Embed Size (px)
Citation preview

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
CHAPTER 59 ■ NEUROGENIC SHOCKSUSANNE MUEHLSCHLEGEL � DAVID M. GREER
Neurologically injured patients, regardless of the nature of theinjury, frequently experience hypotension and shock. Neuro-genic shock refers to a neurologically mediated form of circu-latory system failure that can occur with acute brain, spinalcord, or even peripheral nerve injuries. In this chapter, wewill explain the epidemiology, pathophysiology, clinical pre-sentation, and management strategies for this special form ofshock.
Contrary to common belief, neurogenic shock is not a sin-gle entity due to one single pathologic mechanism. The termis sometimes used in nonneuroscience intensive care units toexplain hypotension occurring in any brain-injured patient,but neurogenic shock should be considered only after systemiccauses of shock have been carefully ruled out. Just like othercritically ill patients, neurologically ill patients are prone to de-veloping systemic conditions, such as dehydration, congestiveheart failure, acute blood loss, sepsis, pericardial tamponade,or massive pulmonary embolism.
SUBTYPES OFNEUROGENIC SHOCK
Once other systemic reasons for shock have been ruled out,neurogenic shock should be considered. Three mechanisms canlead to neurogenic shock (Fig. 59.1):
■ Vasodilatory (distributive) shock from autonomic distur-bance with interruption of sympathetic pathways, with as-sociated parasympathetic excitation, which causes profoundvasodilatation and bradycardia, as seen in spinal cord injuryor diseases of the peripheral nervous system (Guillain-Barresyndrome)
■ Cardiogenic shock, as frequently seen in subarachnoid hem-orrhage (SAH) with stunned myocardium after a cate-cholamine surge or ischemic stroke, especially those involv-ing the right insula
■ Hypopituitarism/adrenal insufficiency.
Although some subtypes of neurogenic shock occur morefrequently with certain disease entities—for example, car-diogenic neurogenic shock after SAH, vasodilatory neuro-genic shock with spinal cord injury—significant overlap ex-ists between different disease entities (intracerebral hemorrhage[ICH], SAH, traumatic brain injury [TBI], ischemic stroke), andone cannot establish a firm rule by which neurogenic shock oc-curs. Interestingly, only some patients with neurologic injuriesexperience true neurogenic shock, and it remains difficult topredict in whom this will be seen.
INCIDENCE OFNEUROGENIC SHOCK
Due to the small number of prospective epidemiologic studies,it is difficult to establish the natural incidence of neurogenicshock. In a retrospective review of cervical spinal cord injuries,Bilello et al. (1) reported a 31% incidence of neurogenic shockwith hypotension and bradycardia after high cervical spinalcord injury (C1–C5) and 24% after low cervical spinal cordinjury (C6–C7).
Cardiogenic neurogenic shock has been studied foremost inSAH and ischemic stroke. Banki et al. (2) prospectively studiedthe incidence of left ventricular (LV) dysfunction with transtho-racic echocardiography (TTE) in the first 7-day period afterSAH in 173 patients. Thirteen percent had a normal ejectionfraction (EF) but had regional wall motion abnormalities thatdid not correlate with coronary artery territories, and 15% hadan LVEF of less than 50%. Others report a 9% incidence ofLV wall motion abnormalities, resulting in hypotension requir-ing vasopressor therapy, as well as pulmonary edema in most(80%) of these patients (3). The spectrum of injury can rangefrom mild to severe systolic dysfunction—the latter defined asan EF less than 30%. Polick et al. (4) observed LV abnormalitieson TTE in 4 of 13 patients (31%) studied within 48 hours ofSAH. Resolution of these neurologically mediated wall motionabnormalities is usually seen (2,3,5).
The third subtype of neurogenic shock, adrenal insuffi-ciency, has been studied primarily in traumatic brain injury.In the largest study to date, adrenal insufficiency occurred inabout 50% of patients and led to hypotension in 26% (6). Al-though it has been documented in other cases of acute braininjury, the exact incidence and relationship to outcome is notclear (7).
PATHOPHYSIOLOGY OFNEUROGENIC SHOCK
Vasodilatory Neurogenic Shock
This variation of neurogenic shock is commonly seen withspinal cord injuries and Guillain-Barre syndrome (acute de-myelinating peripheral neuropathy) but also with traumaticbrain injuries, large hemispheric ischemic strokes, and intra-cerebral hemorrhages. The hallmark of vasodilatory neuro-genic shock is the combination of bradycardia with fluctuatingblood pressures and heart rate variability due to interruption ofsympathetic output and excitation of parasympathetic fibers.
The sympathetic fibers originate in the hypothalamus, giv-ing rise to neurons projecting to autonomic centers in the
925

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
926 Section VI: Shock States
Vasodilatory Shock due to autonomic dysfunction with unopposed vagal tone • Bradycardia, hypotension • Seen in cervical and upper thoracic spinal cord injury
Neuroendocrine Shock due to pituitary or adrenergic dysfunction after CNS injury • Hypotension poorly responsive to vasopressor therapy • Seen in TBI, SAH, hypothalamic stroke
Cardiogenic Shock due to stunned myocardium after catecholamine surge • Tachycardia, hypotension • ↓CO, ↑CVP, ↑PCWP, • Seen in SAH, ischemic stroke involving the insula, TBI
Neurogenic Shock
FIGURE 59.1. Neurogenic shock consists of three pathomechanisms. CNS, central nervous system; CO,cardiac output; CVP, central venous pressure; PCWP, pulmonary capillary wedge pressure; SAH, sub-arachnoid hemorrhage; TBI, traumatic brain injury.
brainstem—the periaqueductal gray matter in the midbrain, theparabrachial regions in the pons, and the intermediate reticu-lar formation located in the ventrolateral medulla. From here,neurons project to nuclei in the spinal cord. The sympatheticpreganglionic neurons originate in the intermediolateral cellcolumn within the spinal cord gray matter between T1 andL2 and are therefore called the thoracolumbar branches. Fromhere, they exit the spinal cord and project to 22 pairs of paraver-tebral sympathetic trunk ganglia next to the vertebral column.The main ganglia within the sympathetic trunk are the cervi-cal and stellate ganglia. The adrenal medulla receives pregan-glionic fibers and thus is equivalent to a sympathetic ganglion.Blood pressure control depends on tonic activation of the sym-pathetic preganglionic neurons by descending input from thesupraspinal structures (8).
The parasympathetic nervous system consists of cranialand sacral aspects. The cranial subdivision originates from theparasympathetic brainstem nuclei of cranial nerves III, VII, IX,X, and XI. The cranial parasympathetic neurons travel alongthe cranial nerves until they synapse in the parasympatheticganglia in close proximity to the target organ. The sacral sub-division originates in the sacral spinal cord (S2–S4), formingthe lateral intermediate gray zone where preganglionic neuronstravel with the pelvic nerves to the inferior hypogastric plexusand synapse on parasympathetic ganglia within the target or-gans.
Following a spinal cord injury, the sympathetic pathwaysare interrupted with dissociation of the sympathetic supplyfrom higher control below the level of transection (9,10).Parasympathetic fibers are usually spared. This results in auto-nomic hyperreflexia with associated hypertension or hypoten-sion with bradycardia, all observed in human studies as wellas in animal models (10–13). Loss of supraspinal control ofthe sympathetic nervous system leads to unopposed vagal tonewith relaxation of vascular smooth muscles below the level ofthe cord injury, resulting in decreased venous return, decreasedcardiac output, hypotension, loss of diurnal fluctuations of
blood pressure, reflex bradycardia, and peripheral adrenore-ceptor hyperresponsiveness (14). The latter accounts for theexcessive vasopressor response repeatedly seen in this clinicalscenario. The acute phase, also known as spinal shock, morefrequently consists of periods of hypotension. After the acutephase, starting about 2 months after the injury, autonomic dys-reflexia occurs in patients with lesions above T5 (15). This stateis characterized by sympathetically mediated vasoconstrictionin muscular, skin, renal, and presumably gastrointestinal vas-cular beds, induced by afferent peripheral stimulation belowthe level of the lesion. For example, stimuli such as urinarycatheterization, dressing changes, or surgical stimulation canlead to severe blood pressure spikes out of proportion to thestimulus. In Guillain-Barre syndrome, the autonomic dysregu-lation is likely caused by acute demyelination not only of sen-sory and motor fibers, but also of autonomic fibers.
Injury to the brain can also lead to vasodilatory neurogenicshock. Certain cerebral structures, such as the insular cortex,amygdala, lateral hypothalamus, and medulla, have great in-fluence on the autonomic nervous system. Cortical asymme-try is present and is reflected in a higher incidence of tachy-cardia, ventricular arrhythmias, and hypertension with lesionsof the right insula—resulting in loss of parasympathetic in-put and thus sympathetic predominance—and a higher inci-dence of bradycardia and hypotension with injuries to the leftinsula—resulting in a loss of sympathetic input and subsequentparasympathetic predominance (16–18) (Fig. 59.2).
Cardiogenic Neurogenic Shock
This form of neurogenic shock is primarily encountered in SAHand TBI but is also seen in ischemic stroke and intracerebralhemorrhage. Cardiac dysfunction is a well-known complica-tion of ischemic and hemorrhagic stroke, first described over50 years ago (19). It is most often recognized on the elec-trocardiogram (ECG) as arrhythmias, QRS, ST-segment, and
P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
Chapter 59: Neurogenic Shock 927
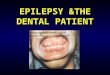
FIGURE 59.2. Example of a right ischemic stroke resulting in ventric-ular arrhythmias and cardiogenic shock. A 61-year-old man presentswith sudden onset of left hemiparesis affecting his face and arm, left-sided neglect, and a left hemianopia. He presented outside of any acutetreatment window and did not undergo thrombolysis. He was admittedto the neurointensive care unit (NICU) for close monitoring of his car-diac and respiratory function. The noncontrast head CT shows a rightmiddle cerebral artery stroke and incidental hemorrhagic conversion.Electrocardiogram on admission showed diffuse T-wave inversion inall leads. Telemetry monitoring revealed frequent premature ventricu-lar complexes and intermittent nonsustained ventricular tachycardia of4 to 8 beats for the first 72 hours after stroke onset. His systolic bloodpressure on admission was elevated at 190 mm Hg but then droppedto 85 mm Hg several hours after admission to the NICU, requiringvasopressor support for 2 days. Troponin T levels were elevated in theemergency room and peaked at 12 hours after stroke onset. Echocar-diogram showed global hypokinesis and no regional wall motion ab-normalities. No other causes for shock were found, so that the strokeinvolving the right insula was the most likely cause. The shock slowlyresolved over 72 hours, and vasopressor infusion was weaned off suc-cessfully. A repeat echocardiogram 2 weeks later showed resolution ofthe abnormalities.
T-wave abnormalities (20,21). Studies of SAH and cardiac in-jury have shown that the severity of SAH is an independent pre-dictor of cardiac injury, supporting the hypothesis that cardiacneurogenic shock is a neurally mediated process (22). Basedon the similarities observed between pheochromocytoma cri-sis and SAH, the cardiovascular changes have been linked to acatecholamine surge.
This hypothesis has been confirmed by many studies. Pa-tients with SAH can have a threefold increase in norepinephrinelevels that are sustained for 10 days or longer after SAH butthat normalize after the acute phase of injury (23). In an animalmodel, an increase in plasma catecholamines after experimen-tal SAH causes specific lesions on electron microscopy within
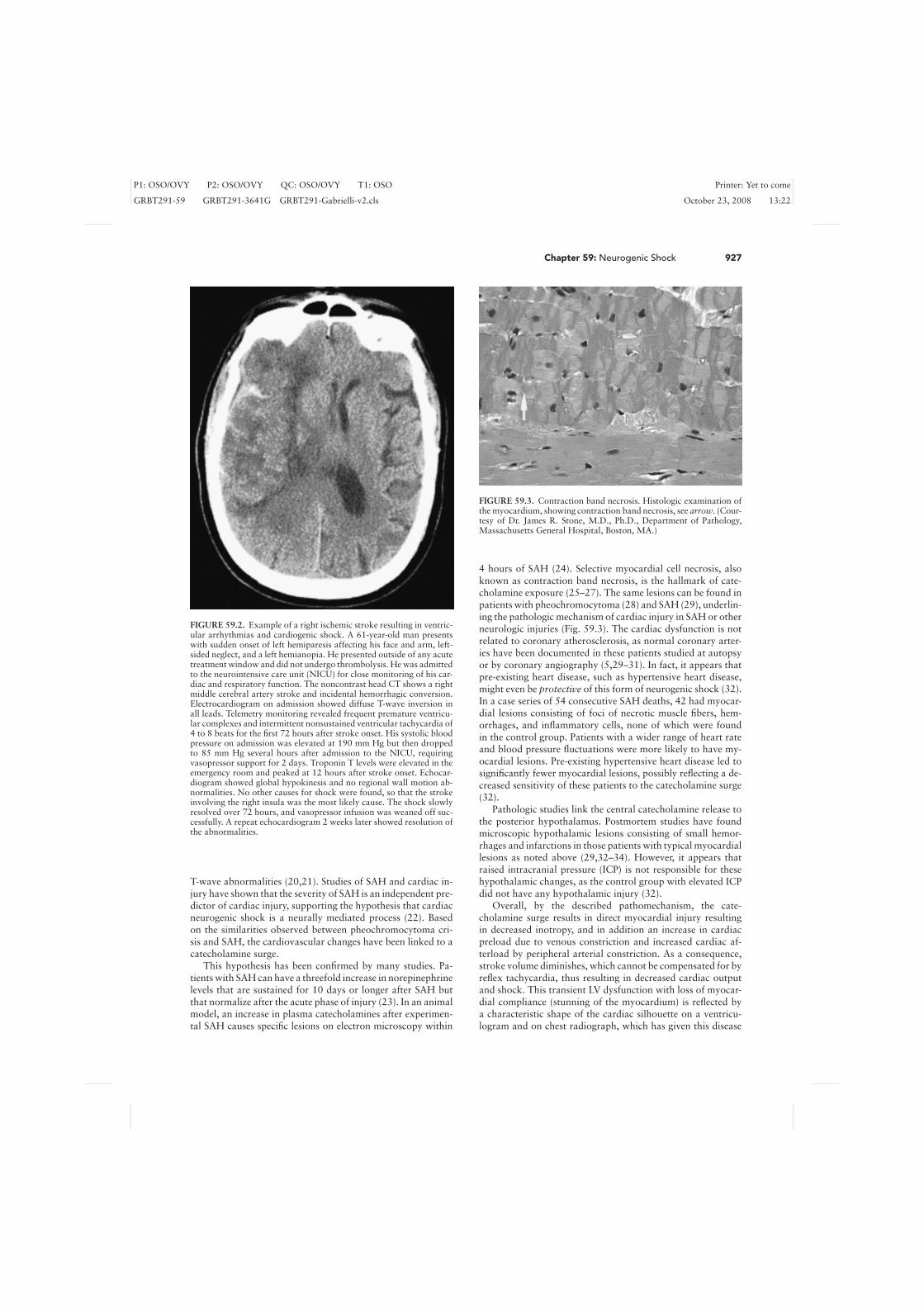
FIGURE 59.3. Contraction band necrosis. Histologic examination ofthe myocardium, showing contraction band necrosis, see arrow. (Cour-tesy of Dr. James R. Stone, M.D., Ph.D., Department of Pathology,Massachusetts General Hospital, Boston, MA.)
4 hours of SAH (24). Selective myocardial cell necrosis, alsoknown as contraction band necrosis, is the hallmark of cate-cholamine exposure (25–27). The same lesions can be found inpatients with pheochromocytoma (28) and SAH (29), underlin-ing the pathologic mechanism of cardiac injury in SAH or otherneurologic injuries (Fig. 59.3). The cardiac dysfunction is notrelated to coronary atherosclerosis, as normal coronary arter-ies have been documented in these patients studied at autopsyor by coronary angiography (5,29–31). In fact, it appears thatpre-existing heart disease, such as hypertensive heart disease,might even be protective of this form of neurogenic shock (32).In a case series of 54 consecutive SAH deaths, 42 had myocar-dial lesions consisting of foci of necrotic muscle fibers, hem-orrhages, and inflammatory cells, none of which were foundin the control group. Patients with a wider range of heart rateand blood pressure fluctuations were more likely to have my-ocardial lesions. Pre-existing hypertensive heart disease led tosignificantly fewer myocardial lesions, possibly reflecting a de-creased sensitivity of these patients to the catecholamine surge(32).
Pathologic studies link the central catecholamine release tothe posterior hypothalamus. Postmortem studies have foundmicroscopic hypothalamic lesions consisting of small hemor-rhages and infarctions in those patients with typical myocardiallesions as noted above (29,32–34). However, it appears thatraised intracranial pressure (ICP) is not responsible for thesehypothalamic changes, as the control group with elevated ICPdid not have any hypothalamic injury (32).
Overall, by the described pathomechanism, the cate-cholamine surge results in direct myocardial injury resultingin decreased inotropy, and in addition an increase in cardiacpreload due to venous constriction and increased cardiac af-terload by peripheral arterial constriction. As a consequence,stroke volume diminishes, which cannot be compensated for byreflex tachycardia, thus resulting in decreased cardiac outputand shock. This transient LV dysfunction with loss of myocar-dial compliance (stunning of the myocardium) is reflected bya characteristic shape of the cardiac silhouette on a ventricu-logram and on chest radiograph, which has given this disease
P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
928 Section VI: Shock States
A C
B
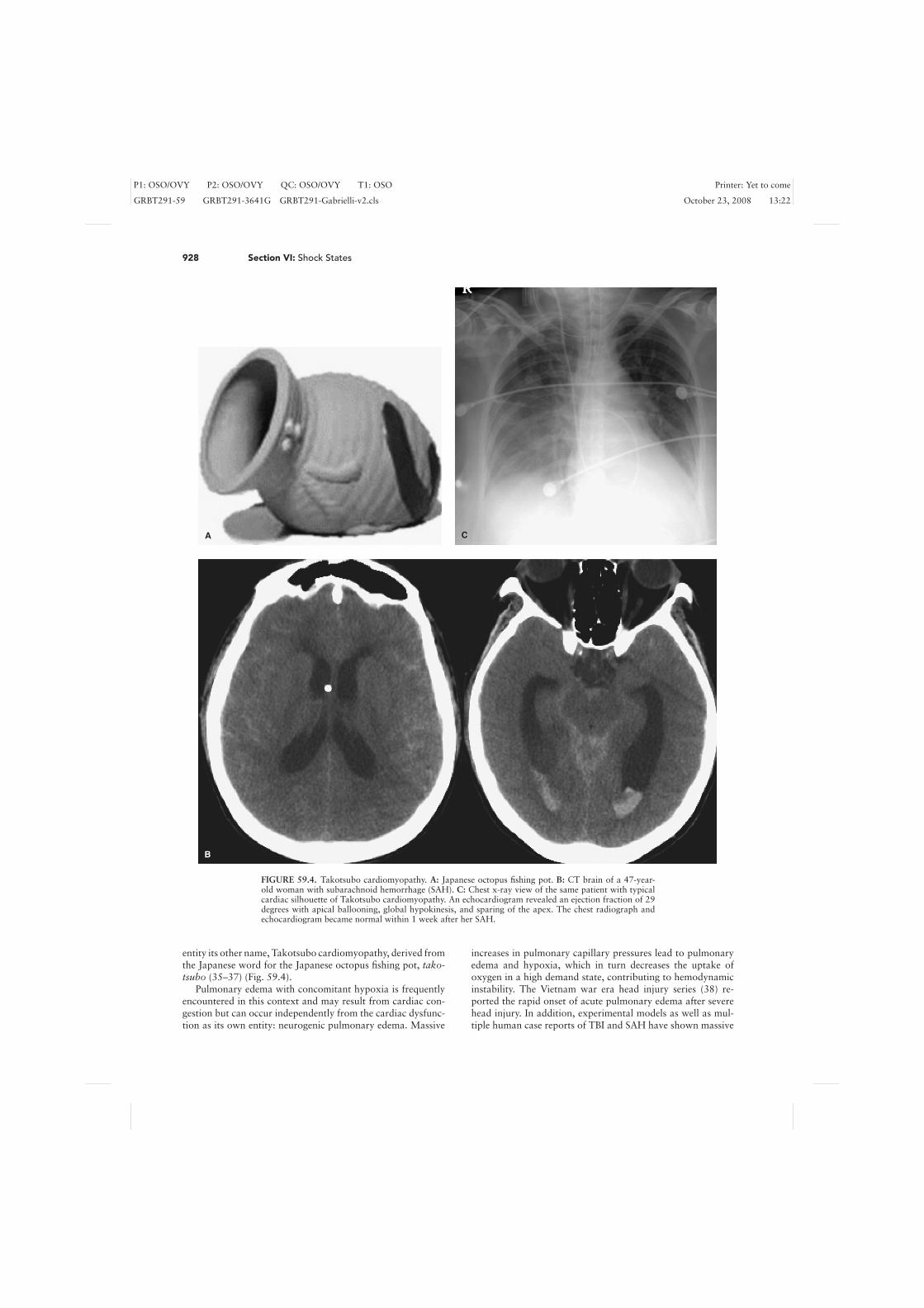
FIGURE 59.4. Takotsubo cardiomyopathy. A: Japanese octopus fishing pot. B: CT brain of a 47-year-old woman with subarachnoid hemorrhage (SAH). C: Chest x-ray view of the same patient with typicalcardiac silhouette of Takotsubo cardiomyopathy. An echocardiogram revealed an ejection fraction of 29degrees with apical ballooning, global hypokinesis, and sparing of the apex. The chest radiograph andechocardiogram became normal within 1 week after her SAH.
entity its other name, Takotsubo cardiomyopathy, derived fromthe Japanese word for the Japanese octopus fishing pot, tako-tsubo (35–37) (Fig. 59.4).
Pulmonary edema with concomitant hypoxia is frequentlyencountered in this context and may result from cardiac con-gestion but can occur independently from the cardiac dysfunc-tion as its own entity: neurogenic pulmonary edema. Massive
increases in pulmonary capillary pressures lead to pulmonaryedema and hypoxia, which in turn decreases the uptake ofoxygen in a high demand state, contributing to hemodynamicinstability. The Vietnam war era head injury series (38) re-ported the rapid onset of acute pulmonary edema after severehead injury. In addition, experimental models as well as mul-tiple human case reports of TBI and SAH have shown massive

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
Chapter 59: Neurogenic Shock 929
Peripheral vasoconstriction
Pulmonary venous constriction
Acute Brain Injury TBI, SAH, ICH,ischemic stroke
Catecholamine Surge
α α + β β
↑Pulmonary capillary pressure
Neurogenic pulmonary edema
LVEF ↓
Shock↑Pulmonary permeability
Damaged endothelium
Platelet aggregation
Microemboli
Myocardial contraction band necrosis
FIGURE 59.5. Summary of the pathophysiology ofthe cardiogenic type of neurogenic shock. ICH, in-tracerebral hemorrhage; LVEF, left ventricular ejectionfraction; SAH, subarachnoid hemorrhage; TBI, trau-matic brain injury.
sympathetic discharges as the primary cause of neurogenic pul-monary edema (39–41). Figure 59.5 summarizes the patho-physiology of cardiogenic neurogenic shock.
Overall, cardiac neurogenic shock, with or without neuro-genic pulmonary edema, is usually transient, with resolutionwithin several days to 2 weeks (2–4). Prevention of secondarybrain injury from hypoxia and decreased cerebral perfusionpressures should be the focus of care in the management ofthis neurally mediated complication.
Neuroendocrine Neurogenic Shock
Insufficiency of the hypothalamic-pituitary-adrenal axis hasbeen recognized as an important cause for shock. Inappropri-ately reduced release of cortisol in stress situations can leadto decreased systemic vascular resistance, reduced cardiac con-tractility, hypovolemic shock, or hyperdynamic shock that canmimic septic shock. Secondary adrenal insufficiency due toinjury to the hypothalamic-hypopituitary feedback loop cancause neuroendocrine neurogenic shock. Acute brain injury,particularly TBI and SAH, can commonly lead to injury ofthe hypothalamus, pituitary gland, or the connecting structures(42). Cohan et al. (6) revealed that adrenal insufficiency aftertraumatic brain injury occurred in about half of all patientsand led to a significantly higher rate of hypotension in thesepatients. Most cases of adrenal insufficiency developed within4 days of injury. Importantly, the authors defined adrenal insuf-ficiency using a low random serum cortisol value and highlightthe fact that an increase in the cortisol level after a stimula-tion test does not rule out the presence of adrenal insufficiency.This issue is particularly relevant in TBI patients, in whomthe hypothalamus and the pituitary gland are the likely af-fected organs, and the adrenal glands might well be expectedto mount an appropriate response when stimulated. When rec-
ognized, primary and secondary adrenal insufficiency can easilybe treated.
In septic shock, a low-dose vasopressin infusion has beenshown to successfully restore blood pressure in hypotensionrefractory to standard catecholamine therapy (43,44). Recentstudies in SAH have shown that endogenous vasopressin serumlevels are elevated during the first 2 days after SAH but decreaseto subnormal levels after 4 days (45–47). Arginine vasopressinsupplementation in SAH at low dose (0.01–0.04 units/min)has been studied in only one single retrospective study (48).The role of vasopressin in the setting of neurogenic shock re-mains to be studied in a prospective manner, but the changes inendogenous vasopressin levels might indicate neuroendocrinechanges in neurogenic shock and potential new treatment op-tions in SAH.
CLINICAL MANIFESTATIONS
Figure 59.6 illustrates the clinical manifestations and symp-toms seen in neurogenic shock. Acutely, vasodilatory neuro-genic shock presents with a “warm and dry” hemodynamicprofile. The patient is hypotensive and frequently bradycardic;however, the peripheral vessels are dilated, leading to warmlimbs and a normal capillary refill time. Central venous pres-sure (CVP) is normal or low, and systemic venous resistance(SVR) is always low. Stroke volume and cardiac output arelow due to the unopposed vagal tone. When a spinal cord in-jury is present, a difference in smooth muscle and vasculaturetone can be observed between the body parts above and belowthe level of the injury. For example, in an injury at thoracic level7 (T7), normal upper limb perfusion might be observed, whilevasodilatation below T7 leads to warm and dry lower extrem-ities. Orthostatic hypotension without reflex tachycardia onchanging from a supine to an upright position—by standing

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
930 Section VI: Shock States
Dry, warm skin Bradycardia Hypotension SVR ↓ SV and CO ↓ CVP ↓ or normal
Dry, or wet, cold skin Tachycardia (rarely bradycardia) Hypotension SVR ↑ SV and CO ↓ CVP ↑ or normal PCWP ↑ EDVI ↑ Chest radiograph “Takotsubo”-shaped heart Echo LVEF ↓ with global or segmental wall motion abnormalities different from coronary artery territory May have pulmonary edema May have cardiac enzyme leak
Hypotension not responsive to vasopressors SVR ↓ SV and CO ↓ CVP ↓ or normal
Vasodilatory Neurogenic Shock
Cardiogenic Neurogenic Shock
Neuroendocrine Neurogenic Shock
FIGURE 59.6. Clinical manifestations ofthe different types of neurogenic shock.CO, carbon monoxide; CVP, central venouspressure; EDVI, end-diastolic volume index;PCWP, pulmonary capillary wedge pressure;SV, stroke volume; SVR, systemic vascularresistance.
or with reverse Trendelenburg position—is common. Whentreating this form of neurogenic shock with a vasopressor infu-sion (such as phenylephrine or other pressors), extreme cautionshould be applied, as vasopressor hypersensitivity can lead tosevere rebound hypertension, which can be difficult to control.
Cardiogenic neurogenic shock manifests as hypotension andtachycardia, with bradycardia seen rarely. Peripheral vessels areoften vasoconstricted, leading to a high SVR and cold and wetskin. Vascular filling, as measured by CVP, pulmonary capil-lary wedge pressure (PCWP), and end-diastolic volume index(EDVI), is normal or high, with low stroke volumes and car-diac output due to global myocardial dysfunction. Leaking ofcardiac enzymes—troponin, creatine kinase (CK), CK-MB—may be seen, but frequently the peak levels are not as high asone would find in myocardial infarction. It is difficult to estab-lish a cutoff value that differentiates stunned myocardium frommyocardial infarction with atherosclerotic coronary artery dis-ease. A retrospective study in SAH measuring troponin-I levelshas reported an appropriate cutoff value to be 2.8 ng/mL (49),whereas CK-MB did not help differentiate between the twokinds of myocardial injury. Higher levels of troponin shouldraise the suspicion of true myocardial infarction, and ECG andechocardiography correlation is important.
Neuroendocrine neurogenic shock presents with hypoten-sion that does not respond well to vasopressor infusion. Hemo-dynamic signs of this category of neurogenic shock are lowCVP, SVR, stroke volume, and cardiac output. Low baselinecortisol levels are the hallmark. A cosyntropin stimulation testfrequently leads to an appropriate increase in the cortisol level,which does not rule out the presence of neuroendocrine neu-rogenic shock, as the adrenal gland is usually not the primarilyaffected organ (6). For this reason, we do not find any clini-cal utility in this test when neuroendocrine neurogenic shockis suspected. Resolution of the hypotension with the use ofhydrocortisone clinically confirms the presence of this shockform.
DIAGNOSTIC CONSIDERATIONS
In any case of hypotension and shock in the neurointensivecare unit, systemic causes for shock must be ruled out first.Especially in the paralyzed patient (for example, one with ahigh spinal cord injury), recognition of other life-threateninginjuries can be quite difficult. Signs of hypovolemic shock maybe absent, even in a patient with profound internal bleed-ing, because of the absence of sympathetic tone below thelevel of injury. The usual pallor from vasoconstriction and re-flex tachycardia might also be absent. The patient may evenbe bradycardic while continuing to bleed. For the same rea-son, signs of peritoneal irritation may be absent in patientswith abdominal injuries. The reported incidence of pulmonaryembolism (PE) varies tremendously in the neurocritical carepatient population—ranging from 0.5% to 20% in ischemicstroke (50,51), to 1% in intracranial hemorrhage (52), to 8.4%in brain tumors (53)—and there are only limited data duringthe acute phase of subarachnoid hemorrhage, traumatic braininjury, and spinal cord injury (51). Interestingly, according tothe study by Skaf et al. (50) using the National Hospital Dis-charge Survey, the incidence of PE did not change in patientswith ischemic and hemorrhagic stroke between 1979 and 2003.However, the death rate from PE in the subgroup with ischemicstroke decreased, likely due to an increased use of antithrom-botic prophylaxis over the last 20 years (54). Additionally,over the last two decades, the methods of PE detection haveimproved immensely—for example, pulmonary CT-angiogramversus nucleotide scan—and autopsy studies report additionalasymptomatic cases. In our opinion, the true incidence of PE isunderestimated. Pulmonary embolism should always be con-sidered in cases of refractory shock. If profound hypotensionis present, or hypotension becomes progressive, reasons otherthan neurogenic shock should be suspected and thoroughlyruled out.

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
Chapter 59: Neurogenic Shock 931
Every patient should undergo serial ECGs, serial cardiacenzyme measurements, and a chest radiograph. As previouslymentioned, pulmonary edema and neurocardiogenic injurymay occur together or separately, making chest x-ray films im-portant diagnostic tools. In particular, one should look for pul-monary vascular congestion and evaluate the size and shape ofthe cardiac silhouette. Hemodynamic monitoring with continu-ous blood pressure and central venous pressure (CVP) monitor-ing with an arterial line and central venous line (CVL) shouldbe undertaken. Blood pressure measurements should be donecontinuously with an arterial line. Arteriosclerosis of the up-per extremities is common and should be kept in mind eitherwhen there is a large discrepancy between right- and left-sidedpressures or when the clinical appearance of the patient doesnot match the readings from the arterial line. Central venousaccess is key for determining CVP and for the administrationof fluids and medications, especially vasopressors. The site ofthe placement of the central venous line (CVL) may play animportant role in the management of shock in a neurologicallyinjured patient. Subclavian vein catheters are the preferred sitein patients with elevated intracranial pressure (ICP), as thereis a theoretical risk of venous stasis within the internal jugularvein with venous congestion and higher risk for venous sinusthrombosis, which could result in increased ICP (55). In ad-dition, trauma patients frequently have cervical spine injuriesand require cervical collars, making the internal jugular veinaccessible only with difficulty.
In patients with cardiogenic neurogenic shock, more exten-sive hemodynamic monitoring may be necessary with eithernoninvasive cardiac monitoring devices or a pulmonary arterycatheter (PAC). Echocardiography is very important to under-standing the etiology of shock. In most cases, a transthoracicechocardiogram is sufficient. The typical echocardiographicappearance is that of apical ballooning, which results fromglobal hypokinesis sparing the apex (56). This part of the heartis devoid of sympathetic nerve terminals, supporting the hy-pothesis that cardiac injury in SAH is neurally mediated by asympathetic storm. Segmental wall motion abnormalities notconforming to distinct coronary artery territories is anothercharacteristic echocardiographic finding. However, myocardialinfarction from ischemic coronary disease is frequently seen inbrain-injured patients, just as in any critically ill patient, andshould always be ruled out first as a cause of shock. In the set-ting of fever and shock, blood cultures must be obtained and thepatient appropriately covered with antibiotics until the culturesyield results. However, older and immunosuppressed patientsmay not mount an appropriate febrile response, and thus sep-sis should still be considered in these patients even when theyare afebrile, especially in the setting of a rising white blood cell(WBC) count. Cerebral spinal fluid cultures are very impor-tant in the neurointensive care unit, with antibiotic coverageof potential central nervous system CNS infections, especiallyin patients after head trauma with skull fracture or sinus dis-ease, after instrumentation of the head or spinal canal, or inimmunocompromised patients. Placement of intracranial pres-sure measurement devices do not contribute to the diagnosticworkup of shock, but they are important tools in the manage-ment of neurogenic shock, such as when the goal mean arterialpressure (MAP) is being titrated to the cerebral perfusion pres-sure. Finally, adrenal insufficiency should always be considered.Random serum cortisol levels should be obtained in the earlystages of shock, keeping in mind that in some forms of brain
injury, low random serum cortisol levels, and thus adrenal in-sufficiency, may be encountered for several days after injury (6).
Many neurologically injured patients, especially those withspinal cord injuries, receive steroids while in the neurointen-sive care unit. The doses administered may be high enough toalter the result of a random serum cortisol level, but often thedose is not enough to treat true adrenal insufficiency appro-priately. In these cases, one could either empirically treat withhigher doses of steroids that also treat adrenal insufficiency—hydrocortisone, with or without fludrocortisone—or, keepingthe potential adverse effects of steroids in acute injury in mind,one could withhold the administration of steroids for 12 hours,then obtain a random cortisol level and resume steroid treat-ment right after the blood draw. However, hypotension is fre-quently severe enough that immediate treatment is warranted,and withholding steroids often is not an option. Dexametha-sone, which is frequently used in the neurointensive care unit, isthe steroid that interferes the least with the cortisol assay aftera corticotropin stimulation test and therefore allows for such atest. In cases of high suspicion, a random cortisol level is oftenpreferred because of its simplicity. The cortisol level should bedrawn immediately before the steroid dose. However, given thelack of mineralocorticoid activity of dexamethasone, changingto hydrocortisone with or without fludrocortisone is recom-mended when adrenal insufficiency is suspected.
MANAGEMENT
Two important reasons for early and proactive treatment ofpatients in neurogenic shock are as follows:
1. Prevention of secondary brain injury from hypoxia and hy-potension
2. The fact that neurogenic shock, especially cardiogenic andneuroendocrine forms, is easily treatable and transient, withpotentially good outcomes despite the moribund appearanceof the patient in the acute phase.
Identifying patients at risk has been very difficult, but at leastin SAH it appears that poor neurologic grade, age older than30 years, and ventricular repolarization abnormalities are riskfactors for neurogenic shock (57).
Once the diagnosis of neurogenic shock has been establishedand the pathophysiology (subtype) has been understood, treat-ment tailored to the specific subtype is initiated. In all cases,euvolemia is of utmost importance and must be achieved beforeany other treatment can be successful. In general, vasopressortreatment as a continuous infusion is initiated and titrated to agoal MAP and cerebral perfusion pressure (CPP). As an impor-tant management tool, an intracranial pressure measurementdevice is very helpful, allowing the indirect measurement ofCPP. We recommend a goal CPP of greater than or equal to 65mm Hg. The optimal CPP is not known. Data regarding theminimum tolerable CPP comes from TBI patients, in whomthe ICP is often elevated. Several studies have suggested an im-proved outcome when CPP is maintained at greater than 70 mmHg (58,59). Other studies using physiologic measurements,such as cerebral blood flow and brain tissue PO2 (PbtO2), indi-cate that adverse changes do not occur unless the CPP is below50 to 60 mm Hg (60,61).
Vasodilatory neurogenic shock can be difficult to treat.In general, vagal tone predominates; however, in this state,

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
932 Section VI: Shock States
patients frequently have peripheral α-adrenoceptor hyperre-sponsiveness, limiting the use of norepinephrine, epinephrine,ephedrine, and phenylephrine. In fact, sympathomimeticsshould be avoided as they can lead to severe blood pressurefluctuations. Since arginine vasopressin (AVP) does not affectα- or β-adrenergic receptors, but acts on V1 receptors, AVPmay have an advantage over catecholamines or phenylephrinein this form of neurogenic shock. It has not been studied in neu-rogenic shock, however, and it remains unclear whether AVPmay have adverse effects on neurologically ill patients. Thisconcern is based on animal studies indicating that vasopressinmay promote the development of vasospasm in SAH, and indi-rect experimental studies showing a reduction in brain edemawith vasopressin antagonists. No prospective human study hasbeen undertaken to confirm or dismiss this concern, and theonly retrospective study on the use of vasopressin in SAH didnot show any of these potentially adverse effects (48). In addi-tion to vasopressors, a temporary demand pacemaker and/oratropine may be required in cases of refractory bradycardiaand hypotension.
In cardiogenic neurogenic shock, some form of inotropicsupport may be necessary, either in the form of a dobutamine,milrinone, or norepinephrine infusion. Dopamine is generallyavoided because of its proarrhythmic properties. Dobutamineand milrinone also have vasodilatory effects, frequently lead-ing to more hypotension, requiring additional therapy with anα-receptor agonist, such as phenylephrine or norepinephrine.Afterload increases in the former, and tachycardia in the latter,might be limiting factors and need careful monitoring. Cardiacoutput monitoring may be undertaken with the guidance ofa PAC. Beta-blockade is usually not recommended. In neuro-genic cardiogenic shock, coronary artery disease is typically notpresent, and compensatory tachycardia is necessary to main-tain cardiac output. Afterload reduction with cautious use ofangiotensin-converting enzyme (ACE) inhibitors should be at-tempted, but further hypotension must be avoided to main-tain tenuous cerebral perfusion pressures. Short-acting agentsshould be used whenever possible. Repeating an echocardio-gram several days after the initial one is recommended to moni-tor the progression/resolution of cardiac dysfunction. The needfor an intra-aortic balloon pump to mechanically reduce after-load and improve coronary perfusion pressure may be consid-ered, albeit rarely used.
Once diagnosed, neuroendocrine neurogenic shock fromprimary, or more often secondary, adrenal insufficiency istreated with steroid replacement therapy. We use the same dos-ing as in adrenal insufficiency in septic shock: hydrocortisone,50 mg intravenously every 6 hours. As previously discussed,a cortisol stimulation test is usually not helpful, and empirictreatment after a random cortisol level should be initiated.
SUMMARY
Neurogenic shock is not a single entity, but rather is composedof three subtypes and pathophysiologies: vasodilatory, cardiac,and neuroendocrine. Other causes of hypotension should beruled out first, prior to making the diagnosis of neurogenicshock. In most cases, neurogenic shock is transient and re-versible, making this entity very treatable. Diagnosis and treat-ment should be tailored to the subtype of neurogenic shock.Maintenance of cerebral perfusion pressures is the key prin-
ciple of management to prevent secondary brain injury andimprove outcome.
References
1. Bilello JF, Davis JW, Cunningham MA, et al. Cervical spinal cord injury andthe need for cardiovascular intervention. Arch Surg. 2003;138(10):1127–1129.
2. Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distri-bution, and rate of recovery of left ventricular systolic dysfunction in patientswith subarachnoid hemorrhage. J Neurosurg. 2006;105(1):15–20.
3. Mayer SA, Lin J, Homma S, et al. Myocardial injury and left ventricularperformance after subarachnoid hemorrhage. Stroke. 1999;30(4):780–786.
4. Pollick C, Cujec B, Parker S, et al. Left ventricular wall motion abnormal-ities in subarachnoid hemorrhage: an echocardiographic study. J Am CollCardiol. 1988;12(3):600–605.
5. Kono T, Morita H, Kuroiwa T, et al. Left ventricular wall motion abnor-malities in patients with subarachnoid hemorrhage: neurogenic stunned my-ocardium. J Am Coll Cardiol. 1994;24(3):636–640.
6. Cohan P, Wang C, McArthur DL, et al. Acute secondary adrenal insuf-ficiency after traumatic brain injury: a prospective study. Crit Care Med.2005;33(10):2358–2366.
7. Dimopoulou I, Tsagarakis S, Douka E, et al. The low-dose corticotropinstimulation test in acute traumatic and non-traumatic brain injury: incidenceof hypo-responsiveness and relationship to outcome. Intensive Care Med.2004;30(6):1216–1219.
8. Calaresu FR, Yardley CP. Medullary basal sympathetic tone. Ann Rev Phys-iol. 1988;50:511–524.
9. Osborn JW, Taylor RF, Schramm LP. Determinants of arterial pressure afterchronic spinal transection in rats. Am J Physiol. 1989;256(3 Pt 2):R666–673.
10. Maiorov DN, Weaver LC, Krassioukov AV. Relationship between sympa-thetic activity and arterial pressure in conscious spinal rats. Am J Physiol.1997;272(2 Pt 2):H625–631.
11. Osborn JW, Taylor RF, Schramm LP. Chronic cervical spinal cord injury andautonomic hyperreflexia in rats. Am J Physiol. 1990;258(1 Pt 2):R169–174.
12. Krassioukov AV, Weaver LC. Episodic hypertension due to autonomic dysre-flexia in acute and chronic spinal cord-injured rats. Am J Physiol. 1995;268(5Pt 2):H2077–2083.
13. Sutters M, Wakefield C, O’Neil K, et al. The cardiovascular, endocrine andrenal response of tetraplegic and paraplegic subjects to dietary sodium re-striction. J Physiology. 1992;457:515–523.
14. Teasell RW, Arnold JM, Krassioukov A, et al. Cardiovascular consequencesof loss of supraspinal control of the sympathetic nervous system after spinalcord injury. Arch Phys Med Rehab. 2000;81(4):506–516.
15. Karlsson AK. Autonomic dysfunction in spinal cord injury: clinical presen-tation of symptoms and signs. Prog Brain Res. 2006;152:1–8.
16. Lane RD, Wallace JD, Petrosky PP, et al. Supraventricular tachycardia inpatients with right hemisphere strokes. Stroke. 1992;23(3):362–366.
17. Oppenheimer SM, Gelb A, Girvin JP, et al. Cardiovascular effects of humaninsular cortex stimulation. Neurology. 1992;42(9):1727–1732.
18. Zamrini EY, Meador KJ, Loring DW, et al. Unilateral cerebral inac-tivation produces differential left/right heart rate responses. Neurology.1990;40(9):1408–1411.
19. Burch GE, Meyers R, Abildskov JA. A new electrocardiographic patternobserved in cerebrovascular accidents. Circulation. 1954;9(5):719–723.
20. Davies KR, Gelb AW, Manninen PH, et al. Cardiac function in aneurysmalsubarachnoid haemorrhage: a study of electrocardiographic and echocardio-graphic abnormalities. Br J Anaesth. 1991;67(1):58–63.
21. Macrea LM, Tramer MR, Walder B. Spontaneous subarachnoid hemorrhageand serious cardiopulmonary dysfunction–a systematic review. Resuscita-tion. 2005;65(2):139–148.
22. Tung P, Kopelnik A, Banki N, et al. Predictors of neurocardiogenic injuryafter subarachnoid hemorrhage. Stroke. 2004;35(2):548–551.
23. Naredi S, Lambert G, Eden E, et al. Increased sympathetic nervous ac-tivity in patients with nontraumatic subarachnoid hemorrhage. Stroke.2000;31(4):901–906.
24. Elrifai AM, Bailes JE, Shih SR, et al. Characterization of the cardiac effectsof acute subarachnoid hemorrhage in dogs. Stroke. 1996;27(4):737–741;discussion 741–732.
25. Cowan MJ, Giddens WE Jr, Reichenbach DD. Selective myocardial cellnecrosis in nonhuman primates. Arch Pathol Lab Med. 1983;107(1):34–39.
26. Baroldi G, Mittleman RE, Parolini M, et al. Myocardial contraction bands.Definition, quantification and significance in forensic pathology. Intern JLegal Med. 2001;115(3):142–151.
27. Todd GL, Baroldi G, Pieper GM, et al. Experimental catecholamine-inducedmyocardial necrosis, I: morphology, quantification and regional distributionof acute contraction band lesions. J Molecular Cell Cardiol. 1985;17(4):317–338.
28. Kline IK. Myocardial alterations associated with pheochromocytomas. AmJ Pathol. 1961;38:539–551.

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
Chapter 60: Anaphylactic Shock 933
29. Hammermeister KE, Reichenbach DD. QRS changes, pulmonary edema, andmyocardial necrosis associated with subarachnoid hemorrhage. Am Heart J1969;78(1):94–100.
30. Koskelo P, Punsar S, Sipilae W. Subendocardial haemorrhage and E.C.G.Changes in Intracranial Bleeding. Br Med J. 1964;1(5396):1479–1480.
31. Smith RP, Tomlinson BE. Subendocardial haemorrhages associated with in-tracranial lesions. J Pathol Bacteriol. 1954;68(2):327–334.
32. Doshi R, Neil-Dwyer G. A clinicopathological study of patients following asubarachnoid hemorrhage. J Neurosurg. 1980;52(3):295–301.
33. Crompton MR. Hypothalamic lesions following the rupture of cerebral berryaneurysms. Brain. 1963;86:301–314.
34. Greenhoot JH, Reichenbach DD. Cardiac injury and subarachnoid hemor-rhage. A clinical, pathological, and physiological correlation. J Neurosurg.1969;30(5):521–531.
35. Kawai S, Suzuki H, Yamaguchi H, et al. Ampulla cardiomyopathy (‘Tako-tsubo’ cardiomyopathy)–reversible left ventricular dysfunction: with ST seg-ment elevation. Jpn Circ J. 2000;64(2):156–159.
36. Akashi YJ, Nakazawa K, Sakakibara M, et al. Reversible left ventricular dys-function “takotsubo” cardiomyopathy related to catecholamine cardiotoxi-city. J Electrocardiol. 2002;35(4):351–356.
37. Akashi YJ, Nakazawa K, Sakakibara M, et al. The clinical features of takot-subo cardiomyopathy. QJM 2003;96(8):563–573.
38. Simmons RL, Martin AM Jr, Heisterkamp CA 3rd, et al. Respiratory insuffi-ciency in combat casualties, II: pulmonary edema following head injury. AnnSurg. 1969;170(1):39–44.
39. Pender ES, Pollack CV Jr. Neurogenic pulmonary edema: case reports andreview. J Emerg Med. 1992;10(1):45–51.
40. Hoff JT, Nishimura M, Garcia-Uria J, et al. Experimental neurogenic pul-monary edema, 1: the role of systemic hypertension. J Neurosurg. 1981;54(5):627–631.
41. Lang SA, Maron MB, Signs SA. Oxygen consumption after massive sympa-thetic nervous system discharge. Am J Physiol. 1989;256(3 Pt 1):E345–351.
42. Aimaretti G, Ambrosio MR, Di Somma C, et al. Traumatic brain injuryand subarachnoid haemorrhage are conditions at high risk for hypopitu-itarism: screening study at 3 months after the brain injury. Clin Endocrinol.2004;61(3):320–326.
43. Dunser MW, Wenzel V, Mayr AJ, et al. Management of vasodilatory shock:defining the role of arginine vasopressin. Drugs. 2003;63(3):237–256.
44. Landry DW, Levin HR, Gallant EM, et al. Vasopressin pressor hypersensi-tivity in vasodilatory septic shock. Crit Care Med. 1997;25(8):1279–1282.
45. Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrationsof natriuretic peptides and antidiuretic hormone after subarachnoid hemor-rhage. Stroke. 1994;25(11):2198–2203.
46. Huang WD, Yang YM, Wu SD. Changes of arginine vasopressin in el-
derly patients with acute traumatic cerebral injury. Chinese J Traumatol.2003;6(3):139–141.
47. Barreca T, Gandolfo C, Corsini G, et al. Evaluation of the secretory patternof plasma arginine vasopressin in stroke patients. Cerebrovasc Dis. 2001;11(2):113–118.
48. Muehlschlegel S, Dunser MW, Gabrielli A, et al. Arginine vasopressinas a supplementary vasopressor in refractory hypertensive, hypervolemic,hemodilutional therapy in subarachnoid hemorrhage Neurocrit Care. 2007;6(1):3–10.
49. Bulsara KR, McGirt MJ, Liao L, et al. Use of the peak troponin valueto differentiate myocardial infarction from reversible neurogenic left ven-tricular dysfunction associated with aneurysmal subarachnoid hemorrhage.J Neurosurg. 2003;98(3):524–528.
50. Skaf E, Stein PD, Beemath A, et al. Venous thromboembolism in patients withischemic and hemorrhagic stroke. Am J Cardiol. 2005;96(12):1731–1733.
51. Hamilton MG, Hull RD, Pineo GF. Venous thromboembolism in neuro-surgery and neurology patients: a review. Neurosurgery. 1994;34(2):280–296; discussion 296.
52. Maramattom BV, Weigand S, Reinalda M, et al. Pulmonary complicationsafter intracerebral hemorrhage. Neurocrit Care. 2006;5(2):115–119.
53. Brisman R, Mendell J. Thromboembolism and brain tumors. J Neurosurg.1973;38(3):337–338.
54. Skaf E, Stein PD, Beemath A, et al. Fatal pulmonary embolism and stroke.Am J Cardiol. 2006;97(12):1776–1777.
55. Stephens PH, Lennox G, Hirsch N, et al. Superior sagittal sinus thrombosisafter internal jugular vein cannulation. Br J Anaesth. 1991;67(4):476–479.
56. Zaroff JG, Rordorf GA, Ogilvy CS, et al. Regional patterns of left ventricularsystolic dysfunction after subarachnoid hemorrhage: evidence for neurallymediated cardiac injury. J Am Soc Echocardiogr. 2000;13(8):774–779.
57. Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers ofabnormal left ventricular wall motion in acute subarachnoid hemorrhage.J Neurosurg. 1995;83(5):889–896.
58. Eisenberg HM, Frankowski RF, Contant CF, et al. High-dose barbituratecontrol of elevated intracranial pressure in patients with severe head injury.J Neurosurg. 1988;69(1):15–23.
59. Narayan RK, Kishore PR, Becker DP, et al. Intracranial pressure: to moni-tor or not to monitor? A review of our experience with severe head injury.J Neurosurg. 1982;56(5):650–659.
60. Bullock R, Chesnut RM, Clifton G, et al. Guidelines for the manage-ment of severe head injury. Brain Trauma Foundation. Eur J Emerg Med.1996;3(2):109–127.
61. Czosnyka M, Guazzo E, Iyer V, et al. Testing of cerebral autoregulationin head injury by waveform analysis of blood flow velocity and cerebralperfusion pressure. Acta Neurochirurgica. 1994;60:468–471.
CHAPTER 60 ■ ANAPHYLACTIC SHOCKMEGHAVI S. KOSBOTH � ERIC S. SOBEL
Anaphylaxis is severe, has a rapid onset, and is potentiallyfatal—a systemic allergic reaction that occurs after contact withan allergy-causing substance (1,2). Activation of mast cell andbasophil populations by either IgE-dependent (i.e., anaphylac-tic reactions) or IgE-independent (i.e., anaphylactoid reactions)mechanisms results in the release of multiple mediators capableof altering vascular permeability and vascular and bronchialsmooth muscle tone, as well as recruiting and activating in-flammatory cell cascades. Because the clinical presentationsof anaphylactic and anaphylactoid reactions are indistinguish-able, they will be referred to as anaphylaxis for the purposesof this chapter. Initial sequelae, which occur within minutesto an hour after exposure to an inciting stimulus, include gen-
eralized hives, tachycardia, flushing, pruritus, faintness, and asensation of impending doom. Dermatologic (i.e., urticaria andangioedema), respiratory (i.e., dyspnea, wheeze, stridor, bron-chospasm, and hypoxemia), and gastrointestinal (i.e., abdom-inal distension, nausea, emesis, and diarrhea) manifestationsare common. Involvement of the cardiovascular and respira-tory systems may result in potentially life-threatening manifes-tations, such as cardiovascular collapse caused by vasodilationand capillary leak, myocardial depression, myocardial ischemiaand infarction, and atrial fibrillation (3). Prompt recogni-tion and effective intervention are essential to preventthe fatal manifestations of anaphylactic and anaphylactoidreactions.

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
934 Section VI: Shock States
INCIDENCE
The incidence of anaphylaxis is difficult to determine accuratelydue to underdiagnosis and underreporting. In the United States,fatal anaphylaxis causes 500 to 1,000 fatalities per year andaccounts for 1% of emergency department visits (1,4,5). Theanaphylaxis rate was found to be 21 per 100,000 person-yearsin a study of nonhospitalized individuals in Olmsted County,Minnesota, between 1983 and 1987 (6). A subsequent analysisof the General Practice database in the United Kingdom notedthe incidence to be 8.4 per 100,000 person-years (7). An epi-demiologic study involving 481,752 individuals suggested thathospitalized patients are at increased risk of anaphylaxis, butthese reactions are rarely fatal (8).
ETIOLOGY
The most common causes of anaphylaxis include insect stings,foods, drugs, and physical factors/exercise. Idiopathic anaphy-laxis (where no causative agent is identified) accounts for upto two thirds of patients referred to allergy/immunology spe-cialty clinics (9,10). Foods such as shellfish, eggs, nuts, and milkaccount for one third of food-induced anaphylactic episodes(10–13) (Tables 60.1 and 60.2). There is a syndrome of food-dependent, exercise-induced anaphylaxis (FDEIA) that devel-ops only if food is ingested prior to exercise or exertion (14).Seafood, nuts, celery, wheat, and grains have been implicatedas allergens in this syndrome. It is important to note that thesefoods are tolerated by the patient in the absence of exertion(14,15).
Anaphylactic reactions to stings or bites of various insects,such as members of the order Hymenoptera (yellow jackets,bees, wasps, hornets, and saw flies) are commonly reported.A positive venom skin test along with a systemic reaction tothe insect sting predicts a 50% to 60% risk of reaction tofuture stings (16). Medications can cause anaphylactic (IgE-mediated) and anaphylactoid (non–IgE-mediated) reactions.Previous exposure to drugs is required for IgE production andanaphylactic reactions, but anaphylactoid reactions can occurupon first administration. Penicillin is one of the most com-mon causes of anaphylaxis, with 1 to 5 per 10,000 courseswith penicillin resulting in allergic reactions and 1 in 50,000to 1 in 100,000 courses with a fatal outcome (17–19). Non-steroidal anti-inflammatory drugs (NSAIDs) and aspirin are thesecond most common class of drugs implicated in anaphylaxis(20). Some hypersensitivity reactions will occur with differ-ent NSAID agents, while others are specific to a single drug(21).
With widespread adoption of universal precautions againstinfections, latex allergy has become a significant problem. Thedevelopment of low-protein, powder-free gloves has been asso-ciated with reduction in occupational-contact urticaria causedby latex rubber gloves (22). Despite this, latex allergy is stilla concern since latex is found in gloves, catheters, and tubing(23–25). Iodinated radiocontrast media can cause anaphylaxis;however, life-threatening reactions are rare (26). A history of aprevious reaction to radiocontrast media, asthma or atopic dis-ease, treatment with β-blockers, and cardiovascular disease arerisk factors for developing anaphylaxis to radiocontrast media(27–29).
CLINICAL MANIFESTATIONS
The clinical syndromes associated with systemic anaphylac-tic and anaphylactoid reactions represent medical emergen-cies, as they are associated with a rapid, critical destabi-lization of vital organ systems. These syndromes, which are,again, clinically indistinguishable, may become rapidly fatalif appropriate therapy is not instituted immediately. Initialsymptoms can appear within seconds to minutes but maybe delayed by as much as 1 (or rarely more) hour after ex-posure to an inciting agent (30), and are often nonspecific(31). These symptoms include tachycardia, faintness, cuta-neous flushing, urticaria, diffuse or palmar pruritus, and asensation of impending doom (32). Of these, generalized ur-ticaria is the most common, occurring in approximately 90%of patients (Table 60.3) (33,34). Subsequent manifestations in-dicate involvement of the cutaneous, gastrointestinal, respira-tory, and cardiovascular systems. Involvement of the cardio-vascular and respiratory systems is responsible for the fatal
TABLE 60.1
ETIOLOGIC AGENTS FOR ANAPHYLAXIS (IGE-MEDIATED)
HAPTENSβ-Lactam antibioticsSulfonamidesNitrofurantoinDemethylchlortetracyclineStreptomycinVancomycinLocal anestheticsOthers
SERUM PRODUCTSγ -GlobulinImmunotherapy for
allergic diseasesHeterologous serum
FOODSNuts (peanuts, brazil
nuts, hazelnuts,cashews, pistachios,almonds, soy nuts)
ShellfishBuckwheatEgg whiteCottonseedCow’s milkCornPotatoRiceLegumesCitrus fruitsChocolateOthers
VENOMStinging insects, particularly
Hymenoptera, fire ants, deerflies, jelly fish, kissing bugs(triatoma), and rattlesnakes
HORMONESInsulinAdrenocorticotropic hormoneThyroid-stimulating hormone
ENZYMESChymopapainL-Asparaginase
MISCELLANEOUSSeminal fluidOthers
Boldface: Relatively common causesModified from Austen KF. Systemic anaphylaxis in man. JAMA. 1965;192:108; and Kaliner M. Anaphylaxis. NER Allergy Proc. 1984;5:324.

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
Chapter 60: Anaphylactic Shock 935
TABLE 60.2
ETIOLOGIC AGENTS FOR ANAPHYLACTOID REACTIONS
COMPLEMENT-MEDIATED REACTIONS ARACHIDONIC ACID MODULATORSBlood Nonsteroidal anti-inflammatory drugsSerum Tartrazine (possible)PlasmaPlasmate (but not albumin) IDIOPATHICImmunoglobins Most common conclusion after thorough evaluation
NONIMMUNOLOGIC MAST CELL ACTIVATORS UNKNOWNOpiates and narcotics SulfitesRadiocontrast media OthersDextransNeuromuscular blocking agents THERMOREGULATORY MECHANISM
Cold temperature, exercise
Boldface: Relatively common causes.Adapted from Kaliner M. Anaphylaxis. NER Allergy Proc. 1984;5:324.
complications of anaphylactic/anaphylactoid reactions. Anunsettling sensation—including hoarseness, dysphonia, ordyspnea—may precede acute upper airway obstruction sec-ondary to laryngeal edema. Other pulmonary manifestationsinclude acute bronchospasm, intra-alveolar pulmonary hemor-rhage, bronchorrhea, and a noncardiogenic, high permeability–type pulmonary edema (17,35). Tachycardia and syncope mayprecede the development of hypotension and frank cardiovas-cular collapse (36,37). Anaphylactic shock occurs as a con-sequence of diminished venous return secondary to systemicvasodilation and intravascular volume contraction caused bycapillary leak. Although transient increases in cardiac outputmay occur at the onset of anaphylaxis, hemodynamic param-eters later reveal decreases in cardiac output, systemic vascu-lar resistance, stroke volume, pulmonary artery occlusion, andcentral venous pressures (38–44). In addition, the acute onset
of a lactic acidosis and diminished oxygen consumption havebeen noticed after an anaphylactoid reaction (45). Other po-tentially serious cardiovascular manifestations are myocardialischemia and acute myocardial infarction, atrioventricular andintraventricular conduction abnormalities such as prolongedPR interval, transient left bundle branch block, and supraven-tricular arrhythmias such as atrial fibrillation. Severe, but re-versible, myocardial depression also has been reported (37).Hematologic manifestations, such as disseminated intravascu-lar coagulation and hemoconcentration secondary to volumecontraction, also may complicate anaphylactic and anaphy-lactoid reactions (32). Gastrointestinal manifestations includenausea, bloating, abdominal cramps, and diarrhea.
In 1% to 20% of patients, there is a recurrence of symptomsafter a period of recovery, termed biphasic anaphylaxis (46). Inmost cases, the symptoms recurred 1 to 8 hours after the initial
TABLE 60.3
CLINICAL MANIFESTATIONS OF ANAPHYLACTIC AND ANAPHYLACTOID REACTIONS
System Symptom Frequency Sign/clinical manifestation
RESPIRATORY 60%–80%Upper Dyspnea, dysphonia, cough, “lump in
throat”Upper airway obstruction caused by laryngeal
edema and spasm; bronchorrheaLower Dyspnea, cyanosis Noncardiogenic pulmonary edema, bronchospasm,
acute hyperinflation, alveolar hemorrhageCARDIOVASCULAR Palpitations, faintness, weakness 20% Shock, tachycardia, capillary leak, syncope,
supraventricular arrhythmias, conductiondisturbances, myocardial ischemia and infarction
CUTANEOUS Flushing, pruritus, rash 90% Urticaria, angioedema, diaphoresisGASTROINTESTINAL Abdominal pain, bloating, cramps,
nausea30% Emesis, diarrhea, hepatosplenic congestion; rarely
hematemesis and bloody diarrheaNEUROLOGIC Dizziness, disorientation, hallucinations,
headache, feeling of impending doom5%–10% Syncope, lethargy, seizures
NASAL Pruritus, sneezing 16%–20% Rhinorrhea, nasal congestionOCULAR Conjunctival pruritus, periorbital edema 10%–15% Conjunctival suffusion, lacrimation
HEMATOLOGIC Hemoconcentration, DIC
DIC, disseminated intravascular coagulation.

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
936 Section VI: Shock States
presentation, although there have been reports of recurrenceup to 72 hours later. There were no features of the primaryresponse that predicted the occurrence of a secondary response(47).
DIAGNOSIS
The diagnosis of anaphylaxis is established on the basis of clin-ical grounds alone because expedient institution of appropri-ate therapy is mandatory. These diagnoses should be consid-ered when typical multisystem manifestations occur in a directtemporal relationship with exposure to an inciting agent. Re-cently, the National Institute of Allergy and Infectious Diseases(NIAID) and the Food Allergy and Anaphylaxis Network(FAAN) proposed clinical criteria for the diagnosis of anaphy-laxis (Fig. 60.1) (1). Because of the multisystem nature of ana-phylactic and anaphylactoid reactions, the list of differentialdiagnoses that must be considered is extensive. Diagnostic pos-sibilities include cardiac dysrhythmias, myocardial infarction,distributive or hypovolemic shock, vasovagal syncope, asthma,pulmonary embolism, upper airway obstruction secondary toingestion of a foreign body, hypoglycemia, and the carcinoidsyndrome (Table 60.4).
Demonstration of acute elevations of markers specific tomast cell activation such as histamine and tryptase have beenproposed to help confirm the diagnosis of anaphylaxis (48,49).However, in a series of 97 patients presenting to an emergencydepartment and given the diagnosis of anaphylaxis, only 42%were found to have elevated plasma histamine levels, and 24%had increased plasma tryptase levels (50). Skin testing or serumantibody tests can help demonstrate the presence of IgE againsta specific allergen. Skin testing should be delayed for up to 4weeks to allow the dermal mast cells to replenish intracellularmediators (51).
PATHOPHYSIOLOGY
The systemic manifestations of anaphylactic and anaphylac-toid reactions represent sequelae that result from the release
of inflammatory mediators by mast cells and basophils. Theclassic anaphylactic response occurs through allergen-inducedcrosslinking of IgE tightly bound to the high-affinity FcεR1 re-ceptor constitutively expressed by mast cells (52). Release ofhistamine from preformed mast cell granules seems to be theprimary pathophysiologic mediator, resulting in systemic va-sodilation, increased vascular permeability, bronchoconstric-tion, pruritus, and increased mucus production. However,a number of other preformed mediators are released, in-cluding heparin, serotonin, and mast cell proteases such aschymase and tryptase (53). In addition, other important medi-ators of anaphylaxis are generated by the metabolism of mem-brane phospholipids. Activation of the 5-lipoxygenase path-way results in synthesis of leukotrienes, including leukotrienesC4, D4, E4 (termed the slow-reacting substance of anaphy-laxis), and B4. Leukotrienes C4, D4, and E4, along with theintermediary products 5-hydroxyeicosatetraenoic acid and 5-hydroperoxyeicosatetraenoic acid, elicit increases in vascularpermeability and bronchoconstriction, whereas leukotriene B4
possesses eosinophil and neutrophil chemotactic properties.Activation of the cyclooxygenase pathway leads to the produc-tion of prostaglandin D2, which produces bronchoconstriction.Platelet-activating factor is also newly synthesized by activatedmast cells and can result in bronchoconstriction, increased vas-cular permeability, platelet aggregation, and neutrophil chemo-taxis. It also leads to further production of platelet-activatingfactor through stimulation of nuclear factor (NF)-κB, a posi-tive feedback mechanism involving the cytokines interleukin-1(IL-1) and tumor necrosis factor (TNF)-α, and contributesto a biphasic pattern seen in some patients (54). Combined,these primary mediators then facilitate the production of a di-verse number of secondary mediators by platelets, neutrophils,eosinophils, and other cells, resulting in activation of the com-plement, coagulation, and fibrinolytic pathways (55).
Many of these mediators have complicated effects, and theirrelative roles in mediating anaphylaxis in vivo have been dif-ficult to evaluate. Mouse models of anaphylaxis using strainswith targeted deletions of specific mediators have been usefulin elucidating the importance of different effector molecules,such as the leukotrienes (56–58), and in identifying regulatory
Acute onset of illness (minutes to few hours)
Exposure to likely allergen for that patient
Exposure to known allergen for that patient
No known exposure to allergen
Skin or mucosal involvement and either:
Respiratory compromise
Reduced BP or associated signs Respiratory compromise
Reduced BP or associated signs
Persistent gastrointestinal symptoms
or
or both
Skin and/or mucosal involvement
At least two of the following: Reduced BP or associated signs
FIGURE 60.1. Clinical criteria for di-agnosing anaphylaxis. Fewer signs arerequired for diagnosis as the historyof allergen exposure becomes morecertain. Signs or symptoms of skininvolvement: Generalized hives, pru-ritus, or flushing. Signs of mucosalinvolvement: Swollen lips, tongue,and/or uvula. Signs of respiratorycompromise: Dyspnea, wheeze, bron-chospasm, stridor, reduced peak expi-ratory flow, and/or hypoxemia. Def-inition of reduced blood pressure(BP): Adults—systolic BP less than90 mm Hg or greater than 30% de-crease from that person’s baseline;children—systolic BP less than 70 mmHg from 1 month to 1 year, less than(70 mm Hg + [2 × age]) from 1to 10 years, and less than 90 mmHg from 11 to 17 years or associ-ated signs: Hypotonia, syncope, in-continence. Persistent gastrointesti-nal symptoms: Crampy abdominalpain and vomiting.

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
Chapter 60: Anaphylactic Shock 937
TABLE 60.4
DIFFERENTIAL DIAGNOSIS OF ANAPHYLAXIS
FLUSH SYNDROME POSTPRANDIAL COLLAPSECarcinoid Airway foreign bodyPheochromocytoma Monosodium glutamate ingestionPeri-postmenopausal hot flushes SulfiteMedullary carcinoma of thyroid Scombroid fish poisoningRed man syndrome (vancomycin)
MISCELLANEOUSHYPOTENSION Panic attacksSeptic shock Systemic mastocytosisHemorrhagic shock Basophilic leukemiaCardiogenic shock Hereditary angioedemaHypovolemic shock Hyper-IgE syndromeVasovagal reaction
RESPIRATORY DISTRESSStatus asthmaticusAirway foreign bodyEpiglottitisPulmonary embolismAsthma and COPD exacerbationVocal cord dysfunction
IgE, immunoglobulin E; COPD, chronic obstructive pulmonary disorder.
pathways, such as IL-10 (59), but have also provided somesurprises that may lead to clinically useful information. For ex-ample, mice with targeted deletions of either the high-affinityFcεR1 receptor or IgE, not surprisingly, had a markedly de-creased susceptibility to IgE-mediated anaphylaxis (53,60).This pathway can also be blocked with targeted deletion ofhistamine receptor 1 and, to a lesser extent, platelet-activatingfactor (52,53). However, such mice also revealed the presenceof an alternate IgE-independent pathway of anaphylaxis (61).This pathway was mediated largely through platelet-activatingfactor, which was triggered by the binding of IgG to Fcγ RIIIreceptors present on macrophages (52,62). Like the classic IgE-mediated pathway, this alternative pathway required prior ex-posure to antigen, but differed in that much higher concentra-tions of antigen were required. The importance of this pathwayin humans is as yet unclear (52). However, the administrationof biologic agents, such as the anti-TNF antibody infliximab,has been reported to cause an IgE-independent anaphylacticresponse (63), and may be an example of this alternative path-way. The use of these biologic agents is expected to continueto increase.
MANAGEMENT
The clinician must have a high index of suspicion for ana-phylactic and anaphylactoid reactions because they require aprompt clinical diagnosis and a rapid therapeutic response. Be-cause anaphylactic and anaphylactoid reactions both representsequelae of mast cell and basophil degranulation, the thera-peutic approaches to these disorders are identical. Initial at-tention should be given to assessment and stabilization of thepulmonary and cardiovascular manifestations of anaphylaxis,because these are the major causes of death.
Epinephrine is the mainstay of initial management andshould be administered immediately. It decreases mediator syn-thesis and release by increasing intracellular concentrationsof cyclic adenosine monophosphate (cAMP) and antagonizesmany of the adverse actions of the mediators of anaphylaxis(41). Aqueous epinephrine, 0.01 mg/kg (maximum dose 0.5mg) administered intramuscularly every 5 to 15 minutes asnecessary to control symptoms and maintain blood pressure, isrecommended (41,64). The participants of the NIAID/FAANsymposium concluded that the intramuscular administration ofepinephrine in the anterior lateral thigh is preferred over sub-cutaneous injection (1,2). In cases of severe laryngospasm orfrank cardiovascular collapse, or when there is an inadequateresponse to subcutaneous epinephrine administration and fluidresuscitation, intravenous epinephrine is an option. There isno established dosage regimen for intravenous epinephrine inanaphylaxis, but suggested dosages are 5 to 10 μg bolus (0.2μg/kg) for hypotension and 0.1 to 0.5 mg in the setting ofcardiovascular collapse (1,2,65). When epinephrine is admin-istered IV, the clinician should be aware of the potential ad-verse consequences of severe tachycardia, myocardial ischemia,hypertension, severe vasospasm, and gangrene—the latterwhen infused by peripheral venous access (66).
Blood pressure measurements should be obtained fre-quently, and an indwelling arterial catheter should be insertedin cases of moderate to severe anaphylaxis. High-flow oxygengiven via endotracheal tube or a nonrebreather mask shouldbe administered to patients experiencing hypoxemia, respira-tory distress, or hemodynamic instability (1,2). Orotrachealintubation may be attempted if the airway obstruction com-promises effective ventilation despite pharmacologic interven-tion; however, attempts may be unsuccessful if laryngeal edemais severe. If endotracheal intubation is unsuccessful, then ei-ther needle-catheter cricothyroid ventilation, cricothyrotomy,

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
938 Section VI: Shock States
or surgical tracheostomy is required to maintain an adequateairway. Clinicians must be familiar with at least one of thesetechniques in the event that endotracheal intubation cannot beaccomplished. It has been suggested that inhaled β2-agonistssuch as albuterol may be useful for bronchospasm refrac-tory to epinephrine (1,2,67). Patients should be placed in therecumbent position, with lower extremities elevated to in-crease fluid return centrally, thereby increasing cardiac out-put (68). Airway protection should be ensured in the event ofvomiting.
Antihistamines (H1 and H2 antagonists) are consideredsecond-line treatment for anaphylaxis (1,2). They are useful inthe treatment of symptomatic urticaria-angioedema and pruri-tus. Recent studies suggest that treatment with a combinationof H1 and H2 antagonists is more effective in attenuating thecutaneous manifestations of anaphylaxis than H1 antagonistsalone (50,69). Diphenhydramine hydrochloride (25 to 50 mgIV or IM for adults and 1 mg/kg, up to 50 mg, for children)and ranitidine (50 mg IV over 5 minutes) are commonly usedin this setting. If hypotension persists despite administration ofepinephrine and H1 and H2 blockers, aggressive volume resus-citation should be instituted. Up to 35% of the blood volumemay extravasated in the first 10 minutes of a severe reaction,with subsequent reduction in blood volume due to vasodilata-tion, causing distributive shock (70). Persistent hypotensionmay require multiple fluid boluses (10 to 20 mL/kg under pres-sure) as well as colloid and crystalloid infusions (1,2). Vaso-pressors such as norepinephrine, vasopressin, Neo-Synephrine,or even metaraminol may be useful in persistent hypotension(31).
There have been no placebo-controlled trials evaluating theefficacy of corticosteroids in anaphylaxis, but their contribu-tion in other allergic diseases has led to their inclusion in ana-phylactic management. Due to their slow onset of action, theyare not useful in acute management. However, it has been sug-gested that they may prevent protracted or biphasic reactions(67,71). The usual dose is 100 to 250 mg of hydrocortisone IVevery 6 hours (39).
The management of anaphylaxis in a patient receiving β-antagonist medications, such as β blockers, represents a spe-cial circumstance in which the manifestations of anaphylaxismay be exceptionally severe (72). β Blockade increases medi-ator synthesis and release, as well as end-organ sensitivity. Inaddition, β-blockade antagonizes the beneficial β-mediated ef-fects of epinephrine therapy, thereby resulting in unopposedα-adrenergic and reflex vagotonic effects: vasoconstriction,bronchoconstriction, and bradycardia. Therapy of anaphylaxisoccurring in patients receiving β-antagonist drugs, however, issimilar to that of other patients. In addition, atropine may beuseful for heart block and refractory bronchospasm, whereasglucagons—which increase cAMP levels through a β-receptor–independent mechanism—have been reported to reverse thecardiovascular manifestations of anaphylaxis in patients re-ceiving β-antagonists (72). Glucagon can be administered asa 1- to 5-mg (20–30 μg/kg with maximum dose of 1 mg inchildren) intravenous infusion over 5 minutes, followed by aninfusion of 5 to 15 μg/minute titrated to a clinical response(1,2). Furthermore, these patients may require extended peri-ods of observation because of the long duration of action ofmany β-antagonist medications.
An emergent evaluation for the inciting etiologic agent mustaccompany initial therapeutic interventions. After the etiologic
agent is identified, the clinician should attempt to prevent fur-ther access to the circulation or limit further absorption. In-fusions of possible etiologic agents should be stopped and thecontents saved for analysis. If a Hymenoptera sting is respon-sible, the stinger should be removed. Small amounts of localepinephrine—0.1 to 0.2 mL of a 1:1,000 solution—should beinjected next to a subcutaneous or intramuscular injection sitethat is dispersing the inciting agent. A tourniquet also shouldbe placed proximal to the injection site and pressure applied toocclude venous return. After successful pharmacologic therapy,the tourniquet may be cautiously removed and the patient care-fully observed for recurrent adverse sequelae. In cases wherethe offending agent was ingested, consideration may be givento insertion of a nasogastric tube to perform gastric lavage andgastric instillation of activated charcoal.
THERAPEUTIC PEARLS
1. Rapidly assess and maintain the airway, breathing, andcirculation. If airway obstruction is imminent, performendotracheal intubation; if unsuccessful, consider needle-catheter cricothyroid ventilation, cricothyrotomy, or tra-cheostomy. Patients in anaphylactic shock should be placedin a recumbent position with the lower extremities ele-vated, unless precluded by shortness of breath or vomiting.
2. Remove the inciting agent (i.e., remove Hymenopterastinger) and follow with an intramuscular epinephrine in-jection in the anterior lateral thigh. Consider gastric lavageand administration of activated charcoal if the incitingagent was ingested.
3. Administer aqueous epinephrine, 0.01 mg/kg (maximumdose, 0.5 mg) intramuscularly every 5 to 15 minutes asnecessary for controlling symptoms and maintaining bloodpressure.
4. Establish intravenous access for hydration and providesupplemental oxygen.
5. Administer histamine antagonists to block vasodilation,capillary leak, and shock (H1 blockade, 25–50 mg ofdiphenhydramine IV or IM for adults, and 1 mg/kg—upto 50 mg—for children; H2 blockade, 50 mg of ranitidineIV).
6. Administer vasopressors for persistent hypotension andtitrate to a mean arterial pressure of 60 mm Hg.
7. Consider aggressive fluid resuscitation with multiple fluidboluses (10–20 mL/kg under pressure), including colloidas well as crystalloid, in patients who remain hypotensivedespite epinephrine.
8. Administer inhaled β2-agonists such as albuterol for bron-chospasm refractory to epinephrine (73).
9. Consider corticosteroid therapy for protracted anaphy-laxis or to prevent biphasic anaphylaxis (1.0–2.0 mg/kgmethylprednisolone IV every 6 hours). Oral prednisone at1.0 mg/kg, up to 50 mg, may be used for milder attacks.Corticosteroids are not effective therapy for the acute man-ifestations of anaphylaxis.
10. Consider glucagon administration (1–5 mg IV over 1minute, then 1–5 mg/hour in a continuous infusion) inthe setting of prior β-blockade because of its positive in-otropic and chronotropic effects mediated by a β-receptor–independent mechanism.

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
Chapter 60: Anaphylactic Shock 939
11. Prevent recurrent episodes by avoidance of the incit-ing agent, desensitization, or premedication with cortico-steroids and H1 and H2 blockade.
12. Admission to the intensive care unit is warranted forinvasive monitoring with arterial and pulmonary arterycatheters, electrocardiography, pulse oximetry, and fre-quent arterial blood gas measurements.
OBSERVATION
An observation period should be considered for all patients fol-lowing treatment of an anaphylactic reaction. On the basis ofclinical data available to date, the NIAID/FAAN symposiumrecommends that observation periods be individualized on thebasis of severity of initial reaction, reliability of the patient,and access to care. A reasonable time would be 4 to 6 hoursfor most patients, with prolonged observation or hospital ad-mission for severe or refractory symptoms and patients withreactive airway disease (1,2).
FOLLOW-UP, MANAGEMENT,AND PREVENTION
The ideal method for managing severe systemic anaphylacticand anaphylactoid reactions is by preventing their occurrence.Persons with a known sensitivity should avoid re-exposure tothe inciting etiologic agents. Patients who have experiencedrespiratory or cardiovascular symptoms of anaphylaxis shouldreceive self-injectable epinephrine for use if anaphylaxis devel-ops. These patients should also have an emergency action plandetailing its use and follow-up management (1,2). If a precip-itating allergen is known or identified, patients should receiveinformation about avoiding it in the future, prior to their dis-charge from the emergency facility. They should be encouragedto obtain prompt follow-up with their primary care physicianas well as an allergist (1,2).
IMPLICATIONS AND OUTCOME
Anaphylactic/anaphylactoid reactions represent important, po-tentially reversible, acute respiratory and cardiovascular emer-gencies. Although the optimal management method is that ofprevention, prompt diagnosis and institution of therapy arecrucial after these reactions have been initiated in order to pre-vent the fatal cardiovascular and pulmonary manifestations.Factors associated with improved survival include the sensitiv-ity of the person to the inciting agent, the duration betweenthe exposure and the onset of symptoms (short latency periodsare associated with more severe manifestations), the route anddose of the offending agent (larger doses and parenteral ad-ministration are associated with more severe manifestations),and the interval between onset of symptoms and subsequentdiagnosis and institution of appropriate therapy (74). Optimalmanagement of acute systemic reactions includes appropriatepharmacologic intervention, support of pulmonary and cardio-vascular function, and removal of the offending agent. Expedi-tious institution of these measures helps to reduce the morbidityand mortality associated with these potentially life-threateningsyndromes.
SUMMARY
1. Anaphylactic reactions represent type I immune responsesmediated by IgE bound to mast cells or basophils. Com-mon inciting agents include β-lactam antibiotics and Hy-menoptera stings. Other common causes include foods, lo-cal anesthetics, and serum products.
2. Anaphylactoid reactions represent IgE-independent activa-tion of mast cells or basophils, with resultant degranula-tion and mediator release. Common inciting agents includeiodinated radiocontrast media, neuromuscular depolariz-ing agents, and opiates, all of which induce direct mastcell activation; nonsteroidal anti-inflammatory agents act-ing through cyclooxygenase inhibition; and blood productsacting through complement activation.
3. A history of a previous reaction to radiocontrast media,asthma or atopic disease, treatment with β-blockers, andcardiovascular disease are risk factors for developing ana-phylaxis to radiocontrast media (27–29).
4. The differential diagnosis of anaphylactic and anaphylac-toid reactions includes cardiac arrhythmias, myocardial in-farction and cardiogenic shock, distributive or hypovolemicshock, vasovagal syncope, asthma, pulmonary embolism,upper airway obstruction secondary to a foreign body, vocalchord dysfunction, hypoglycemia, carcinoid syndrome, sys-temic mastocytosis, hereditary angioedema, and leukemiawith excess histamine production.
5. Epinephrine is the initial drug of choice for the man-agement of anaphylactic or anaphylactoid reactions. H1-and H2-blocking agents also should be administered. Cor-ticosteroids are not effective for the acute managementof anaphylactic or anaphylactoid reactions, but may pre-vent biphasic anaphylaxis or attenuate prolonged reactions.Glucagon may be used for persistent hypotension in patientstaking β-blockers.
References
1. Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposiumon the definition and management of anaphylaxis: summary report–secondNational Institute of Allergy and Infectious Disease/Food Allergy and Ana-phylaxis Network symposium. J Allergy Clin Immunol. 2006;117:391–397.
2. Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposiumon the definition and management of anaphylaxis: summary report–secondNational Institute of Allergy and Infectious Disease/Food Allergy and Ana-phylaxis Network symposium. Ann Emerg Med. 2006;47:373–380.
3. Brown SG. Anaphylaxis: clinical concepts and research priorities. EmergMed Australas. 2006;18:155–169.
4. Clark S, Long AA, Gaeta TJ, et al. Multicenter study of emergency depart-ment visits for insect sting allergies. J Allergy Clin Immunol. 2005;116:643–649.
5. Neugut AI, Ghatak AT, Miller RL. Anaphylaxis in the United States: aninvestigation into its epidemiology. Arch Intern Med. 2001;161:15–21.
6. Yocum MW, Butterfield JH, Klein JS, et al. Epidemiology of anaphylaxis inOlmsted County: a population-based study. J Allergy Clin Immunol. 1999;104:452–456.
7. Peng MM, Jick H. A population-based study of the incidence, cause, andseverity of anaphylaxis in the United Kingdom. Arch Intern Med. 2004;164:317–319.
8. An epidemiologic study of severe anaphylactic and anaphylactoid reactionsamong hospital patients: methods and overall risks. The International Col-laborative Study of Severe Anaphylaxis. Epidemiology. 1998;9:141–146.
9. Webb LM, Lieberman P. Anaphylaxis: a review of 601 cases. Ann AllergyAsthma Immunol. 2006;97:39–43.
10. Tang AW. A practical guide to anaphylaxis. Am Fam Physician. 2003;68:1325–1332.
11. Thong BY, Cheng YK, Leong KP, et al. Anaphylaxis in adults referred to a

P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet to come
GRBT291-59 GRBT291-3641G GRBT291-Gabrielli-v2.cls October 23, 2008 13:22
940 Section VI: Shock States
clinical immunology/allergy centre in Singapore. Singapore Med J. 2005;46:529–534.
12. Novembre E, Cianferoni A, Bernardini R, et al. Anaphylaxis in children:clinical and allergologic features. Pediatrics. 1998;101:E8.
13. Kaliner MA. Anaphylaxis. NER Allergy Proc. 1984;5:324.14. Chong SU, Worm M, Zuberbier T. Role of adverse reactions to food in
urticaria and exercise-induced anaphylaxis. Int Arch Allergy Immunol. 2002;129:19–26.
15. Dohi M, Suko M, Sugiyama H, et al. Food-dependent, exercise-induced ana-phylaxis: a study on 11 Japanese cases. J Allergy Clin Immunol. 1991;87:34–40.
16. Graham DM, McPherson H, Lieberman P. Skin testing in the evaluation ofHymenoptera allergy and drug allergy. Immunol Allergy Clin North Am.2001;21:301–320.
17. Delage C, Irey NS. Anaphylactic deaths: a clinicopathologic study of 43cases. J Forensic Sci. 1972;17:525–540.
18. Joint Task Force on Practice Parameters, American Academy of Allergy,Asthma and Immunology, American College of Allergy, Asthma and Im-munology, and the Joint Council of Allergy, Asthma and Immunology. The di-agnosis and management of anaphylaxis. J Allergy Clin Immunol. 1998;101:S465–S528.
19. Idsoe O, Guthe T, Willcox RR, et al. Nature and extent of penicillin side-reactions, with particular reference to fatalities from anaphylactic shock.Bull World Health Organ. 1968;38:159–188.
20. Brown AF, McKinnon D, Chu K. Emergency department anaphylaxis: a re-view of 142 patients in a single year. J Allergy Clin Immunol. 2001;108:861–866.
21. Stevenson DD. Approach to the patient with a history of adverse reac-tions to aspirin or NSAIDs: diagnosis and treatment. Allergy Asthma Proc.2000;21:25–31.
22. Allmers H, Schmengler J, John SM. Decreasing incidence of occupationalcontact urticaria caused by natural rubber latex allergy in German healthcare workers. J Allergy Clin Immunol. 2004;114:347–351.
23. Schwartz HA, Zurowski D. Anaphylaxis to latex in intravenous fluids. JAllergy Clin Immunol. 1993;92:358–359.
24. Mitsuhata H, Horiguchi Y, Saitoh J, et al. An anaphylactic reaction to topicalfibrin glue. Anesthesiology. 1994;81:1074–1077.
25. Laxenaire MC, Mata-Bermejo E, Moneret-Vautrin DA, et al. Life-threatening anaphylactoid reactions to propofol (Diprivan). Anesthesiology.1992;77:275–280.
26. Katayama H, Yamaguchi K, Kozuka T, et al. Adverse reactions to ionicand nonionic contrast media. A report from the Japanese Committee on theSafety of Contrast Media. Radiology. 1990;175:621–628.
27. Greenberger PA, Halwig JM, Patterson R, et al. Emergency administrationof radiocontrast media in high-risk patients. J Allergy Clin Immunol. 1986;77:630–634.
28. Enright T, Chua-Lim A, Duda E, et al. The role of a documented allergicprofile as a risk factor for radiographic contrast media reaction. Ann Allergy.1989;62:302–305.
29. Bush WH, Swanson DP. Acute reactions to intravascular contrast media:types, risk factors, recognition, and specific treatment. AJR Am J Roentgenol.1991;157:1153–1161.
30. Inomata N, Osuna H, Yanagimachi M, et al. Late-onset anaphylaxis tofermented soybeans: the first confirmation of food-induced, late-onset ana-phylaxis by provocation test. Ann Allergy Asthma Immunol. 2005;94:402–406.
31. Brown SG. Cardiovascular aspects of anaphylaxis: implications for treatmentand diagnosis. Curr Opin Allergy Clin Immunol. 2005;5:359–364.
32. Smith PL, Kagey-Sobotka A, Bleecker ER, et al. Physiologic manifestationsof human anaphylaxis. J.Clin Invest. 1980;66:1072–1080.
33. Kemp SF, Lockey RF, Wolf BL, et al. Anaphylaxis. A review of 266 cases.Arch Intern Med. 1995;155:1749–1754.
34. Kemp SF, Lockey RF. Anaphylaxis: a review of causes and mechanisms. JAllergy Clin Immunol. 2002;110:341–348.
35. Carlson RW, Schaeffer RC Jr, Puri VK, et al. Hypovolemia and permeabilitypulmonary edema associated with anaphylaxis. Crit Care Med. 1981;9:883–885.
36. Simon MR. Anaphylaxis associated with relative bradycardia. Ann Allergy.1989;62:495–497.
37. Raper RF, Fisher MM. Profound reversible myocardial depression after ana-phylaxis. Lancet. 1988;1:386–388.
38. Wasserman SI. The heart in anaphylaxis. J Allergy Clin Immunol. 1986;77:663–666.
39. Nicolas F, Villers D, Blanloeil Y. Hemodynamic pattern in anaphylactic shockwith cardiac arrest. Crit Care Med. 1984;12:144–145.
40. Serafin WE, Austen KF. Mediators of immediate hypersensitivity reactions.N Engl J Med. 1987;317:30–34.
41. Perkin RM, Anas NG. Mechanisms and management of anaphylactic shocknot responding to traditional therapy. Ann Allergy. 1985;54:202–208.
42. Silverman HJ, Van Hook C, Haponik EF. Hemodynamic changes in humananaphylaxis. Am J Med. 1984;77:341–344.
43. Moss J, Fahmy NR, Sunder N, et al. Hormonal and hemodynamic profile ofan anaphylactic reaction in man. Circulation. 1981;63:210–213.
44. Hanashiro PK, Weil MH. Anaphylactic shock in man. Report of two caseswith detailed hemodynamic and metabolic studies. Arch Intern Med. 1967;119:129–140.
45. Fawcett WJ, Shephard JN, Soni NC, et al. Oxygen transport and haemody-namic changes during an anaphylactoid reaction. Anaesth Intensive Care.1994;22:300–303.
46. Brazil E, MacNamara AF. “Not so immediate” hypersensitivity–the dangerof biphasic anaphylactic reactions. J Accid Emerg Med. 1998;15:252–253.
47. Lieberman P. Biphasic anaphylactic reactions. Ann Allergy Asthma Immunol.2005;95:217–226.
48. Bochner BS, Lichtenstein LM. Anaphylaxis. N Engl J Med. 1991;324:1785–1790.
49. Yocum MW, Khan DA. Assessment of patients who have experienced ana-phylaxis: a 3-year survey. Mayo Clin Proc. 1994;69:16–23.
50. Lin RY, Schwartz LB, Curry A, et al. Histamine and tryptase levels in patientswith acute allergic reactions: an emergency department-based study. J AllergyClin Immunol. 2000;106:65–71.
51. Weiss ME, Adkinson NF. Immediate hypersensitivity reactions to penicillinand related antibiotics. Clin Allergy. 1988;18:515–540.
52. Finkelman FD, Rothenberg ME, Brandt EB, et al. Molecular mechanismsof anaphylaxis: lessons from studies with murine models. J Allergy ClinImmunol. 2005;115:449–457.
53. Strait RT, Morris SC, Yang M, et al. Pathways of anaphylaxis in the mouse.J Allergy Clin Immunol. 2002;109:658–668.
54. Choi IW, Kim YS, Kim DK, et al. Platelet-activating factor-mediated NF-kappaB dependency of a late anaphylactic reaction. J Exp Med. 2003;198:145–151.
55. Kaplan AP, Joseph K, Silverberg M. Pathways for bradykinin formation andinflammatory disease. J Allergy Clin Immunol. 2002;109:195–209.
56. Goulet JL, Snouwaert JN, Latour AM, et al. Altered inflammatory responsesin leukotriene-deficient mice. Proc Natl Acad Sci USA. 1994; 91:12852–12856.
57. Haribabu B, Verghese MW, Steeber DA, et al. Targeted disruption of theleukotriene B(4) receptor in mice reveals its role in inflammation and platelet-activating factor-induced anaphylaxis. J Exp Med. 2000;192:433–438.
58. Kanaoka Y, Maekawa A, Penrose JF, et al. Attenuated zymosan-inducedperitoneal vascular permeability and IgE-dependent passive cutaneous ana-phylaxis in mice lacking leukotriene C4 synthase. J Biol Chem. 2001;276:22608–22613.
59. Mangan NE, Fallon RE, Smith P, et al. Helminth infection protects mice fromanaphylaxis via IL-10-producing B cells. J Immunol. 2004;173:6346–6356.
60. Dombrowicz D, Brini AT, Flamand V, et al. Anaphylaxis mediated througha humanized high affinity IgE receptor. J Immunol. 1996;157:1645–1651.
61. Oettgen HC, Martin TR, Wynshaw-Boris A, et al. Active anaphylaxis inIgE-deficient mice. Nature. 1994;370:367–370.
62. Miyajima I, Dombrowicz D, Martin TR, et al. Systemic anaphylaxis in themouse can be mediated largely through IgG1 and Fc gammaRIII. Assess-ment of the cardiopulmonary changes, mast cell degranulation, and deathassociated with active or IgE- or IgG1-dependent passive anaphylaxis. J ClinInvest. 1997;99:901–914.
63. Cheifetz A, Mayer L. Monoclonal antibodies, immunogenicity, and associ-ated infusion reactions. Mt Sinai J Med. 2005;72:250–256.
64. Project Team of the Resuscitation Council (UK). Emergency medical treat-ment of anaphylactic reactions. Resuscitation. 1999;41:93–99.
65. Hepner DL, Castells MC. Anaphylaxis during the perioperative period.Anesth Analg. 2003;97:1381–1395.
66. Taneli Vayrynen MJ, Luurila HO, Maatta TK, et al. Accidental intravenousadministration of racemic adrenaline: two cases associated with adverse car-diac effects. Eur J Emerg Med. 2005;12:225–229.
67. Lieberman P, Kemp SF, Oppenheimer J, et al. The diagnosis and manage-ment of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol.2005;115(Suppl 2):S483–S523.
68. Boulain T, Achard JM, Teboul JL, et al. Changes in BP induced by passiveleg raising predict response to fluid loading in critically ill patients. Chest.2002;121:1245–1252.
69. Simons FE. Advances in H1-antihistamines. N Engl J Med. 2004;351:2203–2217.
70. Fisher MM. Clinical observations on the pathophysiology and treatment ofanaphylactic cardiovascular collapse. Anaesth Intensive Care. 1986;14:17–21.
71. Sampson HA, Mendelson L, Rosen JP. Fatal and near-fatal anaphylacticreactions to food in children and adolescents. N Engl J Med 1992;327:380–384.
72. Thomas M, Crawford I. Best evidence topic report. Glucagon infusion inrefractory anaphylactic shock in patients on beta-blockers. Emerg Med J.2005;22:272–273.
73. Sampson HA. Anaphylaxis and emergency treatment. Pediatrics. 2003;111:1601–1608.
74. Sheffer AL. Anaphylaxis. J Allergy Clin Immunol. 1985;75:227–233.