Embed Size (px)
Citation preview

The Unique Case of The Niemann-Pick Type C Cholesterol Storage Disorder
Andrés D. Klein1, Alejandra R. Alvarez2, Silvana Zanlungo3,4
1Department of Biological Chemistry, Weizmann, Institute of Science, Rehovot 76100, Israel; 2Laboratorio de
Señalización Celular, Departamento de Biología Celular, Facultad de Ciencias Biológicas, Pontificia Universidad
Católica de Chile, Alameda #340, Santiago, 8331150, Chile.; 3Departamento de Gastroenterología, Facultad de
Medicina, Pontificia Universidad Católica de Chile, Marcoleta #364, Santiago, 8330024, Chile, 4FONDAP “Center for
Genome Regulation” (CGR), Santiago, Chile
Corresponding author: A.D. Klein, Department of Biological Chemistry, Weizmann Institute of Science, Rehovot 76100, Israel; Tel:(972)-8-9342704; Email, [email protected], Silvana Zanlungo, Departamento de Gastroenterología, Facultad de Medicina, Pontificia Universidad Católica de Chile, Marcoleta #364, Santiago, 8330024, Chile
Introduction
Niemann-Pick type C disease (NPC) is a lysosomal storage disease in which lipids, mainly unesterified cholesterol, accumulate in the late endosomal/lysosomal (LE/Lys) compartment.(1-3) Patients can present with liver damage and most frequently show progressive neurodegeneration and succumb to early death.(1-4) The prevalence of NPC is low; recent estimates place the incidence rate at 1:120,000 live births.(4)
NPC is an autosomal recessive genetic disease caused by mutations in either one of the genes encoding the NPC1 and NPC2 proteins, (4-6) which are involved in the transport of unesterified cholesterol from the LE/Lys compartment to the rest of the cell. A variety of mutations have been described in NPC patients, with a marked correlation between the loss of NPC1 or NPC2 function and the severity of the disease symptoms.(7, 8) In total, 95% of NPC cases arise from mutations of the gene that codes for the NPC1 protein (4). The NPC1 protein is located in an LE/Lys compartment,(9-11) and several of its structural characteristics are associated with cholesterol transport. Of its 13 transmembrane domains, 5 domains form the sterol-sensing domain (SSD), similar to that of proteins such as HMG-CoA reductase, which is involved in cholesterol metabolism.(5, 11-13) In addition, the N-terminal domain (NTD) is located on the protein’s luminal N-terminus and is capable of binding the 3β-hydroxyl end of the cholesterol molecule with high affinity.(14-16)Mutations in the NPC2 gene are responsible for the remaining 5% of NPC cases. NPC2 is a small, soluble lysosomal glycoprotein that specifically binds cholesterol with nanomolar affinity (6, 17) by recognizing the iso-octyl side chain and leaving the 3β-hydroxyl end of the cholesterol molecule exposed.(16, 18) It has also been shown that the NPC2 protein can considerably accelerate cholesterol transport from and between membranes via the direct interaction of NPC2 with the membrane (19) and/or with NPC1.(16, 20) The NPC2 protein is located in the LE/Lys compartment but is also secreted into the epididymis, milk, plasma, and bile.(6, 17, 21, 22) Several studies have suggested that NPC2 has biological functions in bodily fluids.(22-24)
Abstract
Niemann-Pick type C disease (NPC) is a neuro-visceral lysosomal cholesterol storage disorder that arises from loss-of-function mutations in either
the NPC1 or NPC2 genes. Both genes code for proteins involved in lysosomal cholesterol efflux.NPC is often diagnosed in early childhood, with patients typically displaying cerebellar ataxia, difficulties in speaking and swallowing, and progressive dementia. Unfortunately, to date, there is no curative treatment for this devastating and fatal disorder, although several symptomatic manifestations of NPC are treatable. In this review, we discuss the cell biology of the disease, clinical aspects, diagnostic approaches, and current and potential therapeutic strategies against NPC.
Ref: Ped Endocrinol Rev. 2014; 12(Suppl 1):166-175
Key Words: Lysosomal storage disorders, Niemann-Pick type C, cholesterol, therapeutic approaches, diagnostic
Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014166
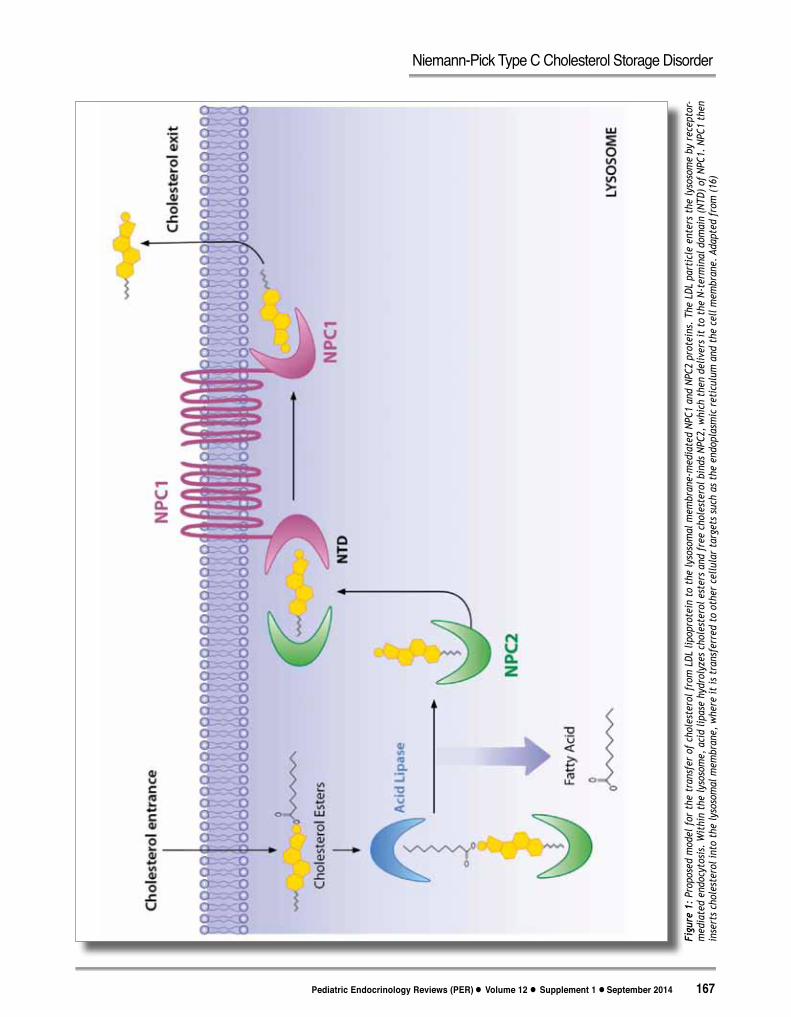
Figu
re 1
: Pro
pose
d m
odel
for
the
tra
nsfe
r of
cho
lest
erol
fro
m L
DL li
popr
otei
n to
the
lyso
som
al m
embr
ane-
med
iate
d NP
C1 a
nd N
PC2
prot
eins
. Th
e LD
L pa
rtic
le e
nter
s th
e ly
soso
me
by r
ecep
tor-
med
iate
d en
docy
tosi
s. W
ithi
n th
e ly
soso
me,
aci
d lip
ase
hydr
olyz
es c
hole
ster
ol e
ster
s an
d fr
ee c
hole
ster
ol b
inds
NPC
2, w
hich
the
n de
liver
s it
to
the
N-te
rmin
al d
omai
n (N
TD)
of N
PC1.
NPC
1 th
en
inse
rts
chol
este
rol i
nto
the
lyso
som
al m
embr
ane,
whe
re it
is t
rans
ferr
ed t
o ot
her
cellu
lar
targ
ets
such
as
the
endo
plas
mic
ret
icul
um a
nd t
he c
ell m
embr
ane.
Ada
pted
fro
m (1
6)
167 Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014
Niemann-Pick Type C Cholesterol Storage Disorder
The current model of cholesterol transport as mediated by the NPC1 and NPC2 proteins is shown in Figure 1. Interestingly, dysfunction in either NPC1 or NPC2 not only results in the abnormal storage of unesterified cholesterol but also affects the storage of other lipids, such as lactosylceramide, glucosylceramide, and the GM2 and GM3 gangliosides in the LE/Lys compartments.(25-28) Although studies have not found a direct linkage between the NPC1 or NPC2 proteins and glycolipid transport in mammalian cells, the current view is that NPC proteins may affect the trafficking and metabolism of other lipids through the endocytic pathway.(3, 29-32) The accumulation of these other types of lipids, in addition to cholesterol, may have a role in the pathogenesis of NPC.
Clinical Aspects
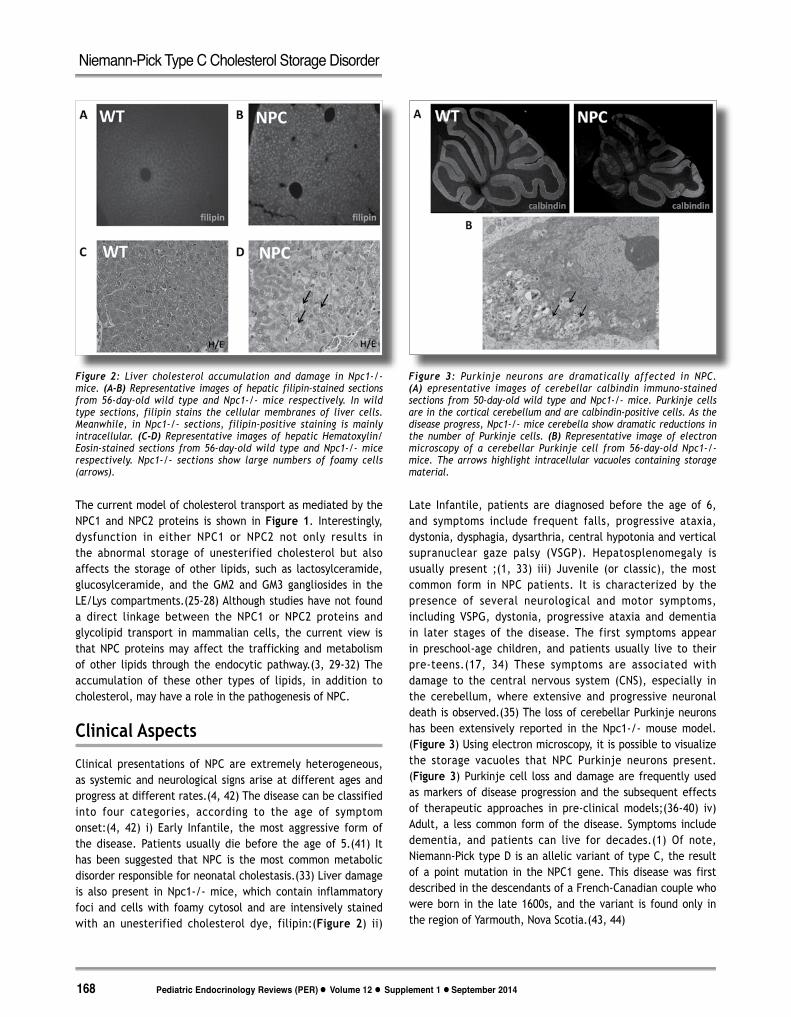
Clinical presentations of NPC are extremely heterogeneous, as systemic and neurological signs arise at different ages and progress at different rates.(4, 42) The disease can be classified into four categories, according to the age of symptom onset:(4, 42) i) Early Infantile, the most aggressive form of the disease. Patients usually die before the age of 5.(41) It has been suggested that NPC is the most common metabolic disorder responsible for neonatal cholestasis.(33) Liver damage is also present in Npc1-/- mice, which contain inflammatory foci and cells with foamy cytosol and are intensively stained with an unesterified cholesterol dye, filipin:(Figure 2) ii)
Late Infantile, patients are diagnosed before the age of 6, and symptoms include frequent falls, progressive ataxia, dystonia, dysphagia, dysarthria, central hypotonia and vertical supranuclear gaze palsy (VSGP). Hepatosplenomegaly is usually present ;(1, 33) iii) Juvenile (or classic), the most common form in NPC patients. It is characterized by the presence of several neurological and motor symptoms, including VSPG, dystonia, progressive ataxia and dementia in later stages of the disease. The first symptoms appear in preschool-age children, and patients usually live to their pre-teens.(17, 34) These symptoms are associated with damage to the central nervous system (CNS), especially in the cerebellum, where extensive and progressive neuronal death is observed.(35) The loss of cerebellar Purkinje neurons has been extensively reported in the Npc1-/- mouse model.(Figure 3) Using electron microscopy, it is possible to visualize the storage vacuoles that NPC Purkinje neurons present.(Figure 3) Purkinje cell loss and damage are frequently used as markers of disease progression and the subsequent effects of therapeutic approaches in pre-clinical models;(36-40) iv) Adult, a less common form of the disease. Symptoms include dementia, and patients can live for decades.(1) Of note, Niemann-Pick type D is an allelic variant of type C, the result of a point mutation in the NPC1 gene. This disease was first described in the descendants of a French-Canadian couple who were born in the late 1600s, and the variant is found only in the region of Yarmouth, Nova Scotia.(43, 44)
Figure 2: Liver cholesterol accumulation and damage in Npc1-/- mice. (A-B) Representative images of hepatic filipin-stained sections from 56-day-old wild type and Npc1-/- mice respectively. In wild type sections, filipin stains the cellular membranes of liver cells. Meanwhile, in Npc1-/- sections, filipin-positive staining is mainly intracellular. (C-D) Representative images of hepatic Hematoxylin/Eosin-stained sections from 56-day-old wild type and Npc1-/- mice respectively. Npc1-/- sections show large numbers of foamy cells (arrows).
Figure 3: Purkinje neurons are dramatically affected in NPC. (A) epresentative images of cerebellar calbindin immuno-stained sections from 50-day-old wild type and Npc1-/- mice. Purkinje cells are in the cortical cerebellum and are calbindin-positive cells. As the disease progress, Npc1-/- mice cerebella show dramatic reductions in the number of Purkinje cells. (B) Representative image of electron microscopy of a cerebellar Purkinje cell from 56-day-old Npc1-/- mice. The arrows highlight intracellular vacuoles containing storage material.
Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014168
Niemann-Pick Type C Cholesterol Storage Disorder

Diagnosis
The age at which clinical manifestations present themselves ranges from perinatal to adulthood. Neonatal cholestasic liver disease with splenomegaly can occur in NPC. Over time, hepatosplenomegaly disappears and organomegaly is not critical in juvenile or adult cases. A key diagnostic symptom that a patient suffers from juvenile or adult NPC disease is the abnormal presence of VSGP. Cognitive decline is also invariably present in these groups of NPC patients. An accurate diagnosis of NPC requires the identification of the many clinical symptoms of each disease subtype. Confirmation of the diagnosis involves cell biology imaging, biochemical analysis and genetic testing.The gold standard method to diagnose NPC involves culturing skin fibroblasts from the patient and staining the cells with filipin.(Figures 4A & 4B) Filipin is a fluorescent polyene that specifically binds free cholesterol. If the cells show perinuclear filipin-positive organelles, which are a hallmark
of the disease, then it is extremely likely that the patient suffers from NPC.(Figure 4B) However, a confirmatory test to measure the LDL-induced rate of cholesteryl ester formation is highly recommended. Fibroblasts from NPC patients will show a null or very low rate of cholesterol esterification. This assay is mandatory in cases where filipin staining is moderately positive. A negative filipin staining excludes NPC as the diagnosis. If possible, electron microscopy of the fibroblasts is helpful to observe the characteristic lipid inclusions present in the disease. The final confirmation is established by sequencing the patient’s NPC1 and NPC2 genes. If mutations are identified in either of these genes, then it is extremely likely that the patient exhibits NPC.The lack of non-invasive tests for NPC has been a major problem in diagnosing the disease. Recently, Ory and colleagues identified several products of non-enzymatic cholesterol oxidation, specifically 7-ketocholesterol (7-KC), 7b-hydroxycholesterol (7b-HC), and cholestane-3b,5a,6b-triol (3b,5a,6b-triol), that are sensitive and specific blood biomarkers for NPC. The blood levels of these oxysterols correlate with disease progression in NPC-afflicted mice, cats and humans. The levels of these oxysterols were reduced when NPC cats were treated with the experimental drug hydroxypropyl-β-cyclodextrin (HPCD).(45) These oxysterols have been reported to be cytotoxic in vitro.(46) The origin of these oxysterol species in NPC still remains unclear.
The levels of other markers such as galectin-3, a pro-inflammatory molecule, and cathepsin D, a lysosomal aspartic protease, are increased in the blood of NPC mice. Their plasma levels are also reduced after treatment with HPCD.(47) Galectin-3 and cathepsin D have the potential to become other NPC blood biomarkers, although they are less-specific markers for NPC because their levels have also been found to be elevated in other pathologies.(47)
Current Treatments
Unfortunately, to date, there is no curative treatment for this devastating and fatal disorder. However, several symptomatic manifestations of NPC are treatable. For instance, cataplexy, which is the sudden and transient loss of muscle tone, is often controlled using tricyclic antidepressants such as amitriptyline or clomipramine.(48) To treat seizures, anti-epileptic drugs are used.(49) Insomnia can be treated with melatonin. Dystonia and tremors can be alleviated with anticholinergic beta-blocking and antispasmodic drugs. Physical therapy is suggested to reduce tremors and improve coordination and muscle control. The neuropsychiatric manifestations of NPC can be treated in a manner similar to that of schizophrenia, with thioridazine or haloperidol,(50) although these drugs can result in undesirable side effects, such as increased
Figure 4: (A) Laboratory diagnostic steps. For a description please refer to the “Diagnosis” section of the manuscript. (B) Filipin staining of unesterified cholesterol in NPC human fibroblasts. Fluorescent filipin staining for unesterified cholesterol in control and NPC1 fibroblasts. Control fibroblasts showed minimal filipin-stained intracellular unesterified cholesterol. In contrast, NPC fibroblasts showed numerous filipin-stained, unesterified, cholesterol-containing intracellular inclusions. Filipin staining of cultured skin fibroblasts is considered a key test for NPC diagnosis.(4)
169 Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014
Niemann-Pick Type C Cholesterol Storage Disorder

tremors and dyskinesia. Another drug that can be used to treat seizure-induced psychotic symptoms is valproate.(51) Interestingly, valproate has histone deacetylase inhibitory (HDAC) properties. It has been shown that HDACs can both reduce cholesterol accumulation in human NPC fibroblasts (52) and induce neuronal differentiation in neural stem cells derived from NPC mice.(53)More details in NPC diagnoses and treatment can be found in Wraith et al. 2009 (4) and Patterson et al. 2011,(42) which contain, respectively, the first and second international recommendations for the clinical management of NPC.Miglustat (N-butyldeoxynojirimycin; NB-DNJ; Zavesca®, Actelion Pharmaceuticals Ltd) is currently approved in the European Union (EU), USA, and several other countries for the treatment of patients with Gaucher disease. In January 2009, the EU Commission extended Miglustat’s indication to include the treatment of progressive neurological manifestations in patients with NPC. This extension was followed by authorization in Brazil and South Korea.(42, 54) Miglustat is a small iminosugar molecule that acts as a competitive inhibitor of the glucosylceramide synthase enzyme, which catalyzes the first committed step in glycosphingolipid (GSL) synthesis.(55, 56) Miglustat can cross the blood–brain barrier,(57) and the Walkley group showed that treatment of NPC mouse and cat models with the drug reduces GSL accumulation and cellular pathology in the brain, delays the onset of neurological symptoms and increases survival.(58) In human patients, Miglustat treatment reduces pathological lipid storage, improves endosomal uptake and normalizes lipid trafficking in B lymphocytes circulating in peripheral blood.(59)Clinical experience with Miglustat is increasing, and a number of cohort studies and case reports/series have been published that demonstrate the stabilization of key neurological manifestations.(42) (60, 61) Furthermore, findings from clinical experience studies have since confirmed these therapeutic effects.(42)The most frequently reported adverse events observed with Miglustat treatment included mild or moderate diarrhea, flatulence, weight loss and tremors.(42) These effects tended to decrease over time on continued therapy and can be managed as previously described.(42) The recommended dose of Miglustat for adult and adolescent patients with NPC is 200 mg t.i.d. Dosing in patients aged 4–12 years should be adjusted according to the calculated body surface area.(4, 42)
Future Approaches
Unfortunately, while enzyme replacement therapy (ERT) has proven to be useful for other lysosomal diseases, it is not successful for the treatment of lysosomal disorders with CNS compromises, due to the inability of the recombinant
enzyme to cross the blood-brain barrier. In addition, ERT is unlikely to work in cases of mutations in NPC1 since it is a membrane bound molecule and not a secretory enzyme like NPC2. Accordingly, until an effective approach with gene therapy is developed for NPC, an approach from multiple fronts may be the most useful strategy for slowing the progression of this disease. In addition, these approaches can be used in combination with Miglustat treatment. Thus, it is of great interest to pursue new therapeutic targets for the disease and to study the effects of cholesterol-lowering, anti-inflammatory, and anti-apoptotic drugs. In addition, there are continuing investigations into potential antioxidants and drugs that improve handling of intracellular calcium, chemical chaperones and HDAC inhibitors.Among the cholesterol-binding agents, hydroxypropyl-β-cyclodextrin (HPCD) appears to be the most promising drug.(62) However, it is necessary to consider recent data suggesting limited or no blood-brain barrier penetration of HPCD following its systemic administration.(63) Treatment of NPC mice with HPCD was found to significantly reduce neurodegeneration and hepatic disease and increase lifespans in Npc1-/- mice, suggesting a potential therapeutic approach for the treatment of individuals with NPC.(64) Although the mechanisms by which HPCD mediates these beneficial effects are still unknown, liberated cholesterol is known to flow into the cytosolic ester pool, thereby suppressing sterol synthesis and reducing the expression of macrophage-associated inflammatory genes. The sequestered cholesterol is excreted as bile acid, thereby suggesting that HPCD acutely reverses the lysosomal transport defect observed in NPC. However, although HPCD has displayed promising effects on the brain and liver, it has had little to no effect on lung dysfunction, which constitutes another important issue in NPC pathology.(65) Nonetheless, the positive data on cyclodextrin as reported in animal models have encouraged its application as a potential treatment in NPC patients. Indeed, the effects of HPCD in two patients with NPC have been reported, and although HPCD did not improve the neurological deficits in either patient, its administration was partially effective in improving hepatosplenomegaly and CNS dysfunction, especially during the first 6 months of treatment.(66) Moreover, in 2011, the European Medicines Agency (EMA) granted orphan drug status to HPCD and designated HPCD as a potential treatment for NPC. In addition, the National Institutes of Health (NIH), in collaboration with the Therapeutics for Rare and Neglected Diseases Program (TRND), announced that they are developing a clinical trial utilizing cyclodextrin for NPC patients. This clinical trial is now open and ongoing. The goal of the Phase 1 trial is to establish a safe and biochemically effective dosing regimen of HPCD for the treatment of human NPC1 subjects.In the search for other potential treatments for NPC, Platt and colleagues evaluated the effects of non-steroidal anti-inflammatory drugs (NSAIDs) on the Npc1-/- mouse model
Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014170
Niemann-Pick Type C Cholesterol Storage Disorder
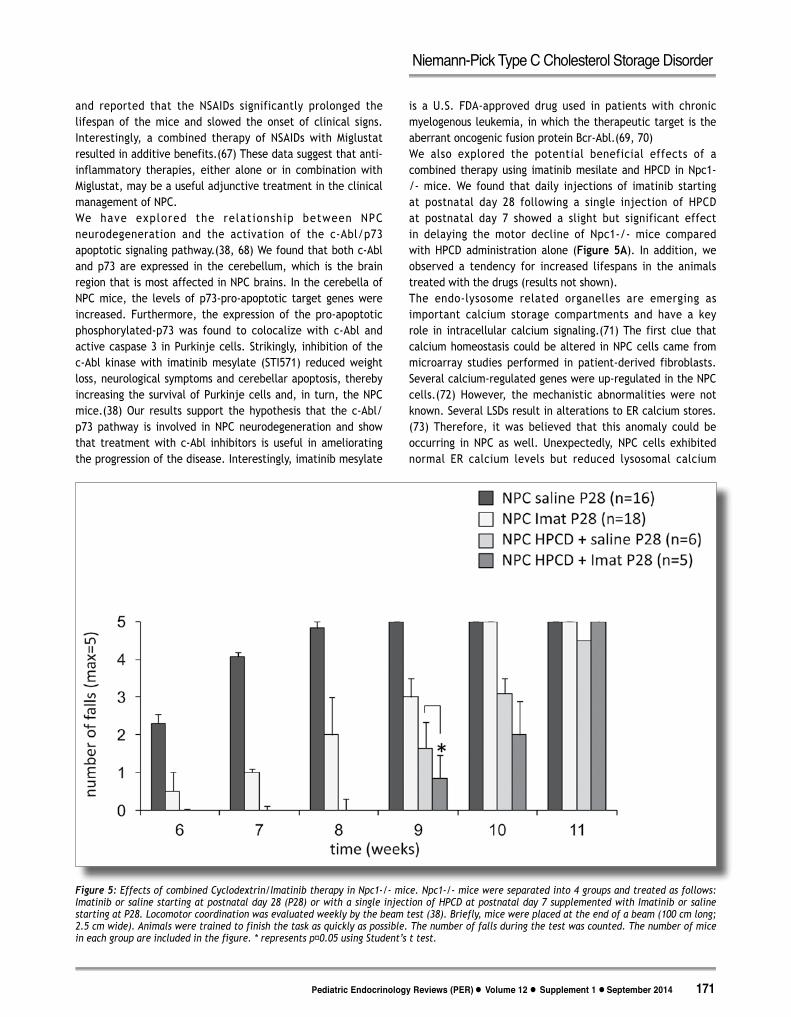
and reported that the NSAIDs significantly prolonged the lifespan of the mice and slowed the onset of clinical signs. Interestingly, a combined therapy of NSAIDs with Miglustat resulted in additive benefits.(67) These data suggest that anti-inflammatory therapies, either alone or in combination with Miglustat, may be a useful adjunctive treatment in the clinical management of NPC.We have explored the relat ionship between NPC neurodegeneration and the activation of the c-Abl/p73 apoptotic signaling pathway.(38, 68) We found that both c-Abl and p73 are expressed in the cerebellum, which is the brain region that is most affected in NPC brains. In the cerebella of NPC mice, the levels of p73-pro-apoptotic target genes were increased. Furthermore, the expression of the pro-apoptotic phosphorylated-p73 was found to colocalize with c-Abl and active caspase 3 in Purkinje cells. Strikingly, inhibition of the c-Abl kinase with imatinib mesylate (STI571) reduced weight loss, neurological symptoms and cerebellar apoptosis, thereby increasing the survival of Purkinje cells and, in turn, the NPC mice.(38) Our results support the hypothesis that the c-Abl/p73 pathway is involved in NPC neurodegeneration and show that treatment with c-Abl inhibitors is useful in ameliorating the progression of the disease. Interestingly, imatinib mesylate
is a U.S. FDA-approved drug used in patients with chronic myelogenous leukemia, in which the therapeutic target is the aberrant oncogenic fusion protein Bcr-Abl.(69, 70) We also explored the potential beneficial effects of a combined therapy using imatinib mesilate and HPCD in Npc1-/- mice. We found that daily injections of imatinib starting at postnatal day 28 following a single injection of HPCD at postnatal day 7 showed a slight but significant effect in delaying the motor decline of Npc1-/- mice compared with HPCD administration alone (Figure 5A). In addition, we observed a tendency for increased lifespans in the animals treated with the drugs (results not shown).The endo-lysosome related organelles are emerging as important calcium storage compartments and have a key role in intracellular calcium signaling.(71) The first clue that calcium homeostasis could be altered in NPC cells came from microarray studies performed in patient-derived fibroblasts. Several calcium-regulated genes were up-regulated in the NPC cells.(72) However, the mechanistic abnormalities were not known. Several LSDs result in alterations to ER calcium stores.(73) Therefore, it was believed that this anomaly could be occurring in NPC as well. Unexpectedly, NPC cells exhibited normal ER calcium levels but reduced lysosomal calcium
Figure 5: Effects of combined Cyclodextrin/Imatinib therapy in Npc1-/- mice. Npc1-/- mice were separated into 4 groups and treated as follows: Imatinib or saline starting at postnatal day 28 (P28) or with a single injection of HPCD at postnatal day 7 supplemented with Imatinib or saline starting at P28. Locomotor coordination was evaluated weekly by the beam test (38). Briefly, mice were placed at the end of a beam (100 cm long; 2.5 cm wide). Animals were trained to finish the task as quickly as possible. The number of falls during the test was counted. The number of mice in each group are included in the figure. * represents p˂0.05 using Student’s t test.
171 Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014
Niemann-Pick Type C Cholesterol Storage Disorder

stores.(28) Calcium release from the lysosome is required for the last step of fusion between endosomes and lysosomes, and this process is impaired in NPC cells.(74) When NPC1-deficient CHO cells were treated with thapsigargin, thereby compensating for the reduced calcium release from the acidic compartment, elevated cytosolic calcium levels were observed, and the NPC cellular phenotype was corrected. However, when LE/Lys calcium was chelated in wild type cells, a phenocopy of the NPC cellular alterations occurred, including defects in endocytic transport and lipid storage.(28) Recently, it was determined that the alterations in lysosomal calcium stores in NPC are due to the inhibition of Mucolipin transient receptor potential channel 1 (TRPML1), which is the principal calcium channel transporter of the lysosome.(75)Targeting calcium homeostasis as a therapeutic option to treat NPC appears to be acting in several ways, in addition to restoring intracellular trafficking. For instance, targeting endoplasmic reticulum (ER) calcium levels using ryanodine receptor (RyR) antagonists increases the steady-state levels of NPC1 I1061T protein (NPC1I1061T), thereby promoting the trafficking of the mutated NPC1 protein to lysosomes, where it shows some activity.(76) Interestingly, the NPC1 I1061T mutation is present in 15-20% of NPC patients,(7, 77, 78) suggesting that RyR antagonists could be useful in a number of NPC patients. In cells derived from NPC1-deficient mice, the increase of extracellular calcium levels enhances the effects of HPCD by stimulating lysosomal exocytosis.(79) In addition, it was recently demonstrated that δ-tocopherol reduced lysosomal cholesterol accumulation, increased cholesterol efflux, and alleviated pathological phenotypes in NPC1 fibroblasts; this was correlated with an intracellular Ca(2+) response and subsequent enhancement of lysosomal exocytosis.(80)NPC patients exhibit decreased antioxidant capacity (expressed as Trolox equivalents) and reduced Coenzyme Q10 in their serum, which indicates a decrease in antioxidant defenses.(81) In addition, as mentioned previously, the plasma of NPC patients exhibited an increase in cholesterol oxidation products.(45) Our group and others have shown a buildup of α-TOH in the endosomal/lysosomal system that may result in decreased bioavailability and impaired antioxidant function of vitamin E in NPC, thereby contributing to the disease pathogenesis.(32, 82) Therefore, oxidative stress has been proposed as a part of the pathogenesis and progression of NPC, and antioxidant molecules have been proposed as potential therapeutic drugs.(83) Currently, there is a clinical protocol to test the safety and efficacy of the N-Acetyl Cysteine (NAC) antioxidant in NPC patients at the National Institutes of Health Clinical Center in the U.S.A. (http://clinicaltrials.gov/ct2/show/NCT00975689). NAC is a powerful antioxidant that acts by increasing cellular glutathione levels,(84-86) and it has been approved by the FDA for treatment of acetaminophen
poisoning and reducing the stickiness of mucus secretions in patients with cystic fibrosis. U18-treated neurons incubated with NAC showed a decrease in apoptosis,(68) and we reported a decrease in foamy cells on the livers of 7-week-old Npc1-/- mice after 2 weeks of treatment with NAC.(87) Although the acute uses of NAC have demonstrated clear beneficial effects, its chronic use for the treatment of oxidative damage diseases is controversial.(88) Interestingly, 4 weeks of NAC treatment was able to partially suppress phenotypes in both antisense-induced (NPC1ASO) and Npc1-/- genetic mouse models, confirming that oxidative stress is relevant to the progression of NPC.(89) However, translation of NAC treatments to patients is not as straightforward as it seems, because a short-term NAC administration therapeutic trial to NPC1 patients showed no significant effects on oxidative stress other than moderate improvement of the fraction of reduced CoQ10, suggesting limited efficacy of NAC therapy.(89)Chemical chaperones have been proposed as a therapy for modulating aberrant protein folding in several lysosomal disorders.(90) In cultured cells, the folding enhancement of the NPC1 I1061T mutation with chemical chaperones resulted in improvement of trafficking of NPC1 to the LE/Lys compartment and reduced the NPC phenotype.(91) The possibility of treating other mutations with this approach requires further research.Another approach that explored different therapeutic approaches for NPC involves the use of small HDAC inhibitors. Maxfield and colleagues found that treatment of NPC human fibroblasts with small HDAC inhibitors of the 1 and 2 families led to a dramatic correction of the NPC phenotype in cells containing one or two copies of the NPC1 I1061T mutation.(52) This effect was associated with increased NPC1 expression. Interestingly, HDAC inhibition was also identified as a candidate therapy for NPC in yeast.(92) The potential use of HDAC inhibitors for NPC is enhanced because several of the inhibitors can cross the blood-brain barrier and are currently in phase III clinical trials for several types of cancer. Finally, it seems that combined interventions to slow disease progression are the most promising therapies for NPC. Indeed, the combined therapy HPCD/Miglustat has shown to be additive in pre-clinical NPC models.(93)
Concluding Remarks
NPC is a unique neuro-visceral storage disorder with no effective treatments available. Several approaches have attempted to delay the disease progression. However, despite years of studying the genetic, molecular, and cellular bases of NPC, little is known of the events that lead from the intra-lysosomal buildup of lipids to cell dysfunction and pathology. Increasing evidence indicates that treatment with the glycosphingolipid synthesis inhibitor Miglustat has
Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014172
Niemann-Pick Type C Cholesterol Storage Disorder

important therapeutic effects in NPC patients. Therefore, Miglustat and antioxidant-, anti-apoptotic-, anti-inflammatory, and calcium homeostasis-restoring drugs may eventually be used in combination with other pharmacological strategies that restore the proper metabolism and transport of lipids, especially cholesterol, in NPC patients.
Acknowledgements
We acknowledge the involvement of many colleagues who have participated in our studies and contributed with helpful discussions. We wish to especially thank the Ara Parseghian Research Medical Foundation for their support at the beginning of our research on NPC. A.D. Klein is support by the UK Gaucher Association and by the Dean’s Fellowship from the Weizmann Institute of Science. Our current work is supported by Fondo Nacional de Desarrollo Científico y Tecnológico, FONDECYT, grants #1080221 (to A.A.) and #1110310 (to S.Z.) and Fondo Nacional de Desarrollo de Areas Prioritarias, FONDAP, project number 15090007, Center for Genome Regulation (CGR ) to SZ.Andrés D. KleinAlejandra R. Alvarez Silvana Zanlungo
References1. Patterson MC, Vanier MT, Suzuki K, Morris JA, Carsteu E, Neufeld
EB, Blanchette-Mackie J, PGl P. Niemann-Pick disease type C. A lipid trafficking disorder. In: Scriver CR BA, Sly WS, Valle D, ed. The metabolic and molecular basis of inherited disease. New York: Mulencer Hill, 2001
2. Sturley SL, Patterson MC, Balch W, Liscum L. The pathophysiology and mechanisms of NP-C disease. Biochim Biophys Acta 2004;1685:83-87
3. Karten B, Peake KB, Vance JE. Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim Biophys Acta 2009;1791:659-670
4. Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, Pineda M, Sedel F, Topcu M, Vanier MT, Widner H, Wijburg FA, Patterson MC, Grp N-CGW. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Molecular Genetics and Metabolism 2009;98:152-165
5. Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF, 3rd, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O'Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 1997;277:228-231
6. Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000;290:2298-2301
7. Park WD, O'Brien JF, Lundquist PA, Kraft DL, Vockley CW, Karnes PS, Patterson MC, Snow K. Identification of 58 novel mutations in Niemann-Pick disease type C: correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum Mutat 2003;22:313-325
8. Verot L, Chikh K, Freydiere E, Honore R, Vanier MT, Millat G. Niemann-Pick C disease: functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin Genet 2007;71:320-330
9. Frolov A, Srivastava K, Daphna-Iken D, Traub LM, Schaffer JE, Ory DS. Cholesterol overload promotes morphogenesis of a Niemann-Pick C (NPC)-like compartment independent of inhibition of NPC1 or HE1/NPC2 function. J Biol Chem 2001;276:46414-46421
10. Ko DC, Gordon MD, Jin JY, Scott MP. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol Biol Cell 2001;12:601-614
11. Ory DS. The niemann-pick disease genes; regulators of cellular cholesterol homeostasis. Trends Cardiovasc Med 2004;14:66-72
12. Ioannou YA. The structure and function of the Niemann-Pick C1 protein. Mol Genet Metab 2000;71:175-181
13. Liscum L. Niemann-Pick type C mutations cause lipid traffic jam. Traffic 2000;1:218-225
14. Infante RE, Abi-Mosleh L, Radhakrishnan A, Dale JD, Brown MS, Goldstein JL. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J Biol Chem 2008;283:1052-1063
15. Infante RE, Radhakrishnan A, Abi-Mosleh L, Kinch LN, Wang ML, Grishin NV, Goldstein JL, Brown MS. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem 2008;283:1064-1075
16. Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009;137:1213-1224
17. Ory DS. Niemann-Pick type C: a disorder of cellular cholesterol trafficking. Biochim Biophys Acta 2000;1529:331-339
18. Xu S, Benoff B, Liou HL, Lobel P, Stock AM. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J Biol Chem 2007;282:23525-23531
19. Storch J, Xu Z. Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim Biophys Acta 2009;1791:671-678
20. Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A 2008;105:15287-15292
21. Okamura N, Kiuchi S, Tamba M, Kashima T, Hiramoto S, Baba T, Dacheux F, Dacheux JL, Sugita Y, Jin YZ. A porcine homolog of the major secretory protein of human epididymis, HE1, specifically binds cholesterol. Biochim Biophys Acta 1999;1438:377-387
22. Klein A, Amigo L, Retamal MJ, Morales MG, Miquel JF, Rigotti A, Zanlungo S. NPC2 is expressed in human and murine liver and secreted into bile: potential implications for body cholesterol homeostasis. Hepatology 2006;43:126-133
23. Yamanashi Y, Takada T, Yoshikado T, Shoda J, Suzuki H. NPC2 regulates biliary cholesterol secretion via stimulation of ABCG5/G8-mediated cholesterol transport. Gastroenterology 2011;140:1664-1674
24. Shi XZ, Zhong X, Yu XQ. Drosophila melanogaster NPC2 proteins bind bacterial cell wall components and may function in immune signal pathways. Insect Biochem Mol Biol 2012;42:545-556
25. Liscum L, Klansek JJ. Niemann-Pick disease type C. Curr Opin Lipidol 1998;9:131-135
26. Liscum L, Ruggiero RM, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann-Pick type C fibroblasts. J Cell Biol 1989;108:1625-1636
27. Liscum L, Sturley SL. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim Biophys Acta 2004;1685:22-27
28. Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 2008;14:1247-1255
173 Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014
Niemann-Pick Type C Cholesterol Storage Disorder

29. Puri V, Watanabe R, Dominguez M, Sun X, Wheatley CL, Marks DL, Pagano RE. Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat Cell Biol 1999;1:386-388
30. Chevallier J, Chamoun Z, Jiang G, Prestwich G, Sakai N, Matile S, Parton RG, Gruenberg J. Lysobisphosphatidic acid controls endosomal cholesterol levels. J Biol Chem 2008;283:27871-27880
31. Zhou S, Davidson C, McGlynn R, Stephney G, Dobrenis K, Vanier MT, Walkley SU. Endosomal/lysosomal processing of gangliosides affects neuronal cholesterol sequestration in Niemann-Pick disease type C. Am J Pathol 2011;179:890-902
32. Yevenes LF, Klein A, Castro JF, Marin T, Leal N, Leighton F, Alvarez AR, Zanlungo S. Lysosomal vitamin E accumulation in Niemann-Pick type C disease. Biochim Biophys Acta 2012;1822:150-160
33. Group N-CGW, Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, Pineda M, Sedel F, Topcu M, Vanier MT, Widner H, Wijburg FA, Patterson MC. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab 2009;98:152-165
34. Pentchev PGV, M.T.; Suzuki, K. y Patterson, M.C. The Metabolic and Molecular Bases of Inherited Disease. In: Scriver CRB, A.L., Sly, W.S, y Valle, D., ed. New York: Mc-Graw Hill, 1995; 2625-2639
35. Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta 2004;1685:48-62
36. Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat Med 2004;10:704-711
37. Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet 2005;1:81-95
38. Alvarez AR, Klein A, Castro J, Cancino GI, Amigo J, Mosqueira M, Vargas LM, Yevenes LF, Bronfman FC, Zanlungo S. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J 2008;22:3617-3627
39. Yu T, Shakkottai VG, Chung C, Lieberman AP. Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum Mol Genet 2011;20:4440-4451
40. Lopez ME, Klein AD, Dimbil UJ, Scott MP. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J Neurosci 2011;31:4367-4378
41. Vanier MT. Niemann-Pick type C disease. Orphanet J Rare Dis 2010;5:16
42. Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F, Group N-CGW. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab 2012;106:330-344
43. Greer WL, Riddell DC, Byers DM, Welch JP, Girouard GS, Sparrow SM, Gillan TL, Neumann PE. Linkage of Niemann-Pick disease type D to the same region of human chromosome 18 as Niemann-Pick disease type C. Am J Hum Genet 1997;61:139-142
44. Greer WL, Riddell DC, Gillan TL, Girouard GS, Sparrow SM, Byers DM, Dobson MJ, Neumann PE. The Nova Scotia (type D) form of Niemann-Pick disease is caused by a G3097-->T transversion in NPC1. Am J Hum Genet 1998;63:52-54
45. Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, Olzeski D, Sidhu R, Dietzen DJ, Fu R, Wassif CA, Yanjanin NM, Marso SP, House J, Vite C, Schaffer JE, Ory DS. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med 2010;2:56ra81
46. Lund EG, I. B. Role of oxysterols in the regulation of cholesterol homeostasis: a critical evaluation. Accounts of Chemical Research 1995;28:241-249
47. Cluzeau CV, Watkins-Chow DE, Fu R, Borate B, Yanjanin N, Dail MK, Davidson CD, Walkley SU, Ory DS, Wassif CA, Pavan WJ, Porter FD. Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum Mol Genet 2012;21:3632-3646
48. Kandt RS, Emerson RG, Singer HS, Valle DL, Moser HW. Cataplexy in variant forms of Niemann-Pick disease. Ann Neurol 1982;12:284-288
49. M.C. Patterson MTV, K. Suzuki, J.A. Morris, E. Carstea, E.B. Neufeld, J.E., Blanchette-Mackie PGP. Niemann-Pick disease type C: a lipid trafficking disorder. In: C. R. Scriver WSS, D. Valle, ed. The Metabolic and Molecular Basis of Inherited Disease. New York,
NY, USA,: Mulencer Hill, 2001; 3611-363450. Josephs KA, Van Gerpen MW, Van Gerpen JA. Adult onset Niemann-
Pick disease type C presenting with psychosis. J Neurol Neurosurg Psychiatry 2003;74:528-529
51. Walterfang M, Kornberg A, Adams S, Fietz M, Velakoulis D. Post-ictal psychosis in adolescent Niemann-Pick disease type C. J Inherit Metab Dis 2010
52. Pipalia NH, Cosner CC, Huang A, Chatterjee A, Bourbon P, Farley N, Helquist P, Wiest O, Maxfield FR. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc Natl Acad Sci U S A 2011;108:5620-5625
53. Kim SJ, Lee BH, Lee YS, Kang KS. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem Biophys Res Commun 2007;360:593-599
54. Perez-Poyato MS, Pineda M. New agents and approaches to treatment in Niemann-Pick type C disease. Curr Pharm Biotechnol 2011;12:897-901
55. Platt FM, Neises GR, Dwek RA, Butters TD. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J Biol Chem 1994;269:8362-8365
56. Butters TD, Mellor HR, Narita K, Dwek RA, Platt FM. Small-molecule therapeutics for the treatment of glycolipid lysosomal storage disorders. Philos Trans R Soc Lond B Biol Sci 2003;358:927-945
57. Treiber A, Morand O, Clozel M. The pharmacokinetics and tissue distribution of the glucosylceramide synthase inhibitor miglustat in the rat. Xenobiotica 2007;37:298-314
58. Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol 2001;11:1283-1287
59. Lachmann RH, te Vruchte D, Lloyd-Evans E, Reinkensmeier G, Sillence DJ, Fernandez-Guillen L, Dwek RA, Butters TD, Cox TM, Platt FM. Treatment with miglustat reverses the lipid-trafficking defect in Niemann-Pick disease type C. Neurobiol Dis 2004;16:654-658
60. Di Rocco M, Dardis A, Madeo A, Barone R, Fiumara A. Early miglustat therapy in infantile Niemann-Pick disease type C. Pediatr Neurol 2012;47:40-43
61. Perez-Poyato MS, Gordo MM, Marfa MP. Initiation and discontinuation of substrate inhibitor treatment in patients with Niemann-Pick type C disease. Gene 2012;506:207-210
62. Sturley SL, Patterson MC, Pentchev P. Unraveling the sterol-trafficking defect in Niemann-Pick C disease. Proc Natl Acad Sci U S A 2009;106:2093-2094
63. Wang MS, Boddapati S, Sierks MR. Cyclodextrins promote protein aggregation posing risks for therapeutic applications. Biochem Biophys Res Commun 2009;386:526-531
64. Vance JE, Peake KB. Function of the Niemann-Pick type C proteins and their bypass by cyclodextrin. Curr Opin Lipidol 2011;22:204-209
65. Muralidhar A, Borbon IA, Esharif DM, Ke W, Manacheril R, Daines M, Erickson RP. Pulmonary function and pathology in hydroxypropyl-beta-cyclodextin-treated and untreated Npc1/ mice. Mol Genet Metab 2011;103:142-147
66. Matsuo M, Togawa M, Hirabaru K, Mochinaga S, Narita A, Adachi M, Egashira M, Irie T, Ohno K. Effects of cyclodextrin in two patients with Niemann-Pick Type C disease. Mol Genet Metab 2012
Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014174
Niemann-Pick Type C Cholesterol Storage Disorder

67. Smith D, Wallom KL, Williams IM, Jeyakumar M, Platt FM. Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol Dis 2009;36:242-251
68. Klein A, Maldonado C, Vargas LM, González M, Robledo F, Perez de Arce K, Muñoz FJ, Hetz C, Alvarez A, Zanlungo S. Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiology of Disease 2011;41:209-218
69. Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov 2002;1:493-502
70. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561-566
71. Morgan AJ, Platt FM, Lloyd-Evans E, Galione A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J 2011;439:349-374
72. Reddy JV, Ganley IG, Pfeffer SR. Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS One 2006;1:e19
73. Lloyd-Evans E, Platt FM. Lysosomal Ca(2+) homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011;50:200-205
74. Kaufmann AM, Krise JP. Niemann-Pick C1 functions in regulating lysosomal amine content. J Biol Chem 2008;283:24584-24593
75. Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, Xu H. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 2012;3:731
76. Yu T, Chung C, Shen D, Xu H, Lieberman AP. Ryanodine receptor antagonists adapt NPC1 proteostasis to ameliorate lipid storage in Niemann-Pick type C disease fibroblasts. Hum Mol Genet 2012;21:3205-3214
77. Davies JP, Ioannou YA. Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J Biol Chem 2000;275:24367-24374
78. Millat G, Marcais C, Rafi MA, Yamamoto T, Morris JA, Pentchev PG, Ohno K, Wenger DA, Vanier MT. Niemann-Pick C1 disease: the I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am J Hum Genet 1999;65:1321-1329
79. Chen FW, Li C, Ioannou YA. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One 2010;5:e15054
80. Xu M, Liu K, Swaroop M, Porter FD, Sidhu R, Finkes S, Ory DS, Marugan JJ, Xiao J, Southall N, Pavan WJ, Davidson C, Walkley SU, Remaley AT, Baxa U, Sun W, McKew JC, Austin CP, Zheng W. delta-Tocopherol Reduces Lipid Accumulation in Niemann-Pick Type C1 and Wolman Cholesterol Storage Disorders. J Biol Chem 2012;287:39349-39360
81. Fu R, Yanjanin NM, Bianconi S, Pavan WJ, Porter FD. Oxidative stress in Niemann-Pick disease, type C. Mol Genet Metab 2010;101:214-218
82. Ulatowski L, Parker R, Davidson C, Yanjanin N, Kelley TJ, Corey D, Atkinson J, Porter F, Arai H, Walkley SU, Manor D. Altered vitamin E status in Niemann-Pick type C disease. J Lipid Res 2011;52:1400-1410
83. Vazquez MC, Balboa E, Alvarez AR, Zanlungo S. Oxidative stress: a pathogenic mechanism for Niemann-Pick type C disease. Oxid Med Cell Longev 2012;2012:205713
84. Arakawa M, Ito Y. N-acetylcysteine and neurodegenerative diseases: Basic and clinical pharmacology. Cerebellum 2007 1-7
85. Atkuri KR, Mantovani JJ, Herzenberg LA. N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 2007;7:355-359
86. Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther 2008;8:1955-1962
87. Vazquez MC, del Pozo T, Robledo FA, Carrasco G, Pavez L, Olivares F, Gonzalez M, Zanlungo S. Alteration of gene expression profile in Niemann-Pick type C mice correlates with tissue damage and oxidative stress. PLoS ONE 2011;6:e28777
88. Aitio ML. N-acetylcysteine -- passe-partout or much ado about nothing? Br J Clin Pharmacol 2006;61:5-15
89. Fu R, Wassif CA, Yanjanin NM, Watkins-Chow DE, Baxter LL, Incao A, Liscum L, Sidhu R, Firnkes S, Graham M, Ory DS, Porter FD, Pavan WJ. Efficacy of N-acetylcysteine in phenotypic suppression of mouse models of Niemann-Pick disease,type C1. Hum Mol Genet 2013;22:3508-3523.
90. Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009;78:959-991
91. Gelsthorpe ME, Baumann N, Millard E, Gale SE, Langmade SJ, Schaffer JE, Ory DS. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J Biol Chem 2008;283:8229-8236
92. Munkacsi AB, Chen FW, Brinkman MA, Higaki K, Gutierrez GD, Chaudhari J, Layer JV, Tong A, Bard M, Boone C, Ioannou YA, Sturley SL. An "exacerbate-reverse" strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J Biol Chem 2011;286:23842-23851
93. Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 2009;4:e6951
175 Pediatric Endocrinology Reviews (PER) n Volume 12 n Supplement 1 n September 2014
Niemann-Pick Type C Cholesterol Storage Disorder





























