Embed Size (px)
Citation preview

British Journal of Plastic Surgery (1984) 31, 412416 0 1984 The Trustees of British Association of Plastic Surgeons
Congenital crania-facial dysmorphosis associated with Ito’s syndrome (incontinentia pigmenti achromians): a case report
M. STRICKER, M. MELEY, J. F. CHASSAGNE and J. BEUREY
Department of Plastic and Maxillo-facial Surgery, Hapital Central, Nancy and the Department of Dermatology, Hbpital Fournier, Nancy, France
Summary-The authors describe a child who presented multiple congenital malformations affecting the crania-facial region and the extremities associated with a rare skin lesion (incontinentia pigmenti achromians).
The discovery of a congenital crania-facial malformation demands a careful search for other associated defects. These usually involve the cardiovascular and locomotor systems but in our clinical practice we recently encountered a young child with a rare dermatological disorder.
Case report
A young boy, aged 4, was referred with “scars” over the right eyelids and the centre of the forehead (Fig. 1) but with no history of any specific cause or injury. On examination it was clear that these lesions, one of which extended from the hair margin to the tip of the nose, were not scars in the accepted sense but rather a manifestation of a median dysraphia or a hemifacial atrophy.
In this child, born of healthy parents, after a normal pregnancy, various malformations were observed at the age of 8 months. These included:
G)
(ii)
(iii)
(iv)
All
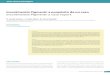
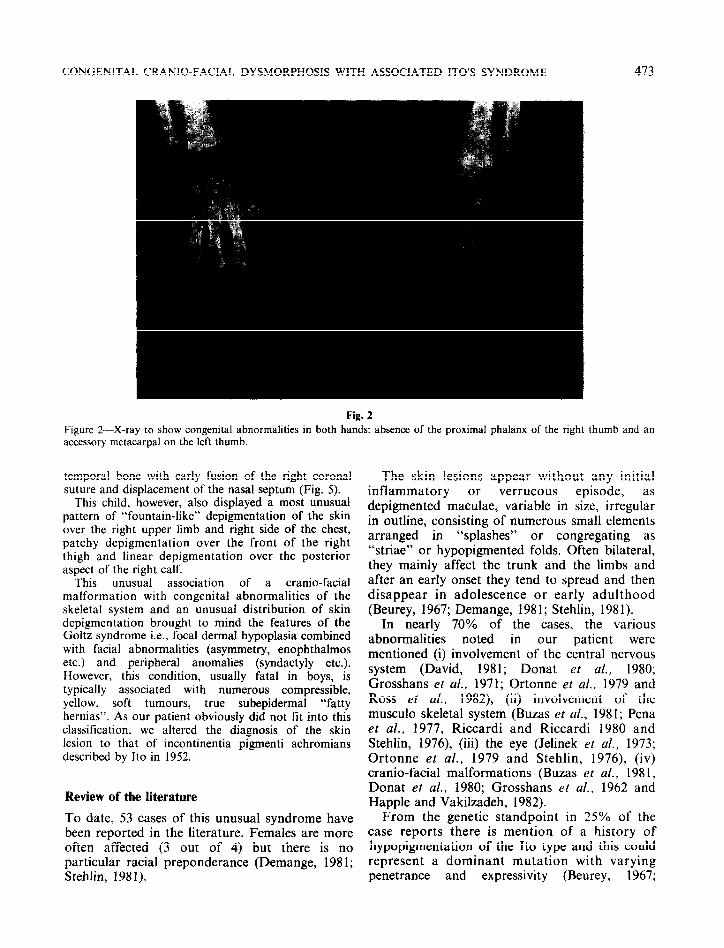
Cranio-facial lesions: telecanthus, right plagiocephaly, right enophthalmos and orbital dystopia. Derangement of dental occlusion comprising an anterior open bite with lingual interposition. Examination of the right eye revealed amblyopia with a postero-inferior subscapular opacity of the lens. Locomotor skeletal lesions: skeletal X-rays (Fig. 2) showed a supernumerary bone alongside the first left metacarpal and absence of the first phalanx of the right thumb that was replaced by two separate bony fragments. Radiological examination of the pelvis showed bilateral coxa valga with many femoral irregularities (Figs. 3,4).
these findings supported the diagnosis of a metaphysio-epiphyseal dysplasia with a right-sided predominance.
Fig. 1
Figure l-Face view to show telecanthus, right sided plagiocephaly, right enopthalmos and orbital dystopia. Note the grooves over the forehead and the right eyelids.
Tomography and CAT scans of the skull showed no abnormality of the brain but there was obvious orbital asymmetry spreading into the petrous part of the
412
CONGENITAL CRANIO-FACIAL DYSMORPHOSIS WITH ASSOCIATED ITO’S SYNDROME 473
Figul
Fig. 2
-e 2-X-ray to show congenital abnormalities in both hands: absence of the proximal phalanx of the right thumb accessory metacarpal on the left thumb.
temporal bone with early fusion of the right coronal suture and displacement of the nasal septum (Fig. 5).
This child, however, also displayed a most unusual pattern of “fountain-like” depigmentation of the skin over the right upper limb and right side of the chest, patchy depigmentation over the front of the right thigh and linear depigmentation over the posterior aspect of the right calf.
This unusual association of a crania-facial malformation with congenital abnormalities of the skeletal system and an unusual distribution of skin depigmentation brought to mind the features of the Goltz syndrome i.e., focal dermal hypoplasia combined with facial abnormalities (asymmetry, enophthalmos etc.) and peripheral anomalies (syndactyly etc.). However, this condition, usually fatal in boys, is typically associated with numerous compressible, yellow. soft tumours, true subepidermal “fatty hernias”. As our patient obviously did not tit into this classification, we altered the diagnosis of the skin lesion to that of incontinentia pigmenti achromians described by Ito in 1952.
Review of the literature
To date, 53 cases of this unusual syndrome have been reported in the literature. Females are more often affected (3 out of 4) but there is no particular racial preponderance (Demange, 1981; Stehlin, 1981).
and an
The skin lesions appear without any initial inflammatory or verrucous episode, as depigmented maculae, variable in size, irregular in outline, consisting of numerous small elements arranged in “splashes” or congregating as “striae” or hypopigmented folds. Often bilateral, they mainly affect the trunk and the limbs and after an early onset they tend to spread and then disappear in adolescence or early adulthood (Beurey, 1967; Demange, 1981; Stehlin, 1981).
In nearly 70% of the cases, the various abnormalities noted in our patient were mentioned (i) involvement of the central nervous system (David, 1981; Donat et al., 1980; Grosshans et al., 1971; Ortonne et al., 1979 and Ross et al., 1982), (ii) involvement of the musculo skeletal system (Buzas et al., 1981; Pena et al., 1977, Riccardi and Riccardi 1980 and Stehlin, 1976), (iii) the eye (Jelinek et al., 1973; Ortonne et al., 1979 and Stehlin, 1976) (iv) crania-facial malformations (Buzas et al., 198 1, Donat et al., 1980; Grosshans et al., 1962 and Happle and Vakilzadeh, 1982).
From the genetic standpoint in 25% of the case reports there is mention of a history of hypopigmentation of the Ito type and this could represent a dominant mutation with varying penetrance and expressivity (Beurey, 1967;
BRITISH JOURNAL OF PLASTIC SURGERY
Fig. 3
Figures 3 extremities
an of
d &X-rays to show bilateral coxa valga with metaphyso-epiphyseal abnormalities at the upper the femur.
Fig. 4
and lo lwer

CONGENITAL CRANIO-FACIAL DYSMORPHOSIS WITH ASSOCIATED ITO’S SYNDROME 475
Fig. 5
Figure 5-CAT scan of the skull to show orbital asymmetry and deviation of nasal septum.
Demange, 198 1, Dotson and Raimer, 198 1 and Ito, 1957).
Histologically, the affection is characterised by a decrease in the number of melanocytes in the basal cell layer, a vacuolation of the keratinocytes with a degeneration of the epidermal cells on light microscopy, the dermis being free from specific abnormalities.
Electron microscopy studies allow one to specify the ultrastructural individuality (Cram and Fukuyama, 1974; Morohashi et al., 1977; Morahashi et al., 1981; Mordlung et al., 1977; Ortonne and Perrot, 1980 and Stoebmer and Grosshans, 1970) of this dyschromia through the presence of dyskeratosic keratinocytes, rare melanocytes and melanosomes, which though small and scanty are responsible for an inadequate production of melanin although the mechanism of synthesis is unaffected.
In the case of the young child on whom this report is based, the facial “dysraphic” stigmata will be kept under review and no immediate treatment is indicated. As for the crania-facial anomalies it is likely that attention will be limited to the management of the plagiocephaly.
To help in the classification and understanding of unusual congenital lesions, the more important references in the dermatological literature are given in full.
References
Fhrey, J. (1967). Hypoplasie dermique en sires et dysplasie m&so-ectodermique. Bulletin de la Soc@tP FranGaise de Dermatologie et Syphiligraphie, 74, 7.
Bums, J. W., Sina, B. and Buruett, J. W. (1981). Hypomelanosis of lto: report of a case and review of the literature. Journal of the American Academy of Dermatology, 4, 195.
Cram, D. L. and Fukuyama, K. (1974). Unilateral systematized hypochromia naevus. Archives of Dermatology (Chicago) 109,416.
David, T. J. (1981). Hypomelanosis of lto: a neurocutaneous syndrome. Archives of Diseases in Childhood, !%, 798.
Demange, I. (1981). Hypomdlanose de Ito: incontinentiu pigmenti achromians. Th&e Mtdecine, Nancy.
Donat. J. F.. WaIsworth. D. M. and Turk. L. L. (19801. Focal , ~ I
cerebral atrophy in ‘incontinentia pigmenti achromians. American Journal of Diseases of Children, 134, 709.
Dotson, A. D. and Raimer, S. S. (1981). lncontinentia pigmenti achromians. International Journal qf Dermatology (Philadelphia), 20, 357.
Feurman, E. J. and Wolf, R. (1982). lncontinentia pigmenti achromians (systematized depigmented nevus). Hautarzt (Berlin) 33, 159.
Goltz, R. W., Peterson, W. C., GorlIn, R. J. and Raujts, H. G. (1962). Focal dermal hypoplasia. Archives of Dermatology, 86, 708.
Grosshans, E. M. Stoebner, P., Bergoend, H. and Stoll, C. (1971). lncontinentia pigmenti achromians (lto). Etude clinique et histo-pathologique. Dermatologica. (Basel), 142, 65.
Happle, R. and VakiIzadeh, F. (1982). Hamartomatous dental cusps in hypomelanosis of lto. Clinical Genetics (Copenhagen), 21,65.
Ito, M. (1957). Studies on melanin Xl incontinentia pigmenti anchromians. A singular case of naevus depigmentus systematicus bilateralis. Tokoshima Journal of E.uperimental Medicine, 65, Supplement, 5.
JeIlnek, J., Bart, R. and SchIff, G. (1973). Hypomelanosis of lto. Archives of Dermatology, 107, 596.
Kukolicb, M. K., Althaus, B. W., Freeman, M. V. and Lewandowski, R. C.(l980). Hypomelanosis of lto with triphalangeal thumbs. Journal of Medical Genetics (London), 17, 151.
Morohashi, M., Haahimoto, K., Goodman, T. F., Newton, D. E. and De Rist, T. (1977). Ultrastructural studies of vitiligo, Vogt Koyanagi syndrome and incontinentia pigmenti achromians. Archives of Dermatology, 113, 755.
Morohashi, M. Maeda, T., Takaheshi, S. and Igarashi, R. (1981). Ultrastructure of incontinentia pigmenti achromians with special reference to melanocytes and nerve endings. Journal of Dermatology (Tokyo) 8, 401.
MordIung, J. J., Klaus, S. N. and Giw, J. (1977). Hypomelanosis of lto. Acta Dermato- Venereologica (Stockholm), 57, 262.
Ortonne, J. P., Coiffet, J. and Floret, D. (1979). Hypom&lanose de lto: B propos d’un cas. Annales de Dermatologie et de V&&eologie (Paris), 106, 47.
Ortoane, J. P. and Perrot, H. (1980). Idiopathic guttate hypomelanosis. Ultrastructural study. Archives of Dermatology, 116, 664.
Pena, L., Ruiz Maldonado, R., Tamayo, L., Astengo Osuna, C. and Gonzales Meodoza, A. (1977). lncontinentia pigmenti achromians. International Journal of Dermatology. 16, 194.

476 BRITISH JOURNAL OF PLASTIC SURGERY
Pinol, &pade, J., Romaguera, C. and Mascara, J. M. (1968). Incontinentia pigmenti achromians. Medicina Cutanea Ibero-Latino-Americana. 3, 281.
Riecardi, V. M. and Riccardi, S. L. (1980). Hypomelanosis of Ito and ectrodactyly (letter). Clefr Palate Journal, 17, 337.
Roberts, D. L. and Byrne, J. P. (1981). Incontinentia pigmenti achromians and morphoea: a case report. Clinical Experimental Dermatology (Oxford), 307, 10.
Ross, D. L., Liwnicz, B. H., Chun, R. W. and Gilbert, E. (1982). Hypomelanosis of Ito (incontinentia pigmenti achromians). A clinico-pathologic study: macrocephaly and gray matter heterotopias. Neurology (NY) 32, 1013.
Savall, R. Ferrandiz, C., Ferrer, I. and Peyri, J. (1980). Idiopathic guttate hypomelanosis. British Journal of Dermatology, 103, 635.
Schwartz, M. F., Esterly, N. B., Fretzin, D. F. Pergament, E. and Rosenfeld, T. H. (1977). Incontinentia pigmenti achromians: a neurocutaneous syndrome. Journal of Pediatrics, 90, 236.
StebIin, M. J. (1976). Incontinentia pigmenti. Th&e Mkdecine, Nancy.
Stoebmer, P. and Grosshans, E. M. (1970). Incontinentia pigmenti achromians (Ito). Etude ultrastructurale. Archiv fir Klinische und Experimentelle Dermatologie.
The Authors
M. Strlcker, MD., M. Meley, MD., J. F. Chassagne, MD, Service de Chirurgie Plastique et Maxillo-faciale, Hapital Central, 29, av. de Lattre de Tassigny, 54037 Nancy CBdex, France.
J. Beurey, MD, Service de Dermatologie, Hapita Fournier, Quai de la Bataille, Nancy, France.
Requests for reprints to: M. Stricker, MD, HBpital Central, 29, av. de Lattre de Tassigny, 54037 Nancy Cidex, France.