Embed Size (px)
Citation preview

1
à plusieurs électrons
Table des matières :
Introduction ____________________________________________________________________ 2
1) Généralités___________________________________________________________________ 2 a) Position du problème_________________________________________________________________ 2 b) Cas de l'atome d'hydrogène____________________________________________________________ 3
2) L'approximation du champ central : vision qualitative des atomes à plusieurs électrons __ 4 a) Principe de la méthode _______________________________________________________________ 4 b) Niveaux d'énergie de l'atome___________________________________________________________ 5 c) Configurations électroniques des atomes : irrégularité dans le remplissage des couches. Les coefficients de Slater : une première approche _________________________________________________________ 7
3) La méthode de Hartree-Fock et le champ "self-consistent" __________________________ 9 a) Position du problème_________________________________________________________________ 9 b) Evaluation de E[Φ] _________________________________________________________________ 10 c) Minimalisation de E[Φ] ; détermination des équations de Hartree _____________________________ 12 d) Contenu physique des équations de Hartree-Fock ; Champ self-consistent ; Théorème de Koopman __ 15 e) Propriétés des potentiels et des fonctions d'onde de Hartree-Fock ; vérification de la légitimité de l'approximation ______________________________________________________________________ 16 f) Retour sur la méthode des coefficients de Slater ; comparaison des résultats _____________________ 17 g) Conclusion________________________________________________________________________ 18
4) Corrections à l'approximation du champ central. Couplage L-S _____________________ 19 a) Rappel sur l'hamiltonien d'un atome à plusieurs électrons ___________________________________ 19 b) Traitement perturbatif du couplage de Russel-Saunders (L-S) ________________________________ 20 c) Classification énergétique des termes; règles de Hund; schéma d'éclatement des termes____________ 23
Conclusion ____________________________________________________________________ 24
Annexe 1: résolution de l'atome d'hydrogène _______________________________________ 25
Annexe 2: compléments sur la méthode des coefficients de Slater ______________________ 30
Annexe 3: caractère sphérique du potentiel de Hartree-Fock pour un atome à sous couches pleines ________________________________________________________________________ 33
Annexe 4: Orbitales atomiques du néon ____________________________________________ 36
Annexe 5: détermination du potentiel central dans le modèle statistique de Thomas-Fermi_ 38
Annexe 6: Classification périodique des éléments ____________________________________ 44
Bibliographie __________________________________________________________________ 45

2
Introduction Il est intéressant d'étudier les atomes, et en particulier leur structure électronique, afin de
pouvoir expliquer et prédire leur comportement lorsqu'ils engagent une liaison chimique avec d'autres atomes, ou bien leurs propriétés magnétiques, leur couleur. Cependant comme il s'agit de systèmes à plus de deux corps en interaction électrostatique (noyau et Z électrons), la résolution analytique exacte des atomes à plusieurs électrons est impossible. En effet, comme l'hamiltonien du système comprend un terme d'interaction coulombienne entre les électrons, le potentiel dépend fortement de l'état des différents électrons. De plus, ce terme est loin d'être petit devant les autres termes de l'hamiltonien, ce qui implique qu'on ne peut pas le traiter en perturbation. Il s'agit donc d'utiliser une méthode variationnelle.
La point de départ de toutes les méthodes de résolution de l'équation de Schrödinger pour les atomes à plusieurs électron s'appelle l’approximation du champ central. L'idée de base de cette approximation est que chacun des électrons se meut dans un potentiel sphérique qui prend en compte l'attraction du noyau et les contributions des autres électrons, et donc que le problème se réduit à un problème de Z particules indépendantes dans un potentiel sphérique effectif. Nous allons tous d'abord nous intéresser aux diverses propriétés de ce potentiel et en déduire un certain nombre de résultats qui peuvent être compris sans le déterminer de manière précise; puis nous nous intéresserons à diverses méthodes qui permettent l'étude de ce potentiel, en particulier la méthode de Hartree, élaborée en 1928 et généralisée en 1930 par Slater et Fock afin que les résultats respectent le principe de Pauli. Nous conclurons par les corrections à apporter aux résultats issus de l'approximation du champ central.
1) Généralités
a) Position du problème
Considérons un atome à Z électrons. Il est constitué d'un noyau de masse Nm et de charge
Zq+ , et de Z électrons de charge Cq 1910.6,1 −−=− et de masse kevme 511= .
La masse du noyau étant très supérieure à la masse des électrons, on peut tout de suite se placer dans un modèle où le noyau est fixe et où seuls les électrons sont mobiles. En négligeant les effets relativistes et les effets de couplage avec les spins des noyaux et des électrons, l'hamiltonien décrivant le mouvement s'écrit :
∑ ∑ ∑= = < −
+−=Z
i
Z
i ji jiie
i
RR
e
R
Ze
m
PH
1 1
222
2, avec
0
2
4πεq
e =
Les deux premiers termes correspondent respectivement au terme d'énergie cinétique des
électrons (qui ont été numérotés arbitrairement de 1 à Z) et au terme d'énergie potentielle d'interaction entre le noyau et chacun des électrons. Le troisième terme, quant à lui, correspond à l'énergie potentielle d'interaction électrostatique entre les divers électrons.
On remarque tout de suite que, si ce troisième terme n'apparaissait pas dans l'hamiltonien, on pourrait aisément déterminer les fonctions d'ondes et les énergies solutions de l'équation
Ψ=Ψ EH (1). En effet les énergies ne sont que la somme des énergies des Z électrons placés
dans le potentiel coulombien r
Ze2− et les fonctions d'ondes le produit tensoriel des Z

3
fonctions d'onde correspondantes, qui serait bien entendu antisymétrisé pour satisfaire au principe de Pauli.
C'est donc bien le dernier terme qui est responsable de l'impossibilité à résoudre de manière analytiquement exacte l'équation (1).
On pourrait alors envisager de traiter ce terme en perturbation de l'hamiltonien ne comportant que les deux premiers termes, mais une rapide évaluation du rapport ρ entre le
troisième et le second terme montre que, en considérant que RRRR iji ≈≈− , on a :
( )
2
12
1
Z
ZZ −≈ρ , terme qui vaut
4
1 pour Z=2 et qui tend vers
2
1 pour les Z grands. Il
paraît donc difficile de considérer ce terme comme une perturbation, et les résultats obtenus de cette manière seraient probablement très éloignés de la réalité. Il s'agit donc de trouver une méthode d'approximation plus fine.
b) Cas de l'atome d'hydrogène
Avant d'aller plus loin, il semble approprié d'étudier le comportement de l'atome
d'hydrogène, dont on peut à priori déterminer les états stationnaires et les énergies qui leurs sont associées.
L'hamiltonien de l'atome d'hydrogène s'écrit :
r
e
m
PH
eH
22
2−= ou, ce qui revient au même :
r
e
mH
eH
22
2−∆−= h
(1H)
La résolution exacte de l'équation aux valeurs propres Ψ=Ψ EH H est donnée dans
l'annexe 1. Nous nous contenterons ici d'examiner les résultats obtenus. Au cours de la résolution, trois nombres quantiques apparaissent : - n : nombre quantique radial, tel que 1≥n - l : nombre quantique azimutal, tel que 10 −≤≤ nl - m : nombre quantique magnétique, tel que lml +≤≤− Par ailleurs, on peut considérer deux nombres quantiques supplémentaires, le spin s de
l'électron et sa projection sur l'axe z, tels que 2
1=s et
−∈
2
1;
2
1sm .
Le traitement de ce problème donne directement le diagramme d'énergie de l'atome
d'hydrogène, c'est à dire les valeurs propres correspondant aux états stationnaires de l'hydrogène :
2n
EE I
n −= , avec eVE I 6,13= , ce qui peut être représenté par le diagramme 1.

4
2) L'approximation du champ central : vision qualitative des atomes à plusieurs électrons Nous avons vu au paragraphe 1)a) les difficultés liées aux interactions entre électrons et la
nécessité de trouver une approximation plus fine que le traitement en perturbation du terme
∑< −ji ji RR
e2
(interactions électron/électron).
Une approche qualitative du problème, connue sous le nom d’approximation du champ central, permet d'obtenir certains résultats, en particulier une idée, qualitative certes, mais assez exacte, du spectre énergétique des atomes.
Cette vision étant semi-classique, on utilisera ici des termes de position des électrons et autres notions qui n'auraient pas de sens dans un traitement purement quantique.
a) Principe de la méthode
Considérons un électron quelconque d'un atome polyélectronique. En première
approximation, on peut supposer que cet électron "voit" le potentiel électrostatique des autres
électrons comme un écrantage du potentiel créé par le noyau r
ZeV
2
= . Dans le cadre de cette
approximation, on peut donc dire que l'électron n'est plus soumis à un potentiel total
∑≠ −
+=ij jii RR
e
R
ZeV
22
, mais à un potentiel effectif ( )ic RV appelé aussi potentiel central.
0 -0,85
1s
2s 2p
3s 3p 3d
4s 4d 4f 4p
-1,51
-3,4
-13,6
E (eV)
Diagramme 1: Niveaux d'énergie de l'atome d'hydrogène. On remarque la dégénérescence accidentelle de ces niveaux, c’est-à-dire que leur énergie ne dépendent que du nombre quantique radial n. Il est intéressant de noter que cette dégénérescence se lève dans le cas des atomes à plusieurs électrons.

5
Ceci constitue bien entendu une approximation, étant donné que le mouvement de l'électron i influence le mouvement des autres électrons, et donc le potentiel ( )ic RV , et que
s'il se trouve au voisinage immédiat d'un autre électron, le potentiel auquel il est soumis ne peut plus être considéré comme central.
Cependant — et cette idée est d'autant plus valable en mécanique quantique du fait de la délocalisation spatiale des électrons — l'idée d'un potentiel moyen apparaît ainsi naturellement.
Ces considérations amènent donc naturellement à écrire l'hamiltonien sous la forme :
( ) WRVm
PH
Z
iic
e
i +
+= ∑
=1
2
2, où ( )∑∑∑
=<=
−−
+−=Z
iic
ji ji
Z
i i
RVRR
e
R
ZeW
1
2
1
2
Si le potentiel central ( )ic RV est bien choisi, W jouera le rôle d'une perturbation de
( )∑=
+=
Z
iic
e
i RVm
PH
1
2
0 2.
On voit tout de suite que la diagonalisation de 0H revient à un problème de Z électrons
indépendants dans un potentiel central : c'est donc un problème de particules indépendantes. Nous verrons plus tard le problème de l'existence et de la détermination d'un tel potentiel.
b) Niveaux d'énergie de l'atome
Bien que la détermination du potentiel ( )ic RV constitue un problème assez ardu, on peut
tout de même se faire une idée de son comportement asymptotique. En effet, en raison de la symétrie sphérique du problème dans le cadre de l'approximation
du champ central, le théorème de Gauss nous permet d'affirmer que :
- pour r grand, l'électron est soumis à un potentiel r
erVc
2
)( ≈
- pour r petit, l'électron est soumis à un potentiel r
ZerVc
2
)( ≈ .
Ces considérations nous donnent l'allure du potentiel ( )rVc (diagramme 2)
r
e2
−r
Vc(r)
r
Ze2
−
Diagramme 2: allure du potentiel Vc(r). On a aussi représenté ses asymptotes en 0 et à l'infini

6
De plus, ces considérations permettent de se faire une première idée du spectre d'énergie
des atomes. Comme ( )rVc n'est plus en r
1, les énergies dépendent non seulement de n , mais
aussi de l . Cependant, comme ( )rVc est toujours un potentiel central, les énergies ne
dépendent toujours pas de m . On obtient donc des niveaux d'énergies lnE , caractérisés par deux nombres quantiques et
dont la dégénérescence s'écrit ( )122, += lg ln en tenant compte du spin de l'électron qui peut
toujours prendre deux valeurs
±=
2
1sm .
On peut par ailleurs ébaucher quelques règles de succession de ces niveaux : - les énergies lnE , croissent avec n pour une valeur de l donnée.
- les énergies lnE , croissent avec l pour une valeur de n donnée.
En fait, ces deux lois se généralisent par la règle empirique de Klechkovski : - l'énergie croît selon les valeurs croissantes de ( )ln + - En cas d'égalité, on prend la plus basse valeur de n Cette règle empirique permet d'élaborer le diagramme 3:
( )ln
E ln
+,
n=1 n=2 n=3 n=4 n=6 n=5 n=7
1s
2s
2p
3s
3p
3d 4s
4p
4d
4f
5s
5d
5p
6s
6d
6p
7s 5f
(2)
(2)
(2)
(2)
(2)
(2)
(2)
(6)
(6)
(6)
(6)
(6)
(10)
(10)
(10)
(10)
(14)
(14)
Diagramme 3: niveaux d'énergie d'un atome à plusieurs électrons. On remarque la levée de la dégénérescence accidentelle observée dans le cas de l'atome d'hydrogène. On remarque également que certains niveaux ont des énergies très proches, et que leurs positions relatives risquent de changer avec Z.

7
Ce schéma est purement qualitatif, et il n'a aucune prétention à respecter quelque échelle
d'énergie que ce soit. Cependant, il donne une première idée de l'enchaînement des niveaux d'énergie des atomes.
Par ailleurs, on peut également avoir une petite idée des fonctions d'onde à l'ordre zéro en
perturbation de ( )∑=
+=
Z
iic
e
i RVm
PH
1
2
0 2. En effet on voit immédiatement que, comme pour
l'atome d'hydrogène, [ ] [ ] 0,, 200 == LHLH
r et donc, par séparation des variables, on aura des
fonctions d'onde du type : ( )
sS mm
llnmmln YrR χθ ϕ=Ψ ,)(,,, , c'est à dire que l'on retrouve la même dépendance
angulaire et de spin que pour l'atome d'hydrogène. Seule "l’expansion" radiale (en fait la densité de probabilité de présence de l'électron à une distance r ) du noyau est modifiée.
c) Configurations électroniques des atomes : irrégularité dans le remplissage des couches. Les coefficients de Slater : une première approche
La première application de ces résultats, en particulier du diagramme 3, est de déterminer
la configuration électronique dans l'état fondamental des atomes. Il suffit en fait de remplir les couches successives avec les Z électrons de l'atome en respectant le principe de Pauli (deux électrons ne peuvent avoir les mêmes quatre nombres quantiques smmln ,,, ).
On en déduit par exemple que la configuration électronique de l'hélium dans son état fondamental est 21s , où :
- 1 désigne la valeur de n - s désigne la valeur de l par les notations spectroscopiques
( ...,:3,:2,:1,:0 fldlplsl ==== )
- 2 désigne le nombre d'électrons dans cette couche (ensemble des états individuels ayant la même énergie)
En allant plus loin dans le tableau périodique1, on trouve par exemple que le carbone
( 6=Z ) a la configuration électronique : 222 221 pss L'ensemble des configurations électroniques des atomes est donné dans la classification
périodique des éléments1. Cependant, il est évident que dans le cadre de l'approximation du champ central, les
énergies ainsi que l'ordre des couches vont dépendre fortement de Z , étant donné que le potentiel )(rVc auquel est soumis l'électron dépend des interactions entre électrons, en
particulier pour les niveaux d'énergies proches comme s4 et d3 ou bien f4 , d5 et s6 .
1 Cf. annexe 6

8
On peut alors utiliser une méthode simple, qui consiste à dire que le potentiel )(rVc s'écrit
r
eZrV eff
c
2
)( = , où en fait on remplace, pour un électron i , l'ensemble physique des
interactions attraction par le noyau/répulsion par les autres électrons par l'ensemble virtuel d'une attraction par un noyau ne comprenant que effZ protons, avec ZZ eff < , les autres
électrons exerçant un effet électrique d'écran. On définit donc, pour chaque électron ij ≠ , une constante d'écran ijσ qui caractérise son
action répulsive vis-à-vis de l'électron i et on a la relation :
∑−=j ijeff ZiZ σ)( .
Le calcul des ijσ se fait grâce au tableau empirique suivant, appelé table des coefficients
de Slater :
Contribution des autres électrons à la constante d'écran Couche n
Origine de l'électron considéré
Couches <n-1
Couche =n-1 s, p d f
Couches >n
s ou p 1,00 0,85 0,35 0 0 0 d 1,00 1,00 1,00 0,35 0 0 f 1,00 1,00 1,00 1,00 0,35 0
De plus, comme ce modèle revient à remplacer un atome polyélectronique par une
superposition d'atomes hydrogénoïdes avec un noyau comportant effZ protons, on associe à
chaque orbitale iχ l'énergie 2
2
*n
ZKE eff
i −= , avec eVK 6,13−= et nn =* si ]3,1[∈n ,
7,3* =n si 4=n , 4* =n si 5=n , 2,4* =n si 6=n . Grâce à ces coefficients, on peut rendre compte dans ce modèle : - que pour 19=Z , on remplit bien la couche s4 avant la couche d3 . En effet, l'énergie
d'un électron placé sur la couche s4 dans l'atome de potassium vaut eV8,4− alors que placé sur la couche s3 son énergie vaut eV5,1− . De même pour l'atome de calcium ( 20=Z ), on trouve comme énergies : eVdeVs 04,6:3;0,8:4 −− .
- qu'à partir de 21=Z , la couche d3 se stabilise en se remplissant. En effet, on trouve comme énergie dans la configuration électronique 12 34 ds les énergies :
eVdeVs 6,13:3;94,8:4 −− : c'est donc la couche d3 qui a la plus basse énergie. Par contre, ce modèle ne rend pas compte de l'anomalie du remplissage électronique pour
24=Z (configuration 15 4,3 sd au lieu de 24 4,3 sd ). En fait ceci semble normal puisque ce modèle ne prend pas en compte l'énergie d'appariement des électrons alors que c'est ce phénomène qui intervient ici (l'énergie nécessaire pour apparier les deux électrons de la couche s4 est supérieure à celle nécessaire pour placer un électron supplémentaire sur la couche d3 , avec les effets d'écrantage que cette configuration implique sur l'énergie de cette couche).

9
De plus, et ceci témoigne bien de ses limites, ce modèle ne lève pas totalement la dégénérescence accidentelle de l'hydrogène puisqu'il traite de manière identique les sous couches s et p1.
3) La méthode de Hartree-Fock et le champ "self-consistent" Cette méthode est une méthode plus élaborée qui prend en compte des considérations
physiques similaires à l'approximation du champ central. Le point de départ de cette méthode est de considérer en effet que chaque électron est
soumis à un potentiel qui prend en compte l'attraction du noyau écranté par la répulsion due aux autres électrons. Par conséquent, chaque électron du système polyélectronique sera décrit par sa propre fonction d'onde et sera donc dans un état énergétique donné.
Cette méthode, proposé par Hartree, consiste en une méthode d'itération originale fondée sur l'auto-cohérence (self-consistency) du champ électronique. Cependant, la méthode de Hartree ne satisfaisait pas au principe d'exclusion de Pauli, c'est à dire que la fonction d'onde totale des électrons devait être antisymétrique. La généralisation de cette méthode a en fait été apportée par Slater et Fock en 1930, et c'est cette généralisation que nous allons présenter.
a) Position du problème
On commence par considérer l'hamiltonien du système qui s'écrit encore :
21 HHH += où,
∑=
=Z
iihH
11
i
re
i r
Ze
mh
i
22
2−∆−= h
et
∑=<
=Z
ji ijrH
12
1,
ji
ijrr
r−
= 1
On retrouve bien ici les termes du 1)a) : le premier terme représente une somme de Z
hamiltoniens à un corps identiques, qui correspondent à la somme de l'énergie cinétique et de l'énergie potentielle de chaque électron (encore une fois numérotés arbitrairement de 1à Z), et
le second terme la somme de ( )
2
1−ZZ hamiltoniens à deux corps identiques qui
correspondent aux interactions électron-électron. On va ici partir d'une méthode variationnelle, et donc nous allons pour cela prendre une
fonction d'essai Φ .
1 Cf. annexe 2

10
Nous pouvons d'ores et déjà dire que, en vertu de la nature de la méthode variationnelle, que si l'on considère l'énergie fondamentale d'un atome 0E , on aura [ ] ΦΦ=Φ< HEE0 .
Afin de satisfaire directement au principe de Pauli, nous prendrons la fonction d'essai sous
la forme d'un déterminant de Slater :
( )
( ) ( ) ( )( )
( ) ( ) ( )
..
...
.....
.....
....
..
!
1,....,,
2
111
21
ZZZ
Z
quququ
qu
quququ
Zqqq
υβα
α
υβα
=Φ
La fonction sera donc automatiquement antisymétrique. Ici les nombres υβα ....,,, .
représentent un jeu de nombre quantique ( )smmln ,,, . Par ailleurs, on impose les conditions
d'orthonormalisation λµµλµλ δ== ∫ ∗ dququuu )( , ce qui implique que Φ sera également
orthonormée.
b) Evaluation de E[Φ]
Réécrivons Φ sous une forme plus compacte :
( ) ( ) ( ) ( )[ ]
H
Zu
AZ
quququZ
Φ=
−=Φ ∑!
....1!
121
σβα
σ σ
où HΦ est le simple produit tensoriel des Z états individuels des électrons :
( ) ( ) ( )ZuH quququ ....21 βα=Φ
et où A est l'opérateur d'antisymétrisation :
( )∑ −=σ
σ σ1!
1
ZA .
Cet opérateur est hermitique et est un projecteur, ce qui signifie que AA =2 . De plus, les deux hamiltoniens 1H et 2H sont invariants dans l'échange des électrons, c'est
à dire que [ ] 2,1∈∀= pHH pp σσ , et donc commutent avec A .
Ces remarques nous permettent immédiatement de calculer l'énergie [ ]ΦE :
[ ] ΦΦ+ΦΦ=Φ 21 HHE .

11
Le premier terme se calcule aisément :
HH
HH
HH
AHZ
AHZ
AAHZH
ΦΦ=
ΦΦ=
ΦΦ=ΦΦ
1
21
11
!
!
!
On en déduit ensuite que :
( )
∑∑
∑∑
=
ΦΦ=
ΦΦ−=ΦΦ
=
=
λλλ
σ
σ σ
)()(
1
1
11
iii
N
iHiH
Z
iHiH
quhqu
h
hH
On a utilisé ici le fait que les ih étaient des hamiltoniens à un corps et donc que seul la
permutation identité ne faisait pas apparaître dans HiH h ΦΦ σ des termes en
( ) ( )∫ =∗ 0dqququ λµ si µλ ≠ et ensuite le fait que dans les termes qui restent tous les termes
qui n'avaient pas la coordonnée i donnaient par intégration 1 (on somme ensuite sur λ car tous les hamiltoniens ih sont identiques et on a attribué à chaque électron arbitrairement noté
i un état λ ). En définissant alors )()( iii quhquI λλλ = comme la valeur moyenne de l'hamiltonien ih
dans l'état λu , on obtient que
∑=ΦΦλ
λIH1
En fait, λI représente la somme de l'énergie cinétique de l'électron i dans l'état λ et de
l'énergie potentielle due à l'attraction du noyau. Elle correspond en ce sens à une énergie hydrogénoïde.
Le second terme, lui, est un peu plus compliqué, mais se traite de la même manière. On a également :
HH AHZH ΦΦ=ΦΦ 12 ! .
Et par conséquent :
( )
( )∑
∑∑
=<
=<
Φ−Φ=
ΦΦ−=ΦΦ
N
jiHij
ijH
Z
jiH
ijH
Pr
rH
1
12
11
11
σ
σ σ
où ijP est l'opérateur permutation qui interchange les coordonnées (spatiales et de spin) des
électrons i et j .

12
Ensuite, en effectuant le même raisonnement que pour le premier terme, on obtient que :
( ) ( ) ( ) ( ) ( ) ( ) ( ) ( )∑
−=ΦΦ
µλλµµλµλµλ
,2
11ji
ijjiji
ijji ququ
rququququ
rququH
La somme sur λ et µ se faisant sur les ( )
2
1−ZZ paires d'orbitales.
Définissons alors le terme direct :
( ) ( ) ( ) ( )jiij
ji ququr
ququJ µλµλλµ1= , qui représente la valeur moyenne de
l'interaction entre l'électron i dans l'orbitale λ et l'électron j dans l'orbitale µ . Nous introduisons également le terme d'échange :
( ) ( ) ( ) ( )jiij
ji ququr
ququK λµµλλµ1= , qui est l'élément de matrice de l'interaction
ijr
1
entre les deux états ( ) ( )ji ququ µλ et ( ) ( )ji ququ λµ obtenu en interchangeant les électrons i et
j . On remarque immédiatement que λµJ et λµK sont réels et sont symétriques en λ et µ .
On a donc le résultat suivant :
[ ] [ ]∑∑∑ −+=Φλ µ
λµλµλ
λ KJIE2
1
c) Minimalisation de E[Φ] ; détermination des équations de Hartree
Maintenant que nous avons une expression de E[Φ], nous devons utiliser le fait que cette
énergie est stationnaire, et donc qu'elle est minimale par rapport aux variations des orbitales atomiques λu , qui sont soumises aux 2Z conditions d'orthonormalisation. Nous introduisons
donc 2Z multiplicateurs de Lagrange que nous noterons λµε . L'équation aux variations s'écrit
alors : 0=− ∑∑
λ µλµλµδεδ uuE
Avant de procéder à l'analyse de cette relation et de ces conséquences, intéressons-nous
aux coefficients λµε .
L'équation aux variations montre clairement que ∗= λµλµ εε , et donc que les 2Z
multiplicateurs de Lagrange peuvent être considérés comme une matrice hermitienne Ξ , et donc diagonalisable.

13
Ecrivons alors la transformation unitaire qui permet de diagonaliser Ξ : 1’ −Ξ=Ξ UU , où ( )λµUU = est une matrice carrée unitaire.
Si l'on veut trouver l'expression des orbitales dans cette nouvelle base, il faut écrire la
relation : ∑=
µµµλλ uUu’ .
Le nouveau déterminant de Slater utilisé comme fonction d'essai s'écrira alors : ( )Φ=Φ Udet’ . Or, comme U est unitaire, on a 1det =U , donc la fonction d'essai ne dépend pas de la
base λ , µ choisie. On peut donc directement prendre la fonction d’essai Φ dans la base où Ξ est diagonale, ce qui simplifie l'équation aux variations à :
0=− ∑λ
λλλδδ uuEE , où les λE sont tels que ( )λµλδE=Ξ’ , c'est-à-dire qu'ils
représentent les valeurs propres de la matrice Ξ . En utilisant alors l'expression de [ ]ΦE et les définitions de λµλµλ KJI ,, , on obtient un
système d'équations integro-différentielles :
( ) ( ) ( ) ( )
( ) ( ) ( ) ( ) υβαµλλλµµ
λµ
λµ
µµλ
,....,,,1
1
2
22
==
−
+
−∆−
∑∫
∑∫
∗
∗
iijjij
j
ijjij
jii
re
quEqudqqur
qu
qudqqur
ququr
Ze
m i
h
Ce système d'équation est connu sous le nom d'équations de Hartree-Fock. Comme la
sommation sur jq représente une sommation sur les variables d'espace et de spin, on peut, en
posant ( ) ( ) λχλλ
Smii ruqu,2/1
=
et en utilisant la relation d'orthonormalisation des fonctions de spin,
µλµλ δχχssss mmmm ,,2/1,2/1
=
réduire les équations de Hartree-Fock à la partie spatiale des orbitales :
( ) ( ) ( ) ( )
( ) ( ) ( ) ( ) υβαµλδ λλµµ
λµ
λµ
µµλ
µλ ,....,,,1
1
2
,
22
==
−
+
−∆−
∑ ∫
∑∫
∗
∗
iijjij
jmm
ijjij
jii
re
quEqudqqur
qu
rurdrur
rurur
Ze
m
SS
i
rrrrrh

14
On peut formaliser ces équations de manière plus compacte en définissant l'opérateur
direct :
( ) ( ) ( ) ( )id
jjij
jid rVdqqu
rquqV
rµµµµ ≡= ∫ ∗ 1
qui est simplement la répulsion électrostatique de l'électron j placé dans l'orbitale µ , ainsi que l'opérateur d'échange :
( ) ( ) ( ) ( ) ( )ijjij
jiiex qudqqf
rquqfqV µµµ
= ∫ ∗ 1
, où ( )iqf est une fonction quelconque.
Pour ( ) ( )ii quqf λ= , on a alors :
( ) ( ) ( ) ( ) ( )
( ) ( ) ( )
( ) ( ) µµλ
µµλ
χδ
χδ
λµ
µλµ
µλµλµ
SSS
SSS
miiex
mm
mijjij
jmm
ijjij
jiiex
rurV
rurdrur
ru
qudqqur
ququqV
,2/1,
,2/1,
1
1
rr
rrrr
=
=
=
∫
∫
∗
∗
où on a défini de manière similaire un opérateur d'échange qui agit uniquement sur les variables spatiales :
( ) ( ) ( ) ( ) ( )ijjij
jiiex rurdrf
rrurfrV
rrrrrrµµµ
= ∫ ∗ 1
Finalement, les équations de Hartree-Fock s'écrivent :
( ) ( ) ( )iiiex
id
ir
e
quEquqVrVr
Ze
m i λλλµ
µµ
µ =
−+−∆− ∑∑ )(
2
22rh
Enfin, en définissant les potentiels direct et d'échange : ( ) ( )∑=
µµ id
id rVr
rV et ( ) ( )∑=
µµ iex
iex qVqV
ainsi que le potentiel global de Hartree-Fock :
( ) ( ) ( )iex
id
ii qr
r
Zeq VVV −+−= r
2
les équations de Hartree-Fock prennent la forme étonnamment simple :
( ) ( ) ( )iiire
quEquqm i λλλ =
+∆− V
2
2h

15
d) Contenu physique des équations de Hartree-Fock ; Champ self-consistent ; Théorème de Koopman
On a vu que les équations se réduisaient à une expression extrêmement simple, et qui
ressemble au premier abord à une triviale équation aux valeurs propres pour chacune des fonctions d'onde ( )iquλ . En fait, cette équation n'est pas une équation aux valeurs propres car
le potentiel ( )iqV dépend directement des autres fonctions d'ondes. En fait, pour résoudre le
système d'équation intégro-différentielles de Hartree-Fock, on procède par itérations successives. On intuite des fonctions d'ondes individuelles )1()1()1( ,...,, υβα uuu et on calcule par ce
biais le potentiel ( )iq)1(V . On réinjecte cette expression dans le système que l'on résout, ce
qui nous donne une autre série d'orbitales )2()2()2( ,...,, υβα uuu , ce qui nous donne un autre
potentiel ( )iq)2(V et ainsi de suite. On itère la procédure jusqu'à obtenir un potentiel
( )in q)(V qui est identique au potentiel de l'étape précédente ( )i
n q)1( −V . Le potentiel ainsi
obtenu est connu sous le nom de champ "self-consistent" (ou auto-cohérent). Bien que les équations de Hartree-Fock ne soient pas des "vraies" équations aux valeurs
propres, on peut tout de même interpréter l'hamiltonien ( )ire
HF qm
hi
V+∆−= rh
2
2
comme
l'opérateur énergie d'un électron dans l'état λu . En effet, si l'on regarde d'un peu plus près les
définitions du potentiel direct et du potentiel d'échange, on s'aperçoit aisément que: ( ) ( ) ( ) ( )ii
exii
d quqVquqV λλλλ =
En reprenant l'expression originale des équations de Hartree-Fock, on s'aperçoit alors qu'il
n'y a pas d'auto-contribution ( µλ = ) énergétique au potentiel. On peut donc alors définir les potentiels direct et d'échange modifiés de la manière
suivante: ( ) ( )∑
≠
=λµ
µλ id
id rVr
rrV et ( ) ( )∑
≠
=λµ
µλ iex
id qVqV
de telle manière que l'équation de Hartree-Fock va s'écrire:
( ) ( ) ( ) ( )iiiex
id
ir
e
quEquqrr
Ze
m i λλλλλ =
−+−∆− VV
rhr
22
2
On s'aperçoit alors qu'en plus du terme d'énergie cinétique et du terme d'énergie
d'attraction nucléaire, l'hamiltonien HFh contient un terme dλV qui représente le potentiel
moyen dû aux 1−Z autres électrons, et un terme en exλV qui prend en compte les effets
d'échange entre l'état λu et les autres états occupés par les électrons.
Si on interprète les résultats de cette façon, la quantité λE prend bien la signification d'une
valeur propre d'un système à un électron indépendant. Rappelons que c'est là la base de l'approximation du champ central.

16
Pour donner une signification plus précise à l'énergie λE , on peut écrire, en utilisant la
forme première des équations de Hartree-Fock, que: ∑ −+=
µλµλµλλ KJIE , ce qui donne, en sommant sur tous les λ :
ΦΦ+ΦΦ=
−+= ∑ ∑∑∑
21 2 HH
KJIEλ λ µ
λµλµλλ
λ
par définition des opérateurs énergie nucléaire, énergie directe et énergie d'échange. On s'aperçoit alors que: [ ] ΦΦ−=Φ ∑ 2HEE
λλ , et on s'aperçoit, non sans inquiétude
d'ailleurs, que l'énergie totale n'est pas la somme des énergies individuelles. Ceci est dû au fait que lorsque l'on somme les énergies individuelles des électrons, les énergies d'échange sont comptées deux fois. Il faut donc retirer le terme ΦΦ 2H de l'expression de [ ]ΦE .
Maintenant, imaginons que l'électron λ soit retiré du système à Z électrons (il peut s'agir de la première ionisation d'un atome, par exemple Na). Si l'on regarde l'expression de [ ]ΦE déterminer au paragraphe 3)b), et en considérant que les fonctions d'ondes des électrons restants sont les mêmes dans les systèmes à Z et 1−Z électrons, on a:
[ ] λµ
λµλµλ EKJIEE ZZ =−+=− ∑−1
La quantité λE représente donc approximativement l'énergie nécessaire pour ôter un
électron de l'orbitale λu , soit en fait l'énergie d'ionisation de l'électron λ . Ce résultat est
connu sous le nom de théorème de Koopman. Cette valeur, cependant, ne peut pas être la valeur exacte, puisque nous n'avons pas tenu compte du réarrangement des orbitales résultant du départ de l'électron λ . Par ailleurs, il est important de noter que λE n'est pas
nécessairement un majorant de l'énergie d'ionisation, bien que ZE et 1−ZE le soient, car nous avons pris la différence de ces deux termes.
e) Propriétés des potentiels et des fonctions d'onde de Hartree-Fock ; vérification de la légitimité de l'approximation
Regardons à présent les équations de Hartree-Fock simplifiée. La première chose que l'on
remarque est que, pour un état donné, tous les électrons se trouvent soumis au même potentiel. Ceci vient déjà corroborer l'approximation du champ central : on retrouve ici l'idée d'un potentiel moyen qui ne dépendrait que de l'état dans lequel se trouve les électrons.
Par ailleurs, on peut également démontrer que, pour les atomes ou ions qui ont toutes leurs sous couches remplies (He, Li+, Be, B+, C2+, Ne, etc.), le potentiel de Hartree-Fock est sphérique1, et donc que l'on vérifie la validité de l'approximation du champ central après le traitement variationnel, en tout cas pour ces atomes. Par ailleurs, comme en général on s'intéressera à l'état fondamental des atomes, on peut considérer que tous les atomes auront un potentiel de Hartree sensiblement sphérique, car ils n'auront qu'une seule sous couche non remplie, et l'écart à la sphère sera négligeable, et ceci sera d'autant plus vrai que l'atome aura un grand nombre d'électrons.
On vérifie par ailleurs que les fonctions d'onde correspondant à une énergie donnée sont toutes orthonormées. 1 La démonstration de cette propriété est donnée en annexe 3.

17
Nous avons déjà indiqué que la résolution des équations de Hartree-Fock se faisait par itérations successives par la méthode du champ auto-cohérent. De fait, à chaque étape, les équations doivent être résolue numériquement et le résultat final n'est alors que l'ensemble des valeurs numériques de la partie radiale des orbitales de Hartree-Fock (les parties angulaires sont toujours les mêmes, à savoir les vecteurs propres des opérateurs 2L et zL , les ( )ϕ,θm
lY ).
Cependant, pour des raisons pratiques évidentes, on préfère manipuler des fonctions analytiques. Pour cette raison, on a posé comme une base convenable de calcul une famille de fonctions, connues sous le nom d'orbitales de Slater, dont la forme générale est:
( )ϕ= −− ,1 θχ α ml
rnnlm YeNr où N est une constante d'intégration qui vaut
( )( )!2
2 2
1
nN
n+
= α
Tout d'abord, on peut aisément s'apercevoir que pour r grand devant 0a , les orbitales de
Slater se comportent de la même manière que les fonctions d'onde de l'atome d'hydrogène, ce qui est tout à fait normal, puisque, conformément à ce qui a été déjà dit dans le cadre de l'approximation du champ central, un électron loin du noyau voit Z charges positives écrantées par 1−Z charges négatives (les autres électrons), et donc qu'il se retrouve dans un
potentiel central en r
1.
On cherche alors les orbitales de Hartree sous la forme:
∑=
=Z
iii rcru
1
)()(rr χ .
La résolution des équations de Hartree-Fock consiste alors, en ce qui concerne les orbitales, à déterminer les coefficients ic . On trouvera l'exemple des solutions avec un tel
traitement pour l'atome de néon dans l'annexe 4.
f) Retour sur la méthode des coefficients de Slater ; comparaison des résultats
Les paragraphes 3)d) et 3)e) interprétant les équations de Hartree-Fock ainsi que les
propriétés du potentiel de Hartree-Fock et des fonctions d'ondes issues de la méthode du champ self-consistent peuvent se résumer ainsi:
- le comportement des électrons dans un atome polyélectronique peut être considéré comme un comportement de particules indépendantes placées dans un potentiel central.
C'est en effet le sens de l'expression de l'hamiltonien ( )ire
HF qm
hi
V+∆−= rh
2
2
, à partir
du moment où l'on trouve que le potentiel ( )iqV est central.
- Les fonctions d'onde issues de cette méthode se comportent, pour des valeurs de r suffisamment grande, comme les fonctions d'onde d'un système hydrogènoïde.
- Pour un état donné, les électrons sont tous soumis au même potentiel central. - L'énergie λE associée à un état donné est l'énergie d'un seul électron indépendant, et
correspond approximativement à l'énergie nécessaire pour enlever cet électron de l'atome.
Toutes ces considérations tendent en fait à légitimer ce qui semblait au départ une
approximation grossière, à savoir considérer que les électrons d'un atome polyélectronique

18
sont des particules indépendantes soumis à un potentiel de la forme r
eZ eff2
, où effZ dépend,
comme le potentiel de Hartree-Fock, de l'état dans lequel se trouvent les 1−Z autres électrons, et que les énergies des états seraient données par la loi générale des systèmes
hydrogènoïdes 2
2
n
ZEE effI
nl −= 1. Il s'agit en fait maintenant de vérifier la concordance des
résultats numériques obtenus par rapport à la réalité physique. On donne dans le tableau ci-dessous les valeurs de l'énergie totale d'un système à plusieurs
électrons:
Modèle de H.F. Modèle de Slater Système atomique
Eexact
EHF ∆E/E ESl ∆E/E He -2,904 -2,862 0,014 -2,723 0,062 Be -14,667 -14,573 0,006 -14,273 0,027 B+ -24,349 -24,238 0,005 -23,798 0,023 Ne -128,93 -128,55 0,003 -127,35 0,012
Comparaison des énergies obtenues par les modèles de H.F. et de Slater avec les énergies réelles. Les énergies sont données en u.a. ( 2/1;1 −==== Ie Eem h ). Elles correspondent à l'énergie totale de l'atome
à savoir ∑=ln lnElnqE
, ,, . Par ailleurs, l'énergie exacte est celle de l'atome non-relativiste.
Au vu de ces résultats, on peut faire un certain nombre de commentaires: - on retrouve bien le fait que l'énergie obtenue par la méthode de Hartree-Fock est
supérieure à l'énergie exacte, du fait que la méthode est une méthode variationnelle. - Etant donné que le potentiel de Hartree-Fock est un potentiel moyenné sur tous les
électrons, la méthode doit être d'autant plus valable que le nombre d'électrons est élevé, et c'est ce qui se dessine dans le tableau.
- Le modèle (plus grossier) de Slater donne des résultats extrêmement satisfaisants (6% au maximum pour He), bien que l'erreur soit environ cinq fois plus élevée que celle commise dans le modèle de Hartree Fock.
On peut tirer comme conclusions de ces diverses remarques: - que la méthode des coefficients de Slater est légitimée dans son principe par
l'interprétation des équations de Hartree-Fock. - que cette méthode, bien que procédant d'une approximation apparemment extrêmement
grossière, donne des résultats relativement satisfaisants.
g) Conclusion
Le traitement des atomes polyélectroniques par la méthode de Hartree-Fock donne, au vu
des valeurs numériques données dans le paragraphe précédent, des résultats extrêmement satisfaisants quant à la position des niveaux d'énergie de l'atome. Par ailleurs, en ce qui concerne les fonctions d'onde, le traitement opéré dans le cadre du modèle de Z électrons indépendants montre que l'on peut considérer la fonction d'onde totale du système comme un 1 On pourra se reporter à l'annexe 2 pour visualiser la forme du potentiel donné par la méthode de Slater, ainsi que l'évolution des énergies des niveaux en fonction de Z.

19
produit tensoriel antisymétrisé de fonctions d'ondes individuelles correspondant à un électron donné et que, de plus, l'énergie associée à cet état correspond également à l'énergie d'un électron indépendant placé dans un potentiel effectif (le potentiel de Hartree-Fock). Enfin, la décomposition des fonctions d'onde sur les orbitales de Slater montre que l'état individuel d'un électron est toujours caractérisé par les quatre nombres quantiques smmln ,,, et donc que l'on
peut bien décrire l'état d'un atome polyélectronique par sa configuration électronique. C'est d'ailleurs ainsi que l'on peut construire pas à pas la classification périodique des éléments en donnant les configuration des divers atomes.
Les deux méthodes ont par ailleurs conduit à la détermination du potentiel )(rVc qui avait
été amené au paragraphe 2)a): dans la méthode des coefficients de Slater il s'agit d'un potentiel discret1, et dans la méthode de Hartree-Fock il correspond exactement au potentiel de Hartree2. Il existe également d'autres méthodes de détermination de ce potentiel3. Cependant, on a vu au paragraphe 2)a) qu'en introduisant cette notion de potentiel central, il apparaissait un terme qui pouvait être traité en perturbation de l'hamiltonien. C'est ce que nous allons traiter à présent.
4) Corrections à l'approximation du champ central. Couplage L-S
a) Rappel sur l'hamiltonien d'un atome à plusieurs électrons
On a vu au paragraphe 2)a) que l'on pouvait écrire l'hamiltonien d'un atome à Z électrons
sous la forme:
( ) 11
2
2HRV
m
PH
Z
iic
e
i +
+= ∑
=
, où ( )∑∑∑=<=
−−
+−=Z
iic
ji ji
Z
i i
RVRR
e
R
ZeH
1
2
1
2
1
et que si le potentiel central ( )ic RV est bien choisi, 1H jouera le rôle d'une perturbation de
( )∑=
+=
Z
iic
e
i RVm
PH
1
2
0 2.
Pour le potentiel )(rVc , on peut par exemple prendre le potentiel de Hartree V , ou tout
autre potentiel central.
1 On peut tout de même en tracer l'allure générale en interpolant sur les valeurs discrète. C'est le traitement que l'on a effectué dans l'annexe 2. 2 Et non pas le potentiel de Hartree-Fock, qui comprend le potentiel d'échange et donc dépend des variables de spin. On peut ici se reporter à l'annexe 3 où l'on retrouve les équations radiales de Hartree ainsi que le potentiel radial, qui servent de support simplifié pour effectuer les calculs du champ self-consistent. 3 Il existe par ailleurs la méthode de Fermi-Dirac qui prend en compte des considérations semi-classiques et statistiques (voir annexe 5).

20
b) Traitement perturbatif du couplage de Russel-Saunders (L-S)
L'hamiltonien que nous allons donc étudier s'écrit alors: 10 HHH += , et nous allons traiter l'hamiltonien 1H en perturbation de 0H 1.
Signalons tout de même que nous n'étudierons ici les divers couplage que qualitativement,
c'est à dire que nous nous contenterons de regarder l'éclatement des états non perturbés issus de l'étude de 0H ainsi que les termes qui apparaissent. Nous ne calculerons donc pas
quantitativement les énergies séparant les divers états excités. Avant de se lancer dans des calculs hasardeux, il convient (comme toujours…) de regarder
les symétries du problème. En effet la première étape du calcul consiste à diagonaliser l'opérateur 1H dans l'état des électrons issu de 0H .
Définissons les opérateurs moment cinétique orbital total et moment cinétique de spin total :
∑∑
=
=
ii
ii
SS
LL
rr
rr
Comme 0H n’agit que sur r , il commute évidemment avec iL
r et iS i ∀
r, et donc avec L
r
et Sr
. En ce qui concerne 1H , on montre que bien qu'il ne commute pas avec les moments
cinétiques orbitaux individuels des électrons, il commute par contre avec Lr
, et bien entendu avec S
r puisqu'il n'agit que sur les variables spatiales.
On a donc la relation de commutation, avec 10’ HHH += :
[ ] [ ] 0,’,’ == LHSHrr
Or les opérateurs zz SSLL ,,, 22 génèrent l'espace des fonctions d'ondes de 0H , et, grâce
aux relations de commutation établie ci-dessus, on voit que les opérateurs
zz SSLLHH ,,,,, 2210 forment un E.C.O.C., et donc que les valeurs propres de ’H vont être
caractérisées par les valeurs propres des opérateurs 2L et 2S uniquement, car comme, d'après les relations de commutations, 1H commute également avec ±L et ±S , l'énergie d'écart avec
le niveau non perturbé ne dépend ni de LM , ni de SM .
Chaque niveau d'énergie va donc donner naissance à des niveaux repérés par les valeurs de L et S , nommés termes, et qui sont notés )(12 LS Α+ , où ( )LΑ est la lettre correspondant à la valeur de L . La dégénérescence de ces niveaux est toujours bien entendu donnée par l'ensemble des valeurs que peuvent prendre LM et SM , soit ( )( )1212 ++ SL .
1 Ce traitement en perturbation n'est valable que pour les atomes à bas Z. En effet, lorsque Z devient trop élevé, on doit tenir compte des effets de couplage spin orbite (j-j), et la structure fine de l'atome apparaît comme le terme prépondérant dans les perturbations. Nous ne nous étendrons pas ici sur ce phénomène.

21
Afin de déterminer les diverses valeurs de L et de S données par un état, on doit utiliser
les règles d'addition des moments cinétiques. Par exemple, si 21 LLLrrr
+= (resp. 21 SSSrrr
+= ),
alors les valeurs propres L (resp. S ) de l'opérateur 2L (resp. 2S ) seront données par:
212121
212121
......,1,
......,1,
ssssssS
llllllL
−−++=
−−++=
En fait, pour des niveaux à plus de deux électrons, on commence par combiner deux électrons, puis on combine le terme obtenu avec un autre électron, et ainsi de suite jusqu'à avoir combiné tous les électrons. Remarquons tout de même qu'il est inutile de s'occuper dans ce traitement des électrons qui sont sur des couches pleines, car, comme
0==== ∑∑i
sSi
iL imMmM , le seul terme qui pourra être issu d'une telle couche est S1
(dégénérescence égale à un), et le couplage d'un tel terme avec n'importe quel électron donne un terme similaire avec l'état de cet électron (en effet 0=L et 0=S pour S1 ).
Nous nous limiterons donc à considérer des atomes possédant des couches incomplètes. Comme l'indique la remarque précédente, nous n'avons qu'à considérer les électrons qui ne se trouvent pas sur des couches pleines pour déterminer les valeurs possibles du couple ( )SL, .
Trois cas se présentent alors: Electrons appartenant à des sous couches différentes (électrons non-équivalents) Dans ce cas, deux électrons distincts ne peuvent avoir le même jeu de nombres quantiques,
et donc le principe de Pauli sera automatiquement satisfait. Nous allons illustrer la technique de détermination des diverses valeurs possibles pour le couple ( )SL, par deux exemples:
Configuration np, n’p:
Nous avons 121 == ll et 2
121 == ss , et donc 2,1,0=L et 1,0=S . Les termes possibles
sont donc: DPSDPS 333111 ,,,,,
Remarquons que nous retrouvons la dégénérescence totale du système: en effet, la
dégénérescence d'un état np, n’p est donnée par ( )( )( )( ) 3622121212 2121 =++++= ssllG , et les dégénérescences des divers termes sont:
- 1:1 =gS
- 3:1 =gP
- 5:1 =gD
- 3:3 =gS
- 9:3 =gP
- 15:3 =gD Et on a bien : 361593531 =+++++ .

22
Configuration np, n’d:
Ici nous avons 11 =l , 22 =l et toujours 2
121 == ss , ce qui donne 3,2,1=L et 1,0=S .
Les termes possibles sont donc: FDPFDP 333111 ,,,,,
Et on retrouve ici aussi la dégénérescence totale qui vaut 60=g Electrons appartenant à la même couche (électrons équivalents) Contrairement à ce qui se passe pour des électrons non-équivalents, ici le principe de Pauli
impose la contrainte d'antisymétrisation qui va interdire certaines valeurs du couple ( )SL, .
Par exemple on voit que pour l'état fondamental de l'atome d'hélium 21s , qui n'est pas dégénéré, la technique d'addition des moments cinétique fait apparaître un terme S1 et un terme S3 , ce dernier étant évidemment à exclure pour conserver la dégénérescence totale du niveau.
Pour les niveaux à deux électrons équivalents, on peut montrer que la restriction imposée
par le principe de Pauli se réduit à ne conserver que les termes tels que SL + pair. Cependant, il n'existe aucune règle de ce type pour les niveaux comportant plus de deux électrons équivalents. On doit donc utiliser une méthode plus élaborée qui prend en compte les valeurs de ∑=
iiL mM et de ∑=
isS i
mM . Il s'agit d'une méthode qui se fonde sur les
propriétés des moments cinétiques Lr
et Sr
, en particulier le fait que l'on a LL MLM ≤≤− et
SS MSM ≤≤− . En déterminant touts les états quantiques possibles des électrons dans la
sous couche considéré, on trouve des séries de valeurs de LM et SM que l'on associera à des
valeurs de L et de S . Et comme lors de la détermination des états quantiques possibles, on a pris en compte le principe de Pauli, tous les termes possibles et tolérés par ce principe — et seulement ceux-là — apparaîtront.
Cette méthode s'appliquant au cas par cas et étant de plus très fastidieuse (il faut lister et
classer tous les états quantiques possibles, ce qui fait par exemple 120 états pour une couche d3) nous nous contenterons ici d'en donner les résultats:
Configuration
ns 2S ns2 1S
np np5 2P np2 np4 1S, 1D 3P
np3 2P, 2D 4S np6 1S
nd nd9 2D nd2 nd8 1S, 1D, 1G 3P, 3F nd3 nd7 2P, 2D, 2F, 2G, 2H 4P, 4F nd4 nd6 1S, 1D, 1F, 1G, 1I 3P, 3D, 3F, 3G, 3H 5D
nd5 2S, 2P, 2D, 2F, 2G, 2H, 2I 4P, 4D, 4F, 4G 6S nd10 1S
Termes issus des configurations (nl)k, avec l=0,1,2

23
Au regard de ces résultats, on remarque plusieurs choses: - pour une couche pleine, il n'y a qu'une seule distribution possibles des électrons, et on a
obligatoirement un unique terme S1 .
- on voit que l'éclatement des termes est le même pour une couche ( )knl que pour une
couche ( ) ( ) klnl −+122 , ce qui revient à dire que les termes possibles pour une couche à k électrons sont les mêmes que pour une couche où il manque k électrons (c'est-à-dire à k trous).
- Pour une couche à moitié remplie, on remarque qu'il existe une configuration des électrons où l'on obtient la valeur maximale de SM . Cette configuration n'existe que
pour 0=LM , et donc donne un terme sphérique Sl 22 + . Cette propriété prend toute son importance avec les règles de Hund (voir plus loin) qui indiquent que c'est cet état qui est l'état fondamental. Autrement dit une couche à moitié remplie possède elle aussi un état fondamental sphérique. Par ailleurs, ceci peut expliquer l'irrégularité du remplissage des couches pour Z=24 ( 5134 ds au lieu de 42 34 ds ), car la couche 53d fait apparaître un terme S6 de très basse énergie, et il est possible que l'énergie totale du système soit plus basse dans cette configuration que dans la configuration "normale" qui ne fait apparaître qu'un terme D5 , d'énergie plus haute que S6 .
Electrons équivalents et non équivalents: Pour traiter ce problème, il suffit juste de déterminer en premier lieu les termes issus du
couplage des électrons équivalents. Ensuite, on obtient les termes définitifs en utilisant les règles d'addition des moments cinétiques évoqués dans le premier cas.
c) Classification énergétique des termes; règles de Hund; schéma d'éclatement des termes
Nous avons donc obtenus par couplage L-S les termes qui proviennent de la correction à
l'hamiltonien 0H , mais nous ne savons pour l'instant rien sur les positions relatives en énergie
de ces différents termes. Des calculs peuvent être menés en calculant la valeur moyenne de
1H dans la configuration de base de 0H :
( ) SLSL MSMLlnlnHMSMLlnlnSL ,,,....;,;,,,,....;,;,, 221112211=δ , où le ket
SL MSMLlnln ,,,....;,;, 2211 est bien entendu antisymétrisé.
Cependant, on peut appliquer, pour les états fondamentaux des atomes et, plus généralement, les états à électrons équivalents1, les règles de Hund, établies empiriquement, qui s'énoncent ainsi:
- le terme possédant la plus forte valeur de S pour une configuration donnée a la plus basse énergie, et l'énergie des autres termes croît quand S décroît.
- Pour une valeur donnée de S, le termes qui a la plus grande valeur de L est le terme de plus basse énergie.
1 Ce cas peut sembler très restrictif. Cependant, comme on l'a vu dans l'annexe 3 et au paragraphe 3)e), les résultats les plus probants sont donnés pour les atomes s'écartant très peu de la sphéricité du potentiel, c'est-à-dire les atomes n'ayant qu'une sous couche incomplète et donc ne possédant que des électrons équivalents.

24
Ces considérations nous permettent donc d'avoir une représentation, qualitative certes, mais conforme à la réalité, de la structure énergétique réelle1 des atomes polyélectronique.
Conclusion Nous avons donc passé en revue les difficultés liées au fait que les atomes
polyélectroniques étaient des systèmes à plus de deux corps en interaction, et donc qu'on ne pouvaient trouver de solution analytique exacte. C'est donc dans le cadre de l'approximation du champ central que nous avons pu déterminer des solutions et en particulier une description assez proche de la réalité des niveaux énergétiques des atomes polyélectronique. Ce modèle, comme nous l'avons vu, implique que les électrons sont placés dans un potentiel effectif qui prend en compte l'attraction du noyau et la répulsion du nuage électronique. Physiquement, cette approximation revient exactement à considérer le problème comme un problème de Z électrons indépendants. Ce modèle nous a permis, dans un premier temps, d'effectuer des conjonctures qualitatives sur la structure électronique des atomes qui permet, entre autre, de construire le tableau périodique des éléments. Ensuite, des calculs plus poussés nous ont permis de déterminer la forme du potentiel central, tout d'abord de manière discrète (méthode des coefficients de Slater), puis de manière numérique et continue (méthode de Hartree et méthode de Thomas-Fermi). Nous avons alors trouvé des résultats, très proches de la réalité, et qui légitimaient à posteriori l'approximation du champ central. Cependant, ces résultats devaient encore être modifiés pour inclure dans l'étude les corrections à apporter à ce modèle, et c'est ce que nous avons fait en étudiant le couplage de Russel-Saunders.
Les résultats de cette étude ont énormément d'applications. Tout d'abord, ils permettent de comprendre (partiellement car les effets de structure fine et de structure hyperfine n'ont pas été traités), le spectre des atomes. Des calculs simples (le modèle des coefficients de Slater peut suffire) nous permettent de vérifier la cohérence de ce modèle en calculant des grandeurs telles que l'énergie d'ionisation, les rayons ioniques… Par ailleurs, ils nous renseignent sur les énergies des niveaux et donc sur la manière dont ces atomes vont engager des liaisons chimiques entre eux pour constituer des molécules. Enfin, la classification périodique des éléments — dont on a légitimé la construction — nous permet de mieux comprendre les similitudes de propriétés pour des atomes à priori totalement différents (mais situés dans la même colonne et donc ayant une structure électronique similaire). L'étude détaillée des atomes polyélectroniques revêt donc dans cette perspective une importance pratique fondamentale.
1 Sans tenir compte des effets de structure fine et hyperfine.

25
Annexe 1: résolution de l'atome d'hydrogène Il s'agit donc ici de résoudre l'équation aux états stationnaires:
Ψ=Ψ
−∆− E
r
e
me
22
2
h (1H)
1) Séparation des variables On peut montrer que l'opérateur ∆ peut s'écrire sous la forme:
22
2
2
21
r
Lr
rr h−
∂∂=∆ , où L
r est l'opérateur moment cinétique orbital de l'électron.
L'équation aux états stationnaires (1H) devient donc:
Ψ=Ψ
−+
∂∂− E
r
e
rm
L
r
r
rm ee
2
2
2
2
22
2
.1
2
h
Comme L
r n'agit que sur les variables angulaires en symétrie sphérique, il est clair que
l'on a les quatre relations de commutation: [ ][ ] 0,
0,2 =
=
LH
LH
H
H
r
On peut donc trouver des fonctions propres communes à 2, LH H et zL (projection
selon l'axe z du vecteur Lr
). Les fonctions propres communes à 2L et zL sont les harmoniques
sphériques ( )ϕ,θmlY , qui vont contenir toute la dépendance en θ et ϕ de la fonction d'onde
Ψ . On va donc chercher Ψ sous la forme: ( ) ( )ϕ=ϕΨ ,)(,, θθ mlYrRr .
On injecte cette expression dans l'équation (1H) et on obtient, après simplification par ( )ϕ,θm
lY ,
( )
)(.)()(2
1)(.1
2
2
2
2
2
22
rRErRr
erR
rm
ll
r
rRr
rm ee
=−++∂
∂− hh (2H)
On observe déjà que pour chaque valeur de l il y aura une nouvelle équation à résoudre.
Aussi nous allons noter les fonctions radiales )(rRl . Par ailleurs, il faut bien entendu que ces
fonctions obéissent à la condition de normalisation
ldrrrRl ∀=∫∞
1)(0
22.
26
2) Résolution de l'équation radiale; détermination de E et de Ψ pour les états liés d'énergie E<0
Posons à présent )(.)( rRrrul = l'équation (2H) devient:
( ))(.)(
2
1)(
2
2
2
2
2
22
ruErur
e
rm
ll
r
ru
m ll
e
l
e
=
−++
∂∂
− hh (3H)
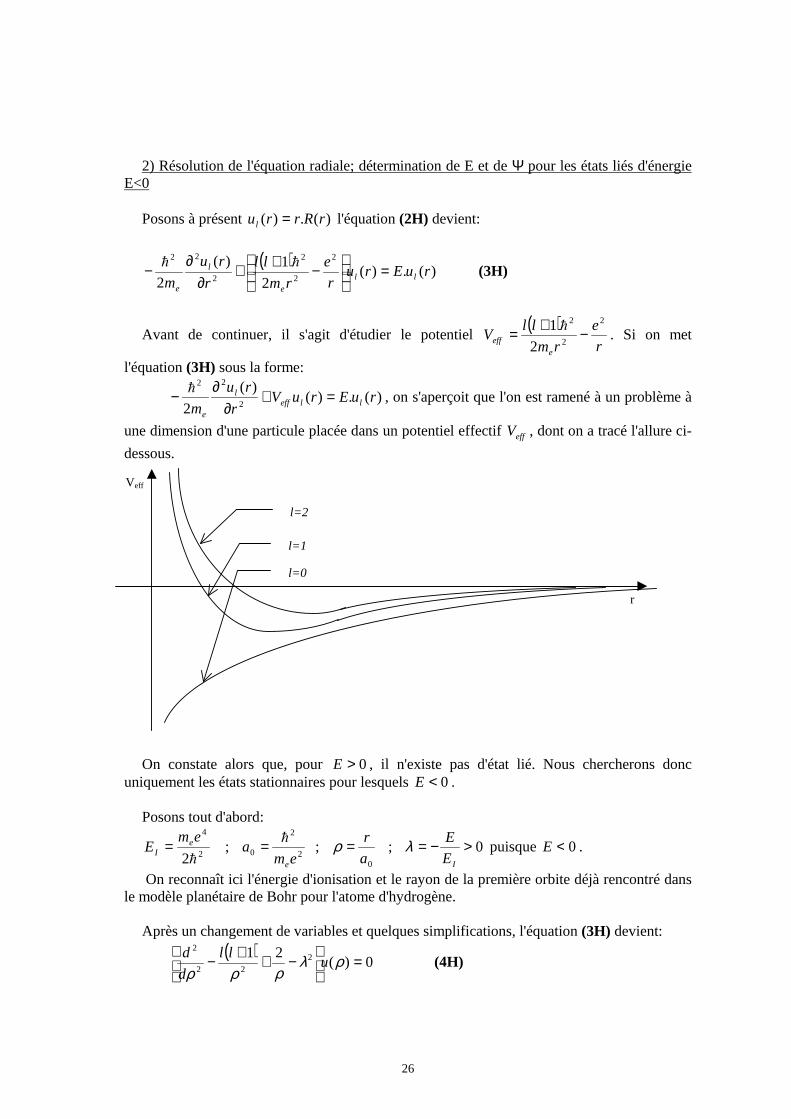
Avant de continuer, il s'agit d'étudier le potentiel ( )
r
e
rm
llV
eeff
2
2
2
2
1 −+= h. Si on met
l'équation (3H) sous la forme:
)(.)()(
2 2
22
ruEruVr
ru
m lleffl
e
=+∂
∂− h
, on s'aperçoit que l'on est ramené à un problème à
une dimension d'une particule placée dans un potentiel effectif effV , dont on a tracé l'allure ci-
dessous. On constate alors que, pour 0>E , il n'existe pas d'état lié. Nous chercherons donc
uniquement les états stationnaires pour lesquels 0<E . Posons tout d'abord:
0;;;2 0
2
2
02
4
>−====Ie
eI E
E
a
r
ema
emE λρh
h puisque 0<E .
On reconnaît ici l'énergie d'ionisation et le rayon de la première orbite déjà rencontré dans le modèle planétaire de Bohr pour l'atome d'hydrogène.
Après un changement de variables et quelques simplifications, l'équation (3H) devient:
( )
0)(21 2
22
2
=
−++− ρλ
ρρρu
ll
d
d (4H)
Veff
r
l=2
l=1
l=0

27
Si ∞→ρ , l'équation (4H) apparaît sous la forme limite 022
2
=− ud
ud λρ
, dont les solution
mathématiques sont les fonctions λρ±Ae , mais dont les seules solutions physiquement acceptables pour des raisons évidentes de convergences sont les λρ−Ae .
Nous allons donc chercher ( )ρu sous la forme:
)()( ρρ λρ feu −= . En injectant cette expression dans l'équation (4H), on obtient:
( )0)(
122
22
2
=
+−+− ρρρρ
λρ
fll
d
d
d
d (5H)
Il s'agit alors d'écrire )(ρf sous la forme d'un développement en série entière:
∑=q
s cf ρρρ)(
Revenons ici à la condition de normalisation ldrrrRl ∀=∫∞
1)(0
22.
Pour que celle-ci soit vérifiée, il faut que 0)0( =u , car si on prend 0,)0( ≠= KKu alors
r
KrrR
2
0
22)( ≈ et donc n'est pas intégrable en 0.
En prenant en compte ce résultat, on voit que 0>S , car 0=S entraînerait 00 =c , ce qui
rendrait la définition du développement en série absurde et 0<S ferait diverger )0(f . On calcule alors:
( ) ( )
( )( ) ( )∑
∑−+
−+
−++=
+=
q
qSq
q
qSq
qSqScd
fd
qScd
df
22
2
1
1 ρρ
ρρ
En injectant ces expressions dans l'équation (5H), il vient alors: ( )( ) ( )[ ]∑ =+−++−−++ −+−+−+−+
q
qSq
lSq
qSq
qSq cllcqScqSqSc 0)1(221 2112 ρρρλρ
et il suffit d'annuler tous les coefficients de la série entière, par unicité du développement
en série entière. On a donc pour le terme en 2−Sρ , ( ) 0)1(1 =+−+ llSS car 00 ≠c . On obtient alors deux
solutions lS −= et 1+= lS , seule la seconde étant à conserver en raison de la contrainte sur le signe de S .
On a donc finalement 1+= lS

28
Pour les autres termes, on a la relation de récurrence: ( )( ) ( )[ ] ( )[ ] 11211 −−+=+−+++ qq cqlllqlqlc λ , soit, en arrangeant un peu:
( )( ) qqlq
lq
c
cq
q
q λλ 2
12
12
1
→++
−+= ∞→−
Etant donné le comportement de la série ainsi définie (c'est-à-dire le même que la série
entière correspondant à λρ2e ), on doit admettre que la série est tronquée à partir d'une valeur kq = telle que kqcq ≥∀= 0 et 01 ≠−kc .
Pour que cette condition soit satisfaite, il faut que ( ) 01 =−+ λlk , donc nlk
11 =+
=λ ,
avec lkn += entier strictement positif (car 00 ≠c )
On a donc les énergies des états stationnaires de l'atome d'hydrogène, puisque IEE 2λ−= :
*,2
INnn
EE I ∈−=
On peut également dire que pour une valeur donnée de n , on peut avoir n valeurs de l , et
que pour une valeur de l on a 12 +l valeurs pour m , ce qui nous permet de calculer aisément
la dégénérescence d'un niveau d'énergie nE : ( )∑−
=
=+=1
0
22122n
ln nlg , en tenant également
compte du spin des électrons. 3) Calcul des fonctions d'onde: On a donc bien des niveaux d'énergie caractérisés par un seul nombre quantique n et des
fonctions d'ondes caractérisées elles par quatre nombres quantiques smmln ,,, .
On peut d'ailleurs calculer ces fonctions d'ondes: Pour la première, ( 0,0 == ln ), on a 0=k et donc 0.)( cf ρρ = .
On en déduit directement que ρρρ −= ecu .)( 0 et donc que 0
0
0)( a
r
ea
crR
−= .
Avec la condition de normalisation, on obtient que 0
30
0,1
1)( a
r
ea
rR−
=π
.
En combinant ces solutions avec les fonctions propres ( )ϕ,θmlY de 2L , on obtient les
fonctions d'ondes associées aux états stationnaires de l'atome d'hydrogène.

29
On trouvera dans le tableau ci-dessous les fonctions d’onde pour 2,1=n :
Niveau 1s 0
30
0,0,1
1 a
r
ea
−=Ψ
π
Niveau 2s 02
030
0,0,2 21
8
1 a
r
ea
r
a
−
−=Ψ
π
Niveau 2p
ϕ−
−=Ψ ia
r
eea
r
a.sin
8
102
030
1,1,2 θπ
θπ
cos4
102
030
0,1,2a
r
ea
r
a
−=Ψ
ϕ−−
+=Ψ ia
r
eea
r
a.sin
8
102
030
1,1,2 θπ
On peut également représenter les dépendances angulaires des fonctions d'onde: on obtient
une surface délimitant la zone où la probabilité de présence de l'électron est supérieure à une constante donnée.
Dépendance angulaire des fonctions d'ondes de l'atome d'hydrogène. On a en fait ici représenté des surfaces délimitant la zone de l'espace où l'électron à un certaine probabilité de se trouver (en général 9/10)
Dépendance radiale des fonctions d'ondes de l'atome d'hydrogène. En fait ces fonctions n’indiquent pas grand chose. Il faudrait plutôt tracer la densité de probabilité, à savoir D(r)=r2R2(r)

30
Annexe 2: compléments sur la méthode des coefficients de Slater
1) Un exemple de calcul de l'énergie d'un niveau. Nous avons donné au paragraphe 2)c) les énergies des niveaux 4s et 3d pour les atomes de
potassium (Z=19), de calcium (Z=20) et de scandium (Z=21), et nous allons ici montrer comment nous avons procédé.
On rappelle tout d'abord le tableau des coefficients de Slater :
Contribution des autres électrons à la constante d'écran Couche n
Origine de l'électron considéré
Couches <n-1
Couche =n-1 s, p d f
Couches >n
s ou p 1,00 0,85 0,35 0 0 0 d 1,00 1,00 1,00 0,35 0 0 f 1,00 1,00 1,00 1,00 0,35 0
La configuration électronique de l'atome de potassium est donnée par le tableau périodique
des éléments (Cf. annexe 6): K (Z=20): 1s22s22p63s23p64s1 Dans ce modèle, on peut donc dire que l'électron situé sur la couche 4s "voit" : - les deux électrons situés sur la couche 1s et dont la constante d'écran vaut 1 - les deux électrons situés sur la couche 2s et dont la constante d'écran vaut 1 - les six électrons situés sur la couche 2p et dont la constante d'écran vaut 1 - les deux électrons situés sur la couche 3s et dont la constante d'écran vaut 0,85 - les six électrons situés sur la couche 3p et dont la constante d'écran vaut 0,85 L'énergie du niveau 4s pour l'atome de potassium vaut donc:
( )
eV
Zn
Ei
iss
80,4
)85,0685,02161212(197,3
6,136,13 2
2
2
,424
−=
×+×+×+×+×−−=
−−= ∑∗ σ
Et on peut calculer de même ce qui se passe si l'électron est situé sur la couche 3d, c'est à
dire pour la configuration électronique 1s22s22p63s23p63d1. En effet, cet électron voit: - les deux électrons situés sur la couche 1s et dont la constante d'écran vaut 1 - les deux électrons situés sur la couche 2s et dont la constante d'écran vaut 1 - les six électrons situés sur la couche 2p et dont la constante d'écran vaut 1 - les deux électrons situés sur la couche 3s et dont la constante d'écran vaut 1 - les six électrons situés sur la couche 3p et dont la constante d'écran vaut 1 Soit une énergie
( )
eV
Zn
Ei
idd
51,1
)118(199
6,136,13 22
,323
−=
×−−=
−−= ∑σ

31
2) Variation avec Z des énergies et positions relatives des sous couches En effectuant les mêmes calculs pour toutes les couches dans tous les atomes, on peut
tracer le diagramme d'évolution de l'énergie des couches en fonction de Z.
On remarque tout de suite sur ce diagramme que les positions relatives des couches en
énergie ne correspondent pas à celle donnée par la règle de Klechkowsky. En effet, on voit que les couches nd passent en dessous des couches ( )sn 1+ dès qu'elles commencent à se remplir (interversions signalées par é). Ceci généralise ce que nous avions déjà remarqué pour les couches 4s et 3d.
Ensuite, on remarque que toutes les énergies ont une tendance asymptotique hydrogènoïde, en se sens que pour Z suffisamment grand, l'effet d'écran va devenir négligeable devant Z et
l'énergie d'un niveau sera alors donné par: 2
2
n
ZEn ≈ . Ceci est assez compréhensible car
lorsque le nombre de proton devient grand, l'écrantage créé par les électrons situés entre un électron donné et le noyau va se faire de plus en plus faible.
-200,00
-180,00
-160,00
-140,00
-120,00
-100,00
-80,00
-60,00
-40,00
-20,00
0,00
0,00 10,00 20,00 30,00 40,00 50,00 60,00
En
erg
ie (
eV)
Diagramme d'évolution de l'énergie des couches en fonction de Z. Pour effectuer le calcul de l'énergie des couches non remplies, on a considéré l'état excité de l'atome où un électron se trouve dans cet état.
Z
1s 3s et 3p
3d 4s et 4p
4d
5s et 5p
2s et 2p

32
3) Visualisation du potentiel effectif de Slater: Le modèle de Slater n'étant qu'une superposition d'atomes hydrogènoïdes, on peut lui
appliquer les résultats du modèle de Bohr pour l'atome d'hydrogène généralisés aux atomes hydrogènoïdes. On peut donc définir la "distance" d'une sous couche au noyau (en mécanique quantique, cette distance correspond à celle où la probabilité de présence d'un électron dans cet état est maximale), et on peut donc tracer les variations du potentiel effectif en posant que
2effV
E = . On obtient ainsi le graphique ci-dessous, sur lequel on a également tracé les
potentiels de l'atome d'hydrogène et d'un atome hydrogènoïde à Z électrons.
On retrouve alors le graphique qualitatif que nous avions ébauché au paragraphe 2)b), à
savoir une courbe intermédiaire entre la courbe r
eV
2
−= (comportement pour ∞→r ) et la
courbe r
ZeV
2
−= (comportement pour 0→r ) : le modèle de Slater est donc extrêmement
proche de l'approximation du champ central, qui sert de base à tous les modèles d'étude des atomes polyélectroniques.
0 50 100 150 200 250 300 350
Potentiel effectif dans le modèle des coefficients de Slater

33
Annexe 3: caractère sphérique du potentiel de Hartree-Fock pour un atome à sous couches pleines
Problème du traitement dans le cas général; équations de Hartree Il s'agit ici de démontrer le caractère sphérique (c'est à dire indépendant des variablesθ et
ϕ ) du potentiel de Hartree-Fock dans le cas d'atomes ou d'ions qui ne possèdent que des couches pleines.
Afin de démontrer ce résultat qui justifie le traitement par la méthode de H.F., nous devons poser que les orbitales spatiales sont de la forme:
( ) ( )ϕ= − ,)( 1 θmlnlnlm YrPrru
r, où ( ) )(. rRrrP nlnl =
On se souvient que le potentiel de Hartree-Fock V contient un terme (central) en ir
Ze 2
− ,
un terme de potentiel direct dV et un terme d'échange exV . Dans le cas d'une sous couche complète ( )’’ln , il vient:
( )
( ) ( )∫ ∑
∑ ∫
−=
−=
Ωϕ=
=
’
’’
2’’
2
’’
’
’’
2
’’’’’
,1
2
12
l
lmjjjj
ml
ijjln
l
lmj
ijjmln
dln
ddrYr
rP
rdr
ru
θ
rrV
le facteur 2 venant du fait qu'un même état est occupé par deux électrons de spin opposés. De plus, on sait d'après les propriétés des harmoniques sphériques que:
( )π
θ4
1’2,
’
’’
2’’
+=ϕ∑−=
lY
l
lmjj
ml
Et on en déduit: ( ) ( )∫ Ω+= jj
ijjln
dln ddr
rrP
l 1
4
1’22
2
’’’’ πV
L'intégration sur la partie angulaire se fait selon la méthode classique en exploitant le fait
que ( ) ( ) ( )∑ ∑
∞
=
+
−=
∗+
>
< ϕϕ+
=0
1,,
)(12
41
l
l
lmjj
mlii
mll
l
ij
YYr
r
lrθθπ
, où ( ) ( )jiji rrrrrr ,max;,min == ><
En réinjectant ceci dans l'expression précédente, on obtient:
( ) ( ) ( ) ( ) ( )∫ ∑ ∑ Ωϕϕ+
+=∞
=
+
−=
∗+
>
<jj
l
l
lmjj
mlii
mll
l
jlndln ddrYY
r
r
lrP
l
01
2
’’’’ ,,)(12
4
4
1’22 θθπ
πV
( ) ( ) ( ) ( ) ( )∫∑ ∑∫ ϕϕϕ+
+= ∗∞
= −=+
>
<jjjj
mlii
ml
l
l
lml
l
jlnjdln ddYY
lr
rrPdr
l θθθππ
,,12
4
)(4
1’22
01
2
’’’’V

34
Et, en se souvenant que π4
100 =Y , on obtient:
( ) ( ) ( ) ( ) ( ) ( ) ( )∫∑ ∑∫ ϕϕϕϕ+
+= ∗∗∞
= −=+
>
<jjjj
mljjii
ml
l
l
lmjl
l
jlnjdln ddYYY
lr
r
rrPdr
l θθθθππ
,,,12
4
)(4
1’22 0
0
2
3
0
21
2
’’’’V
Ici on voit directement qu'en raison des propriétés d'orthonormalisation des harmoniques
sphériques, l'intégration sur les variables angulaires ( )jj ϕ,θ est non nulle que si 0== ml
D'où le résultat:
( ) ( )∫>
+= 22
’’’’)(
11’22 jljlnj
dln r
rrPdrlV , expression qui est clairement indépendante des
variables angulaires ( )jj ϕ,θ . On en déduit directement que, comme il est la somme de tous
les contributions directes de chaque couche, dV est à symétrie sphérique. Intéressons-nous à présent au potentiel exV . De la même manière que pour le potentiel
direct, il vient pour une sous couche complète :
( ) ( )[ ]
( ) ),(
),()(1
),()(,
’’’,’
1
’
’’
’’’,’
1
iim
lilni
l
lmjjjj
mljnl
ijjj
mljlnii
mlinli
exn’l’
YrPr
ddrYrPr
YrPYrPri
ϕ×
Ωϕϕ=ϕ
−
−=
∗∗− ∑ ∫θ
θθθV
On développe alors ijr
1 de la même manière que pour le potentiel dV et en utilisant les
propriétés d'orthonormalisation des harmoniques sphériques et les coefficients de Clebsch-Gordan, on trouve:
( ) ( )[ ] ( )
( ) ),(
)()(000’12
11’2,
’’’,’
1
’
’10 ’’
21
iim
lilni
ll
llLjjnlL
L
jlniim
linliex
n’l’
YrPr
drrPr
rrPLll
LlYrPr
i
ϕ×
++=ϕ
−
+
−=+
>
<∞ ∗− ∑ ∫θ
θV
On voit alors que lorsque ce potentiel d'échange agit sur un état ’’ln , et en particulier sur la
partie angulaire ’’m
lY , il donne un résultat proportionnel à cette même partie angulaire, le
coefficient de proportionnalité étant quant à lui indépendant des variables angulaires. L’action du potentiel d'échange ne dépend donc pas des variables θ et ϕ et est donc à symétrie sphérique. Le potentiel d'échange global n'étant qu'une somme sur tous les états de potentiels de cette forme, il est lui aussi sphérique.
De ces conjonctures on tire donc que le potentiel de Hartree-Fock V est central.

35
Pour les atomes qui possèdent des (en général un seule d'ailleurs) couches insaturées, on réécrit l'équation de Hartree-Fock:
( ) ( ) ( ) ( )iiiex
id
ir
e
quEquqrr
Ze
m i λλλλλ =
−+−∆− VV
rhr
22
2
et on néglige le potentiel d'échange devant les potentiels électrostatiques nucléaire et
direct. On obtient alors:
( ) ( ) ( )iiid
ir
e
quEqurr
Ze
m i λλλλ =
+−∆− rh
r V22
2
Par ailleurs, on sait que le potentiel dV ne dépend que des variables spatiales, et que tous
les termes de l'hamiltonien obtenu sont indépendants des variables de spin. De plus, en se servant une nouvelle fois de l'approximation du champ central, on peut en fait moyenner le potentiel dV sur les variables angulaires et obtenir les équations radiales de Hartree:
( ) ( ) ( ) ( )inlnlinli
d
iii
i
ie
rPErPrr
Ze
r
ll
dr
rd
rm=
+−
+−− λV2
22
22 1.1
2
h
Ici, on voit de manière très claire qu'on peut obtenir des solutions par une méthode
variationnelle en utilisant comme fonction d'essai un simple produit tensoriel (non antisymétrisé) d'orbitales atomiques. L’absence du potentiel d'échange dans les équations de Hartree ne satisfont donc pas au principe d'antisymétrie du système imposé par le principe d'exclusion de Pauli.
Il est également intéressant de noter que, dans ces équations, le potentiel dépend de l'état dans lequel se trouve l'électron, et donc que, contrairement à ce qu'on avait obtenu lors de l'interprétation des équations de Hartree-Fock, pour un état donné, les électrons ne sont plus tous soumis au même potentiel. Ceci aura pour conséquence directe que les orbitales de Hartree ne seront pas orthonormées.

36
Annexe 4: Orbitales atomiques du néon dans son état fondamental développées en orbitales de Slater
Il s'agit juste ici de donner un exemple de résultat obtenu en considérant que pour un
atome, les orbitales spatiales de Hartree-Fock sont données par:
( ) ( )∑=
=Z
iii rcru
1
rr χ
On trouve les solutions suivantes:
( ) ( )654
3210
011
1
01999,000551,000064,0[
00058,004899,093717,0,
χχχχχχθ
++−++=ϕ= − YrPru ss
( ) ( )654
3210
021
2
13871,030910,066899,0[
18620,000635,023093,0,
χχχχχχθ
−++++−=ϕ= − YrPru ss
( ) ( ) 1098710
121
2 01872,032933,053338,021799,0, χχχχθ +++=ϕ= − YrPru pp
avec:
( )( )( )( )( )( )( )( )( )( )ϕ=
ϕ=
ϕ=
ϕ=
ϕ=
ϕ=
ϕ=
ϕ=
ϕ=
ϕ=
−
−
−
−
−
−
−
−
−
−
,
,
,
,
,
,
,
,
,
,
01
13464,91010
01
48489,499
01
38168,288
01
45208,177
00
79242,766
00
82530,455
00
86423,244
00
96184,133
00
56590,1522
00
48486,911
θχθχθχθχθχθχθχθχθχ
θχ
YreN
YreN
YreN
YreN
YreN
YreN
YeN
YreN
YeN
YeN
r
r
r
r
r
r
rr
r
r
r
où les constantes de normalisation sont données par, si ( )ϕ= −− ,1 θχ α m
lrn
nlm YeNr ,
( )
( )!2
2 2
1
nN
n+
= α
Les fonctions radiales pour le néon sont représentés à la figure 3.1. Il est important de noter
que l'on connaît également les fonctions d'onde angulaire, qui sont celles de l'hygrogène (cf. annexe 1).
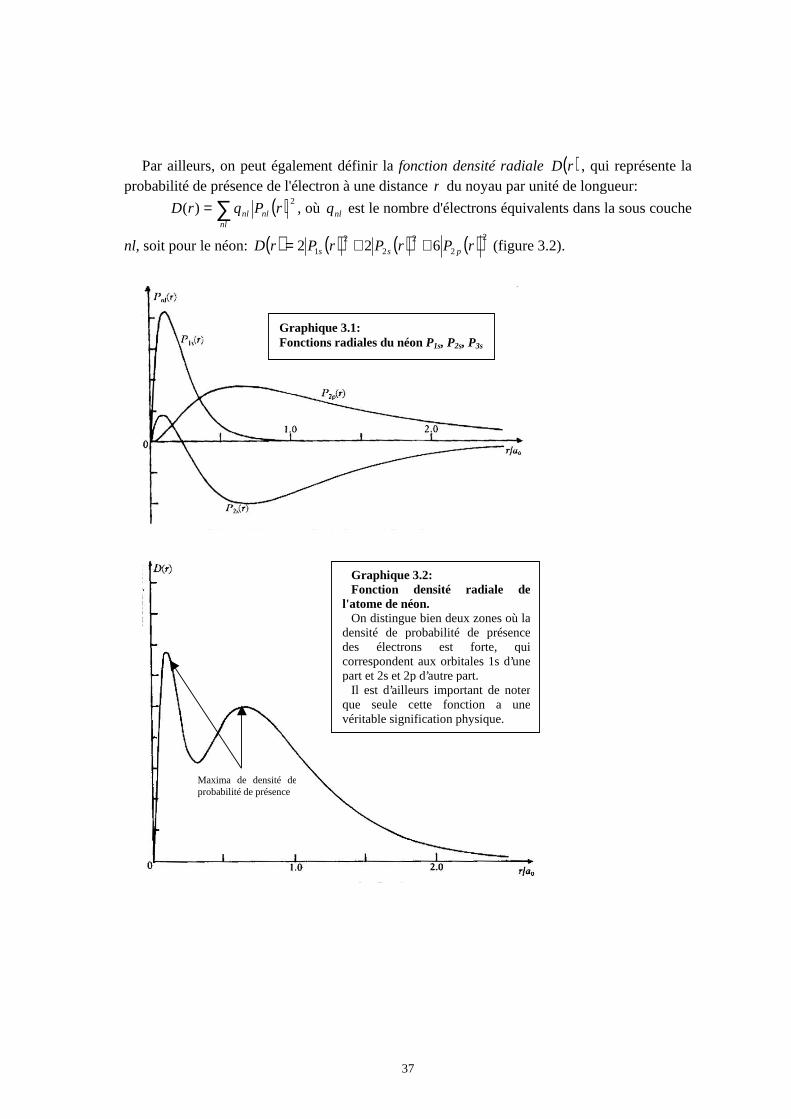
37
Par ailleurs, on peut également définir la fonction densité radiale ( )rD , qui représente la probabilité de présence de l'électron à une distance r du noyau par unité de longueur:
( ) 2)( rPqrD nl
nlnl∑= , où nlq est le nombre d'électrons équivalents dans la sous couche
nl, soit pour le néon: ( ) ( ) ( ) ( ) 2
2
2
2
2
1 622 rPrPrPrD pss ++= (figure 3.2).
Graphique 3.1: Fonctions radiales du néon P1s, P2s, P3s
Graphique 3.2: Fonction densité radiale de
l'atome de néon. On distingue bien deux zones où la
densité de probabilité de présence des électrons est forte, qui correspondent aux orbitales 1s d’une part et 2s et 2p d’autre part.
Il est d’ailleurs important de noter que seule cette fonction a une véritable signification physique.
Maxima de densité de probabilité de présence

38
Annexe 5: détermination du potentiel central dans le modèle statistique de Thomas-Fermi
1) Rappel sur le gaz d’électrons de Fermi Avant d’analyser la théorie développée par Thomas et Fermi pour l’état fondamental d’un
atome, il est utile considérer le problème plus simple du gaz d’électrons de Fermi. Ce système est défini comme un grand nombre N électrons libres contenus dans un grand cube de côté L. Conséquemment, chaque électron se meut de manière indépendante dans un potentiel constant (et nous le prendrons nul) dans le cube et infini à l’extérieur. L’équation de Schrödinger pour chaque électron dans ce système s’écrit:
( ) ( )rErzyxm
rrh =
∂∂+
∂∂+
∂∂− ψ
2
2
2
2
2
22
2 à l’intérieur du cube, et 0=ψ à l’extérieur.
Les solutions de cette équation sont obtenus en y séparant les variables et en résolvant l’équation linéaire à coefficients constants obtenue. Elles sont données par:
( )
= z
L
ny
L
nx
L
nCr zyx
nnn zyx
πππψ sinsinsin,,
r où
3
8
LC = est une constante de
normalisation et où les zyx nnn ,, sont des entiers positifs1. Les énergies correspondantes sont
données par:
( ) 22
22222
2
22
22n
mLnnn
mLE zyx
hh ππ =++= , où 2222zyx nnnn ++= .
Nous remarquons ici qu’un même niveau d’énergie peut être obtenu par plusieurs combinaisons des nombres zyx nnn ,, , et est généralement dégénéré.
Par ailleurs, comme les électrons ont un spin ½, nous devons multiplier les solutions
spatiales par les fonctions de spin sm,2/1χ , avec
2
1±=sm . Les fonctions d’ondes individuelles
des électrons s’écrivent alors: ( )
szxxszxx mnnnmnnn r ,2/1,,,,, χψψ r= et les états quantiques associés sont donc définis par la
donnée de quatre nombres quantiques. En raison du fait que les espaces entre les divers niveaux d’énergie est petit devant les
dimensions caractéristiques du système macroscopique considéré, on peut passer d’une description discrète des énergies à une description continue. On peut donc introduire la notion de densité d’états ( )ED , définie comme le nombre d’états quantiques électroniques par unité d’énergie. En conséquence, dEED )( est le nombre d’états dont l’énergie est comprise entre E et dEE + .
Pour déterminer ( )ED , il nous faut nous placer dans l’espace des zyx nnn ,, .
1 Une valeur nulle du triplet (nx, ny, nz) conduit à la solution triviale et impossible 0=ψ , et des valeurs
négatives de ces entiers conduisent strictement aux mêmes solutions que pour les entiers positifs.

39
Comme les zyx nnn ,, sont positifs, nous
ne nous intéresserons qu’ à un huitième de l’ espace. Nous voyons (figure 5.1), qu’ un état caractérisé par le triplet zyx nnn ,, occupe
un cube unitaire. Par ailleurs, pour des grandes valeurs du triplet zyx nnn ,, , le
nombre total d’ état d’ énergie inférieure à une valeur donnée E est sensiblement égal au volume d’ un huitième de la sphère de rayon
222zyx nnnn ++= . Le nombre d’ état ainsi
considéré est donc donné par: 33
3
1
3
4
8
12 nnN S ππ == , où le facteur 2
provient de la dégénérescence due au spin. En utilisant alors la valeur de l’ énergie et en posant VL =3 , on obtient:
2
32
3
22
2
3
1VE
mN S
=hπ
En différenciant cette expression, il vient:
dEVEm
dEEDdN S2
12
3
22
2
2
1)(
==hπ
,
d’ où on tire directement
2
12
3
22
2
2
1)( VE
mED
=hπ
On peut montrer que ces résultats restent valides quel que soit le volume macroscopique
choisi, à condition que les dimensions caractéristiques de ce volume restent très grandes devant la distance entre deux niveaux énergétiques.
Si on se réfère au principe de Pauli, la fonction d’ onde totale décrivant ce système est un produit tensoriel antisymétrisé des fonctions d’ ondes déterminées par la résolution de
l’ équation de Schrödinger. L’ énergie totale correspondante est la somme des énergies individuelles. Si on admet que le système est dans son état fondamental (c’ est-à-dire que le gaz d’ électrons de Fermi est à T=0), l’ énergie la plus basse est obtenue quand les Z électrons remplissent tous les états d’ énergie inférieure à une certaine énergie, appelée énergie de Fermi EF, les orbitales restantes étant vides. Ceci est illustré par la figure 5.2.
Ceci nous donne un moyen d’ évaluer
l’ énergie EF, car celle-ci doit être telle que:
( )∫= FEdEEDN
0
EF E 0
D(E)
Figure 5.2: densité d'état D(E). Les états occupés dans l'état fondamental sont représenté par la partie ombrée.
nz
ny
nx
Figure 5.1: représentation de l'espace des n

40
Et en intégrant on obtient:
( )3
22
2
32
ρπm
EF
h= , où V
N=ρ représente la densité électronique.
Nous pouvons d’ ailleurs remarquer que l’ énergie totale d’ un gaz électronique de Fermi dans son état fondamental est donné par:
F
E
tot NEdEEEDEF
5
3)(
0== ∫
L’ énergie moyenne d’ un électron est donc FEE5
3= .
Il est enfin commode d’ introduire la notion de vecteur d’ onde défini par:
( )zyx kkkk ,,=r
où on a zyxnL
k ,,,2 == λπ
λλ .
On peut ici exprimer l’ énergie en fonction de kr
:
m
kE
2
22r
h=
En faisant le même raisonnement que dans l’ état des n, on peut écrire que dans l’ état
fondamental du gaz, tous les états qui ont une valeur de kr
inférieure à une valeur Fk sont
occupée, tandis que les autres sont vides. Conséquemment, en considérant le huitième de sphère correspondant et en n’ oubliant pas la multiplicité de spin, on obtient:
323
1FVkN
π= , soit ( )3
123 ρπ=Fk .
On peut également dire qu’ à la surface de cette sphère, l’ énergie est l’ énergie de Fermi, et donc que:
22
2 FF km
Eh= .
2) Le modèle de Thomas-Fermi pour les ions et atomes polyélectroniques Dans ce modèle, on traite les N électrons du système comme un gaz d’ électrons de Fermi
dans son état fondamental, localisé dans une région de l’ espace par un potentiel central ( )rV qui s’ annule à l’ infini. On suppose que le potentiel est sensiblement constant sur la longueur d’ onde de Broglie des électrons, ce qui implique la présence d’ un grand nombre d’ électrons dans un volume où le potentiel peut être considéré comme constant, et donc que l’ on peut appliquer le modèle statistique de Fermi. Le but de ce modèle est de calculer le potentiel ( )rV
et la densité électronique ( )rρ .
L’ énergie totale d’ un électron peut être écrite comme ( )rVm
p +2
2
, et cette énergie ne peut
être positive car sinon l’ électron s’ échapperait à l’ infini. Comme l’ énergie cinétique maximale d’ un électron dans un gaz d’ électrons de Fermi est l’ énergie de Fermi FE , on peut directement écrire:
( )rVEE F +=max

41
Il est évident que maxE ne peut dépendre de r, car sinon les électrons se regrouperaient tous
dans la région où maxE a la plus petite valeur. De plus, on sait que maxE doit être négatif ou
nul. On peut donc écrire:
( ) [ ])(2
max22 rVE
mrkF −=
h ainsi que ( ) ( )[ ]2
3
max
2
3
22
2
3
1rVE
mr −
=hπ
ρ
On remarque que ρ s’ annule pour ( ) maxErV = , et nous devons fixer 0=ρ pour la région
interdite maxEV > , puisque s’ il n’ en était pas ainsi, on aurait une énergie cinétique maximale
0<FE .
Posons ( ) ( )rVe
r1−=φ le potentiel électrostatique et
e
Emax0 −=φ une constante positive.
Si on pose ensuite ( ) ( ) 0φφ −=Φ rr , on voit que ( )rρ et ( )rΦ sont reliés par:
( ) ( )[ ]
<Φ
≥ΦΦ
=
00
02
3
12
32
3
22re
mr hπρ
Une seconde relation entre ( )rρ et ( )rΦ peut être obtenue en considérant que les causes
du potentiel électrostatique ( )rφ sont: - la charge considérée comme ponctuelle du noyau Ze , localisée à l’ origine - la distribution électrique due aux N électrons1. Si on considère que la densité de charge ( )reρ− des électrons est continue, on peut utiliser
l’ équation de Poisson de l’ électrostatique:
( ) ( )[ ] ( )re
dr
rrd
rr ρ
ε 02
21 =Φ=∆Φ
En utilisant ces deux relations et en y éliminant ρ , on obtient que:
( )[ ] ( )[ ]
<Φ
≥ΦΦ
=Φ
00
02
31 2
32
3
20
22
2re
me
dr
rrd
r hεπ
On peut déjà dire que ( )0
0 4lim
πεZe
rrr
=Φ→
, étant donné que pour r petit, le potentiel est
uniquement dû au noyau, et que, puisque les N électrons sont supposés être localisés dans un
sphère de rayon 0r ( qui est donné par l’ équation 0=Φ ), on doit avoir ( )∫= 0
0
24r
drrrN ρπ .
1 Comme on considère indifféremment des atomes ou des ions, on n'a pas obligatoirement Z=N

42
Pour simplifier les équations ci-dessus, on pose:
bxr = et ( ) ( )xZe
rr χπε 04
=Φ , avec ( ) 3
1
03
1
0
3
7
3
2
8853,0
2
3 −−≈= ZaZab
π et où 0a est le rayon
de la première orbite de Bohr. L’ équation différentielle sur Φ s’ écrit donc, avec ces variables sans dimensions,
<
≥=
00
01 2
3
2
2
χ
χχχx
dx
d, équations connues sous le nom d’équations de Thomas-Fermi.
En plus, on a les relations:
<
≥
=
00
04
2
3
3
χ
χχπρ xb
Z
ainsi que la conditions aux limites en 0=r , qui s’ écrit ( ) 10 =χ
Il est clair que χ possède au moins un zéro dans +IR . Soit 0x la position de ce zéro.
D’ après la discussion précédente, nous avons b
rx 0
0 = , où 0r est la « borne » du système. On
note par ailleurs que χ est du signe de xx −0 .
Il est curieux de voir que l'équation de Thomas-Fermi est une équation universelle, qui ne
dépend ni de Z, ni de constantes physiques telles que me,,h qui ont été éliminées par les changements de variables. Nous voyons également qu'il s'agit d'une équation du second ordre non linéaire.
La solution de l'équation de Thomas-Fermi pour 0xx > est directement donnée par:
( ) ( )0xxCx −=χ , où C est une constante négative. Nous remarquons alors
immédiatement que pour une valeur finie de 0x , on ne peut avoir C nul puisque cette
situation conduirait à la solution triviale et physiquement inacceptable 0=χ .
Comme nous pouvons aisément déterminer χ pour 0xx > , les solutions de l'équation de
Thomas-Fermi seront complètement déterminées par la donnée de χ pour 0xx < . On peut
déjà remarquer que comme la condition en 0=x ne donne qu'une contrainte, il existe une infinité de solutions à cette équation. Il est cependant évident que ces équations doivent être concaves. On peut donc distinguer deux type de solutions1:
1. une solution qui admet l'axe des x comme asymptote à l'infini ( ∞=0x )
2. une solution qui s'annule pour une valeur finie 0xx =
1 On ne s'intéressera ici qu'au premier type de solutions, qui concerne les atomes neutres, les seuls qui nous intéressent ici.

43
L’interprétation physique des ces deux solutions est donnée par la condition de normalisation:
[ ] 0
0
0
0
0
0
2
3
’
’’
x
x
x
xZ
dxxZ
dxxZN
χχ
χ
χ
−=
=
=
∫∫
En utilisant la condition en 0=x et le fait que ( ) 00 =xχ on obtient que:
( )Z
ZNxx
−=00 ’χ .
Si on considère donc un atome neutre (c'est-à-dire pas un ion), on obtient que ( ) 0’ 0 =xχ ,
et donc directement que ∞=0x . On voit donc que la solution du type 1 correspond aux
atomes neutres. De plus, on voit que dans le modèle de Thomas-Fermi, un atome neutre n'admet pas de bornes. Conséquemment, on introduit pour les atomes neutres une seconde condition aux limites qui est ( ) 0=∞χ .
On obtient donc une fonction universelle ( )xχ , qui caractérise tous les atomes neutres. L'allure de cette fonction, obtenue par intégration numérique, est donnée dans le graphique 5.3.
En utilisant cette fonction, on peut remonter au potentiel central ( )rV . En remarquant que
pour un atome neutre, 00 =φ , et en effectuant les changements de variables dans l'autre sens,
on voit que:
( ) χπε r
ZerV
0
2
4−=
Par ailleurs, on peut calculer que 588,1)0(’ −=χ , et donc que le potentiel central dans le
modèle de Thomas-Fermi peut se développer comme:
( )
++−= ...794,1
4 0
3
4
0
2
a
Z
r
ZerV
πε, et l'on peut interpréter le premier terme comme le
terme d'attraction nucléaire, alors que le second, qui est répulsif, vient de la contribution des électrons.
0
0,2
0,4
0,6
0,8
1
1,2
0 10 20 30 40 50 60
χ
x
Graphique 5.3: fonction universelle χ(x).On voit bien ici que la fonction est concave, et qu'elle s'annule à l'infini, ce qui indique que dans le modèle de Thomas-Fermi, les atomes neutres ne sont pas bornés.

44
Annexe 6: Classification périodique des éléments

45
Bibliographie & Claude Cohen-Tannoudji, Bernard Diu, Franck Laloë, Mécanique quantique t. I & II,
Hermann, Coll. Enseignement des sciences, 1996.
& B.H. Bransden and C.J. Joachain, Physics of atoms and molecules, Longman Scientific
and Technical, New-York, 1992.
& Albert Messiah, Mécanique quantique t.2, Dunod, 1995.
& Mitchel Weissbluth, Atoms and Molecules, Student edition, Academic Press, New-
York, 1978.
& Jean-Claude Mallet, Roger Fournier, Cours de chimie, Dunod, 1995.
& René Didier, Chimie générale, Technique et documentation - Lavoisier, Paris, 1988.
& Physique quantique résumé de cours de J.P. Marsault.
& Le complément VI.A du polycopié de M. Klein.





![[Jean-Christophe de Mestral] L'Atome Vert(BookZZ.org)](https://img.pdfslide.net/doc/110x75/55cf941c550346f57b9fb37c/jean-christophe-de-mestral-latome-vertbookzzorg.jpg)























