Embed Size (px)
Citation preview

Tritium contamination of hematopoietic stem cells alters long-term hematopoietic
reconstitution
Fabio Di Giacomo1, Christine Granotier2, Vilma Barroca1, David Laurent1, François D.
Boussin2, Daniel Lewandowski1, Yannick Saintigny1 and Paul-Henri Romeo1, *
1 Laboratoire de recherche sur la Réparation et la Transcription dans les cellules Souches,
2 Laboratoire de Radiopathologie,
UMR INSERM-Paris VII-Paris XI U967, Institut de Radiobiologie Cellulaire et Moléculaire,
Commissariat à l'Énergie Atomique (CEA), Direction des Sciences du Vivant (DSV), 92265
Fontenay-aux-Roses
Running Title: Effects of Tritium on hematopoietic stem cells
*Correspondence: [email protected]
Conflict of interest statement. None declared.

Di Giacomo, F., Granotier, C., Barroca, V., Laurent, D., Boussin, F.D., Lewandowski, D.,
Saintigny, Y. and Romeo, P.H. Tritium contamination of hematopoietic stem cells alters long-
term hematopoietic reconstitution. Radiat. Res.
Abstract
In vivo effects of manufactured tritium from nuclear weapon testing, nuclear power plant, and
industry or research are poorly documented. Here, we contaminate mouse hematopoietic stem
cells with low, medium and high concentrations of [3H] thymidine and characterize the
biological properties of these contaminated hematopoietic stem cells in vitro and in vivo.
Proliferation, viability and double strand breaks were dependent of the [3H] thymidine dose
used for contamination but the in vitro myeloid differentiation of hematopoietic stem cells
was not affected by any doses of [3H] thymidine contamination. Transplantation of
contaminated hematopoietic stem cells into lethally irradiated mice compromised their long-
term capacity of hematopoietic reconstitution even after low doses of [3H] thymidine
contamination and their differentiation potential into B-lymphoid lineage only after medium
and high doses of [3H] thymidine contamination. Finally, competition experiments showed a
diminished capacity of hematopoietic stem cells contaminated with medium or high doses of
[3H] thymidine to reconstitute hematopoiesis. These results indicate that contaminations of
hematopoietic stem cells with doses of tritium that do not result in death of these cells have
long term effects on hematopoiesis.

Introduction
Tritium is naturally produced in the atmosphere by the interaction of high-energy
cosmic radiation with oxygen and nitrogen and by ternary fission in geological formations.
Since 1954, a significant portion of tritium in the atmosphere has resulted from the release of
large amounts of manufactured tritium into the environment from nuclear weapon testing,
nuclear power plant, and industrial or research uses of tritiated compounds. Much of the
tritium that remains in the environment exists as tritiated water (HTO) but it can also be found
as organically bound tritium (OBT). Both OBT and HTO may contaminate organisms after
ingestion or inhalation and OBT can then be incorporated into numerous biochemical
compounds (1). Because of the low disintegration energy, tritium’s biological effects cannot
come from external exposure but from integration of OBT into tissue resulting in an in situ
chronic auto-irradiation of the contaminated cells. Consequently, the energy deposition is
concentrated in the sub-cellular compartment in which the OBT is incorporated.
Little information is available on long-term biological effects of contamination by OBT
of living animals but the characterization of somatic stem cells present in most adult tissues
has now opened a new field of research on the long-term effects of OBT in living animals.
The adult somatic stem cells are tissue specific, can remain quiescent, undergo apoptosis, self-
renew or differentiate into progenitors (2). In addition, the adult somatic stem cells can
reconstitute long term biological functions of an injured organ from which they are specific
(3-5). Thus, contamination of somatic stem cells by OBT can be used to study the long-term
effects of contamination of a specific tissue or organ by OBT in living animals. The best-
characterized mammalian adult somatic stem cells are the hematopoietic stem cells (HSCs).
HSCs are the only cells in the hematopoietic system that can self-renew for life and that can
produce life-long complete hematopoietic reconstitution after transplantation in a γ-lethally
irradiated recipient mouse. In vivo, the maintenance and development of HSCs in the bone

marrow are dependent on the hematopoietic stem cell niche through niche-regulating
pathways that can protect HSCs from endogenous and exogenous genotoxic stresses. Finally,
HSCs can be purified close to homogeneity making their use for experimental research easy.
Of all tissues, the hematopoietic system is the most radiosensitive and hematopoietic
stem cells have already been used to study the effects of γ-irradiation on their biological
properties both in vitro and in vivo. The bone marrow failure induced by γ-irradiation is due to
HSCs but not progenitors DNA damages (6-8) and at the molecular level, γ-irradiation
induces DNA double-strand breaks whose repair is delayed in HSCs (9) and HSCs exhibited
limiting DNA repair during ageing (7, 8). The p53 signaling is a critical pathway that
responds to ionizing radiation by regulating proliferation, DNA repair and survival (10, 11).
Puma (p53 up-regulated mediator of apoptosis) is a direct p53 target gene that is essential for
hematopoietic cell death triggered by ionizing radiation and recently deletion of Puma has
been shown to specifically protect hematopoietic stem cells against high-dose γ-irradiation
(12, 13). In contrast to γ-irradiation, no data are available on the effects of tritium
contamination on the biological properties of HSCs as studies have been limited to
contaminations of transformed hematopoietic cell lines (14). This study showed that DNA
incorporation of tritiated thymidine influenced in a dose dependent manner cell proliferation
and viability in most of the cell lines studied but did not document the effects of tritium
contamination on differentiation and/or self-renewal.
To analyze the effects of OBT on hematopoietic stem cells, we have contaminated HSCs with
different doses of [3H] thymidine and studied the effects of these contaminations both in vitro
and in vivo. Short term effects of HSCs contamination by [3H] thymidine were studied in vivo
by primary transplantations and competitive experiments and long-term effects of [3H]
thymidine contamination was studied in vivo using secondary transplantations.

Results
Efficient contamination of HSC by [3H] thymidine
A hematopoietic cell population enriched in hematopoietic stem cells (the c-kit+, lin-,
Sca+ (KLS) hematopoietic cells) was purified by cell sorting from C57Bl/6 mice bone
marrow. The KLS hematopoietic cells were grown in a culture medium containing [3H]
thymidine concentrations that ranged from 0.37 to 37.03 kBq/ml during twenty-four hours
and the amount of [3H] thymidine incorporated into DNA was determined. A dose dependent
incorporation of [3H] thymidine was found in the contaminated KLS hematopoietic cells
without any DNA saturation (Figure 1A). These [3H] thymidine incorporations in KLS cells
DNA can be due to few cells highly contaminated or to many cells contaminated with few
molecules of [3H] thymidine. Autoradiography was used to analyze [3H] thymidine
incorporation into DNA of individual KLS hematopoietic cells and showed that, at low
concentration of [3H] thymidine (0.74 kBq/ml) almost 50% of the KLS cells were positive for
radioactive DNA with an average of two (1-5) silver grains per cell (Figure 1B and Figure
1C). This percentage of positive KLS cells increased with [3H] thymidine concentration
reaching 88% of positive KLS cells with an average of up to 23 silver grains per cell when a
[3H] thymidine concentration of 7.4 kBq/ml was used (Figure 1B and Figure 1C). Finally, a
detailed analysis of the number of silver grains showed heterogeneity in the number of silver
grains present in the KLS cells even when a [3H] thymidine concentration of 7.4 kBq/ml was
used (Figure 1C) with around 10% of KLS cells without any [3H] thymidine incorporation.
These results showed that [3H] thymidine could be incorporated into the DNA of KLS cells
even at low doses of contamination and that the KLS cells is heterogeneous in term of [3H]
thymidine incorporation into DNA, heterogeneity that could not be suppress even at high
concentration of [3H] thymidine.

Proliferation and viability of KLS hematopoietic cells contaminated with low or high
doses of [3H] thymidine
Irradiation of the nucleus induced DNA damages that can arrest cell cycle and
proliferation and/or induce apoptosis. To evaluate the genotoxic effect of [3H] thymidine
contamination on KLS cells, cell proliferation and viability of contaminated KLS cells were
determined. KLS were grown in culture medium containing a concentration of 3.7, 7.4, 14.8
or 37.03 kBq/ml of [3H] thymidine and proliferation and apoptosis were determined after 24
and 48 hours of culture. At the concentration of 3.7 kBq/ml of [3H] thymidine, cell
proliferation was only lightly affected after 24 hours incubation but, after 48 hours of culture,
a two-fold difference of proliferation was detected between cells cultured in the presence of
3.7 kBq/ml of [3H] thymidine and controls (Figure 2A). Increasing [3H] thymidine
concentration resulted in a two-fold (resp. five-fold) reduced proliferation after 24 hours and a
five-fold (resp. ten-fold) reduction after 48 hours of culture in a medium containing a [3H]
thymidine concentration of 7.4 kBq/ml (resp.14.8 kBq/ml) and no detectable proliferation
when a [3H] thymidine concentration of 37.03 kBq/ml was used in the culture medium
(Figure 2A). We finally studied apoptosis of KLS cells grown in culture medium containing
3.7 or 14.8 kΒq /ml of [3H] thymidine and showed than 20% of apoptotic cells could be
detected after 24 hours of incubation with these two concentrations of [3H] thymidine whereas
45% (resp.55%) of apoptotic cells were found after 48 hours of culture of KLS cells in culture
medium containing a concentration of [3H] thymidine of 3.7 kBq/ml (resp. 14.8 kBq/ml)
(Figure 2B). Taken together, these results show that a dose dependent effect of [3H] thymidine
on proliferation of KLS hematopoietic cells, a small effect of [3H] thymidine incorporation on
apoptosis of this hematopoietic population after 24 hours of culture and a significant effect of

[3H] thymidine incorporation on apoptosis of this hematopoietic population after 48 hours of
culture.
Contamination with [3H] thymidine induces formation of γ-H2AX Foci
Since [3H] thymidine incorporation into DNA can induce double strand breaks (DSBs)
in immortalized cells (15), we analyzed the formation of γ-H2AX foci after incorporation of
[3H] thymidine into DNA of KLS cells at non-lethal doses. After generation of DSBs, H2AX
is phosphorylated (γ-H2AX) and is detectable as nuclear foci at the damaged sites (16) in a
linear relationship with the number of induced DSBs (17). The percentages of cells with γ-
H2AX foci increased significantly as a function of incorporated [3H] thymidine but required
at least a [3H] thymidine concentration of 3.7 kBq/ml in the culture medium to be significant
(Figure 3A). We then determined the mean number of γ-H2AX foci per cell after [3H]
thymidine incorporation and showed that the median number of γ-H2AX foci per KLS cell
increased as a function of [3H] thymidine concentration present in the culture medium (Figure
3B). Foci form immediately after irradiation, after which they disappear within few hours due
to DSB repair. In the present work, γ-H2AX foci were analyzed after 24 hours of culture in
the presence of [3H] thymidine and thus, the frequencies of γ-H2AX foci detected resulted
from the countervailing influences of DSB repair (focus extinction) and the generation of new
DSBs (focus formation) at the time of monitoring. Taken together, these data indicate that
[3H] thymidine incorporation into DNA of KLS cells could induce DSBs in these cells.
Myeloid potential of KLS hematopoietic cells contaminated with low or high doses of
[3H] thymidine
Colony Forming Unit Assay (CFU) was performed to study the effects of [3H]
thymidine contamination on KLS cells differentiation into the myeloid lineage. 300 purified

KLS cells were cultured on a semi-solid methylcellulose media after 24 hours incubation in
medium containing [3H] thymidine concentrations of 3.7 and 14.8 kBq/ml. After 7 days of
culture in methylcellulose, the number and quality of the myeloid colonies obtained were
analyzed. No significant difference in the total number of colonies could be detected (Figure
4A). Qualitatively, we only found a small but not significant decrease of CFU-GEMM and a
small increase of CFU-GM when KLS cells were previously grown in the presence of [3H]
thymidine concentration of 14.8 kBq/ml and no difference when KLS cells were previously
grown in the presence of [3H] thymidine concentration of 3.7 kBq/ml (Figure 4B). These
results indicated that the myeloid progeny of contaminated KLS cells is not (resp. little)
altered by low (resp.high) dose of [3H] thymidine contamination.
Hematopoietic reconstitution with KLS hematopoietic cells contaminated with low or
high doses of [3H] thymidine
Lethally γ-irradiated (11Gy) mice expressing the CD45.1 antigen (CD45.1+) were
transplanted with 20,000 KLS expressing the CD45.2 antigen (CD45.2+) and cultured during
24 hours in media containing three different concentrations of [3H] thymidine (1.48, 3.7 or
14.8 kBq/ml). Long-term reconstituted hematopoiesis was studied 4 months after
transplantation. Whatever levels of contamination, [3H] thymidine contaminated KLS cells
were able to reconstitute long-term hematopoiesis in lethally irradiated mice as shown by
more than 90% of hematopoietic cells expressing the CD45.2 antigen in the reconstituted
hematopoieisis (data not shown). This result indicated that contamination of HSCs by [3H]
thymidine didn’t alter their reconstitution abilities. Contamination with 14.8 kBq/ml was of
high interest, as less than 10% of KLS cells were not contaminated when a concentration of
7.4 kBq/ml (i.e. two times less than 14.8 kBq/ml) of [3H] thymidine was used in the KLS cells
culture. This result indicated that at most 2,000 KLS cells used for transplantation were not

contaminated at this [3H] thymidine concentration, a number of KLS cells that could not
restore hematopoiesis after lethal γ-irradiation (our personal data). Thus hematopoietic
reconstitution by KLS grown for 24 hours in medium containing a concentration of [3H]
thymidine of 14.8 kBq/ml is partly due to [3H] thymidine contaminated KLS cells. The
different mature hematopoietic cell populations derived from the [3H] thymidine
contaminated KLS cells were not affected as no difference in mature B and T lymphocytes,
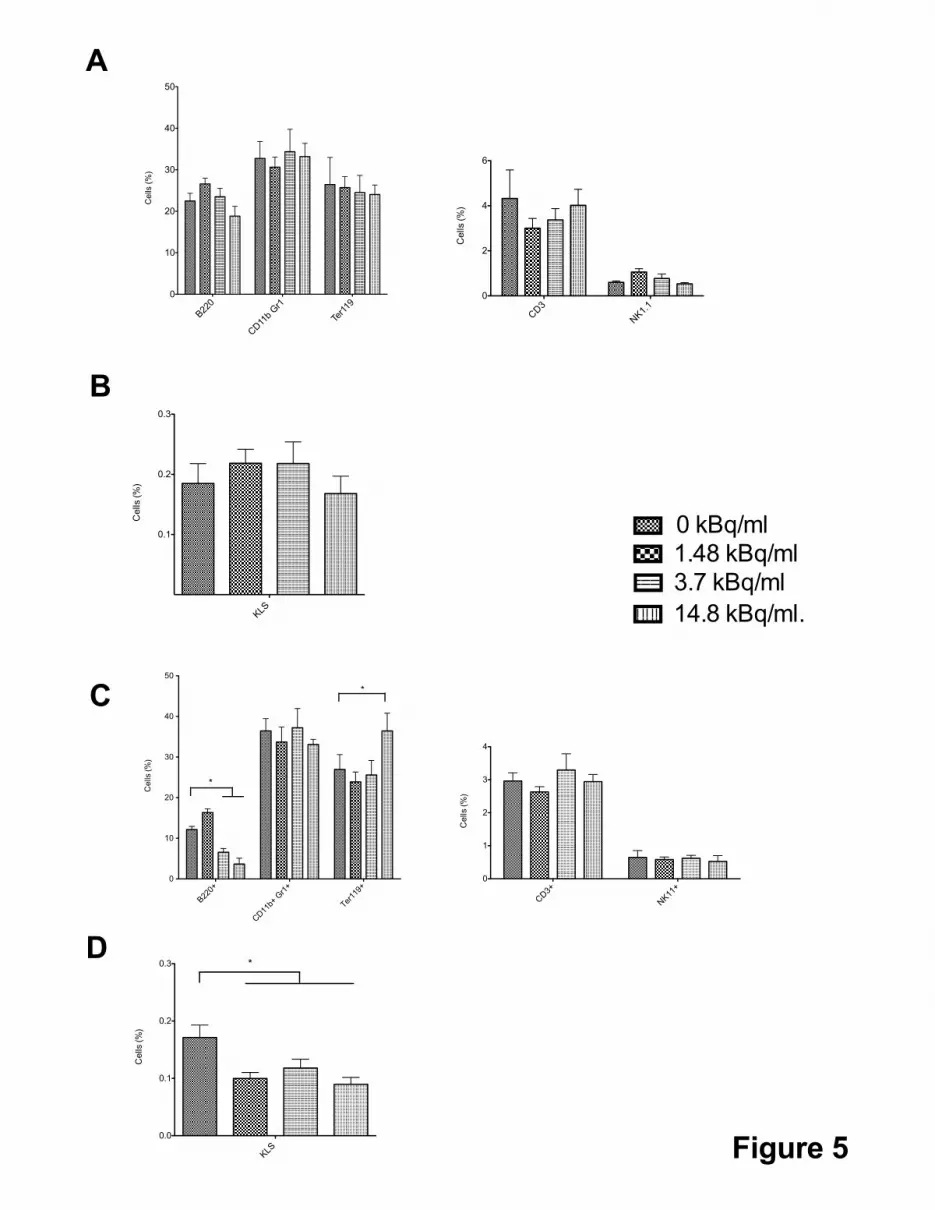
monocytes/granulocytes and red blood cells could be evidenced (Figure 5A). Finally, we
could not detect any difference in the percentage of KLS cells in bone marrow of each mice
group (Figure 5B), indicating that [3H] thymidine contaminated HSC could reconstitute long-
term hematopoiesis.
To document the long-term capacity of hematopoietic reconstitution of KLS cells
contaminated with [3H] thymidine, 500,000 donor CD45.2+ bone marrow cells were isolated
from transplanted mice and injected into lethally irradiated CD45.1+ recipient mice for a
secondary transplantation. All transplanted mice survived (data not shown) indicating that
hematopoietic stem cells retained their hematopoietic reconstitution activity. Analysis of the
different mature hematopoietic populations of the secondary transplanted mice revealed a two
fold decrease of the B lymphocyte populations in mice reconstituted with bone marrow
coming from KLS cells initially contaminated with [3H] thymidine concentration of 3.7 and
14.8 kBq/ml together with a small but significant increase of red blood cells when a [3H]
thymidine concentration of 14.8 kBq/ml was initially used for KLS cells contamination and
no change in the other mature hematopoietic cells (Figure 5C). Analysis of KLS cells of these
reconstituted mice showed a 1.5 fold decrease of the KLS population whatever dose of [3H]
thymidine concentration was initially used for contamination (Figure 5D). These results
indicated that initial contamination of KLS cells with [3H] thymidine compromised their long-

term capacity of hematopoietic reconstitution and their differentiation potential in the B-
lymphoid lineage.
[3H] thymidine contaminated KLS are less efficient than non contaminated KLS cells to
reconstitute hematopoiesis
We finally determined the functional properties of [3H] thymidine contaminated KLS
hematopoietic cells in a competitive repopulation assay. 5000 KLS expressing the CD45.2
antigen were cultured for 24 hours in a medium containing [3H] thymidine (0, 1.48, 3.7 and
14.8 kBq/ml) and co-transplanted with 500,000 non-contaminated bone marrow cells
expressing the CD45.1 antigen in lethally irradiated mice. As KLS cells represent around
0.2% of total bone marrow cells, the number of CD45.2 and CD45.1 hematopoietic cells used
for this experiment resulted in a 1/5 ratio of CD45.1+ and CD45.2+ KLS cells at the time of
transplantation. Four months after transplantation, the CD45.1/CD45.2 chimerism was
determined in the reconstituted bone marrow. Mice transplanted with non-contaminated KLS
cells and mice reconstituted with KLS cells contaminated with a [3H] thymidine concentration
of 1.48 kBq/ml displayed a CD45.2/CD45.1 ratio of 6.8 and 5.1 that is the ratio of
CD45.2/CD45.1 chimerism when these cells were transplanted (Figure 6). This ratio
decreased when CD45.2 KLS cells were contaminated with [3H] thymidine concentration of
3.7, 7.4 and 14.8 kBq/ml reaching a CD45.2/CD45.1 ratio of 2.8 when concentrations of 7.4
or 14.8 kBq/ml of [3H] thymidine were used for initial KLS contamination. These results
indicated that contamination of KLS cells with [3H] thymidine diminished their hematopoietic
reconstitution capacities when compared to non-contaminated KLS cells.

Discussion
Hematopoietic cells, including both hematopoietic stem cells (HSCs) and
hematopoietic progenitors, are highly sensitive to γ-irradiation. Numerous studies have
documented the cellular and molecular effects of γ-irradiation on hematopoiesis in vitro and
in vivo (18-20) but no study has been done on the effects of tritium on primary hematopoietic
cells. In a first step for such research, we studied the biological consequences of incorporation
of [3H] thymidine into the DNA of primary hematopoietic cells. Since the average energy of
3H β particle corresponds to a range that is smaller than the diameter of mammalian cells,
[3H] thymidine incorporated into DNA will affect the nucleus more directly than γ-irradiation
and the continuous release of [3H] thymidine energy to DNA that is in close proximity might
induce DNA damages during the whole life of the contaminated cells. In this article, we
analyzed the in vitro and in vivo effects of incorporation of [3H] thymidine on a hematopoietic
cell population enriched in Hematopoietic Stem Cells (HSCs), the c-kit+/Lin-/Sca1+ (KLS)
cell population.
The [3H] thymidine incorporation into DNA of KLS cells was dose dependent and
contamination with concentrations of [3H] thymidine as low as 0.74 kBq/ml results in
contamination of 50% of the KLS cells with an average of two molecules of [3H] thymidine
incorporated into the contaminated cells whereas more than 90% of the KLS cells were
contaminated with an average of more than 25 silver grains corresponding to [3H] thymidine
incorporated into DNA when contamination was performed with a 10 times higher
concentration of [3H] thymidine. Cell proliferation and viability showed that contamination
with [3H] thymidine induced a dose dependent decrease (resp. increase) in the proliferation
(cell death) process. When a [3H] thymidine concentration of 7.4 kBq/ml was used, a dramatic
decrease of cell proliferation together with an increase of apoptotic cells was observed after

24 or 48 hours of culture. This result is in sharp contrast with the results obtained on
hematopoietic cell lines as incubation with a concentration of 7.4 kBq/ml of [3H] thymidine
scarcely affects cell proliferation and only slightly influenced cell viability even after three
days of culture (14). Assuming that the same amount of [3H] thymidine is incorporated in
primary hematopoietic cells and in hematopoietic cell lines, this result indicated that primary
hematopoietic cells might be less effective in DNA repair after the DNA damages induced by
the incorporation of [3H] thymidine into DNA and/or that hematopoietic cell lines can
proliferate even with DNA damages, a property that might be linked to a deficient p53
pathway in these transformed hematopoietic cell lines.
[3H] thymidine is a continuous source of internal radiation and may introduce mutations and
damages in the progeny of contaminated KLS. However, no significant differences were
found in the myeloid hematopoietic colonies generated by contaminated KLS even with
concentrations of [3H] thymidine that results in multiple [3H] thymidine incorporated in most
if not all the KLS cells. Indeed, little variations in the total numbers and in the composition of
the colonies were observed, contaminated KLS showing a decrease in the total number of
colonies and a slight increase in the percentage of immature CFU-GEMM colonies followed
by a decrease in CFU-GM colonies compared to control KLS cells. When KLS cells are
irradiated with high doses of γ-irradiation, a dramatic decrease of myeloid and lymphoid
potentials of these cells has been reported (21) and our data indicated that [3H] thymidine
incorporated into DNA might have a different effect on the myeloid and lymphoid progeny of
KLS cells (see below). In addition, as γ-irradiation could generate signals from cell
membranes in addition to nuclear signals whereas [3H] thymidine contamination only
generates nuclear signals, our data might indicate effects of cell membranes damages on the
differentiation potentials of KLS cells. Another explanation of the absence of effect of [3H]
thymidine incorporation into the DNA of KLS cells on the cloning capacity and myeloid

potential of KLS cells might be that the colonies obtained in semi-solid methylcellulose assay
comes from less than 40% of the KLS cells and might originated from immature progenitors
that are very efficient in DNA repair (9, 21). Thus, the apoptosis observed might be due to
death of committed hematopoietic progenitors whereas immature progenitors and
hematopoietic stem cells might be protected from the effects of [3H] thymidine into their
DNA.
Transplantation of KLS cells contaminated with [3H] thymidine into lethally irradiated mice,
did not compromise their hematopoietic recovery even when contamination was performed
with concentrations of [3H] thymidine that results in more than 80% of KLS cells that have
incorporated [3H] thymidine in their DNA indicating again a very different effect of γ-
irradiation and [3H] thymidine contamination of hematopoietic stem cells. As γ-irradiation can
induce premature aging of KLS (8), we performed secondary transplantation to monitor any
effect of [3H] thymidine contamination of KLS cells on premature senescence. After this
secondary transplantation, the KLS compartment of the transplanted mice was significantly
decreased whatever concentrations of [3H] thymidine used for contamination and significant
variations in the B and red cell population was observed but only after contamination with
high doses of [3H] thymidine. These results are in line with previous results obtained after γ-
irradiation of KLS cells (8, 22) and suggests that low [3H] thymidine contamination doses
doesn’t induce cell death and impaired hematopoietic reconstitution but could accelerate a cell
senescence process. Finally, competition experiments showed that [3H] thymidine
contamination of KLS cells decreased their capacity to restore hematopoiesis when compared
to non-contaminated KLS cells suggesting that homing and/or retention of these contaminated
KLS cells might be altered.
In conclusion, the results shown in this article indicated that [3H] thymidine contamination of
a cell population enriched in hematopoietic stem cells has significant effects on the biological

properties of the contaminated stem cells that did not died but might display (i) defect in bone
marrow homing and/or (ii) premature senescence. As much of the tritium that remains in the
environment exists as tritiated water (HTO), contamination of KLS cells with tritiated water
will definitively indicate the effects of tritium on somatic stem cells.

Materials and methods
Mice
Eight- to twelve-weeks old C57Bl/6-Ly5.2 (CD45.2) mice were obtained from Charles
River Laboratories (l’Arbresle, France) and used as donors for HSCs cells. All recipient mice
had a CD45.1 genetic background and were bred under pathogen-free conditions in the iRCM
animal facility (C.E.A. Commisariat à l’Energie Atomique, Fontenay aux Roses, France).
Recipient mice have been lethally irradiated with 11Gy using a 137Cs irradiator. Approval for
animal care was received from Services Vétérinaires de la Santé et de la Production Animale
delivered by the Ministère de l’Agriculture, France.
Flow cytometry analysis
Bone marrow (BM) cells were flushed from both tibias and femurs of donor
C57Bl/6Ly5.2 mice and treated with a 0.75% NH4Cl (Sigma-Aldrich, St Louis, MO) to
eliminate erythrocytes. BM cells were stained with antibodies conjugated to fluorescein
isothiocyanate (FITC), phycoerythrin (PE), phycoerythrin-Cyanine 7 (PE-Cy 7) and
Allophycocyanin (APC) (all from Becton Dickinson Biosciences Pharmingen [BD], San
Diego, CA): CD45.1 (A20), CD45.2 (104), B220 (RA3-6B2), CD3 (145-2C11), NK1.1
(PK136), CD11b (M1/70), Gr-1 (RB6-8C5), and TER-119 (TER-119), cKit (2B8), Sca-1
(E13-161.7). Lineage cells were labeled using a byotin-conjugated Lineage cocktail (Miltenyi
Biotech, Bergisch Gladbach, Germany). Biotinylated antibodies were revealed with
streptavidin-phycoerythrin-cyanine-7. Stained cells were analyzed using a FACSCalibur
cytometer and CellQuest software (BD).
Purification of KLS cells by Flow Cytometry
KLS cell population was labeled using Sca1, cKit and Lineage cocktail markers. The

cKit+Lin-Sca1+ (KLS) population was isolated using a MOFLO high-speed cell sorter (Dako,
Glostrup, Denmark).
[3H] thymidine incorporation into DNA
Analysis of [3H] thymidine incorporation into DNA was performed using liquid
scintillation counting. DNA of KLS cells coming from contaminated medium was
precipitated using Trichloroacetic Acid. The radioactive pellet was dissolved in 5 ml of
scintillation fluid (Ecolite, ICN Biomedicals), counted (TriCARB 1900CA, Packard
Instruments, Meridien, CT) and the incorporated radioactivity was expressed in disintegration
per minute (d.p.m.).
Autoradiography
KLS cells were cultured for 24h in a medium with different concentrations of [3H]
thymidine. Then the cells were washed and plated on polysine slides (Kindler, Freiburg,
Germany). Slides were dipped into Kodak NTB2 nuclear emulsion diluted 2:3 with distilled
water at 42°C, dried for 2 hours at room temperature, exposed at 4°C with desiccant for 7
days in a dark box and finally developed by successive baths at 13°C in Kodak developer D-
19 for 4 min 30 sec, in 2% acetic acid stop solution for 30 sec and in Kodak fixer for 10 min.
Slides were counterstained with Mayers’s hemalun (Merck) and mounted with Eukitt (Fluka).
Silver grains found on nuclei were counted under a microscope (Olympus AX70) to assess the
level of tritiated thymidine incorporation.
γ-H2AX Immunostaining
KLS cells were cultured for 24 hours in a medium with different concentration of
[3H] thymidine. KLS cells were seeded on polylysine-coated slides and incubated at 37°C for

10 minutes. Cells were immediately fixed using 1% PFA aqueous solution (E.M.S. 15714)
for 10 min and permeabilized using 0.2% Triton X-100 solution for 5 min (Sigma Aldrich
93443). Staining of γ-H2AX foci was done with antiphospho-histone H2A.X (Ser139), clone
JBW301 antibody at a 1/200 dilution (Millipore, Billerica, MA). After washing, cells were
stained with a secondary antibody at a 1/400 dilution (Alexa fluor 488 goat anti-mouse IgG
antibody, Invitrogen A11001) and 4,6-diamidino-2-phenylindole (Vector Laboratories,
Burlingame, CA). Foci were quantified by fluorescence microscopy using a Leica TCS SPE
confocal imaging microscope with an ACS APO 40X oil objective.
KLS cell culture, [3H] thymidine contamination and proliferation
Sorted KLS cells were maintained into liquid suspension culture in presence of
cytokine cocktails to stimulate proliferation. Cultures were maintained at 37ºC, 5% CO2 and
95% humidity. Cell Media contained IMDM (GIBCO), 10% Fetal Bovin Serum (FBS,
GIBCO) and 1% PSG antibiotic mix (100-U/mL penicillin, 100-g/mL streptomycin, and 2-
mmol/L L-glutamine; Invitrogen, Carlsbad, CA). The cytokine combination (STEMCELL
technologies) included 100 ng/mL murine SCF, 100 ng/mL murine flt3 ligand (FL), 10 ng/mL
murine thrombopoietin (TPO), 20 ng/mL murine IL-3, and 10 ng/mL human IL-6. [3H]
thymidine (3245-3260 GBq/mmol) was purchased from New England Nuclear (Boston, MA,
USA). KLS cells were cultured at the initial concentration of 50 000 cells/ml in a medium
containing different concentrations of [3H] thymidine. After 24 and 48 hours of culture the
total number of live cells was determined after Trypan blue staining.
Apoptosis assay
Annexin V (Becton Dickinson Biosciences Pharmingen [BD], San Diego, CA) was used in
conjunction with 7-Amino-Actinomycin (7-AAD) to identify apoptotic cells by FACS

analysis. Annexin V was used according to manufacturer protocol.
Colony Forming Unit (CFU) Assay
CFU assay (MethoCult 03434, StemCell Technologies) was performed according to
manufacturer’s instructions. Briefly 300 KLS cells were added to 2 ml of complete Methocult
and were seeded to three 35 mm culture dishes. Culture dishes were transferred to an
incubator at 37 ºC, 5% CO2 and 95% humidity and the CFU were enumerated after 7 days in
culture. Cultures were characterized for the presence of myeloid and multi-potential CFU.
Myeloid CFU include Colony-Forming Unit-Granulocyte (CFU-G), Colony-Forming Unit-
Macrophage (CFU-M) and Colony-Forming Unit-Granulocyte, Macrophages (CFU-GM).
Multi-potential CFU includes Colony-Forming Units with mixed populations of erythroid and
myeloid cells (CFU-GEMM).
Bone marrow transplantation
For non-competitive long-term reconstitution assay, recipient mice were lethally γ-
irradiated (11 Gy) 24 hours before transplantation and treated with antibiotic in drinking
water (Baytril 10%; Bayer AG, Leverkusen, Germany) for at least 4 weeks. Twenty thousands
KLS cells cultured for 24h in a medium with different concentration of [3H] thymidine were
injected via the retro-orbital vein into lethally irradiated recipients. The transplanted mice
were analyzed 4 months after transplantation. Five hundreds thousands bone marrow cells
were then isolated from transplanted mice and injected into lethally irradiated recipient mice
for a secondary transplantation. The bone marrow was analyzed 4 months later.
For competitive long-term reconstitution assay, five thousands KLS-CD45.2 cultured for 24h
in a medium with different concentration of [3H] thymidine together with five hundreds
thousands non-contaminated BM-CD45.1 cells were simultaneously injected via the retro-

orbital vein into lethally irradiated mice. The transplanted mice were analyzed 4 months later.
Data analysis and statistics
Values are presented as the mean, median or cell number ± SD. Statistical
comparisons between groups were done using the Student’s t-test, p < 0.05 and p < 0.001
were considered statistically significant (*) and highly statistically significant (**).
Acknowledgments
The authors declare that they have no conflict of interest. We acknowledge Pierre
Fouchet and Zahra Kadri for helpful discussions. We are grateful to the staff of the iRCM
animal facility for excellent support in mouse studies and to Benjelloun H., Deschamps N.
and Baijer J. of the iRCM cytometry platform for excellent support in FACS and cell sorting
experiments. Fabio di Giacomo and Vilma Barroca are supported by fellowships from Marie
Curie Research fellowship from the EU fp6 program ‘Eurythron’ MRTN-CT-2004-005499
and from Inserm. This project was supported by grants from EDF, ARC (3710), Inserm and
CEA/DSV.

References
1. M. Saito, M. R. Ishida and C. C. Travis, Dose-modification factor for accumulated dose to
cell nucleus due to protein-bound 3H. Health Phys 56, 869-874 (1989).
2. F. M. Watt and B. L. Hogan, Out of Eden: stem cells and their niches. Science 287, 1427-
1430 (2000).
3. S. Kale, A. Karihaloo, P. R. Clark, M. Kashgarian, D. S. Krause and L. G. Cantley, Bone
marrow stem cells contribute to repair of the ischemically injured renal tubule. J Clin Invest 112,
42-49 (2003).
4. D. Orlic, J. Kajstura, S. Chimenti, F. Limana, I. Jakoniuk, F. Quaini, B. Nadal-Ginard, D.
M. Bodine, A. Leri and P. Anversa, Mobilized bone marrow cells repair the infarcted heart,
improving function and survival. Proc Natl Acad Sci U S A 98, 10344-10349 (2001).
5. R. Poulsom, S. J. Forbes, K. Hodivala-Dilke, E. Ryan, S. Wyles, S. Navaratnarasah, R.
Jeffery, T. Hunt, M. Alison, et al., Bone marrow contributes to renal parenchymal turnover and
regeneration. J Pathol 195, 229-235 (2001).
6. N. Dainiak, Hematologic consequences of exposure to ionizing radiation. Exp Hematol
30, 513-528 (2002).
7. A. Nijnik, L. Woodbine, C. Marchetti, S. Dawson, T. Lambe, C. Liu, N. P. Rodrigues, T.
L. Crockford, E. Cabuy, et al., DNA repair is limiting for haematopoietic stem cells during ageing.
Nature 447, 686-690 (2007).
8. D. J. Rossi, D. Bryder, J. Seita, A. Nussenzweig, J. Hoeijmakers and I. L. Weissman,
Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age.
Nature 447, 725-729 (2007).
9. M. Milyavsky, O. I. Gan, M. Trottier, M. Komosa, O. Tabach, F. Notta, E. Lechman, K. G.
Hermans, K. Eppert, et al., A Distinctive DNA Damage Response in Human Hematopoietic Stem

Cells Reveals an Apoptosis-Independent Role for p53 in Self-Renewal. Cell Stem Cell. Epub
ahead
10. A. M. Carr, Cell cycle. Piecing together the p53 puzzle. Science 287, 1765-1766 (2000).
11. A. Hirao, Y. Y. Kong, S. Matsuoka, A. Wakeham, J. Ruland, H. Yoshida, D. Liu, S. J.
Elledge and T. W. Mak, DNA damage-induced activation of p53 by the checkpoint kinase Chk2.
Science 287, 1824-1827 (2000).
12. L. Shao, Y. Sun, Z. Zhang, W. Feng, Y. Gao, Z. Cai, Z. Z. Wang, A. T. Look and W. S. Wu,
Deletion of proapoptotic Puma selectively protects hematopoietic stem and progenitor cells
against high-dose radiation. Blood 115, 4707-4714.
13. W. S. Wu, S. Heinrichs, D. Xu, S. P. Garrison, G. P. Zambetti, J. M. Adams and A. T.
Look, Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma.
Cell 123, 641-653 (2005).
14. M. Yanokura, K. Takase, K. Yamamoto and H. Teraoka, Cell death and cell-cycle arrest
induced by incorporation of [3H]thymidine into human haemopoietic cell lines. Int J Radiat Biol
76, 295-303 (2000).
15. Y. Saintigny, S. Roche, D. Meynard and B. S. Lopez, Homologous recombination is
involved in the repair response of mammalian cells to low doses of tritium. Radiat Res 170, 172-
183 (2008).
16. J. A. Aten, J. Stap, P. M. Krawczyk, C. H. van Oven, R. A. Hoebe, J. Essers and R.
Kanaar, Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome
domains. Science 303, 92-95 (2004).
17. K. Rothkamm and M. Lobrich, Evidence for a lack of DNA double-strand break repair in
human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A 100, 5057-5062 (2003).

18. H. Yu, H. Shen, Y. Yuan, R. XuFeng, X. Hu, S. P. Garrison, L. Zhang, J. Yu, G. P. Zambetti
and T. Cheng, Deletion of Puma protects hematopoietic stem cells and confers long-term survival
in response to high-dose gamma-irradiation. Blood 115, 3472-3480.
19. Y. Hirabayashi, M. Matsuda, T. Matumura, H. Mitsui, H. Sasaki, T. Tukada, S. Aizawa, K.
Yoshida and T. Inoue, The p53-deficient hemopoietic stem cells: their resistance to radiation-
apoptosis, but lasted transiently. Leukemia 11 Suppl 3, 489-492 (1997).
20. E. A. Komarova, R. V. Kondratov, K. Wang, K. Christov, T. V. Golovkina, J. R. Goldblum
and A. V. Gudkov, Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic
injury, but protects from gastro-intestinal syndrome in mice. Oncogene 23, 3265-3271 (2004).
21. M. Mohrin, E. Bourke, D. Alexander, M. R. Warr, K. Barry-Holson, M. M. Le Beau, C. G.
Morrison and E. Passegue, Hematopoietic Stem Cell Quiescence Promotes Error-Prone DNA
Repair and Mutagenesis. Cell Stem Cell. Epub ahead
22. Y. Wang, B. A. Schulte and D. Zhou, Hematopoietic stem cell senescence and long-term
bone marrow injury. Cell Cycle 5, 35-38 (2006).

Figure Legends
Figure 1: [3H] thymidine incorporation into KLS DNA. KLS cells were purified by cell sorting
from bone marrow of healthy C57Bl/6 mice and cultured for 24 hours in a medium containing the
indicated concentrations of [3H] thymidine.
A. Dose dependent incorporation of [3H] thymidine into the DNA of KLS cells. DNA from
1000 KLS cells contaminated with the indicated concentrations of [3H] thymidine was TCA
precipitated and the amount of precipitated [3H] thymidine was counted using a liquid
scintillator counter. Data are shown as disintegration per minute (d.p.m.) (Mean ± SEM, n=5; p≤
0.05).
B. Percentage of KLS cells that have incorporated [3H] thymidine in their DNA. Silver grains
corresponding to [3H] thymidine incorporated into DNA of KLS cells were detected after
autoradiography on contaminated KLS cells. Data are expressed as percentage of KLS cells that
contained silver grains (Mean ± SEM, n=5, *p < 0.05).
C. Average number of incorporated [3H] thymidine per KLS cell. Data are expressed as average
number of silver grains per KLS cell (Mean ± SEM, n=5, **p < 0.001).
Figure 2: Proliferation and apoptosis of KLS cells grown for 24 and 48 hours in liquid medium
containing different concentrations of [3H] thymidine.
A. KLS cells were purified by cell sorting from bone marrow of healthy C57Bl/6 mice and
cultured in medium containing the indicated concentrations of [3H] thymidine. KLS cells were
cultured at an initial concentration of 50,000 cells/ml in 100μl of medium and contaminated with
different concentrations of [3H] thymidine (0; 3.7; 7.4; 14.8 and 37.03 kBq/ml). After 24 and 48

hours of culture, total cell number was measured. Results are expressed as cell number (Mean ±
SEM, n=5, *p < 0.05).
B. Percentage of apoptotic cells after 24 and 48 hours of culture of KLS cells in medium
containing various concentrations of [3H] thymidine. KLS cells were cultured in a medium
containing two different concentrations of [3H] thymidine (0; 3.7 and 14.8 kBq/ml). Apoptosis
was monitored by AnnexinV and 7-AAD staining and FACS analysis. Results are expressed as
percentage of apoptotic cells (Mean ± SEM, n=5, *p < 0.05 **p < 0.001).
Figure 3: γ-H2AX foci induced by [3H] thymidine incorporation into KLS DNA. KLS cells
were grown for 24 hours in a medium containing the indicated concentrations of [3H] thymidine,
washed, fixed and Double Strand Breaks (DSBs) were monitored by H2AX immunostaining.
A. Percentage of KLS cells containing at least one γ-H2AX focus as a function of concentrations
of [3H] thymidine in the medium where KLS cells were cultured for 24 hours. For each [3H]
thymidine concentration, a minimum of 100 nuclei was scored (Mean ± SEM, n=3, *p < 0.05).
B. Mean number of γ-H2AX foci per cell as a function of concentrations of [3H] thymidine in the
medium where KLS cells were cultured for 24 hours. For each [3H] thymidine concentration, a
minimum of 50 cells containing γ-H2AX foci were selected and the number of γ-H2AX foci in
each individual cells was scored (Mean ± SEM, n=3, *p < 0.05).
Figure 4: Colony Forming Unit (CFU) Assay on KLS cells previously cultured for 24 hours in
liquid medium containing different concentrations of [3H] thymidine. KLS were cultured for 24
hours in a medium containing the indicated concentration of [3H] thymidine (0; 3.7 and 14.8
kBq/ml). 100 KLS were then seeded on a semi-solid methylcellulose layer and hematopoietic
colonies were counted one week later.

A. Total number of myeloid and multipotential CFU colonies. Data are expressed as number of
colonies (Mean ± SEM, n=5)
B. Percentages of myeloid lineage committed colonies. Myeloid CFU include Colony Forming
Unit-Granulocyte (CFU-G), Colony Forming Unit-Macrophage (CFU-M) and Colony Forming
Unit-Granulocytes and Macrophages (CFU-GM). Multi-potential CFU includes Colony Forming
Units with mixed populations of erythroid and myeloid cells (CFU-GEMM). Data are expressed
as percentage of colonies (Mean ± SEM, n=5).
Figure 5:
A and B. Characterization of bone marrow hematopoietic cell populations of mice lethally
irradiated and transplanted with KLS cells. KLS cells were cultured for 24 hours in a medium
containing the indicated concentrations of [3H] thymidine (0; 1.48; 3.7 and 14.8 kBq/ml). 20,000
KLS were then transplanted in mice lethally irradiated at 11Gy. Mice were sacrificed 4 months
after engraftment and bone marrow cells were labeled to identify the hematopoietic
subpopulations. A. Percentage of bone marrow B-cells (B220+), Neutrophils (CD11b+ Gr1+) and
erythroid cells (Ter119+) (left panel) and T-Lymphocytes (CD3+) and Natural Killer cells
(NK1.1+) (right panel), after transplantation of contaminated KLS cells into lethally irradiated
mice (Mean ± SEM, n=5). B. Percentage of Hematopoietic Stem cells (KLS) after
transplantation of contaminated KLS cells into lethally irradiated mice (Mean ± SEM, n=5).
C and D. 500,000 bone marrow cells isolated 4 months after transplantation into lethally
irradiated mice of 20,000 KLS cells contaminated with different amounts of [3H] thymidine were
engrafted in lethally irradiated mice. Mice were sacrificed 4 months after this secondary
transplantation and bone marrow cells were characterized to identify the hematopoietic
subpopulations. C. Percentage of bone marrow B-cells (B220+), Neutrophils (CD11b+ Gr1+) and
erythroid cells (Ter119+) (left panel) and T-Lymphocytes (CD3+) and Natural Killer cells

(NK1.1+) (right panel), after secondary engraftment (Mean ± SEM, n=5, *p < 0.05). D.
Percentage of Hematopoietic Stem cells (KLS) after secondary transplantation into lethally
irradiated mice (Mean ± SEM, n=5, *p < 0.05).
Figure 6: Bone marrow CD45.2/CD45.1 chimerism four months after transplantation, in mice
lethally irradiated, of 5,000 KLS-Ly5.2 cultured for 24 hours in medium containing the indicated
concentrations of [3H] thymidine (0; 1.48; 3.7; 7.4 and 14.8 kBq/ml) and 500,000 Ly-5.1
(CD45.1+) bone marrow cells. Mice were sacrificed 4 months after transplantation and bone
marrow chimerism was determined by FACS analysis. Data are expressed as ratio
CD45.2/CD45.1 (Mean ± SEM, n=5, *p < 0.05) and 5 indicated the initial ratio of transplanted
KLS cells.

0
100
200
300
1000
2000
3000
4000
5000
0.37 0.74 1.48 7.4 14.8 37.03
kBq/ml
3.7
d.p.m./1000cells
0
20
40
60
80
100
*
0.74 1.48 3.7 7.4
kBq/ml
Silvergrain-positivecells(%)
0
10
20
30
40
**
**
**
0.74 1.48 3.7 7.4
kBq/ml
Averagenumberofsilvergrain/cell
Figure 1
A
B
C

0h 24h
48h
0
20000
40000
60000
80000
0 kBq/ml3.7 kBq/ml
7.4 kBq/ml
37.03 kBq/ml14.8 kBq/ml
5000
*
*
*
*
*
*
Cellnumber
24h
48h
0
20
40
60
80
0 kBq/ml3.7 kBq/ml14.8 kBq/ml*
**
Apoptoticcells(%)
A
B
Figure 2

0
20
40
60
80
100
0 0.74 1.48 3.7 14.8 37.03
kBq/ml
�-H2AXfoci-positivecells(%)
0
5
10
15
20
0 0.74 1.48 3.7 14.8 37.03
kBq/ml
Averagenumberof�-H2AXfoci/cell
Figure 3
A
B
*
*

0
20
40
60
0 14.83.7
kBq/ml
Numberofcolonies
CFU-G
CFU-M
CFU-GM
CFU-GEMM
0
10
20
30
40
500 kBq/ml3.7 kBq/ml14.8 kBq/ml
Colonies(%)
A
B
Figure 4
B22
0
CD11
b Gr1
Ter1
190
10
20
30
40
50
Cells
(%)
KLS
0.1
0.2
0.3
Cells
(%)
B22
0+
CD11
b+Gr1
+
Ter11
9+0
10
20
30
40
50
*
*
Cells
(%)
KLS
0.0
0.1
0.2
0.3 *
Cells
(%)
A
B
C
D
Figure 5
CD3
NK1.
10
2
4
6
Cells
(%)
CD3+
NK11
+0
1
2
3
4
Cells
(%)
0 kBq/ml
1.48 kBq/ml
3.7 kBq/ml
14.8 kBq/ml.

0
2
4
6
8
0 1.48 3.7 7.4 14.8
5
*
Figure 6
kBq/ml
RatioCD45.2/CD45.1





























