Embed Size (px)
Citation preview

1
TITLE PAGE
Full title: The tyrosine kinase receptor RET interacts in vivo with AIP to alter survivin availability.
Authors’ names and institutions:
Manuela Vargiolu
Unità di Genetica Medica, Policlinico Universitario S. Orsola-Malpighi, Bologna, Italy
Daniela Fusco
Unità di Genetica Medica, Policlinico Universitario S. Orsola-Malpighi, Bologna, Italy
Ivana Kurelac
Unità di Genetica Medica, Policlinico Universitario S. Orsola-Malpighi, Bologna, Italy
Dietmar Dirnberger
Bio3/Bioinformatics and Molecular Genetics (Faculty of Biology), University of Freiburg, Germany.
Ralf Baumeister
Bio3/Bioinformatics and Molecular Genetics (Faculty of Biology),Center for Biochemistry and
Molecular Cell Research (Faculty of Medicine), Center for Systems Biology (ZBSA) and FRIAS,
School of Life Sciences (LIFENET), Albert-Ludwigs-University Freiburg, Schänzlestr. 1, D79104
Freiburg i. Brsg., Germany
Isabella Morra
Department of Histopathology, Ospedale Infantile Regina Margherita, Torino, Italy
Antonio Melcarne
Department of Neurosurgery, A.S.O. CTO-CRF-M.Adelaide, Torino, Italy
Roberto Rimondini
Dept. of Pharmacology, University of Bologna, Via Irnerio 48 Bologna, Italy
J Clin Endocrin Metab. First published ahead of print April 14, 2009 as doi:10.1210/jc.2008-1980
Copyright (C) 2009 by The Endocrine Society

2
Giovanni Romeo
Unità di Genetica Medica, Policlinico Universitario S. Orsola-Malpighi, Bologna, Italy
Elena Bonora
Unità di Genetica Medica, Policlinico Universitario S. Orsola-Malpighi, Bologna, Italy
Abbreviated title (max 40 chrs): AIP interacts with RET
Word count excluding abstract, figure captions, ref: 3599
Name and address of person to whom reprint requests should be addressed: Giovanni Romeo
Any grants or fellowships supporting the writing of the paper: European Specific Targeted
Research Projects project HERMIONE, University of Turin, University of Bologna, the EC 6th
Framework Network of Excellence LIFESPAN and a program of the German Federal Ministry in
Research and Education.
Author Disclosure Summary: M.V., D.F., I.K., D.D., R.B., I.M., A.M., R.R., G.R., E.B. have nothing
to declare. G.R. received grant support (2006-2009) from EU.
Precis: The identification of the AIP-RET complex represents a starting point to study key cellular
processes involved in RET induced apoptosis.

3
ABSTRACT (230 words)
Context: RET is a tyrosine kinase transmembrane receptor expressed in two main alternative isoforms:
RET9 and RET51. RET transduces a positive signal leading to survival, differentiation or migration in
the presence of its ligand GDNF, whilst in its absence a proapoptotic fragment which initiates a
negative signalling for apoptosis is generated. The signal transduction mechanisms leading to apoptosis
are still unclear.
Objective: In order to shed light on the mechanisms of RET induced apoptosis we searched for novel
interactors of RET51.
Design: The “split ubiquitin yeast two hybrid system” was used with RET51 as bait against a human
brain expression library.
Results: We identified AIP, a co-chaperone recently found mutated in pituitary adenoma patients, as a
novel interactor of RET. We showed that RET interacts specifically with AIP both in mammalian cell
lines and in vivo in the pituitary gland, regardless of the presence of pituitary adenoma specific
mutations. AIP and RET genes were sequenced in 28 pituitary adenoma but no relevant mutation were
found. In addition, we identified the pro-apoptotic domain of RET as responsible for the interaction
with AIP. Finally, we demonstrated that the AIP-RET interaction does not require RET kinase activity
or kinase dependent signal transduction and that it prevents the formation of the AIP-survivin complex.
Conclusions: The identification of the AIP-RET complex represents a starting point to study key
cellular processes involved in RET induced apoptosis.

4
INTRODUCTION
The RET proto-oncogene is the tyrosine kinase receptor for the glial cell line-derived neurotrophic
factor (GDNF) family of ligands. Binding occurs in conjunction with glycosylphosphatidyl inositol-
membrane anchored co-receptors designated GDNF-family receptor-α (GFRα)(1). RET is expressed in
two main isoforms: the short and the long isoforms, namely RET9 and RET51, which differs at the C-
terminal tail(2). Gain- and loss-of-function mutations of RET have been associated respectively with
neoplasia (Multiple Endocrine Neoplasia Type 2A–MEN2A-and 2B–MEN2B-and Familial Medullary
Thyroid Carcinoma–FMTC-) and with Hirschsprung disease, a neurodevelopmental disorder.
RET has a dual role according to the presence or absence of GDNF. In the presence of GDNF, RET
promotes survival, growth and migration of cells such as neurons and epithelial renal cells. The survival
signal transduced by RET is required for maturation of several cell lineages of the peripheral nervous
system, kidney morphogenesis, and spermatogenesis(3). Conversely, in the absence of its ligand RET is
able to induce programmed cell death by releasing a proapoptotic fragment which initiates a negative
signal for apoptosis, as shown in Neuro2A cells and in transient transfected HEK293T(4) and GH4C1
cell lines(5). The precise mechanisms of RET induced apoptosis remain nevertheless unclear.
In order to shed light on the proteins involved in the pathway induced by RET, we used a modified
yeast two hybrid method named the“split ubiquitin system”(6). This method led to the identification of
a novel interactor of RET51 among human brain proteins. We here identify a novel binding partner of
RET, the aryl hydrocarbon receptor interacting protein (AIP), also known as ARA9 or XAP2(7, 8), a
tumor-suppressor protein recently found mutated in pituitary adenoma. We show that RET-AIP
interaction is maintained both in cell lines and in the pituitary gland in vivo, regardless of the presence
of pituitary adenoma specific mutations, possibly altering the cellular apoptotic potential.

5
MATERIALS AND METHODS
Split Ubiquitin Yeast Two Hybrid
The bait construct was based on the previously described vector pPCUP1-ubc9-CRU (6), which is a
yeast–E.coli shuttle vector that carries a CRU locus, a copper-dependent promoter (PCUP1) for bait
expression, and a HIS3 marker. The human coding sequence corresponding to RET51 intracellular
domain excluding the transmembrane segment was amplified by PCR from a human whole brain cDNA
preparation (Clontech) using primers RET51-FW(5’-
AATTGTCGACATGCACTGCTACCACAAGTTTGCCC-3’) and RET51-RV(5’-
AGCGGCCGCACTATCAAACGTGTCCATTA-3’). The RET51 PCR product was used to replace the
Ubc9 insert in pPCUP1-ubc9-CRU, resulting in pPCUP1-Fz1-CRU. The human fetal brain NubI—split
ubiquitin cDNA library was kindly provided by GPC Biotech AG. The cDNA library screening and the
selection of positive clones was performed as previously described (6).
Cell cultures and transfections
HEK293, Neuro2A and SH-SY5Y cells were grown at 37°C with 5% CO2 in DMEM (Celbio)
supplemented with 10% fetal bovine serum (Euroclone), 2 mM L-glutamine (Euroclone), 100 units/ml
penicillin and 100 µg/ml streptomycin (Euroclone). Cells were plated on 60 mm Petri dishes and
transfection was performed with Lipofectamine Reagent (Invitrogen) following the manufacturer's
instructions.
cDNA construct preparation
Total RNA extracted from the SH-SY5Y neuroblastoma cell line using Trizol Reagent (Invitrogen)
was retrotranscribed using SuperScript™III Reverse Transcriptase (Invitrogen). The human AIP coding
sequence was amplified from cDNA by PCR using primers EcoRI_AIP-F(5’-
CCCGAATTCGCCGAAGCAAGTCCG-3’) and XhoI_AIP-R (5’-
GGCTCGAGCATGGGAGAAGATCCC-3’) and cloned into pcDNA3.1myc/His expression vector

6
(Invitrogen). The plasmids pJ7Ω-RET51 (RET51-FL) and pJ7Ω-RET9 (RET9-FL) were kindly
provided by P. Mehlen, (Apoptosis Cancer and Development Laboratory, Lyon, France;(4)) Single base
mutations were introduced into wild-type AIP and RET51 clones using the QuikChange site-directed
mutagenesis kit (Stratagene).
The construct containing tag-free AIP was produced by subcloning the AIP coding sequence from
pcDNA3.1myc/His expression vector (Invitrogen) to pcDNA3.1(+) (Invitrogen) using the primers
EcoRI_AIP-F(5’-CCCGAATTCGCCGAAGCAAGTCCG-3’) and XhoI_ext_AIP(5’-
GGGTGACCTCGAGTCAATGGGAGAAGATCCC-3’).
The deleted constructs RET-TK (aa 1-999) and RET-IUXTA (aa 1-725) were generated by PCR
amplification using primers RET_HindIII_F(5’-GGGATATCCCATGGCGAAGGCGACG-3’),
RET_IUXTA_XbaI_R(5’-GGTCTAGAAGAACCAAGTTCTTCCGAGG-3’), RET_TK_XbaI_R(5’-
GGTCTAGAGCCGCAAACACCGGCCTTTTGTCCG-3’) and cloned into pcDNA3.1/V5-His©
TOPO® TA Expression Vector (Invitrogen). RET-PRO (708-1017) was generated using primers
RET_HindIII_F(5’-GGGATATCCCATGGCGAAGGCGACG-3’) and NotI_PRORET_R(5’-
GCGGCCGCGTCCAAGTAGTCTCTCCT-3’) and cloned into pcDNA3.1myc/His expression vector
(Invitrogen).
All plasmid sequences were verified by direct sequencing using Big Dye v3.1 kit according to the
manufacturer’s instruction and loaded onto a 3730 automated sequencer (Applied Biosystems).

7
Immunoprecipitation assays and Western blot analysis
Cells were lysed in 100 µl of IP Buffer 1X (Sigma-Aldrich) containing protease Inhibitors (Roche
Diagnostics). Rat pituitary glands were homogenized in ice-cold IP Buffer 1X containing protease
Inhibitors (Roche Diagnostics). Cell lysates were incubated with 1 µg of primary antibody. After 4 hrs
20 µl of Protein G beads (Sigma-Aldrich) equilibrated in IP buffer were added and incubated overnight.
Bead pellets were washed 3 times with 200 µl of IP Buffer and resuspended in 40 µl of Laemmli buffer
(8% SDS; 20% glycerol; 125mM Tris HCl, pH 6.8; 0.0025% bromophenol blue; 10% 2-
mercaptoethanol). Immunoprecipitates and total cell lysates were separated by SDS-PAGE and
transferred onto nitrocellulose membrane. Immunodetection and immunoprecipitation were performed
using primary antibodies specific for RET (Ret(C-19) sc-167; Ret(C-20) sc-1290; Ret(H-300) sc-
13104), AIP (XAP2(35-2) sc-59730; XAP2(N-20) sc-27445) from Santa Cruz Biotechnology, for
survivin (GTX10584) from GeneTex and for the myc epitope (R950-25) from Invitrogen. Secondary
antibodies were HRP-conjugate anti-mouse or anti-rabbit (Sigma-Aldrich) HRP-conjugate anti-goat
(DAKO) and AP-conjugate anti-mouse (Invitrogen). Development was performed using ECL system
(Millipore).
Patient material and mutation screening
A total of 28 bioptic samples of sporadic pituitary adenoma were obtained from the Department of
Histopathology of Turin and kept anonymous. DNA was extracted from frozen or paraffin-embedded
pathological pituitary tissue using the NucleoSpin Tissue Kit (Mackerey-Nagel).
The whole AIP and RET coding sequences were analyzed for mutations by direct sequencing using Big
Dye v3.1 and 3730 automated sequencer (Applied Biosystems). The PCR conditions are available
under request. In silico analysis was performed using TargetScan 4.2(9), Mireval(10) and miRBase
v5(11) for miRNA binding site predictions and mfold 3.2 (12)for RNA folding analysis.

8
RESULTS
Identification of AIP as a binding partner of the long isoform of RET–In order to better characterise the
signaling pathway downstream of RET51, we performed a split ubiquitin yeast two-hybrid screening
using the cytoplasmic domain of RET51 as a bait against a human fetal brain cDNA library(6). We
employed the split-ubiquitin system based on the R-URA3p reporter which allowed positive selection
for uracil auxotrophy and negative selection in the presence of the otherwise toxic URA3p-specific
antimetabolite 5-fluoroorotic acid (5-FOA).
We used the full length cDNA of human RET51 to generate the pPCUP1-RET51-CRU bait expression
vector in which full-length RET51 cDNA was expressed as a translational fusion protein to URA3.
However, the transformants with pPCUP1-RET51-CRU did not grow on the appropriate medium. Thus,
the cytoplasmatic portion of RET51 was used as bait and we screened for protein interactors of this
fragment using a poly(T)-primed human fetal brain cDNA-library.
After eliminating false-positive interaction candidates, we isolated 10 URA-auxotrophic and 5-FOA
resistant clones. The 10 positive clones were sequenced, and nucleotide sequence database searches
were performed using BLAST (National Center for Biotechnology Information—NCBI). One of these
clones contained in the prey plasmid the cDNA encoding the entire protein of Aryl Hydrocarbon
Receptor Interacting Protein (NM_003977).
AIP interacts both with RET9 and RET51 in mammalian cell lines–In order to validate the AIP-RET51
interaction observed in yeast we performed a co-immunoprecipitation assay in mammalian cell lines.
Full-length RET51 (RET51-FL)(Fig.1) and AIP myc-tagged were expressed in 293 human embryonic
kidney (HEK) cells (Fig.2A). Cells transfected either with a mock or AIP expressing vector and
RET51-FL were lysed and immunoprecipitated with an antibody specific for the long isoform of RET
(Fig.2A). When the total cell lysates and the corresponding immunoprecipitates were analysed by SDS-
PAGE, the presence of a 39 KDa protein (the expected size of myc-tagged AIP) was detected both in
the total cell lysate and in the lysate immunoprecipitated with anti-RET51 antibody (Fig.2A). The 39
KDa protein was not detected when the mock plasmid was transfected (Fig.2A, third lane) thus

9
indicating that anti-myc antibody specifically detected AIP. Also, when lysates obtained from cells
transfected with the mock plasmid were immunoprecipitated with the anti-RET51 antibody, the 39 KDa
band was absent (Fig.2A, fourth lane). This demonstrated that AIP and RET51 co-immuprecipitated in
HEK293, indicating that AIP was able to bind RET51 in human cells.
To understand whether the AIP-RET51 interaction was also present in cells expressing RET
endogenously, the same experiment was performed in two neuroblastoma cell lines, human SH-SY5Y
and murine Neuro2A (Fig.2C,D). Immunoblotting confirmed the expression of AIP as well as RET in
both neuroblastoma cell lines (Fig. 2C). Neuro2A cells transfected either with a mock plasmid or with
the vector expressing myc-tagged AIP were lysated and immunoprecipitated with the antibody specific
for RET51 (Fig. 2D). As expected, when the cell lysates and the corresponding immunoprecipitates
were analysed by immunoblotting with an anti-myc antibody, the presence of AIP was detected both in
the total cell lysate and in the lysate immunoprecipitated with the anti-RET51 antibody (Fig. 2D, first
and second lane), whereas it was not detected when the mock plasmid was transfected (Fig. 2D, third
and fourth lane). In addition, when the cell lysate of non-transfected Neuro2A were immunoprecipitated
with the anti-RET51 antibody, the presence of endogenous AIP was detected (Fig. 2D, fifth and sixth
lane). Indeed, endogenous AIP was able to immunoprecipitate RET demonstrating that the AIP-RET
interaction is not dependent upon transfection of these proteins. The same results were obtained in
SHSY-5Y (data not shown).
To address the specificity of AIP binding for the long isoform of RET, the same co-
immunoprecipitation assay was performed in HEK293, transfected with full-length RET9 (RET9-FL)
(Fig.1) and myc-tagged AIP. Cells transfected either with a mock or AIP-expressing vector and RET9-
FL were lysated and immunoprecipitated with an antibody specific for RET9 (Fig.2B). Also in this
case, the presence of AIP was detected both in the total cell lysate and in the lysate immunoprecipitated
with the anti-RET9 antibody (Fig.2B, first and second lane), whereas it was not detected when the
mock plasmid was transfected (Fig.2B, third and fourth lane). This indicated that AIP was able to bind
to both RET isoforms in a region likely common to RET9 and RET51.

10
AIP interacts with the proapoptotic domain of RET–To determine the domain of RET involved in
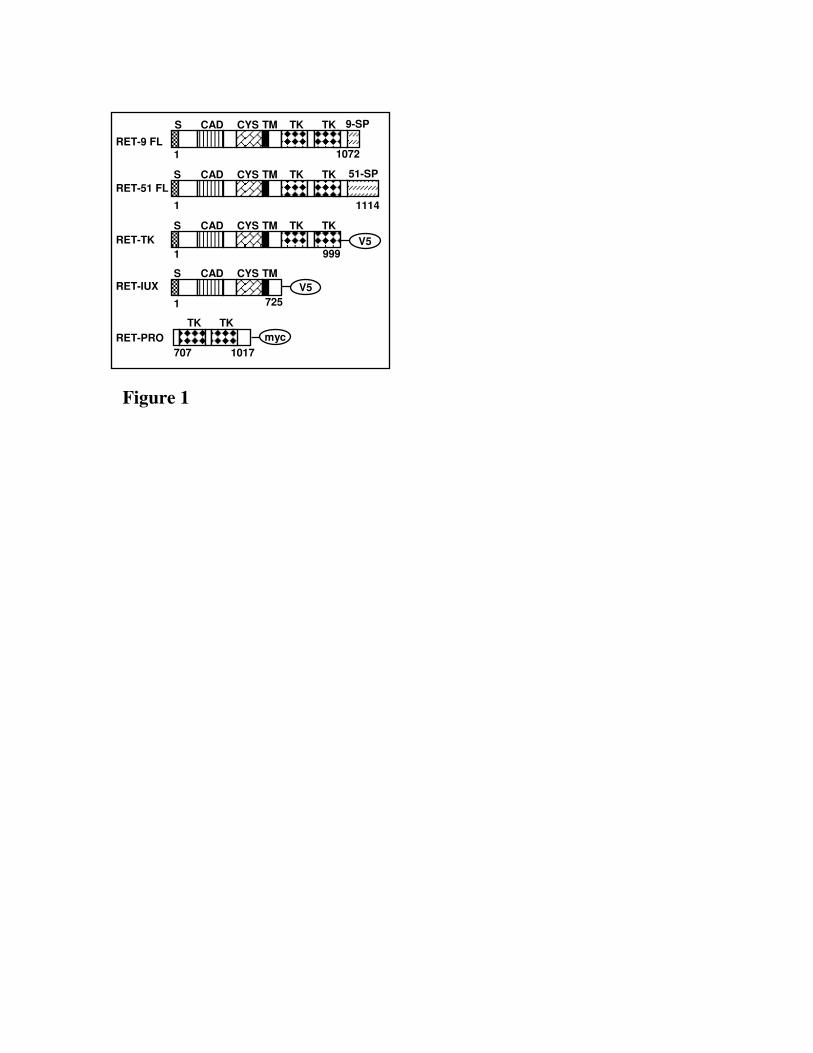
binding to AIP we generated various constructs containing different functional domains of RET. RET-
IUXTA (aa 1-725) included the juxtamembrane domain of RET, known to have a role in regulating the
kinase activity of RET(13), RET-PRO (aa 707-1017) contained the proapoptotic domain of RET tagged
with myc and RET-TK (aa 1-999) encompassed both the juxtamembrane and the tyrosine kinase
domain of RET (Fig.1).
Cells transfected with either a mock or myc-tagged AIP expressing vector and either RET-IUXTA or
RET-TK were lysed and immunoprecipitated with an antibody specific for the N-terminal portion of
RET. AIP was present both in the cell lysate and in the lysate immunoprecipitated with the anti-RET
antibody when RET-TK was expressed, whereas it was not detected in the immunoprecipitates when
the RET-IUXTA plasmid was transfected (Fig.2E). Accordingly, in a reverse experiment using anti-
myc antibody for immunoprecipitation, we could detect RET by immunoblotting only in the
immunoprecipitates of cells expressing RET-TK (Fig.2F).
To further refine the mapping of the AIP-RET interaction, the proapoptotic domain of RET was tested
for its ability to bind AIP. HEK293 transfected with a mock or AIP-expressing vector and either a mock
or RET-PRO myc-tagged vector were lysated and immunoprecipitated with anti-AIP antibody. The
presence of RET-PRO was detected in the immunoprecipitates when AIP was also expressed, whereas
no protein could be revealed in the immunoprecipitates when each of the two mock plasmids were
expressed (Fig.2G). These data demonstrated that the portion of RET interacting with AIP spans from
amino acid 707 to 999, a region common to both isoforms.
AIP and RET interact with each other in vivo in the pituitary gland–The above results indicated that the
AIP-RET interaction was present in cells of different origin. In order to investigate whether this
interaction occurs in vivo, we analysed the rat pituitary, given that RET, GDNF and AIP are expressed
in somatotrophs and AIP is involved in pituitary tumor pathogenesis. To verify the endogenous
interaction between RET and AIP we performed co-immunoprecipitation experiments in lysates
prepared from rat pituitary gland tissue. Analysis of the total lysate by immunoblotting confirmed the

11
expression of both proteins (Fig.3A). Using a specific antibody against AIP, both AIP and RET could
be detected in the immunoprecipitates (Fig. 3B). No unspecific binding of the proteins occurred in a
control experiment using nonspecific IgG (Fig. 3, third lane). These results clearly proved the presence
of a AIP-RET complex in the pituitary gland.
RET phosphorylation status does not affect the AIP-RET interaction–In our experiments we transiently
over-expressed RET, a condition known to result in high levels of RET autophosphorylation in the
absence of ligand stimulation(14, 15). To investigate whether the interaction of RET with AIP relied on
a kinase-dependent signal transduction, we checked the ability of RET to retain the interaction with AIP
when its kinase activity was abolished. For this purpose cells expressing AIP and a mutated form of
RET containing a point mutation of the lysine in position 758 that renders RET kinase inactive (RET51-
DK, K758R)(14) were studied. The presence of AIP was detected in the immunoprecipitates of cells
expressing either RET51-FL or RET51-DK, suggesting that the AIP-RET interaction is not dependent
on RET phosphorylation status (Fig.4A).
RET51 prevents AIP binding to the anti-apoptotic protein survivin – In a previous study transient
transfection of the full-length RET in human embryonic kidney cells was shown to induce apoptosis
(4). AIP also has a role in regulating the cellular apoptotic threshold, directly binding and regulating the
stability of survivin, a member of the IAP (inhibitor of apoptosis) family(16). To investigate the
connection between these two processes, we tested the ability of transiently transfected RET in HEK to
affect the formation of the AIP-survivin complex. Cells over-expressing RET51 were lysed and the
ability of AIP to retain the interaction with survivin was assessed by immunoprecipitating with anti-
survivin antibody (Fig.4B). The presence of AIP in the immunoprecipitates was detected only in
absence of RET. In contrast, the presence of RET abolished the ability of AIP to co-immunoprecipitate
with survivin, demonstrating that RET was sufficient to prevent the formation of the AIP-survivin
complex (Fig.4B).
Pathogenic mutations of RET and AIP do not impair the AIP-RET interaction–Germline mutations of
AIP were recently described in sporadic and familial pituitary adenoma (17). The six more frequent

12
missense mutations found in pituitary adenoma patients were selected, namely R16H, K241E, R271W,
E293G, A299V, R304Q, and introduced in our construct containing AIP (Fig.5A). The resulting
constructs were expressed in HEK293 together with either RET51-FL or RET-9. Their ability to modify
the AIP-RET interaction was tested by immunoprecipitating the total cell lysate with an anti-RET
antibody and immonoblotting for AIP. The presence of all the six mutated AIP proteins was detected in
the lysate immunoprecipitated with an antibody specific for either RET51 (Fig.5B) or RET9 (data not
shown). These data clearly showed that none of these six missense mutations impairs the interaction of
AIP with RET51 or with RET9.To further prove that none of these AIP mutations impaired the
interaction with RET, the ability of the proapoptotic fragment of RET to bind the mutated forms of AIP
was tested. Cells expressing myc-tagged RET-PRO and each of the six mutated forms of AIP were
lysed and immunoprecipitated with the antibody specific for AIP. As expected, immunoblotting for the
myc tag of RET-PRO showed the presence of RET-PRO when immunoprecipitating the lysates of cells
transfected with each mutated form of AIP (Fig.5C). We therefore conclude that AIP pituitary adenoma
predisposing mutations do not impair AIP-RET interaction.
To further address the involvement of the AIP-RET interaction in tumorigenesis, we tested the effect of
RET germline missense mutations associated with medullary thyroid carcinoma and MEN2, a dominant
inherited cancer syndrome that affects neuroendocrine organs(18-20). As AIP is also involved in the
tumorigenesis of endocrine tumours, we assessed whether these mutations have a role in impairing AIP-
RET interaction.
Eight missense mutations predisposing to medullary thyroid carcinoma and mapping in the region of
RET that interact with AIP, namely E768D, V778I, L790F, V804M, Y806C, S891A, M918T and
S922F (Fig.6A), were introduced into the RET51-FL construct. The resulting constructs were expressed
in HEK cells with the full-length wild type AIP. Their ability to modify the AIP-RET interaction was
tested by immunoprecipitating the cell lysate with an anti-RET51 antibody and immonoblotting for
AIP. The presence of AIP proteins was detected in the lysate immunoprecipitated with an antibody

13
specific for RET51 recognizing each of the eight mutated forms of RET tested (Fig.6B). These data
clearly showed that none of these eight RET mutations impaired the interaction of RET51 with AIP.
Mutation screening in Growth Hormone (GH)-secreting pituitary adenoma patients–Germline
mutations of AIP were recently identified in somatotropinoma(17, 21, 22). To test the hypothesis that
AIP-RET interaction may have a role in the tumorigenesis of pituitary adenoma and that the loss of
RET protein may predispose to this neoplasia, we decided to examine whether RET mutations occurred
in pituitary adenoma patients. The complete coding sequence of AIP and RET were sequenced in the
pituitary tissues from 28 somatotropinoma patients. Furthermore, the enhancer-like sequence in intron
1of RET containing the non-coding RET variant rs2435357 strongly associated with a loss-of-function
of RET was also evaluated. One already reported mutation in AIP (R304Q) and two unreported variants
in the 3’UTR of RET (c. 1590*G>A; c.1643*G>C) were detected in two different patients. The two
variants influenced neither miRNA binding sites nor mRNA folding, as predicted by in silico analysis.
Furthermore the two alleles of the non-coding regulatory variant of RET were homogeneously
distributed among the patients.

14
DISCUSSION
In this study we identified the co-chaperone AIP as a novel RET-binding partner and showed the
presence of an AIP-RET complex in cell lines of different origin (human embryonic kidney and
neuroblastoma) and in the pituitary gland in vivo. Familial and sporadic pituitary adenoma was recently
associated to germline heterozygous mutations and large genomic deletions in the AIP gene(17, 21-26).
It is tempting to speculate that the interaction between RET and AIP might have a role in pituitary
adenoma tumorigenesis. It has been shown that GDNF, GFRalpha1, and RET mRNA and protein are
expressed both in the human anterior pituitary gland and in somatotropinoma(27, 28) and that the
proto-oncogene RET has a role in regulating apoptosis in somatotrophs both in vitro and in vivo(5).
Indeed, loss of RET function results in somatotroph hyperplasia, which is interpreted to be caused by
the absence of RET-derived pro-death signals(5). The interaction between RET and AIP, shown for the
first time in this work, suggests a possible synergistic activity of RET and AIP in regulating
somatotroph proliferation and tumorigenesis, thus providing a link between pituitary adenoma and RET
dependence receptor activity.
Conversely, we showed that neither RET-associated mutations found in MEN2 nor AIP-associated
mutations found in pituitary adenoma impair the ability of RET to bind AIP. Consequently the
tumorigenic process likely does not result in an impairment of the AIP-RET complex, but rather the
presence of this complex leads to downstream events. So far, little is known about RET signal
transduction leading to apoptosis. AIP may contribute to this process, given its ability to interact with
survivin, a member of the IAP (inhibitor of apoptosis) family (16). We showed that the expression of
RET prevents AIP from binding survivin. Since the binding of AIP to survivin has been demonstrated
to protect survivin from degradation and that the disruption of this complex enhances cell death(16),
RET induced apoptosis might be achieved by preventing the formation of the AIP-survivin complex.
Further work on the functional role of the AIP-RET complex should shed light on the key cellular
processes involved in RET-induced apoptosis. Interestingly, the proapoptotic domain of RET is
responsible for the interaction with AIP. In addition, AIP-RET interaction does not require RET kinase

15
activity or kinase dependent signal transduction such as RET induced cell death(4), further supporting a
role of AIP in modulating RET-induced apoptosis.
Accordingly, RET may be considered a good candidate for pituitary adenoma predisposition. We
therefore screened pituitary adenoma patients for the presence of intragenic RET mutations but did not
find any significant alteration in this gene. This failure does not necessarily exclude RET as a candidate
gene for this form of neoplasia. Actually, despite the sensitivity of the sequencing analysis system we
are aware that it does not detect changes such as large genomic rearrangements or changes in promoter
methylation, which are known mechanisms for tumor suppressor gene inactivation(29, 30). In addition,
our group of patients consists exclusively of sporadic GH-secreting pituitary adenomas with an average
age of 45 years. Selection of patients with a familial history for pituitary adenoma and with an earlier
onset of the disease could potentially increase the chance of finding RET alterations.
Our results provide the basis for unravelling the biological processes underlying AIP-associated
pituitary adenoma predisposition and shedding light on the mechanisms of RET induced cell death.
Further functional studies are needed to completely elucidate the biological mechanisms involving the
RET-AIP complex.

16
Acknowledgments
We thank all the families who have participated in the study and the clinicians who collaborated in
this study. We thank Dr. Giuseppe Gasparre for critical reading and thoughtful discussion of the
manuscript, Dr Lucia Fiammetta Pennisi for her help in some of the experiments and Prof. Kerry
Rhoden for proofreading the manuscript. This work was supported by grant LSHC-CT-2006-037530
“HERMIONE” from the EU to G.R.. M.V. was supported by a University of Turin PhD fellowship.
D.F. was supported by a post-doctoral fellowship from grant HERMIONE.
R.B. was supported by the EC 6th Framework Network of Excellence LIFESPAN (LSHGCT-2007-
036894, and by the Deutsche Forschungsgemeinschaft (CRC746, CRC780), FRISYS (# 03139219,
the Freiburg Initiative in Systems Biology) and FRIAS LIFENET.

17
REFERENCES
1. Trupp M, Raynoschek C, Belluardo N, Ibanez CF 1998 Multiple GPI-anchored receptors
control GDNF-dependent and independent activation of the c-Ret receptor tyrosine kinase.
Mol Cell Neurosci 11:47-63
2. Tahira T, Ishizaka Y, Itoh F, Sugimura T, Nagao M 1990 Characterization of ret proto-
oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma
cell line. Oncogene 5:97-102
3. Arighi E, Borrello MG, Sariola H 2005 RET tyrosine kinase signaling in development and
cancer. Cytokine Growth Factor Rev 16:441-467
4. Bordeaux MC, Forcet C, Granger L, Corset V, Bidaud C, Billaud M, Bredesen DE,
Edery P, Mehlen P 2000 The RET proto-oncogene induces apoptosis: a novel mechanism
for Hirschsprung disease. Embo J 19:4056-4063
5. Canibano C, Rodriguez NL, Saez C, Tovar S, Garcia-Lavandeira M, Borrello MG,
Vidal A, Costantini F, Japon M, Dieguez C, Alvarez CV 2007 The dependence receptor
Ret induces apoptosis in somatotrophs through a Pit-1/p53 pathway, preventing tumor
growth. Embo J 26:2015-2028
6. Dirnberger D, Messerschmid M, Baumeister R 2008 An optimized split-ubiquitin cDNA-
library screening system to identify novel interactors of the human Frizzled 1 receptor.
Nucleic Acids Res 36:e37
7. Ma Q, Whitlock JP, Jr. 1996 The aromatic hydrocarbon receptor modulates the Hepa 1c1c7
cell cycle and differentiated state independently of dioxin. Mol Cell Biol 16:2144-2150
8. Carver LA, Bradfield CA 1997 Ligand-dependent interaction of the aryl hydrocarbon
receptor with a novel immunophilin homolog in vivo. J Biol Chem 272:11452-11456
9. Lewis BP, Burge CB, Bartel DP 2005 Conserved seed pairing, often flanked by adenosines,
indicates that thousands of human genes are microRNA targets. Cell 120:15-20

18
10. Ritchie W, Theodule FX, Gautheret D 2008 Mireval: a web tool for simple microRNA
prediction in genome sequences. Bioinformatics 24:1394-1396
11. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS 2003 MicroRNA targets in
Drosophila. Genome Biol 5:R1
12. Zuker M 2003 Mfold web server for nucleic acid folding and hybridization prediction.
Nucleic Acids Res 31:3406-3415
13. Melillo RM, Cirafici AM, De Falco V, Bellantoni M, Chiappetta G, Fusco A,
Carlomagno F, Picascia A, Tramontano D, Tallini G, Santoro M 2004 The oncogenic
activity of RET point mutants for follicular thyroid cells may account for the occurrence of
papillary thyroid carcinoma in patients affected by familial medullary thyroid carcinoma. Am
J Pathol 165:511-521
14. Tsui-Pierchala BA, Ahrens RC, Crowder RJ, Milbrandt J, Johnson EM, Jr. 2002 The
long and short isoforms of Ret function as independent signaling complexes. J Biol Chem
277:34618-34625
15. Coulpier M AJ, Ibanez CF 2002 Coordinated activation of autophosphorylation sites in the
RET receptor tyrosine kinase: importance of tyrosine 1062 for GDNF mediated neuronal
differentiation and survival. J Biol Chem 277:1991-1999
16. Kang BH, Altieri DC 2006 Regulation of survivin stability by the aryl hydrocarbon receptor-
interacting protein. J Biol Chem 281:24721-24727
17. Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, Tuppurainen
K, Ebeling TM, Salmela PI, Paschke R, Gundogdu S, De Menis E, Makinen MJ,
Launonen V, Karhu A, Aaltonen LA 2006 Pituitary adenoma predisposition caused by
germline mutations in the AIP gene. Science 312:1228-1230
18. Eng C 1996 Seminars in medicine of the Beth Israel Hospital, Boston. The RET proto-
oncogene in multiple endocrine neoplasia type 2 and Hirschsprung's disease. N Engl J Med
335:943-951

19
19. Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE,
Moore JK, Papi L. 1993 Germ-line mutations of the RET proto-oncogene in multiple
endocrine neoplasia type 2A. Nature 363:458-460
20. Mulligan LM, Ponder BA 1995 Genetic basis of endocrine disease: multiple endocrine
neoplasia type 2. J Clin Endocrinol Metab 80:1989-1995
21. Georgitsi M, De Menis E, Cannavo S, Makinen MJ, Tuppurainen K, Pauletto P, Curto
L, Weil RJ, Paschke R, Zielinski G, Wasik A, Lubinski J, Vahteristo P, Karhu A,
Aaltonen LA 2008 Aryl hydrocarbon receptor interacting protein (AIP) gene mutation
analysis in children and adolescents with sporadic pituitary adenomas. Clin Endocrinol (Oxf)
69:621-627
22. Georgitsi M, Heliovaara E, Paschke R, Kumar AV, Tischkowitz M, Vierimaa O,
Salmela P, Sane T, De Menis E, Cannavo S, Gundogdu S, Lucassen A, Izatt L, Aylwin S,
Bano G, Hodgson S, Koch CA, Karhu A, Aaltonen LA 2008 Large genomic deletions in
AIP in pituitary adenoma predisposition. J Clin Endocrinol Metab 93:4146-4151
23. Yu R, Bonert V, Saporta I, Raffel LJ, Melmed S 2006 Aryl hydrocarbon receptor
interacting protein variants in sporadic pituitary adenomas. J Clin Endocrinol Metab 91:5126-
5129
24. Toledo RA, Lourenco DM, Jr., Liberman B, Cunha-Neto MB, Cavalcanti MG, Moyses
CB, Toledo SP, Dahia PL 2007 Germline mutation in the aryl hydrocarbon receptor
interacting protein gene in familial somatotropinoma. J Clin Endocrinol Metab 92:1934-1937
25. Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, Chahal HS,
Igreja SC, Jordan S, Rowe J, Stolbrink M, Christian HC, Wray J, Bishop-Bailey D,
Berney DM, Wass JA, Popovic V, Ribeiro-Oliveira A, Jr., Gadelha MR, Monson JP,
Akker SA, Davis JR, Clayton RN, Yoshimoto K, Iwata T, Matsuno A, Eguchi K, Musat
M, Flanagan D, Peters G, Bolger GB, Chapple JP, Frohman LA, Grossman AB,
Korbonits M 2008 The role of the aryl hydrocarbon receptor-interacting protein gene in
familial and sporadic pituitary adenomas. J Clin Endocrinol Metab 93:2390-2401

20
26. Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA,
Murat A, Emy P, Gimenez-Roqueplo AP, Tamburrano G, Raverot G, Barlier A, De
Herder W, Penfornis A, Ciccarelli E, Estour B, Lecomte P, Gatta B, Chabre O, Sabate
MI, Bertagna X, Garcia Basavilbaso N, Stalldecker G, Colao A, Ferolla P, Wemeau JL,
Caron P, Sadoul JL, Oneto A, Archambeaud F, Calender A, Sinilnikova O, Montanana
CF, Cavagnini F, Hana V, Solano A, Delettieres D, Luccio-Camelo DC, Basso A,
Rohmer V, Brue T, Bours V, Teh BT, Beckers A 2007 Aryl hydrocarbon receptor-
interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73
families. J Clin Endocrinol Metab 92:1891-1896
27. Urbano AG, Suarez-Penaranda JM, Dieguez C, Alvarez CV 2000 GDNF and RET-gene
expression in anterior pituitary-cell types. Endocrinology 141:1893-1896
28. Japon MA, Urbano AG, Saez C, Segura DI, Cerro AL, Dieguez C, Alvarez CV 2002
Glial-derived neurotropic factor and RET gene expression in normal human anterior pituitary
cell types and in pituitary tumors. J Clin Endocrinol Metab 87:1879-1884
29. Xing M 2007 Gene methylation in thyroid tumorigenesis. Endocrinology 148:948-953
30. Segev DL, Umbricht C, Zeiger MA 2003 Molecular pathogenesis of thyroid cancer. Surg
Oncol 12:69-90

21
FIGURE LEGEND
Fig. 1. Schematic structure of RET and RET deleted constructs used for mapping AIP interaction. S,
signal sequence; CAD, cadherin-related motif; CYS, cysteine-rich region; TM, transmembrane
domain; TK, tyrosine kinase domain; 51-SP, specific region of RET long isoform; 9-SP, specific
region of RET short isoform.
Fig. 2. A) Whole cell lysates prepared from HEK293 cells co-transfected with pcDNA3.1-AIP-
myc/His (lane 3) or pcDNA3.1myc/His (lane 1) and RET51-FL. B) Immunoprecipitation was carried
out with anti-RET9 antibody using lysates prepared from HEK 293 cells co-transfected with
pcDNA3.1-AIP-myc/His (lane 1) or pcDNA3.1myc/His (lane 3) and RET9-FL. C) Whole cell lysates
prepared from Neuro2A and SH-SY5Y and immunoblotted for anti-RET antibody and anti-AIP
antibody. D) Whole cell lysates prepared from Neuro2A cells co-transfected with pcDNA3.1-AIP-
myc/His (lane 1) or pcDNA3.1myc/His (lane 3) and RET51-FL and from Neuro2A not transfected
(lane 5). D) HEK293 cells co-transfected with pcDNA3.1-AIP-myc/His and RET51-FL (lane 1) or
RET51-D707N (lane 3) . E) HEK293 cells co-transfected with pcDNA3.1-AIP (lane 3 and 5) or
pcDNA3.1(+) (lane 1) and RET-PRO (lane 1 and 5) or pcDNA3.1myc/His (lane 3). F) HEK293 cells
co-transfected with pcDNA3.1-AIP-myc/His (lane 1 and 5) or pcDNA3.1myc/His (lane 3 and 7) and
RET-IUX (lane 5 and 7) or RET-TK (lane 1 and 3). G) HEK293 cells co-transfected with pcDNA3.1-
AIP-myc/His (lane 1 and 3) or pcDNA3.1myc/His (lane 5 and 7) and RET-IUX (lane 1 and 5) or
RET-TK (lane 3 and 7). All the western blot were performed with anti-myc antibody except for (F)
and (C) where immunoblotting was carried out with anti- RET antibody.
Fig. 3. A) Protein extracted from rat pituitary gland were immunoblotted for either anti-AIP or anti-
RET antibody. B) Whole lysate prepared from rat pituitary gland were immunoprecipitated with
different antibodies. A fifth of the lysate immunoprecipitated with anti-AIP antibody was loaded in
Lane1. Whole lysate immunoprecipitated with anti-RET antibody and for a not related

22
immunoglobulin were loaded in Lane2 and Lane 3, respectively. Immunoprecipitates were
immunoblotted with anti-AIP antibody.
Fig. 4. A) Immunoprecipitation was carried out with anti-RET51 antibody using lysates prepared
from HEK 293 cells co-transfected with pcDNA3.1-AIP-myc/His (lane 1 and 3) or
pcDNA3.1myc/His (lane 5) and RET51-FL (lane 1 and 5) or RET51-DK (lane 3). B)
Immunoprecipitation was carried out with anti-survivin antibody using lysates prepared from HEK
293 cells transfected with pcDNA3.1-AIP-myc/His alone (lane 1) or together with RET51-FL (lane
3). C) Hypothetical mechanism of RET induced apoptosis. When RET is not expressed, AIP directly
bind survivin, preventing its degradation thus protecting cell from apoptosis. The presence of RET
abolished the ability of AIP to interact with survivin, likely leading to survivin degradation and
consequent cell death.
Fig. 5. A) Schematic figure of AIP. FK506-binding protein (FKBP)–homology region and
tetratricopeptide repeats (TPRs) are shown. The regions necessary for AHR and HSP90 interaction
are shown with black lines and the exon boundaries of the AIP gene are marked with dashed lines.
AIP mutations tested for their ability to impair the interaction with RET are indicated by black
triangles. B) HEK293 cells co-transfected with pcDNA3.1-AIP-myc/His (lane 1 and 5) or
pcDNA3.1myc/His (lane 3) or the mutated constructs of AIP (lane 7-18) and RET51-FL (lane 1 and
3) or the empty vector (Ω) (lane 5). C) HEK293 cells co-transfected with pcDNA3.1-AIP (lane 1 and
5) or pcDNA3.1(+) (lane 3) or the mutated constructs of AIP (lane 7-18) and RET-PRO (lane 1 and 3)
or pcDNA3.1myc/His (lane 5). Mutations tested: lane 7: R16H; lane 9: K241E; lane 11: R271W; lane
13: E293G; lane 15: A299V; lane 18: R304Q. All immunoblottings were performed with anti-myc
antibody.
Fig. 6. A) Schematic structure of RET. The position of the RET missense mutations mapping to the
AIP interacting region are indicated by arrows. B) HEK293 cells co-transfected with pcDNA3.1-AIP-

23
myc/His (lane 1-18) and RET51-FL (lane 1) or the mutated constructs of RET (lane 3-18). Mutations
tested: lane 3: E768D; lane 5: V778I; lane 7: L790F; lane 9: V804M; lane 11: Y806C; lane 13:
S891A; lane 15: M918T; lane 18: S922F. All immunoblottings were performed with anti-myc
antibody.
707
TK TK
1017
myc
1
S CAD CYS TKTM TK
999V5
1
S CAD CYS TM
725
V5
S CAD CYS TKTM TK 51-SP
1 1114
S CAD CYS TKTM TK 9-SP
1 1072RET-9 FL
RET-51 FL
RET-TK
RET-IUX
RET-PRO
Figure 1
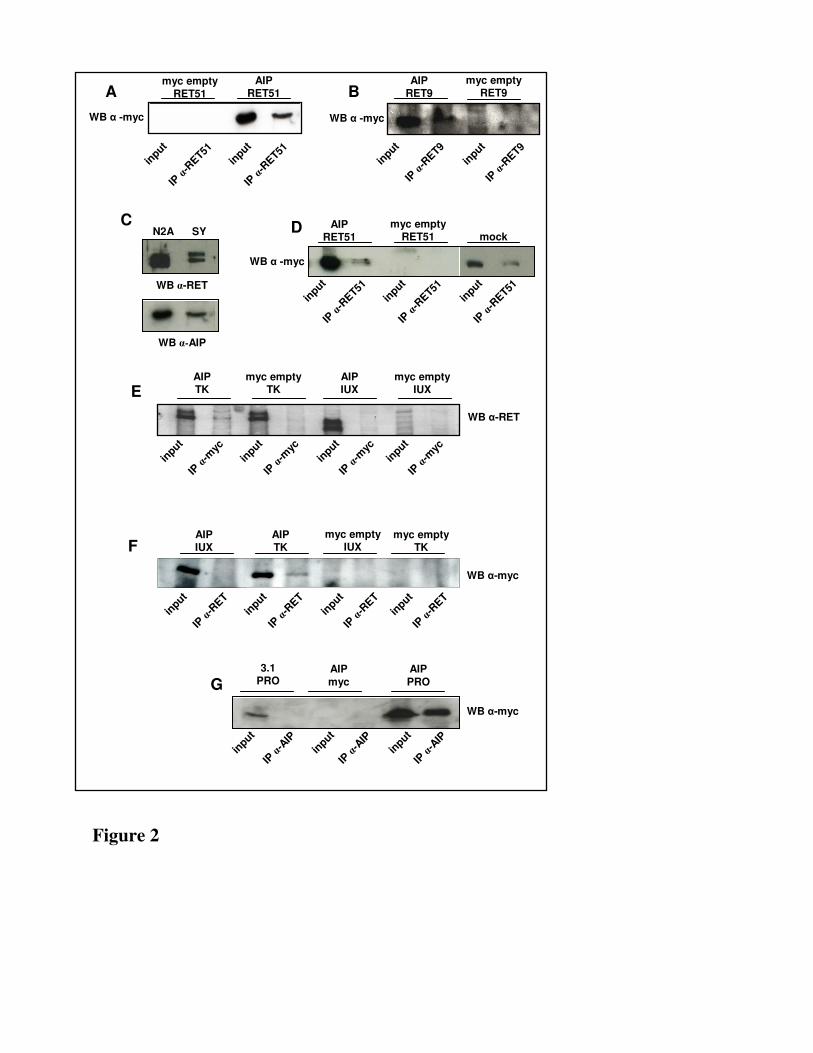
Figure 2
A B
D
E
F
IP α
-RET51
WB α -myc
myc emptyRET9
AIPRET9
AIPRET51
myc emptyRET51
myc emptyRET51
AIPRET51
AIPIUX
myc emptyTK
myc emptyIUX
AIPTK
AIPIUX
myc emptyTK
myc emptyIUX
AIPTK
C
WB α-AIP
WB α-RET
N2A SY
GAIP
myc
AIP
PRO
3.1PRO
WB α -myc
mock
IP α
-RET51
input
input
IP α
-RET9
IP α
-RET9
input
input
IP α
-RET51
IP α
-RET51
input
input
IP α
-RET51
input
IP α
-myc
WB α-RET
IP α
-myc
input
input
IP α
-myc
input
IP α
-myc
input
IP α
-RET
WB α-myc
IP α
-RET
input
input
IP α
-RET
input
IP α
-RET
input
IP α
-AIP
WB α-myc
IP α
-AIP
input
input
IP α
-AIP
input
WB α -myc

Figure 3
WB anti AIP
IP a
nti-AIP
IP a
nti-RET
IP u
nrela
ted Ig
B
A
36 KDa
36 KDa
120 KDa
WB anti AIP WB antiRET
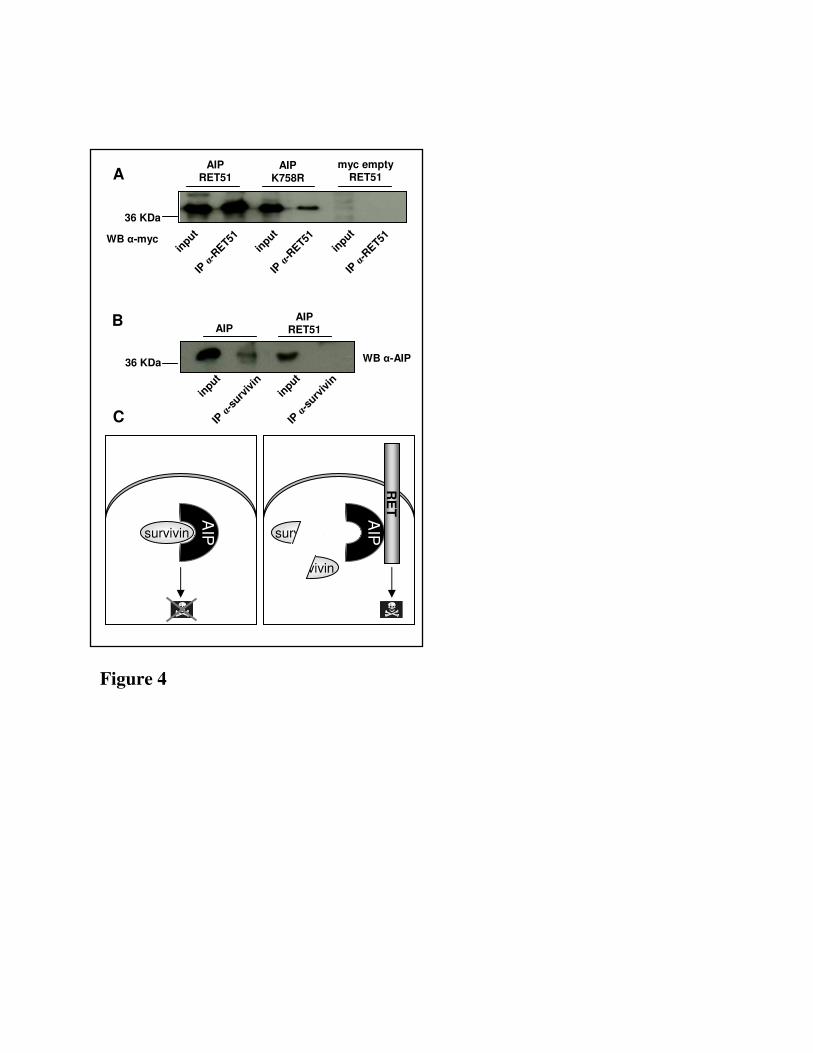
Figure 4
survivin
myc emptyRET51
AIPRET51
AIPK758R
AIPRET51 AIP
B
A
36 KDa
36 KDa
C
survivin
AIP survivin
AIP
RE
TIP
α-s
urviv
in
WB α-AIP
IP α
-surv
ivin
input
input
IP α
-RET51WB α-myc
IP α
-RET51
input
input
IP α
-RET51
input
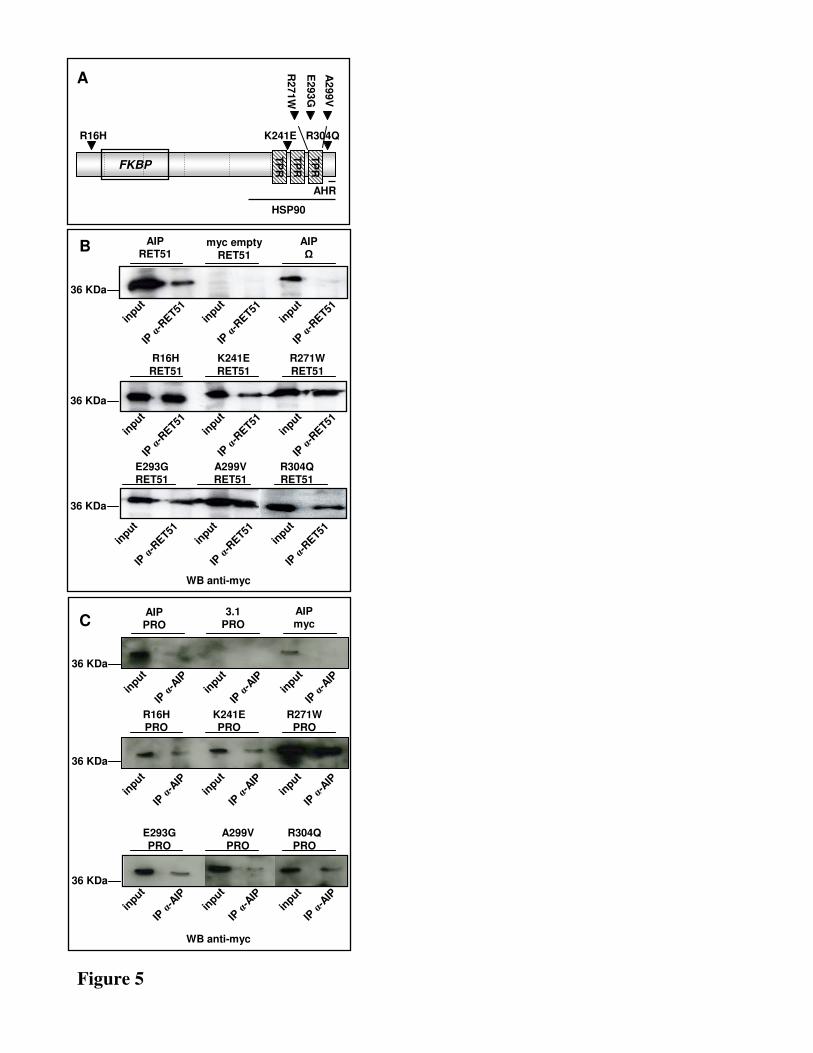
Figure 5
R16H
AHR
HSP90
K241E
E293G
R271W
A2
99V
TP
RFKBP
TP
R
TP
R
R304Q
A
B
C
WB anti-myc
WB anti-myc
A299VRET51
E293GRET51
R304QRET51
R16H
RET51
R271W
RET51
K241E
RET51
AIPΩ
myc empty
RET51
AIPRET51
AIPmyc
3.1PRO
AIPPRO
A299V
PRO
E293G
PRO
R304Q
PRO
R16HPRO
R271WPRO
K241EPRO
36 KDa
36 KDa
36 KDa
36 KDa
36 KDa
36 KDa
IP α
-RET51
IP α
-RET51
input
input
IP α
-RET51
input
IP α
-RET51
IP α
-RET51
input
input
IP α
-RET51
input
IP α
-RET51
IP α
-RET51
input
input
IP α
-RET51
input
IP α
-AIP
IP α
-AIP
input
input
IP α
-AIP
input
IP α
-AIP
IP α
-AIP
input
input
IP α
-AIP
input
IP α
-AIP
IP α
-AIP
input
input
IP α
-AIP
input
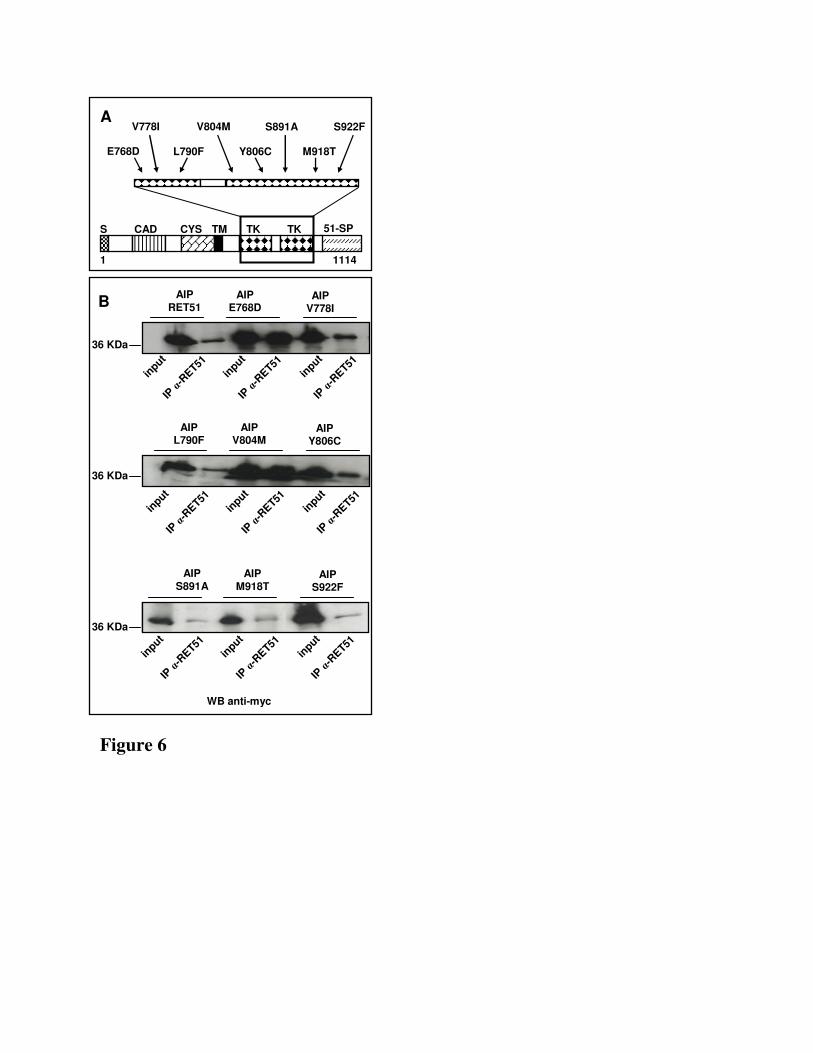
Figure 6
A
B
S CAD CYS TKTM TK 51-SP
1 1114
L790F Y806C
V804MV778I
E768D M918T
S891A S922F
WB anti-myc
AIPL790F
AIP Y806C
AIP V804M
AIPV778I
AIPE768D
AIP
RET51
AIP
M918T
AIP
S891AAIP
S922F
36 KDa
36 KDa
36 KDa
IP α
-RET51
IP α
-RET51
input
input
IP α
-RET51
input
IP α
-RET51
IP α
-RET51
input
input
IP α
-RET51
input
IP α
-RET51
IP α
-RET51
input
input
IP α
-RET51
input




















![e ;mu•] ret) - Valle de Elda](https://img.pdfslide.net/doc/110x75/6337bd26fe1f34a1c300ac50/e-mu-ret-valle-de-elda.jpg)








