Embed Size (px)
Citation preview

Part II => PROTEINS and ENZYMES
§2.3 Protein Structure
§2.3a Secondary Structure
§2.3b Tertiary Structure
§2.3c Quaternary Structure

Section 2.3a:
Secondary Structure

Synopsis 2.3a
- Secondary structure refers to the local ordered conformation of residues within a polypeptide chain
- Many polypeptides adopt such ordered secondary structural elements in the form of so-called α helices or β sheets (or strictly speaking strands!)
- Not all polypeptides or polypeptide segments form regular secondary structure such as α helices or β sheets—such polypeptides are referred to as “structurally disordered” or “intrinsically disordered” proteins
- The majority of amino acid residues (!) in proteins adopt such disordered conformations—whoever said that the 3D structure of a protein is critical to its function must have roamed the protein landscape in the 20th century!!
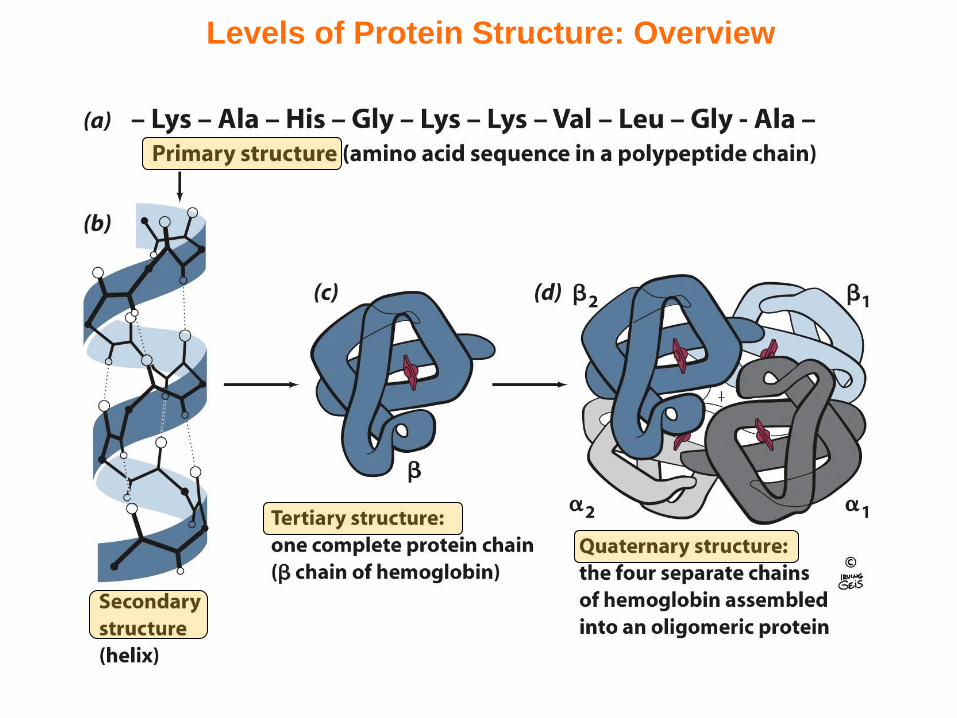
Levels of Protein Structure: Overview

Levels of Protein Structure: Description
Primary (1) StructureThe amino acid sequence of a polypetide chain
Secondary (2) StructureThe local ordered conformation of residues within a polypeptide chain—these are referred to as secondary structure elements such as helices and sheets (or strictly strands!)
Tertiary (3) StructureThe spatial arrangement or organization of various secondary structural elements such as helices and sheets connected via turns and loops into a three-dimensional (3D) fold
Quaternary (4) StructureThe spatial arrangement or organization of individual polypeptide chains with a well-defined 3D fold relative to each other
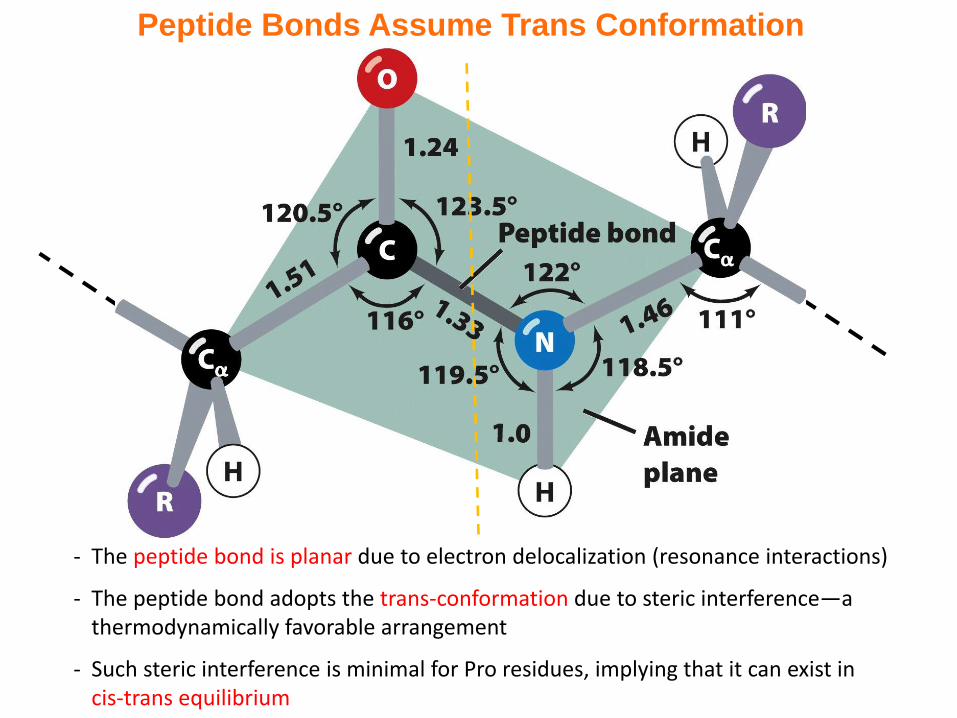
Peptide Bonds Assume Trans Conformation
- The peptide bond is planar due to electron delocalization (resonance interactions)
- The peptide bond adopts the trans-conformation due to steric interference—a thermodynamically favorable arrangement
- Such steric interference is minimal for Pro residues, implying that it can exist in cis-trans equilibrium
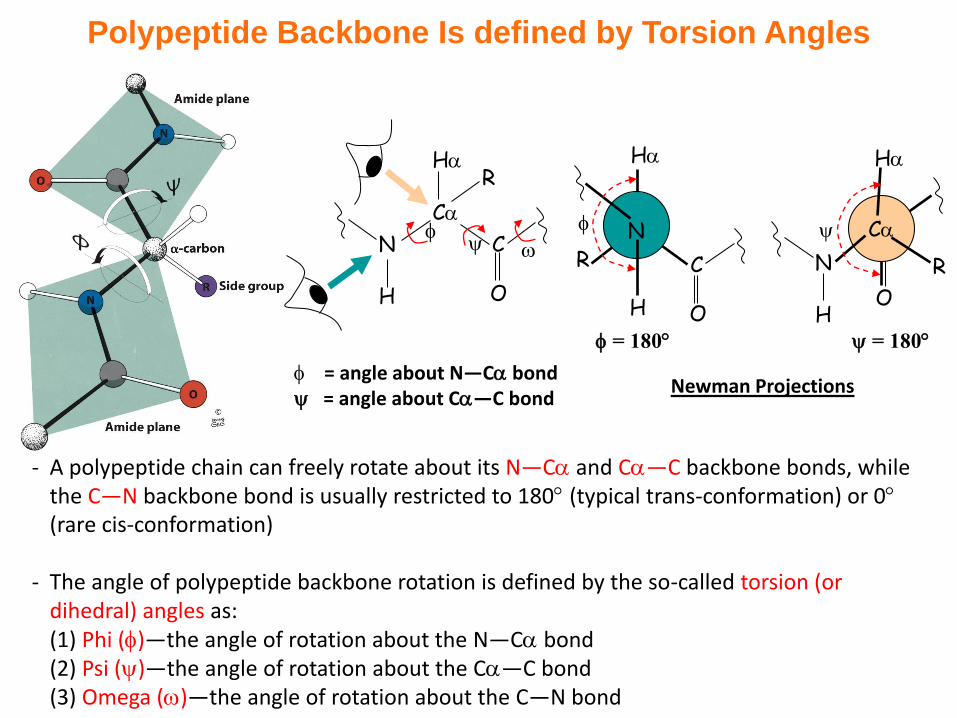
Polypeptide Backbone Is defined by Torsion Angles
- A polypeptide chain can freely rotate about its N—C and C—C backbone bonds, while the C—N backbone bond is usually restricted to 180 (typical trans-conformation) or 0(rare cis-conformation)
- The angle of polypeptide backbone rotation is defined by the so-called torsion (or dihedral) angles as:(1) Phi ()—the angle of rotation about the N—C bond(2) Psi ()—the angle of rotation about the C—C bond(3) Omega ()—the angle of rotation about the C—N bond
= angle about N—C bond = angle about C—C bond
Newman Projections
H
H
R C
O
= 180
NN
H
C
C
O
H
RH
N
HO
= 180
R
C
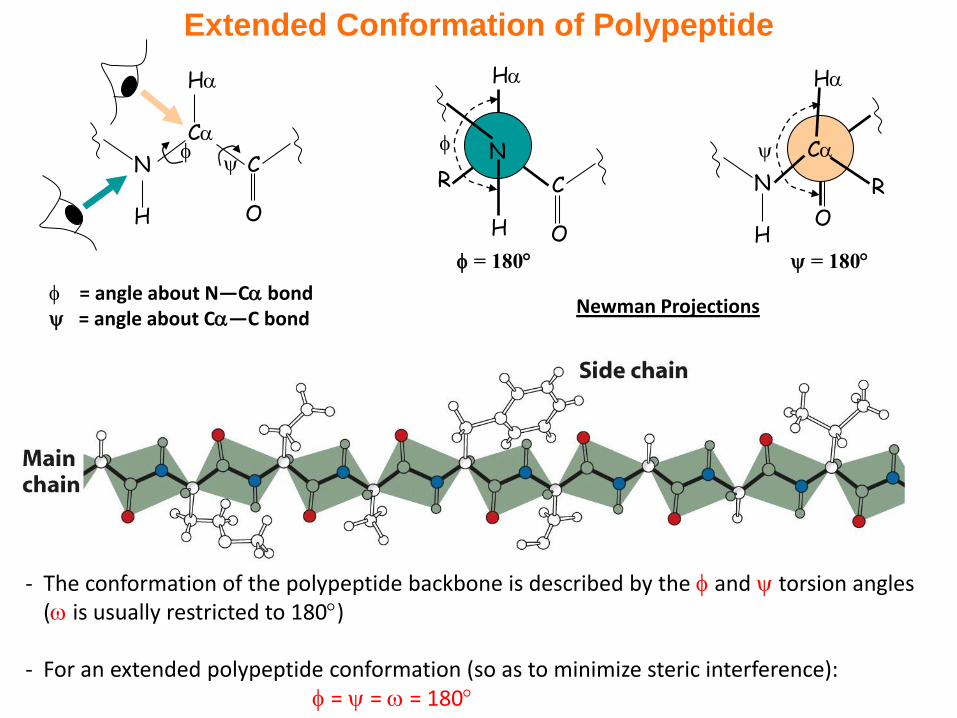
Extended Conformation of Polypeptide
- The conformation of the polypeptide backbone is described by the and torsion angles ( is usually restricted to 180)
- For an extended polypeptide conformation (so as to minimize steric interference): = = = 180
N
H
C
C
O
H
= angle about N—C bond = angle about C—C bond
Newman Projections
H
H
R C
O
= 180
N
H
N
HO
= 180
R
C
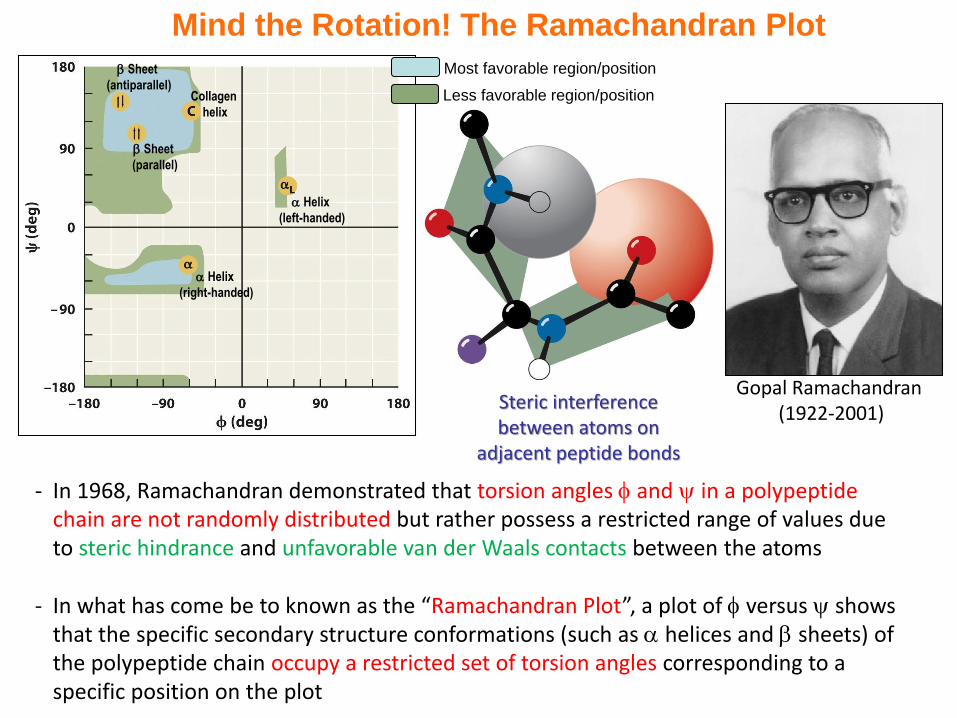
Mind the Rotation! The Ramachandran Plot
- In 1968, Ramachandran demonstrated that torsion angles and in a polypeptide chain are not randomly distributed but rather possess a restricted range of values due to steric hindrance and unfavorable van der Waals contacts between the atoms
- In what has come be to known as the “Ramachandran Plot”, a plot of versus shows that the specific secondary structure conformations (such as helices and sheets) of the polypeptide chain occupy a restricted set of torsion angles corresponding to a specific position on the plot
Steric interference between atoms on
adjacent peptide bonds
Gopal Ramachandran (1922-2001)
Sheet
(antiparallel)
Sheet
(parallel)
Collagen
helix
Helix
(right-handed)
Helix
(left-handed)
Most favorable region/position
Less favorable region/position
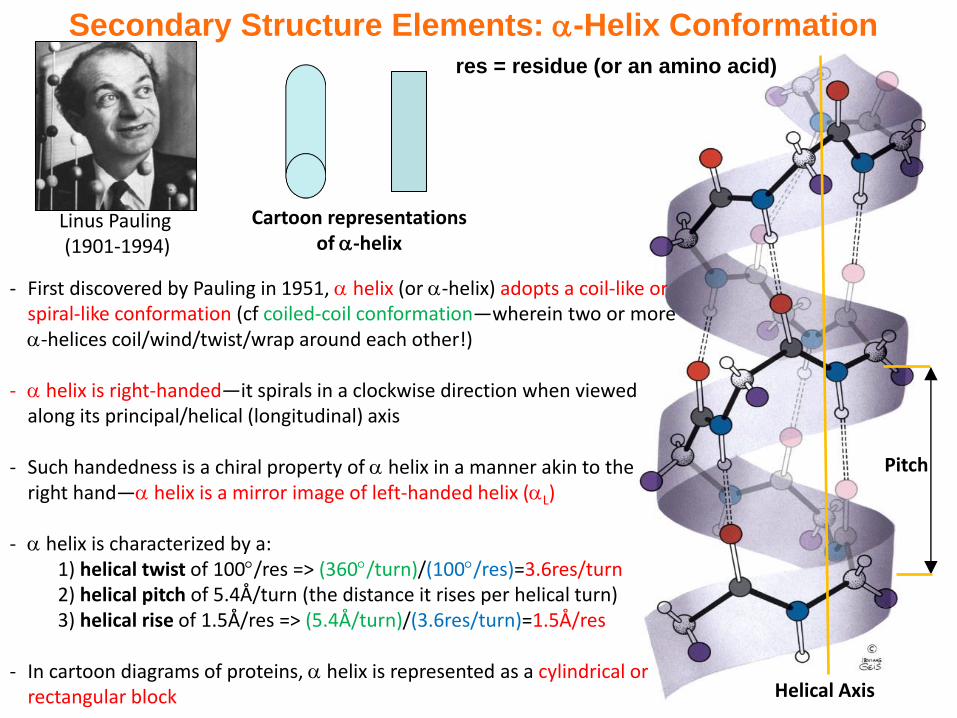
Helical Axis
Pitch
- First discovered by Pauling in 1951, helix (or -helix) adopts a coil-like or spiral-like conformation (cf coiled-coil conformation—wherein two or more -helices coil/wind/twist/wrap around each other!)
- helix is right-handed—it spirals in a clockwise direction when viewed along its principal/helical (longitudinal) axis
- Such handedness is a chiral property of helix in a manner akin to the right hand— helix is a mirror image of left-handed helix (L)
- helix is characterized by a:1) helical twist of 100/res => (360/turn)/(100/res)=3.6res/turn2) helical pitch of 5.4Å/turn (the distance it rises per helical turn)3) helical rise of 1.5Å/res => (5.4Å/turn)/(3.6res/turn)=1.5Å/res
- In cartoon diagrams of proteins, helix is represented as a cylindrical or rectangular block
Secondary Structure Elements: -Helix Conformation
Linus Pauling (1901-1994)
Cartoon representations of -helix
res = residue (or an amino acid)
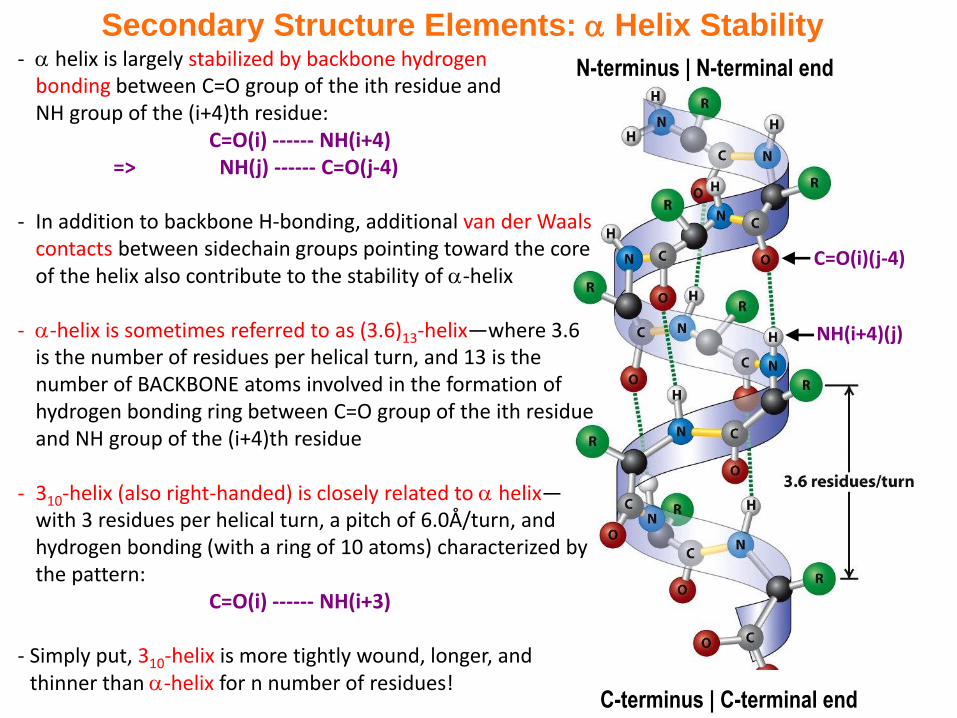
Secondary Structure Elements: Helix Stability
C-terminus | C-terminal end
C=O(i)(j-4)
NH(i+4)(j)
N-terminus | N-terminal end- helix is largely stabilized by backbone hydrogen bonding between C=O group of the ith residue and NH group of the (i+4)th residue:
C=O(i) ------ NH(i+4)=> NH(j) ------ C=O(j-4)
- In addition to backbone H-bonding, additional van der Waals contacts between sidechain groups pointing toward the core of the helix also contribute to the stability of -helix
- -helix is sometimes referred to as (3.6)13-helix—where 3.6 is the number of residues per helical turn, and 13 is the number of BACKBONE atoms involved in the formation of hydrogen bonding ring between C=O group of the ith residue and NH group of the (i+4)th residue
- 310-helix (also right-handed) is closely related to helix—with 3 residues per helical turn, a pitch of 6.0Å/turn, and hydrogen bonding (with a ring of 10 atoms) characterized by the pattern:
C=O(i) ------ NH(i+3)
- Simply put, 310-helix is more tightly wound, longer, and thinner than -helix for n number of residues!
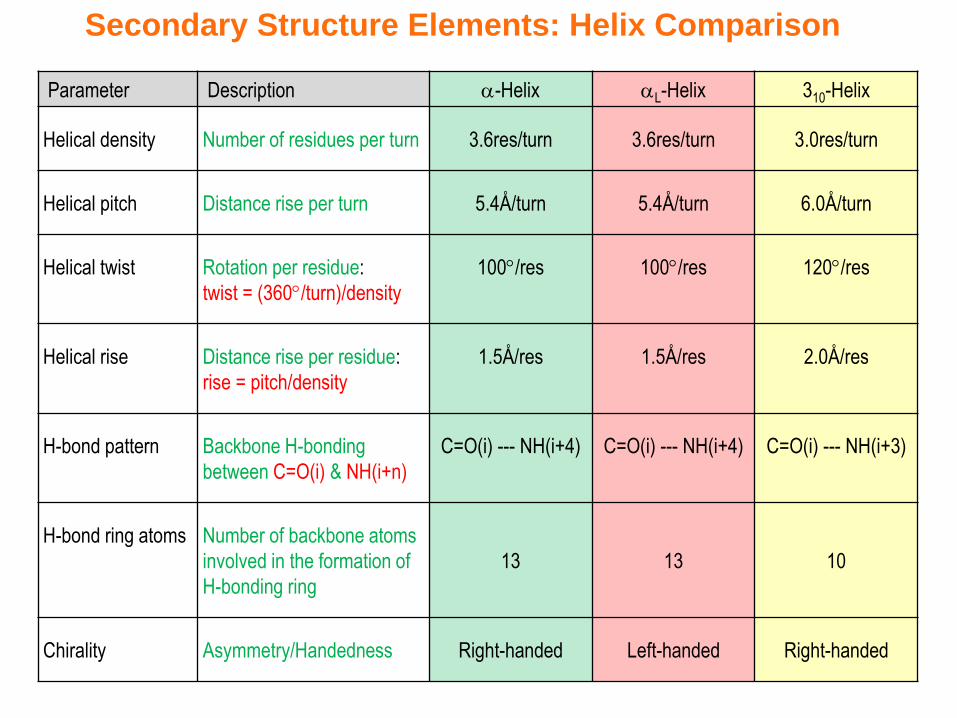
Secondary Structure Elements: Helix Comparison
Parameter Description -Helix L-Helix 310-Helix
Helical density Number of residues per turn 3.6res/turn 3.6res/turn 3.0res/turn
Helical pitch Distance rise per turn 5.4Å/turn 5.4Å/turn 6.0Å/turn
Helical twist Rotation per residue:
twist = (360/turn)/density
100/res 100/res 120/res
Helical rise Distance rise per residue:
rise = pitch/density
1.5Å/res 1.5Å/res 2.0Å/res
H-bond pattern Backbone H-bonding
between C=O(i) & NH(i+n)
C=O(i) --- NH(i+4) C=O(i) --- NH(i+4) C=O(i) --- NH(i+3)
H-bond ring atoms Number of backbone atoms
involved in the formation of
H-bonding ring
13 13 10
Chirality Asymmetry/Handedness Right-handed Left-handed Right-handed
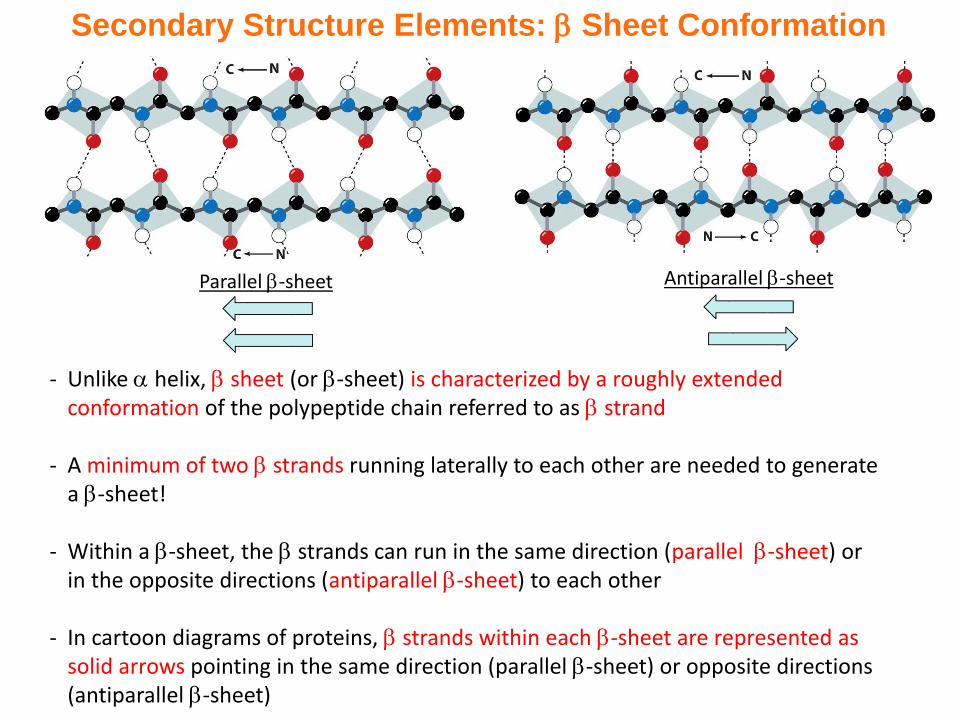
Secondary Structure Elements: Sheet Conformation
- Unlike helix, sheet (or -sheet) is characterized by a roughly extended conformation of the polypeptide chain referred to as strand
- A minimum of two strands running laterally to each other are needed to generate a -sheet!
- Within a -sheet, the strands can run in the same direction (parallel -sheet) or in the opposite directions (antiparallel -sheet) to each other
- In cartoon diagrams of proteins, strands within each -sheet are represented as solid arrows pointing in the same direction (parallel -sheet) or opposite directions (antiparallel -sheet)
Parallel -sheet Antiparallel -sheet
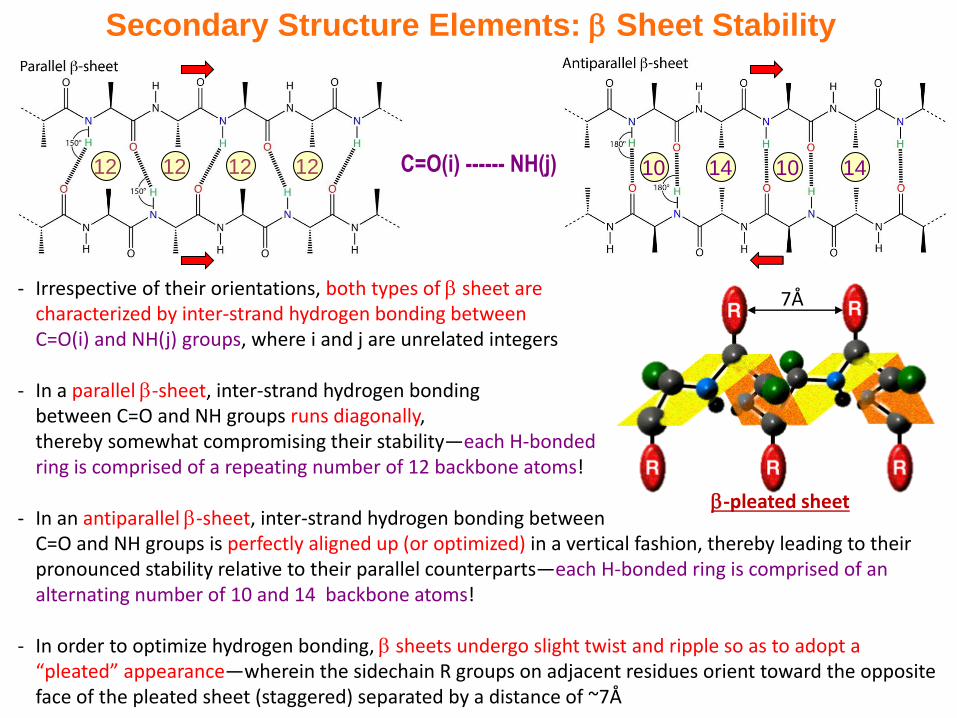
C=O(i) ------ NH(j)
-pleated sheet
7Å
Secondary Structure Elements: Sheet Stability
- Irrespective of their orientations, both types of sheet are characterized by inter-strand hydrogen bonding between C=O(i) and NH(j) groups, where i and j are unrelated integers
- In a parallel -sheet, inter-strand hydrogen bonding between C=O and NH groups runs diagonally, thereby somewhat compromising their stability—each H-bonded ring is comprised of a repeating number of 12 backbone atoms!
- In an antiparallel -sheet, inter-strand hydrogen bonding between C=O and NH groups is perfectly aligned up (or optimized) in a vertical fashion, thereby leading to their pronounced stability relative to their parallel counterparts—each H-bonded ring is comprised of an alternating number of 10 and 14 backbone atoms!
- In order to optimize hydrogen bonding, sheets undergo slight twist and ripple so as to adopt a “pleated” appearance—wherein the sidechain R groups on adjacent residues orient toward the opposite face of the pleated sheet (staggered) separated by a distance of ~7Å
12 12 12 12 14 1410 10
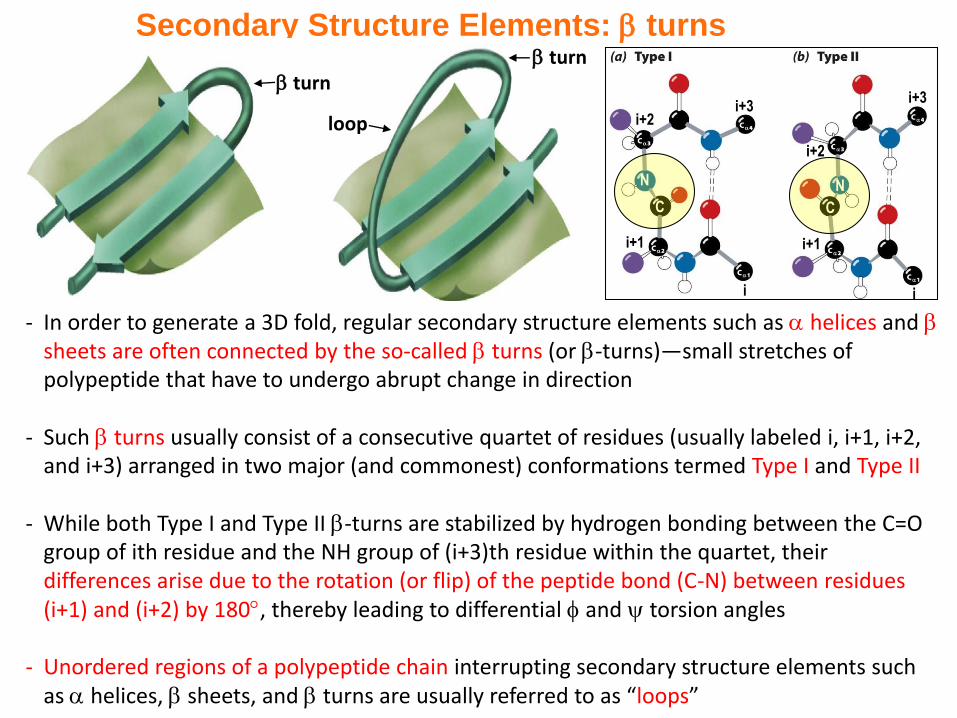
- In order to generate a 3D fold, regular secondary structure elements such as helices and sheets are often connected by the so-called turns (or -turns)—small stretches of polypeptide that have to undergo abrupt change in direction
- Such turns usually consist of a consecutive quartet of residues (usually labeled i, i+1, i+2, and i+3) arranged in two major (and commonest) conformations termed Type I and Type II
- While both Type I and Type II -turns are stabilized by hydrogen bonding between the C=O group of ith residue and the NH group of (i+3)th residue within the quartet, their differences arise due to the rotation (or flip) of the peptide bond (C-N) between residues (i+1) and (i+2) by 180, thereby leading to differential and torsion angles
- Unordered regions of a polypeptide chain interrupting secondary structure elements such as helices, sheets, and turns are usually referred to as “loops”
Secondary Structure Elements: turns
turn
loop
turn
C C
NN
i
i+1
i+2
i+3
i
i+1
i+2i+3
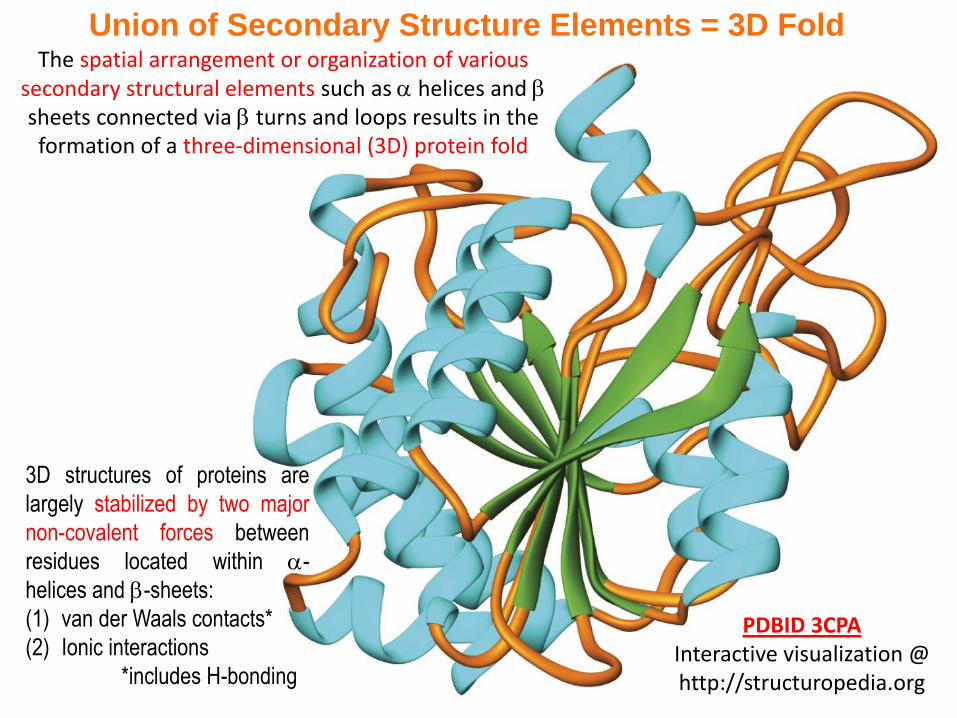
Union of Secondary Structure Elements = 3D FoldThe spatial arrangement or organization of various
secondary structural elements such as helices and sheets connected via turns and loops results in the formation of a three-dimensional (3D) protein fold
PDBID 3CPAInteractive visualization @ http://structuropedia.org
3D structures of proteins are
largely stabilized by two major
non-covalent forces between
residues located within -
helices and -sheets:
(1) van der Waals contacts*
(2) Ionic interactions
*includes H-bonding

- Describe the four levels of protein structure. Do all proteins exhibit all four levels?
- Explain why the conformational freedom of peptide bonds is limited
- Summarize the features of the following secondary structure elements: α helix, parallel sheet, and antiparallel β sheet
- Visualize the structure of PDBID 3CPA @ http://structuropedia.org
Exercise 2.3a
Section 2.3b:
Tertiary Structure

- Many proteins (but not all!) adopt an highly-ordered three-dimensional (3D) shape or structure in order to become biologically active
- Knowledge of protein structure is de rigueur to understanding how it works (its biological function)
- X-ray crystallography (XRC) and nuclear magnetic resonance (NMR) reign supreme as experimental methods to determine 3D protein structures at atomic level—while XRC requires protein crystals, NMR is conducted on proteins in their natural aqueous environment
- While protein sequence is the primary determinant of its 3D structure (or shape), two proteins with distinct sequences can also adopt similar 3D folds (convergent evolution)—though rare, two proteins with very similar sequences may also adopt distinct 3D folds (divergent evolution)
- In 3D structure, apolar residues are usually found in the core or interior of the protein, while polar and charged residues point outwards onto the surface. Why?!
- Protein structures are deposited into the online protein databank (PDB) @ http://rcsb.org
- Protein structures can be interactively visualized and rendered online @ http://structuropedia.org
Synopsis 2.3b

Levels of Protein Structure: Description
Primary (1) StructureThe amino acid sequence of a polypetide chain
Secondary (2) StructureThe local ordered conformation of residues within a polypeptide chain—these are referred to as secondary structure elements such as helices and sheets (or strictly strands!)
Tertiary (3) StructureThe spatial arrangement or organization of various secondary structural elements such as helices and sheets connected via turns and loops into a three-dimensional (3D) fold
Quaternary (4) StructureThe spatial arrangement or organization of individual polypeptide chains with a well-defined 3D fold relative to each other
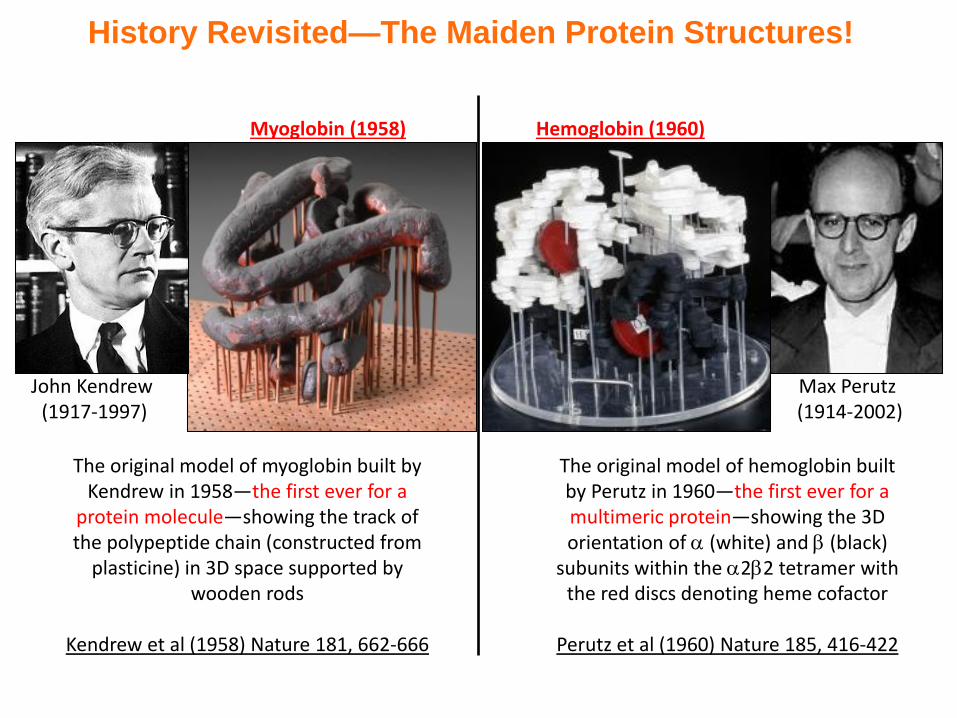
History Revisited—The Maiden Protein Structures!
John Kendrew (1917-1997)
The original model of myoglobin built by Kendrew in 1958—the first ever for a
protein molecule—showing the track of the polypeptide chain (constructed from
plasticine) in 3D space supported by wooden rods
Kendrew et al (1958) Nature 181, 662-666
Myoglobin (1958)
Max Perutz (1914-2002)
The original model of hemoglobin built by Perutz in 1960—the first ever for a multimeric protein—showing the 3D orientation of (white) and (black)
subunits within the 22 tetramer with the red discs denoting heme cofactor
Perutz et al (1960) Nature 185, 416-422
Hemoglobin (1960)
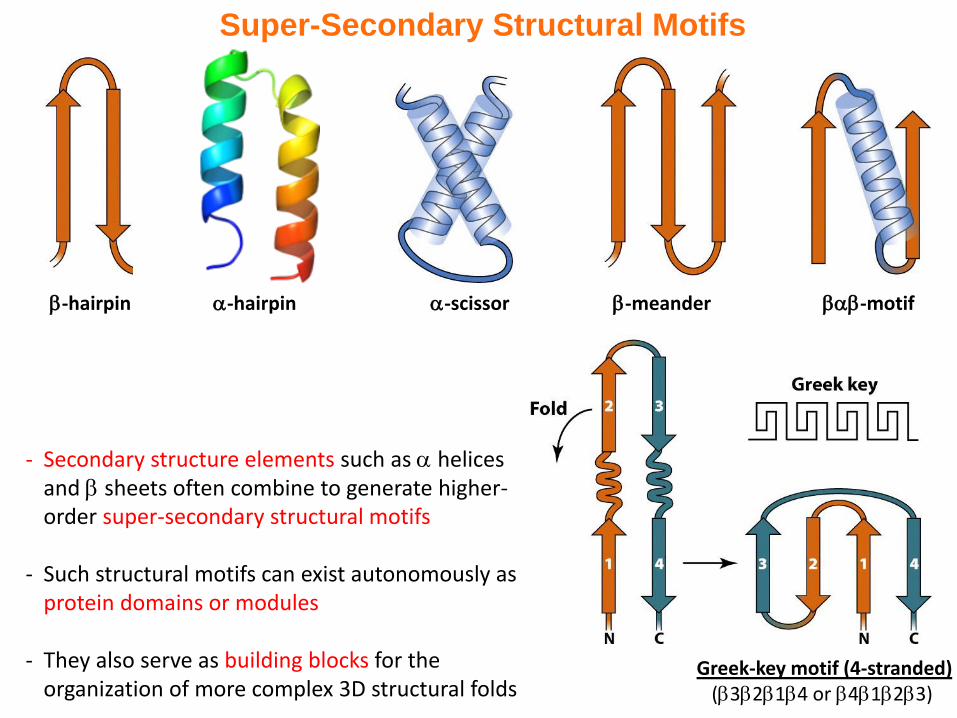
Super-Secondary Structural Motifs
- Secondary structure elements such as helices and sheets often combine to generate higher-order super-secondary structural motifs
- Such structural motifs can exist autonomously as protein domains or modules
- They also serve as building blocks for the organization of more complex 3D structural folds
-motif-hairpin -scissor-hairpin -meander
Greek-key motif (4-stranded)(3214 or 4123)
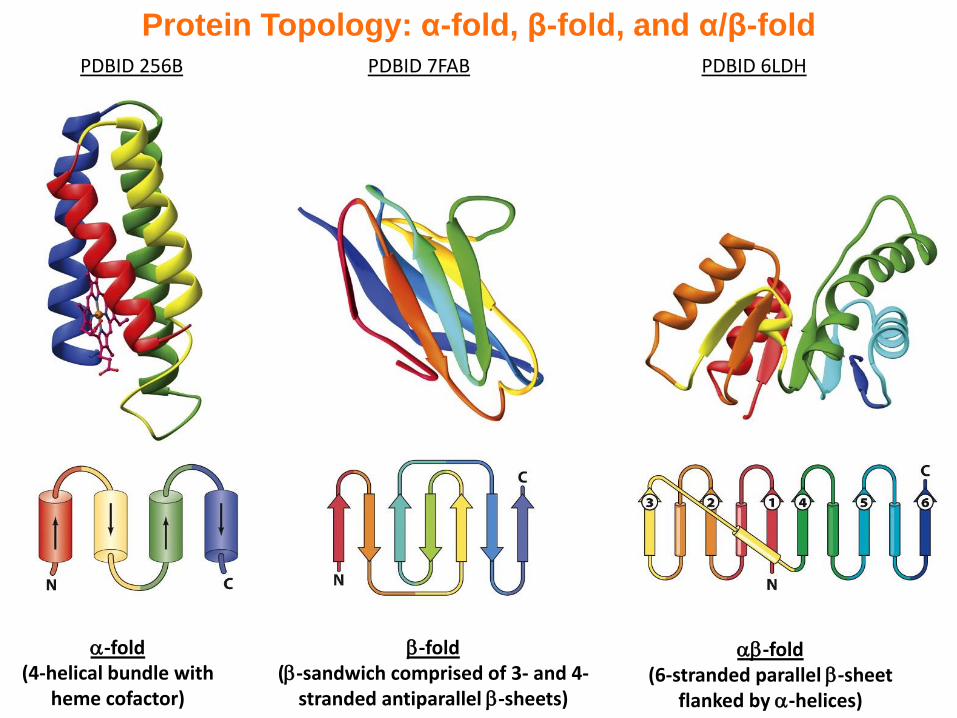
Protein Topology: α-fold, β-fold, and α/β-fold
-fold(4-helical bundle with
heme cofactor)
PDBID 256B
-fold(-sandwich comprised of 3- and 4-
stranded antiparallel -sheets)
PDBID 7FAB
-fold(6-stranded parallel -sheet
flanked by -helices)
PDBID 6LDH
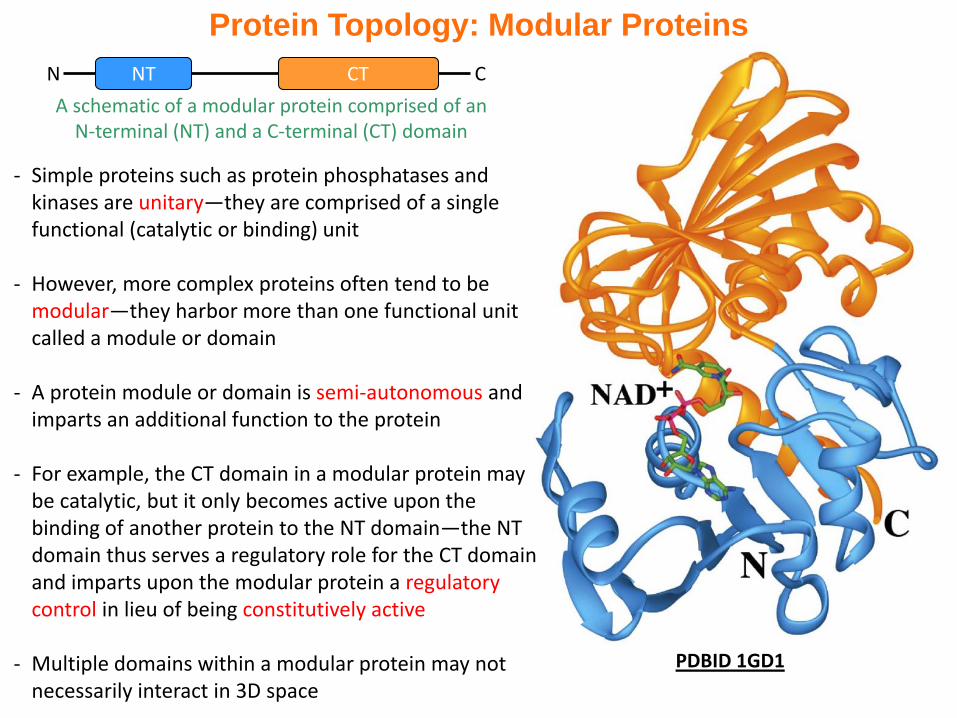
Protein Topology: Modular Proteins
PDBID 1GD1
- Simple proteins such as protein phosphatases and kinases are unitary—they are comprised of a single functional (catalytic or binding) unit
- However, more complex proteins often tend to be modular—they harbor more than one functional unit called a module or domain
- A protein module or domain is semi-autonomous and imparts an additional function to the protein
- For example, the CT domain in a modular protein may be catalytic, but it only becomes active upon the binding of another protein to the NT domain—the NT domain thus serves a regulatory role for the CT domain and imparts upon the modular protein a regulatory control in lieu of being constitutively active
- Multiple domains within a modular protein may not necessarily interact in 3D space
A schematic of a modular protein comprised of an N-terminal (NT) and a C-terminal (CT) domain
N CCTNT
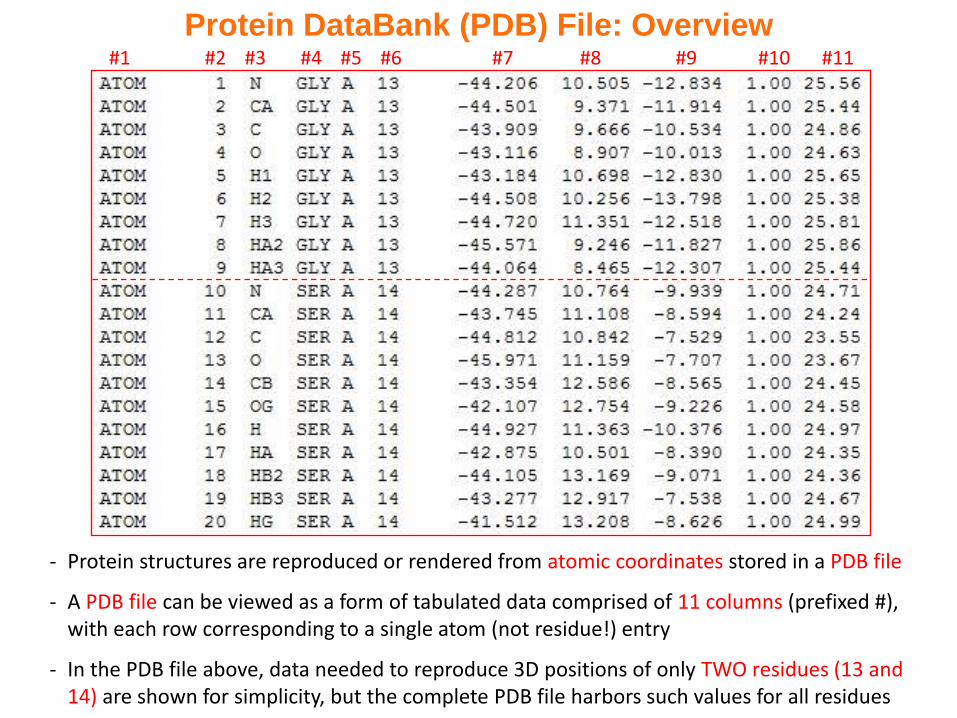
#1 #2 #3 #4 #5 #6 #7 #8 #9 #10 #11
Protein DataBank (PDB) File: Overview
- Protein structures are reproduced or rendered from atomic coordinates stored in a PDB file
- A PDB file can be viewed as a form of tabulated data comprised of 11 columns (prefixed #), with each row corresponding to a single atom (not residue!) entry
- In the PDB file above, data needed to reproduce 3D positions of only TWO residues (13 and 14) are shown for simplicity, but the complete PDB file harbors such values for all residues
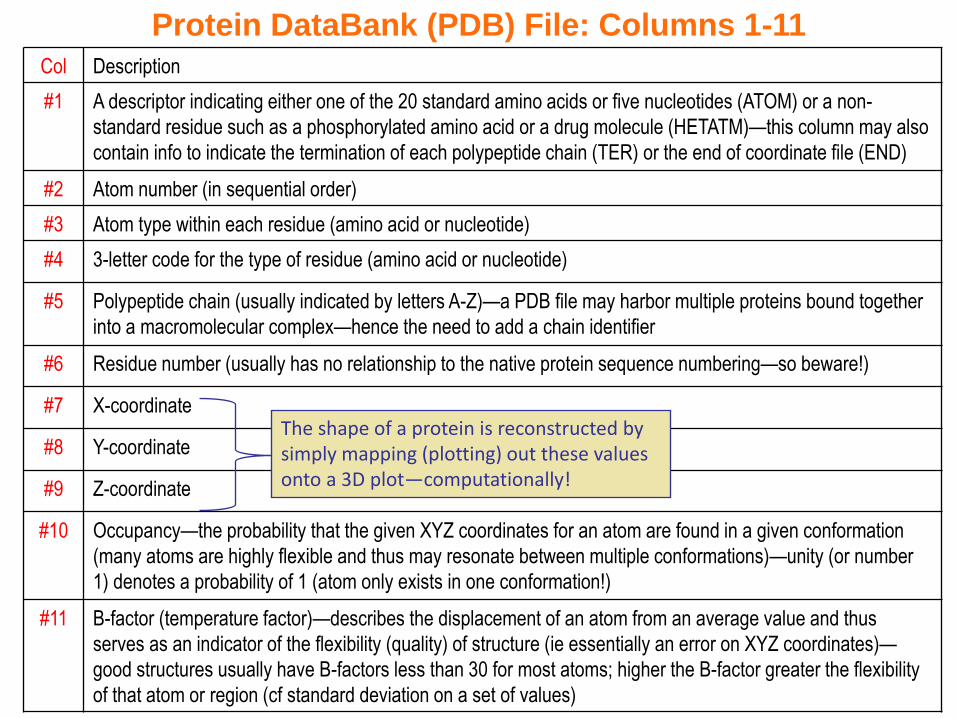
Col Description
#1 A descriptor indicating either one of the 20 standard amino acids or five nucleotides (ATOM) or a non-
standard residue such as a phosphorylated amino acid or a drug molecule (HETATM)—this column may also
contain info to indicate the termination of each polypeptide chain (TER) or the end of coordinate file (END)
#2 Atom number (in sequential order)
#3 Atom type within each residue (amino acid or nucleotide)
#4 3-letter code for the type of residue (amino acid or nucleotide)
#5 Polypeptide chain (usually indicated by letters A-Z)—a PDB file may harbor multiple proteins bound together
into a macromolecular complex—hence the need to add a chain identifier
#6 Residue number (usually has no relationship to the native protein sequence numbering—so beware!)
#7 X-coordinate
#8 Y-coordinate
#9 Z-coordinate
#10 Occupancy—the probability that the given XYZ coordinates for an atom are found in a given conformation
(many atoms are highly flexible and thus may resonate between multiple conformations)—unity (or number
1) denotes a probability of 1 (atom only exists in one conformation!)
#11 B-factor (temperature factor)—describes the displacement of an atom from an average value and thus
serves as an indicator of the flexibility (quality) of structure (ie essentially an error on XYZ coordinates)—
good structures usually have B-factors less than 30 for most atoms; higher the B-factor greater the flexibility
of that atom or region (cf standard deviation on a set of values)
Protein DataBank (PDB) File: Columns 1-11
The shape of a protein is reconstructed by simply mapping (plotting) out these values onto a 3D plot—computationally!

- Why do turns and loops most often occur on the protein surface?
- Which side chains usually occur on a protein’s surface? In its interior?
- Describe some of the common protein structural motifs
- Summarize the types of information provided in a PDB file
- Why is it useful to compare protein structures in addition to protein sequences?
- Visualize the structures with PDBIDs 256B, 7FAB and 6LDH @ http://strucuropedia.org
Exercise 2.3b

Section 2.3c:
Quaternary Structure

Synopsis 2.3c
- Many proteins harbor quaternary structure—they are composed of multiple polypeptide chains (or subunits), usually arranged symmetrically
- Subunits (also called monomers) may be identical (homomers) or non-identical (heteromers)
- Subunits usually associate non-covalently in a process termed “multimerization” or “oligomerization”—however, not all multimers or oligomers are functionally relevant
- The physiologically-relevant multimeric form of a protein is called biological unit (or biological assembly)—for example, the functional form of hemoglobin is a tetramer comprised of four chains (designated 22)
- Commonly observed biological units constituted from individual subunits (monomers) are dimers, trimers, tetramers, hexamers and octamers:
homodimers / heterodimershomotrimers / heterotrimershomotetramers / heterotetramers
- Protein oligomerization confers many advantages—eg enhanced stability, greater regulatory control, attaining allosteric/cooperative behavior, functional versatility, and signal amplification

Primary (1) StructureThe amino acid sequence of a polypetide chain
Secondary (2) StructureThe local ordered conformation of residues within a polypeptide chain—these are referred to as secondary structure elements such as helices and sheets
Tertiary (3) StructureThe spatial arrangement or organization of various secondary structural elements such as helices and sheets connected via turns and loops into a three-dimensional (3D) fold
Quaternary (4) StructureThe spatial arrangement or organization of individual polypeptide chains with a well-defined 3D fold relative to each other
Levels of Protein Structure: Description
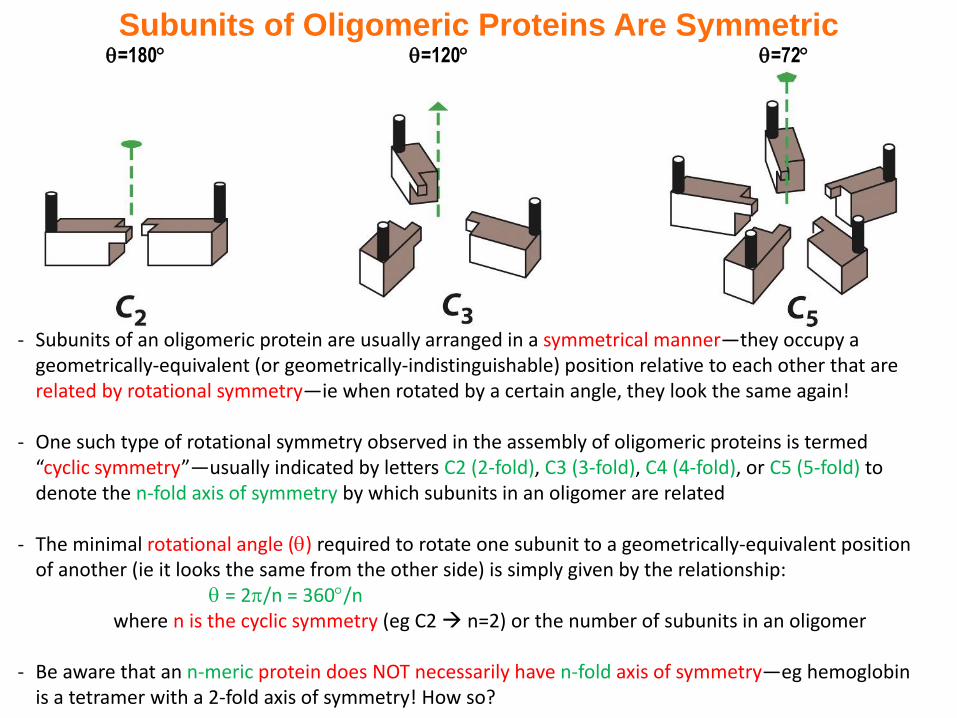
=180 =120 =72
Subunits of Oligomeric Proteins Are Symmetric
- Subunits of an oligomeric protein are usually arranged in a symmetrical manner—they occupy a geometrically-equivalent (or geometrically-indistinguishable) position relative to each other that are related by rotational symmetry—ie when rotated by a certain angle, they look the same again!
- One such type of rotational symmetry observed in the assembly of oligomeric proteins is termed “cyclic symmetry”—usually indicated by letters C2 (2-fold), C3 (3-fold), C4 (4-fold), or C5 (5-fold) to denote the n-fold axis of symmetry by which subunits in an oligomer are related
- The minimal rotational angle () required to rotate one subunit to a geometrically-equivalent position of another (ie it looks the same from the other side) is simply given by the relationship:
= 2/n = 360/nwhere n is the cyclic symmetry (eg C2 n=2) or the number of subunits in an oligomer
- Be aware that an n-meric protein does NOT necessarily have n-fold axis of symmetry—eg hemoglobin is a tetramer with a 2-fold axis of symmetry! How so?
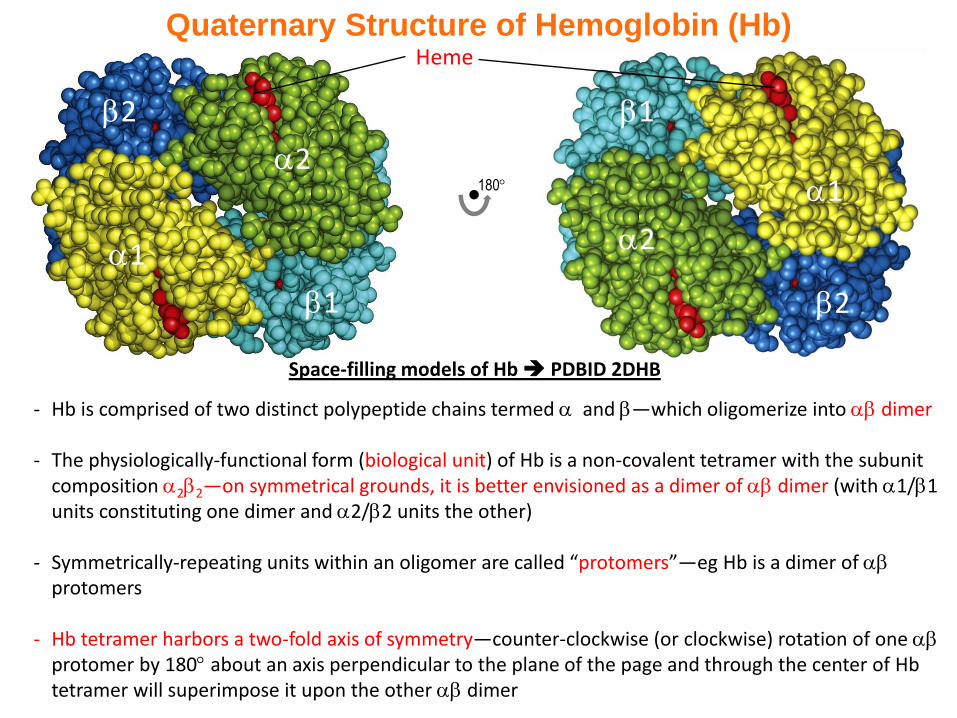
Quaternary Structure of Hemoglobin (Hb)
- Hb is comprised of two distinct polypeptide chains termed and —which oligomerize into dimer
- The physiologically-functional form (biological unit) of Hb is a non-covalent tetramer with the subunit composition 22—on symmetrical grounds, it is better envisioned as a dimer of dimer (with 1/1 units constituting one dimer and 2/2 units the other)
- Symmetrically-repeating units within an oligomer are called “protomers”—eg Hb is a dimer of protomers
- Hb tetramer harbors a two-fold axis of symmetry—counter-clockwise (or clockwise) rotation of one protomer by 180 about an axis perpendicular to the plane of the page and through the center of Hbtetramer will superimpose it upon the other dimer
2
1
1
2
1
2
1
2
180
Heme
Space-filling models of Hb PDBID 2DHB

- List the advantages of multiple subunits in proteins
- What axis of symmetry are the following protein oligomers likely to harbor? Dimer, trimer, and tetramer?
- Visualize hemoglobin structure (PDBID 1GZX) @ http://strucuropedia.org:
(1) Import the tetramer with PDBID(2) Color-code the subunits or chains A/1 (yellow), B/1 (cyan),
C/2 (green), and D/2 (blue)(3) Rotate the tetramer by 180 in the plane of the page (Z-plane)
to observe a two-fold axis of symmetry(4) Display the bound oxygen molecule to each monomer
Exercise 2.3c





























