fold and this effect could be completely blocked by the TRPV4 antagonist (Figure 20A,
20B). The TRPV4 agonist also induced a 2 fold increase in IL-1α and MDC that was
abolished by TRPV4 antagonism (Figure 20C, 20D). Other cytokines were measured
without significant effect (IL-12p70, IL-17A, IL-18, IL-1β, MCP-1, TNF-α).
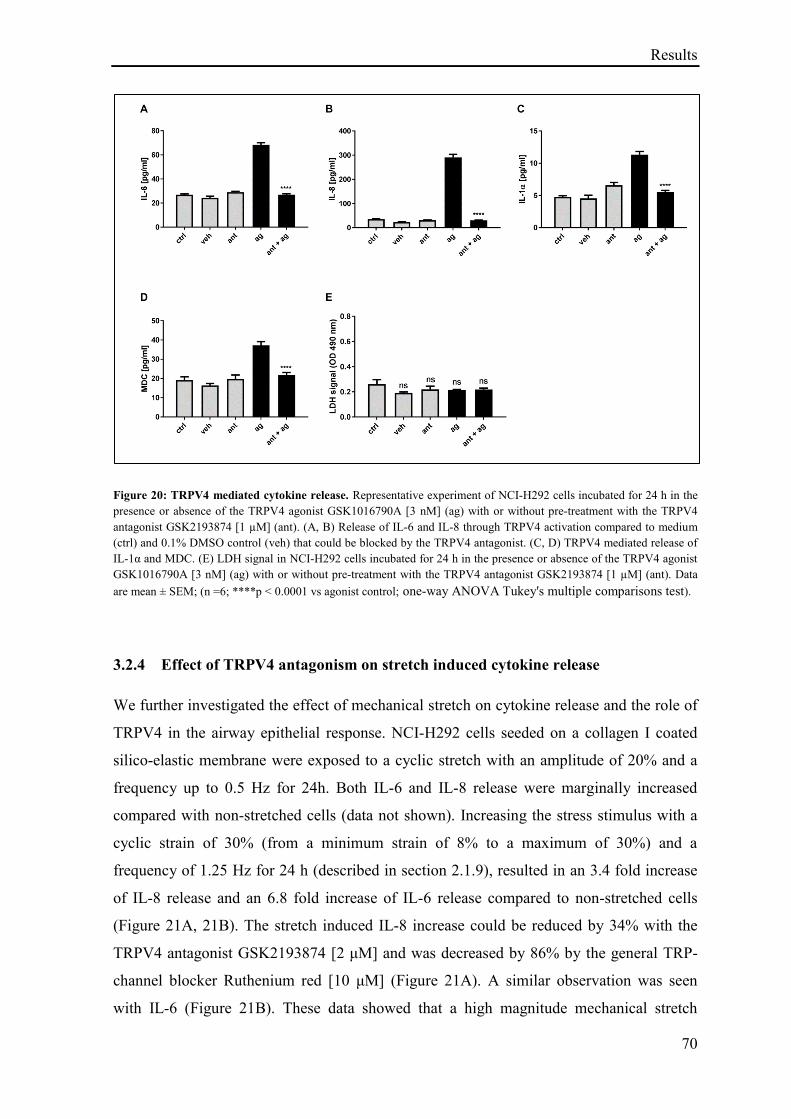
Results
70
Figure 20: TRPV4 mediated cytokine release. Representative experiment of NCI-H292 cells incubated for 24 h in the
presence or absence of the TRPV4 agonist GSK1016790A [3 nM] (ag) with or without pre-treatment with the TRPV4
antagonist GSK2193874 [1 µM] (ant). (A, B) Release of IL-6 and IL-8 through TRPV4 activation compared to medium
(ctrl) and 0.1% DMSO control (veh) that could be blocked by the TRPV4 antagonist. (C, D) TRPV4 mediated release of
IL-1α and MDC. (E) LDH signal in NCI-H292 cells incubated for 24 h in the presence or absence of the TRPV4 agonist
GSK1016790A [3 nM] (ag) with or without pre-treatment with the TRPV4 antagonist GSK2193874 [1 µM] (ant). Data
are mean ± SEM; (n =6; ****p ˂ 0.0001 vs agonist control; one-way ANOVA Tukey's multiple comparisons test).
3.2.4 Effect of TRPV4 antagonism on stretch induced cytokine release
We further investigated the effect of mechanical stretch on cytokine release and the role of
TRPV4 in the airway epithelial response. NCI-H292 cells seeded on a collagen I coated
silico-elastic membrane were exposed to a cyclic stretch with an amplitude of 20% and a
frequency up to 0.5 Hz for 24h. Both IL-6 and IL-8 release were marginally increased
compared with non-stretched cells (data not shown). Increasing the stress stimulus with a
cyclic strain of 30% (from a minimum strain of 8% to a maximum of 30%) and a
frequency of 1.25 Hz for 24 h (described in section 2.1.9), resulted in an 3.4 fold increase
of IL-8 release and an 6.8 fold increase of IL-6 release compared to non-stretched cells
(Figure 21A, 21B). The stretch induced IL-8 increase could be reduced by 34% with the
TRPV4 antagonist GSK2193874 [2 μM] and was decreased by 86% by the general TRP-
channel blocker Ruthenium red [10 μM] (Figure 21A). A similar observation was seen
with IL-6 (Figure 21B). These data showed that a high magnitude mechanical stretch
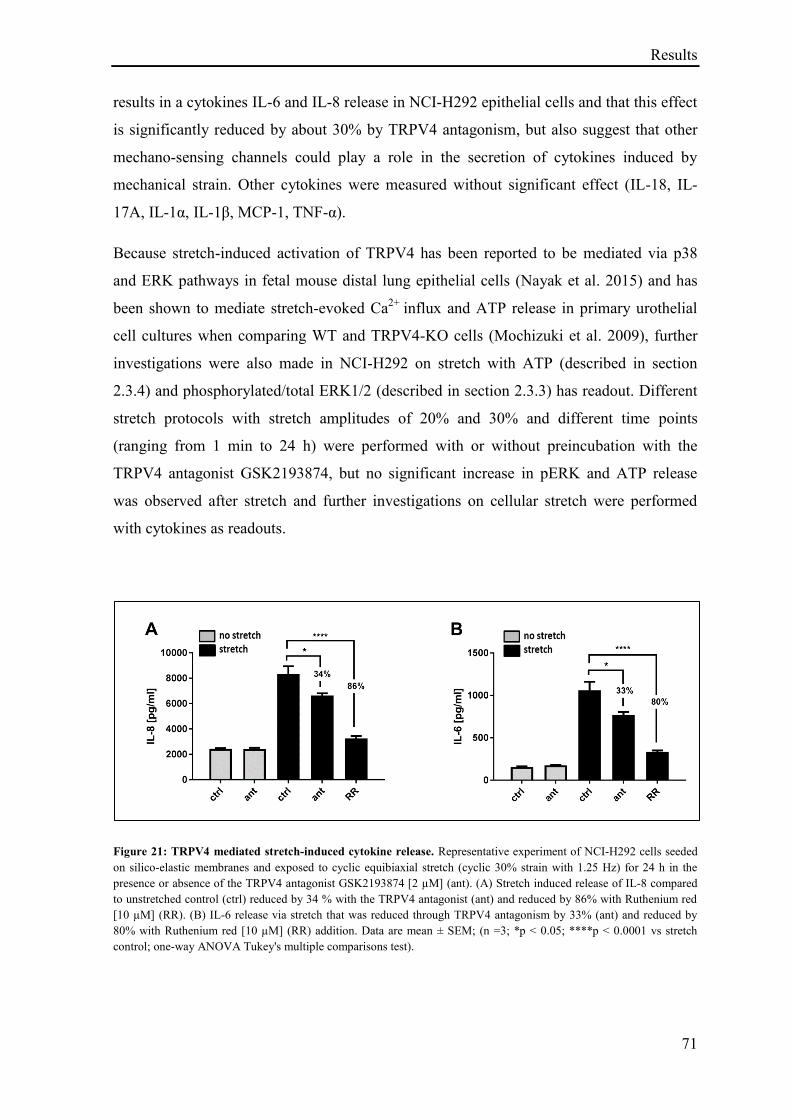
Results
71
results in a cytokines IL-6 and IL-8 release in NCI-H292 epithelial cells and that this effect
is significantly reduced by about 30% by TRPV4 antagonism, but also suggest that other
mechano-sensing channels could play a role in the secretion of cytokines induced by
mechanical strain. Other cytokines were measured without significant effect (IL-18, IL-
17A, IL-1α, IL-1β, MCP-1, TNF-α).
Because stretch-induced activation of TRPV4 has been reported to be mediated via p38
and ERK pathways in fetal mouse distal lung epithelial cells (Nayak et al. 2015) and has
been shown to mediate stretch-evoked Ca2+
influx and ATP release in primary urothelial
cell cultures when comparing WT and TRPV4-KO cells (Mochizuki et al. 2009), further
investigations were also made in NCI-H292 on stretch with ATP (described in section
2.3.4) and phosphorylated/total ERK1/2 (described in section 2.3.3) has readout. Different
stretch protocols with stretch amplitudes of 20% and 30% and different time points
(ranging from 1 min to 24 h) were performed with or without preincubation with the
TRPV4 antagonist GSK2193874, but no significant increase in pERK and ATP release
was observed after stretch and further investigations on cellular stretch were performed
with cytokines as readouts.
Figure 21: TRPV4 mediated stretch-induced cytokine release. Representative experiment of NCI-H292 cells seeded
on silico-elastic membranes and exposed to cyclic equibiaxial stretch (cyclic 30% strain with 1.25 Hz) for 24 h in the
presence or absence of the TRPV4 antagonist GSK2193874 [2 µM] (ant). (A) Stretch induced release of IL-8 compared
to unstretched control (ctrl) reduced by 34 % with the TRPV4 antagonist (ant) and reduced by 86% with Ruthenium red
[10 µM] (RR). (B) IL-6 release via stretch that was reduced through TRPV4 antagonism by 33% (ant) and reduced by
80% with Ruthenium red [10 µM] (RR) addition. Data are mean ± SEM; (n =3; *p ˂ 0.05; ****p ˂ 0.0001 vs stretch
control; one-way ANOVA Tukey's multiple comparisons test).

Results
72
3.2.5 TRPV4 mediated regulation of genes in the Human cAMP / Calcium Signaling
PathwayFinder
Pathway analysis following a TRPV4-mediated increase in intracellular calcium
concentration were performed. The effect of the TRPV4 agonist 4α-PDD on NCI-H292
human lung epithelial cells was examined in the Human cAMP / Calcium Signaling
PathwayFinder (described in section 2.3.6). This data represent a data set of 3 samples per
group pooled to one sample each group, after exposure of the cells to the TRPV4 agonist
and after two washing steps. The purified RNA concentration from the control group was
142.3 ng/ml, 116 ng/ml for the TRPV4 antagonist GSK2193874 treated group, 90.35 ng/ml
for the TRPV4 agonist 4α-PDD treated group and 104.1 ng/ml for the group pretreated
with the TRPV4 antagonist GSK2193874 and afterwards treated with 4α-PDD. The 4α-
PDD treated group indicated a loss of cells during agonist exposure and we already
identified it as a cytotoxic effect (monitored by LDH-release) induced by 4α-PDD [10 µM]
after 24 h. Nevertheless the same amount of purified RNA from each group had to further
be used for cDNA preparation, that normalised somehow the loss in cells observed during
experimental procedure. Interestingly the gene regulation of Protein phosphatase 1,
regulatory (inhibitor) subunit 15A (PPP1R15A) showed an about 45 fold upregulation
compared to the control group after TRPV4 agonist exposure with 4α-PDD, that was
nearly completely blocked by preincubation with the TRPV4 antagonist GSK2193874 [1
µM] (Table 2).
Table 2: Regulation of genes in the Human cAMP / Calcium Signaling PathwayFinder compared to control in
NCI-H292. Cells preincubated for 2 h in presence or absence of the TRPV4 antagonist GSK2193874 [1 µM] (Ant) and
afterward incubated in presence or absence of the TRPV4 agonist 4α-PDD [10 µM] for 24 h.
Up-Down Regulation
(Fold regulation in expression comparing to control group)
Gene Symbol Control GSK Ant
4α-PDD
GSK Ant / 4α-PDD
ADRB1 1 -1.071 1.5746 2.8442
AHR 1 -1.1942 -1.1227 -1.0217
AMD1 1 -1.2466 -3.4248 -2.535
Continuation of table 2 on page 73
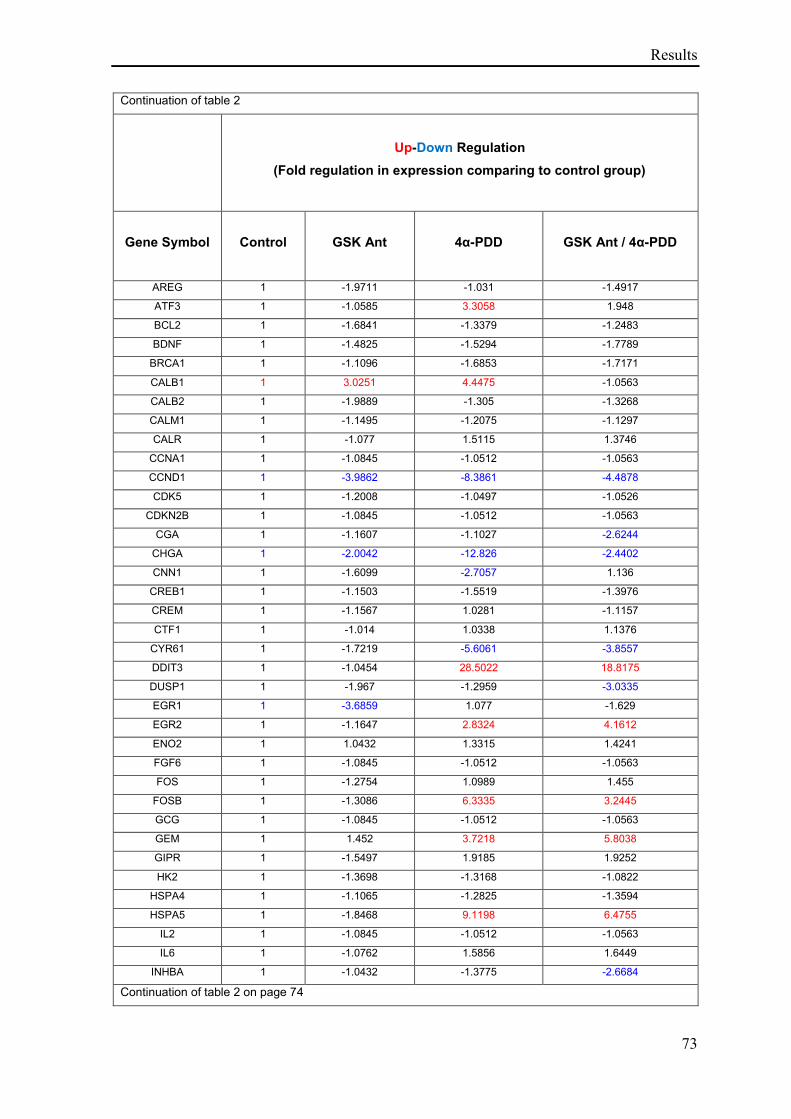
Results
73
Continuation of table 2
Up-Down Regulation
(Fold regulation in expression comparing to control group)
Gene Symbol Control GSK Ant
4α-PDD
GSK Ant / 4α-PDD
AREG 1 -1.9711 -1.031 -1.4917
ATF3 1 -1.0585 3.3058 1.948
BCL2 1 -1.6841 -1.3379 -1.2483
BDNF 1 -1.4825 -1.5294 -1.7789
BRCA1 1 -1.1096 -1.6853 -1.7171
CALB1 1 3.0251 4.4475 -1.0563
CALB2 1 -1.9889 -1.305 -1.3268
CALM1 1 -1.1495 -1.2075 -1.1297
CALR 1 -1.077 1.5115 1.3746
CCNA1 1 -1.0845 -1.0512 -1.0563
CCND1 1 -3.9862 -8.3861 -4.4878
CDK5 1 -1.2008 -1.0497 -1.0526
CDKN2B 1 -1.0845 -1.0512 -1.0563
CGA 1 -1.1607 -1.1027 -2.6244
CHGA 1 -2.0042 -12.826 -2.4402
CNN1 1 -1.6099 -2.7057 1.136
CREB1 1 -1.1503 -1.5519 -1.3976
CREM 1 -1.1567 1.0281 -1.1157
CTF1 1 -1.014 1.0338 1.1376
CYR61 1 -1.7219 -5.6061 -3.8557
DDIT3 1 -1.0454 28.5022 18.8175
DUSP1 1 -1.967 -1.2959 -3.0335
EGR1 1 -3.6859 1.077 -1.629
EGR2 1 -1.1647 2.8324 4.1612
ENO2 1 1.0432 1.3315 1.4241
FGF6 1 -1.0845 -1.0512 -1.0563
FOS 1 -1.2754 1.0989 1.455
FOSB 1 -1.3086 6.3335 3.2445
GCG 1 -1.0845 -1.0512 -1.0563
GEM 1 1.452 3.7218 5.8038
GIPR 1 -1.5497 1.9185 1.9252
HK2 1 -1.3698 -1.3168 -1.0822
HSPA4 1 -1.1065 -1.2825 -1.3594
HSPA5 1 -1.8468 9.1198 6.4755
IL2 1 -1.0845 -1.0512 -1.0563
IL6 1 -1.0762 1.5856 1.6449
INHBA 1 -1.0432 -1.3775 -2.6684
Continuation of table 2 on page 74
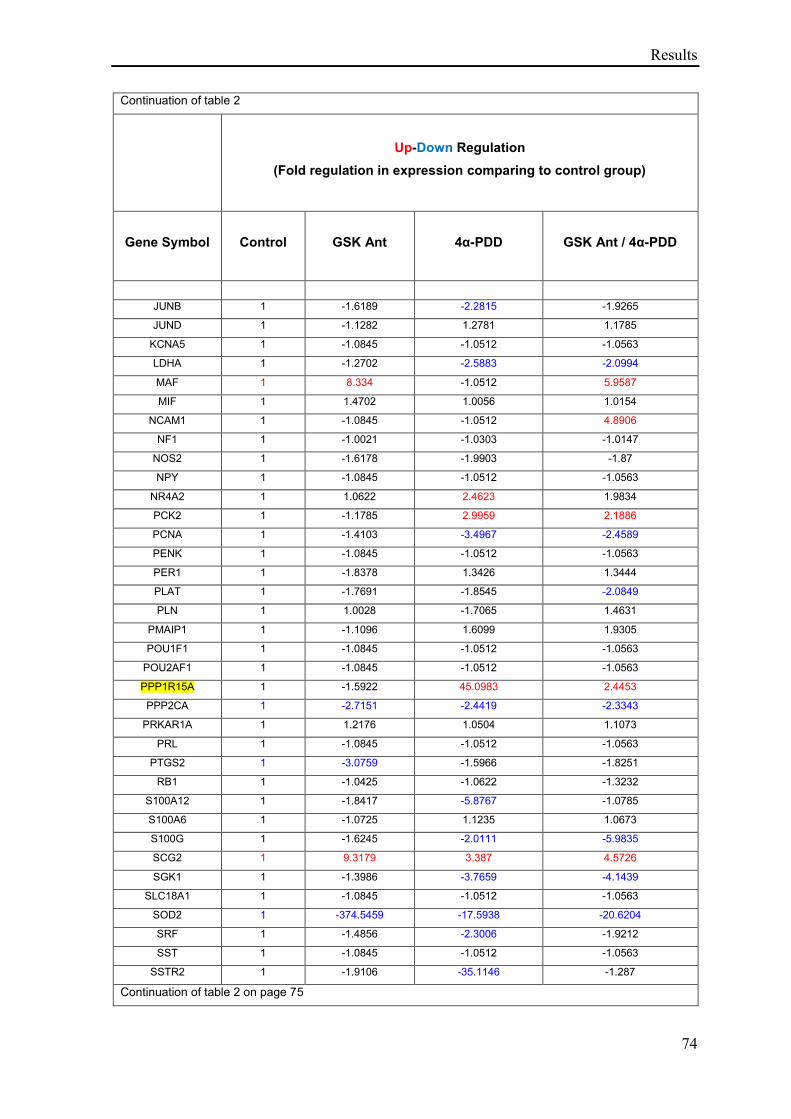
Results
74
Continuation of table 2
Up-Down Regulation
(Fold regulation in expression comparing to control group)
Gene Symbol Control GSK Ant
4α-PDD
GSK Ant / 4α-PDD
JUNB 1 -1.6189 -2.2815 -1.9265
JUND 1 -1.1282 1.2781 1.1785
KCNA5 1 -1.0845 -1.0512 -1.0563
LDHA 1 -1.2702 -2.5883 -2.0994
MAF 1 8.334 -1.0512 5.9587
MIF 1 1.4702 1.0056 1.0154
NCAM1 1 -1.0845 -1.0512 4.8906
NF1 1 -1.0021 -1.0303 -1.0147
NOS2 1 -1.6178 -1.9903 -1.87
NPY 1 -1.0845 -1.0512 -1.0563
NR4A2 1 1.0622 2.4623 1.9834
PCK2 1 -1.1785 2.9959 2.1886
PCNA 1 -1.4103 -3.4967 -2.4589
PENK 1 -1.0845 -1.0512 -1.0563
PER1 1 -1.8378 1.3426 1.3444
PLAT 1 -1.7691 -1.8545 -2.0849
PLN 1 1.0028 -1.7065 1.4631
PMAIP1 1 -1.1096 1.6099 1.9305
POU1F1 1 -1.0845 -1.0512 -1.0563
POU2AF1 1 -1.0845 -1.0512 -1.0563
PPP1R15A 1 -1.5922 45.0983 2.4453
PPP2CA 1 -2.7151 -2.4419 -2.3343
PRKAR1A 1 1.2176 1.0504 1.1073
PRL 1 -1.0845 -1.0512 -1.0563
PTGS2 1 -3.0759 -1.5966 -1.8251
RB1 1 -1.0425 -1.0622 -1.3232
S100A12 1 -1.8417 -5.8767 -1.0785
S100A6 1 -1.0725 1.1235 1.0673
S100G 1 -1.6245 -2.0111 -5.9835
SCG2 1 9.3179 3.387 4.5726
SGK1 1 -1.3986 -3.7659 -4.1439
SLC18A1 1 -1.0845 -1.0512 -1.0563
SOD2 1 -374.5459 -17.5938 -20.6204
SRF 1 -1.4856 -2.3006 -1.9212
SST 1 -1.0845 -1.0512 -1.0563
SSTR2 1 -1.9106 -35.1146 -1.287
Continuation of table 2 on page 75
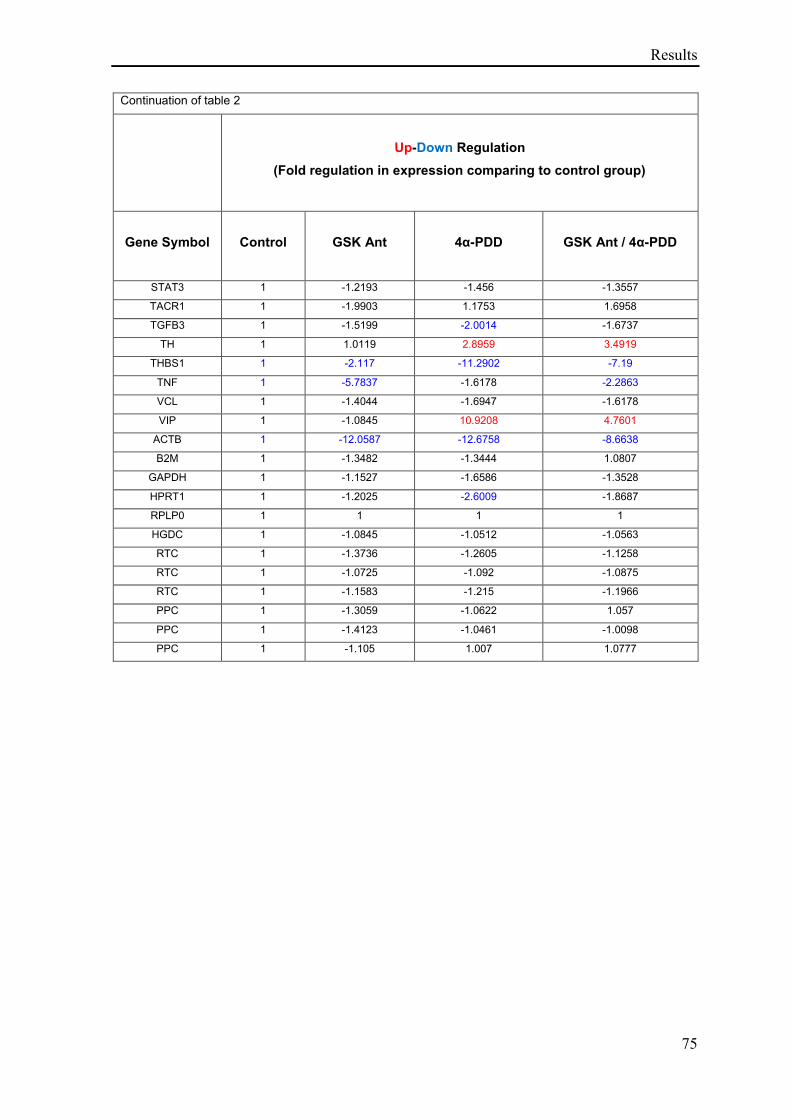
Results
75
Continuation of table 2
Up-Down Regulation
(Fold regulation in expression comparing to control group)
Gene Symbol Control GSK Ant
4α-PDD
GSK Ant / 4α-PDD
STAT3 1 -1.2193 -1.456 -1.3557
TACR1 1 -1.9903 1.1753 1.6958
TGFB3 1 -1.5199 -2.0014 -1.6737
TH 1 1.0119 2.8959 3.4919
THBS1 1 -2.117 -11.2902 -7.19
TNF 1 -5.7837 -1.6178 -2.2863
VCL 1 -1.4044 -1.6947 -1.6178
VIP 1 -1.0845 10.9208 4.7601
ACTB 1 -12.0587 -12.6758 -8.6638
B2M 1 -1.3482 -1.3444 1.0807
GAPDH 1 -1.1527 -1.6586 -1.3528
HPRT1 1 -1.2025 -2.6009 -1.8687
RPLP0 1 1 1 1
HGDC 1 -1.0845 -1.0512 -1.0563
RTC 1 -1.3736 -1.2605 -1.1258
RTC 1 -1.0725 -1.092 -1.0875
RTC 1 -1.1583 -1.215 -1.1966
PPC 1 -1.3059 -1.0622 1.057
PPC 1 -1.4123 -1.0461 -1.0098
PPC 1 -1.105 1.007 1.0777

Results
76
3.2.6 Effect of stretch on macrophages cytokine release
The results on stretch induced cytokine release suggested, that the effect is only partially
modulated by TRPV4 in NCI-H292 and other stretch activated channels and/or integrins
may play a role, but also led to the question, if stronger effector cells exist regarding
TRPV4 mediated stretch-induced cytokine release. We further asked the question, if
macrophages as a major source of cytokine secretion and adhering to lung cells (Tao and
Kobzik 2002) could be the stronger effector cells regarding TRPV4 mediated cytokine
release.
To answer this question, human monocytes were seeded on silico-elastic membranes and
differentiated with GM-CSF (M1 subtype) or M-CSF (M2 subtype) for seven days
(described in section 2.1.10). Macrophages were exposed to the same equibiaxial stretch
protocol (cyclic 30% strain with 1.25 hz) as for the lung epithelial cells, but significant
cytokine release was observed after 36 h and 48 h (described in section 2.1.9). The
mechanical stretch induced stress on the M1 macrophages resulted in a significant increases
in IL-1α (4.3-fold; 48h; Figure 22A), IL-1β (3.2-fold; 48h; Figure 22B), IL-8 (1.5-fold;
48h; Figure 22C), IL-6 (1.5-fold; 48h; Figure 22D), and MCP-1 (2.2-fold; 36h, Figure
22E) compared to unstretched cells. All cytokine release was abolished, by the TRPV4
antagonist GSK2193874 [2 μM]. M2 macrophages showed an about 3 fold increase in
MCP-1 after 36 h and an about 2 fold increase of TNF-α after 48h stretch, that were both
blocked by TRPV4 antagonism with GSK2193874 [2 µM] (Figure 22F, 22G) but no
significant increases in IL-1α, IL-1β, IL-6 or IL-8. Other cytokines were measured without
significant effect (IL-10, IL-12p70, IL-2 and MDC).
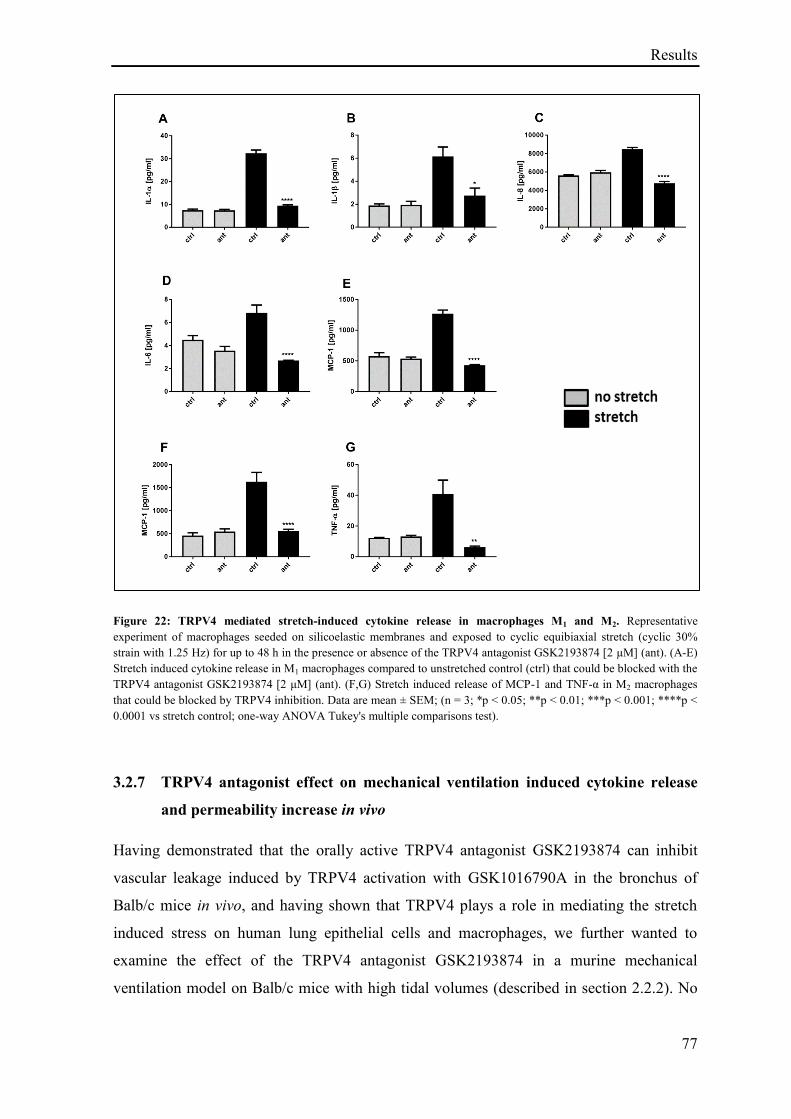
Results
77
Figure 22: TRPV4 mediated stretch-induced cytokine release in macrophages M1 and M2. Representative
experiment of macrophages seeded on silicoelastic membranes and exposed to cyclic equibiaxial stretch (cyclic 30%
strain with 1.25 Hz) for up to 48 h in the presence or absence of the TRPV4 antagonist GSK2193874 [2 µM] (ant). (A-E)
Stretch induced cytokine release in M1 macrophages compared to unstretched control (ctrl) that could be blocked with the
TRPV4 antagonist GSK2193874 [2 μM] (ant). (F,G) Stretch induced release of MCP-1 and TNF-α in M2 macrophages
that could be blocked by TRPV4 inhibition. Data are mean ± SEM; (n = 3; *p ˂ 0.05; **p ˂ 0.01; ***p ˂ 0.001; ****p ˂
0.0001 vs stretch control; one-way ANOVA Tukey's multiple comparisons test).
3.2.7 TRPV4 antagonist effect on mechanical ventilation induced cytokine release
and permeability increase in vivo
Having demonstrated that the orally active TRPV4 antagonist GSK2193874 can inhibit
vascular leakage induced by TRPV4 activation with GSK1016790A in the bronchus of
Balb/c mice in vivo, and having shown that TRPV4 plays a role in mediating the stretch
induced stress on human lung epithelial cells and macrophages, we further wanted to
examine the effect of the TRPV4 antagonist GSK2193874 in a murine mechanical
ventilation model on Balb/c mice with high tidal volumes (described in section 2.2.2). No

Results
78
increase in cytokine release or protein concentration in BALF was observed, when mice
were subjected to a tidal volume (TV) of 20 ml/kg. However, we observed a 23.3 fold
increase of KC/GRO level (Figure 23A) and an IL-6 release that was 15.3 fold higher
(Figure 23B) after mechanical ventilation with 30 ml/kg TV compared to control group
(6.5 ml/kg TV). Furthermore a tidal volume of 30 ml/kg ventilation resulted in a 2.6 fold
increased protein concentration in BALF compared to the normal ventilated control group
(Figure 23C). All were significantly blocked with the TRPV4 inhibitor GSK2193874.
Additionally LDH release was measured in the BALF of mice lungs after mechanical
ventilation showing an increase in LDH signal after a 30 ml/kg TV ventilation compared to
the non-ventilated control group, but that could not be inhibited by TRPV4 antagonism
with GSK2193874 (Figure 23D). Lung elastance and resistance was also measured during
assay procedure showing an elevated but not significant increase in resistance and
elastance in the 30 ml/kg TV ventilated group compared to the 6.5 ml/kg TV control, that
was absent in the 30 ml/kg TV group pretreated with the TRPV4 antagonist GSK2193874
(Figure 23E, 23F). Other cytokines were measured without significant increase (IFN-γ, IL-
1β, IL-2, IL-4, IL-5, IL-10, IL-12p70 and TNF-α.)
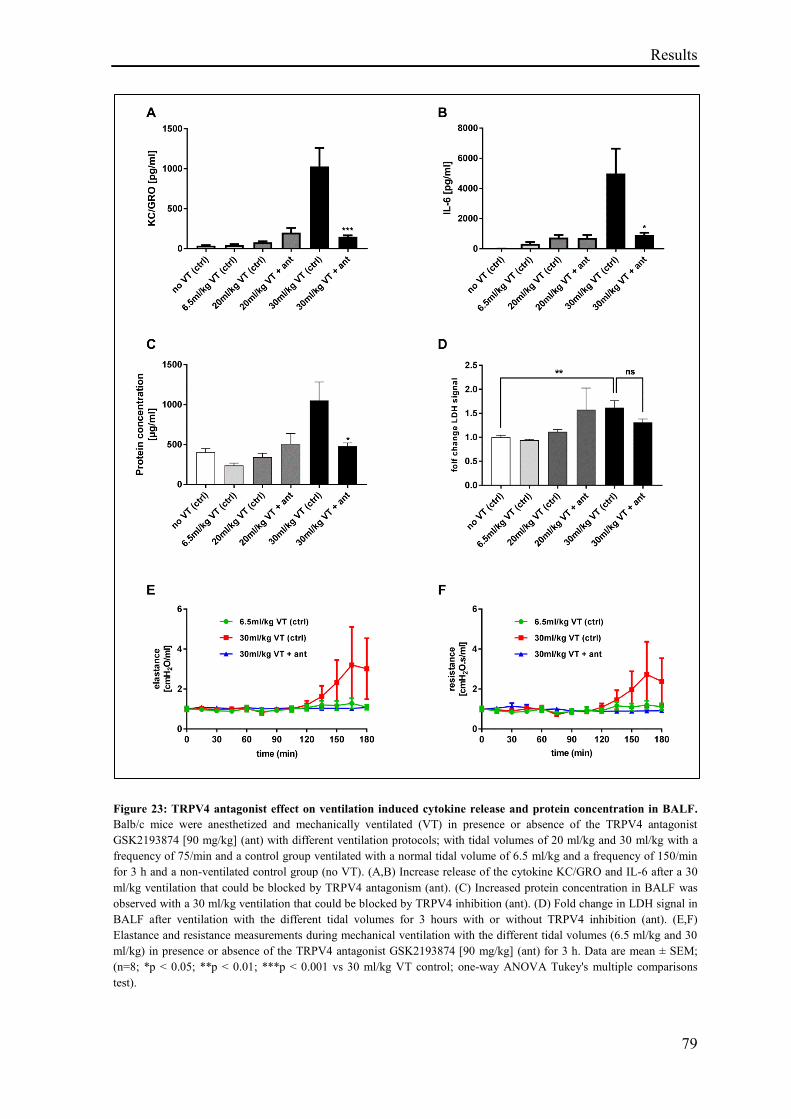
Results
79
Figure 23: TRPV4 antagonist effect on ventilation induced cytokine release and protein concentration in BALF.
Balb/c mice were anesthetized and mechanically ventilated (VT) in presence or absence of the TRPV4 antagonist
GSK2193874 [90 mg/kg] (ant) with different ventilation protocols; with tidal volumes of 20 ml/kg and 30 ml/kg with a
frequency of 75/min and a control group ventilated with a normal tidal volume of 6.5 ml/kg and a frequency of 150/min
for 3 h and a non-ventilated control group (no VT). (A,B) Increase release of the cytokine KC/GRO and IL-6 after a 30
ml/kg ventilation that could be blocked by TRPV4 antagonism (ant). (C) Increased protein concentration in BALF was
observed with a 30 ml/kg ventilation that could be blocked by TRPV4 inhibition (ant). (D) Fold change in LDH signal in
BALF after ventilation with the different tidal volumes for 3 hours with or without TRPV4 inhibition (ant). (E,F)
Elastance and resistance measurements during mechanical ventilation with the different tidal volumes (6.5 ml/kg and 30
ml/kg) in presence or absence of the TRPV4 antagonist GSK2193874 [90 mg/kg] (ant) for 3 h. Data are mean ± SEM;
(n=8; *p ˂ 0.05; **p ˂ 0.01; ***p ˂ 0.001 vs 30 ml/kg VT control; one-way ANOVA Tukey's multiple comparisons
test).

Discussion
80
4 Discussion
This work investigated on the role of the cation channel TRPV4 in endothelial/epithelial
barrier integrity and represents a step by step approach to assess TRPV4 antagonists as
potential drug for the treatment of patients with the need of mechanical ventilation. We
first studied pharmacological activation and inhibition on endothelial cells, using two
widely used selective activators of TRPV4 and a selective potent and orally active TRPV4
blocker GSK2193874 on in vitro permeability assays. Afterwards TRPV4 activation and
inhibition on vascular permeability was investigated in vivo with the TRPV4 activator
GSK1016790A and the TRPV4 channel blocker GSK2193874 given orally.
After compound characterisation in in vitro and in vivo models of permeability, the next
part of the thesis investigated on the pathophysiological role of TRPV4 activation via a
physical stimulus. Therefore TRPV4 activation via cell stretch and the resulting
pathological cellular response was investigated in human bronchial lung epithelial cells
and macrophages in vitro with the TRPV4 antagonist GSK2193874 and afterwards the
effect of the orally given TRPV4 blocker GSK2193874 was investigated on a murine
disease related model of VILI using mechanical ventilation with high tidal volumes.
4.1 Role of TRPV4 in regulating endothelial membrane integrity
To investigate TRPV4 biology and its role in regulating the endothelial membrane
integrity, we used two reported selective activators of TRPV4, GSK1016790A and 4α-
Phorbol 12,13-didecanoate (4α-PDD) (Thorneloe et al. 2008, Willette, Bao et al. 2008)
and a potent and selective TRPV4 blocker GSK2193874 (Thorneloe et al. 2012).
During method establishment initial TER improvement was observed, when HUVECs
were cultured 24 hours under more physiological condition (1% O2, 5% CO2 at 37°C), here
namely hypoxia, in prior to TER measurement. Through this effect it was possible to
establish a model with an assay window high enough to test compound effect on TER in
HUVECs in the cellZscope. Under this assay procedure a dose-dependent decrease in TER
was observed after addition of the TRPV4 agonist GSK1016790A. We also observed that
the cytokines TNF-α [100 ng/ml] and IL-1β [10 ng/ml] induced a reduction in TER after
compound addition. It is reported that hypoxia can induce a large variety of biological
active agents in endothelial cells, e.g. vascular endothelial growth factor (VEGF) (Namiki

Discussion
81
et al. 1995), that has been further implicated to mediate loss of trans-epithelial resistance
(TER) (Ghassemifar et al. 2006). Furthermore Hypoxia-induced hyperpermeability of rat
glomerular endothelial cells has been reported, involving hypoxia-inducible factor-2α
(HIF-2α), to mediate changes in the expression of occludin and ZO-1 inducing
permeability (Luo et al. 2018).
However, in our model hypoxia improved initial TER in HUVECs and we concluded that
this assay procedure 1% O2, 5% CO2 at 37°C represented more physiological conditions
for human umbilical vein endothelial cells rather than hypoxic conditions, leading to an
improvement of initial TER, that remained constant during electrical resistance
measurement in the cellZscope. This observation led to the question, if in general cells
should be cultured under more physiological O2 conditions, e.g. culturing lung epithelial
cells and intestine derived cells under separate conditions. A thought that is shared in the
literature and nicely reviewed (Carreau et al. 2011), evaluating the consequences of
physioxia on cells and importantly emphasizing the discrepancy between in vivo and in
vitro tissues and cells oxygen status, which can have significant impact on experimental
outcome. Pointing out, that the values corresponding to the physioxia of different tissues
are ranging between 11% and 1% O2, whereas current in vitro experimentations are usually
performed in 19.95% O2, an artificial context as far as oxygen balance is concerned,
concluding that most of the experiments performed in so-called normoxia might be
misleading (Carreau et al. 2011). In this review the pO2 in umbilical vein blood, that is in
direct contact to human umbilical vein endothelial cells, is also mentioned to be normally
between 20 and 30 mmHg (≈ 2.6-3.9% O2) (Gluckman et al. 1989, Carreau et al. 2011),
indicating that 1% O2 should represent more physiological levels of oxygen (physioxia) in
HUVECs than the so-called normoxia (19.5% O2) in the incubator.
TRPV4 agonism with GSK1016790A on the endothelial cell layer resulted in a strong
decrease in TER and increase in permeability, which could be dose-dependently blocked
by the TRPV4 antagonist. In contrast, 4α-Phorbol 12,13-didecanoate (4α-PDD) reduced
TER only approximately 5 h after agonism and only with a high concentration of 4α-PDD
[10 µM] and could not be blocked by the TRPV4 antagonist.
Further investigations in vitro showed that the effects on TER correlated with the
intracellular calcium influx measurement over time in HUVECs. Whilst GSK1016790A
resulted in a direct, significant and strong increase in intracellular calcium concentration in

Discussion
82
a dose-dependent manner, high concentrations of 4α-PDD induced a small, late-onset
increase of intracellular calcium concentration. However, the agonist effect could be
blocked by TRPV4 inhibition with GSK2193874. These data suggest that GSK1016790A
induced TER reduction is mediated via TRPV4 in an intracellular calcium-influx
dependent manner in HUVECs, whereas 4α-PDD-mediated TER reduction may be
independent of TRPV4 activation, although we cannot exclude the possibility that
GSK2193874 and 4α-PDD do not share a competitive binding site. Interestingly, the effect
of the TRPV4 agonist GSK1016790A on TER and intracellular calcium influx could not
only be blocked by preincubation with the TRPV4 antagonist, but it was also possible to
reverse the TRPV4 activation on both calcium influx and TER reduction by administration
of the antagonist after agonist addition within a certain time range. These data suggest that
not only GSK2193874 is capable of displacing GSK1016790A from its binding site, but
that reduction in endothelial barrier permeability can be reversed if the intracellular
calcium concentrations fall within a certain time frame (approximately 30 minutes in these
experiments). Efficacy of TRPV4 inhibition, when administered after TRPV4 activation,
was also reported in vivo in a murine model of chemically induced acute lung injury, where
protection from the acute lung injury response to intra-tracheal instillation of hydrochloric
acid was attenuated by administration of TRPV4 inhibitors (GSK2220691 and
GSK2337429A) 30 min after IT HCL (Balakrishna et al. 2014). Beyond this limited time
window, the loss of barrier permeability appears more permanent and that additional, non-
calcium-dependent processes are responsible.
In these studies, we used HUVECs as a cellular test system. However, we also replicated
selected findings in primary epithelial cells.
Furthermore, these data are consistent with the ex vivo findings in which TRPV4 in murine
isolated lungs regulates vascular permeability and its activation, whether via physical
stimuli such as mechanical stress or with pharmacological tools leads to an increase
endothelial and epithelial permeability in an intracellular calcium-influx dependent manner
(Alvarez et al. 2006, Hamanaka et al. 2007, Jian et al. 2008, Yin et al. 2008). It has also
been reported, that 4α-PDD activity on Ca2+
influx and whole-cell currents in human
embryonic kidney (HEK) cells is approximately 300 fold less potent than GSK1016790A
and had only a weak ability to contract bladder strips compared to GSK1016790A.
Furthermore 4α-PDD has been reported to be less selective compared to GSK1016790A
(Thorneloe et al. 2008, Thorneloe et al. 2017), that is consistent with our experimental

Discussion
83
observations. Additionally the exclusivity of 4α-PDD for TRPV4 has been put in question,
by the fact that it can activates mouse DRG neurons independently of TRPV4, by the fact
that it stimulated a dose-dependent increase in [Ca2+]i in neurons from WT and TRPV4-
KO mice, with the proportion of responding neurons and magnitude of increase unaffected
by the genotype (Alexander et al. 2013).
After having demonstrated that TRPV4 activation with the agonist GSK1016790A led to a
permeability increase in vitro and that this effect can be inhibited by the selective and
orally active TRPV4 antagonist GSK2193874 in endothelial cells, further investigations on
TRPV4 activation were performed in murine vascular permeability in vivo models. The
dye Evans blue given intravenously (i.v.) was used to monitor vascular leakage in different
organs after protein leakage induction with the TRPV4 agonist GSK1016790A. Results of
our in vivo investigations on TRPV4 activation in murine vascular permeability models are
consistent with prior findings, demonstrating that activation of TRPV4 with
GSK1016790A produced acute circulatory collapse and failure of the pulmonary
microvascular permeability barrier in rats and mice (Willette et al. 2008). The TRPV4
activators 4α-PDD has also been reported to increased lung endothelial permeability in a
dose-dependent manner in isolated rat lung, that was absent in TRPV4-/- rats (Alvarez et
al. 2006). More importantly our results extent prior finding by demonstrating for the first
time, that lung permeability increase in the bronchus of Balb/c mice induced by the
selective TRPV4 activator GSK1016790A can be inhibited by the selective TRPV4
activator GSK2193874 given orally.
We also questioned the link between pharmacological activation of TRPV4 and the
corresponding functional observations on barrier integrity when there is no affirmed signal
transduction pathway that can be followed to substantiate such a link. We hypothesized
that such effects may also be caused by cytotoxicity. Interestingly, we observed differential
cytotoxic effects in endothelial cells induced by the two TRPV4 agonists at concentrations
within the pharmacological range. 4α-PDD showed a time-dependent release of lactate
dehydrogenase, a cytotoxicity marker released by damaged cells, beginning after 8 h and
reaching a maximum after 12 h, which could not be blocked with the TRPV4 antagonist
GSK2193874. Necrosis was confirmed with a DNA-intercalating dye, but was preceded by
an increase in phosphatidylserine on the outer leaflet of the cell membranes, indicating an
apoptotic process followed by secondary necrosis, which was apparently independent of
TRPV4, again suggesting a possible off-target mechanism in HUVECs. In contrast

Discussion
84
activation of TRPV4 with the agonist GSK1016790A in HUVECs lead to a rapid
concentration-dependent increase in both LDH release and DNA dye intercalation within
the first hour, even with a concentration of the agonist in the low nanomolar range, that
could completely be blocked with the TRPV4 antagonist. Furthermore this effect was
dependent upon extracellular calcium. No cytotoxic effect occurred, when cells were
incubated with GSK1016790A [100 nM] in HBSS in the absence of calcium. Live cell
imaging showed that within the first few minutes after TRPV4 activation with
GSK1016790A cellular swelling and blebbing occurred, followed by apparent bursting of
the plasma membrane. Cellular swelling and blebbing has also been reported in the
literature (Alvarez et al. 2006) following lung exposure to TRPV4 agonists resulting in a
loss of barrier function. Because of the chronological relationship between
GSK1016790A-mediated increases in intracellular calcium concentrations, the cellular
swelling and cytotoxicity, we speculate that this effect maybe a consequence of rapid water
entry into the cell following the rapid increase and high concentrations of intracellular
calcium.
The physiological Ca2+
concentration in extracellular biologic fluids (and media mimicking
these conditions) ranges from 1.6 to 2 mM, in contrast the cytosolic free Ca2+
concentration is kept by cells around 100 nM producing an extremely large
electrochemical gradient between extracellular and intracellular Ca2+
concentrations,
meaning that for a cell at rest the [Ca2+
] is ~ 20.000 times lower in the cytoplasm than
outside the cell (Berridge et al. 2003, Clapham 2003, Bootman 2012). TRP channels
modulate the cations flux through plasma membranes down their electrochemical
gradients, thereby playing an important role in raising the free intracellular Ca2+
concentration (Pedersen et al. 2005, Bootman 2012). TRPV4 has been implicated in the
control of regulatory volume decrease (RVD), a regulatory response to cell swell of cells
exposed to hypotonic solutions, that is normally associated with changes in intracellular
calcium concentrations (Arniges et al. 2004). TRPV4 has been shown to provide the Ca2+
signal, required to activate further Ca2+
potassium channel and the subsequent RVD in
epithelial cells and also interacts with aquaporins to control RVD in astrocytes (Arniges et
al. 2004, Benfenati et al. 2011, Jo et al. 2015). This is an important observation suggesting,
that a disruption of cell volume regulation may have crucial consequences for cell
signalling, barrier integrity and cell viability (Benfenati et al. 2011). Finally TRPV4–AQP4
interactions have been promoted to constitute a molecular system that fine-tunes astroglial

Discussion
85
volume regulation by integrating osmosensing, calcium signaling, and water transport and,
when over-activated, triggers pathological cell swelling (Jo et al. 2015). These prior
finding support our speculation that pharmacological activation of TRPV4, leads to a
permanent opening of TRPV4 channels in HUVECs, that we do not expect to mimic the
situation in real life when activated by a physiological trigger, and led to an extreme
calcium-influx followed by water entry, disturbing cell volume regulation and leading to
the observed excessive cell swelling and the followed disruption of the cell membrane.
Alternatively, it has been suggested that intracellular calcium mediates expression of
ligands that bind to and activate death receptors such as CD95 (Kass and Orrenius 1999),
although within the time frame of these experiments, it seems unlikely that transcriptional
changes could occur. Another possibility is, that mitochondria may respond to an apoptotic
Ca2+
signal by the selective release of cytochrome c or through enhanced production of
reactive oxygen species and opening of an inner mitochondrial membrane pore (Kass and
Orrenius 1999). Our findings are supported by the observations of Olivan-Viguera et al.
(2018), who showed in parallel similar TRPV4 mediated cytotoxic effects on melanoma
cells and keratinocytes.
Taken together we have shown that the effects of pharmacological TRPV4 activation on
TER and vascular permeability assays in vitro resulted in permeability increase. We further
demonstrated that TRPV4 activation with GSK1016790A led to lung permeability increase
in vivo in the bronchus of Balb/c mice and that this effect could be inhibited by
preincubation with the orally active TRPV4 antagonist GSK2193874. Additionally we
explained the functional effects of TRPV4 activation on TER with different cytotoxic
effects induced by two widely-published TRPV4 agonists in endothelial cells. We
conclude that in this test system in HUVECs, 4α-PDD may not be a selective activator of
TRPV4 and mediates TER reduction via apoptosis. In contrast GSK1016790A selectively
activates TRPV4, but that TER reduction is also a consequence of cellular necrosis, during
which the cells swell led to membrane disruption and collapse of the cells. Cell death plays
an important role in regulating barrier integrity and TRPV4 mediated cytotoxicity in
endothelial cells, but also in epithelial cells, is poorly described in the literature and we
believe that these findings add significant context to many reported and further studies
concerning the role of TRPV4 in endothelial and epithelial barrier-function.

Discussion
86
4.2 Role of TRPV4 in stretch induced pathological cellular response
TRPV4 is a force-sensitive Ca2+
-permeable cation channel expressed in many pulmonary
tissues and cells including bronchiolar and alveolar epithelia, alveolar macrophages and the
endothelium (Alvarez et al. 2006, Hamanaka et al. 2010, Yin and Kuebler 2010) and has
been shown to be a particularly promising candidate for the initiation of the acute calcium-
dependent permeability increase during ventilation in isolated mouse lungs (Hamanaka et
al. 2007). Therefore in the second part of this study we investigated the potential of a
TRPV4 inhibitor for the improvement of mechanical ventilation induced pathological
response of lung cells, using the TRPV4 antagonist GSK2193874 in both in vitro and in
vivo models of pathophysiological cell stretch.
Prior to the investigations on cell stretch, compound characterisation was performed on
human lung epithelial cells (NCI-H292), which confirmed a TRPV4 agonism evoked Ca2+
response with GSK1016790A and 4α-PDD, that could be concentration-dependently
reduced and blocked by TRPV4 antagonism with GSK2193874. NCI-H292 also showed an
extension-evoked Ca2+
response after uniaxial cell stretch, which was significantly reduced
by 43% with TRPV4 inhibition. This is consistent with previously reported data in which
TRPV4 mediated stretch-evoked Ca2+
influx contributes to the increase in membrane
permeability due to lung over-distention following high PIP ventilation (Hamanaka et al.
2007) and in mice primary urothelial cells, where the Ca2+
increase was partially reduced
in TRPV4-KO compared to WT cells during stretch stimuli (Mochizuki et al. 2009). In this
study we firstly showed, that stretch evoked Ca2+
influx in human lung epithelial cells and
the involvement of TRPV4. Also the increase in Ca2+
concentration could not be
completely blocked by TRPV4 antagonism leading to the conclusion, that the TRPV4
independent calcium influx could be regulated by other mechanosensory channels and
systems, perhaps with different thresholds, that might play a role on the initial calcium
response of the lung epithelium to extension, e.g. TRPV2 has also been demonstrated to
participates in strain-induced Ca2+
entry in rat primary alveolar type II (ATII) cells (Fois et
al. 2012).
In our experiments, cells had to be stretched with a magnitude of 80% within 400 ms in
order to induce TRPV4 activation. One possible reason for the need of such large stretch
amplitudes is, that in these experiments the mechanical strain system stretches cells in a
uniaxial direction, compared to the multidirectional extension in the mechanically

Discussion
87
ventilated lung in vivo. Another explanation may be, that these cellular experiments were
performed at room temperature (due to technical reasons), whereas in the lung, the
epithelial temperature, particularly in the lower airways, is likely to be higher, which may
impact the heat-sensitive TRPV4 channel. The need for large stretch amplitudes for the
TRPV4 mediated strain-evoked calcium entry was also observed on mice primary
urothelial cells under similar conditions (Mochizuki et al. 2009).
VILI has been described as a cellular response to mechanical stress, that includes a rapid
increase in vascular permeability followed by cytokine release (Dreyfuss and Saumon
1998, Dos Santos and Slutsky 2000). Deformation per se can trigger inflammatory
signalling and it is possible, that alveolar epithelial cells may play an active role in
ventilator-induced lung injury (Vlahakis et al. 1999). Mechanical ventilation has been
reported to be able to induce cytokine upregulation in both injured and healthy lungs and
that the underlying mechanisms include cellular responses to stretch with the frequently
involved cytokines IL-8 and probably IL-6, IL-1β and TNF-α, making cytokines good
surrogate endpoints in exploring the pathogenesis and pathophysiology of VILI in
experimental studies (Halbertsma et al. 2005).
We further tested the hypothesis that the initial calcium response to stretch results also in
an inflammatory response in human epithelial cells. TRPV4 activation of epithelial cells
with the synthetic TRPV4 agonist GSK1016790A resulted in a release of the pro-
inflammatory cytokines IL-6 and IL-8 in vitro, that could also be completely blocked by
addition of the TRPV4 antagonist GSK2193874. This is consistent with a more recent
study in fetal mouse distal lung epithelial cells, which demonstrated that TRPV4 may play
an important role in the transduction of mechanical signals in the lung epithelium by
modulating the release of the cytokine IL-6 via p38 and ERK pathways (Nayak et al.
2015).
Cyclical stretching of lung epithelial cells (NCI-H292) in equibiaxial direction also
increased release of the cytokines IL-6 and IL-8 after stretch, that could also be reduced by
about 30% with GSK2193874 and was decreased by about 80% by addition of the general
TRP-channel blocker Ruthenium red, suggesting that this effect is only partially modulated
by TRPV4 and other stretch activated channels and integrins may play a role. It is not
unreasonable, that the applied stretch of 30% in our studies mimic the deformation by the
lung epithelium in situ when mechanically ventilated with recommended ventilator settings

Discussion
88
(Slutsky 1993) during which the lung volume more than doubles. Interestingly the
reduction of the stretch-evoked increase in intracellular calcium on NCI-H292, that was
reduced by 43% with TRPV4 antagonist addition, is similar to the about 30% reduction of
the stretch induced cytokine release via TRPV4 antagonism, suggesting that the size of
changes in Ca2+
influx extrapolate directly to cytokine release.
Furthermore, during simultaneous live cell imaging and uniaxial mechanical strain, we
observed that some cells (NCI-H292) showed a direct increase of intracellular calcium
concentration and in other cells calcium influx began only 10 sec after stretch, leading to
the hypothesis that the directly responding cells to stretch may release a second messenger
activating other cells with some time delay (indirect responders). TRPV4 has been shown
to mediate stretch-evoked Ca2+
influx and also ATP release in primary urothelial cell
cultures when comparing WT and TRPV4-KO cells (Mochizuki et al. 2009) and ATP has
been demonstrated to interact with TRPV4 has a positive modulator of channel activity
(Lorenzo et al. 2008). We therefore questioned, if ATP could be the second messenger
activating the indirect responding cells. Therefor we studied, if stretch induces a TRPV4
mediated ATP release in NCI-H292 in the biaxial cell stretch system with different
protocols and time points with or without preincubation with the TRPV4 antagonist
GSK2193874, but we could not observe significant increases in ATP after stretch under
our assay procedure.
Macrophages are a major source of cytokine secretion and are known to adhere directly to
lung epithelial cells (Tao and Kobzik 2002) and because of this property may also be
exposed to stretch during mechanical ventilation, and have indeed been shown to be
activated by strain in vitro resulting in an increase in IL-8 (Pugin et al. 1998). Therefore we
examined the effect of mechanical stretch on isolated human macrophages in vitro. The
mechanical stretch induced stress on the pro-inflammatory M1, and to a lesser extent in the
tissue remodelling M2, macrophages resulted in an significant increase of the cytokines
IL-1α, IL-1β and the chemokine MCP-1 and also a small but significant increases in IL-6
and -8. Interestingly, and in contrast to the findings in epithelial cells, the stretch induced
increase of the cyto- and chemokine levels could be nearly completely abolished by the
TRPV4 antagonist GSK2193874. Particularly interesting is that IL-1α has been shown to
directly increase vascular endothelial cell permeability in vitro (Royall et al. 1989), and
therefore may play an important role in the stretch induced pulmonary vascular
permeability increase reported during ventilation leading to VILI. Additionally we also

Discussion
89
observed TER reduction induced by IL-1β and TNF-α in HUVECs. More importantly we
have recently shown that macrophage-derived IL-1α and IL-1β promotes permeability
increases in primary human epithelial cells (SAECs) differentiated in air-liquid interface
and additionally shown that IL-1α, IL-1β and TNF-α but not IL-4, IL-13, GM-CSF, M-
CSF or IL-10 are able to promote permeability increase in SAECs also in the absence of
macrophages (Mang et al. 2018). These findings indicate that the TRPV4 mediated stretch
induced release of the cytokines IL-1α and IL-1β from macrophages could not only affect
the vascular endothelium, but also directly act on the lung epithelium permeability increase
leading to edema formation and alveolar flooding during ventilation, that may induce or
aggravate VILI. M1 macrophages may mimic an alveolar phenotype, because they express
PPARγ, that has been shown to be an alveolar macrophage marker (Schneider et al. 2014).
Furthermore, in the hyperinflammatory environment associated with ARDS, macrophages
are more likely to be driven initially towards a pro-inflammatory, rather than a tissue
remodelling phenotype and TRPV4 has also been reported to mediate polarization of
macrophages toward an M1-like phenotype (Misharin et al. 2013, Scheraga et al. 2016,
Scheraga et al. 2017).
However, the signal transduction pathway from a mechanical stimulus resulting in an
inflammatory response remains elusive. One possibility is that TRPV4 inhibitors block
other Ca2+
dependent processes, such as the release of cytokines. TRPV4 has also been
shown to play a role in the transduction of mechanical signals in the distal epithelium by
modulating inflammation (IL-6 secretion) via p38 and ERK pathways (Nayak et al. 2015).
We also investigated in NCI-H292 the effect of stretch on phosphorylated/total ERK1/2 in
the biaxial cell-stretch system with different protocols and time points with or without
preincubation with the TRPV4 antagonist GSK2193874, but no significant increase in
pERK was observed under our assay procedure. Release of inflammatory cytokines IL-6
and -8 after TRPV1 activation has been shown to be mediated through MAPK signalling in
corneal epithelium (Zhang et al. 2007). These two pathways may also be important
downstream activators of TRPV4 in mediating an inflammatory response after stretch.
Having demonstrated that both human lung epithelial cells and macrophages play an active
role in the TRPV4-mediated stretch induced cytokine release, we wanted to evaluate the
effect of the TRPV4 antagonist GSK2193874 in a murine model of mechanical ventilation
similar to other models reported to induce lung injury (Hegeman et al. 2013, Michalick et
al. 2017). LDH release measured in the BALF of mice lungs after mechanical ventilation

Discussion
90
showed an increase in LDH signal after 30 ml/kg tidal volume (TV) ventilation compared
to the non-ventilated control group that could not be significantly reduced by TRPV4
antagonism with GSK2193874. Lung elastance and resistance showed an elevated but not
significant increase in resistance and elastance in the 30 ml/kg TV ventilated group
compared to the 6.5 ml/kg TV control, that was absent in the 30 ml/kg TV group pretreated
with the TRPV4 antagonist GSK2193874. Studies already shown, that both sterile (e.g.,
ventilator-induced stretch) and infectious triggers of ARDS result in stiffening of the lung
tissue (reduced compliance) (Perlman et al. 2011, Meng et al. 2015, Scheraga et al. 2017).
Our data also indicate that a lung stiffening effect may be induced by mechanical
ventilation with high tidal volumes and that this effect might be attenuated by TRPV4
inhibition, when comparing the 30 ml/kg TV ventilated control group and the 30 ml/kg TV
ventilated with GSK2193874 treated group, but the effect was not significant because of a
too high variability. LDH release during mechanical ventilation with high tidal volume
could not be significantly reduced by TRPV4 inhibition in this model system. Although
TRPV4 deficiency or inhibition by HC-067047 has been demonstrated to attenuated
histological features of mice lung injury in a model of VILI (Michalick et al. 2013).
More importantly we investigated the effect of high tidal volume ventilation on lung
cytokine release and permeability increase. Protein infiltration into the lung as a
measurement of barrier dysfunction was chosen for a number of reasons. We had
previously observed that protein infiltration is both quantitative and more sensitive than the
semi-quantitative histological assessments. Alveolar water and alveolar collapse would
have been lost during the fixation and dehydration steps and therefore only interstitial
oedema-related tissue damage would have been observed, which itself is susceptible to
fixation artefacts. We also considered that lung strain during mechanical ventilation is
poorly defined (Protti et al. 2014, Carrasco Loza et al. 2015) and difficult to estimate
because of the heterogeneous local lung susceptibility during MV (Carrasco Loza et al.
2015). Because of the heterogeneous susceptibility between regions of the lung we opted to
use a total pulmonary barrier function index rather than a local one.
The high tidal volume ventilation (30 ml/kg TV) protocol significantly enhanced protein,
IL-6 and KC/GRO concentrations in BALF compared to the normal ventilated group (6.5
ml/kg TV), all of which could be nearly completely blocked with the TRPV4 antagonist.
These data are consistent with the in vitro findings in macrophages, which were almost
entirely TRPV4-dependent, but less so with the in vitro findings in epithelial cells, which

Discussion
91
were only partially dependent on TRPV4. This may suggest, that macrophages may be
stronger effector cells during ventilation contributing to a pathological response compared
to epithelial cells, a hypothesis which is consistent with the findings of others (Frank et al.
2006, Eyal et al. 2007, Hamanaka et al. 2010) and that these effects are TRPV4-dependent.
An important role for alveolar macrophages in mechanical ventilation models has also
been demonstrated by depletion of macrophages in rat lungs using clodronate-filled
liposomes resulting in an attenuation of ventilator-induced lung injury, where high volume
ventilation resulted not only in a activation-associated adhesion of alveolar macrophages,
but also in an increased alveolar protein leak and lung edema formation, that was
attenuated by depletion of macrophages (Frank et al. 2006, Eyal et al. 2007). A more
important investigation linked TRPV4 channels and macrophages in the role of modulating
VILI. In this study the ventilator induced lung injury was markedly attenuated in TRPV4-
KO mice, whereas reintroduction of TRPV4-WT macrophages in TRPV4-KO mice
reconstituted the lung injury response to mechanical ventilation, showing that macrophages
TRPV4 activation plays a crucial role in initiating this injury (Hamanaka et al. 2010). One
study demonstrated that the inflammatory response to particles was amplified by contact-
dependent interactions between alveolar macrophages and epithelial cells (Tao and Kobzik
2002) and it is also possible that a similar interaction occurs during ventilation. The
importance of TRPV4 in preventing VILI was indirectly addressed in another study, in
which inhalation of nanoparticles containing Ruthenium red prevented ventilator damage
and vascular permeability for several days (Jurek et al. 2014). However Ruthenium red is
known to impact calcium handling in cells via effects on other TRP channels (Vincent et
al. 2009, Yamashiro et al. 2010, Takahashi et al. 2011) and non-TRP proteins (Deinum et
al. 1985, Sasaki et al. 1992, Cardoso and De Meis 1993, Yamada et al. 2000) and it
therefore remains a possibility, that the effect of Ruthenium red on ventilator-induced
oedema in this study may be modulated via other TRP channels or via modulation of
downstream intracellular calcium handling (Jurek et al. 2014).
In summary we have shown, that mechanical stretch evoked intracellular Ca2+
influx and
induced the release of pro-inflammatory cytokines that was partially dependent upon
TRPV4 in human lung epithelial cells, but also induced the release of pro-inflammatory
cytokines from M1 macrophages, that was entirely dependent upon TRPV4. In a murine
ventilation model with high tidal volumes, TRPV4 inhibition attenuated pulmonary barrier
permeability increase and pro-inflammatory cytokines secretion. Taken together, these data

Discussion
92
suggest TRPV4 inhibitors may have utility as a prophylactic pharmacological treatment to
improve pathological responses of lung cells exposed to stretch during ventilation and
potentially may have utility in the support of patients receiving mechanical ventilation.
4.3 Summary and clinical relevance
At present time no effective pharmacological treatments exist for ARDS (Matthay et al.
2012) and only patient management strategies exist to ensure gas exchange while
minimizing the risk of VILI such as a protective mechanical ventilation (Matthay et al.
2012). ARDS can arise as a result of infection and can be the consequence of a non-
infectious cause like mechanical ventilation (lung ventilator stretch) (Rezoagli et al. 2017).
Despite the fact that mechanical ventilation (MV) is an important tool for life support of
ARDS patients, it also has the potential to exert pathological mechanical forces on
different lung cells leading to VILI (Slutsky and Imai 2003). There is an urgent need to
identify the molecular mechanism underlying mechanotransduction leading to the
pathological response of the lung during MV and TRPV4 has been shown to be a
particularly promising candidate for the initiation of the acute calcium-dependent
permeability increase during ventilation (Hamanaka et al. 2007).
Therefore this study investigated the potential of a TRPV4 antagonist to improve MV
induced pathological response in lung cells, using the TRPV4 antagonist GSK2193874 in
both self-established in vitro and in vivo models of permeability and afterward in models of
pathophysiological cell stretch. We have shown that pharmacological TRPV4 activation
leads to TER drop in endothelial cells and that the effect can be inhibited by the TRPV4
antagonist GSK2193874. We further demonstrated that TRPV4 activation with
GSK1016790A led to lung permeability increase in vivo in the bronchus of Balb/c mice
and firstly shown that this effect can be inhibited by preincubation with the TRPV4
antagonist GSK2193874 given orally. Additionally we explained the functional effects of
TRPV4 activation on TER with differential cytotoxic effects induced by two widely-
published TRPV4 agonists in endothelial cells.
We further investigated the effect of TRPV4 inhibition on the cellular response to cell-
stretch and firstly shown in human lung epithelial cells, that mechanical stretch evoked
intracellular Ca2+
influx and induced the release of pro-inflammatory cytokines that was

Discussion
93
partially dependent upon TRPV4 and suggest that other stretch activated channels could
play a role. More importantly we firstly shown the stretch-induced release of pro-
inflammatory cytokines from human macrophages, that was entirely dependent upon
TRPV4 and suggest that macrophages may be the stronger effector cells regarding
mechanical stretch induced TRPV4 activation and the pathological cellular response
occurring during MV.
It is not unreasonable that the applied stretch of 30% in our studies mimic the deformation
of the lung epithelium in situ when mechanically ventilated with recommended ventilator
settings (Slutsky 1993), during which the lung volume more than doubles and could be
even higher when considered that lung strain is poorly defined during MV (Protti et al.
2014, Carrasco Loza et al. 2015) and difficult to estimate because of the heterogeneous
local lung susceptibility during ventilation (Carrasco Loza et al. 2015), especially in regard
of lungs from patients with ARDS. Measured inspiratory capacity in lungs of ARDS
patients is reduced to almost one third of the predicted inspiratory capacity (Beitler et al.
2016) pointing to the fact that almost two third of the total lung volume are not
participating in inspiration during MV. Injured regions of the lung will receive smaller
fractions of the total tidal volume from the inspired tidal volumes, e.g. due to alveolar
collapse and fluid extravasation, therefore other lung areas will receive the majority of the
tidal volume leading to massive over-distension of this areas and local damage perhaps
even with protective ventilation strategies (Carrasco Loza et al. 2015, Bellani et al. 2016).
Studies recommended inspired volume of more than 20 ml/kg to induce lung injury in
healthy young mice during ventilation, but also showed that 40 ml/kg ventilation is
accompanying a high mortality of the laboratory animals (Wilson et al. 2012). In regard of
these facts the 30 ml/kg ventilation we used mimic the clinical features and mechanical
forces occurring during VILI without leading to animal mortality during experimental
procedures and is similar to other models reported to induce lung injury (Hegeman et al.
2013, Michalick et al. 2017).
Furthermore MV has been reported to be able to induce cytokine upregulation in both
injured and healthy lungs and that the underlying mechanisms include cellular responses to
stretch with the frequently involved cytokines IL-8 and probably IL-6, IL-1β and TNF-α
making cytokines good surrogate endpoints in exploring the pathogenesis and
pathophysiology of VILI in experimental studies (Halbertsma et al. 2005).

Discussion
94
Interestingly we observed an significant cell stretch induced and TRPV4 mediated increase
in the cytokines mentioned to be linked to ARDS and VILI, with IL-6 and IL-8 in human
lung epithelial cells and IL-1α, IL-1β, IL-6, IL-8 in M1 macrophages and TNF-α in M2
macrophages. In regard of the clinical relevance of our results on lung epithelial cells
exposed to cellular stretch, our data point to the fact that the effect on cytokine release is
partly modulated by TRPV4 in epithelial cells and suggest that other stretch activated
channel, like other TRP channels may play a role, indicated by a near complete blocked
effect on cytokine release with the general TRP channel blocker Ruthenium red. The
suggestion that other stretch activated channels might play a role in lung cell stretch during
mechanical ventilation is supported by others, who point out the increasing evidence for
the importance of stretch-activated ion channels (SACs) in the activation of lung-resident
and inflammatory cells and indicating that the time has come to seriously consider SACs as
new therapeutic targets against VILI and ARDS (Schwingshackl 2016). Also for a clinical
drug proof of concept a general SACs blocker might be inappropriate and negative side
effects difficult to estimate. In contrast regarding the clinical relevance of our data on
stretch induced and TRPV4 mediated cytokine release in macrophages, that was entirely
dependent upon TRPV4, suggest that macrophages are the stronger effector cells.
Importantly is that alveolar macrophages in MV models have been shown to have the
potential to induce VILI by themselves, demonstrated by depletion of macrophages in rat
lungs resulting in an attenuation of VILI monitored by alveolar protein leak and lung
edema formation (Frank et al. 2006, Eyal et al. 2007). Furthermore TRPV4 channels of
macrophages have been linked to modulate VILI by a study demonstrating, that VILI was
attenuated in TRPV4-KO mice, whereas reintroduction of TRPV4-WT macrophages in
TRPV4-KO mice reconstituted the lung injury response to MV (Hamanaka et al. 2010).
Furthermore our data showing the stretch induced and TRPV4 mediated cytokine release
of IL-1α and IL-1β in macrophages can be linked to another study of our group showing
that macrophage-derived IL-1α and IL-1β promotes permeability increases in primary
human epithelial cells differentiated in air-liquid interface (Mang et al. 2018) and could be
a possible explanation for the permeability increase observed during MV in the literature,
but also in our murine model of ventilation, pointing out that macrophage TRPV4
activation plays a crucial role in initiating the lung injury response to MV by promoting a
permeability increase of the lung epithelium through local release of IL-1α, Il-1β but also
TNF-α (M2), making macrophages TRPV4 a very specific and promising new drug target.
We also demonstrated the efficacy of the TRPV4 antagonist GSK2193874 in a murine

Discussion
95
ventilation model with high tidal volumes in vivo, where the orally given TRPV4
antagonist attenuated both pro-inflammatory cytokines secretion and also pulmonary
barrier permeability increase. The oral bioavailability of the TRPV4 antagonist
GSK2193874 (Thorneloe et al. 2012), demonstrated in our murine ventilation model, gives
this compound a key advantage, by the mean that it can potentially be dose repeatedly for
chronic use and to the time points it is required.
Taken together, these data suggest TRPV4 inhibitors, with special attention on the orally
active TRPV4 antagonist GSK2193874, may have utility as a prophylactic
pharmacological treatment to improve pathological responses of lung cells exposed to
stretch during ventilation and potentially may have utility in the support of patients
receiving mechanical ventilation by attenuating permeability increase and cytokine release.
One possibility is that TRPV4 inhibitors could be given prophylactically to patients who
require mechanical ventilation and exhibit certain risk factors for the development of
ARDS such as sepsis, low blood pH, elevated lactate, low albumin, low respiratory
compliance and patient weight (Xiaoming et al. 2008).
4.4 Next steps
In regard of the outcome of this thesis, the following major points would be part of further
investigation on TRPV4:
During experimental procedure the observation was made, that cells reacted differently to
cellular stretch. While some cells responded to stretch in a direct increase of intracellular
calcium concentration, in other cells calcium influx began only after some time delay. This
observation led to the question, if the directly responding cells to stretch may release a
second messenger activating afterwards other cells and would be very interesting to be
further investigated.
We further observed that TRPV4 in lung epithelial cells partly modulated calcium influx
and stretch induced cytokine release and that stretch induced cytokine release was in
contrast nearly completely abolished by the general stretch activated channel (SAC)
blocker Ruthenium red. For a clinical drug proof of concept a general SACs blocker might
be inappropriate and in this concept it would be very important to identify the other
channels, perhaps TRP channels, leading to the observed stretch induced cellular response,

Discussion
96
by co-administration of different selective TRP channel blocker in this system or by
genetic deletion of suspected channels that might play a role. Furthermore TRPV channels
in general have been demonstrated to be activated by specific, largely non-overlapping
temperature ranges, e.g. TRPV4 is activated by temperature ranging between 24 and 38°C,
TRPV1 by temperatures greater than 43°C and TRPV2 by temperature greater than 52°C
(Watanabe, Vriens et al. 2002, Clapham 2003). Except of only two TRP channels
(TRPM4, TRPM5) that are impermeant for calcium, all other TRP channels are Ca2+
permeable (Pedersen et al. 2005, Owsianik et al. 2006) and several members have been
described to be implicated in mechanotransduction such as TRPA1, TRPC1, TRPC3,
TRPC6 , TRPM7, TRPP2 , TRPV1, TRPV2 and TRPV4 (reviewed in Yin and Kuebler
(2010). In this concept it would be very interesting to investigate which TRP channels
react to similar stretch amplitudes in calcium measurement and furthermore if there are
groups, that react to non-overlapping stretch amplitudes for mechanosensation like the
situation reported in temperature sensing.
We further suggested, that macrophages TRPV4 could be the driving force in regard of
stretch induced and TRPV4 mediated cytokine and permeability increase, by the fact that
stretch induced cytokine release in macrophages was entirely dependent upon TRPV4 and
that the involved cytokines IL-1α and Il-1β derived from macrophages have also been
shown by our group to be able to induce lung permeability increase in primary human lung
epithelial cells (Mang et al. 2018). Further investigations in co-culture models with
macrophages and lung epithelial cells on a lung-on-a-chip model (Huh 2015), allowing cell
stretch experiment in regard of permeability increase, could answer the question if
macrophages TRPV4 activated by stretch is the major driving force of epithelial barrier
permeability increase. Therefore comparing the stretch induced permeability increase of
lung epithelial cells alone to the stretch induced permeability increase of co-culture models
of macrophages and epithelial cells with or without TRPV4 inhibition. A major concern of
this study will be, if this method allows amplitudes of cell stretch high enough to induce
permeability increase strong enough to further compare the effect of TRPV4 inhibition.
Another important point regarding stretch induced cytokine release mediated by TRPV4,
would be to identify the possible pathway leading to this cellular response. We already
began to investigate the possible pathway enhanced by TRPV4 mediated calcium influx,
when pharmacologically activated by TRPV4 agonism with 4α-PDD in NCI-H292 cells
using the Human cAMP / Calcium Signaling PathwayFinder. Although 10 µM 4α-PDD

Discussion
97
also induced a cytotoxic effect, cell loss was somehow normalised by the mean, that the
same amount of purified RNA from each group had to be used for cDNA preparation. An
interesting result of this assay was the gene regulation of Protein phosphatase 1, regulatory
(inhibitor) subunit 15A (PPP1R15A) also named DNA damage-inducible protein 34
(GADD34), showing an about 45 fold upregulation compared to the control group after
TRPV4 agonist exposure with 4α-PDD [10µM], that was nearly completely blocked by
preincubation with the TRPV4 antagonist GSK2193874 [1µM]. Interestingly, a study
showed that dextran sodium sulfate (DSS) induced inflammatory responses were
downregulated by GADD34 deficiency, where the expression of pro-inflammatory
mediators such as TNF-α, IL-6, and iNOS/NOS2 was higher in the colons of WT mice
than in GADD34-KO mice (Tanaka et al. 2015). In this concept it would be now very
interesting to further investigate, in a similar way, the pathway leading to a stretch induced
and TRPV4 mediated release of cytokines with the help of RT² Profiler™ PCR Arrays,
especially in macrophages, where we have shown that the stretch induced cytokine release
was entirely dependent upon TRPV4.
Finally suggesting that TRPV4 inhibitors may have utility as a prophylactic
pharmacological treatment for patient with the need for mechanical ventilation, especially
for patient with risk factors for the development of ARDS such as patient with a “hyper-
inflammatory” subphenotype (Thompson et al. 2017), it will be an interesting point to
make further investigations, that may further allow to enclose and identify such patient
populations, that could benefit from a pharmacological treatment with TRPV4 inhibitors.
TRP channels implication in diseases has been shown by their correlation between the
level of channel expression and the disease symptoms, e.g. TRPV1 expression is
considerably increased in the airway nerves of patients exhibiting chronic cough
(Groneberg et al. 2004). In this regard it would be an important point to investigate, if there
is a significant difference in TRPV4 expression between patients with risk factors for
ARDS developing ARDS compared to patients with risk factors for ARDS not developing
ARDS.

Abstract
98
5 Abstract
Acute respiratory distress syndrome (ARDS) is a rapidly progressive form of acute
respiratory failure characterized by severe hypoxemia and noncardiogenic pulmonary
edema and remains a syndrome with a high incidence frequently resulting in death. At
present time no effective pharmacological treatments exist for ARDS and only patient
management strategies exist to ensure gas exchange while minimizing the risk of
ventilation induced lung injury (VILI) such as a protective mechanical ventilation. ARDS
can arise as a result of infection and can be the consequence of a non-infectious cause like
mechanical ventilation. Despite the fact that mechanical ventilation is an important tool for
life support of ARDS patients, it also has the potential to exert pathological mechanical
forces on different lung cells leading to VILI. There is an urgent need to identify the
molecular mechanism underlying mechanotransduction leading to the pathological
response of the lung during mechanical ventilation and the force sensitive calcium
permeable ion channel transient receptor potential vanilloid 4 (TRPV4) has received
specific attention to be a particularly promising candidate for the initiation of the acute
calcium-dependent permeability increase during ventilation.
In this study we investigated the potential for TRPV4 inhibition in a step by step approach
as a treatment of ARDS with particular attention to ventilation induced pathological
response in lung cells, using the selective TRPV4 antagonist GSK2193874 in both self-
established in vitro and in vivo models of permeability and afterwards in models of
pathophysiological cell-stretch. We first investigated on the role of TRPV4 in modulating
membrane barrier integrity and shown that pharmacological TRPV4 activation leads to
transepithelial/transendothelial electrical resistance (TER) drop in endothelial cells and that
the effect can be inhibited by the TRPV4 antagonist GSK2193874. We further
demonstrated that TRPV4 activation with GSK1016790A led to lung permeability increase
in vivo in the bronchus of Balb/c mice and have shown that this effect can be inhibited by
preincubation with the TRPV4 Antagonist GSK2193874 given orally. Additionally we
explained the functional effects of TRPV4 activation on TER with differential cytotoxic
effects induced by two widely-published TRPV4 agonists in endothelial cells and gave
time points for their occurrence and believe that these findings add significant context to

Abstract
99
many reported and further studies concerning the role of TRPV4 in endothelial and
epithelial barrier-function.
We further investigated the effect of TRPV4 inhibition on the cellular response to cell-
stretch and shown in human lung epithelial cells, that mechanical stretch evoked
intracellular Ca2+
influx and induced the release of pro-inflammatory cytokines that was
partially dependent upon TRPV4 and suggest that other stretch activated channels could
play a role. More importantly we firstly shown the stretch-induced release of pro-
inflammatory cytokines from human macrophages that was entirely dependent upon
TRPV4 and suggest that macrophages may be the stronger effector cells regarding
mechanical stretch induced TRPV4 activation and the pathological cellular response
occurring during MV. Interestingly we observed a significant cell-stretch induced and
TRPV4 mediated increase in the cytokines that have been linked to ARDS and VILI. In a
murine ventilation model with high tidal volumes we demonstrated that the orally given
TRPV4 Antagonist GSK2193874 attenuated both pulmonary barrier permeability increase
and pro-inflammatory cytokines secretion.
Taken together, these data suggest TRPV4 inhibitors, with special attention on the orally
active TRPV4 antagonist GSK2193874, may have utility as a pharmacological treatment to
improve pathological responses of lung cells, especially targeting macrophages TRPV4,
exposed to cell-stretch during ventilation and potentially may have utility in the support of
patients receiving mechanical ventilation by attenuating both permeability increase and
cytokine release. This opens the possibility for the use of TRPV4 inhibitors
prophylactically in patients with the need for mechanical ventilation with risk factors for
the development of ARDS such as sepsis, low blood pH, low respiratory compliance,
obesity and with an hyper-inflammatory subphenotype, that may further allow to enclose
and identify patient populations, that could benefit from a pharmacological treatment with
TRPV4 inhibitors.

References
100
6 References
[1] Abràmoff, M. D., P. J. Magalhães and S. J. Ram (2004): Image processing with
ImageJ. Biophotonics Intern 11(7): 36-42.
[2] Alessandri-Haber, N., O. A. Dina, E. K. Joseph, D. Reichling and J. D. Levine
(2006): A transient receptor potential vanilloid 4-dependent mechanism of
hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci
26(14): 3864-3874.
[3] Alexander, R., A. Kerby, A. A. Aubdool, A. R. Power, S. Grover, C. Gentry and A.
D. Grant (2013): 4α-phorbol 12,13-didecanoate activates cultured mouse dorsal
root ganglia neurons independently of TRPV4. Br J Pharmacol 168(3): 761-772.
[4] Alvarez, D. F., J. A. King, D. Weber, E. Addison, W. Liedtke and M. I. Townsley
(2006): Transient receptor potential vanilloid 4-mediated disruption of the alveolar
septal barrier: a novel mechanism of acute lung injury. Circ Res 99(9): 988-995.
[5] Arniges, M., J. M. Fernández-Fernández, N. Albrecht, M. Schaefer and M. A.
Valverde (2006): Human TRPV4 Channel Splice Variants Revealed a Key Role of
Ankyrin Domains in Multimerization and Trafficking. J Biol Chem 281(3): 1580-
1586.
[6] Arniges, M., E. Vazquez, J. M. Fernandez-Fernandez and M. A. Valverde (2004):
Swelling-activated Ca2+ entry via TRPV4 channel is defective in cystic fibrosis
airway epithelia. J Biol Chem 279(52): 54062-54068.
[7] Ashbaugh, D. G., D. B. Bigelow, T. L. Petty and B. E. Levine (2005): Ashbaugh
DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. The
Lancet, Saturday 12 August 1967. Crit Care Resusc 7(1): 60-61.
[8] Balakrishna, S., W. Song, S. Achanta, S. F. Doran, B. Liu, M. M. Kaelberer, Z. Yu,
A. Sui, M. Cheung, E. Leishman, H. S. Eidam, G. Ye, R. N. Willette, K. S.
Thorneloe, H. B. Bradshaw, S. Matalon and S. E. Jordt (2014): TRPV4 inhibition
counteracts edema and inflammation and improves pulmonary function and oxygen
saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol
Physiol 307(2): L158-172.
[9] Bang, S., S. Yoo, T. J. Yang, H. Cho and S. W. Hwang (2012): Nociceptive and
pro-inflammatory effects of dimethylallyl pyrophosphate via TRPV4 activation. Br
J Pharmacol 166(4): 1433-1443.
[10] Baratchi, S., J. G. Almazi, W. Darby, F. J. Tovar-Lopez, A. Mitchell and P.
McIntyre (2016): Shear stress mediates exocytosis of functional TRPV4 channels
in endothelial cells. Cell Mol Life Sci 73(3): 649-666.
[11] Baratchi, S., F. J. Tovar-Lopez, K. Khoshmanesh, M. S. Grace, W. Darby, J.
Almazi, A. Mitchell and P. McIntyre (2014): Examination of the role of transient
receptor potential vanilloid type 4 in endothelial responses to shear forces.
Biomicrofluidics 8(4): 044117.

References
101
[12] Bazzoni, G. (2006): Endothelial tight junctions: Permeable barriers of the vessel
wall. Thromb Haemost 95(1): 36-42.
[13] Becker, D., J. Bereiter-Hahn and M. Jendrach (2009): Functional interaction of the
cation channel transient receptor potential vanilloid 4 (TRPV4) and actin in volume
regulation. Eur J Cell Biol 88(3): 141-152.
[14] Beitler, J. R., R. Majumdar, R. D. Hubmayr, A. Malhotra, B. T. Thompson, R. L.
Owens, S. H. Loring and D. Talmor (2016): Volume delivered during recruitment
maneuver predicts lung stress in acute respiratory distress syndrome. Crit Care
Med 44(1): 91-99.
[15] Bellani, G., J. G. Laffey, T. Pham and et al. (2016): Epidemiology, patterns of care,
and mortality for patients with acute respiratory distress syndrome in intensive care
units in 50 countries. JAMA 315(8): 788-800.
[16] Benfenati, V., M. Caprini, M. Dovizio, M. N. Mylonakou, S. Ferroni, O. P.
Ottersen and M. Amiry-Moghaddam (2011): An aquaporin-4/transient receptor
potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control
in astrocytes. Proc Natl Acad Sci U S A 108(6): 2563-2568.
[17] Berna-Erro, A., M. Izquierdo-Serra, R. V. Sepúlveda, F. Rubio-Moscardo, P.
Doñate-Macián, S. A. Serra, J. Carrillo-Garcia, A. Perálvarez-Marín, F. González-
Nilo, J. M. Fernández-Fernández and M. A. Valverde (2017): Structural
determinants of 5′ ,6′-epoxyeicosatrienoic acid binding to and activation of
TRPV4 channel. Sci Rep 7(1): 10522.
[18] Berridge, M. J., M. D. Bootman and H. L. Roderick (2003): Calcium signalling:
dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4(7): 517-529.
[19] Bezzerides, V. J., I. S. Ramsey, S. Kotecha, A. Greka and D. E. Clapham (2004):
Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol 6: 709.
[20] Birder, L., F. A. Kullmann, H. Lee, S. Barrick, W. de Groat, A. Kanai and M.
Caterina (2007): Activation of urothelial transient receptor potential vanilloid 4 by
4alpha-phorbol 12,13-didecanoate contributes to altered bladder reflexes in the rat.
J Pharmacol Exp Ther 323(1): 227-235.
[21] Bootman, M. D. (2012): Calcium Signaling. Cold Spring Harb Perspect Biol 4(7).
[22] Brewster, J. L., T. de Valoir, N. D. Dwyer, E. Winter and M. C. Gustin (1993): An
osmosensing signal transduction pathway in yeast. Science 259(5102): 1760-1763.
[23] Brohawn, S. G., E. B. Campbell and R. MacKinnon (2014): Physical mechanism
for gating and mechanosensitivity of the human TRAAK K+ channel. Nature
516(7529): 126-130.
[24] Butenko, O., D. Dzamba, J. Benesova, P. Honsa, V. Benfenati, V. Rusnakova, S.
Ferroni and M. Anderova (2012): The Increased Activity of TRPV4 Channel in the

References
102
Astrocytes of the Adult Rat Hippocampus after Cerebral Hypoxia/Ischemia. PLoS
One 7(6): e39959.
[25] Cardoso, C. M. and L. De Meis (1993): Modulation by fatty acids of Ca2+ fluxes in
sarcoplasmic-reticulum vesicles. Biochem J 296 ( Pt 1): 49-52.
[26] Carrasco Loza, R., G. Villamizar Rodríguez and N. Medel Fernández (2015):
Ventilator-Induced Lung Injury (VILI) in Acute Respiratory Distress Syndrome
(ARDS): Volutrauma and Molecular Effects. Open Respir Med J 9: 112-119.
[27] Carreau, A., B. E. Hafny-Rahbi, A. Matejuk, C. Grillon and C. Kieda (2011): Why
is the partial oxygen pressure of human tissues a crucial parameter? Small
molecules and hypoxia. J Cell Mol Med 15(6): 1239-1253.
[28] Cheng, W., C. Sun and J. Zheng (2010): Heteromerization of TRP channel
subunits: extending functional diversity. Protein Cell 1(9): 802-810.
[29] Christensen, A. P. and D. P. Corey (2007): TRP channels in mechanosensation:
direct or indirect activation?. Nat Rev Neurosci 8: 510.
[30] Clapham, D. E. (2003): TRP channels as cellular sensors. Nature 426: 517.
[31] Cuajungco, M. P., C. Grimm, K. Oshima, D. D'hoedt, B. Nilius, A. R.
Mensenkamp, R. J. M. Bindels, M. Plomann and S. Heller (2006): PACSINs bind
to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of
TRPV4. J Biol Chem 281(27): 18753-18762.
[32] Curry, F. R. (2005): Microvascular solute and water transport. Microcirculation
12(1): 17-31.
[33] D'Hoedt, D., G. Owsianik, J. Prenen, M. P. Cuajungco, C. Grimm, S. Heller, T.
Voets and B. Nilius (2008): Stimulus-specific modulation of the cation channel
TRPV4 by PACSIN 3. J Biol Chem 283(10): 6272-6280.
[34] Darby, W. G., M. S. Grace, S. Baratchi and P. McIntyre (2016): Modulation of
TRPV4 by diverse mechanisms. Int J Biochem Cell Biol 78: 217-228.
[35] De Petrocellis, L., P. Orlando, A. S. Moriello, G. Aviello, C. Stott, A. A. Izzo and
V. Di Marzo (2012): Cannabinoid actions at TRPV channels: effects on TRPV3
and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta
Physiol (Oxf) 204(2): 255-266.
[36] Deinum, J., M. Wallin and P. W. Jensen (1985): The binding of Ruthenium red to
tubulin. Biochim Biophys Acta 838(2): 197-205.
[37] Delany, N.S., M. Hurle, P. Facer, T. Alnadaf, C. Plumpton, I. Kinghorn, C. G. See,
M. Costigan, P. Anand, C. J. Woolf, D. Crowther, P. Sanseau , S. N. Tate (2001):
Identification and characterization of a novel human vanilloid receptor-like protein,
VRL-2. Physiol Genomics 4(3): 165-174.

References
103
[38] Dos Santos, C. C. and A. S. Slutsky (2000): Invited Review: Mechanisms of
ventilator-induced lung injury: a perspective. J Appl Physiol 89(4): 1645.
[39] Dreyfuss, D. and G. Saumon (1993): Role of tidal volume, FRC, and end-
inspiratory volume in the development of pulmonary edema following mechanical
ventilation. Am Rev Respir Dis 148(5): 1194-1203.
[40] Dreyfuss, D. and G. Saumon (1998): Ventilator-induced Lung Injury. Am J Respir
Crit Care Med 157(1): 294-323.
[41] Du, J., X. Ma, B. Shen, Y. Huang, L. Birnbaumer and X. Yao (2014): TRPV4,
TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel.
FASEB J 28(11): 4677-4685.
[42] Earley, S., T. J. Heppner, M. T. Nelson and J. E. Brayden (2005): TRPV4 forms a
novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ
Res 97(12): 1270-1279.
[43] Esteban, A., N. D. Ferguson, M. O. Meade, F. Frutos-Vivar, C. Apezteguia, L.
Brochard, K. Raymondos, N. Nin, J. Hurtado, V. Tomicic, M. Gonzalez, J.
Elizalde, P. Nightingale, F. Abroug, P. Pelosi, Y. Arabi, R. Moreno, M. Jibaja, G.
D'Empaire, F. Sandi, D. Matamis, A. M. Montanez and A. Anzueto (2008):
Evolution of mechanical ventilation in response to clinical research. Am J Respir
Crit Care Med 177(2): 170-177.
[44] Everaerts, W., X. Zhen, D. Ghosh, J. Vriens, T. Gevaert, J. P. Gilbert, N. J.
Hayward, C. R. McNamara, F. Xue, M. M. Moran, T. Strassmaier, E. Uykal, G.
Owsianik, R. Vennekens, D. De Ridder, B. Nilius, C. M. Fanger and T. Voets
(2010): Inhibition of the cation channel TRPV4 improves bladder function in mice
and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci U S A
107(44): 19084-19089.
[45] Eyal, F. G., C. R. Hamm and J. C. Parker (2007): Reduction in alveolar
macrophages attenuates acute ventilator induced lung injury in rats. Intensive Care
Med 33(7): 1212-1218.
[46] Fan, H. C., X. Zhang and P. A. McNaughton (2009): Activation of the TRPV4 ion
channel is enhanced by phosphorylation. J Biol Chem 284(41): 27884-27891.
[47] Ferguson, N. D., E. Fan, L. Camporota, M. Antonelli, A. Anzueto, R. Beale, L.
Brochard, R. Brower, A. Esteban, L. Gattinoni, A. Rhodes, A. S. Slutsky, J. L.
Vincent, G. D. Rubenfeld, B. T. Thompson and V. M. Ranieri (2012): The Berlin
definition of ARDS: an expanded rationale, justification, and supplementary
material. Intensive Care Med 38(10): 1573-1582.
[48] Fernandes, J., I. M. Lorenzo, Y. N. Andrade, A. Garcia-Elias, S. A. Serra, J. M.
Fernandez-Fernandez and M. A. Valverde (2008): IP3 sensitizes TRPV4 channel to
the mechano- and osmotransducing messenger 5'-6'-epoxyeicosatrienoic acid. J
Cell Biol 181(1): 143-155.

References
104
[49] Fernandez-Fernandez, J. M., Y. N. Andrade, M. Arniges, J. Fernandes, C. Plata, F.
Rubio-Moscardo, E. Vazquez and M. A. Valverde (2008): Functional coupling of
TRPV4 cationic channel and large conductance, calcium-dependent potassium
channel in human bronchial epithelial cell lines. Pflugers Arch 457(1): 149-159.
[50] Fois, G., O. Wittekindt, X. Zheng, E. T. Felder, P. Miklavc, M. Frick, P. Dietl and
E. Felder (2012): An ultra fast detection method reveals strain-induced Ca(2+)
entry via TRPV2 in alveolar type II cells. Biomech Model Mechanobiol 11(7): 959-
971.
[51] Frank, J. A. and M. A. Matthay (2002): Science review: Mechanisms of ventilator-
induced injury. Crit Care 7(3): 233.
[52] Frank, J. A., C. M. Wray, D. F. McAuley, R. Schwendener and M. A. Matthay
(2006): Alveolar macrophages contribute to alveolar barrier dysfunction in
ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 291(6): L1191-
1198.
[53] Galizia, L., A. Pizzoni, J. Fernandez, V. Rivarola, C. Capurro and P. Ford (2012):
Functional interaction between AQP2 and TRPV4 in renal cells. J Cell Biochem
113(2): 580-589.
[54] Gao, X., L. Wu and R. G. O'Neil (2003): Temperature-modulated diversity of
TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives
through protein kinase C-dependent and -independent pathways. J Biol Chem
278(29): 27129-27137.
[55] Garcia-Elias, A., I. M. Lorenzo, R. Vicente and M. A. Valverde (2008): IP3
receptor binds to and sensitizes TRPV4 channel to osmotic stimuli via a
calmodulin-binding site. J Biol Chem 283(46): 31284-31288.
[56] Garcia-Elias, A., S. Mrkonjić, C. Jung, C. Pardo-Pastor, R. Vicente and M. A.
Valverde (2014): The TRPV4 Channel. In: Nilius B., Flockerzi V. (eds):
Mammalian Transient Receptor Potential (TRP) Cation Channels. Handbook of
Experimental Pharmacology, vol 222. Springer Berlin Heidelberg: 293-319.
[57] Garcia-Elias, A., S. Mrkonjic, C. Pardo-Pastor, H. Inada, U. A. Hellmich, F. Rubio-
Moscardo, C. Plata, R. Gaudet, R. Vicente and M. A. Valverde (2013):
Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4
cytosolic tails enables channel activation by physiological stimuli. Proc Natl Acad
Sci U S A 110(23): 9553-9558.
[58] García-Sanz, N., A. Fernández-Carvajal, C. Morenilla-Palao, R. Planells-Cases, E.
Fajardo-Sánchez, G. Fernández-Ballester and A. Ferrer-Montiel (2004):
Identification of a Tetramerization Domain in the C Terminus of the Vanilloid
Receptor. J Neurosci 24(23): 5307-5314.
[59] Garcia-Sanz, N., P. Valente, A. Gomis, A. Fernandez-Carvajal, G. Fernandez-
Ballester, F. Viana, C. Belmonte and A. Ferrer-Montiel (2007): A role of the

References
105
transient receptor potential domain of vanilloid receptor I in channel gating. J
Neurosci 27(43): 11641-11650.
[60] Geppetti, P. and M. Trevisani (2004): Activation and sensitisation of the vanilloid
receptor: role in gastrointestinal inflammation and function. Br J Pharmacol
141(8): 1313-1320.
[61] Gerstmair, A., G. Fois, S. Innerbichler, P. Dietl and E. Felder (2009): A device for
simultaneous live cell imaging during uni-axial mechanical strain or compression. J
Appl Physiol 107(2): 613.
[62] Gevaert, T., J. Vriens, A. Segal, W. Everaerts, T. Roskams, K. Talavera, G.
Owsianik, W. Liedtke, D. Daelemans, I. Dewachter, F. Van Leuven, T. Voets, D.
De Ridder and B. Nilius (2007): Deletion of the transient receptor potential cation
channel TRPV4 impairs murine bladder voiding. J Clin Invest 117(11): 3453-3462.
[63] Ghassemifar, R., C. M. Lai and P. E. Rakoczy (2006): VEGF differentially
regulates transcription and translation of ZO-1alpha+ and ZO-1alpha- and mediates
trans-epithelial resistance in cultured endothelial and epithelial cells. Cell Tissue
Res 323(1): 117-125.
[64] Girard, B. M., L. Merrill, S. Malley and M. A. Vizzard (2013): Increased TRPV4
Expression in Urinary Bladder and Lumbosacral Dorsal Root Ganglia in Mice with
Chronic Overexpression of NGF in Urothelium. J Mol Neurosci 51(2): 602-614.
[65] Gluckman, E., H. A. Broxmeyer, A. D. Auerbach, H. S. Friedman, G. W. Douglas,
A. Devergie, H. Esperou, D. Thierry, G. Socie, P. Lehn and et al. (1989):
Hematopoietic reconstitution in a patient with Fanconi's anemia by means of
umbilical-cord blood from an HLA-identical sibling. N Engl J Med 321(17): 1174-
1178.
[66] Goldenberg, N. M., K. Ravindran and W. M. Kuebler (2015): TRPV4:
physiological role and therapeutic potential in respiratory diseases. Naunyn
Schmiedebergs Arch Pharmacol 388(4): 421-436.
[67] Groneberg, D. A., A. Niimi, Q. T. Dinh, B. Cosio, M. Hew, A. Fischer and K. F.
Chung (2004): Increased Expression of Transient Receptor Potential Vanilloid-1 in
Airway Nerves of Chronic Cough. Am J Respir Crit Care Med 170(12): 1276-1280.
[68] Guler, A. D., H. Lee, T. Iida, I. Shimizu, M. Tominaga and M. Caterina (2002):
Heat-evoked activation of the ion channel, TRPV4. J Neurosci 22(15): 6408-6414.
[69] Halbertsma, F., M. Vaneker, G. Scheffer and J. Van der Hoeven (2005): Cytokines
and biotrauma in ventilator-induced lung injury: a critical review of the literature.
Neth J Med 63(10): 382-392.
[70] Hamanaka, K., M. Y. Jian, M. I. Townsley, J. A. King, W. Liedtke, D. S. Weber, F.
G. Eyal, M. M. Clapp and J. C. Parker (2010): TRPV4 channels augment
macrophage activation and ventilator-induced lung injury. Am J Physiol Lung Cell
Mol Physiol 299(3): L353-362.

References
106
[71] Hamanaka, K., M. Y. Jian, D. S. Weber, D. F. Alvarez, M. I. Townsley, A. B. Al-
Mehdi, J. A. King, W. Liedtke and J. C. Parker (2007): TRPV4 initiates the acute
calcium-dependent permeability increase during ventilator-induced lung injury in
isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol 293(4): L923-932.
[72] Hegeman, M. A., M. P. Hennus, P. M. Cobelens, A. Kavelaars, N. J. Jansen, M. J.
Schultz, A. J. van Vught and C. J. Heijnen (2013): Dexamethasone attenuates
VEGF expression and inflammation but not barrier dysfunction in a murine model
of ventilator-induced lung injury. PLoS One 8(2): e57374.
[73] Hellmich, U. A. and R. Gaudet (2014): Structural Biology of TRP Channels. In:
Nilius B., Flockerzi V. (eds) Mammalian Transient Receptor Potential (TRP)
Cation Channels. Handbook of Experimental Pharmacology, vol II. Cham, Springer
International Publishing: 963-990.
[74] Henry, C. O., E. Dalloneau, M. T. Perez-Berezo, C. Plata, Y. Wu, A. Guillon, E.
Morello, R. F. Aimar, M. Potier-Cartereau, F. Esnard, C. Coraux, C. Bornchen, R.
Kiefmann, C. Vandier, L. Touqui, M. A. Valverde, N. Cenac and M. Si-Tahar
(2016): In vitro and in vivo evidence for an inflammatory role of the calcium
channel TRPV4 in lung epithelium: Potential involvement in cystic fibrosis. Am J
Physiol Lung Cell Mol Physiol 311(3): L664-675.
[75] Herridge, M. S. (2011): Recovery and long-term outcome in acute respiratory
distress syndrome. Crit Care Clin 27(3): 685-704.
[76] Hoenderop, J. G. J., T. Voets, S. Hoefs, F. Weidema, J. Prenen, B. Nilius and R. J.
M. Bindels (2003): Homo‐ and heterotetrameric architecture of the epithelial Ca2+
channels TRPV5 and TRPV6. EMBO J 22(4): 776-785.
[77] Hoffmann, E. K., I. H. Lambert and S. F. Pedersen (2009): Physiology of Cell
Volume Regulation in Vertebrates. Physiol Rev 89(1): 193-277.
[78] Huh, D. (2015): A human breathing lung-on-a-chip. Ann Am Thorac Soc 12 Suppl
1: S42-44.
[79] Iudin, A., P.K. Korir, J. Salavert-Torres, G.J. Kleywegt & A. Patwardhan (2016):
EMPIAR: A public archive for raw electron microscopy image data. Nature
Methods volume 13, pages 387–388. https://dx.doi.org/10.1038/nmeth.3806
(16.08.2018).
[80] Jia, Y., X. Wang, L. Varty, C. A. Rizzo, R. Yang, C. C. Correll, P. T. Phelps, R. W.
Egan and J. A. Hey (2004): Functional TRPV4 channels are expressed in human
airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 287(2): L272-
278.
[81] Jian, M.-Y., J. A. King, A.-B. Al-Mehdi, W. Liedtke and M. I. Townsley (2008):
High Vascular Pressure–Induced Lung Injury Requires P450 Epoxygenase–
Dependent Activation of TRPV4. Am J Respir Cell Mol Biol 38(4): 386-392.

References
107
[82] Jo, A. O., D. A. Ryskamp, T. T. T. Phuong, A. S. Verkman, O. Yarishkin, N.
MacAulay and D. Križaj (2015): TRPV4 and AQP4 Channels Synergistically
Regulate Cell Volume and Calcium Homeostasis in Retinal Müller Glia. J Neurosci
35(39): 13525-13537.
[83] Jung, C., C. Fandos, I. M. Lorenzo, C. Plata, J. Fernandes, G. G. Gené, E. Vázquez
and M. A. Valverde (2009): The progesterone receptor regulates the expression of
TRPV4 channel. Pflugers Arch 459(1): 105.
[84] Jurek, S. C., M. Hirano-Kobayashi, H. Chiang, D. S. Kohane and B. D. Matthews
(2014): Prevention of ventilator-induced lung edema by inhalation of nanoparticles
releasing ruthenium red. Am J Respir Cell Mol Biol 50(6): 1107-1117.
[85] Kass, G. E. and S. Orrenius (1999): Calcium signaling and cytotoxicity. Environ
Health Perspect 107(Suppl 1): 25-35.
[86] Klausen, T. K., A. Pagani, A. Minassi, A. Ech-Chahad, J. Prenen, G. Owsianik, E.
K. Hoffmann, S. F. Pedersen, G. Appendino and B. Nilius (2009): Modulation of
the transient receptor potential vanilloid channel TRPV4 by 4alpha-phorbol esters:
a structure-activity study. J Med Chem 52(9): 2933-2939.
[87] Kottgen, M., B. Buchholz, M. A. Garcia-Gonzalez, F. Kotsis, X. Fu, M. Doerken,
C. Boehlke, D. Steffl, R. Tauber, T. Wegierski, R. Nitschke, M. Suzuki, A.
Kramer-Zucker, G. G. Germino, T. Watnick, J. Prenen, B. Nilius, E. W. Kuehn and
G. Walz (2008): TRPP2 and TRPV4 form a polymodal sensory channel complex. J
Cell Biol 182(3): 437-447.
[88] Kung, C. (2005): A possible unifying principle for mechanosensation. Nature
436(7051): 647-654.
[89] Lee, E. J., S. H. Shin, S. Hyun, J. Chun and S. S. Kang (2011): Mutation of a
putative S-nitrosylation site of TRPV4 protein facilitates the channel activates.
Animal Cells Syst (Seoul) 15(2): 95-106.
[90] Liao, M., E. Cao, D. Julius and Y. Cheng (2013): Structure of the TRPV1 ion
channel determined by electron cryo-microscopy. Nature 504: 107.
[91] Liedtke, W. (2005): TRPV4 plays an evolutionary conserved role in the
transduction of osmotic and mechanical stimuli in live animals. J Physiol 567(Pt 1):
53-58.
[92] Liedtke, W., Y. Choe, M. A. Martí-Renom, A. M. Bell, C. S. Denis, AndrejŠali, A.
J. Hudspeth, J. M. Friedman and S. Heller (2000): Vanilloid Receptor–Related
Osmotically Activated Channel (VR-OAC), a Candidate Vertebrate Osmoreceptor.
Cell 103(3): 525-535.
[93] Liedtke, W. and J. M. Friedman (2003): Abnormal osmotic regulation in trpv4-/-
mice. Proc Natl Acad Sci U S A 100(23): 13698-13703.

References
108
[94] Lionetti, V., F. A. Recchia and V. M. Ranieri (2005): Overview of ventilator-
induced lung injury mechanisms. Curr Opin Crit Care 11(1): 82-86.
[95] Liu, X., B. C. Bandyopadhyay, T. Nakamoto, B. Singh, W. Liedtke, J. E. Melvin
and I. Ambudkar (2006): A role for AQP5 in activation of TRPV4 by hypotonicity:
concerted involvement of AQP5 and TRPV4 in regulation of cell volume recovery.
J Biol Chem 281(22): 15485-15495.
[96] Loot, A. E., R. Popp, B. Fisslthaler, J. Vriens, B. Nilius and I. Fleming (2008):
Role of cytochrome P450-dependent transient receptor potential V4 activation in
flow-induced vasodilatation. Cardiovasc Res 80(3): 445-452.
[97] Lorenzo, I. M., W. Liedtke, M. J. Sanderson and M. A. Valverde (2008): TRPV4
channel participates in receptor-operated calcium entry and ciliary beat frequency
regulation in mouse airway epithelial cells. Proc Natl Acad Sci U S A 105(34):
12611-12616.
[98] Loukin, S., X. Zhou, Z. Su, Y. Saimi and C. Kung (2010): Wild-type and
brachyolmia-causing mutant TRPV4 channels respond directly to stretch force. J
Biol Chem 285(35): 27176-27181.
[99] Luo, P.-L., Y.-J. Wang, Y.-Y. Yang and J.-J. Yang (2018): Hypoxia-induced
hyperpermeability of rat glomerular endothelial cells involves HIF-2α mediated
changes in the expression of occludin and ZO-1. Braz J Med Biol Res 51(7): e6201.
[100] Ma, X., K. T. Cheng, C. O. Wong, R. G. O'Neil, L. Birnbaumer, I. S. Ambudkar
and X. Yao (2011): Heteromeric TRPV4-C1 channels contribute to store-operated
Ca(2+) entry in vascular endothelial cells. Cell Calcium 50(6): 502-509.
[101] Ma, X., J. Du, P. Zhang, J. Deng, J. Liu, F. F.-Y. Lam, R. A. Li, Y. Huang, J. Jin
and X. Yao (2013): Functional role of TRPV4-KCa2.3 signaling in vascular
endothelial cells in normal and streptozotocin-induced diabetic rats. Hypertension
62(1):134-9.
[102] Ma, X., D. He, X. Ru, Y. Chen, Y. Cai, I. C. Bruce, Q. Xia, X. Yao and J. Jin
(2012): Apigenin, a plant-derived flavone, activates transient receptor potential
vanilloid 4 cation channel. Br J Pharmacol 166(1): 349-358.
[103] Mang, S., A. Braun, N. Pairet and D. J. Lamb (2018): Importance of the IL-1 Axis
in Haemophilus influenzae–stimulated M1 Macrophages Driving Transepithelial
Signaling. Am J Respir Cell Mol Biol 58(3): 412-415.
[104] Martin, T. R. (1999): Lung cytokines and ARDS: Roger S. Mitchell Lecture. Chest
116(1 Suppl): 2s-8s.
[105] Matthay, M. A., L. B. Ware and G. A. Zimmerman (2012): The acute respiratory
distress syndrome. J Clin Invest 122(8): 2731-2740.

References
109
[106] Matthews, B. D., C. K. Thodeti, J. D. Tytell, A. Mammoto, D. R. Overby and D. E.
Ingber (2010): Ultra-rapid activation of TRPV4 ion channels by mechanical forces
applied to cell surface beta1 integrins. Integr Biol (Camb) 2(9): 435-442.
[107] Meduri, G. U., D. Annane, G. P. Chrousos, P. E. Marik and S. E. Sinclair (2009):
Activation and regulation of systemic inflammation in ARDS: rationale for
prolonged glucocorticoid therapy. Chest 136(6): 1631-1643.
[108] Mehta, D. and A. B. Malik (2006): Signaling mechanisms regulating endothelial
permeability. Physiol Rev 86(1): 279-367.
[109] Mendoza, S. A., J. Fang, D. D. Gutterman, D. A. Wilcox, A. H. Bubolz, R. Li, M.
Suzuki and D. X. Zhang (2010): TRPV4-mediated endothelial Ca2+ influx and
vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298(2):
H466-476.
[110] Meng, F., I. Mambetsariev, Y. Tian, Y. Beckham, A. Meliton, A. Leff, M. L.
Gardel, M. J. Allen, K. G. Birukov and A. A. Birukova (2015): Attenuation of
lipopolysaccharide-induced lung vascular stiffening by lipoxin reduces lung
inflammation. Am J Respir Cell Mol Biol 52(2): 152-161.
[111] Michalick, L., L. Erfinanda, U. Weichelt, M. van der Giet, W. Liedtke and W. M.
Kuebler (2017): Transient Receptor Potential Vanilloid 4 and Serum
Glucocorticoid-regulated Kinase 1 Are Critical Mediators of Lung Injury in
Overventilated Mice In Vivo. Anesthesiology 126(2): 300-311.
[112] Michalick, L., M. Mertens, W. Liedtke and W. M. Kuebler (2013): Transient
receptor potential cation channel vanilloid (TRPV) 4 in ventilator-induced lung
injury (VILI). FASEB J 27(1_supplement): 914.912-914.912.
[113] Minke, B. (1977): Drosophila mutant with a transducer defect. Biophys Struct Mech
3(1): 59-64.
[114] Misharin, A. V., L. Morales-Nebreda, G. M. Mutlu, G. R. Budinger and H. Perlman
(2013): Flow cytometric analysis of macrophages and dendritic cell subsets in the
mouse lung. Am J Respir Cell Mol Biol 49(4): 503-510.
[115] Mizuno, A., N. Matsumoto, M. Imai and M. Suzuki (2003): Impaired osmotic
sensation in mice lacking TRPV4. Am J Physiol Cell Physiol 285(1): C96-101.
[116] Mochizuki, T., T. Sokabe, I. Araki, K. Fujishita, K. Shibasaki, K. Uchida, K.
Naruse, S. Koizumi, M. Takeda and M. Tominaga (2009): The TRPV4 cation
channel mediates stretch-evoked Ca2+ influx and ATP release in primary urothelial
cell cultures. J Biol Chem 284(32): 21257-21264.
[117] Montell, C. (2001): Physiology, Phylogeny, and Functions of the TRP Superfamily
of Cation Channels. Sci STKE 2001(90): re1-re1.
[118] Montell, C. (2005): The TRP Superfamily of Cation Channels. Sci STKE
2005(272): re3-re3.

References
110
[119] Montell, C., K. Jones, E. Hafen and G. Rubin (1985): Rescue of the Drosophila
phototransduction mutation trp by germline transformation. Science 230(4729):
1040-1043.
[120] Montgomery, A. B., M. A. Stager, C. J. Carrico and L. D. Hudson (1985): Causes
of mortality in patients with the adult respiratory distress syndrome. Am Rev Respir
Dis 132(3): 485-489.
[121] Moran, M. M. (2018): TRP Channels as Potential Drug Targets. Annu Rev
Pharmacol Toxicol 58(1): 309-330.
[122] Mullin, J. M., N. Agostino, E. Rendon-Huerta and J. J. Thornton (2005): Keynote
review: Epithelial and endothelial barriers in human disease. Drug Discov Today
10(6): 395-408.
[123] Namiki, A., E. Brogi, M. Kearney, E. A. Kim, T. Wu, T. Couffinhal, L. Varticovski
and J. M. Isner (1995): Hypoxia induces vascular endothelial growth factor in
cultured human endothelial cells. J Biol Chem 270(52): 31189-31195.
[124] Narita, K., S. Sasamoto, S. Koizumi, S. Okazaki, H. Nakamura, T. Inoue and S.
Takeda (2015): TRPV4 regulates the integrity of the blood-cerebrospinal fluid
barrier and modulates transepithelial protein transport. FASEB J 29(6): 2247-2259.
[125] Nayak, P. S., Y. Wang, T. Najrana, L. M. Priolo, M. Rios, S. K. Shaw and J.
Sanchez-Esteban (2015): Mechanotransduction via TRPV4 regulates inflammation
and differentiation in fetal mouse distal lung epithelial cells. Respir Res 16(1): 60.
[126] Nilius, B., G. Owsianik, T. Voets and J. A. Peters (2007): Transient Receptor
Potential Cation Channels in Disease. Physiol Rev 87(1): 165-217.
[127] Nilius, B., J. Prenen, U. Wissenbach, M. Bödding and G. Droogmans (2001):
Differential activation of the volume-sensitive cation channel TRP12 (OTRPC4)
and volume-regulated anion currents in HEK-293 cells. Pflugers Arch 443(2): 227-
233.
[128] Nilius, B., T. Voets and J. Peters (2005): TRP Channels in Disease. Sci STKE
2005(295): re8-re8.
[129] Nilius, B., J. Vriens, J. Prenen, G. Droogmans and T. Voets (2004): TRPV4
calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol
286(2): C195-205.
[130] Nuckton, T. J., J. A. Alonso, R. H. Kallet, B. M. Daniel, J. F. Pittet, M. D. Eisner
and M. A. Matthay (2002): Pulmonary dead-space fraction as a risk factor for death
in the acute respiratory distress syndrome. N Engl J Med 346(17): 1281-1286.
[131] Oancea, E., J. T. Wolfe and D. E. Clapham (2006): Functional TRPM7 Channels
Accumulate at the Plasma Membrane in Response to Fluid Flow. Circ Res 98(2):
245-253.

References
111
[132] Olivan-Viguera, A., A. L. Garcia-Otin, J. Lozano-Gerona, E. Abarca-Lachen, A. J.
Garcia-Malinis, K. L. Hamilton, Y. Gilaberte, E. Pueyo and R. Kohler (2018):
Pharmacological activation of TRPV4 produces immediate cell damage and
induction of apoptosis in human melanoma cells and HaCaT keratinocytes. PLoS
One 13(1): e0190307.
[133] Owsianik, G., K. Talavera, T. Voets and B. Nilius (2006): PERMEATION AND
SELECTIVITY OF TRP CHANNELS. Annu Rev Physiol 68(1): 685-717.
[134] Pairet, N., S. Mang, G. Fois, M. Keck, M. Kühnbach, J. Gindele, M. Frick, P. Dietl
and D. J. Lamb (2018): TRPV4 inhibition attenuates stretch-induced inflammatory
cellular responses and lung barrier dysfunction during mechanical ventilation. PLoS
One 13(4): e0196055.
[135] Parker, J. C., C. L. Ivey and J. A. Tucker (1998): Gadolinium prevents high airway
pressure-induced permeability increases in isolated rat lungs. J Appl Physiol 84(4):
1113-1118.
[136] Paulsen, C. E., J.-P. Armache, Y. Gao, Y. Cheng and D. Julius (2015): Structure of
the TRPA1 ion channel suggests regulatory mechanisms. Nature 520: 511.
[137] Pedersen, S. F. and B. Nilius (2007): Transient receptor potential channels in
mechanosensing and cell volume regulation. Methods Enzymol 428: 183-207.
[138] Pedersen, S. F., G. Owsianik and B. Nilius (2005): TRP channels: An overview.
Cell Calcium 38(3): 233-252.
[139] Perlman, C. E., D. J. Lederer and J. Bhattacharya (2011): Micromechanics of
alveolar edema. Am J Respir Cell Mol Biol 44(1): 34-39.
[140] Phelps, C. B., R. R. Wang, S. S. Choo and R. Gaudet (2010): Differential
regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding
site on the ankyrin repeat domain. J Biol Chem 285(1): 731-740.
[141] Phua, J., J. R. Badia, N. K. Adhikari, J. O. Friedrich, R. A. Fowler, J. M. Singh, D.
C. Scales, D. R. Stather, A. Li, A. Jones, D. J. Gattas, D. Hallett, G. Tomlinson, T.
E. Stewart and N. D. Ferguson (2009): Has mortality from acute respiratory distress
syndrome decreased over time?: A systematic review. Am J Respir Crit Care Med
179(3): 220-227.
[142] Protti, A., E. Votta and L. Gattinoni (2014): Which is the most important strain in
the pathogenesis of ventilator-induced lung injury: dynamic or static? Curr Opin
Crit Care 20(1): 33-38.
[143] Pugin, J. (2003): Molecular mechanisms of lung cell activation induced by cyclic
stretch. Crit Care Med 31(4): S200-S206.

References
112
[144] Pugin, J., I. Dunn, P. Jolliet, D. Tassaux, J. L. Magnenat, L. P. Nicod and J. C.
Chevrolet (1998): Activation of human macrophages by mechanical ventilation in
vitro. Am J Physiol 275(6 Pt 1): L1040-1050.
[145] Ramsey, I. S., M. Delling and D. E. Clapham (2006): AN INTRODUCTION TO
TRP CHANNELS. Annu Rev Physiol 68(1): 619-647.
[146] Ranieri, V. M., G. D. Rubenfeld, B. T. Thompson, N. D. Ferguson, E. Caldwell, E.
Fan, L. Camporota and A. S. Slutsky (2012): Acute respiratory distress syndrome:
the Berlin Definition. Jama 307(23): 2526-2533.
[147] Ranieri, V. M., P. M. Suter, C. Tortorella, R. De Tullio, J. M. Dayer, A. Brienza, F.
Bruno and A. S. Slutsky (1999): Effect of mechanical ventilation on inflammatory
mediators in patients with acute respiratory distress syndrome: a randomized
controlled trial. Jama 282(1): 54-61.
[148] Rezoagli, E., R. Fumagalli and G. Bellani (2017): Definition and epidemiology of
acute respiratory distress syndrome. Ann Transl Med 5(14): 282.
[149] Royall, J. A., R. L. Berkow, J. S. Beckman, M. K. Cunningham, S. Matalon and B.
A. Freeman (1989): Tumor necrosis factor and interleukin 1 alpha increase vascular
endothelial permeability. Am J Physiol 257(6): L399.
[150] Rubenfeld, G. D., E. Caldwell, E. Peabody, J. Weaver, D. P. Martin, M. Neff, E. J.
Stern and L. D. Hudson (2005): Incidence and outcomes of acute lung injury. N
Engl J Med 353(16): 1685-1693.
[151] Saliez, J., C. Bouzin, G. Rath, P. Ghisdal, F. Desjardins, R. Rezzani, L. F. Rodella,
J. Vriens, B. Nilius, O. Feron, J. L. Balligand and C. Dessy (2008): Role of
caveolar compartmentation in endothelium-derived hyperpolarizing factor-
mediated relaxation: Ca2+ signals and gap junction function are regulated by
caveolin in endothelial cells. Circulation 117(8): 1065-1074.
[152] Sasaki, T., M. Naka, F. Nakamura and T. Tanaka (1992): Ruthenium red inhibits
the binding of calcium to calmodulin required for enzyme activation. J Biol Chem
267(30): 21518-21523.
[153] Scheraga, R. G., S. Abraham, K. A. Niese, B. D. Southern, L. M. Grove, R. D.
Hite, C. McDonald, T. A. Hamilton and M. A. Olman (2016): TRPV4
Mechanosensitive Ion Channel Regulates Lipopolysaccharide-Stimulated
Macrophage Phagocytosis. J Immunol 196(1): 428-436.
[154] Scheraga, R. G., B. D. Southern, L. M. Grove and M. A. Olman (2017): The Role
of Transient Receptor Potential Vanilloid 4 in Pulmonary Inflammatory Diseases.
Front Immunol 8: 503.
[155] Schneider, C., S. P. Nobs, M. Kurrer, H. Rehrauer, C. Thiele and M. Kopf (2014):
Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical
for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol
15(11): 1026-1037.

References
113
[156] Schwingshackl, A. (2016): The role of stretch-activated ion channels in acute
respiratory distress syndrome: finally a new target? Am J Physiol Lung Cell Mol
Physiol 311(3): L639-L652.
[157] Segond von Banchet, G., M. K. Boettger, C. König, Y. Iwakura, R. Bräuer and H.-
G. Schaible (2013): Neuronal IL-17 receptor upregulates TRPV4 but not TRPV1
receptors in DRG neurons and mediates mechanical but not thermal hyperalgesia.
Mol Cell Neurosci 52: 152-160.
[158] Seminario-Vidal, L., S. F. Okada, J. I. Sesma, S. M. Kreda, C. A. van Heusden, Y.
Zhu, L. C. Jones, W. K. O'Neal, S. Penuela, D. W. Laird, R. C. Boucher and E. R.
Lazarowski (2011): Rho signaling regulates pannexin 1-mediated ATP release from
airway epithelia. J Biol Chem 286(30): 26277-26286.
[159] Shigematsu, H., T. Sokabe, R. Danev, M. Tominaga and K. Nagayama (2010): A
3.5-nm structure of rat TRPV4 cation channel revealed by Zernike phase-contrast
cryoelectron microscopy. J Biol Chem 285(15): 11210-11218.
[160] Shin, S. H., E. J. Lee, J. Chun, S. Hyun and S. S. Kang (2015): Phosphorylation on
TRPV4 Serine 824 Regulates Interaction with STIM1. Open Biochem J 9: 24-33.
[161] Shin, S. H., E. J. Lee, S. Hyun, J. Chun, Y. Kim and S. S. Kang (2012):
Phosphorylation on the Ser 824 residue of TRPV4 prefers to bind with F-actin than
with microtubules to expand the cell surface area. Cell Signal 24(3): 641-651.
[162] Shukla, A. K., J. Kim, S. Ahn, K. Xiao, S. K. Shenoy, W. Liedtke and R. J.
Lefkowitz (2010): Arresting a transient receptor potential (TRP) channel: beta-
arrestin 1 mediates ubiquitination and functional down-regulation of TRPV4. J Biol
Chem 285(39): 30115-30125.
[163] Silversides, J. A. and N. D. Ferguson (2013): Clinical review: Acute respiratory
distress syndrome - clinical ventilator management and adjunct therapy. Crit Care
17(2): 225-225.
[164] Slutsky, A. S. (1993): Mechanical Ventilation. Chest 104(6): 1833-1859.
[165] Slutsky, A. S. and Y. Imai (2003): Ventilator-induced lung injury, cytokines, PEEP,
and mortality: implications for practice and for clinical trials. Intensive Care Med
29(8): 1218-1221.
[166] Slutsky, A. S. and L. N. Tremblay (1998): Multiple system organ failure. Is
mechanical ventilation a contributing factor? Am J Respir Crit Care Med 157(6 Pt
1): 1721-1725.
[167] Smith, P. L., K. N. Maloney, R. G. Pothen, J. Clardy and D. E. Clapham (2006):
Bisandrographolide from Andrographis paniculata activates TRPV4 channels. J
Biol Chem 281(40): 29897-29904.

References
114
[168] Sokabe, T., T. Fukumi-Tominaga, S. Yonemura, A. Mizuno and M. Tominaga
(2010): The TRPV4 channel contributes to intercellular junction formation in
keratinocytes. J Biol Chem 285(24): 18749-18758.
[169] Sokabe, T. and M. Tominaga (2010): The TRPV4 cation channel: A molecule
linking skin temperature and barrier function. Commun Integr Biol 3(6): 619-621.
[170] Sonkusare, S. K., A. D. Bonev, J. Ledoux, W. Liedtke, M. I. Kotlikoff, T. J.
Heppner, D. C. Hill-Eubanks and M. T. Nelson (2012): Elementary Ca2+ signals
through endothelial TRPV4 channels regulate vascular function. Science
336(6081): 597-601.
[171] Stewart, A. P., G. D. Smith, R. N. Sandford and J. M. Edwardson (2010): Atomic
force microscopy reveals the alternating subunit arrangement of the TRPP2-TRPV4
heterotetramer. Biophys J 99(3): 790-797.
[172] Strotmann, R., C. Harteneck, K. Nunnenmacher, G. Schultz and T. D. Plant (2000):
OTRPC4, a nonselective cation channel that confers sensitivity to extracellular
osmolarity. Nat Cell Biol 2: 695.
[173] Strotmann, R., G. Schultz and T. D. Plant (2003): Ca2+-dependent potentiation of
the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin
binding site. J Biol Chem 278(29): 26541-26549.
[174] Sutherasan, Y., M. Vargas and P. Pelosi (2014): Protective mechanical ventilation
in the non-injured lung: review and meta-analysis. Crit Care 18(2): 211.
[175] Suzuki, M., A. Hirao and A. Mizuno (2003): Microtubule-associated [corrected]
protein 7 increases the membrane expression of transient receptor potential
vanilloid 4 (TRPV4). J Biol Chem 278(51): 51448-51453.
[176] Suzuki, M., A. Mizuno, K. Kodaira and M. Imai (2003): Impaired pressure
sensation in mice lacking TRPV4. J Biol Chem 278(25): 22664-22668.
[177] Tabeling, C., H. Yu, L. Wang, H. Ranke, N. M. Goldenberg, D. Zabini, E. Noe, A.
Krauszman, B. Gutbier, J. Yin, M. Schaefer, C. Arenz, A. C. Hocke, N. Suttorp, R.
L. Proia, M. Witzenrath and W. M. Kuebler (2015): CFTR and sphingolipids
mediate hypoxic pulmonary vasoconstriction. Proc Natl Acad Sci U S A 112(13):
E1614-1623.
[178] Takahashi, N., D. Kozai, R. Kobayashi, M. Ebert and Y. Mori (2011): Roles of
TRPM2 in oxidative stress. Cell Calcium 50(3): 279-287.
[179] Tanaka, Y., S. Ito, R. Oshino, N. Chen, N. Nishio and K.-i. Isobe (2015): Effects of
growth arrest and DNA damage-inducible protein 34 (GADD34) on inflammation-
induced colon cancer in mice. Br J Cancer 113(4): 669-679.
[180] Tao, F. and L. Kobzik (2002): Lung Macrophage–Epithelial Cell Interactions
Amplify Particle-Mediated Cytokine Release. Am J Respir Cell Mol Biol 26(4):
499-505.

References
115
[181] Thodeti, C. K., B. Matthews, A. Ravi, A. Mammoto, K. Ghosh, A. L. Bracha and
D. E. Ingber (2009): TRPV4 channels mediate cyclic strain-induced endothelial cell
reorientation through integrin-to-integrin signaling. Circ Res 104(9): 1123-1130.
[182] Thompson, B. T., R. C. Chambers and K. D. Liu (2017): Acute Respiratory
Distress Syndrome. N Engl J Med 377(6): 562-572.
[183] Thorneloe, K. S., M. Cheung, W. Bao, H. Alsaid, S. Lenhard, M.-Y. Jian, M.
Costell, K. Maniscalco-Hauk, J. A. Krawiec, A. Olzinski, E. Gordon, I.
Lozinskaya, L. Elefante, P. Qin, D. S. Matasic, C. James, J. Tunstead, B. Donovan,
L. Kallal, A. Waszkiewicz, K. Vaidya, E. A. Davenport, J. Larkin, M. Burgert, L.
N. Casillas, R. W. Marquis, G. Ye, H. S. Eidam, K. B. Goodman, J. R. Toomey, T.
J. Roethke, B. M. Jucker, C. G. Schnackenberg, M. I. Townsley, J. J. Lepore and R.
N. Willette (2012): An Orally Active TRPV4 Channel Blocker Prevents and
Resolves Pulmonary Edema Induced by Heart Failure. Sci Transl Med 4(159):
159ra148.
[184] Thorneloe, K. S., M. Cheung, D. A. Holt and R. N. Willette (2017): PROPERTIES
OF THE TRPV4 AGONIST GSK1016790A AND the TRPV4 ANTAGONIST
GSK2193874. Physiol Rev 97(4): 1231-1232.
[185] Thorneloe, K. S., A. C. Sulpizio, Z. Lin, D. J. Figueroa, A. K. Clouse, G. P.
McCafferty, T. P. Chendrimada, E. S. Lashinger, E. Gordon, L. Evans, B. A.
Misajet, D. J. Demarini, J. H. Nation, L. N. Casillas, R. W. Marquis, B. J. Votta, S.
A. Sheardown, X. Xu, D. P. Brooks, N. J. Laping and T. D. Westfall (2008): N-
((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-
1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide
(GSK1016790A), a novel and potent transient receptor potential vanilloid 4
channel agonist induces urinary bladder contraction and hyperactivity: Part I. J
Pharmacol Exp Ther 326(2): 432-442.
[186] Tiruppathi, C., G. U. Ahmmed, S. M. Vogel and A. B. Malik (2006): Ca2+
signaling, TRP channels, and endothelial permeability. Microcirculation 13(8):
693-708.
[187] Todaka, H., J. Taniguchi, J. Satoh, A. Mizuno and M. Suzuki (2004): Warm
temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an
essential role in thermal hyperalgesia. J Biol Chem 279(34): 35133-35138.
[188] Tremblay, L. N. and A. S. Slutsky (1998): Ventilator-induced injury: from
barotrauma to biotrauma. Proc Assoc Am Physicians 110(6): 482-488.
[189] Ueda, T., M. Shikano, T. Kamiya, T. Joh and S. Ugawa (2011): The TRPV4
channel is a novel regulator of intracellular Ca2+ in human esophageal epithelial
cells. Am J Physiol Gastrointest Liver Physiol 301(1): G138-147.
[190] Uhlén, M., L. Fagerberg, B. M. Hallström, C. Lindskog, P. Oksvold, A.
Mardinoglu, Å. Sivertsson, C. Kampf, E. Sjöstedt, A. Asplund, I. Olsson, K.
Edlund, E. Lundberg, S. Navani, C. A.-K. Szigyarto, J. Odeberg, D. Djureinovic, J.
O. Takanen, S. Hober, T. Alm, P.-H. Edqvist, H. Berling, H. Tegel, J. Mulder, J.

References
116
Rockberg, P. Nilsson, J. M. Schwenk, M. Hamsten, K. von Feilitzen, M. Forsberg,
L. Persson, F. Johansson, M. Zwahlen, G. von Heijne, J. Nielsen and F. Pontén
(2015): Tissue-based map of the human proteome. Science 347(6220). Human
Protein Atlas available from www.proteinatlas.org:
https://www.proteinatlas.org/ENSG00000111199-TRPV4/tissue (16.08.2018).
[191] Umbrello, M., P. Formenti, L. Bolgiaghi and D. Chiumello (2017): Current
Concepts of ARDS: A Narrative Review. Int J Mol Sci 18(1): 64.
[192] Venkatachalam, K. and C. Montell (2007): TRP Channels. Annu Rev Biochem
76(1): 387-417.
[193] Villalta, P. C., P. Rocic and M. I. Townsley (2014): Role of MMP2 and MMP9 in
TRPV4-induced lung injury. Am J Physiol Lung Cell Mol Physiol 307(8): L652-
L659.
[194] Vincent, F., A. Acevedo, M. T. Nguyen, M. Dourado, J. DeFalco, A. Gustafson, P.
Spiro, D. E. Emerling, M. G. Kelly and M. A. Duncton (2009): Identification and
characterization of novel TRPV4 modulators. Biochem Biophys Res Commun
389(3): 490-494.
[195] Vlahakis, N. E. and R. D. Hubmayr (2003): Response of alveolar cells to
mechanical stress. Curr Opin Crit Care 9(1): 2-8.
[196] Vlahakis, N. E., M. A. Schroeder, A. H. Limper and R. D. Hubmayr (1999): Stretch
induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol 277(1 Pt
1): L167-173.
[197] Voets, T., J. Prenen, J. Vriens, H. Watanabe, A. Janssens, U. Wissenbach, M.
Bödding, G. Droogmans and B. Nilius (2002): Molecular Determinants of
Permeation through the Cation Channel TRPV4. J Biol Chem 277(37): 33704-
33710.
[198] Volynets, V., A. Reichold, G. Bardos, A. Rings, A. Bleich and S. C. Bischoff
(2016): Assessment of the Intestinal Barrier with Five Different Permeability Tests
in Healthy C57BL/6J and BALB/cJ Mice. Dig Dis Sci 61(3): 737-746.
[199] Vriens, J., G. Owsianik, A. Janssens, T. Voets and B. Nilius (2007): Determinants
of 4α-Phorbol Sensitivity in Transmembrane Domains 3 and 4 of the Cation
Channel TRPV4. J Biol Chem 282(17): 12796-12803.
[200] Vriens, J., H. Watanabe, A. Janssens, G. Droogmans, T. Voets and B. Nilius
(2004): Cell swelling, heat, and chemical agonists use distinct pathways for the
activation of the cation channel TRPV4. Proc Natl Acad Sci U S A 101(1): 396-
401.
[201] Wang, Y., X. Fu, S. Gaiser, M. Kottgen, A. Kramer-Zucker, G. Walz and T.
Wegierski (2007): OS-9 regulates the transit and polyubiquitination of TRPV4 in
the endoplasmic reticulum. J Biol Chem 282(50): 36561-36570.

References
117
[202] Watanabe, H., J. B. Davis, D. Smart, J. C. Jerman, G. D. Smith, P. Hayes, J. Vriens,
W. Cairns, U. Wissenbach, J. Prenen, V. Flockerzi, G. Droogmans, C. D. Benham
and B. Nilius (2002): Activation of TRPV4 channels (hVRL-2/mTRP12) by
phorbol derivatives. J Biol Chem 277(16): 13569-13577.
[203] Watanabe, H., J. Vriens, J. Prenen, G. Droogmans, T. Voets and B. Nilius (2003):
Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4
channels. Nature 424(6947): 434-438.
[204] Watanabe, H., J. Vriens, S. H. Suh, C. D. Benham, G. Droogmans and B. Nilius
(2002): Heat-evoked activation of TRPV4 channels in a HEK293 cell expression
system and in native mouse aorta endothelial cells. J Biol Chem 277(49): 47044-
47051.
[205] Webb, H. H. and D. F. Tierney (1974): Experimental pulmonary edema due to
intermittent positive pressure ventilation with high inflation pressures. Protection
by positive end-expiratory pressure. Am Rev Respir Dis 110(5): 556-565.
[206] Wegierski, T., K. Hill, M. Schaefer and G. Walz (2006): The HECT ubiquitin
ligase AIP4 regulates the cell surface expression of select TRP channels. EMBO J
25(24): 5659-5669.
[207] White, J. P. M., M. Cibelli, L. Urban, B. Nilius, J. G. McGeown and I. Nagy
(2016): TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol Rev 96(3):
911.
[208] Willette, R. N., W. Bao, S. Nerurkar, T.-l. Yue, C. P. Doe, G. Stankus, G. H.
Turner, H. Ju, H. Thomas, C. E. Fishman, A. Sulpizio, D. J. Behm, S. Hoffman, Z.
Lin, I. Lozinskaya, L. N. Casillas, M. Lin, R. E. L. Trout, B. J. Votta, K. Thorneloe,
E. S. R. Lashinger, D. J. Figueroa, R. Marquis and X. Xu (2008): Systemic
activation of the transient receptor potential vanilloid subtype 4 channel causes
endothelial failure and circulatory collapse: Part 2. J Pharmacol Exp Ther 326(2):
443-452.
[209] Wilson, M. R., B. V. Patel and M. Takata (2012): Ventilation with ‘clinically-
relevant’ high tidal volumes does not promote stretch-induced injury in the lungs of
healthy mice. Crit Care Med 40(10): 2850-2857.
[210] Wilson, S. R., A. M. Nelson, L. Batia, T. Morita, D. Estandian, D. M. Owens, E. A.
Lumpkin and D. M. Bautista (2013): The ion channel TRPA1 is required for
chronic itch. J Neurosci 33(22): 9283-9294.
[211] Wissenbach, U., M. Bödding, M. Freichel and V. Flockerzi (2000): Trp12, a novel
Trp related protein from kidney. FEBS Lett 485(2-3): 127-134.
[212] Xia, Y., Z. Fu, J. Hu, C. Huang, O. Paudel, S. Cai, W. Liedtke and J. S. K. Sham
(2013): TRPV4 channel contributes to serotonin-induced pulmonary
vasoconstriction and the enhanced vascular reactivity in chronic hypoxic
pulmonary hypertension. Am J Physiol Cell Physiol 305(7): C704-C715.

References
118
[213] Xiaoming, J., A. Malhotra, M. Saeed, R. G. Mark and D. Talmor (2008): Risk
Factors for Acute Respiratory Distress Syndrome in Patients Mechanically
Ventilated for Greater Than 48 Hours. Chest 133(4): 853-861.
[214] Xu, F., E. Satoh and T. Iijima (2003): Protein kinase C-mediated Ca2+ entry in
HEK 293 cells transiently expressing human TRPV4. Br J Pharmacol 140(2): 413-
421.
[215] Yamada, A., O. Sato, M. Watanabe, M. P. Walsh, Y. Ogawa and Y. Imaizumi
(2000): Inhibition of smooth-muscle myosin-light-chain phosphatase by Ruthenium
Red. Biochem J 349 Pt 3: 797-804.
[216] Yamashiro, K., T. Sasano, K. Tojo, I. Namekata, J. Kurokawa, N. Sawada, T.
Suganami, Y. Kamei, H. Tanaka, N. Tajima, K. Utsunomiya, Y. Ogawa and T.
Furukawa (2010): Role of transient receptor potential vanilloid 2 in LPS-induced
cytokine production in macrophages. Biochem Biophys Res Commun 398(2): 284-
289.
[217] Yiangou, Y., P. Facer, N. H. Dyer, C. L. Chan, C. Knowles, N. S. Williams and P.
Anand (2001): Vanilloid receptor 1 immunoreactivity in inflamed human bowel.
Lancet 357(9265): 1338-1339.
[218] Yin, J., J. Hoffmann, S. M. Kaestle, N. Neye, L. Wang, J. Baeurle, W. Liedtke, S.
Wu, H. Kuppe, A. R. Pries and W. M. Kuebler (2008): Negative-feedback loop
attenuates hydrostatic lung edema via a cGMP-dependent regulation of transient
receptor potential vanilloid 4. Circ Res 102(8): 966-974.
[219] Yin, J. and W. M. Kuebler (2010): Mechanotransduction by TRP channels: general
concepts and specific role in the vasculature. Cell Biochem Biophys 56(1): 1-18.
[220] Yin, J., L. Michalick, C. Tang, A. Tabuchi, N. Goldenberg, Q. Dan, K. Awwad, L.
Wang, L. Erfinanda, G. Nouailles, M. Witzenrath, A. Vogelzang, L. Lv, W. L. Lee,
H. Zhang, O. Rotstein, A. Kapus, K. Szaszi, I. Fleming, W. B. Liedtke, H. Kuppe
and W. M. Kuebler (2016): Role of Transient Receptor Potential Vanilloid 4 in
Neutrophil Activation and Acute Lung Injury. Am J Respir Cell Mol Biol 54(3):
370-383.
[221] Zhang, F., H. Yang, Z. Wang, S. Mergler, H. Liu, T. Kawakita, S. D. Tachado, Z.
Pan, J. E. Capó-Aponte, U. Pleyer, H. Koziel, W. W. Y. Kao and P. S. Reinach
(2007): Transient receptor potential vanilloid 1 activation induces inflammatory
cytokine release in corneal epithelium through MAPK signaling. J Cell Physiol
213(3): 730-739.
[222] Zheng, X., N. S. Zinkevich, D. Gebremedhin, K. M. Gauthier, Y. Nishijima, J.
Fang, D. A. Wilcox, W. B. Campbell, D. D. Gutterman and D. X. Zhang (2013):
Arachidonic acid-induced dilation in human coronary arterioles: convergence of
signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry. J Am Heart
Assoc 2(3): e000080.

Acknowledgement
119
Acknowledgement
“We” in this thesis indicates several people made this work possible and contributed to its
outcome.
Firstly, I would like to express my sincere thanks to my supervisor Dr. David Lamb for the
continuous support and guidance during my doctorate and for giving me the opportunity to
conduct my doctoral thesis work at Boehringer Ingelheim Pharma GmbH & Co. KG in
collaboration with the University of Ulm. Thank you for supporting my ideas and
encouraging my research with your positive way of thinking, helping me to focus on the
essential goals and also for reading and making suggestions to this written work.
I extent my thanks to my collaborators and further supervisors Prof. Dr. Paul Dietl and
Prof. Dr. Manfred Frick from the Institute of General Physiology at the University of Ulm
for the welcome during my research visit, for offering me lab space for experiments and
for your guidance and mentorship during my thesis. In this team I would also like to give
special thanks to Dr. Giorgio Fois for his technical advice and sharing knowledge during
my research visit.
Many thanks also goes to whole the members of the department of Immunology and
Respiratory Diseases Research of Boehringer Ingelheim, giving me the opportunity to join
their team, giving me access to the laboratory and research facilities, helping me to deepen
and expand my understanding of biology in health and disease by sharing their knowledge
and making it possible to achieve my PhD in such a great team. In this team I would like to
give special thanks to my fellow lab mates Samuel Mang, Ingrid Christ, Martina Keck,
Tobias Kiechle, Nadine Laufhäger, Melanie Kühnbach and Julia Gindele for technical
advice and help and for being such good companions contributing to an excellent
atmosphere not only at work.
Although they already know, I would like to thank my parents and my brothers for always
supporting me throughout my thesis and my life in general.
Last but not the least, I would like to thank my friends, my rugby and fitness mates for
changing my mind, when I needed to. Finally I want to thank my love Debora, for
everything in the last years.





























