Embed Size (px)
Citation preview

Toward reduction in animal sacrificefor drugs: Molecular modeling ofMacaca fascicularis P450 2C20 forvirtual screening of Homo sapiensP450 2C8 substrates
Biotechnology andApplied Biochemistry
Francesco Rua,1 Giovanna Di Nardo,1,2 Sheila J. Sadeghi,1 and Gianfranco Gilardi1,2 ∗
1Department of Life Sciences and Systems Biology, University of Torino, Torino, Italy2Centre for Research and Development of Diffractometric Crystallography, University of Torino, Torino, Italy
Abstract.Macaca fascicularis P450 2C20 shares 92% identity withhuman cytochrome P450 2C8, which is involved in themetabolism of more than 8% of all prescribed drugs. To date,only paclitaxel and amodiaquine, two substrate markers of thehuman P450 2C8, have been experimentally confirmed as M.fascicularis P450 2C20 drugs. To bridge the lack of informationon the ligands recognized by M. fascicularis P450 2C20, in thisstudy, a three-dimensional homology model of this enzymewas generated on the basis of the available crystal structure ofthe human homologue P450 2C8 using YASARA. The resultsindicated that 90.0%, 9.0%, 0.5%, and 0.5% of the residues ofthe P450 2C20 model were located in the most favorable,
allowed, generously allowed, and disallowed regions,respectively. The root-mean-square deviation of the C-alphasuperposition of the M. fascicularis P450 2C20 model with theHomo sapiens P450 2C8 was 0.074 A, indicating a very highsimilarity of the two structures. Subsequently, the 2C20 modelwas used for in silico screening of 58 known P450 2C8substrates and 62 inhibitors. These were also docked in theactive site of the crystal structure of the human P450 2C8. Theaffinity of each compound for the active site of bothcytochromes proved to be very similar, meaning that the fewkey residues that are mutated in the active site of the M.fascicularis P450 do not prevent the P450 2C20 fromrecognizing the same substrates as the human P450 2C8.
c© 2012 International Union of Biochemistry and Molecular Biology, Inc.Volume 59, Number 6, November/December 2012, Pages 479–489 •E-mail: [email protected]
Keywords: animal drug testing, cytochrome P450, drug metabolism,molecular modeling, in silico screening
1. IntroductionUnited States Food and Drug Administration and the EuropeanMedicines Agency demand severe preclinical studies on animalsbefore a new drug can be deemed safe for human administra-tion. Macaca fascicularis represents the most important nonhu-man primate used as an animal model, thanks to its close evo-lutionary relationship to humans compared to other mammals[1–3]. One approach to reducing animal testing has been thedevelopment of electrochemical platforms capable of measur-ing the electrocatalytic signal of the immobilized enzyme [4],[5].Alternatively, the binding of potential substrates or inhibitorscan be carried out by in silico methodology using the three-dimensional (3D) structure or model of the relevant enzyme asa tool for screening drugs/inhibitors.
Abbreviations: CYP, cytochrome P450; M. fascicularis, Macaca fascicularis.∗Address for correspondence: Gianfranco Gilardi, PhD, Department of Life Sciences andSystems Biology, University of Torino, Via Accademia Albertina 13, 10123, Torino, Italy.Tel.: +39 011 6704593; Fax: +39 011 6704643; e-mail: [email protected] 29 July 2012; accepted 4 October 2012DOI: 10.1002/bab.1051Published online 18 December 2012 in Wiley Online Library(wileyonlinelibrary.com)
To date, the ortholog genes of the majority of the humancytochromes P450 (CYPs) have been identified in M. fasciculariswith a high sequence identity (between 90% and 99%). Theyinclude CYP1A1, CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6,CYP2E1, and CYP3A4/5/7 [1],[6], and several studies have re-ported similar expression levels when compared to their hu-man counterpart [7],[8]. On the basis of sequence similarities,it is often assumed that macaque P450 enzymes turn over thesame substrates recognized by the equivalent human enzyme,but it is known that differences in few amino acids can sig-nificantly affect substrate specificities [9],[10]. Furthermore, or-thologous enzymes do not automatically allow comparison ofcatalytic specificities between species [11] because gene dupli-cation events can occur during evolution that might produce par-alogs, pseudogenes, and truncated gene fragments [12], leadingto a possible different P450 metabolism profile [13],[14].
In contrast to the vast information available on drugmetabolism by human CYPs, little structural and functionalinformation is available on the P450 enzymes from M. fas-cicularis, with particular reference to the CYP2C family thathas been generated in mammals by gene duplication, lead-ing to a different number of isoforms in each animal species[11],[12].
479

The human CYP2C subfamily is composed of four members(CYP2C8, CYP2C9, CYP2C18, and CYP2C19) that are involvedtogether in the metabolism of more than 25%–30% of clini-cal drugs [9],[15]. To date, five active members of CYP2C havebeen identified in M. fascicularis (CYP2C18, CYP2C20, CYP2C43,CYP2C45, and CYP2C76) [16],[17]. Except for CYP2C76, whichdoes not have any ortholog in humans [16], all these enzymespresent a high sequence identity with the human P450 counter-parts belonging to the 2C subfamily.
Furthermore, preliminary catalytic data available in theliterature [16],[18] propose the M. fascicularis CYP2C subfamilyas a model for the corresponding human enzymes even if anextensive analysis of the ability of macaque CYP2C members tometabolize human drugs is still needed.
Human P450 2C8 is the key member of the CYP2C sub-family. Because of its large active site [19], it is involved in themetabolism of more than 50 clinical drugs [9],[15], including an-timalarial agents such as amodiaquine [20] and chloroquine [21],thiazolidinedione antidiabetic drugs such as troglitazone [19],rosiglitazone [22], and pioglitazone [23], statins such as ceriva-statin [24] and atorvastatin [25], opioids such as morphine [26],loperamide [24], and R-ibuprofen [27].
Macaca fascicularis P450 2C20 shares 92% sequenceidentity with human P450 2C8 [16] and, to date, only pacli-taxel and amodiaquine have been validated as substrates ofmacaque P450 2C20 [4],[16]. As in silico modeling of M. fasci-cularis P450 2C20 and docking calculations can represent animportant source for predicting the binding of a drug, in thiswork, we present the results of a 3D homology model of M. fas-cicularis P450 2C20 calculated using the crystal structure of thehuman P450 2C8 as a template [19],[28]. This model is validatedand used for in silico screening of the drugs metabolized byhuman P450 2C8.
2. Materials and methods2.1. Sequence analysisAn analysis of the percentage identity existing between M.fascicularis P450 2C20 and the main Homo sapiens CYPslisted on the website drnelson.uthsc.edu/cytochromeP450.html has been carried out by the European Bioinformatics Insti-tute ClustalW program available at www.npsa-pbil.ibcp.fr. Thesame program was also used for the multiple alignment of aminoacid sequences of the human P450 2C8 and M. fascicularis P4502C20.
2.2. Molecular modelingHomology modeling was used to build a 3D model of M. fasci-cularis P450 2C20. The structure of the human P450 2C8 (ProteinData Bank entry 2NNJ) at 2.28 A resolution [19], cocrystallizedwith the inhibitor felodipine, was used as a template to build themodel. The amino acid sequence of the P450 2C20 was uploadedin YASARA [29]. The quality of the P450 2C20 model was thenvalidated with PROCHECK, analyzing the Ramachandran plot[30].
The model of P450 2C20 was then used for in silico dockingexperiments to predict the affinity of M. fascicularis P450 2C20
for the substrates typical of the human P450 2C8. AutoDock 4.0(Scripps Research Institute, La Jolla, CA, USA) [31], embeddedinto the YASARA Structure package, was used to dock 58 sub-strates and 62 inhibitors registered on the Drug Bank database[32]. Results of the docking of the ligands to the receptor werecarried out with 15 × 15 × 15 A3 cell around the active site.Results of the docking runs of the ligand to the proteins weresorted by binding energy, where more positive energies indi-cate stronger binding. A t-test analysis on the binding energiesof both the proteins for the same substrate library was carriedout using the SigmaPlot 11.0 program (Systat, London, UK).
3. Results and discussion3.1. Model construction and validationMacaca fascicularis P450 2C20 shares 92% identity with humanP450 2C8 and >79% identity with the other main human CYPs(Table 1). This makes M. fascicularis P450 2C20 a good candi-date for interspecies comparisons to human P450 2C8. To thisdate, no 3D structure of M. fascicularis P450 2C20 is available,preventing in silico analysis of binding of potential substratesor inhibitors. For this reason, the amino acid sequence of theP450 2C20 (Fig. 1) was used by YASARA software [29] as input tosearch for the best template for building a homology model. Thecrystal structure of the human P450 2C8 cocrystallized with theinhibitor felodipine with a resolution limit of 2.28 A [19] (ProteinData Bank code 2NNJ), which resulted in the best template forbuilding the model shown in Fig. 2A.
The N-terminal region of P450 2C20 was not constructedbecause the coordinates of the corresponding region of hu-man P450 2C20 were not determined in the P450 2C8 crystalstructure [19]. The M. fascicularis P450 model and the humanP450 2C8 crystal structure present an almost complete super-imposition of polypeptide backbone, with a root-mean-squaredeviation of 0.074 A taking into account the Calpha. The hemecofactor was positioned in the core of the protein with the Fe co-ordinated to the sulfur of Cys435. The Fe–S distance predictedis 2.3 A. This residue aligns with Cys435 of the human P450 2C8that is known to be the residue involved in heme coordinationof the crystal structure.
The model was validated using PROCHECK [30]; the Ra-machandran plot (Fig. 2B) revealed that 90.0%, 9.0%, 0.5%,and 0.5% of the residues of the P450 2C20 model are locatedin the most favorable, allowed, generously allowed, and disal-lowed regions, respectively. The four amino acids found in thedisallowed region of the Ramachandran plot are Gln184, Arg377,Ser429, and Ile476. These amino acids are located in loop re-gions between the E and F helices, theβ2–1 andβ2–2 strands, K′′
and L helices, and β2–1 and β2–2 strands, respectively. It shouldbe noted that these residues correspond to the same residues inhuman P450 2C8 and also that the X-ray structure of this enzymeshows its location in disallowed regions of the Ramachandranplot. The PROCHECK G-factor value of −0.12 obtained for themodel also indicated a high quality of the constructed model.Together, the ProSA (Fig. 2C) and PROCHECK results revealedthat the optimized model was satisfactory and thus consideredreliable for the subsequent structural/functional studies.
480 Biotechnology and Applied Biochemistry

Table 1Amino acid identity of M. fascicularis P450 2C20 shared withH. sapiens counterparts
M. fascicularis P450 2C20
H. sapiens Strong WeakP450 2C8 Identity similarity similarity
P450 2C8 92.04 4.90 1.22P450 2C19 78.57 12.45 3.47P450 2C9 78.16 11.63 5.10P450 2C18 75.31 12.65 4.49P450 2E1 55.06 21.46 7.69P450 2A13 50.13 24.90 8.10P450 2B6 50.10 23.46 8.35P450 2F1 49.70 20.40 10.71P450 2A6 48.99 25.71 8.10P450 2A7 48.58 25.51 8.70P450 2S1 42.86 24.00 9.10P450 2D6 40.08 22.24 9.62P450 2J2 38.93 22.13 10.08P450 2W1 36.44 26.52 8.91P450 2R1 33.80 25.45 11.53P450 2U1 32.66 24.40 7.34P450 1A1 29.40 24.37 10.44P450 1B1 26.92 23.26 11.17P450 1A2 26.78 23.31 11.18P450 17A1 25.29 26.25 11.28P450 4V2 22.24 25.61 12.52P450 3A5 21.88 23.63 13.87P450 3A4 21.57 23.33 14.12P450 3A7 20.43 24.75 13.36P450 27B1 20.27 21.83 15.59P450 3A43 20.24 24.95 13.75P450 46 20.23 23.93 13.04P450 26C1 19.63 19.63 12.66P450 4X1 19.31 23.36 11.58P450 26A1 19.10 2.44 12.87P450 27C1 18.66 20.89 9.13P450 7B1 18.30 22.64 10.75P450 4A11 18.18 23.67 10.80P450 39A1 18.11 21.73 12.88P450 4F22 18.10 23.88 11.38P450 7A1 18.04 20.15 14.01P450 26B1 17.98 22.47 13.11P450 4F12 17.84 24.48 11.57P450 4F2 16.89 24.1 11.01P450 11A1 16.76 22.79 13.56P450 4F11 16.29 24.43 11.93P450 51 15.73 21.91 12.92P450 4F8 15.53 25.95 11.93P450 4B1 15.06 23.05 11.71
3.2. Key amino acid residuesCompared to most human CYPs [28], the crystal structure ofP450 2C8 exhibits a relatively large active-site cavity (1,438 A3)where several drugs, such as paclitaxel (Mr: 853.9) [33] and theinhibitor montelukast (Mr: 586.2) [34] can be accommodated.The volume of the cavity of human P450 2C8 active site is twicethat of the rabbit P450 2C5 structure (645 A3) [35], thanks to
Fig. 1. Multiple sequence alignment of M. fascicularis P4502C20 (2C20Mf) and H. sapiens P450 2C8 (2C8Hs). Solidlines below the sequences indicate regions of the sixputative substrate recognition sites (SRS) of P450 2C20.Identical residues (red) are indicated by an asterisk (*),similar residues by two dots (high similarity, green) or onedot (low similarity, blue). Alignment was obtained using theClustalW program (www.npsa-pbil.ibcp.fr).
a difference in two key amino acids, Ser114 and Ile476. Thesetwo amino acids are conserved in M. fascicularis P450 2C20and allow the active-site cavity to extend farther from the heme[28]. The active-site cavity volume of P450 2C20 is similar tothe human P450 2C8, with approximately 50 amino acids thatcan potentially contact the substrates [28]. The analysis of suchresidues shows that most of them are conserved in M. fascicu-laris P450 2C20 but, out of the nine substitutions found, five arenot conserved, as listed in Table 2. These residues are: Ile99 inthe B–B′ loop, Leu100 in the helix B′, Thr202 and Thr209 in thehelix F, and Ile386 in the β1–4 sheets. Conserved and noncon-served amino acids of M. fascicularis P450 2C20 correspondingto residues that can potentially contact substrates in the humanP450 2C8 are shown in Fig. 3.
The importance of some of these amino acids in humanP450 2C8 has been already demonstrated through site-directedmutagenesis studies [36].
Toward reduction in animal sacrifice for drugs 481
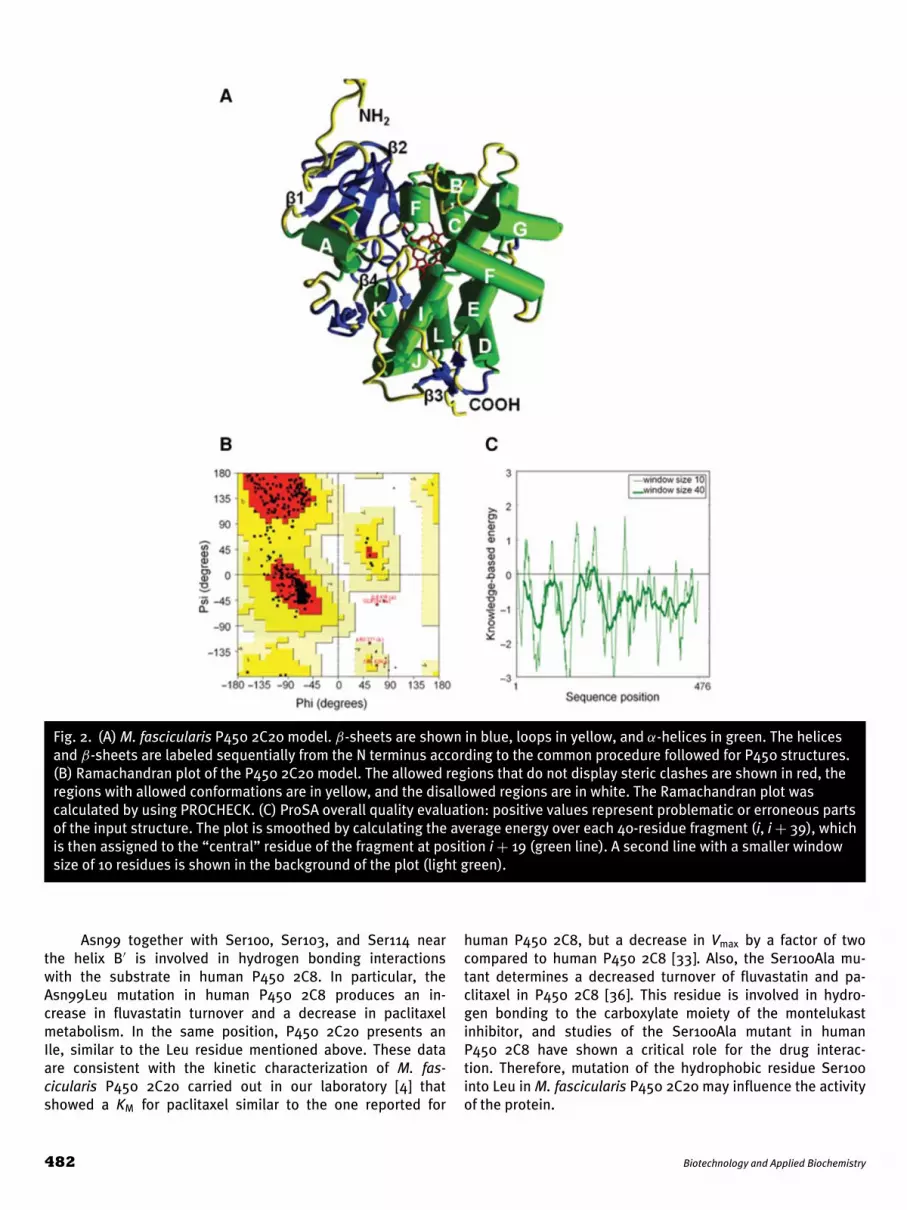
Fig. 2. (A) M. fascicularis P450 2C20 model. β-sheets are shown in blue, loops in yellow, and α-helices in green. The helicesand β-sheets are labeled sequentially from the N terminus according to the common procedure followed for P450 structures.(B) Ramachandran plot of the P450 2C20 model. The allowed regions that do not display steric clashes are shown in red, theregions with allowed conformations are in yellow, and the disallowed regions are in white. The Ramachandran plot wascalculated by using PROCHECK. (C) ProSA overall quality evaluation: positive values represent problematic or erroneous partsof the input structure. The plot is smoothed by calculating the average energy over each 40-residue fragment (i, i + 39), whichis then assigned to the “central” residue of the fragment at position i + 19 (green line). A second line with a smaller windowsize of 10 residues is shown in the background of the plot (light green).
Asn99 together with Ser100, Ser103, and Ser114 nearthe helix B′ is involved in hydrogen bonding interactionswith the substrate in human P450 2C8. In particular, theAsn99Leu mutation in human P450 2C8 produces an in-crease in fluvastatin turnover and a decrease in paclitaxelmetabolism. In the same position, P450 2C20 presents anIle, similar to the Leu residue mentioned above. These dataare consistent with the kinetic characterization of M. fas-cicularis P450 2C20 carried out in our laboratory [4] thatshowed a KM for paclitaxel similar to the one reported for
human P450 2C8, but a decrease in Vmax by a factor of twocompared to human P450 2C8 [33]. Also, the Ser100Ala mu-tant determines a decreased turnover of fluvastatin and pa-clitaxel in P450 2C8 [36]. This residue is involved in hydro-gen bonding to the carboxylate moiety of the montelukastinhibitor, and studies of the Ser100Ala mutant in humanP450 2C8 have shown a critical role for the drug interac-tion. Therefore, mutation of the hydrophobic residue Ser100into Leu in M. fascicularis P450 2C20 may influence the activityof the protein.
482 Biotechnology and Applied Biochemistry

Fig. 3. (A) Molecular structure of the inhibitor felodipine(drug bank code DB01023). Views of felodipine docked inthe active site of (B) the structure of human P450 2C8(Protein Data Bank file 2NNJ) and (C) the 3D model of M.fascicularis P450 2C20. Amino acids interacting with thesubstrate are shown in yellow; the heme is in red.
Table 2Amino acids involved in the substrate interaction in H.sapiens P450 2C8 and M. fascicularis P450 2C20; conservedsubstitutions are shown in bold and nonconserved in bolditalic
Secondarystructure motif
Human P4502C8dH
MacaqueP450 2C20dH
Helix A Ile50 Ile50Phe54 Phe54
β1–1 Phe69 Phe69β1–2 Asn72 Asn72
Ile74 Val74B–B′ loop Arg97 Arg97
Gly98 Gly98Asn99 Ile99
Helix B′ Ser100 Leu100Ile102 Ile102Ser103 Ser103Ile106 Ile106Thr107 Thr107
B′–C loop Ile113 Ile113Ser114 Ser114
Helix F Arg200 Arg200Phe201 Phe201Asn202 Thr202Asn204 Asn204Phe205 Phe205Leu208 Leu208Asn209 Thr209
Helix F′ Ile213 Ile213Gln214 Gln214Asn217 Asn217Asn218 Asn218
Helix G Val233 Leu233Asn236 Asn236Val237 Val237Thr240 Thr240Arg241 Lys241Arg297 Arg297Thr301 Thr301
Helix I Gly289 Gly289Ala292 Ala292Asp293 Asp293Val296 Val296Ala297 Ala297Thr301 Thr301
K-β1–3 loop leu361 leu361Val362 Val362Thr364 Thr364Gly365 Gly365val366 val366Pro367 Pro367
β1–4 Thr386 Ile386Met388 Met388
Sheet β4 Gly475 Gly475Ile476 Ile476Val477 Ile477
Toward reduction in animal sacrifice for drugs 483

Table 3P450 2C8 substrate binding energy values for M. fascicularis P450 2C20 model and human P450 2C8 crystal structure (ProteinData Bank file 2NNJ); the orientation of the poses in the docked structures is indicated with (+++) for high, (++) medium, and(+) low similarity
Drug bank Similarity of the ligand P450 2C8 binding P450 2C20 bindingcode pose in the active site energy (kcal/mol) energy (kcal/mol) Substrate Reference
DB00661 + 14.4 16.2 Verapamil [37]DB00231 ++ 10.8 10.2 Temazepam [38]DB00448 ++ 10.2 10.9 Lansoprazole [21]DB00918 +++ 9.8 9.9 Almotriptan [15]DB01198 +++ 9.8 9.3 Zopiclone [39]DB01124 +++ 9.7 9.5 Tolbutamide [40]DB00857 +++ 9.6 9.9 Terbinafine [41]DB00619 +++ 9.4 9.1 Imatinib [42]DB00818 +++ 9.3 10.0 Propofol [43]DB01037 ++ 9.3 8.8 Selegiline [44]DB00412 ++ 9.2 9.7 Rosiglitazone [45]DB00564 +++ 9.1 8.8 Carbamazepine [46]DB00586 + 9.1 8.0 Diclofenac [47]DB00250 +++ 9.0 8.8 Dapsone [48]DB00295 + 9.0 10.3 Morphine [26]DB00333 +++ 8.9 8.4 Methadone [49]DB00370 +++ 8.9 9.0 Mirtazapine [50]DB00197 +++ 8.9 8.9 Troglitazone [51]DB01120 +++ 8.8 8.6 Gliclazide [52]DB00850 ++ 8.8 9.4 Perphenazine [53]DB00755 +++ 8.8 8.6 Tretinoin [55],[56]DB00439 +++ 8.7 9.0 Cerivastatin [50]DB00495 +++ 8.7 8.7 Zidovudine [56]DB00281 +++ 8.6 9.5 Lidocaine [57]DB00531 +++ 8.5 8.3 Cyclophosphamide [58]DB00836 +++ 8.5 8.6 Loperamide [59]DB01050 ++ 8.3 7.7 Ibuprofen [22]DB01645 ++ 8.1 8.8 Genistein [60]DB01229 ++ 8.1 8.9 Paclitaxel [33]DB01132 ++ 8.0 8.5 Pioglitazone [23]DB00604 +++ 7.9 7.7 Cisapride [61]DB00972 ++ 7.8 8.1 Azelastine [62]DB00921 +++ 7.8 7.5 Buprenorphine [63]DB01218 ++ 7.8 7.2 Halofantrine [64]DB00214 ++ 7.7 7.1 Torasemide [65]DB00343 +++ 7.4 7.3 Diltiazem [66]DB01181 ++ 7.4 7.7 Ifosfamide [67]DB00912 ++ 7.4 7.0 Repaglinide [68]DB00440 ++ 7.4 7.5 Trimethoprim [69]DB00613 +++ 7.3 7.4 Amodiaquine [70]DB00788 +++ 7.3 7.5 Naproxen [71]DB01095 ++ 7.2 6.5 Fluvastatin [49]DB00201 +++ 7.1 7.0 Caffeine [72]DB00321 +++ 6.7 6.5 Amitriptyline [73]DB00338 +++ 6.7 6.8 Omeprazole [74]DB00622 +++ 6.6 6.9 Nicardipine [15]DB00675 +++ 6.5 6.8 Tamoxifen [44]DB00363 +++ 6.2 6.2 Clozapine [75]DB01076 +++ 5.8 5.6 Atorvastatin [25]DB00682 +++ 5.8 6.1 Warfarin [76]DB00829 +++ 5.6 5.4 Diazepam [77]DB00641 +++ 5.5 5.5 Simvastatin [78]DB01118 +++ 5.2 5.2 Amiodarone [79]DB01435 +++ 4.9 5.1 Antipyrine [80]DB00608 ++ 4.7 5.1 Chloroquine [57]DB00749 ++ 4.4 5.0 Etodolac [81]DB00347 +++ 4.4 4.5 Trimethadione [82]
484 Biotechnology and Applied Biochemistry

Table 4P450 2C8 inhibitor binding energy values for M. fascicularis P450 2C20 model and human P450 2C8 crystal structure (ProteinData Bank file 2NNJ); the orientation of the poses in the docked structures is indicated with (+++) for high, (++) medium, and(+) low similarity
Drug bank Similarity of the ligand P450 2C8 binding P450 2C20 bindingcode pose in the active site energy (kcal/mol) energy (kcal/mol) Substrate Reference
DB01229 + 16.2 14.4 Paclitaxel [20]DB00471 ++ 13.7 12.4 Montelukast [20]DB00796 + 12.2 9.9 Candesartan [20]DB00227 + 11.3 9.9 Lovastatin [20]DB00549 + 11.3 6.1 Zafirlukast [20]DB00641 ++ 10.9 10.2 Simvastatin [20]DB01136 ++ 10.8 10.0 Carvedilol [20]DB00997 ++ 10.8 11.4 Doxorubicin [83]DB01026 +++ 10.8 10.6 Ketoconazole [84]DB00481 +++ 10.5 10.2 Raloxifene [20]DB01149 ++ 10.3 11.0 Nefazodone [20]DB01076 ++ 10.2 10.8 Atorvastatin [85]DB00678 ++ 10.2 11.2 Losartan [20]DB00342 ++ 10.2 10.9 Terfenadine [20]DB01095 +++ 9.9 9.6 Fluvastatin [86]DB00603 +++ 9.9 9.4
Medroxyprogesterone[20]
DB00620 +++ 9.9 9.6 Triamcinolone [20]DB00197 +++ 9.9 9.8 Troglitazone [87]DB00918 +++ 9.8 9.9 Almotriptan [15]DB00764 ++ 9.8 10.5 Mometasone [20]DB01039 +++ 9.7 9.9 Fenofibrate [20]DB01234 +++ 9.6 9.7 Dexamethasone [20]DB04216 +++ 9.5 9.8 Quercetin [88]DB00959 +++ 9.3 9.2
Methylprednisolone[20]
DB00421 ++ 9.3 9.9 Spironolactone [20]DB00977 +++ 9.2 9.0 Ethinyl Estradiol [20]DB01062 +++ 9.2 8.6 Oxybutynin [20]DB00990 +++ 9.1 9.0 Exemestane [20]DB00455 +++ 9.0 8.4 Loratadine [20]DB01112 + 8.9 7.2 Cefuroxime [20]DB01029 +++ 8.9 8.8 Irbesartan [20]DB00468 +++ 8.8 8.9 Quinine [89]DB00381 +++ 8.7 9.1 Amlodipine [20]DB01132 +++ 8.6 8.8 Pioglitazone [90]DB00675 +++ 8.6 8.5 Tamoxifen [91]DB00938 ++ 8.5 8.2 Salmeterol [20]DB01129 ++ 8.4 8.9 Rabeprazole [20]DB00783 +++ 8.3 8.8 Estradiol [20]DB00412 +++ 8.3 8.5 Rosiglitazone [90]DB01115 +++ 8.2 8.1 Nifedipine [20]DB00897 ++ 8.2 8.7 Triazolam [89]DB00758 ++ 8.1 7.5 Clopidogrel [20]DB01241 +++ 8.0 7.9 Gemfibrozil [78]DB00482 +++ 7.8 8.4 Celecoxib [20]DB01104 ++ 7.6 8.0 Sertraline [20]DB00580 +++ 7.6 7.3 Valdecoxib [20]DB01023 +++ 7.5 8.2 Felodipine [20]DB00257 +++ 6.9 6.9 Clotrimazole [20]DB00683 +++ 6.7 6.8 Midazolam [89]DB00752 + 6.1 5.0 Tranylcypromine [20]DB00440 +++ 5.4 5.6 Trimethoprim [92]
Toward reduction in animal sacrifice for drugs 485

Table 5P450 2C8 suicide inhibitor binding energy values for M. fascicularis P450 2C20 model and human P450 2C8 crystal structure(Protein Data Bank file 2NNJ); the orientation of the poses in the docked structures is indicated with (+++) for high, (++)medium, and (+) low similarity
Drug bank Similarity of the ligand P450 2C8 binding P450 2C20 bindingcode pose in the active site energy (kcal/mol) energy (kcal/mol) Substrate Reference
DB01118 +++ 9.5 9.7 Amiodarone [93]DB00661 ++ 9.5 8.6 Verapamil [93]DB00622 +++ 9.1 9.4 Nicardipine [93]DB00343 +++ 9.0 8.7 Diltiazem [93]DB01388 ++ 8.6 9.4 Mibefradil [93]DB01241 +++ 8.0 7.9 Gemfibrozil [93]DB01023 ++ 7.5 8.2 Felodipine [93]DB00540 +++ 7.1 7.5 Nortriptyline [93]DB00472 ++ 7.0 7.3 Fluoxetine [93]DB01151 ++ 6.7 7.1 Desipramine [93]DB00951 ++ 5.5 4.9 Isoniazid [93]DB00780 +++ 5.4 5.2 Phenelzine [93]
Other important substitutions have been found in regionsof the protein that are not directly involved in substrate binding.One of these is Ile359, which corresponds to Ser359 in humanP450 2C8.
Mutant Ser359Ile in human protein [36] has been shownnot to have any effect on paclitaxel metabolism.
Other nonconserved differences between human andmacaque protein are Ile123Thr, Glu204Val, Thr374Ile, andAsp462Ala, but to date, there are no experimental data on theirimportance to substrate metabolism.
A conserved substitution is found at position 241 (Arg inH. sapiens, Lys in M. fascicularis). This amino acid does notbelong to the active-site cavity, but it is involved in the bindingof anionic substrates in the structure of the human P450 2C8[28] and has been shown to reorient to stabilize the negativelycharged functional groups [19].
3.3. Ligand dockingA library of compounds was built for virtual screening of bindingof these compounds to P450 2C20. The library is composed of:(i) 58 known substrates of human P450 2C8 (Table 3), (ii) 50known P450 2C8 inhibitors (Table 4), and (iii) 12 known P4502C8 suicide inhibitors (Table 5). These molecules were testedthrough local docking experiments into the active site of the M.fascicularis P450 2C20 model.
According to Melet et al. [36], computer-simulated dock-ing of P450 2C8 indicates that the large active-site cavity mayaccommodate the substrate in different orientations; neverthe-less, for the inhibitor felodipine (Fig. 3A), comparable poseswere detected in both the active site of human P450 2C8 (Fig. 3B)and M. fascicularis P450 2C20 (Fig. 3C). In any case, for all themolecules tested, the complexes chosen were those with thehighest binding energy that also shared the same orientation inhuman P450 2C8 and M. fascicularis P450 2C20.
Furthermore, as a proof of the predictive validity of the insilico approach, the orientation of the felodipine docked in the
active site of human P450 2C8 was quasi-identical to that of thestructure of human P450 2C8 cocrystalized with the inhibitor.
The large size of the active-site cavity of the two enzymesand the flexibility of the ligands allow for the presence of mul-tiple orientations of the bound molecules, as also observed inthe literature for many ligands, for example, in the case of flu-vastatin docked in the core of P450 2C8 [36].
For this reason, Tables 2–4 categorize the ligands in threegroups based on the similarity of the orientations found in theactive sites of two enzymes. Overall, the two enzymes displaya similar behavior in the way in which they bind their ligands.Although it is difficult to predict the nature of the productsfrom the in silico data, it is reasonable to expect that humanand M. fascicularis lead to the same or very similar products.It is also interesting to note that the highest degree of similar-ity in the orientations is shown by the data on the substrates(Table 3).
Carbon 4 of the dichlorophenyl ring of felodipine dockedin both the structures (Fig. 3) is positioned toward the hemeiron with a distance of 4.66 and 4.99 A for P450 2C8 and P4502C20, respectively. A weak torsion of around 3 A of the rest ofthe inhibitor structure has been noticed in M. fascicularis P450,probably because of the presence of mutated amino acids in theactive site in comparison to human protein. In particular, Schochet al. [19] have noted some deformation in the P450 2C8 crystalrelative to the original substrate-free structure, especially inthe vicinity of Ile476. This is because the conservative mutationVal477Ile between human and macaque is close to this residueand can influence the pose.
To compare the resulting binding modes and energies tothe ones of human P450 2C8, the same compounds present inthe ligand library were docked into the active site of humanP450 2C8.
Among these binding modes, the complex of protein–ligand bearing the highest binding energy calculated by YASARAwas considered. The binding energy outputs obtained from theanalysis of the P450 2C20 model and P450 2C8 structure were
486 Biotechnology and Applied Biochemistry

Fig. 4. Correlation between the binding energy output of M.fascicularis 2C20 versus H. sapiens P450 2C8 for (A) 58known substrates, (B) 50 known inhibitors, and (C) 12known suicide inhibitors of human P450 2C8. Predictionintervals for each regression line with a 95% confidencewere calculated with Sigma plot and are shown in red.
compared to determine whether both proteins can potentiallybind the same substrates and inhibitors (Fig. 4).
The energy of binding to human P450 2C8 found for eachgroup of compounds was plotted against the energy of bindingto the model of M. fascicularis P450 2C20. The resulting plotshows a linear trend well contained within the regression linesobtained applying a prediction interval with a confidence of95%.
From this analysis, zafirlukast (a leukotriene receptor an-tagonist) was found to be an outlier among the inhibitors. Thepose inside the active sites of the two proteins was indeed dif-ferent. In the case of P450 2C20, the inhibitor was found tointeract with residues of the B′-helix and β1–4 sheets. As forthe human P450 2C8, additional interactions were also presentwith residues of the F and I helices. These interactions are be-lieved to be responsible for the different orientations in the twoenzymes.
The binding energies of P450 2C20 model and P450 2C8for zafirlukast were 11.3 and 6.1 kcal/mol, respectively.
In conclusion, M. fascicularis represents the most impor-tant nonhuman primate species used in drug research becauseof its evolutionary closeness to humans, but to date there isvery little data on the substrates recognized by macaque P450enzymes.
Macaca fascicularis P450 2C20 represents a good modelcandidate of human P450 2C8, which is involved in the detoxifi-cation of more than 50 pharmaceutical drugs. Because in silicomethods are currently used in drug discovery for ligand screen-ing and profiling, M. fascicularis P450 2C20 has been modeledon the basis of the available crystal structure of human P4502C8 and used to predict the affinity for substrates and inhibitorsin the human counterpart. The model used is of good quality andno evident structural differences have been detected betweenP450 2C8 and P450 2C20. Both human and macaque homologueprotein showed a similar affinity for the same substrates, whichmeans that these proteins share a high sequence homology andstructural similarity that result in the ability to bind the samedrugs. Few amino acid substitutions were found in the activesite of M. fascicularis P450 2C20 that can account for the dif-ferent binding of a few specific compounds, such as substratezafirlukast.
Finally, the availability of a reliable 3D model of M. fas-cicularis P450 2C20, together with an in vitro electrochemicalplatform for testing its catalysis [4], will contribute to reducinganimal testing in this primate.
AcknowledgementsThe authors wish to acknowledge financial supportfrom the CIPE programme—Ricerca Industriale e SviluppoPrecompetitivo—Call 2006—Project: CYPTECH: Engineering al-ternative methods for P450 testing in animals and humans.Applications to drug metabolism. Regione Piemonte Italy.
References[1] Ebeling, M., Kung, E., See, A., Broger, C., Steiner, G., Berrera, M., Heckel,
T., Iniguez, L., Albert, T., Schmucki, R., Biller, H., Singer, T., and Certa, U.(2011) Genome Res. 21, 1746–1756.
Toward reduction in animal sacrifice for drugs 487

[2] Iwasaki, K., and Uno, Y. (2009) Xenobiotica 39, 578–581.[3] Ferguson, B., Street, S. L., Wright, H., Pearson, C., Jia, Y., Thompson, S.
L., Allibone, P., Dubay C. J., Spindel, E., and Norgren, R. B. Jr. (2007) BMCGenomics, 7, 8–43.
[4] Rua, F., Sadeghi, S. J., Castrignano, S., Di Nardo, G., and Gianfranco G.(2012) J. Inorg. Biochem, 117, 277–284.
[5] Sadeghi, S. J., Fantuzzi, A., and Gilardi, G. (2011) Biochim. Biophys. Acta—Proteins Proteomics. 1814, 237–248.
[6] Evans, W. E., and Relling, M. V. (1999) Science 286, 487–491[7] de Jonge, J., Kurian, S., Shaked, A., Reddy, K. R., Hancock, W., Salomon, D.
R., and Olthoff, K. M. (2009) Am. J. Transplant. 9, 758–772.[8] Shimada, T., Mimura, M., Inoue, K., Nakamura, S., Oda, H., Ohmori, S., and
Yamazaki, H. (1997) Arch. Toxicol. 71, 401–408.[9] Totah, R. A., and Rettie, A. E. (2005) Clin. Pharmacol. Ther. 77, 341–352.
[10] Lindberg, R. L., and Negishi, M. (1989) Nature 339, 632–634.[11] Nelson, D. R., Kamataki, T., Waxman, D. J., Guengerich, F. P., Estabrook,
R. W., Feyereisen, R., Gonzalez, F. J., Coon, M. J., Gunsalus, I. C., Gotoh, O.(1993) Cell Biol. 12, 1–51.
[12] Nelson, D. R., Zeldin, D. C., Hoffman, S. M., Maltais, L. J., Wain, H. M., andNerbedt, D. W. (2004) Pharmacogenetics 14, 1–18.
[13] Bogaards, J. J., van Ommen, B., Wolf, C. R., and van Bladeren, P. J. (1995)Toxicol. Appl. Pharmacol. 132, 44–52.
[14] Stevens, J. C., and Wrighton, S. A. (1993) J. Pharmacol. Exp. Ther. 266,964–971.
[15] Lai, X. S., Yang, L. P., Li, X. T., Liu, J. P., Zhou, Z. W., and Zhou, S. F. (2009)Curr. Drug Metab. 10, 1009–1047.
[16] Uno, Y., Fujino, H., Kito, G., Kamataki, T., and Nagata, R. (2006) Mol.Pharmacol. 70, 477–486.
[17] Uno, Y., Fujino, H., Iwasaki, K., and Utoh, M. (2010) Curr. Drug Metab. 11,142–152.
[18] Uno, Y., Hosaka, S., Matsuno, K., Nakamura, C., Kito, G., Kamataki, T., andNagata, R. (2007) Arch. Biochem. Biophys. 466, 98–105.
[19] Schoch, G. A., Yano, J. K., Sansen, S., Dansette, P. M., Stout, C. D., andJohnson, E. F. (2008) J. Biol. Chem. 20, 17227–17237.
[20] Walsky, R. L., Gaman, E. A., and Obach, R. S. (2005) J. Clin. Pharmacol. 45,68–78.
[21] Kim, K. A., Chung, J., Jung, D. H., and Park, J. Y. (2004) Eur. J. Clin. Pharmacol.60, 575–581.
[22] Daily, E., and Aquilante, C. (2009) Pharmacogenomics 10, 1489–1510.[23] Jaakkola, T., Laitila, J., Neuvonen, P. J., and Backman, J. T. (2006) Basic Clin.
Pharmacol. Toxicol. 99, 44–51.[24] Wang, J. S., and DeVane, C. L. (2003) Drug Metab. Dispos. 31, 742–747.[25] Jacobsen, W., Kuhn, B., Soldner, A., Kirchner, G., Sewing, K. F., Kollman, P.
A., Benet, L. Z., and Christians, U. (2000) Drug Metab. Dispos. 28, 1369–1378.
[26] Projean, D., Morin, P. E., and Tu, T. M. (2003) Xenobiotica 33, 841–854.[27] Chang, S. Y., Li, W., Traeger, S. C., Wang, B., Cui, D., Zhang, H., Wen, B.,
and Rodrigues, A. D. (2008) Drug Metab. Dispos. 36, 2513–2522.[28] Schoch, G. A., Yano, J. K., Wester, M. R., Griffin, K. J., Stout, C. D., and
Johnson, E. F. (2004) J. Biol. Chem. 279, 9497–9503.[29] Krieger, E., Koraimann, G., and Vriend, G. (2002) Proteins 47, 393–402.[30] Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R., and
Thornton, J. M. (1996) J. Biomol. NMR 8, 477–486[31] Goodsell, D. S., Morris, G. M., and Olson, A. J. (1996) J. Mol. Recognit. 9,
1–5.[32] Wishart, D. S., Knox, C., Guo, A. C., Cheng, D., Shrivastava, S., Tzur, D.,
Gautam, B., and Hassan Ali, M. (2008) Nucleic Acids Res. 36 (Databaseissue):D901–D906.
[33] Cresteil, T., Monsarrat, B., Dubois, J., Sonnier, M., Alvinerie, P., andGueritte, F. (2002) Drug Metab. Dispos. 30, 438–445.
[34] VandenBrink, B. M., Foti, R. S., Rock, D. A., Wienkers, L. C., and Wahlstrom,J. L. (2011) Drug Metab. Dispos. 39, 1546–1554.
[35] Williams, P. A., Cosme, J., Sridhar, V., Johnson, E. F., and McRee, D. E.(2000) Mol. Cell 5, 121–131.
[36] Melet, A., Marques-Soares, C., Schoch, G. A., Macherey, A. C., Jaouen,M., Dansette, P. M., Sari, M. A., Johnson, E. F., and Mansuy, D. (2007)Biochemistry 43, 15379–15392.
[37] Tracy, T. S., Korzekwa, K. R., Gonzalez, F. J., and Wainer, I. W. (1999) Br. J.Clin. Pharmacol. 47, 545–552.
[38] Yang, T. J., Shou, M., Korzekwa, K. R., Gonzalez, F. J., Gelboin, H. V., andYang, S. K. (1998) Biochem. Pharmacol. 55, 889–896.
[39] Becquemont, L., Funck-Brentano, C., and Jaillon, P. (1999) Fundam. Clin.Pharmacol. 13, 232–236.
[40] Dansette, P. M., Amar, C., Smith, C., Pons, C., and Mansuy, D. (1990)Biochem. Pharmacol. 39, 911–918.
[41] Vickers, A. E., Sinclair, J. R., Zollinger M., Heitz, F., Glanzel, U., Johanson L.,and Fischer V. (1999) Drug Metab. Dispos. 27, 1029–1038.
[42] Nebot, N., Crettol, S., d’Esposito, F., Tattam, B., Hibbs, D., and Murray M.(2010) Br. J. Pharmacol. 161, 1059–1069.
[43] Guitton, J., Buronfosse T., Desage M., Flinois J. P., Perdrix J. P., Brazier J. L.,and Beaune P. (1992) Br. J. Anaesth. 80, 788–795.
[44] Salonen, J. S., Nyman, L., Boobis, A. R., Edwards, R. J., Watts, P., Lake, B.G., Price, R. J., Renwick, A. B., Gomez-Lechon, M. J., Castell, J. V., Ingelman-Sundberg, M., Hidestrand, M., Guillouzo, A., Corcos, L., Goldfarb, P. S.,Lewis, D. F., Taavitsainen, P., and Pelkonen, O. (2003) Drug Metab. Dispos.31, 1093–1102.
[45] Baldwin, S. J., Clarke, S. E., and Chenery, R. J. (1999) Br. J. Clin. Pharmacol.48, 424–432.
[46] Gu, L., Gonzalez, F. J., Kalow, W., and Tang, B. K. (1992) Pharmacogenetics2, 73–77
[47] Mancy, A., Antignac, M., Minoletti, C., Dijols, S., Mouries, V., Duong, N.T., Battioni, P., Dansette P. M., and Mansuy, D. (1999) Biochemistry 38,14264–14270.
[48] Winter, H. R., Wang, Y., and Unadkat, J. D. (2000) Drug Metab. Dispos. 28,865–868.
[49] Wang, J. S., Neuvonen, M., Wen, X., Backman, J. T., and Neuvonen, P. J.(2002) Drug Metab. Dispos. 30, 1352–1356.
[50] Stormer, E., von Moltke, L. L., Shader, R. I., and Greenblatt, D. J. (2000)Drug Metab. Dispos. 28, 1168–1175 .
[51] Yamazaki, H., Shibata, A., Suzuki, M., Nakajima, M., Shimada, N., Guen-gerich, F. P., and Yokoi, T. (1999) Drug Metab. Dispos. 27, 1260–1266.
[52] Elliot, D. J., Suharjono, M. S., Lewis, B. C., Gillam, E. M., Birkett, D. J., Gross,A. S., and Miners, J. O. (2007) Br. J. Clin. Pharmacol. 64, 450–457.
[53] Olesen, O. V., and Linnet, K. (2000) Br. J. Clin. Pharmacol. 50, 563–571.[54] Marill, J., Cresteil, T., Lanotte, M., and Chabot, G. G. (2000) Mol. Pharmacol.
58, 1341–1348.[55] Eagling, V. A., Howe, J. L., Barry, M. J., and Back, D. J. (1994) Biochem.
Pharmacol. 48, 267–276.[56] Nadin, L., and Murray, M. (1999) Biochem. Pharmacol. 58, 1201–1208.[57] Imaoka, S., Enomoto, K., Oda, Y., Asada, A., Fujimori, M., Shimada, T.,
Fujita, S., Guengerich, F. P., and Funae, Y. (1990) J. Pharmacol. Exp. Ther.255, 1385–1391.
[58] Yu, L., and Waxman, D. J. (1996) Drug Metab. Dispos. 24, 1254–1262.[59] Kim, K. A., Kim, M. J., Park, J. Y., Shon, J. H., Yoon, Y. R., Lee, S. S., Liu, K.
H., Chun, J. H., Hyun, M. H., and Shin, J. G. (2003) Drug Metab. Dispos. 31,1227–1234.
[60] Hu, M., Krausz, K., Chen, J., Ge, X., Li, J., Gelboin, H. L., and Gonzalez, F. J.(2003) Drug Metab. Dispos. 317, 924–931.
[61] Desta, Z., Soukhova, N., Mahal, S. K., and Flockhart, D. A. (2000) DrugMetab. Dispos. 28, 789–800.
[62] Nakajima, M., Nakamura, S., Tokudome, S., Shimada, N., Yamazaki, H.,and Yokoi, T. (1999) Drug Metab. Dispos. 27, 1381–1391.
[63] Hanna, I. H., Krauser, J. A., Cai, H., Kim, M. S., and Guengerich, F. P. (2001)J. Biol. Chem. 276, 39553–39561.
[64] Halliday, R. C., Jones, B. C., Smith, D. A., Kitteringham, N. R., and Park, B.K. (1995) Br. J. Clin. Pharmacol. 40, 369–378.
[65] Miners, J. O., Rees, D. L., Valente, L., Veronese, M. E., and Birkett, D. J.(1995) J. Pharmacol. Exp. Ther. 272, 1076–1081.
[66] Molden, E., Asberg, A., and Christensen, H. (2000) Eur. J. Clin. Pharmacol.56, 575–579.
[67] Chen, C. S., Lin, J. T., Goss, K. A., He, Y. A., Halpert, J. R., and Waxman, D. J.(2004) Mol. Pharmacol. 65, 1278–1285.
[68] Kajosaari, L. I., Laitila, J., Neuvonen, P. J., and Backman, J. T. (2005) BasicClin. Pharmacol. Toxicol. 97, 249–256.
[69] Damsten, M. C., de Vlieger, J. S., Niessen, W. M., Irth, H., Vermeulen, N. P.,and Commandeur, J. N. (2008) Chem. Res. Toxicol. 21, 2181–2187.
[70] Gil, J. P. (2008) Pharmacogenomics 9, 1385–1390.
488 Biotechnology and Applied Biochemistry

[71] Rodrigues, A. D., Kukulka, M. J., Roberts, E. M., Ouellet, D., and Rodgers,T. R. (1996) Drug Metab. Dispos. 24, 126–136.
[72] Picard, N., Cresteil, T., Djebli, N., and Marquet, P. (2005) Drug Metab.Dispos. 33, 689–695.
[73] Venkatakrishnan, K., von Moltke, L. L., and Greenblatt, D. J. (2001) J. Phar-macol. Exp. Ther. 297, 326–337.
[74] Karam, W. G., Goldstein, J. A., Lasker, J. M., and Ghanayem, B. I. (1996)Drug Metab. Dispos. 24, 1081–1087.
[75] Eiermann, B., Engel, G., Johansson, I., Zanger, U. M., and Bertilsson, L.(1997) Br. J. Clin. Pharmacol. 44, 439–446.
[76] Hermans, J. J., and Thijssen, H. H. (1993) Br. J. Pharmacol. 110, 482–490.[77] Ono, S., Hatanaka, T., Miyazawa, S., Tsutsui, M., Aoyama, T., Gonzalez, F.
J., and Satoh, T. (1996) Xenobiotica 26, 1155–1166.[78] Prueksaritanont, T., Ma, B., and Yu, N. (2003) Br. J. Clin. Pharmacol. 56,
120–124.[79] Ohyama, K., Nakajima, M., Nakamura, S., Shimada, N., Yamazaki, H., and
Yokoi, T. (2000) Drug Metab. Dispos. 28, 1303–1310.[80] Engel, G., Hofmann, U., Heidemann, H., Cosme, J., and Eichelbaum, M.
(1996) Clin. Pharmacol. Ther. 59, 613–623.[81] Tougou, K., Gotou H., Ohno, Y., and Nakamura, A. (2004) Xenobiotica 34,
449–461.[82] Tanaka, E., Kurata, N., and Yasuhara, H. (2003) J. Clin. Pharm. Ther. 28,
493–496.
[83] Bun, S. S., Ciccolini, J., Bun, H., Aubert, C., and Catalin, J. (2003) J.Chemother. 15, 266–274.
[84] O’Donnell, C. J., Grime, K., Courtney, P., Slee, D., and Riley, R. J. (2007)Drug Metab. Dispos. 35, 381–385.
[85] Tornio, A., Pasanen, M. K., Laitila, J., Neuvonen, P. J., and Backman, J. T.(2005) Basic Clin. Pharmacol. Toxicol. 97, 104–108.
[86] Fischer, V., Johanson, L., Heitz, F., Tullman, R., Graham, E., Baldeck,J. P., and Robinson, W. T. (1999) Drug Metab. Dispos., 27, 410–416.
[87] Yamazaki, H., Suzuki, M., Tane, K., Shimada, N., Nakajima, M., and Yokoi,T. (2000) Xenobiotica 30, 61–70.
[88] Donato, M. T., Jimenez, N., Castell, J. V., and Gomez-Lechon, M. J. (2004)Drug Metab. Dispos. 32, 699–706.
[89] Ong, C. E., Coulter, S., Birkett, D. J., Bhasker, C. R., and Miners, J. O. (2000)Br. J. Clin. Pharmacol. 50, 573–580.
[90] Sahi, J., Black, C. B., Hamilton, G. A., Zheng, X., Jolley, S., Rose K. A., Gilbert,D., LeCluyse, E. L., and Sinz, M. W. (2003) Drug Metab. Dispos. 31, 439–446.
[91] Mancy, A., Dijols, S., Poli, S., Guengerich, P., and Mansuy, D. (1996) Bio-chemistry 35, 16205–16212.
[92] Wen, X., Wang, J. S., Backman, J. T., Laitila, J., and Neuvonen, P. J. (2002)Drug Metab. Dispos. 30, 631–635.
[93] Polasek, T. M., Elliot, D. J., Lewis, B. C., and Miners, J. O. (2004) J. Pharmacol.Exp. Ther. 311, 996–1007.
Toward reduction in animal sacrifice for drugs 489





























