Embed Size (px)
Citation preview

A multiplex endokrin neoplasia 1-es típusának
klinikai és genetikai vizsgálata
Doktori értekezés
Dr. Balogh KatalinDr. Balogh KatalinDr. Balogh KatalinDr. Balogh Katalin
Semmelweis Egyetem
Klinikai Orvostudományok Doktori Iskola
Témavezető: Dr. Rácz Károly, egyetemi tanár, MTA doktora
Hivatalos bírálók: Dr. Garami Miklós, egyetemi docens, PhD Dr. Kovács László, főorvos, PhD
Szigorlati bizottság elnöke: Dr. Szende Béla, egyetemi tanár, MTA doktora
Szigorlati bizottság tagjai: Dr. Váradi András, tudományos tanácsadó, MTA doktora
Dr. Alföldi Sándor, egyetemi docens, PhD
Budapest, 2006.

2
TARTALOMTARTALOMTARTALOMTARTALOM OLDALOLDALOLDALOLDAL
RÖVIDÍTÉSEK JEGYZÉKE 4.
I. BEVEZETÉS 6.
Irodalmi háttér 8.
I. 1. Klinikum 8.
Definíció és prevalencia 8.
Klinikai tünetek, diagnózis és kezelés 9.
1. Hyperparathyreosis 9.
2. Enteropancreatikus neuroendokrin daganatok 11.
3. Hypophysis daganatok 14.
4. Carcinoid tumorok 15.
5. Mellékvesekéreg dagantok 16.
6. Nem endokrin daganatok 16.
I. 2. Genetika 17.
„Pre-screening” módszerek 26.
1. SSCP 26.
2. DGGE, TGGE és CSGE 28.
3. Ritkábban használt, alternatív módszerek 32.
A családszűrés jelentősége 34.
Szomatikus genetikai eltérések MEN 1-asszociált tumorokban 36.
II. CÉLKITŰZÉSEK 37.
III. AZ ÁLTALUNK VIZSGÁLT BETEGEK 38.
IV. MÓDSZEREK 41.
DNS-izolálás 41.
PCR-reakciók 41.

3
TGGE 42.
DNS-szekvenálás 43.
V. EREDMÉNYEK 44.
A TGGE paraméterek optimalizálása 44
Előszűrés és szekvenálás 53.
Esetbemutatás és családszűrés 54.
VI. MEGBESZÉLÉS 63.
VII. KÖVETKEZTETÉSEK 69.
MAGYAR NYELVŰ ÖSSZEFOGLALÓ 71.
ANGOL NYELVŰ ÖSSZEFOGLALÓ 72.
IRODALOMJEGYZÉK 73.
SAJÁT KÖZLEMÉNYEK LISTÁJA 89.
KÖSZÖNETNYILVÁNÍTÁS 91.

4
RÖVIDÍTÉSEK
ACTH adenokortikotropin
ASO allél-specifikus oligonukleotid hibridizáció
BMP csont morfogenetikus fehérje
(bone morphogenetic protein)
CRH kortikotrop releasing hormon
CSGE konformáció-szenzitív gél elektroforézis
(conformation-sensitive gel electrophoresis)
DGGE denaturáló grádiens gél elektroforézis
(denaturing gradient gel electrophoresis)
DHPLC denaturáló HPLC
DNS dezoxi-ribonukleinsav
FHH familiáris hypercalcaemiás hypocalciuria
FIHP familiáris izolált hyperparathyreosis
F-SSCP fluoreszcens SSCP
GFAP glia fibrilláris savas protein
GH növekedési hormon
GHRH növekedési hormon releasing hormon
GTP guanidin triszfoszfát
HPLC magas felbontású folyadék-kromatográfia
(high-performance liquid chromatography)
IF intermedier filamentum
MEN multiplex endokrin neoplasia
MEN1 multiplex endokrin neoplasia 1 gén
MEN 1 multiplex endokrin neoplasia 1-es típus
MEN 2 multiplex endokrin neoplasia 2-es típus
MIP menin interakciós proteinek
mRNS messenger ribonukleinsav
PCR polimeráz láncreakció

5
RAS rat sarcoma protoonkogén
RFLP restrikciós fragmenthossz polimorfizmus
(restriction fragment length polymorphism)
RT-PCR reverz transzkripciós polimeráz láncreakció
SSCP egyszálú konformáció polimorfizmus
(single strand conformational polymorphism)
TGF-ß transformáló növekedési faktor-béta
TGGE hőmérséklet-grádiens elekroforézis
(temperature gradient gel electrophoresis)
VIP vazoaktív intesztinális peptid
WDHA vízszerű diarrhoea, hypokalaemia, achlorhydria szindróma

6
I. BEVEZETÉS
A multiplex endokrin neoplasia (MEN) szindrómákban az endokrin szervek
daganatai jellegzetes társulás szerint, családon belüli halmozódással fordulnak elő
[Arnold 1999, Brandi és mtsai 2001]. Két fő típusa a MEN 1 és MEN 2 szindróma; az
utóbbin belül további alcsoportok különíthetők el. Az eltérő klinikai megjelenés és a
különböző genetikai háttér ellenére a MEN 1 és MEN 2 szindróma néhány közös
sajátossággal is bír: mindkét szindróma autoszomális domináns öröklődésű, a benignus
vagy malignus daganatok az érintett endokrin szervekben gyakran több gócban
fejlődnek ki és a sporadikus endokrin daganatokhoz képest általában korábbi életkorban
jelentkeznek. [Frank-Raue és mtsai 2005, Tóth és mtsai 1993] A MEN 1 szindróma
(Wermer-kór) (OMIM 131100) három fő komponense a primer hyperparathyreosis
(általában többszörös mellékpajzsmirigy adenoma vagy hyperplasia), az
enteropancreatikus neuroendokrin daganat és a hypophysis adenoma, azonban a
szindrómához előbél carcinoid tumor, mellékvese daganat, valamint nem endokrin
szervek daganatai is társulhatnak (1. táblázat). [Burgess és mtsai 1998] A tünetek
nagymértékben függenek a daganatok lokalizációjától, illetve azok hormontermelő
sajátosságaitól. [Brandi és mtsai 2001]
A MEN 1 szindróma az egészséges populációhoz képest a morbiditás és
mortalitás jelentős növekedésével jár. A betegek 28 %-ában a mortalitás a MEN 1
szindróma részjelenségeire, illetve azok közvetlen szövődményeire vezethető vissza
(gastrinoma esetében gastrointestinális vérzés, insulinoma esetében hypoglycaemia,
stb.). Valószínűnek tartják azonban, hogy a MEN 1 szindrómával közvetlenül nem
összefüggő halálozásban a betegséghez társuló metabolikus és elektrolit eltéréseknek is
jelentős szerepe lehet (pl. a hyperparathyreosis miatt kialakuló hypercalcaemia
myocardialis kalcifikációt, magas vérnyomást, bal kamra hypertrophiát okozhat, sőt az
emelkedett parathormon szint közvetlenül szívizomsejt hypertrophiát is kiválthat
[Schussheim és mtsai 2001, Tsukada és mtsai 2001]).

7
1. táblázat: A MEN 1 szindrómához társuló daganatok relatív gyakorisága*
[Brandi és mtsai 2001]
Endokrin daganatok
� Mellékpajzsmirigy adenoma 90 %
� Enteropancreatikus daganat
o Gastrinoma 40%
o Insulinoma 10%
o Egyéb hormontermelő daganat 2%
o Nem hormontermelő daganat 20%
� Hypophysis daganat
o Prolaktinoma 20%
o GH- és prolaktin-termelő adenoma 5%
o GH-termelő adenoma 5%
o ACTH-termelő adenoma 2%
o TSH-termelő adenoma ritka
o Nem hormontermelő adenoma 5%
� Előbél carcinoid
o Thymus carcinoid 2%
o Bronchus carcinoid 2%
o Gyomor ECL sejt carcinoid 10%
� Mellékvesekéreg
o Primer aldosteronizmus ritka
o Cushing szindróma ritka
o Nem hormontermelő daganat 25%
Nem endokrin daganatok
� Lipoma 30%
� Angiofibroma (arcon) 85%
� Kollagenoma 70%
*40 év feletti életkorban, MEN1 szindrómás betegben
(ECL sejt: entrochromaffin-szerű sejt)

8
A MEN1 gén a 11q13 kromoszómarégióban található tumorszuppresszor gén,
melynek csírasejtes mutációi okozzák a MEN 1 szindrómát [Guru és mtsai 1998.]. Az
elmúlt években hazánkban is egyre több munkacsoport foglalkozott a különböző
öröklődő betegségek hátterében álló genetikai eltérések vizsgálatával, melyek
eredményei fontos szerepet kapnak a klinikumban is; számos esetben a nemzetközi
ajánlások genetikai diagnózisra építve tesznek javaslatot a terápiás stratégiákra, követési
protokollokra. Munkámban a mulitplex endokrin neoplasia 1-es típusának klinikai és
genetikai vonatkozásait foglalom össze, különös tekintettel a nemzetközi ajánlások
hazai megvalósításának lehetőségeire és a munkacsoportunk által végzett genetikai
szűrés révén gyűjtött hazai tapasztalatokra.
Irodalmi háttér
I.1. Klinikum
Definíció és prevalencia
Nemzetközi irodalmi adatok alapján a MEN 1 szindróma prevalenciája
~1/30000 [Roijers és mtsai 2000.]. Hazai adatok nem állnak rendelkezésre, azonban
feltehetően a betegség Magyarországon is a nemzetközi adatokkal megegyező
gyakoriságú. A betegek több mint 95%-ában a betegség első manifesztációi az ötödik
évtizedig megjelennek.
A legegyszerűbb klinikai definíció szerint MEN 1 szindrómáról akkor
beszélünk, ha a 3 fő komponens közül legalább 2 jelen van. MEN 1 szindrómás
családnak tekintjük azt a családot, amelyben legalább egy MEN 1 szindrómás
családtagon kívül legalább egy elsőfokú vérrokon családtagban a 3 fő MEN 1
komponens közül legalább 1 előfordul. Fontos megfigyelés, hogy a mutációk egy része
de novo alakul ki, ezért negatív családi anamnézis nem zárja ki MEN 1 szindróma
lehetőségét [Arnold 1999]. Nemzetközi ajánlások szerint MEN 1 szindróma gyanúja
akkor megalapozott, ha bármely fő lézió fiatal korban (<35év) jelentkezik, vagy
multiplex formában fordul elő, illetve ha egy fő lézió mellett legalább egy MEN 1-
asszociált minor eltérést is találunk [Stratakis és mtsai 2000].

9
Klinikai tünetek, diagnózis és kezelés
1. Hyperparathyreosis
A MEN 1 szindróma leggyakoribb és általában legkorábban kialakuló
komponense. Legtöbbször fiatal felnőttkorban (20-30 éves korban), de néha már
gyermekkorban kialakul. MEN 1 szindrómás családok 40 év feletti érintett tagjaiban a
hyperparathyreosis valószínűsége 90%. A betegek nagy részében előbb-utóbb mind a 4
mellékpajzsmirigyben daganat alakul ki [Pannett és Thakker 1999], de a daganatok
mérete általában egymástól eltérő. A daganatok rendszerint benignus adenomák [Haven
és mtsai 2004]. Becslések szerint a MEN 1 hyperparathyreosisok az összes primer
hyperparathyreosis eset mindössze 1-3%-át teszik ki.
MEN 1 szindrómához társuló hyperparathyreosisban a szérum kalcium és
parathormon koncentrációk közötti negatív szabályozás mértéke különbözik a
sporadikus hyperparathyreosis esetekhez képest. A mellékpajzsmirigy adenomás
sejteken a kalcium-érzékelő receptorok számának csökkenésével magyarázzák, hogy a
megnövekedett szérum kalcium szint hatására létrejövő parathormon szuppresszió
küszöbértéke magasabb érték felé tolódik el. A küszöbérték eltolódás MEN 1
szindrómához társuló hyperparathyreosisban kisebb mértékű, mint sporadikus
hyperparathyreosis eseteiben.
A MEN 1 szindrómához társuló hyperparathyreosis klinikai tünetei
megegyeznek a sporadikus hyperparathyreosis tüneteivel; kalcium-tartalmú vesekövek
alakulnak ki, a hypercalcaemia okozta renális diabetes insipidus miatt polyuria és
polydipsia jelentkezik, a csontok ásványianyag tartalma csökken. Súlyos
hypercalcaemia esetén gyakran alakulnak ki gastrointestinalis tünetek, exsiccosis és
izomtünetek.
A MEN 1 szindrómához társuló hyperparathyreosis diagnózisának alappillérei
az emelkedett szérum kalcium (ionizált kalcium) és emelkedett szérum parathormon
szint [Glascock és mtsai 2002]. A mellékpajzsmirigy daganatok lokalizálására alkalmas
vizsgálatok (ultrahang és Tc99m-sestamibi szcintigráfia, ritkán CT, MRI, kivételesen
PET és invazív vizsgálatok) megegyeznek a sporadikus eseteknél alkalmazható
vizsgálatokkal. A sporadikus esetektől eltérően azonban MEN 1 szindrómához társuló
hyperparathyreosis esetén mind a négy mellékpajzsmirigy feltárása indokolt, ezért a
lokalizációs vizsgálatok túlzott alkalmazására általában nincs szükség. Műtét utáni

10
recidíva esetén azonban a képalkotó vizsgálatok széles körére szükség lehet. [Doherty
2005].
MEN 1 szindrómán kívül más örökletes szindrómák is társulhatnak
hyperparathyreosissal, amelyeket a differenciáldiagnózis során számításba kell venni.
Ezek közé tartozik a MEN 2 szindróma, a familiáris hypercalcaemiás hypocalciuria
(FHH), valamint a familiáris izolált hyperparathyreosis különböző altípusai (FIHPT)
[Corbetta és mtsai 2005, Pannett és mtsai 2003]. Az FHH autoszomális domináns
öröklődésű betegség, a kalcium-érzékelő receptor gén (CasR gén) inaktiváló mutációi
okozzák. A diagnózist a rendszerint tünetmentes hypercalcaemia családon belüli
halmozódásán kívül az emelkedett szérum parathormon szint és a vizeletben a csökkent
kalciumürítés (kalcium/kreatinin clearance arány <0.01) alapozzák meg. Az FIHPT
esetek mintegy 20%-át a MEN1 gén mutációk okozzák. [Villablanca és mtsai 2002] Az
FIHPT 2. altípusában a multiplex cystás mellékpajzsmirigy adenomák állkapocs
fibromával társulnak; a betegséget a parafibromint kódoló HRPT2 gén inaktiváló
mutációi okozzák [Moon és mtsai 2005, Rubin és mtsai 2005]. A mellékpajzsmirigy
daganatok kialakulásában a cyclin D1 (CCND1) és a D-vitamin receptor (VDR) gének
mutációi is szerepet játszhatnak [Haven és mtsai 2004].
A MEN 1 szindrómához társuló hyperparathyreosis műtéti kezelése mind a
választandó időpont, mind a műtét típusa tekintetében eltér a sporadikus
hyperparathyreosis eseteknél alkalmazott eljárástól. Tünetmentes egyéneknél, 3 mmol/l-
nél kisebb szérum kalcium szint esetén a műtét halasztása javasolt. Bár a
megnövekedett szérum kalcium szint növeli a szérum gastrin szintet, a Zollinger-Ellison
szindróma hatásos gyógyszeres kezelési lehetősége miatt a hyperparathyreosishoz
társuló gastrinoma nem jelent egyértelmű indikációt a mellékpajzsmirigy műtét
elvégzésére. A mellékpajzsmirigy műtét során mind a négy mellékpajzsmirigy
feltárásának szükségessége miatt a minimálisan invazív műtéti módszer nem javasolt,
bár egyes munkacsoportok postoperatív követéses vizsgálataik során arra a
következtetésre jutottak, hogy háromnál kevesebb mellékpajzsmirigy érintettsége
esetében a szelektív parathyroidectomia eredményei nem maradnak el a radikálisabb
műtétek eredményeitől [Lee és mtsai 2006]. MEN1 szindrómában a leggyakrabban
alkalmazott eljárás a szubtotális mellékpajzsmirigy eltávolítás (mind a négy
mellékpajzsmirigy feltárása után 3 mellékpajzsmirigy teljes és 1 részleges eltávolítása

11
mintegy 50 mg épnek tűnő mellékpajzsmirigy szövet megőrzésével). Végezhető teljes
parathyroidectomia és a leginkább épnek tűnő mellékpajzsmirigyből szövetdarabkák
autotranszplantációja a nem domináns alkar brachioradiális izmainak rostjai közé.
Ilyenkor a mellékpajzsmirigyszövet krioprezervációja is indokolt, ami műtét utáni tartós
hypoparathyreosis esetén később ismételt beültetést tesz lehetővé [Johnson és mtsai
2005]. MEN 1 szindrómás betegek mellékpajzsmirigy műtéte során részleges
thymectomia elvégzése is ajánlott (részben a thymuson belül elhelyezkedő
mellékpajzsmirigy, részben a thymusban előforduló carcinoid miatt) [Lambert és mtsai
2005]. Az említett műtétek során meghagyott mellékpajzsmirigy-szövetben évekkel
vagy évtizedekkel később recidíva alakulhat ki, ezért egyes szerzők MEN 1 szindrómás
betegekben totális mellékpajzsmirigy eltávolítást (és élethossziglan D-vitamin analóg
kezelést) javasolnak. [Doherty és mtsai 2005] A hyperparathyreosis gyógyszeres
kezelésében perspektívát jelentő kalcium-érzékelő receptor agonisták klinikai vizsgálata
jelenleg folyamatban van.
2. Enteropancreatikus neuroendokrin daganatok
A MEN 1 szindróma második leggyakoribb megjelenési formája, az érintett
esetek 30-75%-ában alakul ki [Pannett és mtsai 1999]. A hasnyálmirigy szigetsejtekből
kiinduló hormontermelő daganatok rendszerint 40 éves kor után, a daganat által termelt
hormonoknak megfelelő szindrómák képében jelentkeznek. A szigetsejtekből kiinduló
daganatok rendszerint multicentrikusak [Hoff és mtsai 2000], méretük a néhány mm-es
adenomától a több cm-es invazív és metasztatikus carcinomáig terjedhet. A pancreas
szigetsejtes daganatok chromogranin A-t, pancreas polypeptidet, glukagont, inzulint,
proinzulint, szomatosztatint, gasztrint, vazoaktív intesztinális peptidet, szerotonint,
calcitonint, GH-releasing peptidet és ACTH-t termelhetnek. Malignus szigetsejtes
daganat 30 éves életkor alatt ritkán fordul elő, a későbbi életkorban azonban
gyakrabban.
Gastrinoma a MEN 1 szindrómás betegek mintegy 30%-ában alakul ki.
Klinikai megfigyelések szerint az összes gastrinoma 25 %-a MEN 1 szindróma
részjelensége. A MEN 1 szindrómához társuló gastrinomák jelentős része a
duodenumban helyezkedik el, ezek gyakran kisméretűek és többszörösek [Asgharian és
mtsai 2004, Anlauf és mtsai 2005]. A pancreas eredetű gastrinomák általában rosszabb

12
prognózisúak: a sporadikusan jelentkező Zollinger-Ellison szindrómához (ZES) képest
MEN 1 szindrómában átlagosan 10 évvel korábban kezdődik a betegség, és a nyelőcső
elváltozások (Barrett oesophagus, stricturák, oesophagitis, hiatus hernia) általában
gyakoribbak és súlyosabbak [Hoffmann és mtsai 2005.]. Nem ritka a malignus daganat,
a felismeréskor a betegek felében daganat-metasztázis mutatható ki. A gastrinoma a
gasztrin túltermelés révén gyomorsav-túltermelést, nehezen gyógyuló, gyakran
többszörös és szövődményekhez vezető pepticus fekélyeket, oesophagitist és hasmenést
okoz (ZES). A diagnózist a megnövekedett gyomorsav szekréció, a nagy szérum
gasztrin szint (rendszerint >115 pmol/l) és a szekretin vagy kalcium infúzió után
túlzottan nagy szérum gasztrinszint növekedés bizonyítják. Bár definitív gyógyulás csak
a gastrinoma teljes műtéti eltávolításától várható, a műtéti kezelés hosszú távú
eredményei (valószínűleg a MEN1 szindrómában gyakori több gócú gastrinoma, vagy a
kis primer daganat esetén is gyakori lokális metasztázisok miatt) gyakran alulmaradnak
a várakozástól. Újabban kiterjesztett műtéti eljárásokat vezettek be, de ezeknek az
eredményeknek az értékeléséhez további vizsgálatok szükségesek. [Doherty 2005] A
gastrinomák gyógyszeres kezelésére a H2 hisztamin-receptor blokkolók és proton-
pumpa gátlók hatásos kiegészítő terápiás lehetőséget jelentenek, használatuk óta a
totális gastrectomia elkerülhetővé vált. Szomatosztatin-analógok is eredményesen
alkalmazhatók; mind a gastrin, mind a gyomorsav szekréciót hatásosan gátolják, és a
MEN 1 szindrómában gyakori gyomor carcinoid regresszióját válthatják ki.
Insulinoma a MEN 1 szindrómás betegek közel 10%-ában fejlődik ki; az összes
insulinoma mintegy 10 %-a MEN 1 szindrómához társul. A MEN 1 szindrómában
kialakuló insulinomák általában multicentrikusak, méretük 1-3 cm és a hasnyálmirigy
bármely részéből kiindulhatnak, 25%-uk malignus. A klinikai tünetek (éhezési
hypoglycaemia neuroglycopenia tüneteivel, hízás) megegyeznek a sporadicus
insulinoma tüneteivel. A diagnózishoz alkalmazható legérzékenyebb teszt a vércukor,
szérum inzulin és C-peptid meghatározással egybekötött éhezési teszt. Az insulinoma
kezelése sebészi; a kisméretű és gyakran multiplex daganatok miatt a műtét előtti
lokalizáció nehéz lehet. A műtéti megoldás a tumor enucleatiótól a totális
pancreatectomiáig terjedhet [Doherty 2005]. Inoperábilis, vagy kiterjedten
metasztatizáló esetekben diétás (gyakori étkezés) és gyógyszeres kezelés alkalmazható

13
(diazoxid, streptozocin). A szomatosztatin-analógok kevésbé hatásosak, mint
gastrinomában.
Glucagonoma ritkán fordul elő; jellegzetes bőrtünetekkel (necroticus migráló
erythema), fogyással, anaemiával, stomatitissel és csökkent glukóz toleranciával járhat.
Leggyakoribb lokalizációja a pancreas farki része; a diagnózis időpontjában az esetek
50 %-ában metasztázis mutatható ki. A diagnózishoz a hyperglucagonaemia bizonyítása
szükséges. Kezelése sebészi; a műtét előtt a betegek állapotának javítására, illetve
inoperábilis esetekben somatostatin-analógok alkalmazhatók. Inoperábilis daganat vagy
metasztázis esetén cytotoxikus kemoterápia is alkalmazható.
VIPoma MEN1 szindrómában ritka; jellemzői a vízszerű diarrhoea,
hypokalaemia és achlorhydria (WDHA szindróma, vagy Verner-Morrison szindróma).
A diagnózist az megnövekedett szérum VIP koncentráció bizonyítja. Gyakran a
pancreas farokban helyezkednek el, sebészi eltávolításuk végleges gyógyulást
eredményezhet. Kiegészítő kezelésként somatostatin-analógok is alkalmazhatók.
Egyéb hormontermelő enteropancreatikus daganatok a termelt hormonnak
megfelelő szindrómákat okozhatják (pl. akromegalia, Cushing szindróma). Kezelésük
sebészi.
Hormonálisan inaktív enteropancreatikus daganatok a MEN 1 szindrómához
társuló összes enteropancreaticus daganat mintegy 30-40 %-át teszik ki. Ezek a
daganatok nem okoznak hormontúltermelés szindrómát, bár ilyen esetekben is a
részletes vizsgálatok kórosan emelkedett hormonszinteket (pl. pancreas polypeptid)
mutathatnak ki. A daganatok gyakran nagy méretűek, sok esetben malignusak és már a
felismeréskor metasztázisok lehetnek jelen. Kezelésük sebészi; MEN 1 szindrómás
betegekben a kisméretű (rendszerint szűrővizsgálattal felismert) hormonálisan inaktív
daganatok esetén azonban a műtéti indikációval kapcsolatban nem alakult ki egységes
álláspont [Lairmore és mtsai 1999].
Az enteropancreatikus neuroendokrin daganatok lokalizálására ultrahang,
endoszkópos ultrahang, angio-CT, angio-MRI és radionuclid szcintigráfiás módszerek
(szomatosztatin-receptor szcintigráfia, pozitron emissziós tomográfia) alkalmazhatók
[Hellman P és mtsai 2005]. Az intraoperatív ultrahang vizsgálat közel teljes
biztonsággal segíti a pancreas endokrin daganatok detektálását, ezért ezek műtéti
megoldásához ennek biztosítása indokolt.

14
Hasnyálmirigy szigetsejtes daganatok esetén gyakori a májmetasztázis, ezekben
az esetekben intervenciós radiológiai eljárások (artéria hepatica embolizáció,
kemoembolizáció, radiofrekvenciás tumor-abláció), illetve kemoterápia (5-fluorouracil,
streptozocin, doxorubicin, dacarbazin) végezhető. A korai és agresszív sebészeti kezelés
megelőzheti a májmetasztázisok kialakulását. [Bartsch és mtsai 2005]
3. Hypophysis daganatok
MEN 1 szindrómás esetek 10-60%-ában fordulnak elő [Dean és mtsai 2000]. A
daganatok kétharmada mikroadenoma, egyharmada makroadenoma. Az esetek 25%-
ában a MEN 1 szindróma első jeleként alakulnak ki. A hypophysis adenomák
valamennyi típusa előfordulhat, ezek közül leggyakoribb a prolactinoma [Verges és
mtsai 2002]. A familiáris prolactinoma hátterében is MEN 1 szindróma állhat [Hoff és
mtsai 2000]. A MEN 1 szindrómában kialakuló akromegáliát ritkán nem GH-termelő
hypophysis daganat, hanem GHRH-termelő carcinoid vagy szigetsejtes tumor okozza.
A MEN 1 szindrómához társuló Cushing szindróma oka szintén többféle lehet (ACTH-
termelő hypophysis adenoma, ectopiás ACTH-termelő carcinoid vagy szigetsejtes
tumor, illetve ectopiás CRH-termelő daganat). Thyreotrop és gonadotrop tumorok ritka
eseteit is leírták [Benito és mtsai 2005, Burgess és mtsai 1996]. A MEN 1 szindrómához
társuló hypophysis daganatok klinikai tünetei megegyeznek a sporadikus hypophysis
daganatok tüneteivel; a hormontermelő daganatok a termelt hormonnak megfelelő
klinikai szindrómákat okozzák (prolactinoma, akromegalia, Cushing kór), illetve mind a
hormontermelő, mind a hormonálisan inaktív daganatok a környező struktúrákon
kompressziós tüneteket válthatnak ki (fejfájás, bitemporális hemianopsia, szemmozgató
izombénulás, hypopituitarismus). A sporadikus esetekhez hasonlóan a hypophysis
daganatok MEN 1 szindrómában is rendszerint jóindulatúak, de igen ritka esetben
carcinoma is előfordulhat [Benito és mtsai 2005].
A MEN 1 szindrómához társuló hypophysis daganatok diagnózisára az MRI
vizsgálat a legérzékenyebb és leggyakrabban alkalmazott módszer. A hypophysis
daganatok hormontermelésének kivizsgálása megegyezik a sporadikus hypophysis
daganatoknál alkalmazott eljárásokkal. Mutáció-hordozó egyénekben a prolactinoma
szűrésére a bazális prolactin szint vizsgálat, a GH-termelő hypophysis adenoma
szűrésére a klinikai tüneteken (akromegalia) kívül szérum IGF-I vizsgálat javasolt

15
[Dreijerink és mtsai 2005]. A MEN 1 szindróma atípusos prolactinoma (Burin) variánsa
esetén az érintett egyénekben a hyperparathyreosis mellett szokatlanul nagy
gyakorisággal alakul ki prolactinoma és carcinoid típusú tumor, míg a gastrinoma igen
ritka [Olufemi és mtsai 1998, Petty és mtsai 1994, Verges és mtsai 2002, Hao és mtsai
2004]. Egy másik, Tasmániában talált, több mint 200 fős családban szintén igen nagy
gyakorisággal fordult elő prolactinoma (76%), míg nem-funkcionáló hypophysis
adenomát az esetek 24%-ában figyeltek meg. Ezt a variációt Tasmán variációként írták
le. [Beckers és mtsai 2003, Burgess és mtsai 2000]
A MEN 1 szindrómás betegekben kialakuló hypophysis daganatok kezelése nem
különbözik a sporadikus hypophysis daganatos esetek kezelésétől. [Doherty 2005]
4. Carcinoid tumorok
MEN 1 szindrómás esetek 5-15 %-ában fordulnak elő [Görtz és mtsai 1999];
kialakulásuk a többi daganathoz képest későbbi életkorban várható. Csaknem minden
esetben előbél eredetűek (thymus, bronchus, gyomor carcinoid). Megfigyelések szerint
mediastinalis lokalizáció esetén a MEN 1 szindrómához társuló carcinoid az elülső, míg
a sporadikus carcinoid a hátsó mediastinumban fordul elő. Jellegzetes a nemi
megoszlás: thymus carcinoid főleg dohányzó férfiakban, míg a bronchus carcinoid főleg
nőkben fordul elő [Snabboon és mtsai 2005]. MEN 1 szindrómában a thymus carcinoid
rendszerint agresszív kórlefolyású, 70%-uk malignus [Ferolla és mtsai 2005]. A gyomor
hisztamin-termelő ECL sejtjeiből származó carcinoid tumorok elsősorban ZES-hez
társulva fordulnak elő, ritkán okoznak hormontúltermelésre visszavezethető endokrin
tüneteket, többségüket hormonálisan nem funkcionáló daganatként ismerik fel és ritkán
malignusak. Carcinoid szindróma MEN 1 szindrómában ritkán alakul ki.
A MEN 1 szindrómához társuló carcinoid tumorokhoz ritkán társul
hormontúltermelés szindróma. A biokémiai vizsgálatok közül a szérum chromogranin A
koncentráció tűnik a legfontosabbnak A MEN 1 szindrómában ritkán előforduló
carcinoid szindróma esetén megnövekedett szerotonin, szerotonin metabolit (5-hidroxi-
indolecetsav), illetve hisztamin szintek mutathatók ki. A bronchus és thymus
carcinoidok ritkán ACTH-t, GHRH-t vagy calcitonint termelhetnek, ilyen esetek
igazolása a megfelelő hormonvizsgálatokkal lehetséges [Baudin és mtsai 1999, Yano és
mtsai 2005]. A gyomor carcinoid kimutatásában a gastroscopiának és az endoszkópos

16
ultrahang vizsgálatnak, a thymus és a bronchus carcinoid lokalizálásában a CT és a
szomatosztatin-receptor szcintigráfiás vizsgálatnak van kiemelt jelentősége [Doherty
2005].
Ritka előfordulásuk miatt kevésbé ismert a MEN 1 szindrómához társuló
carcinoidok optimális kezelési stratégiája.
5. Mellékvesekéreg daganatok
MEN 1 szindrómás betegek 20-40%-ában fordulnak elő. Hyperplasia, nodularis
hyperplasia, adenoma és ritkán carcinoma egyaránt előfordulhat, az elváltozás gyakran
mindkét mellékvesét érinti [Bhuiyan és mtsai 2001]. Többségük hormonálisan inaktív,
ritkán Cushing szindrómával vagy primer aldosteronismussal társul [Guo és Sawicki
2001, Libé és Bertherat 2005]. A MEN 1 szindrómához társuló kétoldali
mellékvesekéreg hyperplasia és nodularis hyperplasia a MEN 1 szindróma egyéb
részjelenségeinek, mint pl. insulinoma vagy ACTH-termelő adenoma másodlagos
következménye is lehet. A daganatok CT vizsgálattal és hormontúltermelés esetén
(Cushing szindróma, primer aldosteronismus) a sporadikus eseteknél alkalmazott
hormonvizsgálatokkal mutathatók ki. MEN 1 szindrómás betegben hormontermelő vagy
malignitásra gyanús mellékvesekéreg daganat esetén javasolt a mellékvese műtét.
6. Nem endokrin daganatok.
MEN 1 szindrómás betegek egyharmadában bőr alatti és viscerális lipomák
alakulnak ki; az elváltozás gyakran multiplex és gyakran érinti a pleurát vagy
retroperitoneumot. Gyakoriak az arc angiofibromái (esetek 40-85%-ában), melyek
legtöbbször a felső ajkon, orron és az arcon helyezkednek el. Kollagenomák az esetek
több mint 70%-ában a felsőtesten, nyakon és vállakon lehetnek jelen. A bőrtumorok
jelenléte segítheti a tünetmentes mutáció-hordozók szűrését [Asgharian és mtsai 2004].
MEN 1 szindrómában multiplex leiomyomák is előfordulhatnak (ureter, nyelőcső,
gyomor, tüdő, uterus és bőr) [Ikota és mtsai 2004].

17
I.2.Genetika
A multiplex endokrin neoplasia 1-es típusáért felelős MEN1 gént 1997-ben
azonosították a 11. kromoszóma hosszú karjának pericentromérás területén (11q13)
[Chandrasekharappa és mtsai 1997, The European Consortium on MEN1 1997]. (1.
ábra).
1. ábra: A MEN1 gén pozícionális klónozása, a gén szerkezete [Guru és mtsai 1997]
1
2
3
4
56
7
8
9
10
1
2
3
4
56
7
8
9
10
Baloldalon a 11-es kromoszóma látható, külön jelölve a hosszú karon a
pericentromérás területen elhelyezkedő 11q13 régió. MEN 1 szindrómás családok

18
”linkage analysis” vizsgálatával szoros kapcsoltságot mutattak ki a 11q13 régió
polimorf DNS markereivel, majd két rekombináns szakasszal ezt a területet a
nagyobb szürke téglalapnak megfelelő részre szűkítették (~2Mb). MEN 1-
asszociált és sporadikus tumorok LOH analízisével a területet a kisebb szürke
téglalap méretűre (~600kb) szűkítették, majd a megfelelő DNS szakaszt
szekvenálták. Számos kandidáns gént fedeztek fel, végül a MEN 1 szindrómás
családokban előforduló mutációk segítségével azonosították a betegségért felelős
gént, melyet MEN1 génnek neveztek el. A MEN1 gén exon-intron szerkezetét és a
kódoló régió sémás ábráját az ábra jobb oldali része mutatja be. A nyíl a
transzkripció irányát jelzi; a ferde vonalakkal kiemelt terület az exonok nem-
kódoló régiójának felelnek meg.
A MEN1 gén egy tumor szuppresszor gén [Guru és mtsai 1998]; 10 exont
tartalmaz, amelyből az első és a 10. exon második fele nem íródik át. A kódoló szakasz
1830 bp méretű [Heppner és mtsai 1999]. Az eddigi vizsgálatok során összesen több
mint 400 mutációt írtak le a gén területén [Pannett és Thakker 1999]; ezek legtöbbje
csonkolt fehérje kialakulásához vezet. A gén területén nincs mutációs “hot spot”, és
eddig genotípus-fenotípus összefüggést sem sikerült felfedezni [Hoff és mtsai 2000,
Kouvaraki és mtsai 2002].
A gén egy 610 aminosavból álló fehérjét kódol, melyet meninnek neveztek el. A
menin nem mutat homológiát az eddig ismert fehérjékkel; interakciói alapján azonban
feltételezhető, hogy szerepet játszik a sejt ciklus, a sejt növekedés és a genom stabilitás
szabályozásában. (2. ábra)
A homozigóta menin-null egerek embrionális korban 10,5 és 12,5 nap között
elpusztulnak, ezért a menin hiányát az élettel összeegyeztethetetlennek tartják.
Heterozigóta gén-kiütött egerek 24%-ában 9 hónapos korra mellékpajzsmirigy adenoma
alakul ki, bár a szérum kalcium szint nem változik. 16 hónapos egerek 26%-ában nagy
hypophysis adenoma, 22 hónapos korukra pedig 28%-ukban pancreas szigetsejt
adenoma alakul ki; utóbbiak nagy része (83%) insulinoma. A heterozigóta gén-kiütött
egerek 20%-ában mellékvesekéreg carcinomát találtak. [Crabtree és mtsai 2001.]
19
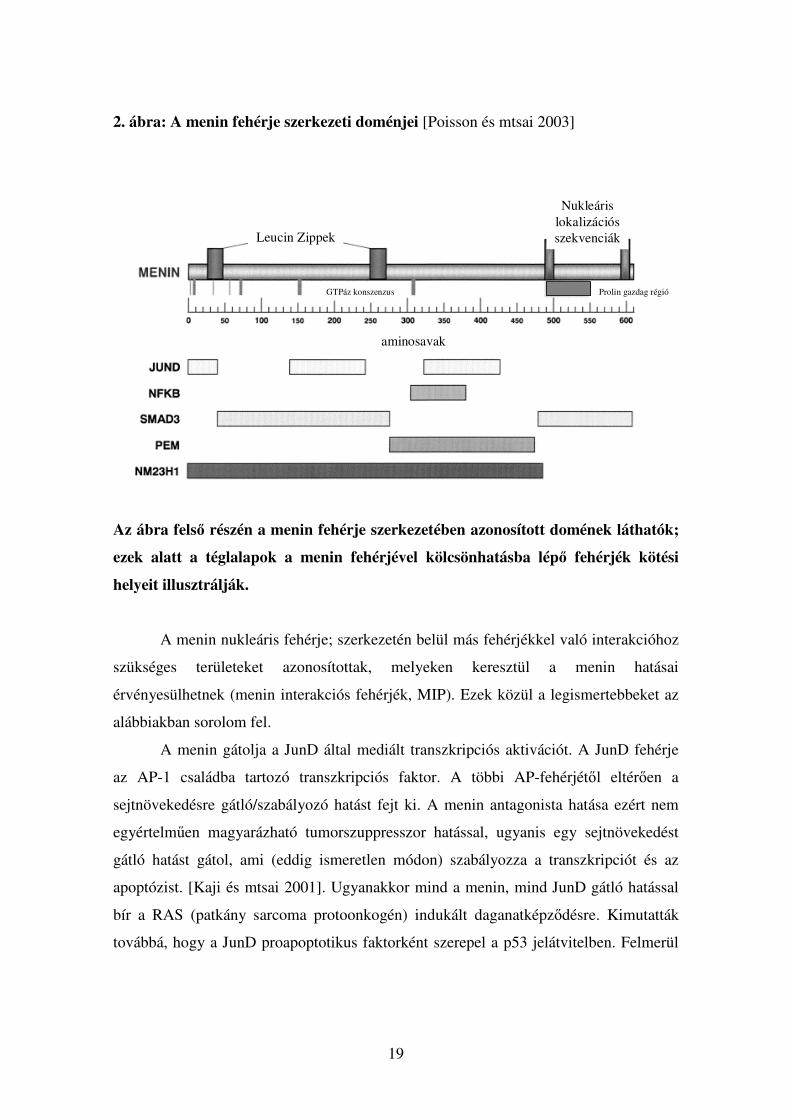
2. ábra: A menin fehérje szerkezeti doménjei [Poisson és mtsai 2003]
Leucin Zippek
Nukleárislokalizációsszekvenciák
aminosavak
Prolin gazdag régióGTPáz konszenzus
Leucin Zippek
Nukleárislokalizációsszekvenciák
aminosavak
Prolin gazdag régióGTPáz konszenzus
Az ábra felső részén a menin fehérje szerkezetében azonosított domének láthatók;
ezek alatt a téglalapok a menin fehérjével kölcsönhatásba lépő fehérjék kötési
helyeit illusztrálják.
A menin nukleáris fehérje; szerkezetén belül más fehérjékkel való interakcióhoz
szükséges területeket azonosítottak, melyeken keresztül a menin hatásai
érvényesülhetnek (menin interakciós fehérjék, MIP). Ezek közül a legismertebbeket az
alábbiakban sorolom fel.
A menin gátolja a JunD által mediált transzkripciós aktivációt. A JunD fehérje
az AP-1 családba tartozó transzkripciós faktor. A többi AP-fehérjétől eltérően a
sejtnövekedésre gátló/szabályozó hatást fejt ki. A menin antagonista hatása ezért nem
egyértelműen magyarázható tumorszuppresszor hatással, ugyanis egy sejtnövekedést
gátló hatást gátol, ami (eddig ismeretlen módon) szabályozza a transzkripciót és az
apoptózist. [Kaji és mtsai 2001]. Ugyanakkor mind a menin, mind JunD gátló hatással
bír a RAS (patkány sarcoma protoonkogén) indukált daganatképződésre. Kimutatták
továbbá, hogy a JunD proapoptotikus faktorként szerepel a p53 jelátvitelben. Felmerül

20
annak a lehetősége is, hogy a JunD két ismert izoformájához a menin nem egyforma
affinitással kötődik [Yazgan és Pfarr 2001, Ikeo és mtsai 2004].
A menin az NF-κB nukleáris faktor család három tagjával lép kölcsönhatásba
(NF-KappaB1, -B2 és a RelA transzkripciós regulátorok). Ezek a fehérjék homo- és
heterodimerek formájában számos gén expresszióját szabályozzák. A menin a JunD-hez
hasonló hatást fejt ki: gátolja az NF-KappaB által szabályozott transzaktivációt. A JunD
és NF-KappaB együttműködését (és direkt kölcsönhatását) patkány hepatocytákban
bizonyították. Kaji és mtsai kimutatták, hogy a menin a TGF-ß jelátvivő rendszerrel a
Smad3 szintjén lép kölcsönhatásba [Kaji és mtsai 2001]. Napjainkra az is ismertté vált,
hogy számos átkapcsolódás van a Smad-mediált TGF-ß jelátviteli rendszer és a RAS
foszforilációs útvonal között. Ez vezet például az AP-1 komplexek aktiválásához,
melyek egyik legfőbb összetevője a JunD, és lehetséges komponense a menin (GTP-áz
aktivitása révén).
A következő fehérje a Pem, melynek meninhez történő direkt kapcsolódását is
kimutatták [Lemmens és mtsai 2001]. A Pem egy homeobox-fehérje, amely elsősorban
a herében expresszálódik és feltehetően transzkripciót szabályozó szereppel bír.
Bár a menin alapvetően nukleáris lokalizációjú fehérje, több munkacsoport
beszámolt a menin citoplazmában való szekvesztrációjáról [Huang és mtsai 1999,
Lopez Egido és mtsai 2002, Wautot és mtsai 2000] Kimutatták, hogy a GFAP (glia
fibrilláris savas protein) az intermedier filamentum (IF) része, melyben a vimentin az
egyik fő alkotóelem. In vivo és in vitro vizsgálatokkal bizonyították a vimentin és a
menin direkt kapcsolódását, ezért a meninnek a citoplazma IF hálózattal való
kölcsönhatása és a menin citoplazmatikus szekvesztrációja is valószínűsíthető. Szintén a
GFAP-tartalmú IF hálózattal és ezáltal indirekt módon a meninnel is kapcsolatot létesít
az Nm23 metasztázis szuppresszor fehérje, amely egyben lehetővé teszi a menin
számára a GTP hidrolízisét azáltal, hogy a menint Ras-közeli GTP-ázokhoz (mint pl. a
Rad) köti [Yaguchi és mtsai 2002].
Ezeken a kapcsolatokon kívül a menin számos transzkripciós faktorral és
fehérjével alakít ki kölcsönhatást. A DNS-repairben résztvevő fehérjék közül, pl. az
RPA [Sukhodolets és mtsai 2003] és a FancD2 [Jin és mtsai 2003] direkt módon
befolyásolja a ciklin dependens kinázok expresszióját [Milne és mtsai 2005]. Továbbá a
menin kettősszálú DNS-hez való közvetlen kötődését is kimutatták [La és mtsai 2004].

21
Ezeken a kölcsönhatásokon keresztül a meninnek szerepet tulajdonítanak a sejt-
növekedés [Karnik és mtsai 2005], illetve sejt-ciklus szabályozásában [Ratineau és
mtsai 2004, Schnepp ás mtsai 2004], valamint a genom stabilitásban [Poisson és mtsai
2003, Agarwal és mtsai 2004.] (3. ábra)
3. ábra: A menin fehérje feltételezhető szerepe és más fehérjékkel való
kölcsönhatásai [Poisson és mtsai 2003]
RAS+ MAP kinázok
Transzkripció
Apoptózis
Sejtnövekedés szabályozásaSejtciklus szabályozásaGenom stabilitásSinapszis plaszticitás
RAS+ MAP kinázok
Transzkripció
Apoptózis
Sejtnövekedés szabályozásaSejtciklus szabályozásaGenom stabilitásSinapszis plaszticitás
Az ábra szemlélteti, hogy bár a meninnek más fehérjékkel való kölcsönhatásai
révén betöltött szerepe még számos ponton tisztázatlan, a menin főbb hatásai már
körvonalazódtak.
A menin in vivo funkciójával kapcsolatban is számos új funkcióra derült fény az
utóbbi időben, az endokrin szövetekben betöltött szabályozó mechanizmusok is egyre
nagyobb részletességgel válnak ismertté. A menin fehérje szerepének vizsgálatakor
Hendy és mtsai MEN 1-asszociált tumorokban kimutatták, hogy agyalapi mirigy

22
sejtekben a menin inaktivációja gátolta a TGF-ß és aktivin jelátvitelt és antagonizálta
azok növekedés-gátló hatását. A menin szerepét mutatták ki a prolaktin expresszió
aktivin-indukálta gátlásában is; ebben a szabályozó mechanizmusban a Smad
fehérjékkel és a Pit-1 transzkripciós faktorral való együttműködés tűnt jelentősnek. A
menin inaktivációja mellékpajzsmirigy sejtekben megszűntette a proliferációt és a PTH-
szekréciót gátló TGF-ß hatást. Kimutatták azt is, hogy MEN 1 szindrómás betegek
mellékpajzsmirigy sejtjeiben a TGF-ß nem képes a sejtek proliferációjának és PTH-
szekréciójának szabályozására. MEN1-null egerek magzataiban cranialis/facialis
hypoplasiát észleltek, ami a menin csontfejlődésben betöltött szerepét is felveti. Menin
fehérje szükséges a multipotens mesenchymális őssejtek osteoblast sejtvonalba történő
differenciálódásához is. A menin fizikailag és funkcionálisan is kapcsolatban áll a
BMP-2 (csont morfogenetikus fehérje-2) szabályozása alatt álló Smad fehérjékkel (pl.
Smad 1 és Smad5) és az osteoblast kulcsszabályozó Runx2 fehérjével is. Ezeknek az
interakcióknak a hatásai megszűnnek, amint az osteoblastok tovább differenciálódnak.
Ekkor a menin a Smad3 fehérjével lép kapcsolatba, ami a Runx2 fehérje TGF-ß által
történő negatív szabályozásához vezet. A menin fehérje a JunD fehérje differenciálódást
serkentő hatásainak gátlása révén is gátolja az osteoblast érést [Hendy és mtsai 2005,
Sowa és mtsai 2003].
A MEN1 gén mutációk többsége a menin fehérje hiányát, vagy rövidebb és ezért
funkcióképtelen menin fehérje képződését okozza [Guru és mtsai 1998, Brandi és mtsai
2001]. Napjainkig több mint 400 MEN 1 szindrómát okozó veleszületett (minden
sejtben jelenlevő, csírasejtes) mutációt írtak le, melyek mintegy 22 %-a nonsense, 56 %-
a deléciós/inserciós, 17 %-a missense és 5 %-a donor-splice mutáció [Pannett és mtsai
1999]. A betegek 5-10 %-ában a mutáció nem a kódoló szakaszon, hanem a promoter
vagy az intronikus szakaszokon található, melyek rutin módszerekkel nem, vagy
nehezen detektálhatók. A mutációk az 1. exon kivételével (amely nem íródik át a
transzláció során [Karges és mtsai 2000]) az összes exont érinthetik [Goebel és mtsai
2000, Kouvaraki és mtsai 2002].

23
A menin tumor szuppresszor fehérje, a betegségre jellemző endokrin daganatok
mindkét allél defektusa esetén alakulnak ki (Knudson-féle ”két ütés” modell). Az
érintett egyénekben típusosan az egyik allél veleszületett mutációján kívül a
daganatszövetben a másik allél szerzett, szomatikus defektusa alakul ki (rendszerint
allél-vesztés; LOH: a heterozigótaság elvesztése) [Agarwal és mtsai 2005]. Szomatikus
MEN1 gén mutációk a MEN 1 szindrómához társuló endokrin daganatokon kívül
sporadikus endokrin daganatokban is előfordulhatnak (VIPomák 50 %-ában,
hypophysis adenomák 40 %-ában, bronchus carcinoidok 25-35 %-ában, gastrinomák 25
%-ában, insulinomák, nem funkcionáló pancreas neuroendokrin tumorok és
mellékpajzsmirigy adenomák 10-20 %-ában, angiofibromák és lipomák 10-17 %-ában)
[Kawamura és mtsai 2005, Libé és mtsai 2005, Miedlich és mtsai 2000]. Családi
anamnézis hiányában is kialakulhat a későbbiekben öröklődő csírasejtes MEN1 mutáció,
ilyen esetekben a betegséget rendszerint de novo mutáció okozza (4. ábra).
4. ábra: A Knudson-féle „két ütés” modell A csírasejtes MEN1 mutációk esetén, familiáris és de novo esetben is, a proband
utódjai öröklik a betegséget okozó genetikai eltérést. Sporadikus MEN 1-asszociált
daganatokban kialakult mutáció esetén azonban csak az adott endokrin őssejt és a
belőle származó endokrin sejtek tartalmazzák majd a betegség kialakulásáért
felelős mutációt, a csírasejtek nem, így ezen daganatok esetében nem kell
öröklődésre számítani. A daganatsejt a normál endokrin sejtből mindhárom
esetben ugyanazon folyamat eredményeként jön létre: egy második „csapás”,
szomatikus mutáció (leggyakrabban allélvesztés) eredményeképpen. A daganatsejt
tehát a MEN1 allélra nézve vagy hemizigóta, vagy compound heterozigóta lesz.

24

25
A MEN1 gén mutációinak vizsgálata munka- és időigényes feladat, mégis
kulcsfontosságú, hiszen a betegek, a betegségre gyanús személyek és mutáció-hordozó
egyének követéskor és ellenőrzésekor a nemzetközi ajánlásokban megfogalmazott
szempontok érvényesítése szükséges. A MEN1 gén mutációk vizsgálatának széles
körűen elfogadott jelenlegi indikációit a 2. táblázat foglalja össze.
2. táblázat: A MEN1 gén mutációanalízisének indikációi [Brandi és mtsai 2001]
A genetikai szűrés indikációi
Index eset:
▪ Klinikailag definiált MEN 1 szindróma
(sporadikus vagy familiáris formában: 2 major lézió, vagy 3 major és/vagy
minor eltérés)
▪ Klinikailag gyanús, vagy atípusos MEN 1 szindróma
2 vagy több MEN 1-tumor; multiplex mellékpajzsmirigy tumor 30 éves kor
alatt; eredményes műtét után kiújuló hyperparathyreosis; gastrinoma vagy
multiplex pancreas szigetsejt tumor bármely életkorban
(Familiaris izolált hyperparathyreosis.)
Ismert MEN1 mutációval rendelkező egyén vérrokon családtagjai:
▪ Minden tünetmentes vérrokonnál
▪ Bármely rokonnál, aki a MEN 1 szindróma bármely jelét mutatja
(megerősítésként)
Klinikailag MEN 1 szindrómás betegben, MEN1 mutáció hiányában :
▪ Mint fent, csak a MEN1 mutációanalízis nem hozzáférhető; abban az esetben is
ez az ajánlás, ha a genetikai szűrés negatív*
* ajánlható haplotípus-analízis, linkage-analízis és klinikai kémiai tesztek.

26
„Pre-screening” módszerek
Az utóbbi időkben a nagy formátumú DNS-szekvenálási módszerek elterjedése
és a különböző mutáció detektáló rendszerek fejlődése a genetikai betegségek
pontosabb diagnózisát teszik lehetővé. Bár a mutációk és más genetikai eltérések
kimutatására napjaink ”gold standard” módszerének az adott gén teljes automata
szekvenálása tekinthető, ez az eljárás költség- és időigényes, sőt nagyobb génszakaszok
deléciója esetén nem minden esetben informatív. A szekvenálás hátrányai miatt számos
módszert dolgoztak ki, melyeket gyakran a szekvenálás előtti szűrőmódszerként („pre-
screening”) alkalmaznak egy adott génen belül a genetikai eltérést tartalmazó
génszakasz (exon) azonosítására. A szűrőmódszerek nagymértékben csökkenthetik a
mutációk kimutatásának költségeit, azonban hasznosságukat döntően megbízhatóságuk
határozza meg. A szűrőmódszerekkel a genetikai eltérések (polimorfizmusok, mutációk)
jelenléte vagy hiánya állapítható meg, ezért megbízható szűrőmódszer alkalmazása
esetén a genetikai eltérést pontosan kimutató DNS szekvenálást nem szükséges az
összes DNS szakaszon (exonon) elvégezni. A szűrőmódszerek családszűrésre is
hasznosak lehetnek, ha a betegségben szenvedő családtagban már korábban kimutattuk
a keresett genetikai eltérést.
A MEN1 gén mutációanalízisére jelenleg használt módszerek a gén összes
exonjának PCR-sokszorosításán alapulnak. A munkacsoportok nagy része a „The
European Consortium on MEN1” által korábban leírt oligonukleotid primereket
használja [The European Consortium on MEN1 1997], bár néhány módszer egyedi
tervezésű primereket igényel.
1. SSCP (egyszálú konformáció polimorfizmus analízis)
Irodalmi adatok alapján a MEN1 gén mutációk szűrésére az SSCP tűnik a
leggyakrabban használt módszernek [Cetani és mtsai 2000, Costa és mtsai 2001, Karges
és mtsai 1998]. A módszer azon alapul, hogy az egyszálú nukleinsavak másodlagos
szerkezetét szekvenciájuk határozza meg, és akár egyetlen nukleotid cseréje a
másodlagos szerkezet olyan változását hozhatja létre, amely megváltoztatja a DNS
szakasz elektroforetikus mobilitását [Orita és mtsai 1989]. Az elektroforézishez nem

27
denaturáló körülmények mellett poliakrilamid gélt alkalmaznak. Nagyméretű PCR
termékek restrikciós emésztése vagy multiplex PCR használata esetében a módszer
multiplex fragmens-szűrésre is lehetőséget ad. Optimális eredményt 150-200 bp méretű
DNS-szakaszokkal lehet elérni. A genetikai eltérés kimutatásának valószínűsége
fordított arányban áll a PCR-termékek méretével; minél hosszabb a termék, annál
kisebb a kimutatási valószínűség. A módszer érzékenysége a gél összetételétől (pl.
glicerin koncentráció) és az elektroforézis alatt biztosított egyenletes hőmérséklettől is
függ (ingadozó hőmérséklet esetén csökken a szenzitivitás). Optimális feltételek mellett
a báziscserék ~80-90%-a mutatható ki SSCP-vel [Perrier és mtsai 2002, Bassett és mtsai
1998]. Az elektroforézis után a DNS-t általában ezüstözéssel (ritkábban
autoradiográfiával) mutatják ki. A MEN1 gén SSCP-vel történő genetikai szűrésének
néhány részletét a 3. táblázat foglalja össze.
3. táblázat: MEN1 gén eltérések szűrése SSCP-vel
N bp T Gél t P Szenz. Ref.
15 200-300 Szobahő 25% MDE 14-16 h 6 W NA [0]
15 200-300 Szobahő 25% MDE 12 h 6-8 W NA [0]
12
18
211
NA Szobahő 0.5x MDE 14-18 h 4 W
76%
75%
[0]
n: fragmens szám, bp: fragmens hossz, T: az elektroforézis hőmérséklete, t:
elektroforézis idő, P: teljesítmény, Szenz.: szenzitivitás, Ref.: irodalmi hivatkozás,
NA: Nincs adat
Park és mtsai 7 nem-rokon családban végzett vizsgálatai során 4 különböző
csírasejtes mutációt azonosítottak, de a 7-es exon Tyr323Stop mutációját nem sikerült
SSCP-vel kimutatniuk, noha DHPLC-vel detektálták ezt a mutációt [Park és mtsai
2003]. Bassett és mtsai 63 család-reprezentáló MEN 1 szindrómás beteg vizsgálata
alapján az SSCP szenzitivitását >85%-nak találták (a Glu388stop mutációt nem sikerült
SSCP-vel kimutatniuk), és a vizsgált esetek 7%-ában álpozitív eredményt kaptak

28
[Bassett és mtsai 1998]. A reagensek összetétele, illetve az elektroforézis körülményei
lényegesen befolyásolhatják az eredményeket; ugyanannak a PCR-terméknek többféle
elektroforetikus környezetben történő vizsgálata javíthatja a mutáció kimutatásának
valószínűségét. A módszer hátrányai közé tartozik, hogy az optimális körülmények
meghatározása empirikus módszerekkel történik. Jelenleg nem áll rendelkezésre olyan
számítógépes szoftver, amelynek segítségével pontosan számíthatóak lennének a
kérdéses paraméterek (gélösszetétel, hőmérséklet).
Kapilláris elektroforézis, standardizálás és automatizálás segítségével a szűrés
ideje lerövidíthető. A fluoreszcens kapilláris elektroforézis SSCP (F-SSCP)
szenzitivitását ~95%-osnak, specificitását ~97%-osnak találták [Mogensen és mtsai
2003]. Az F-SSCP nagy mennyiségű minta feldolgozására is alkalmas, azonban a
készülék költséges beruházást jelent.
2. DGGE (denaturáló grádiens gél elektroforézis), TGGE (hőmérséklet grádiens
gél elektroforézis) és CSGE (konformáció-szenzitív gél elektroforézis)
Ezekkel a módszerekkel poliakrilamid gélen kétszálú DNS fragmentek
elektroforézisét végzik. DGGE esetében a gél fokozatosan növekvő koncentrációban
tartalmaz valamely denaturáló ágenst (urea, vagy formamid). TGGE esetében a
denaturáló ágens (általában urea) konstans koncentrációja mellett a hőmérséklet
növelésével biztosítható a fokozatos denaturáció. CSGE esetében pedig konstans
koncentrációjú denaturáló ágens és állandó hőmérséklet mellett mutatják ki a kétszálú
DNS fragmentek közötti konformáció-eltérést. E módszerek közös sajátossága, hogy a
hagyományos PCR protokollt követően heteroduplex képzésre van szükség. Erre
számos leírást találhatunk az irodalomban:
� denaturálás 94°C-on 3 percig, majd inkubáció 68°C-on 1 órán át, ezt
követően lassú hűtés szobahőig [Cebrián és mtsai 2002]
� denaturálás 94°C-on 5 percig, majd inkubáció 1 órán át szobahőn [Morelli
és mtsai 2000] vagy

29
� denaturálás 94°C-on 5 percig, melyet 35 lassú ciklus követ 94°C-ról
indulva 2°C/1 perc alatt csökkenő hőmérséklettel, majd 1 perces inkubáció
25°C-on [Howell és mtsai 2003]
DGGE és TGGE esetében a mutáns és vad típusú homoduplex és heteroduplex
molekulák a mutáció területén kialakuló ”mismatch” által okozott különböző olvadási
tulajdonságaiknak megfelelően válnak szét. A homoduplex és heteroduplex molekulák
más-más időben kezdődő denaturálódása miatt a korábban denaturáló fragmentek
lemaradnak a gélen. DGGE során a denaturáló ágens koncentrációjának, TGGE során
pedig a használt puffer vagy a gél hőmérsékletének folyamatos növekedése hozza létre a
lineáris denaturációs grádienst. Amint elkezdődik a denaturálódás, az elektroforetikus
mobilitás csökken és a minták haladása a gélen lelassul. A tapasztalatok azt mutatják,
hogy e módszerek esetében az ideális fragment hosszúság kb 1000 bp. Az ún. ”melting”
domének (közel azonos olvadási hőmérsékletű szerkezeti egységek) számát GC-gazdag
régiók („GC-clamp”-ek) segítségével növelhetjük úgy, hogy a primer végére GC-
gazdag szakaszt kötünk, így a „clamp” a keletkező PCR termék megfelelő végére is
rákerül. Ez azért fontos, mert a GC-clampnak megfelelő terület a fragmenten belül igen
magas olvadáspontú szakaszt képez és mivel az összes ”melting” domén analizálható,
kivéve a legmagasabb hőmérsékleten denaturálódót (Tm), a GC-gazdag régión kívül az
adott fragmens minden eleme vizsgálhatóvá válik. A nemzetközi irodalomban olvasható
eredmények azt mutatják, hogy a heteroduplex képzés alapján GC-clampek alkalmazása
esetén csaknem minden mutáció detektálhatóvá válik [Nollau és Wagener 1997]. A
MEN1 gén DGGE-vel történő mutáció analízisének részleteit Morelli és mtsai
ismertették [Morelli és mtsai 2000] Ennek részleteit és eredményeit a 4. táblázat foglalja
össze.
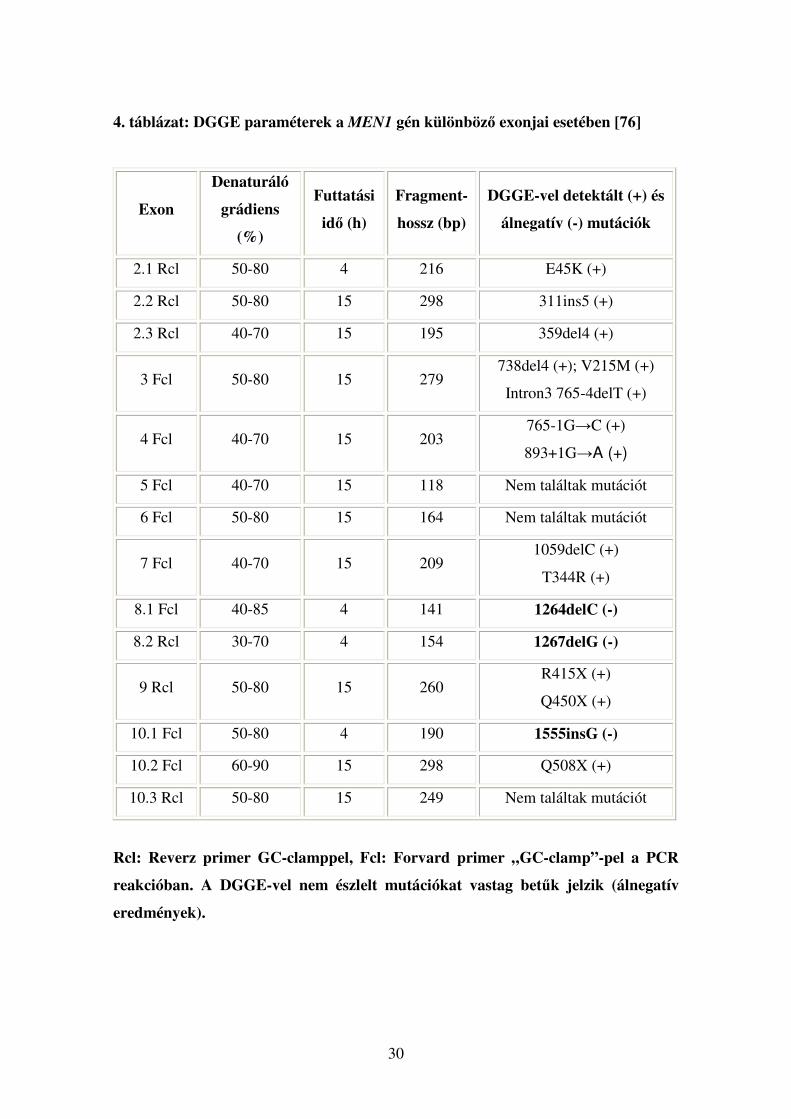
30
4. táblázat: DGGE paraméterek a MEN1 gén különböző exonjai esetében [76]
Exon
Denaturáló
grádiens
(%)
Futtatási
idő (h)
Fragment-
hossz (bp)
DGGE-vel detektált (+) és
álnegatív (-) mutációk
2.1 Rcl 50-80 4 216 E45K (+)
2.2 Rcl 50-80 15 298 311ins5 (+)
2.3 Rcl 40-70 15 195 359del4 (+)
3 Fcl 50-80 15 279 738del4 (+); V215M (+)
Intron3 765-4delT (+)
4 Fcl 40-70 15 203 765-1G→C (+)
893+1G→A (+)
5 Fcl 40-70 15 118 Nem találtak mutációt
6 Fcl 50-80 15 164 Nem találtak mutációt
7 Fcl 40-70 15 209 1059delC (+)
T344R (+)
8.1 Fcl 40-85 4 141 1264delC (-)
8.2 Rcl 30-70 4 154 1267delG (-)
9 Rcl 50-80 15 260 R415X (+)
Q450X (+)
10.1 Fcl 50-80 4 190 1555insG (-)
10.2 Fcl 60-90 15 298 Q508X (+)
10.3 Rcl 50-80 15 249 Nem találtak mutációt
Rcl: Reverz primer GC-clamppel, Fcl: Forvard primer „GC-clamp”-pel a PCR
reakcióban. A DGGE-vel nem észlelt mutációkat vastag betűk jelzik (álnegatív
eredmények).

31
Az előzőekben említett tanulmány során az elekroforézist 15 órán keresztül, 44
V feszültséggel, vagy 4 órán át, 160 V-tal végezték, 58˘C-on, 9%-os akrilamid gélen, a
denaturáló grádiens növekvő koncentrációja mellett. A DGGE szenzitivitását ~82%-
osnak találták; a szűrés során 2 deléciós és egy inzerciós mutáció esetében álnegatív
eredményt kaptak, melyeket végül direkt szekvenálással azonosítottak. A módszer
hátránya, hogy a kémiai denaturáló ágensekkel készülő akrilamid gélek nem mindig,
vagy nem pontosan reprodukálhatóak.
TGGE-vel a fent említett problémák kikerülhetőek. A módszer könnyen
kivitelezhető, költséghatékony és az eredmények jól reprodukálhatóak. További előnye,
hogy a vizsgált DNS fragmentek teoretikus olvadási profiljainak és az elektroforetikus
paraméterek kiszámolásához szoftverek is rendelkezésre állnak, mint pl. a WinMelt
(Bio-Rad, Munich, Germany) vagy a Poland [Steger 1994] (http://www.biophys.uni-
duesseldorf.de/local/POLAND/poland.html). Az elektroforetikus paraméterek
optimalizálásának lépéseit az „Eredmények” című alfejezetben ismertetem.
CSGE esetében állandó koncentrációjú denaturáló ágens (formamid)
alkalmazásával a homo- és heteroduplexek különböző konformációjuk alapján
választhatók szét. A módszer alapelve, hogy az egy-bázisú ”mismatch”-eltérések a
kétszálú DNS-ben konformációs különbségeket hoznak létre, melyek MDE gélen a
hetero- és homoduplexek különböző vándorlása alapján mutathatók ki [Cebrián és mtsai
2002, Cebrián és mtsai 2003]. A MEN1 gén mutációanalízise során CSGE-vel több
fragment együttes futtatása is lehetséges (multiplex PCR amplifikációt követően a 2. +
5.-6. exonok, a 3. + 10. exonok, a 7. + 8. exonok vagy a 9. + 4. exonok). Az 1xMDE
gélek összetétele az alábbi: 15% formamid, 10% ethylen glycol, és 0.6xTBE. Az
elektroforézis 150 V-tal, 20 órán keresztül szobahőn, vagy 7 W-tal 17-20 órán át
0.6xTBE pufferben történik. A módszer előnyei közé tartozik, hogy a MEN1 szekvencia
eltéréseket ~96%-ban mutatja ki (az A541T polimorfizmust ezzel a módszerrel nem
lehetett detektálni). További előnye, hogy a fenti összeállításban a MEN1 gén összes
kódoló régiójának szűrését 4 elektroforézissel el lehet végezni [Cebrián és mtsai 2002,
Cebrián és mtsai 2003]. Az elektroforézis után az eredmények mind ethidium bromidos
festéssel, mind ezüstözéssel vizualizálhatók.

32
3. Ritkábban használt, alternatív módszerek
A heteroduplex analízis nem denaturáló poliakrilamid gélen történik, melynek
során vad típusú és mutáns PCR termékek keverékéből felmelegítéssel, majd re-
annelálással heteroduplexeket képezünk. Az optimális fragmenthoszzúság 200-600 bp.
A módszer szenzitivitását 80%-ra becsülik [Morelli és mtsai 2000, Nollau és mtsai
1997, Agarwal és mtsai 1997].
A denaturáló HPLC (DHPLC; denaturing high-performance liquid
chromatography) alapelve a TGGE módszeréhez hasonló, azonban a kromatográfiás
oszlopról leoldott fragmentek detektálása UV fényben történik és az eredményeket a
chromatogram alapján értékelik. Ez a módszer is heteroduplex-képzést igényel; a
vizsgálati paraméterek optimalizálása WAVEMAKER szoftverrel (Transgenomic,
Omaha, NJ) történik, mellyel a szükséges hőmérséklet minden egyes PCR termékre
nézve meghatározható. A módszer szenzitivitását és specificitását egyaránt közel 100%-
osnak találták. Mogensen és mtsai az eredmények reprodukálhatósága érdekében a
DHPLC analízisre szánt minták -20°C-on történő tárolását ajánlották, ugyanis azt
tapasztalták, hogy a heteroduplexek szobahőn nem stabilak [Mogensen és mtsai 2003].
A módszer gyors és nagyszámú minta analízisét teszi lehetővé. Egyedüli hátrányként
említendő a készülék nagy költsége. A nemzetközi irodalomban Park és mtsai
használták a DHPLC-t MEN1 gén analízisre; a gént 12 részletben amplifikálták, de
további részleteket nem közöltek [Park ás mtsai 2003].
Számos olyan egyéb módszer is ismert, melyek alkalmasak lehetnek, de még
nem használták a MEN1 gén vizsgálatára, mint pl. a cysticus fibrosis vizsgálatára bevált
PNA-DNA hybrid-kötő cyanin festés [Wilhelmsson és mtsai 2002], vagy a dupla GC-
clamp módszer, amelyet eredményesen alkalmaztak az 1-es típusú neurofibromatosis
gén (NF1) mutációk TGGE-vel történő szűrésére [Fahsold és mtsai 2000]. Napjaink
vívmánya a DNS chip módszer, amellyel egyre több gén mutációanalízisére nyílik
lehetőség, melyet a MEN 1 szindróma genetikai szűrésére még nem alkalmazták, noha a
MEN 1 asszociált tumorok génexpressziós mintázatát DNS chip segítségével már több
esetben vizsgálták [Dilley és mtsai 2005, Forsberg és mtsai 2005].
A tapasztalatok alapján a klinikailag MEN 1 szindrómás betegek 10-15%-ában
ezeknek a módszereknek egyikével sem lehet mutációt kimutatni a MEN1 gén kódoló

33
régióban és az exon-intron határokon. Ezért számos erőfeszítés történt a MEN1 gén
promoter régiójának és nem-kódoló szakaszainak, illetve a nagyméretű csírasejtes
deléciók kimutatására. Ezeknek a vizsgálatoknak az eredményei alapján ajánlásokat is
kidolgoztak; az egyik ilyen ajánlás elsőként a MEN1 mRNS RT-PCR analízisét
javasolja, mellyel a splicing mechanizmushoz szükséges intronikus régiók konzervatív
szakaszait érintő rendellenességek is kizárhatók. Megemlíthető, hogy Mutch és
mtsainak 5 esetből 3-ban nem sikerült splicing mutációt felfedni annak ellenére, hogy
más módszerrel kimutatták a kóros splicing termékek jelenlétét [Mutch és mtsai 1999].
Azokban a betegekben, akikben az RT-PCR nem hozott eredményt a haplotípus analízis
ajánlott, mellyel megerősíthetjük, hogy valóban a 11q13 kromoszómarégióban található
génterület felelős a betegség kialakulásáért. A vizsgálattal a polimorf markerek co-
segregációja mutatható ki (pl. PYGM, D11S449, D11S913, D11S4946). Az
intragenikus lókuszokat érintő nagy deléciók mind a korábban részletezett „pre-
screening” módszerekkel, mind a DNS szekvenálással álnegatív eredményt adnak. E
deléciók egy része a gyakori intragenikus polimorfizmusok vizsgálatával is
felfedezhető, ugyanis a polimorfizmusok öröklődését követve a mutáns MEN1 allélek
öröklődése is nyomon követhető [Cavaco és mtsai 2002]. A nagy deléciók pontos
méretének megítélése nehéz, de egy hozzávetőleges méretbecslés ezekkel a
módszerekkel is végezhető. További lehetőségként a Southern-blot analízis említhető. A
genomikus DNS-t különböző restrikciós enzimekkel feldarabolva (pl. KpnI, SmaI, SacI,
SacII), majd Southern-blot vizsgálatot végezve a MEN1 cDNS szegmentek (pl. 152-753
nukleotidok a 2-3. exonokat lefedve, illetve a 1012-1474 nukleotidok a 7-10. exonokat
tartalmazva) vizsgálhatók és denzitometriás analízissel pl. a GAPHD (humán
gliceraldehid-3-foszfát dehidrogenáz) cDNS szegmentek korrigált intenzitásai
összehasonlíthatók [Cavaco és mtsai 2002].
Azokban a családokban, ahol a betegség genetikai hátterét nem sikerül
felderíteni, a mutáció-hordozókra vonatkozó protokoll szerint a klinikai követéses
vizsgálatok alkalmazása ajánlott minden családtag esetében, akik örökölhették a
mutációt.

34
A családszűrés jelentősége
A „pre-screening” módszerek és a direkt szekvenálás családszűrésre is
alkalmazhatók. Ezeken kívül családszűrésre az alábbiakban felsorolt módszerek is
rendelkezésre állnak, melyek alkalmazásához a mutáció előzetes ismerete szükséges.
Az allél-specifikus oligonukleotid hibridizáció (ASO) alapja, hogy a PCR
termékhez allél-specifikus oligonukleotidokat hibridizálunk dot blot (a PCR terméket
blottoljuk a membránon) vagy reverz dot blot módszerrel (számos különböző
oligonukleotid-blot a membránon). Napjainkban leginkább e módszerek mikrotiter
formáit használják.
Allél-specifikus amplifikáció esetén a PCR reakcióban allél specifikus
primereket használunk; a mutáció jelenlétét vagy hiányát a PCR termék képződése vagy
hiánya jelzi.
A családszűrés leggyakrabban használt módszere a restrikciós fragmenthossz
polimorfizmusok vizsgálata (RFLP). Azokban az esetekben bizonyult nagyon
hasznosnak, ahol a genetikai eltérés valamely restrikciós enzim hasítási helyét
változtatja meg. A módszer könnyen kivitelezhető, költséghatékony és automatizálható.
A MEN1 gén területén található mutációk igen nagy számúak, a gén bármely
szakaszán kialakulhatnak. Ha a család klinikai és genetikai szűrése sem tudja feltárni a
betegségben szenvedő egyénben kimutatott mutáció eredetét, de novo mutáció
lehetőségére kell gondolnunk. Ha egy mutáció több olyan családban is fellelhető,
melyek rokonsági kapcsolata nem ismert, haplotípus analízist végezhetünk 11q13
markerekkel az esetleges közös eredet (”founder effect”) felderítésére (pl. D11S956,
D11S1765, D11S480, D11S457, D11S449, D11S913, D11S987 és INT2-telomer
markererrel) [Mutch és mtsai 1999, Agarwal és mtsai 1997].
A genetikai vizsgálat egyik legfőbb haszna, hogy MEN 1 szindrómás családok
tagjaiban a mutáció-hordozó állapot kizárása feleslegessé teszi a további klinikai
szűrővizsgálatokat, míg a mutáció-hordozó állapot genetikai igazolása segítheti a
potenciálisan kialakuló daganatok korai klinikai szűrését. A családszűrésen kívül a
genetikai szűrővizsgálat olyan esetekben is hasznos, amikor klinikai vizsgálatokkal a
betegben csak egyetlen manifesztáció mutatható ki, de bizonyos körülmények a MEN 1

35
szindróma gyanúját vetik fel (pl. familiáris izolált hyperparathyreosis, 30 év alatt 2 vagy
több mellékpajzsmirigy adenoma; recidiváló hyperparathyreosis; gastrinoma vagy
multiplex szigetsejtes daganat bármely életkorban) [Thakker 1998]. Genotípus-
fenotípus összefüggés nem ismert, ezért a mutációt hordozó egyénekben az összes
lehetséges manifesztáció rendszeres klinikai szűrése szükséges. A nemzetközi ajánlások
szerinti követéses vizsgálatokat az 5. táblázat foglalja össze.
5. táblázat: MEN1 gén-mutáció hordozó egyénekben endokrin daganatok
szűrésére javasolt tesztek és követéses vizsgálatok [Dean és mtsai 2000]
Tumor A szűrés kezdetének
javasolt időpontja
Évente javasolt
biokémiai tesztek
3 évente javasolt
képalkotó
vizsgálatok
Mp. ad. 8 éves kor Szérum kalcium,
parathormon -
Gastr. 20 éves kor Szérum gastrin -
Ins. 5 éves kor Vércukor, szérum
insulin -
Egyéb ent.
pancr. tu. 20 éves kor
Tüneteknek
megfelelő hormonok CT vagy MRI
Hypoph. tu. 5 éves kor Szérum prolactin MRI
Carcinoid 20 éves kor - CT

36
Szomatikus genetikai eltérések MEN 1-asszociált tumorokban
A MEN1 gén mutációk nagy része korai fehérje-rövidülést eredményez, és
gyakran okoz gén inaktivációt (Knudson hipotézis szerinti első csapás) [Thakker 1993,
Gou és Sawicki 2001, Agarwal és mtsai 1997, Agarwal és mtsai 2004]. A második
csapás az érintett szövetekben jön létre, általában a vad típusú allél elvesztése miatt
(LOH). MEN 1-es betegek tumorszövetéből származó DNS mintákban az allélvesztés
vizsgálatára a 11q13 szakaszon proximálisan (cen-PYGM, D11S449, D11S913-qter),
vagy intragenikusan (D11S4946) elhelyezkedő polimorf microsatellita lókuszok
használhatók [Teh és mtsai 1998, Cavaco és mtsai 2002].

37
II. CÉLKITŰZÉSEK
A MEN 1 szindróma genetikai vizsgálatát a Semmelweis Egyetem ÁOK II. sz.
Belgyógyászati Klinika és az Élettani Intézet együttműködésével hazánkban elsőként,
2001. szeptemberében vezettük be. Munkámban 32 MEN 1 szindrómás vagy MEN 1-
szerű állapotban szenvedő beteg és családtagjaik klinikai és genetikai vizsgálatával az
alábbi célok megvalósítását tűztem ki:
(1) A MEN1 gén mutációk vizsgálatát számos körülmény nehezíti, melyek
elsősorban a gén nagy méretével és a mutációs ”hot-spot” hiányával
állnak összefüggésben. Ezért munkámban célul tűztem ki az
irodalomban közölt genetikai ”pre-screening” módszerek előnyeinek és
hátrányainak szisztematikus elemzését követően egy megbízható,
könnyen reprodukálható, idő- és költségkímélő szűrőmódszer
bevezetését a MEN1 gén kódoló régióiban előforduló rendellenességek
kimutatásának elősegítésére.
(2) Célkitűzéseim közé tartozott a szűrőmódszerként bevezetett
hőmérsékletgrádiens elektroforézis rendszerek optimális technikai
paramétereinek kidolgozása és az ily módon optimalizált ”pre-
screening” módszerek specificitásának és szenzitivitásának
meghatározása.
(3) A MEN 1 szindrómás és MEN 1-szerű állapotban szenvedő betegekben
a ”pre-screening” vizsgálatot követően a MEN1 gén kódoló szakaszain
előforduló mutációk direkt DNS szekvenálással történő kimutatásával
elemezni kívántam a magyarországi betegek mutációs spektrumát és a
mutációknak az irodalomban közölt adatokhoz való viszonyát.
(4) A MEN 1 szindrómában és MEN 1-szerű állapotban szenvedő
betegekben tanulmányozni kívántam, hogy van-e összefüggés a MEN 1
szindróma tüneteinek száma, típusa, vagy egyéb fenotípus jellemzők és a
MEN1 mutációanalízis hatékonysága között.
(5) MEN1 gén mutációhordozó egyének vérrokon családtagjainak genetikai
szűrésével tanulmányozni kívántam a genetikai vizsgálatra alapozott
családszűrés közvetlen jelentőségét és klinikai értékét.

38
III. AZ ÁLTALUNK VIZSGÁLT BETEGEK
2001 szeptembere óta összesen 32 MEN 1 szindrómás vagy MEN 1-szerű
állapotban szenvedő beteget vizsgáltunk. A betegeket kezelőorvosaik és vagy genetikai
tanácsadáson tájékoztatták a genetikai vizsgálatról és beleegyező nyilatkozatot írtak alá.
A betegek között összesen 19 beteg (59.3%) volt klinikailag biztosan MEN 1
szindrómás, 1 beteg (3.1%) familiáris izolált hyperparathyreosisban szenvedett és 12
betegben (37.5%), klinikailag MEN 1-hez hasonló, MEN 1-szerű állapotot találtunk.
MEN 1–szerű állapot esetében, legalább egy fő és egy nem fő komponens volt
jelen; ebbe soroltuk azokat a betegeket is, akiknél 30 éves kor előtt kialakult multiplex
mellékpajzsmirigy tumorban, vagy rekurrens hyperparathyreosisban vagy familiáris
izolált hyperparathyreosisban szenvedtek. A vizsgált 32 beteg közül 19 MEN 1
szindrómás volt (6 familiáris, 13 sporadikus) és 13 beteg MEN 1-szerű állapotban
szenvedett ( 2 familiáris és 11 sporadikus).
A betegek tüneteit a 6. táblázat foglalja össze; a klinikailag biztosan MEN 1
szindrómás betegek közül 2 betegben találták meg mind a három major léziót (6.25%),
míg 17 betegben (53.13%) a vizsgálatkor két major elváltozás fordult elő. A MEN 1-
szerű állapotban szenvedők közül 2 betegben fiatalkori sporadikus MEN 1 tumor
(6.25%), 2 betegben egyféle MEN 1 daganat mellett a családi anamnézis (6.25%), 9
esetben (28.13%) pedig egy MEN 1 major lézió mellett valamely ismert minor lézió
megjelenése vetette fel a betegség gyanúját.
6. táblázat: MEN 1 daganatok az általunk vizsgált betegekben és a genetikai
vizsgálat eredménye
Indexeset száma
Hyperpara-thyreosis
Pancreas neuroendocrine
tumor
Agyalapi mirigy tumor
Minor MEN 1 lézió A genetikai
vizsgálat eredménye
Familiáris MEN 1 Betegek 2 fő MEN 1 lézióval
1. + + - Mv kéregadenoma Q209X 2. + - + - Nt657insC 3. + - + - A91V 4. + - + - L301R 5. + - + - Nt359del4bp

39
6. + - + - E26X
Sporadikus MEN 1 Betegek 3 fő MEN 1 lézióval
7. + + + - negatív 8. + + + - Nt317ins5bp
Betegek 2 fő MEN 1 lézióval
9. + - + Mv kéregadenoma Negatív 10. - + + - Nt738del4bp 11. + - + - Negatív 12. + - + Mv kéregadenoma Negatív 13. - + + - Negatív 14. + + - Hasfali lipoma C354X 15. + (rekurrens) + - - Negatív 16. + - + - Negatív 17. - + + - Negatív 18. + - + - Negatív 19. + - + - Negatív
Familiáris MEN 1-szerű állapot Betegek 1 fő MEN 1 lézióval
20. + (familiáris) - - - Negatív 21. - + (multiplex) - - Negatív
Sporadikus MEN 1-szerű állapot Betegek 1 fő MEN 1 lézióval
22. + (fiatal kori) - - - G28A 23. - - + Mv kéregadenoma Negatív 24. - + - Mv kéregadenoma Negatív 25. + (fiatal kori) - - - Negatív 26. + - - Mv kéreg carcinoma Negatív 27. - - + Kétoldali mv
kéregadenoma Negatív
28. + - - Carotis glomus tumor Negatív 29. + - - Mv kéregadenoma Negatív 30. + - - Mv angiomyolipoma Negatív 31. + - - Mv kéregadenoma Negatív 32. - - + Mv kéregadenoma Negatív
+: a kezelőorvos a dokumentációban leírta; -: a kezelőorvos a dokumentációban
nem írta le; mv: mellékvese.
A 32 beteg közül primer hyperparathyreosis 24 betegben (75%), pancreas
neuroendokrin daganat 10 betegben (31.25%) és hypophysis daganat 19 betegben

40
(59.38%) fordult elő. A minor léziók közül a leggyakoribb a mellékvesekéreg adenoma
volt (9 betegben: 28.13%). A mutáció azonosítása esetén a genetikai vizsgálatokat a
vérrokon családtagokban is elvégeztük.

41
IV. MÓDSZEREK
DNS-izolálás
EDTA-val vagy citráttal alvadásgátolt teljes perifériás vérből a kereskedelemben
forgalmazott kit-tek segítségével (DNA Isolation Kit for Mammalian Blood, Boehringer
Mannheim, Németország) genomiális DNS-t izoláltam. Az DNS törzsoldatokat a
felhasználásig –80 ˚C-on, a higított mintákat –20 ˚C-on tároltam.
PCR reakciók
A genomiális DNS-ből a MEN1 gén exonjait polimeráz láncreakcióval (PCR)
amplifikáltam. Minden PCR reakcióhoz 200-400 ng DNS-t használtam templát DNS-
ként 50 µl végtérfogatban. A reakció összetétele az alábbi volt: 15 és 35 pmol GC-
clamppel kiegészített és hagyományos (GC-clamp nélküli) oligonukleotid primer,
10mM Tris-HCl, 2,5mM MgCl2, 50mM KCl, 0.2 mmol/l dezoxinukleotid trifoszfát, 2 U
Taq Polymerase (Sigma-Aldrich Corp., Saint Louis, Missouri, USA) és 5% glicerin.
TGGE-vel történő előszűréshez minden primerpár esetében az egyik egy 40 bázis
hosszúságú GC-gazdag régiót (GC-clampet) tartalmazott a futtatás során a teljes
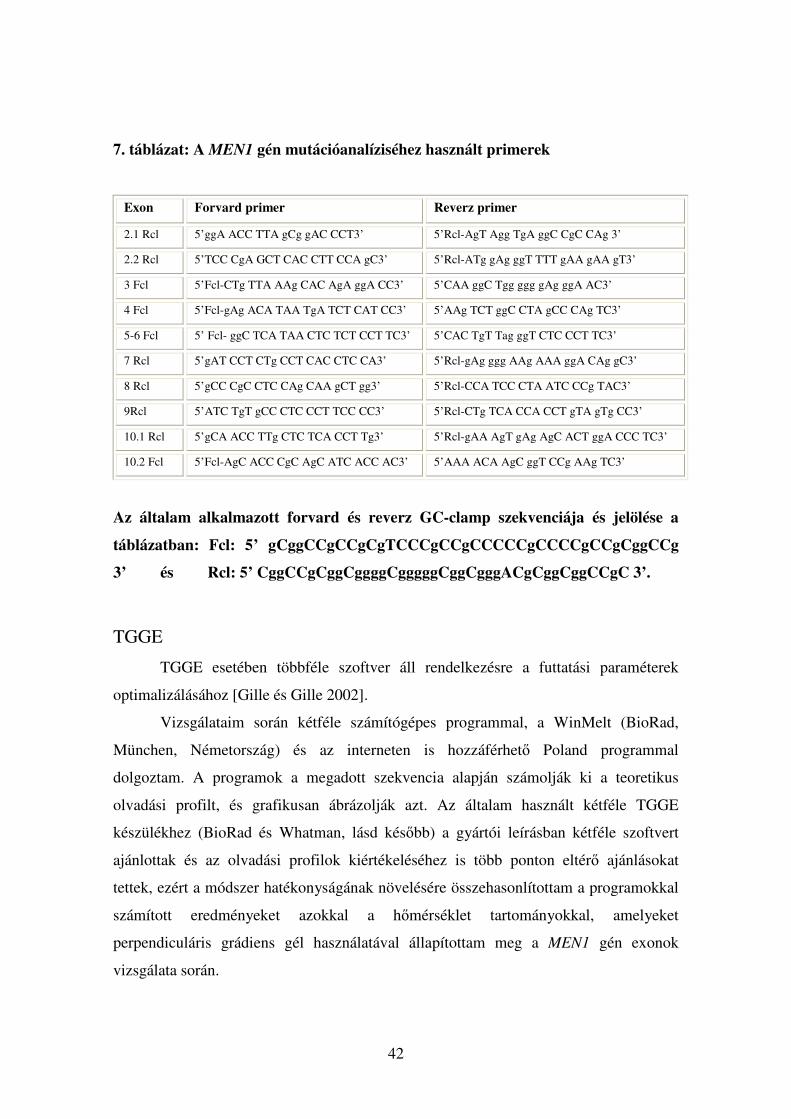
denaturáció megelőzésére. A primereket Morelli és mtsai munkája alapján terveztem
[Morelli és mtsai 2000], a módszer optimalizálásához szükséges minimális
változtatásokkal (7. táblázat). Szekvenáláshoz ugyanezeket a primereket használtam
GC-clamp nélkül, de M13-véggel kiegészítve (lásd később).
A PCR reakció az alábbi lépéseket tartalmazta: kezdeti denaturáció 95 ˚C-on 5
percig, melyet 95 ˚C-on 1 perc denaturáció, 55-65 ˚C-on (a primernek megfelelő
speciális „annealing” hőmérséklet) 1,5 perc anellálás és 72 ˚C-on 1,5 perc elongáció
követett 35 ciklusban. A végső extenzió 72 ˚C-on 5 percig tartott. E hagyományos PCR
protokoll után a TGGE vizsgálathoz heteroduplex képzésre is szükség volt, melyhez az
alábbi módszert választottam: denaturáció 95 ˚C-on 5 percig, majd inkubáció szobahőn
1 órán át [Morelli és mtsai 2000]. A mintákat a mutáció-analízisig -20 ˚C-on tároltam. A
szekvenáláshoz használt PCR termékek esetében nem volt szükség heteroduplex
képzésre, viszont tisztításra igen, amit High Pure PCR Product kit-tel (Roche
Diagnostics, Mannheim, Germany) végeztem.
42
7. táblázat: A MEN1 gén mutációanalíziséhez használt primerek
Exon Forvard primer Reverz primer
2.1 Rcl 5’ggA ACC TTA gCg gAC CCT3’ 5’Rcl-AgT Agg TgA ggC CgC CAg 3’
2.2 Rcl 5’TCC CgA GCT CAC CTT CCA gC3’ 5’Rcl-ATg gAg ggT TTT gAA gAA gT3’
3 Fcl 5’Fcl-CTg TTA AAg CAC AgA ggA CC3’ 5’CAA ggC Tgg ggg gAg ggA AC3’
4 Fcl 5’Fcl-gAg ACA TAA TgA TCT CAT CC3’ 5’AAg TCT ggC CTA gCC CAg TC3’
5-6 Fcl 5’ Fcl- ggC TCA TAA CTC TCT CCT TC3’ 5’CAC TgT Tag ggT CTC CCT TC3’
7 Rcl 5’gAT CCT CTg CCT CAC CTC CA3’ 5’Rcl-gAg ggg AAg AAA ggA CAg gC3’
8 Rcl 5’gCC CgC CTC CAg CAA gCT gg3’ 5’Rcl-CCA TCC CTA ATC CCg TAC3’
9Rcl 5’ATC TgT gCC CTC CCT TCC CC3’ 5’Rcl-CTg TCA CCA CCT gTA gTg CC3’
10.1 Rcl 5’gCA ACC TTg CTC TCA CCT Tg3’ 5’Rcl-gAA AgT gAg AgC ACT ggA CCC TC3’
10.2 Fcl 5’Fcl-AgC ACC CgC AgC ATC ACC AC3’ 5’AAA ACA AgC ggT CCg AAg TC3’
Az általam alkalmazott forvard és reverz GC-clamp szekvenciája és jelölése a
táblázatban: Fcl: 5’ gCggCCgCCgCgTCCCgCCgCCCCCgCCCCgCCgCggCCg
3’ és Rcl: 5’ CggCCgCggCggggCgggggCggCgggACgCggCggCCgC 3’.
TGGE
TGGE esetében többféle szoftver áll rendelkezésre a futtatási paraméterek
optimalizálásához [Gille és Gille 2002].
Vizsgálataim során kétféle számítógépes programmal, a WinMelt (BioRad,
München, Németország) és az interneten is hozzáférhető Poland programmal
dolgoztam. A programok a megadott szekvencia alapján számolják ki a teoretikus
olvadási profilt, és grafikusan ábrázolják azt. Az általam használt kétféle TGGE
készülékhez (BioRad és Whatman, lásd később) a gyártói leírásban kétféle szoftvert
ajánlottak és az olvadási profilok kiértékeléséhez is több ponton eltérő ajánlásokat
tettek, ezért a módszer hatékonyságának növelésére összehasonlítottam a programokkal
számított eredményeket azokkal a hőmérséklet tartományokkal, amelyeket
perpendiculáris grádiens gél használatával állapítottam meg a MEN1 gén exonok
vizsgálata során.

43
DNS-szekvenálás
A TGGE-vel lokalizált genetikai eltéréseket automata szekvenálással
azonosítottuk. A DNS mintákat tisztítás után Big Dye Terminator Cycle-Sequencing kit
segítségével (Applied Biosystems, Foster City, CA, USA) ABI Prism 310 automata
szekvenálóval (Genetic analyser, Applied Biosystems) vagy Cycle-sequencing kit with
7-deaza (Pharmacia Biotech, Uppsala, Svédország) segítségével LiCOR IR2 (Li-COR
Inc., Lincoln, USA) készülékkel szekvenáltuk. Utóbbi esetben a módszer M13-véggel
kiegészített primereket igényelt, melyek szekvenciája forward primer esetében 5’-3’
irányban ”CAC GAC GTT GTA AAA CGA C” szakasszal, reverse primer esetében
pedig 5’-3’ irányban ”GGA TAA CAA TTT CAC ACAGG” szakasszal bővült.

44
V. EREDMÉNYEK
A TGGE paramétereinek optimalizálása
A 8. táblázat adataiból látható, hogy ugyanazon DNS szekvencia esetében a
WinMelt program általában alacsonyabb hőmérséklet értékeket ad meg a Poland
programhoz képest, és mindkét program esetében a számított értékek különböznek a
tapasztalati úton, perpendiculáris futtatás segítségével nyert értékektől. Perpendiculáris
futtatás során egyetlen PCR-termék denaturációját követhetjük végig: a futtatás iránya
és a hőmérséklet grádiens egymásra merőlegesek, így megfigyelhető, hogy az adott
minta a gélben különböző hőmérsékleteken hogyan viselkedik. Láthatóvá válik festés
után, melyik hőmérsékleti ponton kezdődött a denaturáció és milyen hőmérsékleten
válik szét teljesen, azaz két külön szálra a DNS. Itt térnék ki ismét a GC-clampek
alkalmazásának jelentőségére: ha a PCR reakcióban az általunk amlifikált termék egyik
végére egy GC-gazdag régiót készíttetünk a Taq polimerázzal (előre megtervezett GC-
clampes primerek segítségével), a keletkezett PCR-termék azon része, amely ezt a
régiót tartlmazza, sokkal magasabb hőmérsékleti tatományokban fog denaturálódni. Így
az egész fragmens olvadási profilja megváltozik, jobban vizsgálhatóvá válik, azáltal,
hogy nem egy hőmérsékleti ponton, hirtelen egyszálú DNS-re esik szét, hanem lassan,
fokozatosan nyílik fel a DNS kettős spirál, ezáltal az egészen kis területet érintő,
egyetlen nukleotidot érintő mismatch-ek is kimutathatóvá válnak. Ez alapján érthető,
hogy a módszer specificitása és szenzitivitása a megfelelő primer tervezéstől és az
optimális hőmérsékleti tartományoktól kritikus mértékben függ.
Mivel egy adott DNS-szakasz denaturálási tulajdonságai nem csak a
szekvenciától, hanem a DNS koncentrációjától, az alkalmazott pufferek
ionkoncentrációjától is függ, szoftverek segítségével igen nehéz az adott futtatás precíz
körülményeinek in silico modellezése. Sokkal megbízhatóbbnak és
reprodukálhatóbbnak bizonyultak a perpendiculáris futtatás által „mérhető”
(kiszámítható) hőmérsékleti tartományok, mivel itt egy általunk beállított hőmérsékleti
grádiens az általunk használt körülmények (pl. só- és DNS koncentráció) mellett teszi
lehetővé, hogy a hőmérsékleti skálát a denaturációs profilhoz rendelve megállapítsuk a
denaturáció hőmérsékleti kezdő- és vágpontját. Vizsgálataim során azt tapasztaltam,
hogy a perpendiculáris elektroforézis segítségével kiszámított futtatási paraméterek

45
alkalmazásával kiküszöbölhetők a genetikai eltérés kimutatását nehezítő ”zavaros
sávok”-ból eredő problémák, melyeket a WinMelt programmal számított paraméterek
alkalmazásakor több exon esetében tapasztaltunk.
8. táblázat: A MEN1 gén TGGE-vel történő genetikai szűréséhez használt
módszerek hőmérsékleti tartományainak összehasonlítása
Magyarázat a táblázat rövidítéseihez:
Rcl: reverz primer GC-clamppel, Fcl: forvard primer GC-clamppel a PCR
reakcióban; „-„: az adott exon területén nem találtunk mutációt. (+): TGGE-vel
kimutatott mutáció; (-): TGGE-vel mutációt nem sikerült kimutatni.
Eredményeinket direkt szekvenálással ellenőriztük. *Bio-Rad, München,
Germany; **Ref. [Steger 1994]

46
nt1657insC (-)52-56°°°°C59-74°°°°C75-90°CZavaros band-ek58-68°°°°C74-84°CExon 10 Fcl
D418D (+)L432L (+)
60-67°°°°C29-74°°°°C45-90°CD418D (+)L432L (+)
40-67°°°°C56-83°CExon 9 Rcl
C354X (+)60,4-67,2°°°°C46-71°°°°C62-87°CC354X (+)35-65°°°°C51-81°CExon 8 Rcl
-50,2-53,6°°°°C32-74°°°°C48-90°CZavaros band-ek32-64°°°°C48-80°CExon 7 Rcl
-48,4-55,6°°°°C46-74°°°°C62-90°C-52-64°°°°C68-80°CExon 5-6 Fcl
-48,4-57,4°°°°C36-70°°°°C52-86°C-52-64°°°°C68-80°CExon 4 Fcl
nt738del4bp (+)Q209R (+)R171Q (+)
53-60,5°°°°C44-74°°°°C60-90°Cnt738del4bp (+)
Q209R (+)R171Q (+)
46-64°°°°C62-80°CExon 3 Fcl
nt317ins5bp54,8-66°°°°C53-74°°°°C69-90°C-58-63°°°°C74-79°CExon 2.2 Rcl
-46-58°°°°C49-75°°°°C65-91°CZavaros band-ek54-68°°°°C70-84°CExon 2.1 Rcl
Tm perpendicular electroforésis segítségével meghatározva, 8M urea használatával, a szűrés eredményei
Tm Poland software-rel** számolva, 8M
ureával(-16°°°°C)
Tm Poland program-mal
számolva
Tm by Winmelt software-rel*Számítva, 8M urea (-16°°°°C)
használatával, a szűrés eredményei
Tm WinMeltSoftware-rel
számítvaExon
nt1657insC (-)52-56°°°°C59-74°°°°C75-90°CZavaros band-ek58-68°°°°C74-84°CExon 10 Fcl
D418D (+)L432L (+)
60-67°°°°C29-74°°°°C45-90°CD418D (+)L432L (+)
40-67°°°°C56-83°CExon 9 Rcl
C354X (+)60,4-67,2°°°°C46-71°°°°C62-87°CC354X (+)35-65°°°°C51-81°CExon 8 Rcl
-50,2-53,6°°°°C32-74°°°°C48-90°CZavaros band-ek32-64°°°°C48-80°CExon 7 Rcl
-48,4-55,6°°°°C46-74°°°°C62-90°C-52-64°°°°C68-80°CExon 5-6 Fcl
-48,4-57,4°°°°C36-70°°°°C52-86°C-52-64°°°°C68-80°CExon 4 Fcl
nt738del4bp (+)Q209R (+)R171Q (+)
53-60,5°°°°C44-74°°°°C60-90°Cnt738del4bp (+)
Q209R (+)R171Q (+)
46-64°°°°C62-80°CExon 3 Fcl
nt317ins5bp54,8-66°°°°C53-74°°°°C69-90°C-58-63°°°°C74-79°CExon 2.2 Rcl
-46-58°°°°C49-75°°°°C65-91°CZavaros band-ek54-68°°°°C70-84°CExon 2.1 Rcl
Tm perpendicular electroforésis segítségével meghatározva, 8M urea használatával, a szűrés eredményei
Tm Poland software-rel** számolva, 8M
ureával(-16°°°°C)
Tm Poland program-mal
számolva
Tm by Winmelt software-rel*Számítva, 8M urea (-16°°°°C)
használatával, a szűrés eredményei
Tm WinMeltSoftware-rel
számítvaExon
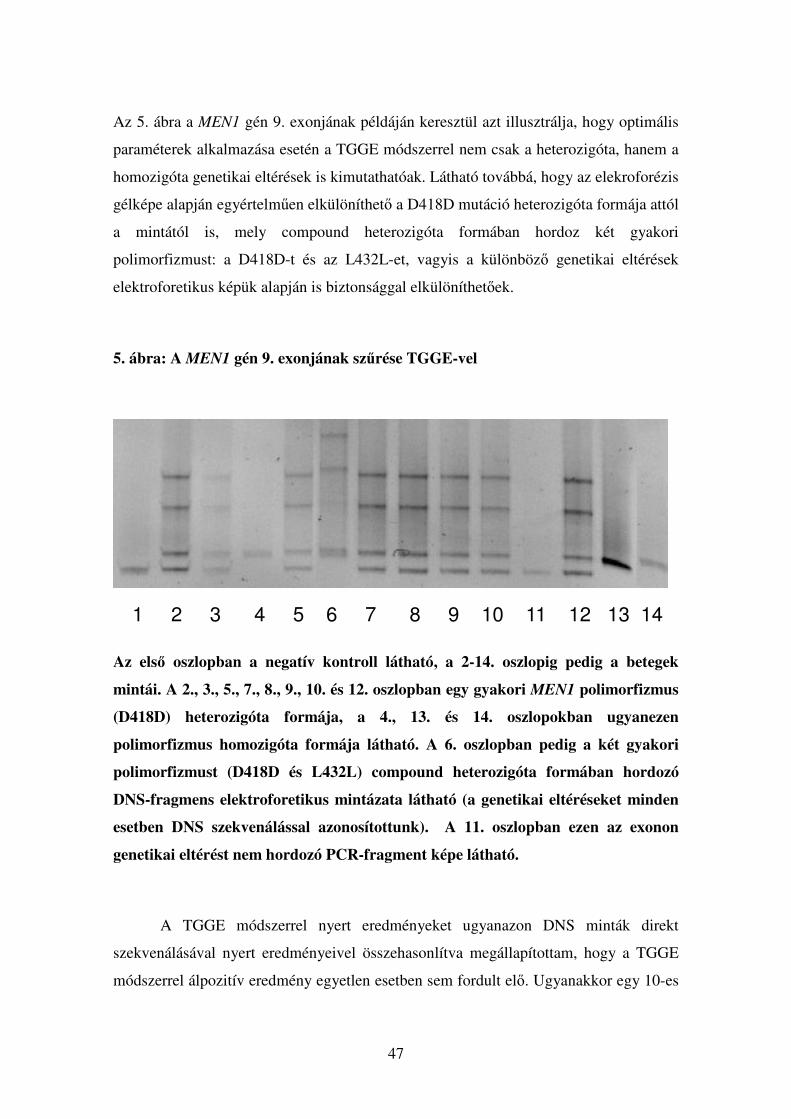
47
Az 5. ábra a MEN1 gén 9. exonjának példáján keresztül azt illusztrálja, hogy optimális
paraméterek alkalmazása esetén a TGGE módszerrel nem csak a heterozigóta, hanem a
homozigóta genetikai eltérések is kimutathatóak. Látható továbbá, hogy az elekroforézis
gélképe alapján egyértelműen elkülöníthető a D418D mutáció heterozigóta formája attól
a mintától is, mely compound heterozigóta formában hordoz két gyakori
polimorfizmust: a D418D-t és az L432L-et, vagyis a különböző genetikai eltérések
elektroforetikus képük alapján is biztonsággal elkülöníthetőek.
5. ábra: A MEN1 gén 9. exonjának szűrése TGGE-vel
Az első oszlopban a negatív kontroll látható, a 2-14. oszlopig pedig a betegek
mintái. A 2., 3., 5., 7., 8., 9., 10. és 12. oszlopban egy gyakori MEN1 polimorfizmus
(D418D) heterozigóta formája, a 4., 13. és 14. oszlopokban ugyanezen
polimorfizmus homozigóta formája látható. A 6. oszlopban pedig a két gyakori
polimorfizmust (D418D és L432L) compound heterozigóta formában hordozó
DNS-fragmens elektroforetikus mintázata látható (a genetikai eltéréseket minden
esetben DNS szekvenálással azonosítottunk). A 11. oszlopban ezen az exonon
genetikai eltérést nem hordozó PCR-fragment képe látható.
A TGGE módszerrel nyert eredményeket ugyanazon DNS minták direkt
szekvenálásával nyert eredményeivel összehasonlítva megállapítottam, hogy a TGGE
módszerrel álpozitív eredmény egyetlen esetben sem fordult elő. Ugyanakkor egy 10-es
1 2 3 4 5 6 7 8 9 10 11 12 13 14

48
exonon található mutációt (nt1657insC) nem sikerült kimutatnom sem TGGE-vel, sem
heteroduplex analízissel annak ellenére, hogy a két program által számított
paramétereken kívül a perpendiculáris futtatás során kapott paramétereket is használtam
(a mutációt végül direkt szekvenálással azonosítottam), több DNS mintából is
megpróbáltam, minden családtagnál, akik az adott mutációt örökölték. Ezen álnegatív
eredmény okát felderíteni nem sikerült. Megemlíthető, hogy a 10-es exonon a TGGE
módszerrel álnegatívnak bizonyuló mutációt Bassett és mtsai SSCP-vel ki tudták
mutatni [Bassett és mtsai 1998].
Egy másik szűrőprogramban, ahol WinMelt-TGGE paramétereket használtak, az
összes mutációt sikerült lokalizálni, azonban 4 esetben álpozitív eredményt kaptak
[Wrocklage és mtsai 2002], feltehetően a kétszálú DNS teljes denaturálása miatt. A két
számítógépes program között azonban nem volt különbség az adott DNS szakaszon
meghatározott melting domének és a GC-clampek helyzetének kiszámításában (6. ábra).
Összefoglalva tehát, saját tapasztalataim megerősítik, hogy a TGGE módszer
sikeres alkalmazásához jelentős segítséget nyújt a PCR primerek szoftverrel történő
megtervezése és az ideális hőmérsékletgrádiens kiszámítása. A jelenleg elérhető
szoftverek használata segítheti annak eldöntését, hogy a forward vagy a reverse primer
tartalmazza-e a GC-clampet. A szofverekkel modellezhető, hogy a keletkező PCR
fragmens 3’ vagy 5’ végére érdemes a GC-gazdag régiót illeszteni: TGGE-vel történő
elektroforézishez az a termék ideális, melyben az egymást követő melting domének
egyre magasabb hőmérsékleti tartományokban denaturálnak, és a legmagasabb
denaturációs hőmérséklet a GC-gazdag régió „olvadásához” szükséges. Így biztosítható,
hogy az általunk vizsgált exon a GC-gazdag régióval elletétes végén kezdjen „felnyílni”
és ez a faág-szerű denaturáció folyamatos legyen a termék teljes hosszában.
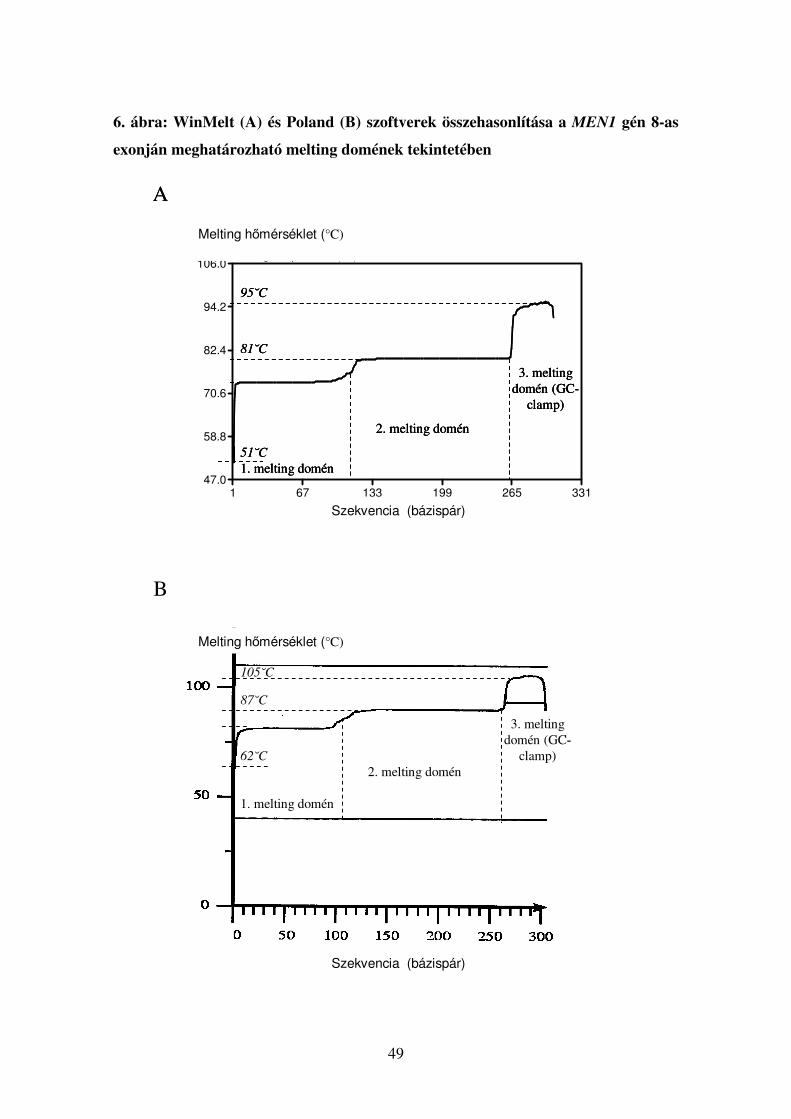
49
6. ábra: WinMelt (A) és Poland (B) szoftverek összehasonlítása a MEN1 gén 8-as
exonján meghatározható melting domének tekintetében
1 67 133 199 265 33147.0
58.8
70.6
82.4
94.2
106.0
Sequence position (bp)
Melting temperature (°C)
MNexon8Rcl
A
1. melting domén
2. melting domén
3. meltingdomén (GC-
clamp)62˘C
87˘C
105˘C
95˘C
81˘C
51˘C
1. melting domén
2. melting domén
3. meltingdomén (GC-
clamp)
Melting hőmérséklet (°C)
Szekvencia (bázispár)
Melting hőmérséklet (°C)
Szekvencia (bázispár)
B
1 67 133 199 265 33147.0
58.8
70.6
82.4
94.2
106.0
Sequence position (bp)
Melting temperature (°C)
MNexon8Rcl
A
1. melting domén
2. melting domén
3. meltingdomén (GC-
clamp)62˘C
87˘C
105˘C
95˘C
81˘C
51˘C
1. melting domén
2. melting domén
3. meltingdomén (GC-
clamp)
Melting hőmérséklet (°C)
Szekvencia (bázispár)
95˘C
81˘C
51˘C
1. melting domén
2. melting domén
3. meltingdomén (GC-
clamp)
Melting hőmérséklet (°C)
Szekvencia (bázispár)
Melting hőmérséklet (°C)
Szekvencia (bázispár)
B

50
A korrekt denaturációs hőmérséklet-tartomány meghatározásához, a teljes
denaturáció, vagy a mutáció detektálási szempontból érdektelen alacsony hőmérsékletek
elkerüléséhez azonban hasznosabbnak tűnik a perpendiculáris elektroforézis használata.
(7. ábra).
7. ábra: A futtatási paraméterek meghatározása perpendiculáris electrophoresis
segítségével [Balogh és mtsai 2004]
34
˘C7
0˘C
52°C 56°C
T1 T2
Lineáris grádienssel emelkedő hőmérséklet
ele
ktr
ofo
rézis
34
˘C7
0˘C
52°C 56°C
T1 T2
Lineáris grádienssel emelkedő hőmérséklet
ele
ktr
ofo
rézis
Kis hőmérsékleten (T1 alatt) a DNS kétszálú molekulaként vándorol; közepes
hőmérsékleten (T1 és T2 között) a DNS egyik oldalán denaturálódás kezdődik (a
másik oldalon a GC-clamp összetartja a két szálat) és a részlegesen denaturált
DNS fokozatosan lelassul. Nagy hőmérsékleten (T2 felett) a DNS irreverzibilisen 2
külön szálra bomlik. A minták analízise csak a T1 és T2 hőmérsékletek között
értékelhető. (Ha a paramétereket WinMelt vagy Poland szoftverekkel számoljuk,
általában T2 feletti hőmérséklet tartományokban futtatjuk a DNS-t).
Munkám során két különböző mutáció detektáló rendszerrel dolgoztam.
Elsőként a DCode Universal Mutation Detection System (Bio-Rad, München,
Németország) készüléket használtam (8. ábra).

51
8. ábra: DCode Universal Mutation Detection System (Bio-Rad, München,
Németország)
A DCode Universal Mutation Detection System készüléken a puffer
hőmérsékletének növelésével érhető el az elekroforézis alatt a gél és a benne lévő DNS
minták melegítése. A futtatási paramétereket kezdetben a készülékhez ajánlott WinMelt
szoftverrel számoltam. Munkám során azonban többször találkoztam értékelési
nehézségeket okozó ”zavaros band”-ekkel.
Az általam használt másik készülék a TGGE standard system (Whatman
Biometra, Göttingen, Németország) volt (9. ábra), melyhez a forgalmazó a Poland
szoftver használatát ajánlotta, azonban az optimális hőmérséklet tartományok
beállításához perpendiculáris futtatásokat is végeztem. A TGGE standard system
készülék egy száraz rendszerű elektroforézis módszert alkalmaz; a gél polybond filmen
keresztül egy teflon bevonatú, szabályozható hőmérsékletű felületen fekszik, melyre az
akrilamid kovalens kötéssel kötődik. A gél két oldalán található pufferterek közötti
összeköttetést szűrőpapírok biztosítják. A készülék lehetővé teszi a gél hőmérsékletének
közvetlen szabályozását, és ezáltal a minták hőmérsékletének pontosabb beállítását.

52
9. ábra: TGGE standard system (Whatman Biometra, Göttingen, Németország)
Az elekroforézis mindkét készüléken 8%-os acrylamid/bisacrylamid (37.5:1)
gélen történik, 8 M urea jelenlétében (az urea csökkenti a DNS olvadási hőmérsékletét),
1xTAE pufferben. A két rendszer közötti különbséget a 9. táblázat foglalja össze.
9. táblázat: A két TGGE rendszer összehasonlítása
DCode (BioRad) TGGE standard system
(Whatman Biometra)
Gélösszetétel
8% acrylamid/bisacryl-
amid, 8M urea
8% acrylamid/bisacryl-
amid, 8M urea,
2% glicerin
Gél mérete 16 cm x 16 cm 8 cm x 8 cm
Gélen vizsgálható minták
száma 16 12
Elektroforézis típusa nedves száraz
Melegítés puffer gél
Szükséges puffermennyiség 7 l/futtatás 0,5 l/futtatás
PCR termék mennyisége 8 µl 1,5-2,5 µl
Futtatási idő 6-9 óra 50 perc
Feszültség 140V 250V
Festés Ethidium bromiddal Ezüst nitráttal

53
Az elektroforézis után a DCode (BioRad) rendszer esetében ethidium bromid
festést követően Fluor-S MultiImager (Bio-Rad) készülékkel fényképet készítettem, a
TGGE standard system használatakor pedig ezüst nitráttal történő festés után
fényképeztem le a géleket.
ELŐSZŰRÉS ÉS SZEKVENÁLÁS
A TGGE módszerrel a 2-es exonon 4 esetben, a 3-as exonon 9 esetben, az 5-6-os
exonokat tartalmazó szakaszon 1 esetben, a 8-as exonon 1 esetben, a 9-es exonon pedig
18 esetben heterozigóta és 3 esetben homozigóta genetikai eltérést találtam. Az összes
DNS minta és a MEN1 gén összes kódoló exonja esetében szekvenálást is végeztem.
10. táblázat. MEN1 gén mutációk és polimorfizmusok az általunk vizsgált
betegekben
Mutáció Polimorfizmus
Exon 2 nt317ins5bp
A91V
G28A
nt359del4bp
E26X*
S145S
Exon 3 nt738del4bp
Q209X
R171Q
Exon 5-6 L301R -
Exon 8 C354X -
Exon 9 - D418D
L432L
Exon 10 nt1657insC** -
A vastag betűkkel jelölt mutációkat a nemzetközi irodalomban korábban még nem
írták le. *:PCR optimalizálási probléma miatt a TGGE analízis nem volt
kivitelezhető, **:TGGE-vel álnegatív eredmény

54
Összesen 10 probandban találtam betegség-okozó MEN1 gén mutációt (2
esetben deléció, 2 esetben inzerció, 3 esetben nonsense és 3 esetben missense mutáció).
A 10 mutáció közül 5 a nemzetközi irodalomban korábban még nem ismertetett, új
mutációnak felelt meg. A mutációkon kívül több ismert, betegséget nem okozó
polimorfizmust is azonosítottam (10. táblázat): heterozigóta formában S145S
polimorfizmust 1 betegben, heterozigóta R171Q polimorfizmust 7 betegben, hetero- és
homozigóta formában a D418D polimorfizmust 18, illetve 3 betegben, és heterozigóta
formában egy betegben az L432L polimorfizmust. Érdekességként említeném meg,
hogy az egyik betegünkben 3 különböző polimorfizmust (R171Q, L432L, D418D) is
találtunk a betegségokozó mutáció mellett (C354X).
A módszertani részben tárgyalt álnegatív eredményű TGGE vizsgálaton kívül
(10-es exon TGGE vizsgálat negatív, de direkt DNS szekvenálással inzerciós mutáció)
egy további esetben a 2-es exon vizsgálatakor GC-clampet tartalmazó primerekkel nem
sikerült kellő mennyiségű és tisztaságú PCR terméket előállítani a pre-screeninghez,
viszont DNS szekvenálással egy új, a nemzetközi irodalomban még nem közölt
nonsense mutációt találtam.
Családvizsgálattal két esetben bizonyítottam, hogy a betegség-okozó MEN1 gén
mutáció de novo alakult ki; további két esetben az egyik szülő korai halála miatt nem
sikerült a mutáció eredetét, esetleges de novo voltát megállapítani. Családszűrést azon
betegek elsőfokú rokonainál végeztem, akiknél sikerült azonosítani a betegség-okozó
mutációt, összesen 6 szimptómás és 15 aszimptomatikus első-fokú családtagnál. Egy
családban a mutációanalízis egy tünetmentes, fiatal gyermekben mutatta ki a mutáció
öröklését, a többi családtagnál a genetikai vizsgálat a klinikai képnek megfelelő
eredményt adott.
ESETBEMUTATÁS, CSALÁDSZŰRÉS
Az első, általunk vizsgált család vizsgálata egy atípusos MEN1 szindrómás
proband genetikai vizsgálatával kezdődött. A fiatal nő 25 éves korában a betegség első
tünete egy nagyméretű (11 cm-es), multiplex nem funkcionáló pancreas neuroendokrin
daganat volt, melyet distális pancreatectomiával távolítottak el (10. ábra). A műtét során
egy 3 cm-es hasfali lipoma is eltávolításra került.

55
10. ábra: MEN 1 szindrómás beteg pancreas neuroendokrin tumorának
makroszkópos és hisztológiai képe
1cm
a, b: HE festésc: synaptophysind: chromogranin Ae: neuron specifikus enolázf: inzulin immunhisztokémia
1cm
a, b: HE festésc: synaptophysind: chromogranin Ae: neuron specifikus enolázf: inzulin immunhisztokémia
A kép bal oldalán látható neuroendokrin pancreas daganatot egy MEN 1
szindrómás fiatal beteg operációja során distális pancreatectomiával távolították
el. A kép jobb oldala a hisztológiai és immunhisztokémiai vizsgálatok eredményeit
illusztrálja; a daganatban endocrin jellegű sejtekből álló nodulusok trabecularis
(100x) (a) és solid (200x) (b) szerkezete ismerhető fel hematoxilin-eosin festéssel. A
daganat synatophysin (400x) (c), chromogranin A (600x) (d) és neuron specifikus
enoláz (400x) (e) pozitivitást mutatott és egy kisméretű nodulusban insulin
jelenlétét is ki lehetett mutatni (400x) (f).
A beteg családi anamnézise ugyan nem utalt MEN 1 szindróma lehetőségére,
azonban a pancreas neuroendokrin daganat multiplex jellege és a társuló lipoma már
ekkor felvethette volna atípusos MEN 1 szindróma diagnózisát. A diagnózist azonban
csak 2 évvel később állapították meg, amikor a MEN 1 szindróma új tüneteként primer
hyperparathyreosist észleltek és mellékpajzsmirigy műtétet végeztek. A genetikai
vizsgálatok a probandban egy új, a nemzetközi irodalomban korábban nem ismertetett
nonsense MEN1 gén mutáció jelenlétét bizonyították (Q354X) (11./a, b ábra). Ez a
mutáció a kodonok szintjén egy glutamint kódoló bázistriplet stop kodonra cserélődését
eredményezi a 8-as exonon, ami egy rövidebb és feltehetően kóros funkciójú fehérje

56
képződésével jár. A betegség-okozó mutáción kívül több polimorfizmust is találtam;
egy arginint glutaminra cserélő polimorfizmust a 3-as exon (R171Q) és két, aminosavat
nem cserélő polimorfizmust a 9-es exon területén (L432L, D418D). A proband
édesanyja és testvére nem hordozták a betegség-okozó mutációt, ezért feltételezhető,
hogy a mutáció de novo keletkezett, vagy a proband ismeretlen okból korán elhunyt
édesapjától öröklődhetett. Az édesanya a probandban észlelt polimorfizmusok közül a
D418D-t heterozigóta formában hordozta, míg a beteg testvérében egyik genetikai
eltérés sem volt jelen (11./c, d ábra).
11. ábra: C354X mutáció szűrése TGGE módszerrel (a), direkt DNS
szekvenálással történő azonosítása (b), a családszűrés eredményei (c) és a
polimorfizmusok vizsgálata (d)
--
++--
a) b)
c)
C
R171Q
d)D418D
D418D, L432L
--
++--
--
++--
--
++--
--
++--
--
++--
a) b)
c)
C
R171Q
d)D418DD418D
D418D, L432L
Atípusos MEN 1 szindrómás beteg családfája: A MEN1 gén mutáció jelenlétét a
ferde vonalakkal kitöltött szimbólum jelzi. ↑↑↑↑ proband; + klinikai tünetek vannak; -
klinikai tünetek nincsenek; † elhunyt; ���� férfi; O nő. A betegség hátterében álló
TGC→→→→TGA mutáció a 354. kodon területén Cys →→→→STOP cserét eredményez.

57
A 12. ábra egy típusos MEN 1 szindrómás családban mutatja be a betegség
öröklődését. A proband a két MEN 1 szindrómás leánytestvér egyike volt, akik
édesanyja klinikailag igazolt MEN 1 szindrómában szenvedett és fiatalon
gyomorvérzésben hunyt el. A probandban és lánytestvérében már fiatal korban primer
hyperparathyreosis alakult ki. A probandban „pre-screening” módszerrel nem lehetett
genetikai eltérést kimutatni, a DNS szekvenálás azonban a két leánytestvérben és a
proband tünetmentes gyermekében a MEN1 gén inzerciós mutációjának jelenlétét
bizonyította (nt1657insC). A mutáció következtében a 10-es exon területén egy citozin
ékelődik be a normál bázissorrendbe, ami a leolvasási keret eltolódását (”frameshift”) és
ettől a ponttól kezdve kóros fehérjeszerkezet, illetve korai STOP kodon kialakulását
okozza. A genetikai vizsgálat eredménye a tünetmentes mutáció-hordozó gyermekben
különösen fontos adatot jelent a követéses vizsgálatok megfelelő tervezéséhez.
12. ábra: MEN 1 család 13. ábra: MEN 1 család 14. ábra: MEN 1 család
(nt1657insC) (nt738del4bp) (Q209X)
++ ++
--
--
++
--
? -
+-
+
+
-++ ++
--
--
++
--
nt1657insC
? -
+-
nt738del4bp
+
+
-
Q209X
++ ++
--
--
++
--
? -
+-
+
+
-++ ++
--
--
++
--
nt1657insC
? -
+-
nt738del4bp
+
+
-
Q209X
A 13. ábrán illusztrált családban a fiatal férfibeteg hypoglycaemia tüneteivel
került kórházi felvételre. Endokrinológiai kivizsgálása során pancreas insulinomát és
prolactin-termelő hypophysis adenomát találtak. Évekkel később primer
hyperparathyreosisa is kialakult. A MEN1 gén mutáció analízis a 3-as exon területén
egy korábban már leírt, 4 bázist érintő deléciós mutációt (nt738del4bp) mutatott ki. A 4
bázis deléciója a kereteltolódás miatt kóros fehérjeszerkezettel jár és korai STOP kodon
kialakulását okozza. A betegben egy gyakori, aminosavat nem cserélő polimorfizmus

58
(D418D) is jelen volt homozigóta formában. A beteg édesanyja klinikailag és
genetikailag is negatívnak bizonyult, a beteg édesapja korán elhunyt (oka ismeretlen). A
beteg testvére nem járult hozzá a genetikai vizsgálathoz, de klinikailag tünetmentes volt,
és más családtagban sem jelentkeztek eddig a MEN1 szindróma tünetei. Ezek alapján
feltételezhető, hogy a mutáció de novo alakult ki.
A 14. ábrán illusztrált családban a proband hyperparathyreosisa évek óta ismert
volt, és a mellékpajzsmirigy műtétet követően recidiváló gyomorfekély irányította a
figyelmet MEN 1 szindróma irányába. Hasi ultrahang vizsgálattal később mellékvese
adenomát is felfedeztek. A proband fiánál szintén hyperparathyreosist diagnosztizáltak,
unokája klinikailag tünetmentes volt. A genetikai vizsgálat során egy nonsense MEN1
gén mutációt találtam, amely a 3-as exon területén egy glutamin cserél STOP kodonra
(Q209X) és ezáltal a 3-as exonnak megfelelően csonkolt fehérje képződését okozza.
Feltehető, hogy a rövid fehérje nem rendelkezik azokkal a szerkezeti egységekkel,
amelyek nélkülözhetetlenek a normális működéséhez. A probandban a D418D
polimorfizmus is jelen volt heterozigóta formában. Mind a mutációt, mind a
polimorfizmust a proband fiában is ki lehetett mutatni. A proband unokája nem örökölte
a mutációt, ezért mentesülhet az élethossziglan tartó követéses vizsgálatok alól.
15. ábra. MEN1 gén nt317ins5bp mutációjának szekvenálási képe és a mutáció
által okozott kereteltolódás szekvenciális alapja
1. allél: Normál A-G-C-C-C-C-G-C-C-C-C-C-G-A-C-C-C-G-C-C
2. allél: Mutáns A-G-C-C-C-C-G-C-C-C-C-G-C-C-C-C-C-G-A-C5 bázispár inzerciója
A G C C C C G C C C C C/G G/C A/C C/C C/C C/G G/G C/A C/C
317.
1. allél: Normál A-G-C-C-C-C-G-C-C-C-C-C-G-A-C-C-C-G-C-C
2. allél: Mutáns A-G-C-C-C-C-G-C-C-C-C-G-C-C-C-C-C-G-A-C5 bázispár inzerciója
A G C C C C G C C C C C/G G/C A/C C/C C/C C/G G/G C/A C/C
317.
1. allél: Normál A-G-C-C-C-C-G-C-C-C-C-C-G-A-C-C-C-G-C-C
2. allél: Mutáns A-G-C-C-C-C-G-C-C-C-C-G-C-C-C-C-C-G-A-C5 bázispár inzerciója
A G C C C C G C C C C C/G G/C A/C C/C C/C C/G G/G C/A C/C
317.
1. allél: Normál A-G-C-C-C-C-G-C-C-C-C-C-G-A-C-C-C-G-C-C
2. allél: Mutáns A-G-C-C-C-C-G-C-C-C-C-G-C-C-C-C-C-G-A-C5 bázispár inzerciója
A G C C C C G C C C C C/G G/C A/C C/C C/C C/G G/G C/A C/C
317.
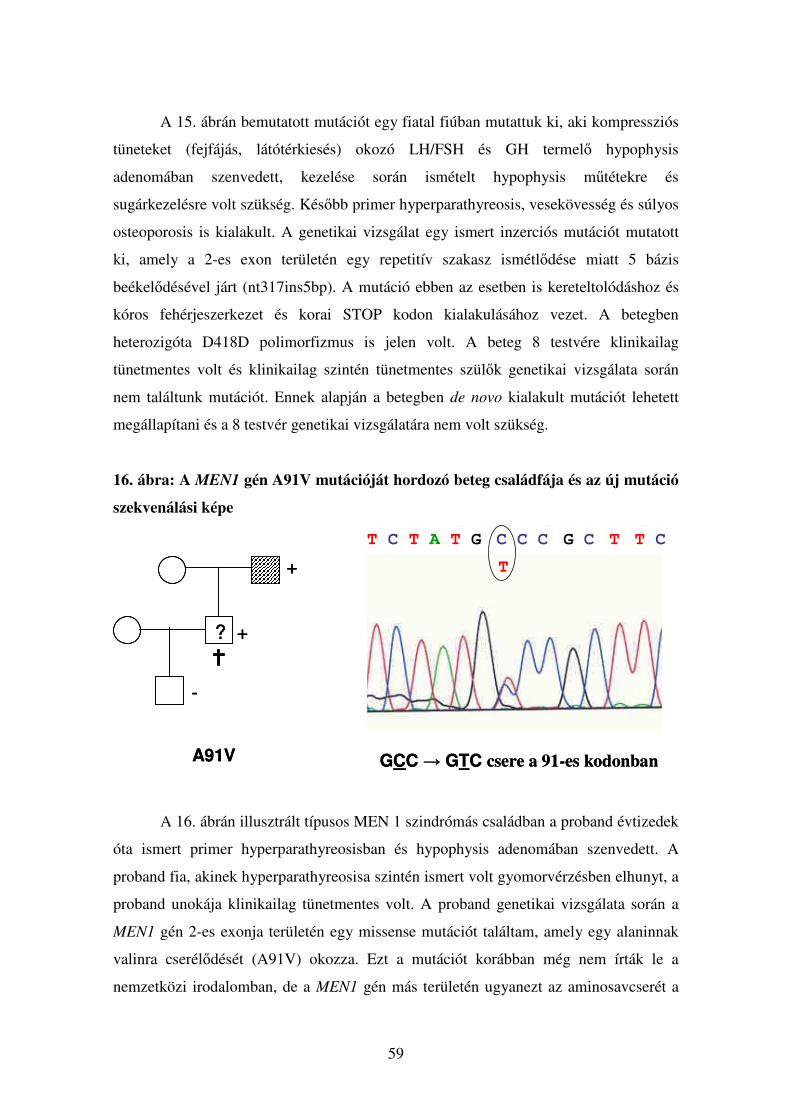
59
A 15. ábrán bemutatott mutációt egy fiatal fiúban mutattuk ki, aki kompressziós
tüneteket (fejfájás, látótérkiesés) okozó LH/FSH és GH termelő hypophysis
adenomában szenvedett, kezelése során ismételt hypophysis műtétekre és
sugárkezelésre volt szükség. Később primer hyperparathyreosis, vesekövesség és súlyos
osteoporosis is kialakult. A genetikai vizsgálat egy ismert inzerciós mutációt mutatott
ki, amely a 2-es exon területén egy repetitív szakasz ismétlődése miatt 5 bázis
beékelődésével járt (nt317ins5bp). A mutáció ebben az esetben is kereteltolódáshoz és
kóros fehérjeszerkezet és korai STOP kodon kialakulásához vezet. A betegben
heterozigóta D418D polimorfizmus is jelen volt. A beteg 8 testvére klinikailag
tünetmentes volt és klinikailag szintén tünetmentes szülők genetikai vizsgálata során
nem találtunk mutációt. Ennek alapján a betegben de novo kialakult mutációt lehetett
megállapítani és a 8 testvér genetikai vizsgálatára nem volt szükség.
16. ábra: A MEN1 gén A91V mutációját hordozó beteg családfája és az új mutáció
szekvenálási képe
+
+?
-
T C T A T G C C C G C T T C
T
GCC → GTC csere a 91-es kodonbanA91V
+
+?
-
+
+?
-
T C T A T G C C C G C T T C
T
GCC → GTC csere a 91-es kodonbanA91V
A 16. ábrán illusztrált típusos MEN 1 szindrómás családban a proband évtizedek
óta ismert primer hyperparathyreosisban és hypophysis adenomában szenvedett. A
proband fia, akinek hyperparathyreosisa szintén ismert volt gyomorvérzésben elhunyt, a
proband unokája klinikailag tünetmentes volt. A proband genetikai vizsgálata során a
MEN1 gén 2-es exonja területén egy missense mutációt találtam, amely egy alaninnak
valinra cserélődését (A91V) okozza. Ezt a mutációt korábban még nem írták le a
nemzetközi irodalomban, de a MEN1 gén más területén ugyanezt az aminosavcserét a

60
nemzetközi irodalomban betegségokozó mutációként tartják számon (pl. A535V,
A242V) [Zhuang és mtsai 1997]. A probandban a 9-es exonon D418D heterozigóta
polimorfizmus is jelen volt. A proband unokája nem örökölte a mutációt.
17. ábra. A MEN1 gén G28A mutációját hordozó beteg családfája és a mutáció
szekvenálási képe
- -
+
C T G G G C C G A
C
G28A GGC → GCC csere a 28-as kodonban
- -
+
- -
+
C T G G G C C G A
C
G28A GGC → GCC csere a 28-as kodonban
A 17. ábrán szemléltetett családban a MEN1 gén mutáció de novo alakult ki. A
fiatal nőbeteg recidíváló primer hyperparathyreosisban szenvedett, édesanyja és
édesapja klinikailag tünetmentes volt. A proband genetikai vizsgálata során a 2-es exon
területén egy glicint alaninra cserélő missense mutációt (G28A) találtam, melyet a
nemzetközi irodalomban eddig még nem írtak le. A mutáció egyik szülőben sem volt
jelen. A MEN1 gén területén glicin-alanin cserét eredményező mutációt még nem írtak
le a nemzetközi irodalomban és a két aminosav szerkezete is hasonló, ezért a mutáció
biztosan betegség-okozó szerepét egyelőre nem tekinthetjük egyértelműen bizonyosnak.
A probandban a 3-as exon területén egy aminosav cserével járó gyakori polimorfizmust
is találtam (R171Q).
A 18. ábrán szemléltetett családban a proband FSH/LH termelő hypophysis
adenomában és primer hyperparathyreosisban szenvedett. A proband két
leánytestvérénél szintén hyperparathyreosist, valamint recidíváló gyomorfekélyt
állapítottak meg. Mindkét szülő elhunyt (az édesapa fiatal korban), betegségeikről
orvosi dokumentáció nem állt rendelkezésre. A proband két leánya közül az egyiknél
hyperparathyreosist és hyperprolactinaemiát állapítottak meg, a másik leány klinikailag
tünetmentes volt. A genetikai vizsgálatok során a MEN1 gén 6-ös exonjának területén
lizin - arginin cserét okozó missense mutációt (L301R) mutattam ki. Ezt a mutációt
korábban még nem közölték a nemzetközi irodalomban, de a MEN1 gén más területén

61
ugyanezen aminosavcserével járó mutációt betegség-okozó mutációnak tekintik (pl.
L22R [Agarwal és mtsai 1997], L89R [Hessmann és mtsai 1998]). A család szűrésével
nyert eredmények a L301R mutáció oki szerepét támasztották alá; a MEN1 szindróma
klinikai tünetei a genetikai eltérést hordozó összes családtagban jelen voltak, míg a
klinikailag tünetmentes családtagokban a genetikai eltérést nem lehetett kimutatni.
18. ábra. A MEN1 gén L301R mutációját hordozó beteg családfája és a mutáció
szekvenálási képe
?
+ + +
+ - -
?
L301R
A C C C T C T A C
G
CTC → CGC csere a 301-es kodonban
?
+ + +
+ - -
? ?
+ + +
+ - -
?
L301R
A C C C T C T A C
G
A C C C T C T A C
G
CTC → CGC csere a 301-es kodonban
A 19. ábra egy primer hyperparathyreosisban és hypophysis adenomában
szenvedő fiatal férfibetegben a MEN1 gén 2-es exonjának területén 4 bázispár
deléciójával járó mutációt (nt359del4bp) szemléltet. A mutáció kereteltolódás révén
kóros fehérjeszerkezet és korai STOP kodon kialakulásához vezet. A beteg klinikailag
tünetmentes édesanyjánál a mutációt nem lehetett kimutatni, a család többi tagjánál a
klinikai adatok gyűjtése és a genetikai szűrés még folyamatban van.

62
19. ábra. A MEN1 gén nt359del4bp mutációjának szekvenálási képe, a mutáció
által okozott kereteltolódás mechanizmusa
A C C T G/A T/T C/C T/A A/T T/C C/G A/A T/C
359.
1. allél: Normál: A C C T G T C T A T C A T
2. allél: Mutáns: A C C T A T C A T C G A C
Négy bázis deléciója
A C C T G/A T/T C/C T/A A/T T/C C/G A/A T/C
359.
1. allél: Normál: A C C T G T C T A T C A T
2. allél: Mutáns: A C C T A T C A T C G A C
Négy bázis deléciója
Az utolsóként felfedezett mutációhordozó családban a családszűrés jelenleg
folyamatban van: a proband a 2-es exon területén az E26X nonsense mutációt hordozza,
mely korábban a nemzetközi irodalomban nem szerpelt. A mutáció a 26-os kodon
területén egy GAG>TAG nukelotid cserét okoz, amely az amionosav sorrendben a 26-
os helyen található glutaminsav helyett kialakuló STOP kodon révén korai terminációt
eredményez. Így ezen esetben egy igen rövid, mindössze 25 aminosavból álló csonkolt
fehérje keletkezik, így a mutáció betegség-okozó szerepe nem kétséges. A fiatal nőbeteg
hyperparathyreosisban szenved, emellett prolactinomát is leírtak nála. Szintén hasonló
manifesztációkkal rendelkező édesapja is mutációhordozónak bizonyult. A többi
családtag szűrése jelenleg folyamatban van.

63
VI. MEGBESZÉLÉS
A MEN 1 szindróma hátterében álló MEN1 gén mutációk vizsgálata igen nagy
kihívás a gén nagy mérete, a lehetséges csírasejtes mutációk nagy száma, a mutációs
”hot spot” hiánya, továbbá a genotípus-fenotípus összefüggések hiánya miatt. A
genetikai eltéréseket tartalmazó gén-szakaszok kimutatására többféle szűrőmódszert
dolgoztak ki, melyek közül munkámban a TGGE módszert használtam. A TGGE
módszerrel kimutatott heterozigóta vagy homozigóta genetikai eltérések
(polimorfizmusok és mutációk) pontos meghatározására direkt DNS szekvenálást
végeztem. A TGGE szűrőmódszerrel szerzett tapasztalatokat az alábbiakban foglalom
össze:
� A módszer érzékenységének növeléséhez az oligonukleotid primerek
tervezésekor a rendelkezésre álló számítógépes szoftver programok
használata ajánlott, melyekkel a melting domének pontosan
megítélhetők.
� Az elektroforézis paramétereinek optimalizálásához viszont egy
experimentális módszer, a perpendiculáris elektroforézis bizonyult a
leghatékonyabbnak.
� Megfigyelések szerint MEN 1 szindrómában szenvedő betegekben a
perifériás vérből származó DNS instabilabb és törékenyebb, mint
egészségesekben. Vizsgálataim során egy beteg DNS mintájában az első
alkalommal sikeres próbálkozást követően a 2-es exon PCR
amplifikálását nem sikerült elvégezni. A betegtől később levett
vérmintából frissen izolált DNS-ből végzett vizsgálat problémamentes
volt.
� A MEN1 gén 10-es exon területén konzekvensen észlelt álnegatív
eredmény miatt ennek az exonnal TGGE módszerrel való vizsgálata nem
ajánlott.
� A 9-es exon területén kétféle gyakori polimorfizmus fordult elő
heterozigóta vagy homozigóta formában (D418D, L432L). A különböző
polimorfizmusok elektroforetikus képe egymástól jól elkülöníthető, ezért

64
ennek az exonnak a vizsgálata során belső kontrollként – a
polimorfizmusokat nem tartalmazó negatív kontroll mintán kívül -
D418D és L432L heterozigóta DNS minták használata is ajánlott.
� A TGGE módszerre alapozott szűréssel nyert eredmények
megerősítéséhez a megfelelő DNS szakasz nukleotid szekvenciájának
meghatározása szükséges.
Megállapítottam, hogy a MEN1 gén leggyakoribb polimorfizmusai a magyar
populációban a nemzetközi adatokhoz hasonló gyakorisággal fordulnak elő. A MEN 1
szindrómás betegekben kimutatott polimorfizmusok betegség-okozó szerepe kizárható,
hiszen nemzetközi irodalmi adatok szerint ezek az egészséges populációban is nagy
gyakorisággal mutathatók ki, aminosavat a menin protein-szekvenciájában nem
cserélnek. Az általunk vizsgált betegek és egészséges hozzátartozóik vizsgálata során
számos esetben találkoztunk polimorfizmusokkal nemcsak heterozigóta, de homozigóta
formában is, és a betegség manifesztációjával semmilyen formában nem korreláltak. A
betegekben kimutatott mutációk oki szerepének értékelésekor többféle szempontot
vettem figyelembe. A hazai MEN 1 szindrómás betegekben kimutatott 10 különböző
mutáció közül 5 mutációt a nemzetközi irodalomban betegség-okozó mutációként
tartottak számon, ezért ezeket a mutációkat – a nemzetközi tapasztalatokat megerősítve
– ennek megfelelően értékeltem: a 2 deléciós és 2 inzerciós mutáció kereteltolódáshoz,
így megváltozott fehérje aminosavsorrendhez és korai STOP kodon kialakulásához
vezet, a nonsense mutáció pedig egy aminosavat STOP kodonra vált, így csonkolt
fehérje kialakulásához vezet. A hazai MEN 1 szindrómás betegekben észlelt további 5
mutáció azonban új, a nemzetközi irodalomban korábban még nem ismertetett
mutációnak felelt meg, ezért ezek értékelésekor nem állt rendelkezésre korábbi
ismeretanyag. A menin fehérje röntgenkrisztallográfiás szerkezete még nem ismert,
ezért a mutációknak a háromdimenziós fehérjeszerkezetre gyakorolt hatásának
számítógépes elemzése jelenleg még nem lehetséges. Továbbá nehézséget jelent, hogy a
sejtosztódásban, genom stabilitásban és sejtciklus szabályozásában betöltött szerepe
[Stalberg és mtsai 2004] ellenére a menin funkciója még nem teljesen ismert. Antimenin
fehérje (ellenanyag) segítségével Cavallari és mtsai már végeztek in situ vizsgálatokat a

65
menin expressziójának meghatározására [Cavallari és mtsai 2003], és a menin GTPáz
aktivitása alapján a GTPáz consensus szekvenciákat érintő mutációk hatása is
meghatározható. A menin karboxi-terminális részének csonkolásával járó mutációk a
nukleoláris szignálok elvesztése miatt a fehérje cytosolban történő felhalmozódásához
vezetnek, ami laboratóriumi módszerekkel szintén kimutatható. Mindezek azonban csak
részben fedik le a mutációk teljes spektrumát, ezért jelenleg még nem áll rendelkezésre
megfelelő módszer, amellyel az összes mutáns fehérje funkciója meghatározhatóvá
válna. Mindezek miatt munkámban a nemzetközi irodalomban még nem közölt új
mutációk betegség-okozó szerepét a mutáció típusának és az érintett egyének
családfájának elemzésére alapoztam. Nonsense mutációk (valamely aminosav STOP
kodonra cserélődése), vagy kereteltolódással járó új mutációk esetében az oki szerep
nyilvánvalónak tekinthető, hiszen a csonkolt fehérje kötőhelyeinek, aktivitási
doménjének és nukleoláris szignáljának elvesztése a fehérje működésének sérülését
okozza. Nehezebben ítélhető meg a missense mutációk szerepe, ugyanis egy aminosav
másikra cserélődése nem feltétlenül jár együtt a fehérje funkció károsodásával. Ilyen
esetekben a mutáció oki szerepének megítélését leginkább a mutáció családon belüli
öröklődésének elemzése segítheti. Ha a klinikai kép egyértelműen követi a családban a
mutáció öröklődésének menetét, az oki szerep nagy valószínűséggel megállapítható.
A MEN1 gén missense és deléciós mutációinak vizsgálata során Yaguchi és
mtsai kimutatták, hogy a MEN 1 szindrómához társuló különböző mutációk a fehérje
stabilitásának csökkenését és ubiquitináció révén a sejt proteosomáiban való gyors
degradálódását okozzák. Ugyanakkor a MEN 1 szindrómás betegekben kimutatott
polimorfizmusokra a gyors degradálódás nem jellemző. Ez a megfigyelés magyarázatot
adhat arra is, hogy noha a missense mutációk a MEN1 génen belül bárhol
kialakulhatnak, genotípus-fenotípus összefüggéseket MEN 1 szindrómában eddig még
nem találtak [Yaguchi és mtsai 2004].
A hazai betegekben talált új mutációk az alábbi doméneken változtatják meg a
menin fehérjeszerkezetét: a G28A mutáció a JunD és nm23 kötőhelyeket kialakító
fehérje doméneket érinti és az első leucin zip szerkezetét is átformálhatja. Az érintett
családban a mutáció és a klinikai fenotípus közötti összefüggés vizsgálatára nem volt
lehetőség, ugyanis a mutáció a probandban de novo alakult ki, akinek leszármazottja
nem volt. A két aminosav szerkezeti hasonlósága ellenére az alaninnak glicinre

66
cserélődését más fehérjék esetében a nemzetközi irodalomban affinitást befolyásoló
tényezőként írták le, azonban a G28A mutáció betegség-okozó szerepét nem
tekinthetjük kétséget kizáróan igazoltnak. Az A91V mutáció a Smad3 és nm23 kötő
doméneket változtatja meg; ebben az esetben a családfa vizsgálat alátámasztotta a
genetikai vizsgálat eredményeit: csak azon családtagokban (és minden ilyen
családtagban) volt megtalálható, akik klinikailag is MEN 1 szindrómások voltak. A
családfa elemzése az L301R mutáció esetében is oki kapcsolatra utalt; ez a mutáció a
GTPáz konszenzus szekvenciát érinti és a menin-PEM és menin-nm23 kapcsolódást
befolyásolhatja. Az E26X és C354X mutációk csonkolt fehérje képződését okozzák; az
első esetben a fehérje mindössze 26 aminosavból áll, míg a második esetben a nukleáris
lokalizációs szignálok és a prolin gazdag régió elvesztése mellett a fehérje szinte
minden kötési tulajdonsága megváltozhat, ezért oki szerepük kétségtelennek tűnik (20.
ábra), és azt a családfa-elemzés is alátámasztotta.
20. ábra: Az általunk talált új mutációk lokalizációja és feltételezett szerepe a
menin szerkezetén belül
G28A
Leucin Zippek
Nukleárislokalizációsszekvenciák
aminosavak
Prolin gazdag régióGTPáz konszenzus
A91V L301R C354X új mutációkE26X
G28A
Leucin Zippek
Nukleárislokalizációsszekvenciák
aminosavak
Prolin gazdag régióGTPáz konszenzus
A91V L301R C354X új mutációkE26X
Nemzetközi adatok alapján nem egyértelmű, hogy a klinikai kritériumok alapján
definiált MEN 1 szindrómás családokban milyen gyakorisággal mutatható ki mutáció a

67
MEN1 gén kódoló régiójában. Egyes vizsgálatok a családok mindössze 5-20%-ában
[Thakker 1998], míg más vizsgálatok ennél lényegesen nagyobb gyakorisággal találtak
mutációt. Egy 2005-ben közzétett brit felmérés összesen 186, a nemzetközi
kritériumrendszer alapján MEN1 genetikai szűrésre ajánlott család-reprezentáló beteg
adatait elemezve megállapította, hogy a betegség-okozó mutációt az esetek 37%-ában
lehetett azonosítani. A vizsgált betegek sporadikus és familiáris csoportokba sorolását
követően kiderült, hogy a familiáris esetekben a 3, 2 és 1 MEN 1-asszociált tumorral
rendelkezők körében kimutatott mutációk gyakorisága 91%, 69% és 29% volt (ebben a
sorrendben), míg a sporadikus esetekben ugyanebben a sorrendben a mutációk
előfordulása 69%, 23% és 0% volt [Ellard és mtsai 2005]. A MEN 1-asszociált léziók
nagy száma tehát növelheti annak a valószínűségét, hogy a vizsgált betegben a MEN1
gén kódoló régiójában mutáció igazolható. Familiáris esetekben a sporadikus esetekhez
képest a MEN1 gén mutációk nagyobb gyakorisággal való kimutathatóságát mások is
megfigyelték [Ohye és mtsai 1999]. Ezeknek az új megfigyeléseknek a tükrében a MEN
1 szindróma genetikai szűrésének definíciói újabb szempontokkal egészülhetnek ki,
amelyek figyelembe veszik a tünetek familiáris vagy sporadikus megjelenését és a MEN
1-asszociált léziók számát.
Összehasonlítva a klinikai adatokat a genetikai eredményekkel elmondhatjuk,
hogy mind a 6, familiáris MEN 1 szindrómás betegben kimutattuk a betegségokozó
mutációt (100%) és a 13 sporadikus MEN 1 szindrómás betegből pedig összesen 3
esetben (13%) volt sikeres a genetikai vizsgálat. A 13 MEN 1-szerű állapotban
szenvedő esetek közül összesen 1 rekurrens izolált primer hyperparathyreosisos
betegben találtunk MEN1 gén mutációt. Továbbá az általunk vizsgált 32 beteg
tekintetében elmondható, hogy a MEN 1-asszociált tumorok száma és a családi
anamnaesis nagy mértékben korrelál a mutációanalízis hetékonyságával. Az általunk
vizsgált 32 beteg közül 10 betegben (31,25%) sikerült a MEN1 gén kódoló régiójában
betegség-okozó mutációt kimutatni. A familiáris csoportban a 2 és 1 MEN 1-asszociált
daganatban szenvedő betegek 100%, és 0%-a, míg a sporadikus esetek körében a 3, 2 és
1 MEN 1-asszociált daganatban szenvedő betegek 50%, 18% és 9%-a volt MEN1 gén
mutáció hordozó (11. táblázat).
Megfigyelhető továbbá, hogy a mellékvesekéreg adenomák igen magy arányban
fordulnak elő MEN 1-szerű állapotban szenvedő esetekben, akár agyalapi mirigy

68
daganattal, vagy primer hyperparathyreosissal, de igen ritka ezen társult tünetek
esetében a MEN1 gén mutáció, ami azt mutathatja, hogy a mellékvese adenomák
prediktív értéke elhanyagolható MEN1 mutációk szempontjából.
11. táblázat: A MEN1 gén mutációk előfordulása és a MEN 1-asszociált daganatok
száma közötti összefüggés az általunk vizsgált betegekben
Familiáris esetek száma Sporadikus esetek száma MEN 1-asszociált
fő tumorok száma N Mutációt hordozó
esetek száma (%) N
Mutációt hordozó
esetek száma (%)
3 - - 2 1 (50)
2 6 6 (100) 11 2 (18)
1 2 0 (0) 11 1 (9)
Összesen 8 6 (75) 23 4 (17%)
Azokban a családokban, amelyekben nem sikerült a betegség hátterében álló
mutációt kimutatni, a genetikai eltérés a szabályozó vagy nem-kódoló régiókban lehet,
de az 5’-szakasz GC-gazdag területének hypermetilációjának oki szerepe sem zárható ki
[Agarwal és mtsai 1997, Bassett és mtsai 1998]. Ezeken kívül a MEN1 gén nagyméretű
deléciója, esetleg más gén hibája is szóba jön [Nahimira és mtsai 1999]. Az elmúlt
évben a 4-es kromoszóma területén egy új gént (MENX) írtak le, amely a ”multiple
endocrine neoplasia-like” szindróma elnevezésű betegséget okozza. Ez az öröklődő
daganatos szindróma a neuroendokrin rendszert érintő, patkányokban az első életévben
kialakuló multiplex tumoros betegséget okozza, melyben az érintett szervek spektruma
a MEN 1- és MEN 2 szindrómák közötti átmenetnek felel meg [Piotrowska és mtsai
2004].

69
VII. KÖVETKEZTETÉSEK
Munkám során összesen 32 MEN 1 szindrómás vagy MEN 1-szerű
szindrómában szenvedő betegben végeztem el a MEN1 gén teljes kódoló régiójának
vizsgálatát. A MEN1 gén nagy mérete, a lehetséges csírasejtes mutációk nagy száma,
illetve a mutációs ”hot spot” hiánya miatt a genetikai eltéréseket tartalmazó gén-
szakaszok kimutatására TGGE szűrőmódszert használtam és a genetikai eltérések
pontos meghatározására direkt DNS szekvenálást végeztem.
(1) Megállapítottam, hogy a rendelkezésre álló számítógépes szoftver programok az
oligonukleotid primerek tervezésekor növelik a TGGE szűrőmódszer
érzékenységét. Ugyanakkor elektroforézis paraméterek optimalizálásához egy
experimentális módszer, a perpendiculáris elektroforézis bizonyult a
leghatékonyabbnak.
(2) Megállapítottam, hogy a TGGE szűrőmódszer a MEN1 gén 10-es exon nt1657ins
mutációja esetében álnegatív eredményt ad, ezért ezzel a módszerrel a 10-es exon
vizsgálata nem ajánlott.
(3) Kimutattam, hogy a MEN1 gén 9-es exon területén előforduló kétféle gyakori
polimorfizmus (D418D, L432L) elektroforetikus képe egymástól jól
elkülöníthető, ezért ennek az exonnak a vizsgálata során belső kontrollként – a
polimorfizmusokat nem tartalmazó mintán kívül - D418D és L432L heterozigóta
DNS minták használata is ajánlott.
(4) A vizsgált betegekben a MEN1 gén kódoló szakaszainak direkt DNS
szekvenálásával megállapítottam, hogy a MEN1 gén leggyakoribb
polimorfizmusai a nemzetközi adatokhoz hasonló gyakorisággal fordulnak elő.
(5) Megállapítottam, hogy a vizsgált 32 MEN 1 szindrómás vagy MEN 1-szerű
állapotban szenvedő beteg közül 10 betegben (31.25%) fordult elő a MEN1 gén
kódoló szakaszán betegség-okozó mutáció, melyek közül 5 mutációt még nem
írtak le a nemzetközi irodalomban (A91V, G28A, E26X, L301R és C354X).
(6) Megállapítottam, hogy MEN 1 szindrómás és MEN 1-szerű állapotban szenvedő
betegekben a MEN1 gén kódoló szakaszán a betegség-okozó mutációk

70
előfordulásának gyakorisága familiáris esetekben nagyobb, mint sporadikus
esetekben. A MEN 1 szindrómára jellemző két, vagy három különböző daganat
társulásával járó esetekben a mutációk nagyobb gyakorisággal mutathatók ki
azokhoz az esetekhez képest, amelyekben csak egyféle MEN 1 szindrómára
jellemző daganat fordul elő.

71
Magyar nyelvű összefoglaló
PhD dolgozatomban a multiplex endokrin neoplasia 1-es típusának (MEN 1)
klinikai és genetikai vizsgálata során összegyűlt eredményeket foglalom össze. A MEN
1 szindróma autoszomális dominánsan öröklődő betegség, amely az agyalapi mirigy, a
mellékpajzsmirigyek és a hasnyálmirigy endokrin tumoraival és egyéb endokrin vagy
nem endokrin daganatokkal jár. Munkám során összesen 32 MEN 1 szindrómás vagy
klinikailag MEN 1 szindrómára gyanús betegben végeztem el a MEN1 gén teljes kódoló
régiójának vizsgálatát. A perifériás vérből izolált genomiális DNS mintákban a MEN1
gén teljes kódoló szakaszán előforduló genetikai eltérések kimutatására
hőmérsékletgrádiens gélelektroforézis (TGGE) szűrőmódszert használtam és a genetikai
eltérések pontos meghatározására direkt DNS szekvenálást végeztem. Megállapítottam,
hogy a rendelkezésre álló számítógépes szoftver programok az oligonukleotid primerek
tervezésekor növelik a TGGE szűrőmódszer érzékenységét, míg az elektroforézis
paraméterek optimalizásához egy experimentális módszer, a perpendiculáris
elektroforézis bizonyult a leghatékonyabbnak. Megállapítottam, hogy a TGGE
szűrőmódszer a MEN1 gén 10-es exon nt1657insC mutációja esetében álnegatív
eredményt ad, ezért ezzel a módszerrel a 10-es exon vizsgálata nem ajánlott.
Kimutattam, hogy a MEN1 gén 9-es exon területén előforduló kétféle gyakori
polimorfizmus (D418D, L432L) elektroforetikus képe egymástól jól elkülöníthető, ezért
ennek az exonnak a szűrővizsgálata során belső kontrollként – a polimorfizmusokat
nem tartalmazó mintán kívül - D418D és L432L heterozigóta DNS minták használata is
ajánlott. A vizsgált betegekben a MEN1 gén kódoló szakaszainak direkt DNS
szekvenálásával megállapítottam, hogy a MEN1 gén leggyakoribb polimorfizmusai a
nemzetközi adatokhoz hasonló gyakorisággal fordulnak elő. Megállapítottam, hogy a
vizsgált 32 beteg közül 10 betegben (31.25%) fordult elő a MEN1 gén kódoló szakaszán
betegség-okozó mutáció, melyek közül 5 mutációt még nem írtak le a nemzetközi
irodalomban (A91V, G28A, E26X, L301R és C354X). Megállapítottam, hogy a vizsgált
betegekben a MEN1 gén kódoló szakaszán a betegség-okozó mutációk előfordulásának
gyakorisága familiáris esetekben nagyobb, mint sporadikus esetekben. A MEN 1
szindrómára jellemző két vagy három különböző daganat társulásával járó esetekben a
mutációk nagyobb gyakorisággal mutathatók ki azokhoz az esetekhez képest,
amelyekben csak egyféle MEN 1 szindrómára jellemző daganat fordul elő.

72
Angol nyelvű összefoglaló
In my PhD thesis I summarized the results of clinical and genetic investigations
of multiple endocrine neoplasia type 1. The MEN 1 syndrome is an autosomal dominant
disorder characterized by endocrinopathies involving the anterior pituitary gland,
parathyroid glands, endocrine pancreas, and other endocrine or non-endocrine tumors. I
have examined the entire coding region of the MEN1 gene in 33 MEN 1 patients and
suspicious cases. Genomic DNA was extracted from peripheral blood for pre-screening
with temporal temperature gradient gel electrophoresis (TGGE); the identification of the
localized genetic alterations was performed by direct DNA sequencing. I have
established, that using the melting behavior-calculating softwares for primer-design
increases the sensitivity of TGGE, while optimizing the electrophoretic parameters
during TGGE has been shown to be the most efficient by perpendicular electrophoresis.
I have found that screening exon 10 of the MEN1 gene is not recommended by TGGE,
because the electrophoresis was false negative in the case of nt1657insC mutation. I
have shown that the electrophoretic bands of two common benign polymorphisms
(D418D and L432L) could be distinguished clearly by TGGE in exon 9, thus, it is
recommended to use not just a negative, but two, D418D and L432L, heterozygous
controls during the electrophoresis of this exon. The most common polymorphisms of
the MEN1 gene, identified by direct DNA sequencing, were found to appear with the
same prevalence as mentioned in the international literature. I have found MEN1
disease-causing mutation in ten cases among the 32 patients with MEN 1 or MEN 1-like
state (31.25%), and five of them were not known in the international literature (A91V,
G28A, E26X, L301R, C354X). I have also established that the presence of a disease-
causing mutation is more frequent in familial cases than in sporadic cases. Furthermore,
in cases with two or three MEN 1-associated lesions the disease-causing mutations are
more frequent than in patients only with one MEN 1-associated tumor.

73
Irodalomjegyzék:
Agarwal SK, Burns AL, Sukhodolets KE, Kennedy PA, Obungu VH, Hickman AB,
Mullendore ME, Whitten I, Skarulis MC, Simonds WF, Mateo C, Crabtree JS, Scacheri
PC, Ji Y, Novotny EA, Garrett-Beal L, Ward JM, Libutti SK, Alexander HR, Cerrato A,
Parisi MJ, Anna-A SS, Oliver B, Chandrasekharappa S, Collins FS, Spiegel AM, Marx
SJ: Molecular Pathology of the MEN1 gene. Ann NY Acad Sci 2004, 1014, 189-198.
Agarwal SK, Kennedy PA, Scacheri PC, Novotny EA, Hickman AB, Cerrato A, Rice
TS, Moore JB, Rao S, Ji Y, Mateo C, Libutti SK, Oliver B, Chandrasekharappa SC,
Burns AL, Collins FS, Spiegel AM, Marx SJ: Menin molecular interactions: Insights
into normal functions and tumorigenesis. Horm Metab Res 2005, 37, 369-374.
Agarwal SK, Kester MB, Debelenko LV, Heppner C, Emmert-Buck MR, Skarulis MC,
Doppman JL, Kim YS, Lubensky IA, Zhuang Z, Green JS, Guru SC, Manickham P,
Olufemi S-E, Liotta LA, Chandrasekhappara SC, Collins FS, Spiegel AM, Burns AL,
Marx SJ: Germline mutations of the MEN1 gene in familial multiple endocrine
neoplasia type 1 and related states. Hum Mol Genet 1997, 6, 1169-1175.
Anlauf M, Perren A, Meyer CL, Schmid S, Saremaslani P, Kruse ML, Weihe E,
Komminoth P, Heitz PU, Kloppel G: Precursor lesions in patients with multiple
endocrine neoplasia type 1-associated duodenal gastrinomas. Gastroenterology 2005,
128(5), 1187-98.
Arnold A: Approach to therapy in multiple endocrine type 1. Endocrinology UpToDate,
Copyright 1999. www.uptodate.com electronic textbook, Szerk: Rose BB, UpToDate
inc, Wellesley, Ma, USA
Arnold A: Clinical manifestations and diagnosis of multiple endocrine neoplasia type 1.
Endocrinology UpToDate, Copyright 1999. www.uptodate.com electronic textbook,
Szerk: Rose BB, UpToDate inc, Wellesley, Ma, USA

74
Asgharian B, Turner ML, Gibril F, Entsuah LK, Serrano J, Jensen RT: Cutaneus tumors
in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas:
Prospective study of frequency and development of criteria with high sensitivity and
specificity for MEN1. J Clin Endocrinol Metab, 2004, 89(11), 5328-5336.
Balogh K, Patócs A, Majnik J és mtsai: Genetic screening for the detection of mutations
responsible for multiple endocrine neoplasia type 1. Mol Genet Metab 2004, 83, 74-81.
Bartsch DK, Fendrich V, Langer P, Celik I, Kann PH, Rothmund M: Outcome of
duodenopancreatic resections in patients with mutiple endocrine neoplasia type 1. Ann
surg 2005, 242(6), 757-766.
Bassett JHD, Forbes SA, Pannett AAJ, Lloyd SE, Christie PT, Wooding C, Harding B,
Besser GM, Edwards CR, Monson JP, Sampson J, Wass JAH, Wheeler MH, Thakker
RV, Characterization of mutations in patients with multiple endocrine neoplasia type 1.
Am J Hum Genet 1998, 62, 232-244.
Baudin E, Bidart J-M, Rougier P, Lazar V, Ruffié P, Ropers J, Ducreux M, Troalen F,
Sabourin J-C, Comoy E, Lasser P, DeBaere T, Schlumberger M: Screening for multiple
endocrine neoplasia type 1 and hormonal production in apparently sporadic
neuroendocrine tumors. J Clin Endocinol Metab 1999, 84(1), 69-75.
Beckers A, Betea D, Valdes Socin H, Stevenaert A: The treatment of sporadic versus
MEN1-related pituitary adenomas. J Int Med 2003, 253, 599-605.
Benito M, Asa SL, LiVolsi VA, West VA, Snyder PJ: Gonadotroph tumor associated
with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab, 2005, 90(1), 570-
574.
Bhuiyan M M R, Sato M, Murao K és mtsai: Differential expression of menin in various
adrenal tumors. Cancer 2001, 92, 1393-1401.

75
Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx
B, Falchetti A, Gheri RG, Libroia A, Lips CJM, Lombardi G, Mannelli M, Pacini F,
Ponder BAJ, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW,
Tomassetti P, Tonelli F, Wells SA, Marx SJ: Guidelines for diagnosis and therapy of
MEN type 1 and type 2. J Clin Endocrinol Metab 2001, 86, 5658-5671.
Burgess JR, Greenaway TM, Sheperd JJ: Expression of the MEN-1 gene in a large
kindred with multiple endocrine neoplasia type 1. J Int Med 1998, 243, 465-470.
Burgess JR, Nord B, David R, Greenaway TM, Paramenswaran V, Larsson C, Sephern
JJ, Teh BT: Phenotype and phenocopy: the relationship between genotype and clinical
phenotype in a asingle large family with mulitple endocrine neoplasia type 1 (MEN1).
Clin Endocrinol 2000, 53, 205-211.
Burgess JR, Shepherd JJ, Parameswaran V, Hoffman L, Greenaway TM: Spectrum of
pituitary disease in multiple endocrine neoplasia type 1 (MEN1): clinical, biochemical,
and radiological features of pituitary disease in a large MEN 1 kindred. J Clin
Endocrinol Metab 1996, 81(7), 2642-6.
Cavaco BM, Domingues R, Bacelar MC, Cardoso H, Barros L, Gomes L, Ruas MMA,
Agapito A, Garrao A, Pannett AAJ, Silva JL, Sobrinho LG, Thakker RV, Leite V:
Mutational analysis of Portugese families with multiple endocrine neoplasia type 1
reveals large deletions. 2002, 56, 465-473.
Cavallari I, D’Agostino DM, Ferro T, Rosato A, Barzon L, Pasquali C, Fogar P,
Theodoropoulou M, Esposito G, Boscaro M, Pagotto U, Tebaldi E, Fallo F, Chieco-
Bianchi L, Ciminale V: In situ analysis of human menin in normal and neoplastic
pancreatic tissues: evidence for differential expression in exocrine and endocrine cells. J
Clin Endoc Metab 2003, 88(8), 3893-3901.
Cebrián A, Ruíz-Llorente S, Cascón A, Osorio A, Martínez-Delgado B, Benítez J,
Robledo M: A rapid and easy method for multiple endocrine neoplasia type 1 mutation

76
detection using conformational-sensitive gel electrophoresis. J Hum Genet 2002, 47,
190-195.
Cebrián A, Ruíz-Llorente S, Cascón A, Pollan M, Diez JJ, Pico A, Telleria D, Benítez J,
Robledo M: Mutational and gross deletion study of the MEN1 gene and correlation with
clinical features in Spanish Patients. J Med Genet 2003, 40e72 Online Mutation Report,
www.jmedgenet.com/cgi/content/full/40/5/e72.
Cetani F, Pardi E, Giovanetti A, Carrai P, Borsari S, Vignali E, Picone A, Cianferotti L,
Miccoli P, Pinchera A, Marcocci C: Six novel MEN1 gene mutations in sporadic
parathyroid tumors. Hum Mut 2000, 16(5), 445-448.
Chandrasekkharappa SC, Guru SC, Manickham P, Olufemi S-E, Collins FS, Emmert-
Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y,
Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C,
Dong Q, Spiegel AM, Burns AL, Marx SJ: Positional cloning of the gene for multiple
endocrine neoplasia type 1. Science 1997, 276, 404-407.
Corbetta S, Vincentini L, Ferrero S, Lania A, Mantovani G, Cordella D, Beck-Peccoz P,
Spada A: Activity and function of the nuclear factor kappaB pathway in human
parathyroid tumors. Endocr Relat Cancer 2005, 12(4), 929-37.
Costa SC, Nascimento LS, Ferriera FJ, Mattos PS, Camara-Lopes LH, Ward LS: Lack
of mutations of exon 2 of the MEN1 gene in endocrine and nonendocrnine sporadic
tumors. Braz J Med Biol Res 2001, 34, 861-865.
Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA,
Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS: A
mouse modell of multiple endocrine neoplasia, type 1, develops multiple endocrine
tumors. Proc Natl Acad Sci USA 2001, 98, 1118-1123.

77
Dean PG, van Heerden JA, Farley DR, Thompson GB, Grant CS, Harmsen WS, Ilstrup
DM: Are patients with multiple endocrine neoplasia type I prone to premature death?
World J Surg 2000, 24, 1437.
Dilley WG, Kalyanamaran S, Verma S, Cobb JP, Laramie JM, Lairmore TC: Global
gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Mol
Cancer 2005, 4(9), doi:10.1186=476-4598-4-9.
Doherty GM: Multiple endocrine neoplasia type 1. J Surg Oncol 2005, 89, 143-150.
Dreijerink KM, van Beek AP, Lentjes EGWM, Post JG, van der Luijt RB, Canninga-
van Dijk M, Lipd CJM: Acromegaly in a multiple endocrine neoplasia type 1 (MEN1)
family with low penetrance of the disease. Eur J Endocrinol 2005, 153, 741-746.
Ellard S, Hattersley AT, Brewer CM, Vaidya B: Detection of an MEN1 gene mutation
depends on clinical features and supports current referral criteria for diagnostic
molecular genetic testing. Clin Endocrinol 2005, 62, 169-175.
Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kücükceylan N, Abdel-Nour
M, Gewies A, Peters H, Kaufmann D, Buske A, Tinschert S, Nürnberg P: Minor lesion
mutational spectrum of the entire NF1 gene does not explain its high mutability but
points to a functional domain upstream of the GAP-related domain. Am J Hum Genet
2000, 66, 790-818.
Ferolla P, Falchetti A, Filosso P, Tomassetti P, Tamburrano G, Avenia N, Daddi G,
Puma F, Ribacchi R, Santeusanio F, Angeletti G, Brandi ML: Thymic Neuroendocrine
Carcinoma (carcinoid) in MEN1 syndrome: the Italian series. J Clin Endocrinol Metab
2005, 91, 2603-2609.
Forsberg L, Björck E, Hashemi J, Zedenius J, Höög A, Farnebo L-O, Reimers M,
Larsson C: Distinction in gene expression profiles demonstrated in parathyroid
adenomas by high-density oligoarray technology. Eur J Endocrinol 2005, 152, 459-470.

78
Frank-Raue K, Rondot S, Hoeppner W, Goretzki P, Raue F, Meng W: Coincidence of
multiple endocrine neoplasia types 1 and 2: mutations in the RET protooncogene and
MEN1 tumor suppressor gene in a family presenting with recurrent primary
hyperparathyroidism. J Clin Endocrinol Metab 2005, 90(7), 4063-4067.
Gille C, Gille A: TGGE-STAR: primer design for melting analysis using PCR gradient
gel electrophoresis. Biotechniques 2002, 32(2), 264-268.
Glascock M, Carty SE: Multiple endocrine neoplasia type 1: fresh perspective on
clinical features and penetrance. Surg Oncol 2002, 11, 143-150.
Goebel SU, Heppner C, Burns AL, Marx SJ, Spiegel AM, Zhuang Z, Lubensky IA,
Gibril F, Jensen RT, Serrano J: Genotype/phenotype correlation of multiple endocrine
neoplasia type 1 gene mutations in sporadic gastrinomas. J Clin Endocrinol Metab
2000, 85(1), 116-123.
Görtz B, Roth J, Krahenmann A, Krijger RR, Muletta-Feurer S, Rütimann K,
Saremaslani P, Speel EJM, Hitz PU, Komminoth P: Mutations and allelic deletions of
the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and
neuroendocrine tumors and not restricted to foregut neoplasms. Am J Pathol 1999, 154,
429-436.
Guo SS, Sawicki MP: Molecular and genetic mechanisms of tumorigenesis in multiple
endocrine neoplasia type 1. Mol Endocrinol 2001, 15, 1653-1664.
Guru SC, Agarwal SK, Manickam P, Olufemi S-E, Crabtree JS, Weisemann JM, Kester
MB, Kim YS, Wang AM, Emmert-Buck MR, Liotta LA, Spiegel AM, Boguski MS,
Roe BA, Collins FS, Marx SJ, Burns L, Chandrasekharappa SC: A transcript for the
2.8-Mb region containing the multiple endocrine neoplasia type 1 locus. Genome Res
1997, 7, 725-735.

79
Guru SC, Goldsmith PK, Burns AL: Menin, the product of the MEN1 gene, is nuclear
protein. Proc Natl Acad Sci 1998, 95, 1630-1634.
Guru SC, Manickam P, Crabtree JS: Identification and characterization of the multiple
endocrine neoplasia type 1 (MEN1) gene. J Int Med 1998, 243, 433-439.
Hao W, Skarulis MC, Simonds WF, Weinstein LS, Agarwal SK, Mateo C, James-
Newton L, Hobbs GR, Gibril F, Jensen RT, Marx SJ: Multiple endocrine neoplasia type
1 variant with frequent prolactinoma and rare gastrinoma. J Clin Endocrinol Metab.
2004, 89(8), 3776-3784.
Haven CJ, Howell VM, Eilers PHC, Dunne R, Takahashi M, van Puijenbroek M, Furge
K, Kievit J, Tan M-H, Fleuren GJ, Robinson BG, Delbridge LW, Philips J, Nelson AE,
Krause U, Dralle H, Hoang-Vu C, Gimm O, Morresu H, Marsh DJ, Teh BT: Gene
expression of parathyrois tumors: molecular subclassification and identification of the
potential malignant phenotype. Cancer Res, 2004, 64, 7405-7411.
Hellman P, Hennings J, Akerstrom G, Skogseid B: Endoscopic ultrasonography for
evaluation of pancreatic tumours in multiple endocrine neoplasia type 1. Br J Surg
2005, 92(12), 1508-12.
Hendy GN, Kaji H, Sowa H, Lebrun J-J, Canaff L: Menin and TGF-ß superfamily
member signaling via the Smad pathway in pituitary, parathyroid and osteoblast. Horm
Metab Res 2005, 37, 375-379.
Heppner C, Reincke M, Agarwal SK, Mora P, Allolio B, Burns AL, Spiegel AM, Marx
SJ: MEN1 gene analysis in sporadic adrenocortical neoplasms. J Clin Endocrinol Metab
1999, 84, 216-219.
Hessmann O, Lindberg D, Skogseid B, Carling T, Hellmann P, Rastad J, Akerstrom G&
Westin G: Mutation of the multiple endocrine neoplasia type 1 gene in nonfamilial,
malignant tumors of the endocrine pancreas. Cancer Res 1998, 58, 377-379.

80
Hoff AO, Cote GJ, Gagel RF: Multiple endocrine neoplasias, Ann Rev Physiol 2000,
62, 377-411.
Hoffmann KM, Gibril F, Entsuah LK, Serrano J, Jensen RT: Patients with MEN1 with
gastrinomas have an increased risk of severe esophageal disease including stricture, and
the premalignant condition, Barett esophagus. J Clin Endocrinol Metab 2005 oct. 25. in
press.
Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, Fleuren GJ, Robinson
BG, Delbridge LW, Philips J, Nelson AE, Krause U, Hammje K, Dralle H, Hoang-Vu
C, Gimm O, Marsh DJ, Morreau H, The BT: HRPT2 mutations are associated with
malignancy in sporadic parathyroid tumours. J Med Genet 2003, 40, 657-663.
Huang SC, Zhuang Z, Weil RJ, Pack S, Wang C, Krutzsch HC, Pham TA, Lubensky
IA: Nuclear/cytoplasmic localization of the multiple endocrine neoplasia type 1 gene
product, menin. Lab Invest 1999, 79, 301-310.
Ikeo Y, Yumita W, Sakurai A, Hashizume K: JunD-Menin interaction regulates c-Jun-
mediated AP-1 transactivation. Endocrine J 2004, 51(3): 333-342.
Ikota H, Tanimoto A, Kometsu H, Ozawa Y, Matsushita H: Ureteral leiomyoma causing
hydronephrosis in type a multiple endocrine neoplasia. Pathol Int 2004, 54(6), 457-9.
Jin S, Mao H, Schnepp R, Sykes SM, Silva AC, D’Andrea AD, Hua X: Menin
associates with FancD2, a protein involved in repair of DNA damage. Cancer Res 2003,
63, 4204-4210.
Johnson SJ, Sheffield EA, McNicol AM: Examination of parathyroid gland specimens.
J Clin Pathol 2005, 58, 338-342.

81
Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN: Inactivation of menin, a Smad-3
interaction protein, blocks transforming growth factor type beta signalling. Proc Natl
Acad Sci 2001, 98, 3837-3842.
Karges W, Jostarndt K, Maier S, Flemming A, Weitz M, Wissmann A, Feldmann B,
Dralle H, Wagner P, Boehm BO: Multiple endocrine neoplasia type 1 (MEN1) gene
mutations in a subset of patients with sporadic and familial primary
hyperparathyroidism target the coding sequence but spare the promoter region. J
Endocrinol 2000, 166, 1-9.
Karges W, Ludwig L, Kessler H, Wissmann A, Wagner PK, Boehm BO: Menin
mutations in the diagnosis and prediction of mutliple endocrine neoplasia type 1. Arch
Surg 1998, 383, 183-186.
Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, Mclean GW, Xiong Y, Meyerson
M, Kim SK: Menin regulates pancreatic islet growth by promoting histone methylation
and expression of genes encoding p27Kip1 and p18INK4c. PNAS 2005, 102(41),
14659-64.
Kawamura J, Shimada Y, Komoto I, Okamoto H, Itami A, Doi R, Fujimoto K, Kosugi
S, Imamura M: Multiple endocrine neoplasia type 1 gene mutations in sporadic
gastrinomas in Japan. Oncol Rep 2005, 14(1), 47-52.
Kouvaraki MA, Lee JE, Saphiro SE, Gagel RF, Sherman SI, Sellin RV, Cote GJ, Evans
DB: Genotype-phenotype analysis in multiple endocrine neoplasia type 1. Arch Surg
2002, 137, 641-647.
La P, Silva AC, Hou Z, Wang H, Schnepp RW, Yan N, Shi Y, Hua X: Direct binding of
DNA by tumor suppressor menin. J Biol Chem. 2004, 279, 49045-49054.

82
Lairmore TC, Chen VY, DeBenedetti MK, Gillanders WE, Norton JA, Doherty GM:
Duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann
Surg 1999, 231(6), 909-918.
Lambert LA, Shapiro SE, Lee JE, Perrier ND, Truong M, Wallace MJ, Hoff AO, Gagel
RF, Evans DB: Surgical treatment of hyperparathyroidism in patients with multiple
endocrine neoplasia type 1. Arch Surg 2005, 140, 374-382.
Lee C-H, Tseng L-M, Chen J-Y, Hsiao H-Y, Yang A-H: Primary hyperparathyroidism
in multiple endocrine neoplasia type 1: individual management with low recurrence
rates. Ann Surg Oncol, 2006. jan. 1. in press.
Lemmens IH, Forsberg L, Pannett AA, Meyen E, Piehl F, Turner JJ, van de Ven WJ,
Thakker RV, Larsson C, Kas K: Menin interacts directly with the homeobox-containing
protein Pem. Biochem Biophys Res Commun 2001, 286, 426-431.
Libé R, Bertherat J: Molecular genetics of adrenocortical tumours, from familial to
sporadic diseases. Eur J Endocrinol 2005, 153, 477-487.
Lopez Egido I, Cunningham J, Berg M, Oberg K, Bongcam-Rudloff E, Gobl A:
Menin’s interaction with Glial Fibrillary Acidic Protein and Vimentin suggests a role
for the intermediate filament network in regulating menin activity. Exp Cell Res 2002,
278, 175.
Miedlich S, Krohn K, Lamesch P Müller A, Paschke R: Frequency of somatic MEN1
gene mutations in monoclonal parathyroid tumours of patients with primary
hyperparathyroidism. Eur J Endocrinol 2000, 143, 47-54.
Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW,
Krankel C, LiVolsi VA, Gibbs D, Hua X, Roeder RG, Meyerson M, Hess JL: Menin
and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors.
PNAS 2005, 102(3), 749-754.

83
Mogensen J, Bahl A, Kubo T, Elanko N, Taylor R, McKenna WJ, Comparison of
fluorescent SSCP and denaturing HPLC analysis with direct sequencing for mutation
screening in hypertrophic cardiomyopathy. J Med Genet 2003, 40, e59.
Moon S-D, Park J-H, Kim E-M, Kim J-H, Han J-H, Yoo S-J, Yoon K-H, Kang M-I,
Lee K-W, Son H-Y, Kang S-K, Oh S-J, Kim K-M, Yoon S-J K, Park J-G, Kim I-J,
Kang HC, Hong S-W, Kim K-R, Cha B-Y: A novel IVS2-1G>A mutation causes
aberrant splicing of the HRPT2 gene in a family with Hyperparathyroidism-Jaw tumor
syndrome. J Clin Endocrinol Metab, 2005, 90(2), 878-883.
Morelli A, Falchetti A, Martineti V, Mark M, Friedman E, Brandi ML: MEN1 gene
mutation analysis in Italian patients with multiple endocrine neoplasia type 1. Eur J
Endocrinol 2000, 142, 131-137.
Multiple endocrine neoplasia, type 1; MEN1. OMIM Entry 131100
Mutch MG, Dilley WG, Sanjurjo F, DeBenedetti MK, Doherty GM, Wells Jr. SA,
Goodfellow PJ, Lairmore TC: Germline mutations in the multiple endocrine neoplasia
type 1 gene: Evidence for frequent splicing defects. Hum Mut 1999, 13, 175-185.
Namihira H, Sato M, Matsubara S, Ohye H, Bhuiyan M, Murao K, Takahara J: No
evidence of germline mutation or somatic deletion of the MEN1 gene in a case of
familial multiple endocrine neoplasia type 1 (MEN1). Endocr J 1999, 46(6), 811-6.
Nollau P, Wagener C: Methods for detection of point mutations: performance and
quality assessment. Clin Chem 1997, 43, 1114-1128.
Ohye H, Sato M, Matsubara S, Miyauchi A, Kishi-Imai K, Murao K, Takahara J: A
novel germline mutation of multiple endocrine neoplasia type 1 (MEN1) gene in a
Japanese MEN1 patient and her daughter. Endocr J 1999, 46(2), 325-9.

84
Olufemi SE, Green JS, Manickam P, Guru SC, Agarwal SK, Kester MB, Dong Q,
Burns AL, Spiegel AM, Marx SJ, Collins FS, Chandrasekharappa SC: Common
ancestral mutation in the MEN1 gene is likely responsible for the prolactinoma variant
of MEN1 (MEN1Burin) in four kindreds from Newfoundland. Hum Mut 1998, 11(4),
264-9.
Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T: Detection of polymorphisms
of human DNA by gel electrophoresis as single-strand conformation polymorphisms.
Proc. Natl. Acad. Sci. USA 1989, 86, 2766-2770.
Pannett AAJ, Kennedy AM, Turner JJO, Forbes SA, Cavaco BM, Bassett JHD,
Cianferotti L, Harding B, Shine B, Flinter F, Maidmetn CGH, Trembath R, Thakker
RV: Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial
isolated primary hyperparathyroidism. Clin Endocrinol 2003, 58, 639-646.
Pannett, AAJ, Thakker, RV: Multiple endocrine neoplasia type 1. Endocrin-Related
Cancer 1999, 6, 449-473.
Park J-H, Kim I-J, Kang H-C, Lee S-H, Shin Y, Kim K-H, Lim S-B, Kang S-B, Lee K-
U, Kim S-Y, Lee M-S, Lee M-K, Park J-H, Moon S-D, Park J-G, Germline mutations
of the MEN1 gene in Korean families with multiple endocrine neoplasia type 1 (MEN1)
or MEN1-related disorders. Clin Genet 2003, 64, 48-53.
Perrier ND, Villablanca A, Larsson C, Wong M, Ituarte P, Teh BT, Clark OH, Genetic
screening for MEN1 mutations in families presenting with familial primary
hyperparathyroidism, World J Surg 2002, 26, 907-913.
Petty EM, Green JS, Marx SJ, Taggart RT, Farid N, Bale AE: Mapping the gene for
hereditary hyperparathyroidism and prolactinoma (MEN1Burin) to chromosome 11q:
evidence for a founder effect in patients from Newfoundland. Am J Hum Genet 1994,
54(6), 1060-6.

85
Piotrowska K, Pellegata NS, Rosemann M, Fritz A, Graw J, Atkinson MJ: Mapping of a
novel MEN-like syndroma locus to rat cheomosome 4. Mamm Genome, 2004, 15(2),
135-41.
Poisson A, Zablewska B, Gaudray P: Menin interacting proteins as clues toward the
understanding of multiple endocrine neoplasia type 1. Cancer Lett 2003, 189, 1-10.
Ratineau C, Bernard C, Poncet G, Blanc M, Josso C, Fontaniere S, Calender A,
Chayvialle JA, Zhang CX, Roche C: Reduction of menin expression enhances cell
proliferation and tumorigenic in intestinal epithelial cells. J Biol Chem, 2004,
279(23):24477-84.
Roijers JFM, de Wit MJ, van der Luijt RB, Ploos van Amstel HK, Höppener JWM, Lips
CJM: Criteria for mutation analysis in MEN I-suspected patients: MEN I case-finding.
Eur J Clinical Invest 2000, 30, 487-492.
Rubin MR, Silverberg SJ: Editorial: HRPT sin parathyroid cancer: A piece of the
puzzle. J Clin Endocrinol Metab 2005, 90(9), 5505-5507.
Schnepp RW, Hou Z, Wang H, Petersen C, Silva A, Masai H, Hua X: Functional
interaction between tumor supressor menin and activatior of S-Phase Kinase. Cancer
Res, 2004, 64, 6791-6796.
Schussheim DH, Skarulis MC, Agarwal SK, Simonds WF, Burns AL, Spiegel AM,
Marx SJ: Multiple endocrine neoplasia type 1: new clinical and basic findings. Trends
Endocrinol Metab 2001, 12, 173-178.
Snabboon T, Plengpanich W, Siriwong S, Wisedopas N, Suwanalaikorn S,
Khovidhunkit W, Shotelersuk V: A novel germline mutation, 1793delG, of the MEN1
gene underlying multiple endocrine neoplasia type 1. Jap J Clin Oncol 2005, 35(5), 280-
282.

86
Sowa H, Kaji H, Canaff L, Hendy GN, Tsukamoto T, Yamaguchi T, Miyazono K,
Sugimoto T, Chihara K: Inactivation of menin, the product of the multiple endocrine
neoplasia type 1 gene, inhibits the commitment of multipotential mesenchymal stem
cells into the osteoblast lineage. J Biol Chem 2003, 278(23): 21058-21069.
Stalberg P, Grimfjard P, Santesson M, Zhou Y, Lindberg D, Gobl A, Öberg K, Westin
G, Rastad J, Wang S, Skogseid B: Transfection of the multiple endocrine neoplasia type
1 gene to a human endocrine pancreatic tumor cell line inhibits cell growth and affects
expression of JunD, δ-loke protein 1/preadipocyte factor-a, proliferating cell nuclear
antigen, and QM/Jif-1. J Clin Endocrinol Metab 2004, 89(5), 2326-2337.
Steger G: Thermal denaturation of double-stranded nucleic acids: prediction of
temperatures critical for gradient gel electrophoresis and polymerase chain reaction,
Nucl Acid Res 1994, 22, 2760-2768.
Stratakis CA, Ball DW: A concise genetic and clinical guide to multiple endocrine
neoplasias and related syndromes. J Pediatr Endocrinol Metab, 2000, 13, 457-465.
Sukhodolets KE, Hickman AB, Agarwal SK, Sukhodolets MV, Obungu VH, Novotny
EA, Crabtree JS, Chandrasekharappa SC, Collins FS, Spiegel AM, Burns AL, Marx SJ:
The 32-kilodalton subunit of Replication Protein A interacts with menin, the product of
the MEN1 tumor supressor gene. Mol Cell Biol. 2003, 23(2), 493-509.
Teh BT, Kyötola S, Farnebo F, Bergman L, Wong FK, Weber G, Haymard N, Larsson
C: Mutation analysis fo the MEN1 gene in multiple endocrine neoplasia type 1, familial
acromegaly and familial isolated hyperparathyroidism. J Clin Endocrinol Metab 1998,
83(8), 2621-2626.
Thakker RV: Editorial: Multiple endocrine neoplasia – syndromes of twentieth century.
J Clin Endocrinol Metab 1998, 83, 2617-2620.

87
Thakker RV: The molecular genetics of the multiple endocrine neoplasia syndromes.
Clin Endocrinol 1993, 38, 1-14.
The European Consortium on MEN1: Identification of the multiple endocrine neoplasia
type 1 (MEN1) gene. Hum Mol Genet 1997, 6, 1177-1183.
Tóth M, Rácz K, Jakab Cs, Gláz E: Multiplex endocrin neoplasiák. Orv Hetil 134(40),
2187-2192.
Tsukada T, Yamaguchi K, Kameya T: The MEN1 gene and associated diseases: An
update. Edocr Pathol 2001, 12(3), 259-274.
Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, Cougard P, Chambe
B, Montvernay C, Calneder A: Pituitary disease in MEN type 1 (MEN1): data from the
France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 2002, 87, 457-465.
Villablanca A, Wassif WS, Smith T, Höog A, Vierimaa O, Kassem M, Dwight T,
Forsberg L, Du Q, Learoyd D, Jones K, Stranks S, Juhlin C, Teh BT, Carling T,
Robinson B, Larsson C: Involvement of the MEN1 gene locus in familial isolated
hyperparathyroidism. Eur J Endocrinol 2002, 147, 313-322.
Wautot V, Khodaei S, Frappart L, Buisson N, Baro E, Lenoir GM, Calender A, Zhang
CX, Weber G: expression analysis of endogenous menin, the product of the multiple
endocrine neoplasia type 1 gene, in cell lines and human tissues. Int J Cancer 2000, 85,
877-881.
Wilhelmsson LM, Nordén B, Mukherjee K, Dulay MT, Zare RN: Genetic screening
using the colour change of a PNA-DNA hybrid-binding cyanine dye. Nucl Acid Res
2002, 30, e3.
Wrocklage C, Gold H, Kackl W, Buchfelder M, Fahlbusch R, Paulus W, Increased
menin expression in sporadic pituitary adenomas. Clin Endocrinol 2002, 56, 589-594.

88
Yaguchi H, Ohkura N, Takahashi M, Nagamura Y, Kitabayashi I, Tsukada T: Menin
missense mutants associated with multiple endocrine neoplasia type 1 are rapidly
degraded via the ubiquitin-proteasome pathway. Mol Cell Biol 2004, 24, 6569-6580.
Yaguchi H, Ohkura N, Tsukada T, Yamaguchi K: Menin, the multiple endocrine
neoplasia type 1 gene product, exhibits GTP-hydrolysing activity in the presence of the
tumor metastasis suppressor nm23. J Biol Chem 2002, 277, 38197-38204.
Yano M, Fukai I, Kobayashi Y, Mizuno K, Konishi A, Haneda H, Suzuki E, Endo K,
Fujii Y: ACTH secreting thymic carcinoid associated with multiple endocrine neoplasia
type 1. Ann Thorac Surg 2006, 81, 366-368.
Yazgan O, Pfarr CM: Differential binding of the Menin tumor suppressor protein to
JunD isoforms. Cancer Res 2001,61, 916-920.
Zhuang Z, Vortmeyer AO, Pack S, Hunag S, Pham TA, Wang C, Park W-S, Agarwal
SK, Debelenko LV, Kester MB, Guru SC, Manickam P, Olufemi S-E, Yu F, Heppner
C, Crabtree JS, Skarulis MC, Venzon DJ, Emmert-Buck MR, Spiegel AM,
Chandrasekharappa SC, Collins FS, Burns AL, Marx SJ, Jensen RT, Liotta LA,
Lubensky IA: Somatic mutations of the MEN1 tumor supressor gene in sporadic
gastrinomas and insulinomas. Cancer Res 1997, 57, 4682-4686.

89
SAJÁT KÖZLEMÉNYEK LISTÁJA
A disszertációhoz kapcsolódó közlemények:
K Balogh, A. Patócs, J Majnik, F Varga, Gy Illyés, L Hunyady, K Rácz. Unusual
presentation of multiple endocrine neoplasia type 1 in a young woman with novel
mutation of the MEN1 gene. J Hum Genet (2004) 49(7): 380-6.
K Balogh, A Patócs, J Majnik, K Rácz, L Hunyady. Genetic methods in the detection of
mutations responsible for multiple endocrine neoplasia type 1 (MEN1). Mol Gen Metab
(2004) 83(1-2): 75-82.
K Balogh, L Hunyady, A Patócs, Z Valkusz, R Bertalan, P Gergics, J Majnik, J Tőke,
M Tóth, N Szűcs, E Gláz, L Fűtő, J Horányi, K Rácz, Z Tulassay. Az 1-es típusú
multiplex endokrin neoplasia klinikai tünetei, diagnózisa és kezelése. A genetikai
vizsgálatok hazai tapasztalatai. Orv Hetil (2005) 146(43): 2191-7.
K Balogh, K Rácz, A Patócs, L Hunyady. Menin interacting proteins: elucidation of
menin function. Trends in Endocrinol & Metab (2006) 17(9): 357-64.
K Balogh, L Hunyady, A Patócs, P Gergics, K Rácz. High prevalence of novel
mutations of the MEN1 gene in Hungarian patients with multiple endocrine neoplasia
type 1. Clin Endocrinol (2006) Közlésre beküldve.
A disszertációtól független közlemények:
M Bor, K Balogh, Á Pusztai, G Tasnádi, L Hunyady. Identification of acute
intermittent porphyria carriers by molecular biologic methods. Acta Pysiol Hung (1999)
86: 147-53.

90
A Patócs, Z Valkusz, P Igaz, K Balogh, M Tóth, I Varga, K Rácz. Segregation of the
V804L mutation and S836S polymorphism of exon 14 of the RET gene in an extended
kindred with familial medullary thyroid cancer. Clin Genet (2003) 63: 219-23.
J Majnik, N Szücs, A Patócs, M Tóth, K Balogh, I Varga, E Gláz, K Rácz. Effect of
Single Doses of Dexamethason and Adrenocorticotrop Hormone Serum Bone Markers
in Healthy Subjects and in Patients with Adrenal Incidentalomas and Cushing’s
Syndrome. J Endocrinol Invest (2004) 27(8): 747-53.
A Patócs, É Karádi, M Tóth, I Varga, N Szücs, K Balogh, J Majnik, E Gláz, K Rácz.
Clinical and biochemical features of sporadic and hereditary pheochromocytomas: an
analysis of 41 cases investigated in a single endocrine center Eur J of Cancer Prev
(2004) 13(5): 403-9.
J Majnik, A Patócs, K Balogh, M Tóth, K Rácz. A rapid and simple method for
detection of Asn363Ser polymorphism of the human glucocorticoid receptor gene. J
Steroid Biochem Mol Biol (2004) 92(5):465-8.
P Gergics, A Patocs, J Majnik, K Balogh, A Szappanos, M Toth, K Racz. Detection of
the Bcl I polymorphism of the glucocoricoid receptor gene by single-tube allele-specific
polymerase chain reaction. J Steroid Biochem Mol Biol (2006) 100(4-5): 161-6.
J Majnik, A Patocs, K Balogh, M Toth, P Gergics, A Szappanos, A Mondok, G
Borgulya, P Panczel, Z Prohaszka, K Racz. Overrepresentation of the N363S variant of
the glucocorticoid receptor gene in patients with bilateral adrenal incidentalomas. J Clin
Endocrinol Metab (2006) 91(7): 2796-9.
V Szabo, K Balogh, I Suveges, K Racz, L Hunyady, ZZ Nagy. The role of lumican and
keratocan genes in persistent subepithelial corneal haze following excimer laser
photorefractive keratectomy. Mol Vis (2006) 12: 597-605.

91
KÖSZÖNETNYILVÁNÍTÁS
Munkám nem lehetett volna eredményes, ha nem segített volna éveken át a II.
Belklinikán és az Élettani Intézetben sok-sok ember, akiknek ezúton szeretnék
köszönetet mondani.
Prof. Dr. Tulassay Zsoltnak, aki programvezetőként lehetővé tette, hogy a II-es
Belklinikán dolgozhassak.
Témavezetőmnek, Prof. Dr. Rácz Károlynak, hogy mindvégig támogatott,
útmutatást adott munkám során, segítette és irányította szakmai fejlődésemet, és mindig
új lendületre sarkallt a nehezebb időszakokban is.
Prof. Dr. Hunyady Lászlónak, az Élettani Intézet igazgatójának, aki már egyetemista
éveim alatt, TDK témavezetőmként mindig állhatatosan, kitartóan mellettem állt, majd
PhD munkám során mindvégig lehetővé tette a molekuláris genetikai vizsgálatok egy
részének elvégzését az Élettani Intézetben.
Prof. Dr. Gláz Editnek, aki tapasztalatát velem megosztva figyelmemet számos
lényegi dologra felhívta, betegei révén számos érdekes, hasznos eset tanúja lehettem.
Mindazon klinikusoknak, akik azzal is segítették munkámat, hogy az általuk
diagnosztizált MEN 1 szindrómás betegeket a genetikai vizsgálatokra a
laboratóriumunkba irányították, és akikkel a közös munka mindvégig örömöt jelentett:
Dr. Fűtő László, Dr. Horányi János, Dr. Kiss Róbert, Dr. Szűcs Nikolette, Dr. Tóth
Miklós, Dr. Valkusz Zsuzsa.
Dr. Patócs Attilának, aki PhD éveim elején segített áthidalni a kezdeti nehézségeket,
közös munkánk során mindvégig szakmai és baráti segítséget nyújtott számomra.
A II. sz. Belgyógyászati Klinika molekuláris biológiai laboratóriumában dolgozó
PhD hallgatóknak: dr. Majnik Juditnak, dr. Gergics Péternek, Dr. Tőke Juditnak, Dr.
Bertalan Ritának, dr. Mondok Ágnesnek és a molekuláris biológiai laboratóriumban
dolgozó laborasszisztensünknek Krauszné Vaczula Máriának szakmai és baráti
támogatásukért.
A II. sz. Belgyógyászati Klinika endokrin munkacsoportja minden dolgozójának a
mindennapok közös munkájáért.

92
Köszönöm az Élettani Intézet laboratóriumában dolgozó PhD hallgatóknak és
asszisztenseknek, hogy már a kezdetektől segítették szakmai fejlődésemet,
megtanítottak a molekuláris biológia alapjaira és sok éven át mindig mindenben lehetett
számítani rájuk.
Végül köszönöm családomnak a legmesszebbekig menő támogatást, kitartásukat és
türelmüket.





























