Embed Size (px)
Citation preview

3. Material und Methoden
3.1 Untersuchte Organismen Die für diese Arbeit verwendeten Kulturen wurden aus der Sammlung von Algenkulturen der
Universität Göttingen (SAG) bezogen. Die genauen Bezeichnungen der Spezies sind in
Tabelle 3-1 aufgeführt.
Spezies Ordnung (sensu LEEDALE 1967)
Stamm
Distigma curvatum PRINGSHEIM 1936 Eutreptiales SAG B 1216-1b
Khawkinea quartana (MOROFF) JAHN et MCKIBBEN 1937 Euglenales SAG 1204-9
Khawkinea sp.1) Euglenales ────
Lepocinclis ovata (PLAYFAIR 1921) CONRAD 1934 Euglenales SAG B 1244-5
Phacus smulkowskianus (ZAKRYS 1986) KUSBER 1998 Euglenales SAG 58.81
Gyropaigne lefèvrei BOURELLY et GEORGES 1951 Rhabdomonadales SAG B 227.80
Rhabdomonas gibba (SKUJA 1939) PRINGSHEIM nom. nud. Rhabdomonadales SAG B 1271-2
Rhabdomonas incurva (FRESENIUS) KLEBS 1883 Rhabdomonadales SAG B 235.80 Tab. 3-1: Übersicht über die in dieser Arbeit untersuchten Organismen. 1) : Isolat aus einer verunreinigten Kultur von Rhabdomonas spiralis.
3.2 Lichtmikroskopie Die lichtmikroskopischen Untersuchungen auf Reinheit der Kulturen wurden an einem
Olympus BH-Mikroskop mit Phasenkontrast durchgeführt. Zur Dokumentation wurde Alfo
Color Film (200 ASA) verwendet.
3.3 Elektronenmikroskopie (TEM) Die elektronenmikroskopischen Untersuchungen wurden an einem HITACHI H 500
Elektronenmikroskop bei einer Beschleunigungsspannung von 100 kV durchgeführt. Die
Dokumentation erfolgte auf Agfa Scientia EM-Film. Von allen untersuchten Arten wurden
mindestens 30 Zellen betrachtet.

Material & Methoden
21
3.3.1 Fixierung, Entwässerung und Einbettung der Proben 103 - 107 Zellen wurden für 1 min bei 2700 x g abzentrifugiert und das Pellet 2 x in EM-Puffer
gewaschen. Die anschließende Fixierung erfolgte für 30 min in 5% Glutaraldehyd in EM-
Puffer, nachfolgend wurde mit 2% Osmiumtetroxid für 30 min nachfixiert. Nach 5 x 5
minütigem Waschen in EM-Puffer folgte eine ansteigende Entwässerungsreihe in Aceton (15,
30, 45, 60, 75, 90%; je 15 min). Danach wurden die Proben 3 x für 10 min in 100% Aceton
belassen und im Anschluß daran in das Epoxyharz Transmit überführt, wobei die Proben
zuerst in Aceton/Transmit (1/1, v/v) für 20 min bei RT stehengelassen wurden. Anschließend
wurden die Proben in Aceton/Transmit im Verhältnis (1/2, v/v) überführt und über Nacht
stehengelassen. Nach Absaugen des Aceton/Transmit-Gemisches und Zugabe von Transmit
(100%) wurde dreimal für 1 h im Exsikkator entgast. Nach Überführen in Kunststoffkapseln
(Beem) polymerisierten die Proben über Nacht in einem Trockenschrank bei 70°C aus.
EM Puffer: 50 mM KH2PO4 / Na2HPO4, 38.8 / 61.2 (v/v), pH: 7.2
3.3.2 Herstellung der Schnitte Ultradünnschnitte einer Dicke von 60 - 80 nm wurden an einem Ultramikrotom (Ultracut E,
Reichert) mit einem Diamantmesser (DuPont) hergestellt. Die Schnitte wurden anschließend
auf 200 mesh Nickelnetzchen (Plano) übertragen und jeweils für 15 s mit Kaliumpermanganat
(5%) und für 30 s mit Bleicitrat (2%) nachkontrastiert.
3.4 RNA Isolierung Wählt man Messenger RNA (mRNA) als Ausgangsmaterial für nachfolgende genetische
Arbeiten, so hat dies zum Vorteil, daß Introns, wie sie in der DNA vorliegen, hier nicht
vorhanden sind. Es gibt dabei die Möglichkeit, die Messenger RNA aus der Gesamt-RNA zu
isolieren, oder direkt Gesamt-RNA als Probenmaterial für eine nachfolgende Reverse
Transkription [! 3.9] einzusetzen. In dieser Reaktion wurden mit Hilfe spezieller Primer [!
3.8.1] die für die beiden Hauptproteine des Paraxonemalstabes (PAR 1 und PAR 2)
kodierenden Bereiche in komplementäre einzelsträngige DNA (cDNA) umgeschrieben.

Material & Methoden
22
3.4.1 Isolierung von Gesamt-RNA Gesamt-RNA wurde über eine Guanidinium-Isothiocyanat-Phenol-Chloroform-Fällung
isoliert (CHOMCZYNSKI & SACCI 1987, verändert nach SAMBROCK et al. 1989). Dafür wurden
50 mg Zellmaterial in flüssigem Stickstoff zermahlen, das Pulver in 2 ml Eppendorf-Gefäßen
mit 200 µl Extraktionspuffer + 20 µl Mercaptoethanol überführt und für 30 s kräftig gemischt.
Danach erfolgten Zugaben von 20 µl 2 M Natriumacetat (pH 4), 200 µl wassergesättigtem
Phenol und 80 µl Chloroform:Isoamylalkohol (24:1), wobei nach Hinzufügen der einzelnen
Chemikalien jeweils für 30 s gut durchmischt wurde. Zu beachten war hierbei, daß alle
Schritte möglichst schnell aufeinander folgten und daß die Zellen während des Mörserns nicht
antauten, da sonst eine Angriffsmöglichkeit für RNasen bestünde. Anschließend wurde für
15 min bei 5000 x g und 4°C abzentrifugiert, die obere wäßrige Phase abgenommen, mit
gleichem Volumen Isopropanol gut durchmischt und bei �20°C über Nacht inkubiert. Nach
Abzentrifugieren bei 4°C für 30 min bei 10000 x g wurde das RNA Pellet in 100 µl
Extraktionspuffer mit 10 µl Mercaptoethanol resuspendiert, dabei war in einigen Fällen ein
kurzes Erwärmen auf maximal 65°C notwendig, um die RNA vollständig zu lösen. Nach
Mischen mit gleichem Volumen Isopropanol, mindestens einstündiger Lagerung bei �20°C
und erneutem Abzentrifugieren bei 4°C für 30 min bei 10000 x g wurden auf das RNA Pellet
100 µl eiskalter 75%iger Ethanol gegeben, sofort wieder dekantiert und der Restalkohol in
einem Trockenschrank bei 30°C verdampft. Das nun weiß aussehende RNA Pellet wurde in
30 µl RNase freiem Wasser resuspendiert, wobei auch hier in einigen Fällen ein maximal
5 minütiges Erwärmen auf 65°C im Wasserbad notwendig war. Nach dem Aliquotieren wurde
die RNA bis zur weiteren Verwendung bei -83°C gelagert.
3.4.1.1 Lösungen zur RNA Isolierung Alle Lösungen wurden in RNase-freien Gefäßen angesetzt: dazu wurde Glasmaterial für die
Dauer von 15 h einer Temperatur von 180°C ausgesetzt und Plastikmaterial vor dem
Autoklavieren für 40 min mit UV Licht einer Wellenlänge von 254 nm bestrahlt. Die
Lösungen wurden ausschließlich mit Diethylpyrocarbonat (DEPC) behandeltem H2O nanopore
angesetzt. Von dieser RNasen hemmenden Substanz wurden 1 ml pro l H2O zugegeben und
vor dem nachfolgendem Autoklavieren für mindestens 6 h gerührt.
Wenn nicht anders beschrieben, wurden Chemikalien der Fa. Roth verwendet. Dieses gilt
ebenso für alle folgenden Protokolle dieser Arbeit.

Material & Methoden
23
• RNA Extraktionspuffer: → Lagerung bei Raumtemperatur
4 M Guanidinium-Isothiocyanat
25 mM Natriumcitrat (aus einer 1 M Stammlösung mit pH 7)
0.5 % Sarcosyl [N-Lauroylsarcosine; Sigma]
direkt vor der RNA Isolierung wird zum Extraktionspuffer hinzugefügt:
0.1 M ß-Mercaptoethanol [Merck] → Lagerung bei 4°C unter Lichtabschluß
• Wassergesättigtes Phenol (pH 4 - 4.5) → Lagerung bei 4°C unter Lichtabschluß
• Chloroform:Isoamylalkohol (24:1) → Lagerung bei 4°C unter Lichtabschluß
• Isopropanol → Lagerung bei 4°C
• 2 M Natriumacetat, pH 4,
DEPC - behandelt und autoklaviert → Lagerung bei 4°C
• 75 % Ethanol → Lagerung bei -20°C
• Wasser, DEPC-behandelt und autoklaviert → Lagerung bei Raumtemperatur
3.4.2 RNA Isolierung mit dem RNeasy Kit Wenn mit weniger als 50 mg nur geringe Mengen an Ausgangsmaterial vorlagen, wurde die
Isolierung von RNA mit dem RNeasy Mini Kit (Qiagen) durchgeführt. Hierbei wird die RNA
mit einem Guanidin-Isothiocyanat Puffer lysiert und homogenisiert, anschließend an eine
Membran aus Silikagel gebunden, gewaschen und mit H2O eluiert.
Auf diese Weise können alle RNA Moleküle, die länger als 200 Nukleotide sind, isoliert
werden. Kleinere RNAs, wie die 5.8 S (~160 nt), die 5 S (~120 nt) und tRNAs (~90 nt)
werden unter den genannten Bedingungen nicht quantitativ gebunden. Da solche RNAs 15-
20% der Gesamt-RNA einer Zelle ausmachen, werden die anderen RNAs auf diese Weise
zugleich angereichert.
Die Isolierung wurde nach Angaben des Herstellers mit dem Protokoll "Isolierung aus
Pflanzen und Pilzen" durchgeführt; als Lysepuffer wurde Puffer RLT verwendet.

Material & Methoden
24
3.5 Elektrophoretische Trennung von RNA Da RNA extrem anfällig für einen Abbau durch die ubiquitär verbreiteten und zudem sehr
stabilen Ribonucleasen ist, müssen bei einer Elektrophorese Puffer- und Gelsysteme
verwendet werden, die RNasen inhibieren. Ferner müssen denaturierende Bedingungen
geschaffen werden, da die einzelsträngige RNA sonst intramolekulare und intermolekulare
Basenpaarungen zwischen kurzen Regionen komplementärer Sequenzen ausbilden würde,
was zu einer Faltung und damit zu einer veränderten Wanderungsgeschwindigkeit im Gel
führen würde. Daher werden zur Denaturierung der RNA Gele verwendet, in denen sowohl
im Probenauftragspuffer, als auch im Gel selber Formaldehyd enthalten ist (LEHRACH et al.
1977), das kovalent mit den Aminogruppen von Adenin, Guanin und Cytosin reagiert und so
die Bildung von Basenpaarungen verhindert. Die Gelelektrophoresen der RNA erfolgten in
Horizontalgelkammern mit einem Gelvolumen von 30 ml und einem maximalen
Probenvolumen von 20 µl. Vor dem Giessen der Gele wurden die Kammern für 60 min mit
3%igem H2O2 befüllt und anschließend mit DEPC-H2O gut gespült. Für ein 1%iges Gel
wurden 0.3 g Agarose mit 25.5 ml DEPC-H2O in der Mikrowelle aufgekocht und 1.5 ml
Formaldehyd sowie 3.0 ml 10x MEA-Puffer (MOPS-EDTA-NaAc) erst nach dem Abkühlen
auf ca. 60°C zugegeben und das Gel gegossen. Das Abkühlen ist erforderlich, um eine
Degradierung der im MEA-Puffer enthaltenen [3-(N-Morpholino)-Propansulfonsäure)] zu
verhindern, die zu einem Verlust der Pufferkapazität führen würde; es vermindert zugleich
auch die Entstehung von Formaldehyddämpfen.
Während des Aushärtens der Gele wurden die Proben auf Eis vorbereitet. Nach Zugabe von
drei Volumeneinheiten RNA-Cocktail zu einer Volumeneinheit RNA (5-25 µg) wurden die
Proben durchmischt, für 10 min bei 65°C denaturiert und bis zum Beladen der Gele auf Eis
gelagert. Der Gellauf erfolgte für 30 min bei 100 V und variabler Stromstärke. Als Laufpuffer
wurde 1 x MEA verwendet. Die Detektion erfolgte wie unter ! 3.7 beschrieben.
3.5.1 Lösungen für die elektrophoretische Trennung von RNA • MEA-Puffer 10 x:
0.2 M MOPS [3-(N-Morpholino)
-Propansulfonsäure)], pH 7.0
(41.8 g / l)
0.01 M EDTA (20 ml 0.5 M EDTA, pH 8.0)
0.05 M Natriumacetat (6.8 g / l)

Material & Methoden
25
Nach Zugabe von ca. 900 ml DEPC-H2O wird der pH-Wertes mit KOH auf 7.0
eingestellt. Nach Auffüllen auf 1000 ml wird das Pufferkonzentrat für 30 min bei 121°C
autoklaviert und anschließend bei RT unter Lichtabschluß gelagert.
• RNA-Ladepuffer 10 x:
0.4 % (w/v) Bromphenolblau (2.5 mg / 10 ml)
0.4 % (w/v) Xylencyanol (2.5 mg / 10 ml)
1 mM EDTA (pH 8.0) (20 µl 0.5 M EDTA / 10 ml)
50 % (v/v) steriles Glycerin (5 ml / 10 ml)
50 % (v/v) DEPC-H2O (5 ml / 10 ml)
Vor der Zugabe von EDTA und Glycerin werden Bromphenolblau und Xylencyanol mit
DEPC-H2O zusammengegeben und über einen Filter mit einer Porenweite von 0.45 µm
abfiltriert. Die Lagerung des Puffers erfolgt bei 4°C.
• RNA-Cocktail [ausreichend zum Auftragen von maximal 195 µl RNA]:
500 µl Formamid
175 µl Formaldehyd
50 µl 10 x MEA
50 µl RNA-Ladepuffer 10 x
5 µl Ethidiumbromid (10 mg / ml)
Dieser Cocktail wird immer frisch angesetzt und bis zu seiner Verwendung unter
Lichtabschluß auf Eis belassen.
3.6 DNA Isolierung Die DNA Isolierungen für die SSU rDNA-Analysen wurden sowohl über klassische Phenol-
Extraktionen [!3.6.1], als auch mit dem DNeasy Mini Plant Kit (Qiagen) [!3.6.2]
durchgeführt.
3.6.1 DNA Isolierung durch Phenol-Chloroform-Isoamylalkohol Extraktion Die DNA Isolierung durch Phenol Extraktion ist eine Methode, die dazu dient, Proteine aus
dem Gesamtzellextrakt der Organismen zu entfernen. Dazu werden Phenol oder Gemische

Material & Methoden
26
aus Phenol und Chloroform zu den Zellen gegeben, gut durchmischt und abzentrifugiert.
Durch die organischen Lösemittel werden die Proteine ausgefällt, während die Nucleinsäuren
sich in den wäßrigen Phasen befinden. RNA wird durch Zugabe von RNasen abgebaut, wobei
die entstehenden Ribonukleotide sich ebenfalls in den wäßrigen Phasen befinden. Die
Trennung der DNA von der Ribonukleotiden erfolgt über eine Ethanolpräzipitation. Ethanol
fällt in Gegenwart von einwertigen Kationen wie Na+ und bei einer Temperatur von -20°C
Nucleinsäuren sehr wirksam aus. Der DNA Niederschlag kann dann nach Abzentrifugieren
wieder in H2O gelöst werden, während die Ribonukleotide nicht präzipitieren und somit von
der DNA getrennt werden können.
Protokoll zur DNA Isolierung:
• 5-10 ml Zellkultur für 5 min bei 5000 x g abzentrifugieren und das Pellet in 0.5 ml TE-
Puffer aufnehmen.
• 0.5 ml Phenol hinzufügen, durch Auf- und Abpipettieren gut durchmischen und für
10 min bei 15000 x g abzentrifugieren.
•
•
obere wäßrige Phase in ein neues Reaktionsgefäß überführen, 0.3 ml Phenol/Chloroform
(1:1) zugeben, gut durchmischen, für 10 min bei 15000 x g abzentrifugieren und den
Reaktionsschritt wiederholen.
obere Phase in neues Reaktionsgefäß überführen, 0.4 ml Chloroform/Isoamylalkohol
(24:1) zugeben, gut durchmischen und für 10 min bei 15000 x g abzentrifugieren.
• obere Phase in neues Reaktionsgefäß überführen, 55 µl 3M Natriumacetat pH 5.2
(Endkonzentration: 0.3M) und 1 ml Isopropanol zugeben, gut durchmischen und für
30 min bei 15000 x g zentrifugieren.
• Pellet in 1 ml 70% Ethanol aufnehmen, für 30 min bei -20°C lagern und anschließend für
5 min bei 15000 x g abzentrifugieren.
• Überstand mit der DNA abpipettieren und für 30 min in einer Vakuumzentrifuge
einengen.
• DNA in 20 µl TE-Puffer aufnehmen, aliquotieren und bei -20°C bis zur weiteren
Verwendung lagern.
Die wäßrigen Phasen sollten mit abgeschnittenen 1000 µl Spitzen abgenommen werden;
dadurch werden Scherkräfte vermindert, die ein Zerstückeln der DNA zur Folge hätten.

Material & Methoden
27
3.6.1.1 Lösungen zur DNA Extraktion
Bei der DNA Isolierung ist darauf zu achten, daß der pH-Wert des Phenols über 7.8 liegt, da
DNA bei niedrigeren pH-Werten zum großen Teil in die organische Phenolphase eindringt
und die Menge an isolierter DNA damit erheblich vermindert wird.
• Phenol Lagerung bei 4°C
• Phenol / Chloroform (24:1) Lagerung bei 4°C, die Lösung ist für 4 Wochen stabil
• Chloroform / Isoamylalkohol (24:1) Lagerung bei 4°C, die Lösung ist für 4 Wochen stabil
• 3 M Natriumacetat, pH 5.2 Lagerung bei 4°C nach 30 minütigem Autoklavieren
• TE-Puffer:
10 mM Tris - HCl (pH 7.4)
1 mM EDTA (pH 8.0)
Lagerung bei 4°C nach 30 minütigem Autoklavieren
3.6.2 DNA Isolierung mit dem DNeasy Kit Ein Teil der DNA Isolierungen erfolgte mit dem DNeasy Plant Mini Kit (Qiagen). Hierfür
wurden zunächst 5 - 10 ml Kultur für 5 min bei 5000 x g abzentrifugiert, die Zellen in
flüssigem Stickstoff durch Mörsern aufgebrochen und anschließend nach dem Protokoll des
Herstellers verfahren. Bei dieser Isolierung werden zuerst Proteine und Polysaccharide
ausgefällt und durch Zentrifugieren über eine "QIAshredder-Säule" zurückgehalten. Der
Durchfluß mit der DNA wird dann unter hohen Salzkonzentrationen an eine Membran aus
Silikagel gebunden, durch mehrere Waschschritte von restlichen Protein-, Polysaccharid- und
Salzanteilen befreit und anschließend mit einem Puffer geringen Salzgehaltes eluiert. Da im
Kit auch RNase A eingesetzt wird, ist gewährleistet, daß keine Ribonukleinsäuren mitisoliert
werden.
3.7 Elektrophoretische Trennung von DNA Die Gelelektrophoresen von DNA wurden in Horizontalgelkammern mit einem Gelvolumen
von 30 ml durchgeführt. Dazu wurden 1 - 1.2 % Agarose mit 1x Tris-Acetat-EDTA (TAE)-
Puffer in der Mikrowelle kurz aufgekocht, nach Abkühlen auf ca. 60°C mit 3 µl einer 1 %igen
Lösung von Ethidiumbromid versetzt und das Gel gegossen. Das Ethidiumbromid dient dabei

Material & Methoden
28
dem späteren Sichtbarmachen der Banden, da es in die DNA interkaliert und unter UV-Licht
fluoresziert. Nach 30 minütigem Auspolymerisieren wurden eine Volumeneinheit DNA mit
0.2 Volumeneinheiten 5 x Ladepuffer versetzt und die DNA für 30 min bei 100 V und
variabler Stromstärke aufgetrennt. Als Laufpuffer wurde 1 x TAE-Puffer verwendet, als
Längenstandard für den Bereich von 100 - 2072 bp ein 100 bp DNA Marker (GibcoBRL). Für
größere DNA-Fragmente, oder um quantitative Aussagen machen zu können, wurde ein 1kb
DNA Marker (GibcoBRL) eingesetzt, der einen Bereich von 500 bp - 12 kb umfaßt. Dieser
Marker enthält 10 % seiner gesamten DNA Menge in der 1636 bp - Bande; er wurde so
verdünnt, daß in 1.25 µl Marker insgesamt 10 ng DNA enthalten waren.
Die Detektion der DNA erfolgte mittels UV- Durchlichtapparatur mit angeschlossener CCD -
Kamera (Intas). Die Prints wurden mit einem Video-Graphic-Printer (Intas, Modell UP
890CE) auf UPP-110HA Superior Density Printing Paper (Sony®) erstellt.
3.7.1 Lösungen für die elektrophoretische Trennung von DNA • TAE-Puffer 50 x:
40 mM Tris 243.3 g / l
1 mM EDTA (Titriplex III) 18.62 g / l
Der pH-Wert wurde mit Eisessig (ca. 70 ml / l) auf 7.8 eingestellt, der Puffer für 30 min
bei 121°C autoklaviert und anschließend bei RT gelagert.
• DNA Ladepuffer 5 x:
100 mM Tris-Acetat pH 7.5
50 mM EDTA (Titriplex III)
15 % (w/v) Ficoll
0.5 % (w/v) SDS
0.05 % (w/v) Bromphenolblau
0.05 % (w/v) Xylencyanol
Der DNA Ladepuffer wurde für 30 min bei 121°C autoklaviert und anschließend bei RT
gelagert.

Material & Methoden
29
3.8 PCR Mit der von MULLIS et al. (1986) entwickelten Methode der Polymerase-Kettenreaktion
(polymerase chain reaction, PCR) werden selektiv bestimmte Abschnitte der DNA
exponentiell amplifiziert. Die eingesetzten Oligonukleotide [Primer] dienen in dieser
Reaktion als Startermoleküle. Der Abschnitt, den die Primer beidseitig flankieren, wird in
jedem Zyklus der Reaktion verdoppelt. Der Zyklus selber besteht aus drei Schritten:
1. Hitzedenaturierung der doppelsträngigen DNA bei 94°C, dabei wird die DNA in
Einzelstränge aufgespalten.
2. Anlagerung der Primer ("Annealing") an die DNA-Einzelstränge bei einer
primerspezifischen Temperatur.
3. Synthese des komplementären DNA-Doppelstranges (Extension) bei der optimalen
Arbeitstemperatur der DNA-Polymerase von 72 - 75°C.
Den Zyklen wird eine 3 minütige Denaturierungsphase vorangestellt, um sicherzustellen, daß
die gesamte eingesetzte DNA in einzelsträngiger Form vorliegt. Am Ende der PCR wird eine
5-10 minütige Extensionsphase angehängt, damit alle PCR Produkte vervollständigt werden
können.
Die Wahl der richtigen Annealingtemperatur ist dabei für das Ergebnis der PCR entscheidend.
Wird sie zu niedrig gewählt, so treten durch unspezifische Primerbindung häufig
Nebenprodukte auf; bei zu hohen Annealingtemperaturen hingegen ist es möglich, daß
überhaupt keine Bindung des Primers an die DNA-Matritze stattfindet, und damit kein PCR
Produkt entstehen kann. Die maximalen zu wählenden Annealingtemperaturen für Primer
berechnen sich nach folgender Formel:
Tm= [4°C x (C+G)+ 2°C x (A+T)]
Tm entspricht dabei der maximalen Annealingtemperatur, die sich aus der Summe der Anzahl
der einzelnen Basen im Primer, die dann mit einem spezifischen Faktor multipliziert werden,
zusammensetzt. Oberhalb dieser Temperaturen "schmelzen" die Primer, eine Bindung an das
Template (DNA oder RNA) ist nicht mehr gewährleistet. Die optimale Annealingtemperatur
muß für jede Zielsequenz empirisch ermittelt werden.
Auf der nächsten Seite sind zwei typische PCR-Protokolle zur Amplifizierung von
Paraxonemalstab-DNA und SSU rDNA aufgeführt.

Material & Methoden
30
PCR Bedingungen: Paraxonemalstab-DNA SSU rDNA Initiale Denaturierung 94°C 3 min 94°C 3 min 30 Zyklen: Denaturierung 94°C 1 min 94°C 1 min Annealing 46-53°C 1 min 48-52°C 1 min Extension 72°C 3 min 72°C 2 min Finale Denaturierung 72°C 10 min 72°C 10 min Schluß (endlos) 4°C 30 min 4°C 30 min
Die PCR wurde mit DNA Polymerasen verschiedener Hersteller durchgeführt, eine genaue
Zusammensetzung der PCR-Ansätze und die optimalen Annealingtemperaturen für die
einzelnen untersuchten Spezies befindet sich in Anhang 1.
3.8.1 Primerdesign Für das Erstellen von Primern wurden mit dem Programm ClustalX (THOMPSON et al. 1997)
Alignments mit bekannten Sequenzen von Paraxonemalstabgenen verschiedener
Trypanosomatiden und Euglena gracilis durchgeführt. Alle dort zu findenden
hochkonservierten Bereiche wurden anschließend nach den Vorgaben von RYCHLIK (1995 a,
b) auf geeignete Primeransatzstellen getestet. Mit Hilfe des Programms OLIGO V 4.0
(RYCHLIK & RHOADS 1989) wurden die neu konstruierten Primer auf ihre Qualität überprüft.
Die Primer PAR 9 (forward) und PAR 6 (reverse) umfassen ein 805 bp großes Fragment der
cDNA für Paraxialstabgene, das sowohl hochkonservierte als auch variable Bereiche enthält.
Als interne Kontrolle innerhalb einer "nested PCR" wurde der forward Primer PAR 5R
gewählt, dieser Primer bildet zusammen mit PAR 6 ein 323 bp großes Fragment, das
innerhalb des 805 bp Bereiches liegt.
Primersequenzen für die Gene des Paraxonemalstabes :
PAR 5R: 5'– GAR ATY GAC CGC AAC ATC –3'
PAR 9: 5'– ATC CAG AAG GCT GAT TTG GA –3'
R = A oder G S = C oder G Y = C oder T
PAR 6: 5'– CAA TCT TSA CCT CCT CYT –3'
Die Primer für die Gene der kleinen ribosomalen Untereinheit wurden freundlicherweise von
Ingo Busse (Universität Bielefeld) zur Verfügung gestellt. Mit den Primern AP 3 (forward)

Material & Methoden
31
(reverse) wurden zur Amplifikation des hinteren Teils der SSU rDNA verwendet. Die
Sequenzen, die mit diesen beiden Primerpaaren erhalten wurden, weisen einen überlappenden
Bereich von ca. 800 bp auf.
Primersequenzen für die SSU rDNA:
AP 3: 5'– TTT CAA GGA CTA AGC CAT GCA –3'
AP 5: 5'– CAA CTG GAG GGC AAG TCT GG –3'
AP 6: 5'– GTT GAG TCA AAT TAA GCC GCA –3'
AP 8: 5'– TCA CCT ACA GCW AAC TTG TTA CGA C–3' W = A oder T
Die Primer wurden von Ark Scientific und MWG Biotech bezogen.
3.9 RT-PCR Dient mRNA oder Gesamt-RNA als Ausgangsmaterial für die PCR, so muß vor der
eigentlichen PCR eine Reverse Transkription [RT] durchgeführt werden. Die Reaktion wird
mit der RNA-abhängigen DNA-Polymerase Reverse Transkriptase durchgeführt, wobei als
Produkt eine zur eingesetzten RNA-Matrize komplementäre Einzelstrang-DNA
[complementary DNA, cDNA] gebildet wird. Diese cDNA wird dann in der nachfolgenden
PCR zur Doppelstrang-DNA ergänzt.
Die RT-PCR wurde entweder mit dem RT-PCR Access System (Promega) [! 3.9.1] oder mit
dem Reverse Transcription System (Promega) [! 3.9.2] durchgeführt.
3.9.1 RT-PCR mit dem Access RT-PCR System Das Access RT-PCR System (Promega) erlaubt die gleichzeitige Durchführung der Reversen
Transkription und der nachfolgenden PCR in einem Ansatz (MILLER & STORTS 1995). In
diesem System werden bereits zu Beginn die spezifischen Primer zugegeben, so daß keine
Gesamtmatrize der RNA erstellt, sondern nur der Bereich aufwärts des näher am 3' Ende der
RNA liegenden Primers in cDNA umgeschrieben wird. Nach dem Denaturieren der RNA und
der Reversen Transkriptase erfolgt in einer PCR die Anlagerung des zweiten Primers an die
einzelsträngige cDNA, die dann in der PCR zum Doppelstrang ergänzt, und nachfolgend
amplifiziert wird.

Material & Methoden
32
einzelsträngige cDNA, die dann in der PCR zum Doppelstrangergänzt, und nachfolgend
amplifiziert wird.
Als Reverse Transkriptase wird in diesem System die AMV RT [Avian Myeloblastosis Virus]
eingesetzt; die Tfl DNA Polymerase stammt aus Thermus flavus.
Die einzusetzende Menge an RNA sollte in einem Bereich von 10 pg bis 1 µg liegen.
RT-PCR Bedingungen: Synthese des ersten cDNA Stranges 48°C 45 min Denaturieren der RNA und der Reversen Transkriptase 94°C 2 min Synthese des zweiten cDNA Stranges und Amplifizieren der DNA 40 Zyklen: • Denaturierung 94°C 1 min
• Annealing 46-53°C 1 min • Extension 72°C 3 min
Finale Denaturierung 72°C 10 min Abkühlen 4°C 30 min
Die RT-PCR wurde nach Angaben des Herstellers durchgeführt, wobei die Volumina für
einen Ansatz jeweils halbiert wurden:
Bestandteile Volumen 1x Endkonzentration Nucleasefreies Wasser X µl AMV/Tfl 5x Reaktionspuffer 5.0 µl 1 x dNTP-Mix (jedes dNTP 10mM) 0.5 µl 0.2 mM Primer (forward) 50 pmol 1.0 µM Primer (reverse) 50 pmol 1.0 µM 25 mM MgSO4 1.0 µl 1.0 mM AMV Reverse Transkriptase (5 U/µl) 0.5 µl 0.1 U/µl Tfl DNA Polymerase (5 U/µl) 0.5 µl 0.1 U/µl RNA Isolat Y µl Endvolumen 25 µl
3.9.2 RT-PCR mit dem Reverse Transcription System Das Reverse Transcription System (Promega, A3500) trennt im Gegensatz zum Access RT-
PCR System den Vorgang der Reversen Transkription und der PCR voneinander. Hier
werden zuerst Oligo-(dT)-Primer an die Poly-(A)-Überhänge der mRNA angeheftet, die
gesamte cDNA synthetisiert, und danach das RNA Template denaturiert. Erst in einer
anschließenden PCR erfolgt die Anlagerung der für den gesuchten Genabschnitt spezifischen
Primer an die einzelsträngige cDNA. Als Reverse Transkriptase wurde AMV RT [Avian
Myeloblastosis Virus] eingesetzt (GOODMAN & MACDONALD 1979).
Dieses System hat den Vorteil, daß mit der vorliegenden cDNA und entsprechenden Primern
verschiedene Gene untersucht werden können. Stehen für die Reverse Transkription

Material & Methoden
33
allerdings nur geringe Mengen an RNA zur Verfügung, und möchte man ein Gen mit geringer
Kopienzahl untersuchen, so besteht der Nachteil darin, daß die Ziel-cDNA für dieses Gen
durch die finale Verdünnung und Aliquotierung nicht in allen Aliquots vorliegt.
Lagen nur geringe Mengen an RNA vor, so wurde die Originalvorschrift derart modifiziert,
daß während der Reversen Transkription die Oligo-(dT)-Primer durch die spezifischen Primer
für das gesuchte Gen ersetzt wurden. modifiziertes Protokoll des Reverse Transcription Systems für einen Ansatz:
Nukleasefreies Wasser 7.75 µl Reverse Transkription 10 x Reaktionspuffer 2.0 µl MgCl2, 25 mM 4.0 µl dNTP-Mix (jedes 10 mM) 2.0 µl Recombinant RNasin®Ribonuclease Inhibitor 0.5 µl Spezifischer Primer forward (50 pM/µl) 1.0 µl Spezifischer Primer reverse (50 pM/µl) 1.0 µl AMV Reverse Transkriptase (20 U/µl) 0.75 µl 1.0 µg RNA 1.0 µl
Die Reverse Transkription wurde 20 min bei einer Temperatur von 48°C durchgeführt, das
RNA Template dann 5 min bei 99°C denaturiert und die cDNA für 5 min bei 4°C gelagert.
Nach der Reaktion wurden 80 µl nucleasefreies H2O in den Ansatz pipettiert, dieser
aliquotiert und die Aliquots bis zur Verwendung in einer PCR bei -20°C gelagert. Als Vorlage
für einen PCR Ansatz wurden 10 µl dieser verdünnten cDNA eingesetzt.
3.10 Extraktion von DNA Fragmenten aus Agarosegelen Extraktionen von DNA Fragmenten aus Agarosegelen wurden immer dann durchgeführt,
wenn in PCR Produkten mehrere ähnlich stark fluoreszierende Banden auftraten, die im
Bereich der erwarteten Größe des gesuchten Produktes lagen. Da in diesem Fall nicht
eindeutig war, in welcher Bande sich ein Teilstück des gesuchten Gens befand, mußte für
weitere Analysen als erstes eine Trennung der Banden durchgeführt werden. Dazu wurde das
gesamte PCR Produkt auf ein 1.2 % iges Agarosegel (! 3.7) aufgetragen und die Laufzeit zur
besseren Auftrennung der eng beieinander liegenden Banden auf 40 bis 50 min erhöht.
Anschließend wurde das Gel unter UV-Licht betrachtet (UVIS, Fa. Desaga), die einzelnen
Banden mit sterilen Skalpellen ausgeschnitten und in vorher abgewogene Eppendorf-Gefäße
überführt. Auf 1 mg Gelstück wurden 5-50 µl H2O pipettiert, diese Mischung zum Lösen der

Material & Methoden
34
Agarose für 5 min in einem Wasserbad bei 65°C erwärmt und gut durchmischt. Die
Identifizierung der Bande, in der sich ein Stück DNA des gesuchten Gens befand, erfolgte
über eine nachfolgend durchgeführte PCR. Dabei wurden zum einen wieder die zuvor
verwendeten Primer eingesetzt und in einem weiteren Ansatz interne Primer, die ein kleineres
Teilstück des Gens umfaßten. Für diese PCR wurden 1-2 µl des DNA-Agarose-H2O
Gemisches eingesetzt.
3.11 Reinigen von PCR Produkten Die PCR Produkte wurden mit dem Qiaquick PCR Purification Kit (Qiagen) oder mit dem
E.Z.N.A.® Cycle-Pure Kit (Peqlab) nach Herstellerangaben gereinigt. Das Prinzip dieser
Reinigung beruht auf einer Bindung der DNA an eine Silikagel-Membran; durch
Waschschritte werden alle anderen Komponenten des PCR Ansatzes entfernt und die
gereinigte DNA kann anschließend in einem definierten Volumen H2O aufgenommen
werden.
3.12 Klonierung Alle Klonierungen wurden mit dem TOPOTM TA Cloning Kit, Version H, (Invitrogen)
durchgeführt, wobei chemisch kompetente Escherichia coli Zellen des Stammes TOP10F'
eingesetzt wurden. Die Plasmidvektoren im Kit (pCR 2.1-TOPO ) liegen linearisiert vor und
besitzen Thymidin Überhänge an den 3' - Enden (= T-tail Vektoren), an die das Enzym Topo-
isomerase I gebunden ist. Die Ligation des PCR Produktes in das Plasmid erfolgt über die
Bindung der von der Taq-Polymerase an die 3' - Enden des amplifizierten PCR Produktes
angehängten Poly (A) - Enden mit den Thymidin-Anhängen des Vektors und wird durch die
Topoisomerase I vermittelt (SHUMAN 1994). Die vom Hersteller empfohlenen Mengen für
einen Ligationsansatz wurden auf die Hälfte reduziert. Wenn die Konzentration des zu
klonierenden PCR Produktes unter 5 ng / µl DNA lag, wurde die Menge des gereinigten PCR
Produktes maximal auf das doppelte Volumen des vom Hersteller empfohlenen Wertes
erhöht.
In der nachfolgenden Transformation werden die Plasmide mittels Hitzeschock in die
Bakterienzellen importiert und anschließend wird deren Stoffwechsel durch Inkubation in
einem Nährmedium aktiviert.

Material & Methoden
35
Diejenigen Bakterien, die Plasmide aufgenommen haben, besitzen jetzt eine durch
β-Lactamase vermittelte Ampicillinresistenz und können über dieses Antibiotikum selektiert
werden.
Nur die Plasmide enthalten das lacZ-Gen, das für die β-Galactosidase kodiert. Die Expression
dieses Gens wird durch das Lactoseanalogon Isopropyl β-D-Thiogalactopyranosid (IPTG)
induziert. Die β-Galactosidase spaltet nachfolgend 5-Bromo-4-Chloro-3-Indolyl β-D-
Galactopyranosid (X-Gal) in den blauen Farbstoff 5-Brom-4-Chlorindigo und in die
Zuckerkomponente Allolactose. Das Einbringen von Fremd-DNA in die Plasmide geschieht
an einer definierten Stelle im lacZ-Gen und führt dazu, daß keine β-Galactosidase mehr
gebildet wird. Dadurch kann keine Spaltung von X-Gal erfolgen, und die Klone sind nicht
angefärbt. Die Kolonien der Bakterien, die ein Plasmid ohne Fremd-DNA aufgenommen
haben, sind dagegen dunkelblau gefärbt.
Pro Klonierung wurden auf zwei LB Amp+ Agarplatten je 40 µl IPTG (20 mg / ml) und X-Gal
(20 mg / ml) ausplattiert, die Platten für 30 min bei 37°C gelagert und anschließend je
50 - 150 µl der Transformationsansätze ausplattiert. Die Platten wurden abschließend für
12-16 h bei 37 °C inkubiert.
3.12.1 Lösungen für die Klonierung • Luria Bertani Medium (LB) (verändert nach SAMBROOK et al. 1989):
1.0 % (w/v) Trypton (Oxoid) 1.0 g / 100 ml
0.5 % (w/v) Hefeextrakt (Oxoid) 0.5 g / 100 ml
0.5 % (w/v) NaCl (Merck) 0.5 g / 100 ml Für LB Agarplatten wird hinzugefügt:
1.5 % (w/v) Agar (AppliChem) 1.5 g / 100 ml
Das LB Medium wird bei 121°C für 20 min autoklaviert und vor der Zugabe von
Ampicillin (Endkonzentration 100 µg / ml) auf 50°C heruntergekühlt, da Ampicillin bei
höheren Temperaturen zerfällt. Zur Herstellung von LB Amp+ Agarplatten sollte aus
diesem Grund ein steriler Magnetrührfisch vorliegen und die Agarlösung unter Rühren
abgekühlt werden, da diese ansonsten bereits partiell auspolymerisiert.
Ampicillin Stammlösung (50 mg / ml H2O) 200 µl / 100 ml
! Lagerung bei �20°C

Material & Methoden
36
• Isopropyl β-D-Thiogalactopyranosid
IPTG (20 mg / ml H2O) ! Lagerung bei �20°C
• 5-Bromo-4-Chloro-3-Indolyl β-D-Galactopyranosid
X - Gal (20 mg / ml Dimethylformamid) ! Lagerung bei �20°C unter Lichtabschluß
3.12.2 Flüssigkulturen Um für weitere Untersuchungen eine ausreichende Menge an amplifizierter DNA zur
Verfügung zu haben, erfolgte der Ansatz von Flüssigkulturen. Dazu wurde in zuvor für 6 h
bei 150°C sterilisierte Reagenzgläser 2 ml LB Medium gegeben, mit autoklavierten
Zahnstochern einige Zellen der zu untersuchenden Klone von den Agarplatten abgenommen
und zusammen mit dem Zahnstocher in das Medium überführt. Die Kulturen wurden
anschließend für 12-16 h bei 37°C und 200 rpm in einem Schüttler (Infors) inkubiert und bis
zur weiteren Verwendung bei 4 °C gelagert.
Um in der nachfolgenden Charakterisierung der Plasmide [! 3.12.3] eindeutige Aussagen
über das Vorhandensein eines Inserts machen zu können, wurden als Negativkontrolle
zusätzlich dunkelblau angefärbte Klone ohne Insert in Flüssigkultur genommen.
3.12.3 Charakterisierung der Plasmide Die Charakterisierung der Plasmide erfolgt durch Untersuchung auf ihre Größe (HILLIS et al.
1996). Mit organischen Lösemitteln werden die Bakterienzellen aufgebrochen, die DNA
durch Zentrifugieren von den anderen Zellbestandteilen abgetrennt und auf einem 1%igen
Agarosegel aufgetrennt. Plasmide mit einkloniertem Insert können aufgrund ihrer Größe von
Plasmiden ohne Insert unterschieden werden. Optional kann ein Längenstandard mit auf dem
Gel aufgetragen werden, wobei zu beachten ist, daß die Vektoren in einer �supercoiled�-
Konformation vorliegen und damit eine größere Laufstrecke zurücklegen als der linear
vorliegende Marker. Mit einiger Erfahrung kann aber trotzdem auf die ungefähre Größe der
einklonierten Fremd-DNA geschlossen werden, und Klone, in denen ein Insert unerwünschter
Größe vorliegt, auf diese Weise ausselektiert werden. Die Existenz solcher Klone beruht
darauf, daß während der PCR in geringen Mengen Nebenprodukte entstehen, deren

Material & Methoden
37
Konzentration allerdings häufig so gering ist, daß eine Detektion dieser Banden unter UV-
Licht nicht möglich ist.
Zur Charakterisierung der Plasmide wurden je 25 µl Flüssigkultur mit 25 µl PCI-Lösung
zusammengegeben und kräftig durchmischt. Nach Abzentrifugieren für 5 min bei 7000 x g
bildeten sich zwei Phasen, eine untere organische Phase mit aufliegenden ausgefällten
Proteinen und eine obere wäßrige Phase, in der sich die Nucleinsäuren befanden. Jeweils 5 -
10 µl der wäßrigen Phase wurden mit 1 - 2 µl 5x DNA-Ladepuffer [! 3.7.1] vermischt und
die Proben auf einem 1%igen Agarosegel aufgetrennt.
• PCI: Phenol : Chloroform : Isoamylalkohol (25 : 24 : 1)
Die Mischung wird stark geschüttelt und kurzzeitig bei 4°C gelagert. Es bilden sich 2
Phasen, wobei sich die wäßrige Phase über der zu verwendenden PCI-Phase befindet.
Die Lösung kann bei 4°C maximal 4 Wochen gelagert werden.
3.12.4 "Colony"-PCR zur Charakterisierung der Inserts Diese Methode wird durchgeführt, um sicherzustellen, daß es sich bei den Inserts auch
wirklich um die gesuchten Gene handelt und nicht um mögliche einklonierte
Verunreinigungen, die zufällig eine identische Größe aufweisen. In einer Colony-PCR
(CPCR) wird nicht die gereinigte DNA, sondern einige Klonzellen aus den Kolonien oder die
aus den Klonen angesetzten Flüssigkulturen als Ausgangsmaterial für die PCR eingesetzt.
Mit autoklavierten Zahnstochern wurden aus dem Zentrum der Kolonien einige Zellen
abgenommen und am Boden der PCR Gefäße abgeschabt. Alternativ wurden 20 µl
Flüssigkultur (! 3.12.2) direkt in ein PCR-Gefäß pipettiert, bei 10000 x g für 1 min
abzentrifugiert und 1 x mit H2O nanopore gewaschen. Dieses Bakterienpellet diente als Vorlage
für die PCR.
Die Amplifizierung wurde mit den Primern durchgeführt, mit denen das PCR Produkt für die
Klonierung erhalten wurde, zusätzlich wurden als Kontrolle interne Primer verwendet.
Unterschiede im Vergleich zu einer PCR mit DNA als Ausgangsmaterial bestehen in einer
verlängerten initialen Denaturierungsphase, die den Aufbruch der Bakterienzellen bewirkt
und in einer Reduzierung der Extensionszeit und Zyklenzahl, da das Template in hohen
Mengen vorliegt. In der Colony-PCR wurden sowohl die Mengen der verwendeten
Chemikalien als auch das Ansatzvolumen reduziert (= 12 µl / Ansatz).
Material & Methoden
38
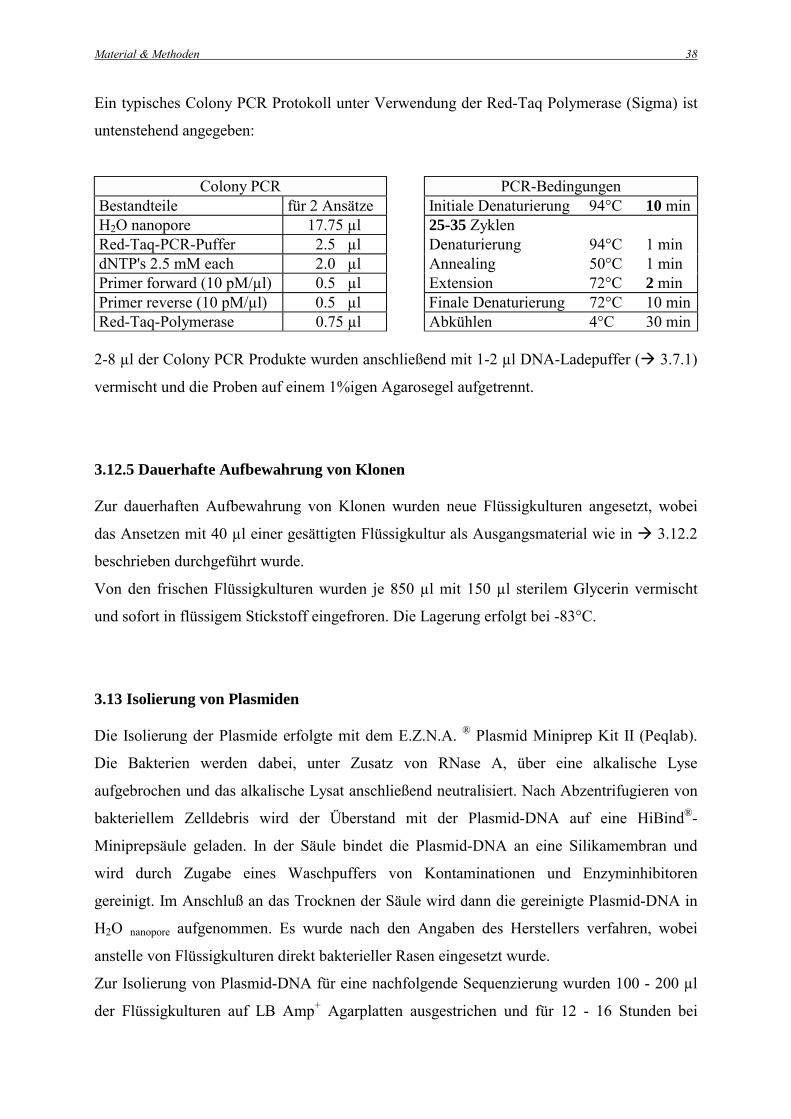
Ein typisches Colony PCR Protokoll unter Verwendung der Red-Taq Polymerase (Sigma) ist
untenstehend angegeben:
Colony PCR PCR-Bedingungen Bestandteile für 2 Ansätze Initiale Denaturierung 94°C 10 min H2O nanopore 17.75 µl 25-35 Zyklen Red-Taq-PCR-Puffer 2.5 µl Denaturierung 94°C 1 min dNTP's 2.5 mM each 2.0 µl Annealing 50°C 1 min Primer forward (10 pM/µl) 0.5 µl Extension 72°C 2 min Primer reverse (10 pM/µl) 0.5 µl Finale Denaturierung 72°C 10 min Red-Taq-Polymerase 0.75 µl Abkühlen 4°C 30 min
2-8 µl der Colony PCR Produkte wurden anschließend mit 1-2 µl DNA-Ladepuffer (! 3.7.1)
vermischt und die Proben auf einem 1%igen Agarosegel aufgetrennt.
3.12.5 Dauerhafte Aufbewahrung von Klonen Zur dauerhaften Aufbewahrung von Klonen wurden neue Flüssigkulturen angesetzt, wobei
das Ansetzen mit 40 µl einer gesättigten Flüssigkultur als Ausgangsmaterial wie in ! 3.12.2
beschrieben durchgeführt wurde.
Von den frischen Flüssigkulturen wurden je 850 µl mit 150 µl sterilem Glycerin vermischt
und sofort in flüssigem Stickstoff eingefroren. Die Lagerung erfolgt bei -83°C.
3.13 Isolierung von Plasmiden Die Isolierung der Plasmide erfolgte mit dem E.Z.N.A. ® Plasmid Miniprep Kit II (Peqlab).
Die Bakterien werden dabei, unter Zusatz von RNase A, über eine alkalische Lyse
aufgebrochen und das alkalische Lysat anschließend neutralisiert. Nach Abzentrifugieren von
bakteriellem Zelldebris wird der Überstand mit der Plasmid-DNA auf eine HiBind®-
Miniprepsäule geladen. In der Säule bindet die Plasmid-DNA an eine Silikamembran und
wird durch Zugabe eines Waschpuffers von Kontaminationen und Enzyminhibitoren
gereinigt. Im Anschluß an das Trocknen der Säule wird dann die gereinigte Plasmid-DNA in
H2O nanopore aufgenommen. Es wurde nach den Angaben des Herstellers verfahren, wobei
anstelle von Flüssigkulturen direkt bakterieller Rasen eingesetzt wurde.
Zur Isolierung von Plasmid-DNA für eine nachfolgende Sequenzierung wurden 100 - 200 µl
der Flüssigkulturen auf LB Amp+ Agarplatten ausgestrichen und für 12 - 16 Stunden bei

Material & Methoden
39
37°C inkubiert. Der gewachsene Bakterienrasen wurde mit einer sterilen Glaspipette
abgeschabt und laut Protokoll weiterverarbeitet.
3.14 Sequenzierung Die Sequenzierungen wurden mit einem Licor Sequenzierer von der Firma IIT Biotech /
Bioservice an der Universität Bielefeld durchgeführt, wobei eine enzymatische, nicht
radioaktive Didesoxy-Sequenzierung modifiziert nach SANGER et al. (1977) zur Anwendung
kam. Zuerst werden hierbei in einer Cycle-Reaktion Kopien der zu sequenzierenden DNA
hergestellt. Zusätzlich befinden sich dabei neben den dNTPs in geringen Mengen ddNTPs im
Reaktionsansatz. Deren Einbau in die DNA führt zum Abbruch der Reaktion, da dann in der
2-Desoxyribose keine 3' OH-Gruppe zur Bildung der nächsten Phosphodiesterbindung mehr
zur Verfügung steht. Die einzelnen ddNTPs sind mit unterschiedlichen Rhodaminen
fluoreszenzmarkiert, deren Strahlung dann während der Sequenzierung gemessen wird.
3.15 Restriktionsspaltungen Die Primer für die Gene, die für die beiden Hauptproteine des Paraxonemalstabes (PAR 1 und
PAR 2) kodieren, stammen aus hochkonservierten Bereichen, die innerhalb dieser beiden
Gene identisch sind. Eine Unterscheidung von par1 und par2 war somit nur mit Hilfe von
Restriktionsspaltungen möglich.
Nachdem von jedem untersuchten Organismus eine für ein PAR-Protein kodierende Sequenz
vorhanden war, wurden die Sequenzdaten aligniert (!3.17.4), um die Sequenz als für PAR 1
oder PAR 2 kodierend einordnen zu können. Anschließend wurden innerhalb des Alignments
konservierte Bereiche gesucht, an denen sich par1 und par2 unterscheiden lassen. Diese
Bereiche wurden dann auf potentielle Restriktionsenzymschnittstellen untersucht.
War von einer Art bereits ein par1 Genstück bekannt, so konnte das Bandenmuster, das sich
durch Spaltung mit einem bestimmten Restriktionsenzym für par1 ergäbe, berechnet werden.
Das mögliche Spaltungsmuster für das von dieser Art noch nicht vorhandenem par2 Gen
konnte anhand der bereits sequenzierten par2 Gene anderer Arten vorhergesagt werden.
Zur Unterscheidung von par1 und par2 wurden die Restriktionsenzyme Alw21I, BbsI, BclI,
BglII, EcoRI, HindIII, HinfI, NspI und PvuII herangezogen. Tabelle 3-2 zeigt eine Übersicht
der einzelnen Enzyme, ihrer spezifischen Schnittstellen sowie der verwendeten Puffer und
Bezugsquellen.
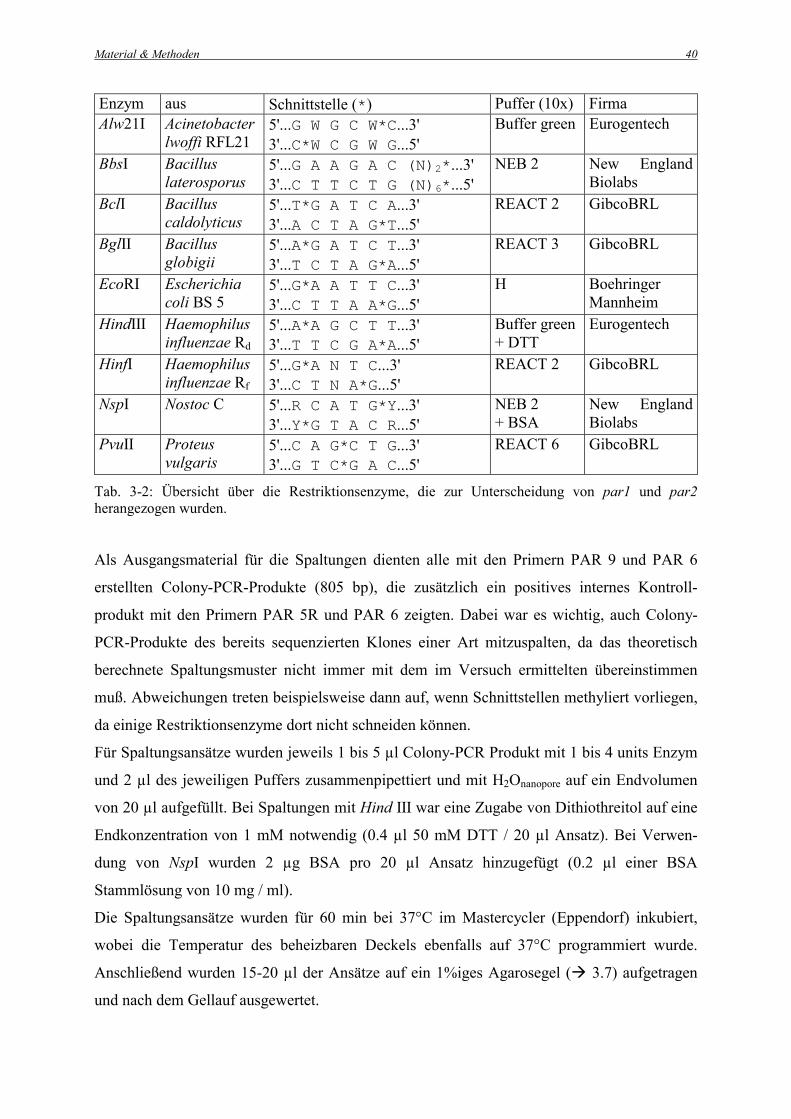
Material & Methoden
40
Enzym aus Schnittstelle (*) Puffer (10x) Firma Alw21I Acinetobacter
lwoffi RFL21 5'...G W G C W*C...3' 3'...C*W C G W G...5'
Buffer green Eurogentech
BbsI Bacillus laterosporus
5'...G A A G A C (N)2*...3' 3'...C T T C T G (N)6*...5'
NEB 2 New England Biolabs
BclI Bacillus caldolyticus
5'...T*G A T C A...3' 3'...A C T A G*T...5'
REACT 2 GibcoBRL
BglII Bacillus globigii
5'...A*G A T C T...3' 3'...T C T A G*A...5'
REACT 3 GibcoBRL
EcoRI Escherichia coli BS 5
5'...G*A A T T C...3' 3'...C T T A A*G...5'
H Boehringer Mannheim
HindIII Haemophilus influenzae Rd
5'...A*A G C T T...3' 3'...T T C G A*A...5'
Buffer green + DTT
Eurogentech
HinfI Haemophilus influenzae Rf
5'...G*A N T C...3' 3'...C T N A*G...5'
REACT 2 GibcoBRL
NspI Nostoc C 5'...R C A T G*Y...3' 3'...Y*G T A C R...5'
NEB 2 + BSA
New England Biolabs
PvuII Proteus vulgaris
5'...C A G*C T G...3' 3'...G T C*G A C...5'
REACT 6 GibcoBRL
Tab. 3-2: Übersicht über die Restriktionsenzyme, die zur Unterscheidung von par1 und par2 herangezogen wurden. Als Ausgangsmaterial für die Spaltungen dienten alle mit den Primern PAR 9 und PAR 6
erstellten Colony-PCR-Produkte (805 bp), die zusätzlich ein positives internes Kontroll-
produkt mit den Primern PAR 5R und PAR 6 zeigten. Dabei war es wichtig, auch Colony-
PCR-Produkte des bereits sequenzierten Klones einer Art mitzuspalten, da das theoretisch
berechnete Spaltungsmuster nicht immer mit dem im Versuch ermittelten übereinstimmen
muß. Abweichungen treten beispielsweise dann auf, wenn Schnittstellen methyliert vorliegen,
da einige Restriktionsenzyme dort nicht schneiden können.
Für Spaltungsansätze wurden jeweils 1 bis 5 µl Colony-PCR Produkt mit 1 bis 4 units Enzym
und 2 µl des jeweiligen Puffers zusammenpipettiert und mit H2Onanopore auf ein Endvolumen
von 20 µl aufgefüllt. Bei Spaltungen mit Hind III war eine Zugabe von Dithiothreitol auf eine
Endkonzentration von 1 mM notwendig (0.4 µl 50 mM DTT / 20 µl Ansatz). Bei Verwen-
dung von NspI wurden 2 µg BSA pro 20 µl Ansatz hinzugefügt (0.2 µl einer BSA
Stammlösung von 10 mg / ml).
Die Spaltungsansätze wurden für 60 min bei 37°C im Mastercycler (Eppendorf) inkubiert,
wobei die Temperatur des beheizbaren Deckels ebenfalls auf 37°C programmiert wurde.
Anschließend wurden 15-20 µl der Ansätze auf ein 1%iges Agarosegel (! 3.7) aufgetragen
und nach dem Gellauf ausgewertet.

Material & Methoden
41
3.16 Phylogenetische Rekonstruktion anhand morphologischer Merkmale Um eine kladistische Analyse anhand morphologischer Merkmale durchführen zu können,
müssen Merkmale gefunden werden, die durch eindeutige Merkmalszustände beschrieben
werden können.
3.16.1 Kladistische Analyse Für eine kladistische Analyse zur phylogenetischen Rekonstruktion werden diese
charakteristischen morphologischen Merkmale in eine Matrix transformiert und mit dem
Programm PAUP*4.0b4a (SWOFFORD 1998) ausgewertet. Dabei werden Apomorphien und
Plesiomorphien nicht definiert, eine Richtung ist somit nicht vorgegeben. Tabelle 3-3 zeigt
die berücksichtigten Merkmale und ihre jeweiligen Zustände:
Merkmale Merkmalszustand a Anzahl Geißeln 1: eine, 2: zwei b Ausprägung Geißeln 1: eine emergent + 1 PAR, 2: zwei emergent + 2 PARs c Stigma 1: vorhanden, 2: nicht vorhanden d Chloroplasten 1: vorhanden, 2: nicht vorhanden e Paramylon 1: vorhanden, 2: nicht vorhanden f Kinetoplast 1: vorhanden, 2: nicht vorhanden g Scroll 1: vorhanden, 2: nicht vorhanden h Pellikula 1: flexibel, 2: rigide, 3: flexibel + Organismen nicht metabol i Ernährungsmodus 1: osmotroph ohne Lichtperzeptionsorgan, 2: osmotroph mit
Lichtperzeptionsorgan, 3: phagotroph, 4: phototroph, 5: parasitär k SSU rDNA-Größe 1: unter 2700 nt, 2: über 2700 nt
Tab. 3-3: Zusammenstellung der Merkmale und ihrer Merkmalszustände, die in eine kladistische Analyse einfließen.
3.17 Phylogenetische Analyse und Rekonstruktion anhand molekularer Merkmale Zur phylogenetischen Rekonstruktion anhand molekularer Merkmale wurden SSU rDNA
Sequenzen und Paraxonemalstabsequenzen herangezogen. Wie in einer morphologischen
Matrix werden hier Nukleotide (Nukleotidpositionen) und ihre Zustände (ATCG─)
homologisiert.

Material & Methoden
42
3.17.1 Wahl der Außengruppe für die phylogenetische Rekonstruktion Phylogenetische Rekonstruktionen anhand molekularer Merkmale führen zu ungewurzelten
Stammbäumen, in denen über die evolutionäre Richtung der Merkmale keine Aussage
gemacht werden kann. Erst durch das sogenannte "Wurzeln" mit einer Außengruppe wird die
Richtung festgelegt. Die gewählte Außengruppe sollte daher von der zu untersuchenden
Innengruppe gut abgrenzbar sein, andererseits aber auch gemeinsame Merkmale mit dieser
aufweisen und somit in nicht zu entfernter Verwandtschaft mit ihr stehen.
Für die phylogenetische Rekonstruktion anhand der kleinen Untereinheit der ribosomalen
RNA wurden als Außengruppenvertreter neben den ebenso wie die Euglenida an der Basis
der Eukaryoten stehenden Physarum polycephalum (Physarida) und Dictyostelium discoideum
(Dictyosteliida) zusätzlich noch Arxiozyma telluris (Fungi), Chilomonas paramecium
(Cryptophyta), Marchantia polymorpha (Viridiplantae), Paramecium tetraurelia (Alveolata),
Proteromonas lacertae (Stramenopila) und Saccharomyces cerevisiae (Fungi) gewählt. Diese
Vertreter haben sich in phylogenetischen Rekonstruktionen als gut geeignet erwiesen (BUSSE
1999, PREISFELD et al. 2000b).
Die Wahl der Außengruppe bei den Analysen der beiden Paraxonemalstabgene erweist sich
als weitaus schwieriger. Paraxonemalstabsequenzen sind nur innerhalb der Euglenozoa
verfügbar. Die Kinetoplastida als Schwestergruppe der Euglenida sind nicht als Außengruppe
geeignet.
3.17.2 Formatierung von Sequenzdaten Die erhaltenen Sequenzdaten wurden im Internet mit Hilfe des BCM Search Launchers
(http://www.hgsc.bcm.tmc.edu/SearchLauncher/, SMITH et al. 1996) unter der Option
"sequence utilities" (http://dot.imgen.bcm.tmc.edu:9331/seq-util/seq-util.html) in ein für
weitere Programme kompatibles "fasta" Format überführt. Hier wurden die Sequenzen
gegebenenfalls auch revers und komplementär umgeschrieben, ebenso erfolgte unter der
Option "6 frame translation" die Transformation der Paraxonemalstabsequenzen in die sechs
möglichen Leseraster.

Material & Methoden
43
3.17.3 Überprüfen von Sequenzen Eine erste Überprüfung der Sequenzdaten erfolgte mit dem Internet Programm BLAST
(Basic local alignment search tool; http://www.ncbi.nlm.nih.gov/BLAST/, ALTSCHUL et al.
1990) des National Center for Biotechnology Information (NCBI), Bethesa, MD, USA. Hier
werden die vom Benutzer eingegebenen Nucleinsäure- oder Proteinsequenzen mit bereits
vorhandenen Sequenzen der Datenbank GenBank des National Center for Biotechnology
Information (NCBI) verglichen, wobei als Resultat Sequenzen mit der höchsten
Übereinstimmung angezeigt werden.
Vor der Eingabe von Daten wurden störende Vektorreste sowie die spezifischen Primer
entfernt. Das Entfernen dieser Oligonukleotide war dabei notwendig, da das Programm
BLAST sonst bereits allein aufgrund der Primersequenzen eine große Ähnlichkeit zum
gesuchten Gen feststellen kann, auch wenn die eigentliche Sequenz nicht der des gesuchten
Gens entspricht.
3.17.4 Alignierung Die Homologisierung der Sequenzen (Alignierung) bildet die Basis der nachfolgenden
phylogenetischen Rekonstruktion. Da falsche Homologisierungen damit falsche
phylogenetische Rekonstruktionen zur Folge haben können, sollten die Alignments
überprüfbar sein. Im Falle der Gene, die für die Paraxonemalstabproteine kodieren, dürfen in
ein korrektes Nukleotid-Alignment keine einzelnen Lücken ("gaps") eingefügt werden, da es
sonst zu Leserasterverschiebungen kommen würde. Allenfalls dürfte in einer
Nukleotidsequenz eine durch drei teilbare Anzahl von gaps direkt hintereinander stehen, was
bedeuten würde, daß im entsprechenden Organismus an dieser Stelle im Protein mindestens
eine Aminosäure fehlte.
Die Sequenzen der kleinen Untereinheit der ribosomalen rRNA sind aufgrund ihrer
Sekundärstruktur sehr gut überprüfbar. Die Sekundärstruktur setzt sich aus variablen, oftmals
aus "loops" bestehenden Bereichen und aus hochkonservierten Helices zusammen. Da die
Sekundärstruktur der SSU rDNA von Euglena gracilis bekannt ist, kann über diesen
Vergleich das Alignment überprüft werden.

Material & Methoden
44
Für die Alignierung und eine nachfolgende phylogenetische Rekonstruktion wurden aus dem
Internet Programm "Entrez" (BENSON et al. 1999) DNA- und Protein- Vergleichssequenzen
heruntergeladen. Die Adressen der Seiten lauten:
http://www.ncbinlm.nih.gov/Entrez/nucleotide.html
http://www.ncbinlm.nih.gov/Entrez/protein.html Die einzelnen Sequenzen der SSU rDNA und der Paraxonemalstabgene sind in den
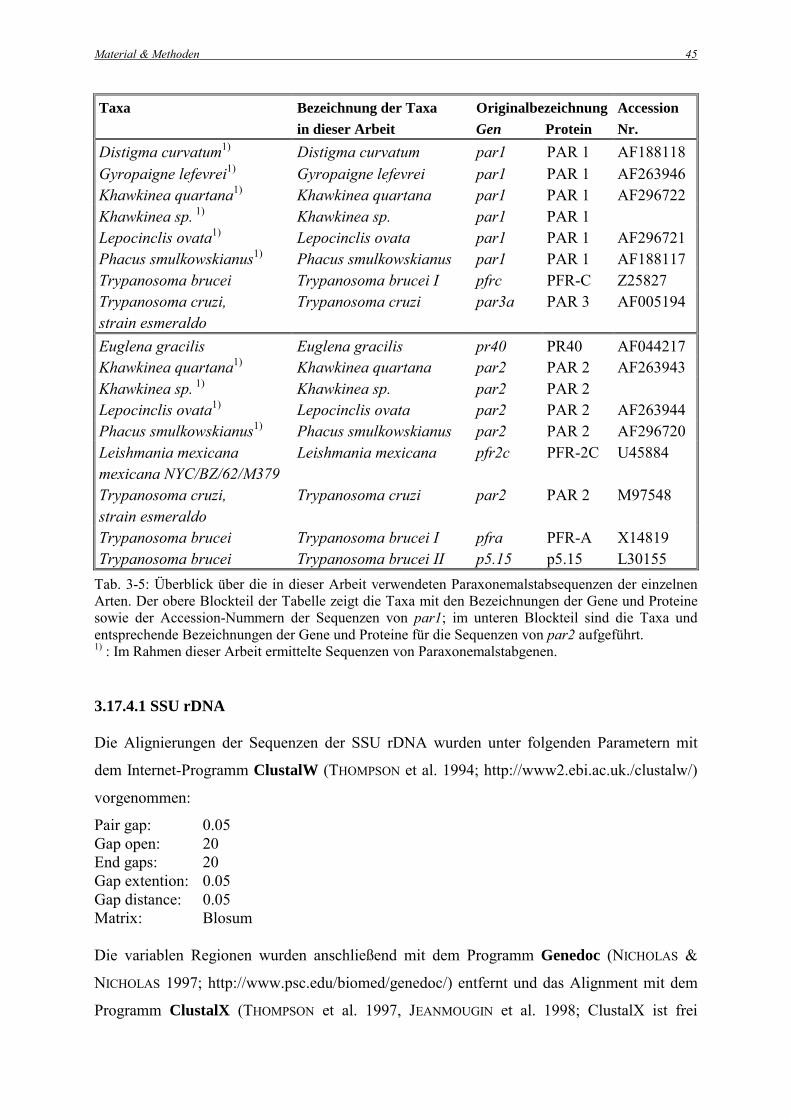
Tabellen 3-4 und 3-5 aufgeführt.
SSU rDNA Accession Nr. Astasia longa
(Euglenida)
AF112871
Distigma curvatum (Euglenida) AF099081 Distigma proteus (Euglenida) AF106036 Euglena gracilis (Euglenida) M12677 Eutreptia viridis (Euglenida) AF157312 Gyropaigne lefèvrei (Euglenida) AF110418 Khawkinea quartana (Euglenida) U84732 Lepocinclis ovata (Euglenida) AF061338 Lepocinclis ovum (Euglenida) AF110419 Peranema trichophorum (Euglenida) U84733 U84734 Petalomonas cantuscygni (Euglenida) U84731 Phacus smulkowskianus (Euglenida) AF119118 Rhabdomonas gibba1) (Euglenida) AF247602 Rhabdomonas incurva1) (Euglenida) AF247601 Rhabdomonas intermedia (Euglenida) AF295020 Rhabdomonas spiralis (Euglenida) AF247599
Bodo caudatus (Kinetoplastida) X53910 Crithidia fasciculata (Kinetoplastida) Y00055 Cryptobia bullocki (Kinetoplastida) AF080224 Leishmania tarentolae (Kinetoplastida) X53916 Trypanosoma brucei (Kinetoplastida) M12676
Arxiozyma telluris (Fungi) Y15849 Chilomonas paramecium (Cryptophyta) L28811 Dictyostelium discoideum (Dictyosteliida) K02641 Marchantia polymorpha (Viridiplantae) X75521 Paramecium tetraurelia (Alveolata) X03772 Physarum polycephalum (Myxomycetes) X13160 Proteromonas lacertae (Stramenopila) U37108 Saccharomyces cerevisiae (Fungi) J01353 M27607
Tab. 3-4: Übersicht der in dieser Arbeit verwendeten Taxa für die phylogenetische Rekonstruktion anhand von Sequenzen der SSU rDNA. 1) : Im Rahmen dieser Arbeit neu ermittelte Sequenzen der SSU rDNA.
Material & Methoden
45
Taxa Bezeichnung der Taxa in dieser Arbeit
Originalbezeichnung Gen Protein
Accession Nr.
Distigma curvatum1) Distigma curvatum par1 PAR 1 AF188118 Gyropaigne lefevrei1) Gyropaigne lefevrei par1 PAR 1 AF263946 Khawkinea quartana1) Khawkinea quartana par1 PAR 1 AF296722 Khawkinea sp. 1) Khawkinea sp. par1 PAR 1 Lepocinclis ovata1) Lepocinclis ovata par1 PAR 1 AF296721 Phacus smulkowskianus1) Phacus smulkowskianus par1 PAR 1 AF188117 Trypanosoma brucei Trypanosoma brucei I pfrc PFR-C Z25827 Trypanosoma cruzi, strain esmeraldo
Trypanosoma cruzi par3a PAR 3 AF005194
Euglena gracilis Euglena gracilis pr40 PR40 AF044217 Khawkinea quartana1) Khawkinea quartana par2 PAR 2 AF263943 Khawkinea sp. 1) Khawkinea sp. par2 PAR 2 Lepocinclis ovata1) Lepocinclis ovata par2 PAR 2 AF263944 Phacus smulkowskianus1) Phacus smulkowskianus par2 PAR 2 AF296720 Leishmania mexicana mexicana NYC/BZ/62/M379
Leishmania mexicana pfr2c PFR-2C U45884
Trypanosoma cruzi, strain esmeraldo
Trypanosoma cruzi par2 PAR 2 M97548
Trypanosoma brucei Trypanosoma brucei I pfra PFR-A X14819 Trypanosoma brucei Trypanosoma brucei II p5.15 p5.15 L30155
Tab. 3-5: Überblick über die in dieser Arbeit verwendeten Paraxonemalstabsequenzen der einzelnen Arten. Der obere Blockteil der Tabelle zeigt die Taxa mit den Bezeichnungen der Gene und Proteine sowie der Accession-Nummern der Sequenzen von par1; im unteren Blockteil sind die Taxa und entsprechende Bezeichnungen der Gene und Proteine für die Sequenzen von par2 aufgeführt. 1) : Im Rahmen dieser Arbeit ermittelte Sequenzen von Paraxonemalstabgenen. 3.17.4.1 SSU rDNA Die Alignierungen der Sequenzen der SSU rDNA wurden unter folgenden Parametern mit
dem Internet-Programm ClustalW (THOMPSON et al. 1994; http://www2.ebi.ac.uk./clustalw/)
vorgenommen:
Pair gap: 0.05 Gap open: 20 End gaps: 20 Gap extention: 0.05 Gap distance: 0.05 Matrix: Blosum Die variablen Regionen wurden anschließend mit dem Programm Genedoc (NICHOLAS &
NICHOLAS 1997; http://www.psc.edu/biomed/genedoc/) entfernt und das Alignment mit dem
Programm ClustalX (THOMPSON et al. 1997, JEANMOUGIN et al. 1998; ClustalX ist frei

Material & Methoden
46
erhältlich unter: ftp://ftp-igbmc.u-strasbg.fr/pub/ClustalX/) unter folgenden Bedingungen
realigniert:
Pairwise Alignment Parameter Multiple Alignment Parameter Gap opening: 10.0 Gap opening: 10.0 Gap extension: 0.5 Gap extension: 0.5 DNA weight matrix: IUB DNA weight matrix: IUB DNA transition weight: 0.5 Eine Nachkorrektur erfolgte nochmals mit dem Programm Genedoc (NICHOLAS & NICHOLAS
1997). Das korrigierte Alignment der SSU rDNA Sequenzen ist in Anhang 2 aufgeführt.
3.17.4.2 Paraxonemalstab-Gene Die erhaltenen Sequenzdateien wurden manuell anhand der Ausdrucke aus den
Sequenzierungen überprüft, gegebenenfalls nachkorrigiert und anschließend mit dem
Progamm ClustalX (THOMPSON et al. 1997, JEANMOUGIN et al. 1998) mit bereits bekannten
Sequenzen von Paraxonemalstabgenen aligniert. Dabei werden die einzelnen Sequenzen nach
Bereichen der größtmöglichen Übereinstimmung untereinander angeordnet (homologisiert).
Eine Nachkorrektur des Alignments erfolgte auch hier mit dem Programm Genedoc
(NICHOLAS & NICHOLAS 1997), in dem Fehler des mit ClustalX erstellten Alignments
behoben werden konnten. Solche Fehler, die zu falschen Homologisierungen führen,
entstehen, wenn zu unterschiedliche oder zu wenige Sequenzen miteinander verglichen
werden. In Folge kann kein generelles Profil für die zu alignierenden Gene aufgebaut werden.
Ebenso ist es mit ClustalX nicht möglich, kurze Sequenzstücke, die aus verschiedenen
Teilbereichen eines Gens stammen, zusammen mit dem gesamten Gen korrekt zu alignieren.
Da mit dem Alignment-Algorithmus nach Bereichen größtmöglicher Übereinstimmung
gesucht wird, werden diese kurzen Stücke selbst dann völlig auseinandergerissen, wenn die
Parameter "gap opening" und "gap extension", mit denen das Strafmaß für ein Einfügen von
Lücken (gaps) im Alignment bestimmt werden kann, hoch eingestellt werden. Die
Alignierungen der PAR Sequenzen wurden mit den folgenden Parametern durchgeführt:
Pairwise Alignment Parameter Multiple Alignment Parameter Gap opening: 15.0 Gap opening: 15.0 Gap extension: 6.66 Gap extension: 6.66 DNA weight matrix: IUB DNA weight matrix: IUB DNA transition weight: 0.5
Das korrigierte Alignment der par-Sequenzen ist in Anhang 3 aufgeführt.

Material & Methoden
47
3.17.5 Rekonstruktion von Stammbäumen Die phylogenetischen Analysen wurden mit den Programmen PAUP*4.0b4a (Phylogenetic
analysis using parsimony *and other methods; SWOFFORD 1998) und TreeCon 1.3b (VAN DE
PEER & DE WACHTER 1997) durchgeführt. Dabei wurden Maximum-Parsimony-, Maximum-
Likelihood- und Distanz-Verfahren eingesetzt. Die erhaltenen Cladogramme wurden
anschließend über das Programm TreeView (PAGE 1996; http://taxonomy.zoology.gla.ac.uk/
rod/treeview.html.) importiert.
3.17.5.1 Maximum-Parsimony Dem Maximum-Parsimony-Verfahren liegt die Annahme zugrunde, daß bei der Deutung
empirischer Befunde nur die einfachsten, sparsamsten Erklärungen wissenschaftlichen
Charakter besitzen (AX 1995). Mit diesem Verfahren wird die aus einem Datensatz zur Er-
stellung eines Stammbaumes erforderliche Anzahl an evolutionären Schritten minimiert. Der
optimale Stammbaum ist damit zugleich auch der �kürzeste� Baum. Mittels einer
heuristischen Suche wird ein möglicher kürzester Baum gefunden, und im Anschluß daran,
durch Austausch von Ästen ("branch swapping") versucht, eine noch sparsamere Topologie
zu erhalten.
Manchmal werden als Resultat mehrere kürzeste Bäume gefunden, was bedeutet, daß die
Anzahl an Schritten identisch ist, die Topologien sich aber unterscheiden. Diese Bäume
können dann gemeinsam in Form eines Consensus-Baumes dargestellt werden. Die in dieser
Arbeit gezeigten Consensus-Bäume wurden unter dem "strict consensus"-Kriterium erstellt,
dabei werden ausschließlich die Gruppierungen angezeigt, die in allen gefundenen kürzesten
Bäumen enthalten sind.
Maximum-Parsimony macht im Gegensatz zu Maximum-Likelihood- und Distanz-Verfahren
keine Annahmen über Evolutionsprozesse, die Daten werden als ungeordnet betrachtet und
Umwandlungen von Nukleotiden sind in jede Richtung erlaubt (FITCH 1971). Die folgenden
Parameter lagen der heuristische Suche nach dem optimalen Baum zugrunde [SSU rDNA und
PAR]: • Gaps are treated as missing • Multistate taxa interpreted as uncertainty • Starting trees obtained via stepwise addition • Addition sequence random, nreps = 100 • Branch-swapping algorithm: Tree-bisection-reconnection (TBR) • Steepest descent option not in effect • �Multrees� option in effect
Material & Methoden
48
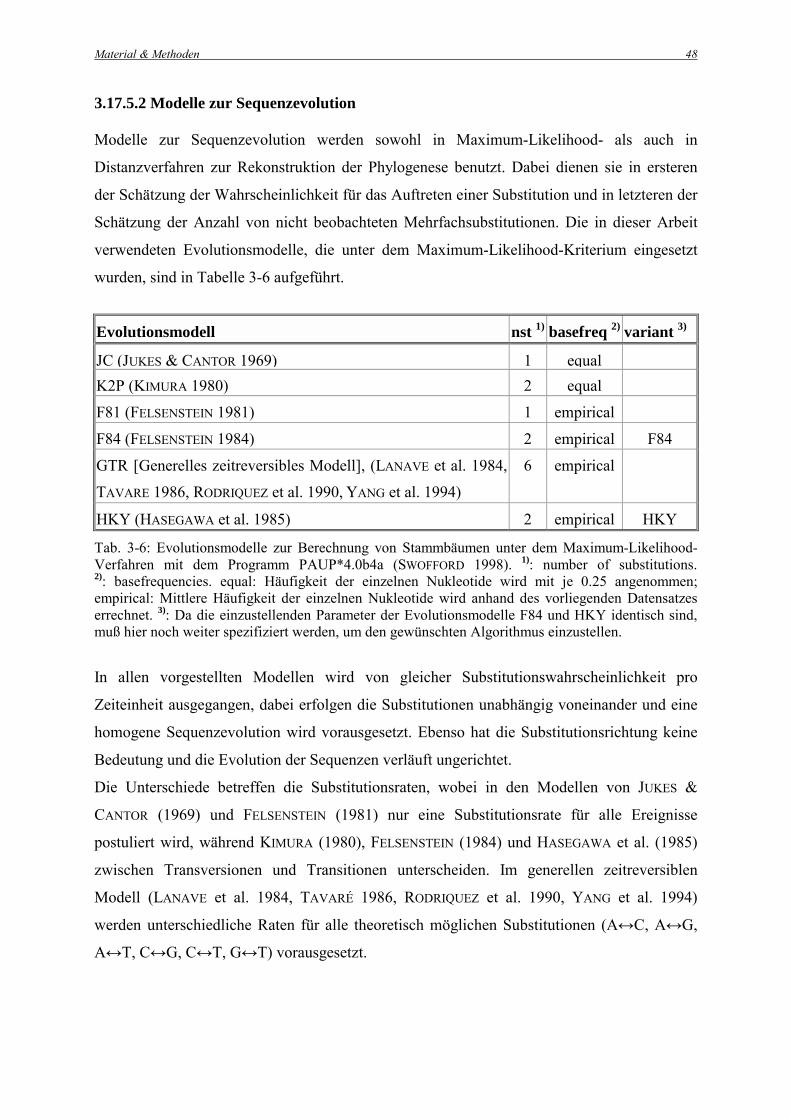
3.17.5.2 Modelle zur Sequenzevolution Modelle zur Sequenzevolution werden sowohl in Maximum-Likelihood- als auch in
Distanzverfahren zur Rekonstruktion der Phylogenese benutzt. Dabei dienen sie in ersteren
der Schätzung der Wahrscheinlichkeit für das Auftreten einer Substitution und in letzteren der
Schätzung der Anzahl von nicht beobachteten Mehrfachsubstitutionen. Die in dieser Arbeit
verwendeten Evolutionsmodelle, die unter dem Maximum-Likelihood-Kriterium eingesetzt
wurden, sind in Tabelle 3-6 aufgeführt.
Evolutionsmodell nst 1) basefreq 2) variant 3)
JC (JUKES & CANTOR 1969) 1 equal K2P (KIMURA 1980) 2 equal
F81 (FELSENSTEIN 1981) 1 empirical
F84 (FELSENSTEIN 1984) 2 empirical F84
GTR [Generelles zeitreversibles Modell], (LANAVE et al. 1984,
TAVARE 1986, RODRIQUEZ et al. 1990, YANG et al. 1994)
6 empirical
HKY (HASEGAWA et al. 1985) 2 empirical HKY Tab. 3-6: Evolutionsmodelle zur Berechnung von Stammbäumen unter dem Maximum-Likelihood-Verfahren mit dem Programm PAUP*4.0b4a (SWOFFORD 1998). 1): number of substitutions. 2): basefrequencies. equal: Häufigkeit der einzelnen Nukleotide wird mit je 0.25 angenommen; empirical: Mittlere Häufigkeit der einzelnen Nukleotide wird anhand des vorliegenden Datensatzes errechnet. 3): Da die einzustellenden Parameter der Evolutionsmodelle F84 und HKY identisch sind, muß hier noch weiter spezifiziert werden, um den gewünschten Algorithmus einzustellen.
In allen vorgestellten Modellen wird von gleicher Substitutionswahrscheinlichkeit pro
Zeiteinheit ausgegangen, dabei erfolgen die Substitutionen unabhängig voneinander und eine
homogene Sequenzevolution wird vorausgesetzt. Ebenso hat die Substitutionsrichtung keine
Bedeutung und die Evolution der Sequenzen verläuft ungerichtet.
Die Unterschiede betreffen die Substitutionsraten, wobei in den Modellen von JUKES &
CANTOR (1969) und FELSENSTEIN (1981) nur eine Substitutionsrate für alle Ereignisse
postuliert wird, während KIMURA (1980), FELSENSTEIN (1984) und HASEGAWA et al. (1985)
zwischen Transversionen und Transitionen unterscheiden. Im generellen zeitreversiblen
Modell (LANAVE et al. 1984, TAVARÉ 1986, RODRIQUEZ et al. 1990, YANG et al. 1994)
werden unterschiedliche Raten für alle theoretisch möglichen Substitutionen (A↔C, A↔G,
A↔T, C↔G, C↔T, G↔T) vorausgesetzt.

Material & Methoden
49
3.17.5.3 Maximum-Likelihood Den Maximum-Likelihood-Verfahren liegen konkrete Evolutionsmodelle zugrunde, die
festlegen, in welchem Rahmen Veränderungen der Sequenzen ablaufen können. Mit dieser
Methode wird die Wahrscheinlichkeit bestimmt, mit der das jeweilige Modell die
beobachteten Daten erzeugt hat, es wird also abgeschätzt, mit welcher Wahrscheinlichkeit
Substitutionen auftraten. Mit Hilfe des jeweiligen Evolutionsmodelles (Tab. 3-6) werden
dabei alle möglichen Baumtopologien erzeugt und der Baum mit dem größten Maximum-
Likelihood (ML)-Wert angezeigt.
3.17.5.4 Distanzverfahren In Distanzverfahren werden jeweils zwei alignierte Sequenzen miteinander verglichen und die
Unterschiede (Substitutionen) gezählt. Die Zahl der beobachteten Unterschiede, dividiert
durch die Gesamtzahl an alignierten Positionen, wird dabei als die beobachtete Distanz (auch:
sichtbare Distanz, p-Distanz) definiert. Für alle möglichen Sequenzpaare werden die
beobachteten Distanzen berechnet, die bei einer Anzahl von y untersuchten Sequenzen gleich
½[y (y─1)] sind. Diese Distanzwerte werden dann in eine Matrix überführt und anhand eines
gewählten Evolutionsmodelles in evolutionäre Distanzen transformiert. In die Rekonstruktion
gehen damit ausschließlich Distanzen ein, die Merkmalszustände werden nicht berücksichtigt.
Mit der Neighbor-Joining Methode, die von SAITOU & NEI (1987) vorgestellt wurde, werden
Bäume gesucht, in denen die Summe aller Ecklängen (= Astlängen) minimiert wird. Das
Prinzip beruht also auf dem Finden von optimalen "Sequenznachbarn", um die Gesamtlänge
des Baumes zu minimieren. Als zwei optimale Sequenznachbarn gelten damit solche, deren
Unterschiede untereinander geringer sind, als zu einer dritten Sequenz. Die Suche danach
beginnt mit einem sternförmigen Baum ohne hierarchische Struktur. Sind von den y
Sequenzen alle ½[y (y─1)] Paare gebildet worden, wird das Paar mit der kleinsten Summe an
Astlängen zueinander festgesetzt und für dieses Paar eine neue mittlere Distanz errechnet.
Aus diesem Wert und den Distanzen der übrigen (y─2) Paare wird eine neue Matrix erstellt
und wiederum das Paar mit der nun kleinsten Summe an Astlängen gesucht, nach diesem
Prinzip wird solange vorgegangen, bis alle Sequenzpaare gefunden sind. Diese Methode setzt
keine ultrametrischen Distanzen voraus, die Taxa dürfen unterschiedliche Evolutionsraten
aufweisen.

Material & Methoden
50
3.17.6 Basenzusammensetzung Extrem unterschiedliche Nukleotidzusammensetzungen führen in phylogenetischen
Rekonstruktionen oftmals zu Stammbäumen, in denen die Taxa eher durch eine ähnliche
Basenzusammensetzung zusammen angeordnet sind, als aufgrund genealogischer
Verwandschaft (OLSEN 1988, HASEGAWA & HASHIMOTO 1993, STEEL et al. 1993, SIMON et
al. 1994). Daher wurde für jede Art die Basenzusammensetzung ermittelt und mit einem χ2-
Test mit dem Programm PAUP*4.0b4a (SWOFFORD 1998) auf signifikante Heterogenität
überprüft.
3.17.7 Substitutionssättigung Bei Mehrfachsubstitutionen an der gleichen Sequenzposition ("multiple hit") erscheinen die
beobachteten Distanzen kleiner als die wirklichen evolutionären Distanzen.
Daher werden zur genauen Ermittlung der Substitutionssättigung Distanzen zwischen zwei
Sequenzen gegen die patristischen Distanzen aufgetragen. Beobachtete Distanzen sind dabei
die Unterschiede (Transversionen und Transitionen) zwischen zwei Sequenzen. Patristische
Distanzen entsprechen der Summe der Astlängen zwischen zwei Taxa in einem Maximum-
Parsimony-Stammbaum. Beim Auftragen der patristischen Distanzen gegen die beobachteten
Distanzen zeigt die Abweichung von der Winkelhalbierenden den Grad der Sättigung an.
Liegen die Werte unterhalb der Winkelhalbierenden, so ist die beobachtete Distanz kleiner als
die wirkliche evolutionäre Distanz, da im Maximum-Parsimony-Baum zur Erklärung der
Daten eine höhere Zahl an Evolutionsschritten angenommen werden muß, als tatsächlich
beobachtet wird.
Die Analysen zur Substitutionssättigung wurden mit dem Programm PAUP*4.0b4a
(SWOFFORD 1998) durchgeführt.
3.17.8 Sequenzdivergenz Große Sequenzdivergenzen, die durch hohe Substitutionsraten einzelner Taxa in der zu
untersuchenden Innengruppe entstehen, führen oftmals zu einem in der angelsächsischen
Literatur als "long branch attraction" bezeichnetem Phänomen (FELSENSTEIN 1978, 1988,
HENDY & PENNY 1989, NEI 1991, SWOFFORD et al. 1996, ZHARKIKH & LI 1993). In
Konsequenz werden solche schnell evolvierenden Taxa durch lange Äste im Stammbaum

Material & Methoden
51
separiert oder an die Basis gestellt, wobei sie im Extremfall der Außengruppe zugeordnet
werden. Die Sequenzdivergenzen wurden über einen paarweisen Vergleich der Taxa mit
folgender Formel errechnet: Sequenzdivergenz = [(Transitionen+Transversionen) x 100] x [Anzahl alignierter Positionen]−1 Die Berechnung der Sequenzdivergenz wurde mit dem Programm PAUP*4.0b4a (SWOFFORD
1998) durchgeführt.
3.17.9 Bootstrap Test Das Bootstrap Verfahren, das von NEYMAN (1971) zum Testen phylogenetischer
Stammbäume vorgeschlagen wurde und von FELSENSTEIN (1985) umgesetzt wurde, gibt
Auskunft über die Wiederfindung der errechneten Stammbäume. Aus der dem Stammbaum
zugrundeliegenden Datenmatrix, die die Gesamtzahl der Merkmale widerspiegelt, werden
dabei nach dem Zufallsprinzip neue Datensätze erstellt, indem solange unabhängige
Einzelstichproben gezogen werden, bis die Gesamtzahl an Einzelstichproben wieder der der
ursprünglichen Matrix entspricht. Dadurch werden einige Merkmale aus dem
Originaldatensatz nicht mitberücksichtigt, während andere mehrfach vorliegen können. Die
anhand dieser neuen Datensätze (Pseudoreplikate, Bootstrapreplikate) erstellten Stammbäume
werden zu einem Consensusbaum zusammengefaßt, in dem prozentual angegeben ist, wie
häufig sich eine bestimmte Aufspaltung in Taxa innerhalb der Pseudoreplikate wiederfinden
läßt.
3.17.10 Decay Index (Bremer Index) Dieses Methode ist eine von BREMER (1988) entwickelte Alternative zum Bootstrap Test
innerhalb des Maximum-Parsimony (MP)-Verfahrens. Die Synonyme Decay-Index
(DONOGHUE et al. 1992) oder Support-Index (SI; BREMER 1994) haben sich in der Literatur
durchgesetzt und werden daher auch in dieser Arbeit verwendet. Über die Ermittlung der
Decay-Indizes wird geprüft, wieviele Schritte zusätzlich in einen MP-Baum eingefügt werden
müssen, um ein bestimmtes Monophylum nicht mehr zu erhalten; somit wird indirekt die
Anzahl der dieses Monophylum stützenden Autapomorphien ermittelt. Ist z.B. nach einer
heuristischen Suche der sparsamste Baum mit 1300 Schritten angegeben und benötigt man

Material & Methoden
52
1300 + n Schritte um eine im sparsamsten Baum vorhandene monophyletische Gruppierung
nicht mehr zu erhalten, wird der Wert n (= Decay Index) als Unterstützung für diese Gruppe
angegeben.
3.17.11 "Concatenated data sets" Bei der unter dem Begriff "concatenated data sets" bekannten Methode werden gleichzeitig
mehrere Datensätze analysiert. Die Theorie dieses Verfahrens beruht darauf, daß Datensätze
einzelner Gene oftmals nicht hinreichend informativ sind, um eine komplette Auflösung der
Taxa innerhalb einer Rekonstruktion zu erreichen. Sind für die zu untersuchende Gruppe von
Organismen mehrere unterschiedliche Gensequenzen bekannt, so müssen diese Sequenzen
innerhalb des Alignments in Blöcken so hintereinander gestellt werden, daß sie wieder
homologisierbar sind. Die concatenated trees wurden mit den Datensätzen der Gene der SSU
rDNA und der beiden par-Gene erstellt. Dabei wurden die drei Einzelalignments in das
Programm BioEdit V4.8.8 (http://www.mbio.ncsu.edu/RNaseP/info/programs/BIOEDIT/
bioedit.html) importiert und dort in Blöcken angeordnet. Nach Umschreiben in ein Nexus-
Format wurde der zusammengesetzte Datensatz innerhalb des Programmes PAUP*4.0b4a
(SWOFFORD 1998) mit Hilfe unterschiedlicher Rekonstruktionsverfahren analysiert. Da in den
drei Blöcken jeweils ein unterschiedlich hoher Informationsgehalt vorlag, mußte vorher eine
Wichtung durchgeführt werden. Dazu wurden für jeden Einzeldatensatz innerhalb des
Programmes PAUP*4.0b4a (SWOFFORD 1998) alle informativen Merkmale bestimmt und in
Relation zueinander gesetzt: Blöcke im zusammengesetzten Datensatz
SSU rDNA
par1
par2
Position
1-984
985-1789
1790-2594 informative Merkmale
191
161
151
151 dividiert durch informative Merkmale
0.791
0.938
1 Anhand dieser Wichtung (Befehlszeile: weights 791:1-984,938:985-1789,1000:1790-2594)
wurde dann mit dem Programm PAUP*4.0b4a (SWOFFORD 1998) eine Rekonstruktion
unter dem Maximum-Parsimony Verfahren durchgeführt. Alle Parsimony uninformativen
Merkmale wurden zuvor aus der Analyse ausgeschlossen.